
Emerging Trends in Allelopathy: A Genetic Perspective for Sustainable Agriculture




Abstract
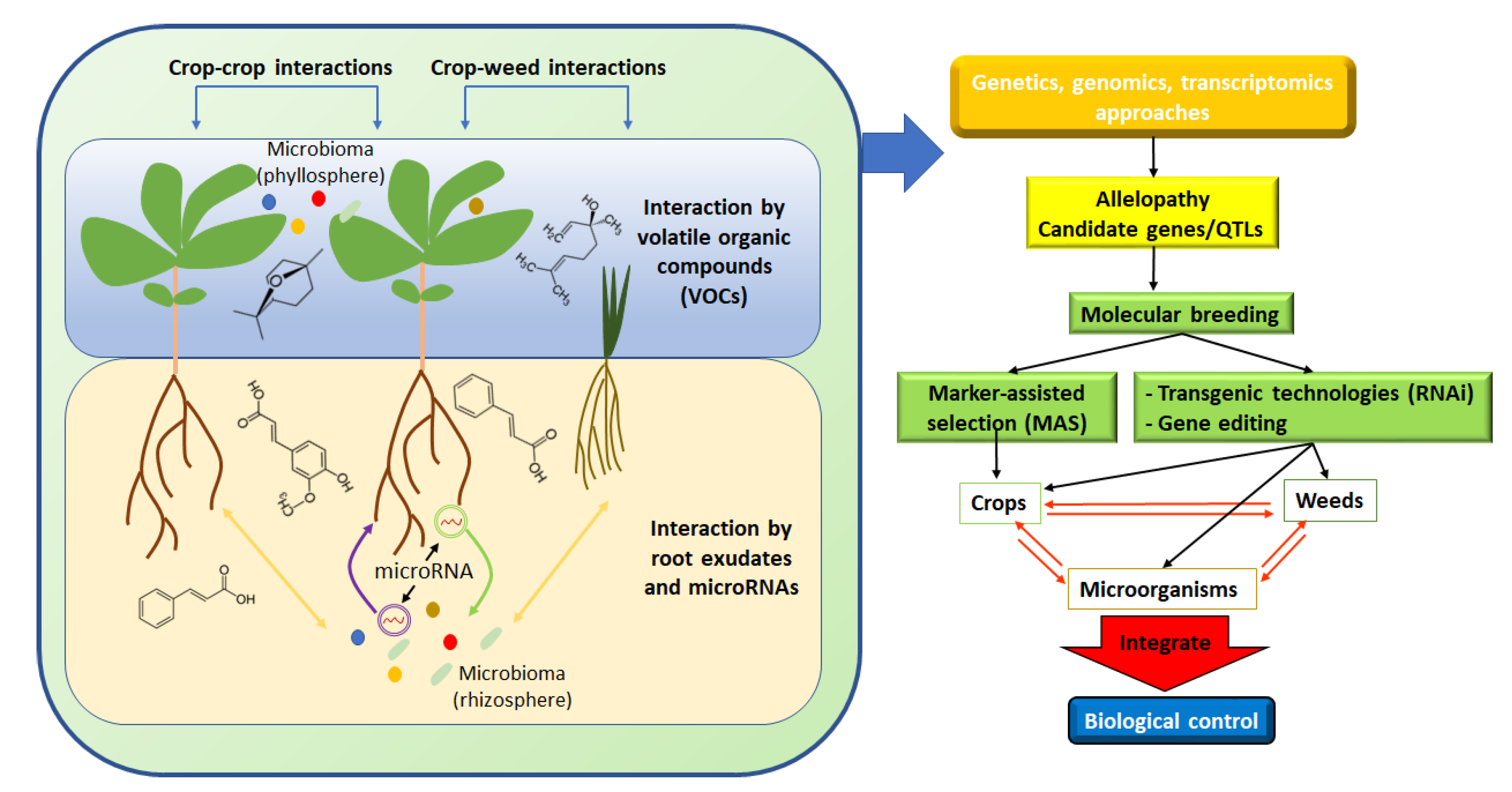
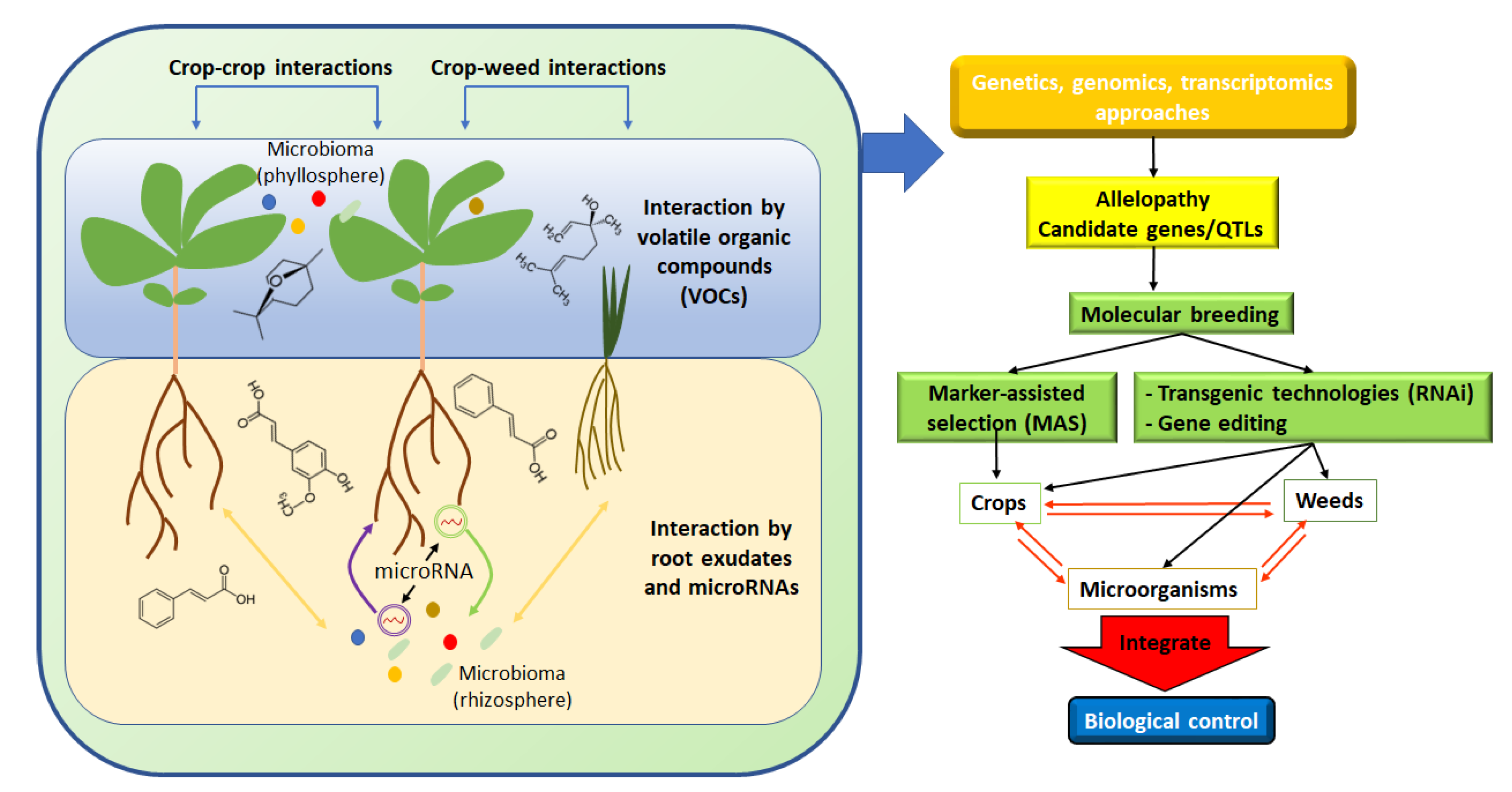
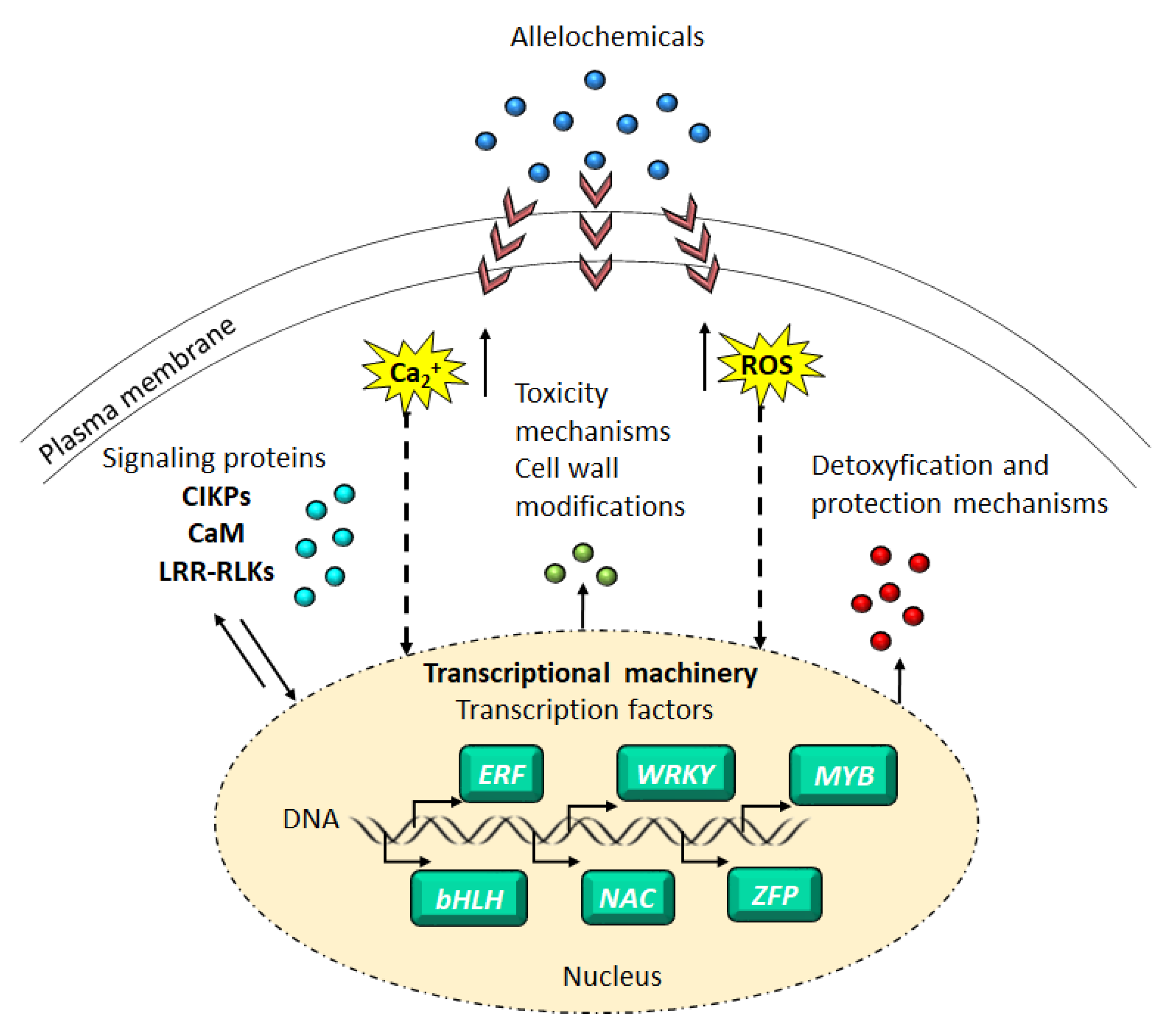
:1. Introduction
2. Genomic Approaches in Allelopathy
2.1. From Metabolite to Gene in Plants
2.2. From Genome to Gene
2.3. Plant Breeding in Allelopathy
2.4. Microorganism in Allelopathy
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Einhellig, F.A. Mechanism of action of allelochemicals in allelopathy. In Allelopathy: Organisms, Processes, and Applications; Inderjit, K.M., Dakshini, M., Einhellig, F.A., Eds.; ACS Symposium Series 582; American Chemical Society: Washington, DC, USA, 1995; pp. 96–116. [Google Scholar]
- Latif, S.; Chiapusio, G.; Weston, L.A. Allelopathy and the role of allelochemicals in plant defence. