
Identification of Advantaged Genes for Lodging Resistance-Related Traits in the Temperate geng Group (Oryza sativa L.) Using a Genome-Wide Association Study


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Phenotypic Investigation
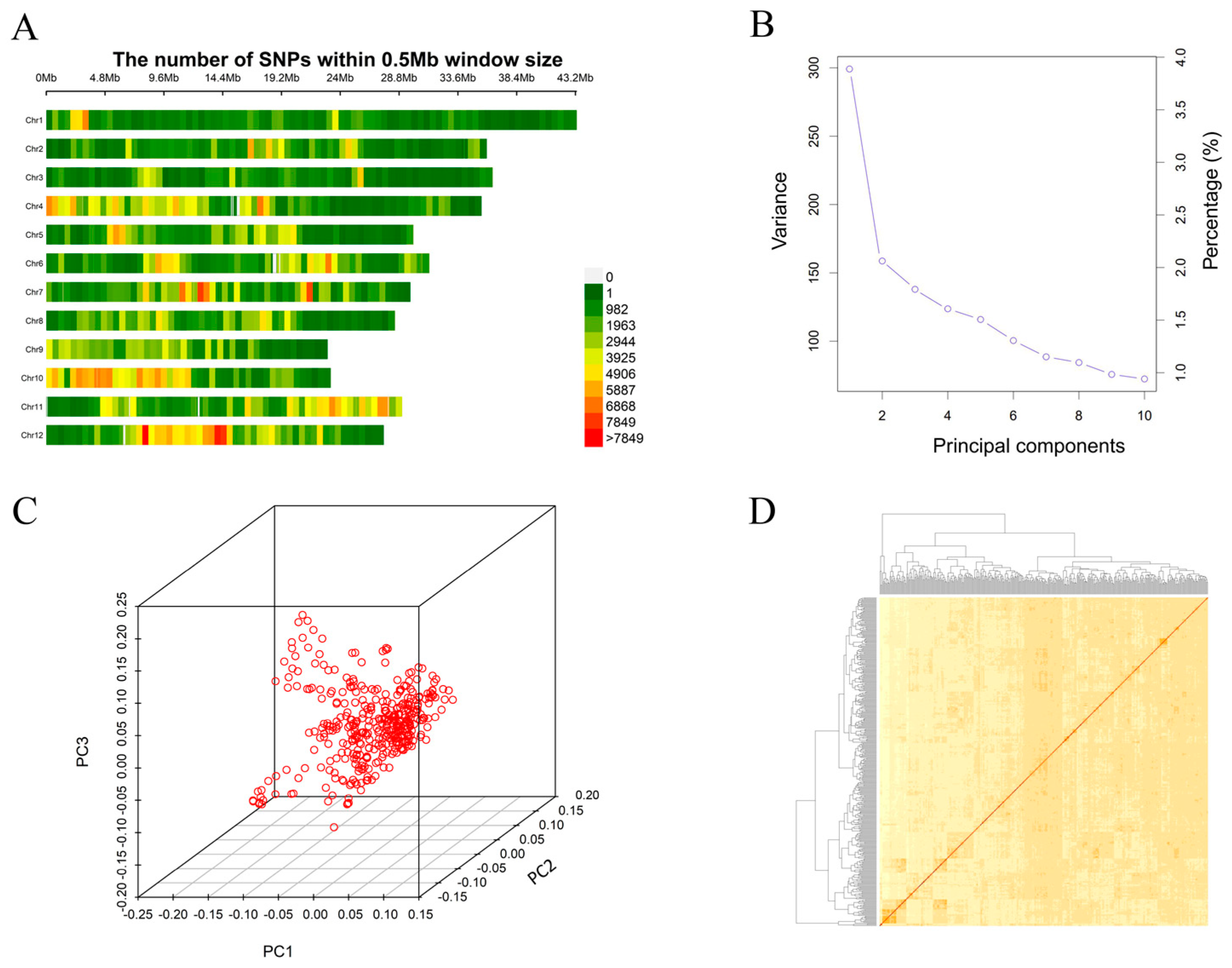
2.2. Genotypic Data
2.3. Genome-Wide Association Study (GWAS)
2.4. Candidate Gene Identification for the Important QTL
3. Results
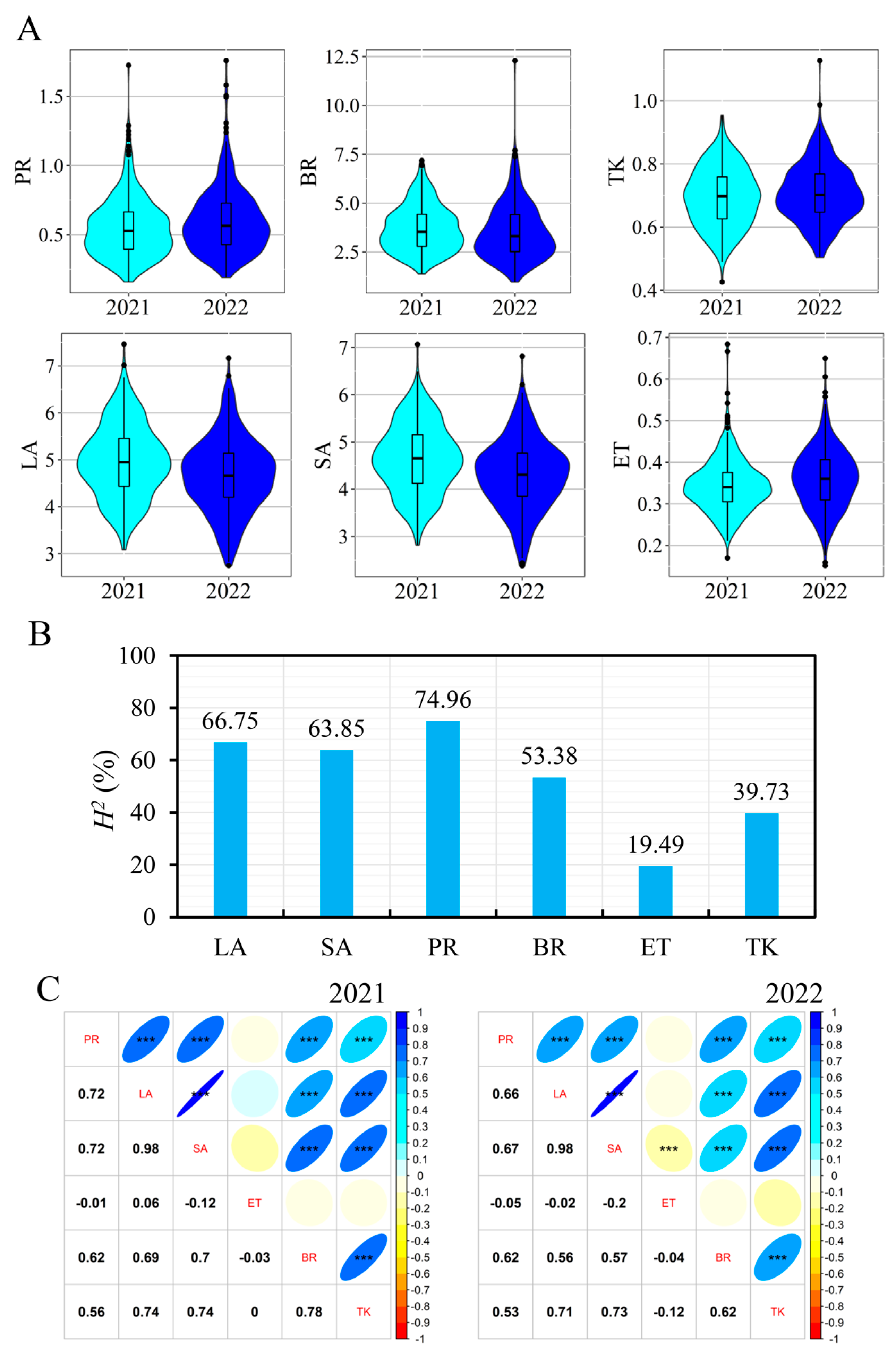
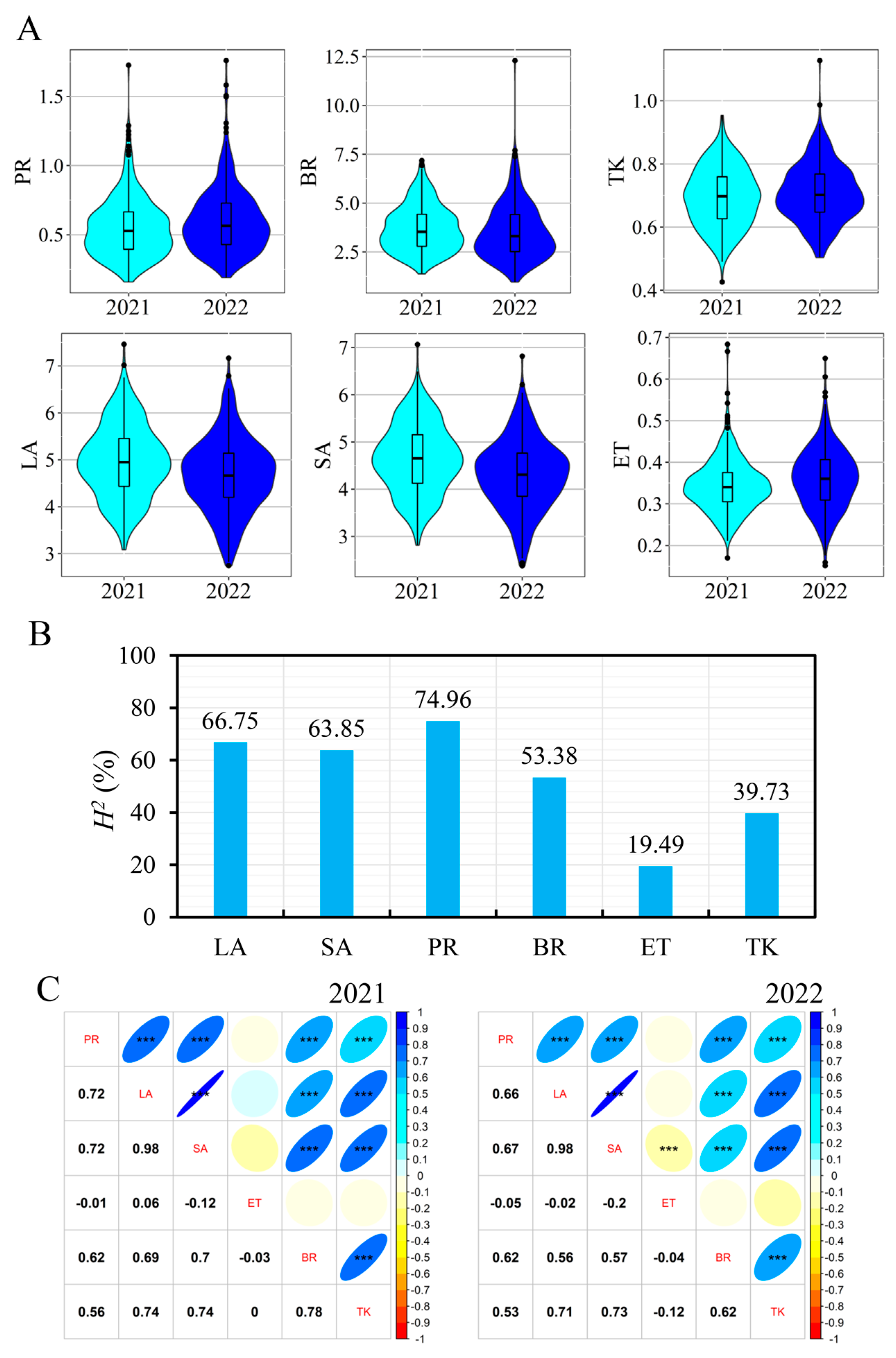
3.1. Phenotypic Date
3.2. Genotypic Data
3.3. Detection of QTL Using GWAS
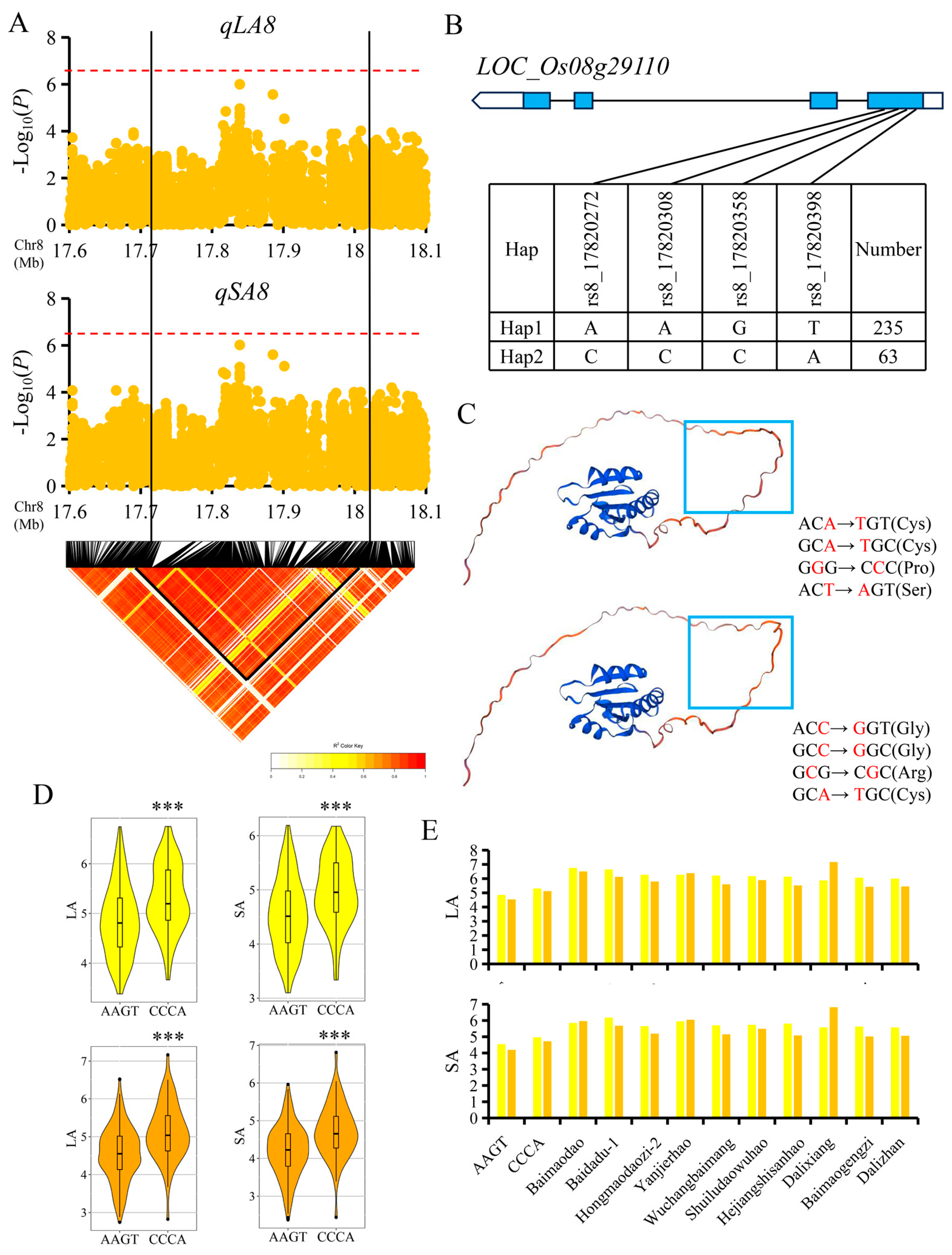
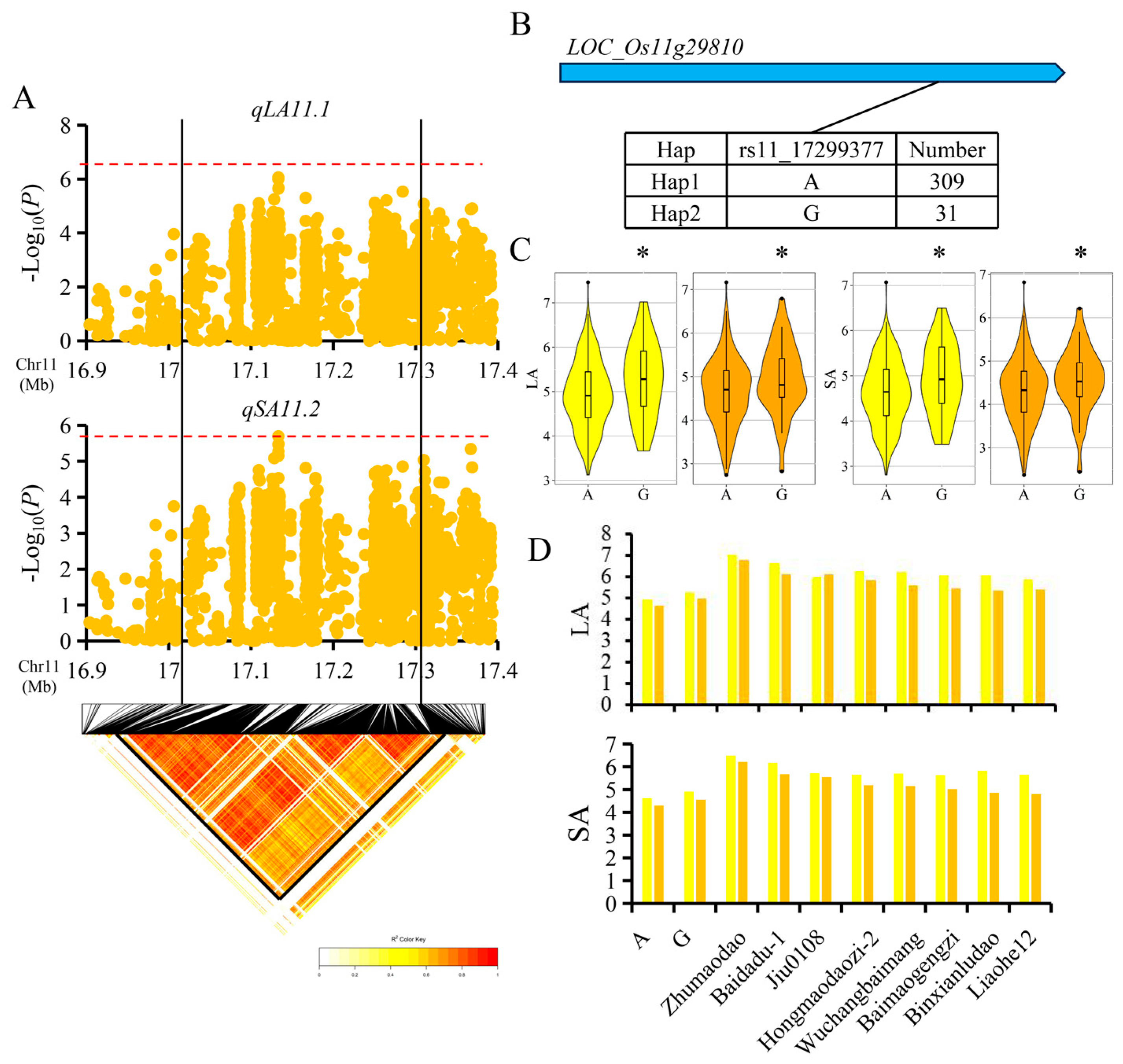
3.4. Candidate Gene Analysis for Important QTL
4. Discussion
4.1. Selection of Test Materials
4.2. Characteristics of Lodging Resistance
4.3. Comparisons of QTL Detected in This Study with Previously Reported QTL
4.4. Candidate Gene Identification for Important QTL
4.5. Application of Gene Pyramiding in Rice Breeding for Lodging Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, T.X.; Shabala, S.; Niu, Y.N.; Cheng, Z.H.; Shabala, L.; Meinke, H.; Venkataraman, G.; Pareek, A.; Xu, J.L.; Zhou, M.X. Molecular mechanisms of salinity tolerance in rice. Crop J. 2021, 9, 506–520. [Google Scholar] [CrossRef]
- Wang, X.; Pang, Y.; Wang, C.; Chen, K.; Zhu, Y.; Shen, C.; Ali, J.; Xu, J.; Li, Z. New candidate genes affecting rice grain appearance and milling quality detected by genome-wide and gene-based association analyses. Front. Plant Sci. 2017, 7, 1998. [Google Scholar] [CrossRef]
- Murchie, E.H.; Pinto, M.; Horton, P. Agriculture and the new challenges for photosynthesis research. New Phytol. 2009, 181, 532–552. [Google Scholar] [CrossRef] [PubMed]
- Berry, P.M.; Sterling, M.; Spink, J.H.; Baker, C.J.; Ennos, A.R. Understanding and Reducing Lodging in Cereals. Adv. Agron. 2004, 84, 217–271. [Google Scholar] [CrossRef]
- Islam, M.S.; Peng, S.; Visperas, R.M.; Ereful, N.; Bhuiya, M.S.U.; Julfiquar, A. Lodging-related morphological traits of hybrid rice in a tropical irrigated ecosystem. Field Crops Res. 2007, 1, 240–248. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Ishimaru, K. Identification and functional analysis of a locus for improvement of lodging resistance in rice. Plant Physiol. 2004, 134, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Ookawa, T.; Aya, K.; Ochiai, Y.; Hirasawa, T.; Ebitani, T.; Takarada, T.; Yano, M.; Yamamoto, T.; Fukuoka, S.; et al. Isolation of a novel lodging resistance QTL gene involved in strigolactone signaling and its pyramiding with a QTL gene involved in another mechanism. Mol. Plant 2015, 8, 303–314. [Google Scholar] [CrossRef]
- Meng, B.; Wang, T.; Luo, Y.; Xu, D.; Li, L.; Diao, Y.; Gao, Z.; Hu, Z.; Zheng, X. Genome-wide association study identified novel candidate loci/genes affecting lodging resistance in rice. Genes 2021, 12, 718. [Google Scholar] [CrossRef]
- Ookawa, T.; Hobo, T.; Yano, M.; Murata, K.; Ando, T.; Miura, H.; Asano, K.; Ochiai, Y.; Ikeda, M.; Nishitani, R.; et al. New approach for rice improvement using a pleiotropic QTL gene for lodging resistance and yield. Nat. Commun. 2010, 1, 132. [Google Scholar] [CrossRef]
- Rashid, M.A.R.; Zhao, Y.; Zhang, H.; Li, J.J.; Li, Z. Nucleotide diversity, Natural variation and evolution of Flexible culm-1 and Strong culm-2 lodging resistance genes in rice. Genome 2016, 59, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Monna, L.; Kitazawa, N.; Yoshino, R.; Suzuki, J.; Masuda, H.; Maehara, Y.; Tanji, M.; Sato, M.; Nasu, S.; Minobe, Y. Positional cloning of rice semidwarfing gene, sd-1: Rice “green revolution gene” encodes a mutant enzyme involved in gibberellin synthesis. DNA Res. 2022, 9, 11–17. [Google Scholar] [CrossRef]
- Su, S.; Hong, J.; Chen, X.; Zhang, C.; Chen, M.; Luo, Z.; Chang, S.; Bai, S.; Liang, W.; Liu, Q.; et al. Gibberellins orchestrate panicle architecture mediated by DELLA-KNOX signalling in rice. Plant Biotechnol. J. 2021, 19, 2304–2318. [Google Scholar] [CrossRef]
- Tu, B.; Tao, Z.; Wang, S.; Zhou, L.; Zheng, L.; Zhang, C.; Li, X.; Zhang, X.; Yin, J.; Zhu, X.; et al. Loss of Gn1a/OsCKX2 confers heavy-panicle rice with excellent lodging resistance. J. Integr. Plant Biol. 2022, 64, 23–38. [Google Scholar] [CrossRef]
- Rong, C.; Liu, Y.; Chang, Z.; Liu, Z.; Ding, Y.; Ding, C. Cytokinin oxidase/dehydrogenase family genes exhibit functional divergence and overlap in rice growth and development, especially in control of tillering. J. Exp. Bot. 2022, 73, 3552–3568. [Google Scholar] [CrossRef]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, Y.; Wei, X.; Li, C.; Wang, A.; Zhao, Q.; Li, W.; Guo, Y.; Deng, L.; Zhu, C.; et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 2011, 44, 32–39. [Google Scholar] [CrossRef]
- Kumar, V.; Singh, A.; Mithra, S.V.; Krishnamurthy, S.L.; Parida, S.K.; Jain, S.; Tiwari, K.K.; Kumar, P.; Rao, A.R.; Sharma., S.K.; et al. Genome-wide association mapping of salinity tolerance in rice (Oryza sativa). DNA Res. 2015, 22, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Zheng, T.; Wang, X.; Wang, Y.; Chen, K.; Wang, S.; Wang, Y.; Xu, J.; Li, Z. QTL mapping and candidate gene analysis of peduncle vascular bundle related traits in rice by genome-wide association study. Rice 2018, 11, 13. [Google Scholar] [CrossRef]
- Rashid, M.A.R.; Atif, R.M.; Zhao, Y.; Azeem, F.; Ahmed, H.G.M.-D.; Pan, Y.; Pan, Y.H.; Li, D.T.; Zhao, Y.; Zhang, Z.Y.; et al. Dissection of genetic architecture for tiller angle in rice (Oryza sativa. L) by multiple genome-wide association analyses. PeerJ 2022, 10, e12674. [Google Scholar] [CrossRef]
- Zhai, L.Y.; Wang, Y.; Yan, A.; Chen, L.Q.; Shao, K.T.; Zhang, W.Z.; Xu, J.L. Genetic bases of flow- and sink-related traits in rice revealed by genome-wide association study. Agronomy 2022, 12, 776. [Google Scholar] [CrossRef]
- Li, X.; Chen, Z.; Zhang, G.; Lu, H.; Qin, P.; Qi, M.; Yu, Y.; Jiao, B.; Zhao, X.; Gao, Q.; et al. Analysis of genetic architecture and favorable allele usage of agronomic traits in a large collection of Chinese rice accessions. Sci. China Life Sci. 2020, 63, 1688–1702. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Li, M.X.; Yeung, J.M.Y.; Cherny, S.S.; Sham, P.C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 2011, 131, 747–756. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Blay, S.; Mcneney, B.; Graham, J. LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. J. Stat. Softw. 2006, 16, 1–9. [Google Scholar] [CrossRef]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.C.; Iwamoto, M.; Abe, T.; et al. Rice Annotation Project Database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol. 2013, 54, e6. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Ren, Y.; Duan, E.; Zhu, X.; Hao, Y.; Zhu, J.; Chen, R.; Lei, J.; Teng, X.; et al. White panicle2 encoding thioredoxin z, regulates plastid RNA editing by interacting with multiple organellar RNA editing factors in rice. New Phytol. 2021, 229, 2693–2706. [Google Scholar] [CrossRef]
- Yin, C.C.; Ma, B.; Collinge, D.P.; Pogson, B.J.; He, S.J.; Xiong, Q.; Duan, K.X.; Chen, H.; Yang, C.; Lu, X.; et al. Ethylene responses in rice roots and coleoptiles are differentially regulated by a carotenoid isomerase-mediated abscisic acid pathway. Plant Cell 2015, 27, 1061–1081. [Google Scholar] [CrossRef]
- Khush, G.S. Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 1997, 35, 25–34. [Google Scholar] [CrossRef]
- Hirano, K.; Ordonio, R.L.; Matsuoka, M. Engineering the lodging resistance mechanism of post-Green Revolution rice to meet future demands. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Atwell, S.; Huang, Y.S.; Vilhjalmsson, B.J.; Willems, G.; Horton, M.; Al, E. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature 2010, 465, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Togawa, E.; Ookawa, T.; Kashiwagi, T.; Madoka, Y.; Hirotsu, N. New target for rice lodging resistance and its effect in a typhoon. Planta 2008, 227, 601–609. [Google Scholar] [CrossRef]
- Hu, H.; Liu, W.; Fu, Z.; Homann, L.; Technow, F.; Wang, H.; Song, C.; Li, S.; Melchinger, A.E.; Chen, S. QTL mapping of stalk bending strength in a recombinant inbred line maize population. Theor. Appl. Genet. 2013, 126, 2257–2266. [Google Scholar] [CrossRef]
- Lijavetzky, D.; Carbonero, P.; Vicente-Carbajosa, J. Genome-wide comparative phylogenetic analysis of the rice and Arabidopsis Dof gene families. BMC Evol. Biol. 2003, 23, 17. [Google Scholar] [CrossRef]
- Ookawa, T.; Nomura, T.; Kamahora, E.; Jiang, M.; Ochiai, Y.; Samadi, A.F.; Yamaguchi, T.; Adachi, S.; Katsura, K.; Motobayashi, T. Pyramiding of multiple strong-culm genes originating from indica and tropical japonica to the temperate japonica rice. Sci. Rep. 2022, 12, 15400. [Google Scholar] [CrossRef]
- Ashikari, M.; Matsuoka, M. Identification, isolation and pyramiding of quantitative trait loci for rice breeding. Trends Plant Sci. 2006, 11, 344–350. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | QTL | Year | Peak SNP | p-Value |
|---|---|---|---|---|
| PR | qPR1 | 2021 | rs1_35016632 | 6.20 × 10−7 |
| qPR2 | 2022 | rs2_24665362 | 1.43 × 10−6 | |
| qPR3 | 2021 | rs3_15388478 | 6.62 × 10−7 | |
| qPR4.1 | 2021 | rs4_30733099 | 4.20 × 10−7 | |
| qPR4.2 | 2021 | rs4_33212260 | 1.36 × 10−6 | |
| qPR5 | 2021 | rs5_3450550 | 8.12 × 10−7 | |
| qPR6.1 | 2021 | rs6_419305 | 1.67 × 10−6 | |
| qPR6.2 | 2021 | rs6_3159124 | 1.88 × 10−6 | |
| qPR6.3 | 2022 | rs6_23316386 | 1.05 × 10−6 | |
| qPR6.4 | 2021 | rs6_30589194 | 1.75 × 10−6 | |
| qPR7.1 | 2021 | rs7_3136495 | 7.89 × 10−7 | |
| qPR7.2 | 2021 | rs7_8819901 | 1.85 × 10−7 | |
| qPR7.3 | 2021 | rs7_29223762 | 5.07 × 10−09 | |
| 2022 | rs7_29070665 | 2.75 × 10−09 | ||
| qPR8 | 2021 | rs8_26330779 | 3.70 × 10−7 | |
| qPR10.1 | 2021 | rs10_15765535 | 2.14 × 10−6 | |
| qPR10.2 | 2021 | rs10_20949494 | 7.55 × 10−7 | |
| qPR11 | 2022 | rs11_21490514 | 9.92 × 10−7 | |
| qPR12 | 2021 | rs12_20273980 | 2.05 × 10−6 | |
| BR | qBR2 | 2021 | rs2_24679318 | 5.23 × 10−7 |
| qBR3 | 2022 | rs3_14974531 | 5.80 × 10−7 | |
| qBR5.1 | 2022 | rs5_15809195 | 1.69 × 10−6 | |
| qBR5.2 | 2022 | rs5_29701630 | 2.06 × 10−6 | |
| qBR7 | 2022 | rs7_400711 | 2.30 × 10−6 | |
| qBR8 | 2022 | rs8_8199660 | 1.16 × 10−6 | |
| qBR10 | 2022 | rs10_799081 | 2.16 × 10−6 | |
| qBR11.1 | 2022 | rs11_21609763 | 4.02 × 10−7 | |
| qBR11.2 | 2022 | rs11_22021365 | 7.35 × 10−8 | |
| qBR11.3 | 2022 | rs11_22931729 | 9.97 × 10−7 | |
| LA | qLA4.1 | 2022 | rs4_8191461 | 1.30 × 10−6 |
| qLA4.2 | 2022 | rs4_10331101 | 1.93 × 10−6 | |
| qLA7.1 | 2022 | rs7_15521491 | 1.22 × 10−6 | |
| qLA7.2 | 2021 | rs7_29282304 | 1.88 × 10−6 | |
| qLA8 | 2022 | rs8_17838906 | 9.98 × 10−7 | |
| qLA11.1 | 2022 | rs11_17133200 | 8.64 × 10−7 | |
| qLA11.2 | 2021 | rs11_26784624 | 2.83 × 10−7 | |
| SA | qSA7.1 | 2022 | rs7_15521491 | 1.51 × 10−6 |
| qSA7.2 | 2021 | rs7_29282304 | 1.43 × 10−6 | |
| qSA8 | 2022 | rs8_17838906 | 9.63 × 10−7 | |
| qSA11.1 | 2021 | rs11_4513337 | 1.99 × 10−6 | |
| qSA11.2 | 2022 | rs11_17133200 | 2.01 × 10−6 | |
| ET | qET1 | 2021 | rs1_2397001 | 1.86 × 10−6 |
| qET4 | 2021 | rs4_11219568 | 7.19 × 10−7 | |
| qET8.1 | 2021 | rs8_2577489 | 3.67 × 10−7 | |
| qET8.2 | 2022 | rs8_9627933 | 2.48 × 10−7 | |
| qET8.3 | 2021 | rs8_16434924 | 8.32 × 10−7 | |
| qET9 | 2021 | rs9_3711177 | 4.62 × 10−7 | |
| qET11 | 2021 | rs11_27735087 | 7.94 × 10−7 | |
| TK | qTK2 | 2021 | rs2_9207667 | 1.48 × 10−6 |
| qTK5 | 2021 | rs5_14052109 | 2.48 × 10−6 | |
| qTK10 | 2021 | rs10_7580846 | 2.33 × 10−6 |
| Haplotype Pyramiding | Count of Panels | Genes | Traits | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wp2 | MHZ5 | OsDof-23 | PR | LA | SA | BR | ||||||
| 2021 | 2022 | 2021 | 2022 | 2021 | 2022 | 2021 | 2022 | |||||
| PHap1 | 2 | + | + | + | 1.190 | 1.671 | 6.071 | 6.773 | 5.760 | 6.432 | 3.853 | 9.783 |
| PHap2 | 13 | + | − | + | 0.642 | 0.598 | 5.451 | 5.068 | 5.083 | 4.611 | 3.493 | 2.878 |
| PHap3 | 10 | − | + | + | 0.784 | 0.851 | 5.189 | 4.765 | 4.852 | 4.440 | 4.554 | 4.193 |
| PHap4 | 42 | + | − | − | 0.600 | 0.657 | 5.232 | 5.017 | 4.899 | 4.648 | 4.059 | 4.453 |
| PHap5 | 6 | − | + | − | 0.644 | 0.636 | 5.146 | 4.857 | 4.912 | 4.579 | 4.361 | 3.610 |
| PHap6 | 32 | − | − | + | 0.695 | 0.707 | 5.232 | 4.878 | 4.873 | 4.499 | 3.796 | 3.464 |
| PHap7 | 164 | − | − | − | 0.490 | 0.535 | 4.746 | 4.443 | 4.456 | 4.104 | 3.446 | 3.300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, L.; Li, D.; Ren, N.; Zhu, S.; Wang, D.; Shen, C.; Chen, K.; Xu, J. Identification of Advantaged Genes for Lodging Resistance-Related Traits in the Temperate geng Group (Oryza sativa L.) Using a Genome-Wide Association Study. Agronomy 2023, 13, 2711. https://doi.org/10.3390/agronomy13112711
Zhai L, Li D, Ren N, Zhu S, Wang D, Shen C, Chen K, Xu J. Identification of Advantaged Genes for Lodging Resistance-Related Traits in the Temperate geng Group (Oryza sativa L.) Using a Genome-Wide Association Study. Agronomy. 2023; 13(11):2711. https://doi.org/10.3390/agronomy13112711
Chicago/Turabian StyleZhai, Laiyuan, Duxiong Li, Ningning Ren, Shuangbing Zhu, Dengji Wang, Congcong Shen, Kai Chen, and Jianlong Xu. 2023. "Identification of Advantaged Genes for Lodging Resistance-Related Traits in the Temperate geng Group (Oryza sativa L.) Using a Genome-Wide Association Study" Agronomy 13, no. 11: 2711. https://doi.org/10.3390/agronomy13112711
APA StyleZhai, L., Li, D., Ren, N., Zhu, S., Wang, D., Shen, C., Chen, K., & Xu, J. (2023). Identification of Advantaged Genes for Lodging Resistance-Related Traits in the Temperate geng Group (Oryza sativa L.) Using a Genome-Wide Association Study. Agronomy, 13(11), 2711. https://doi.org/10.3390/agronomy13112711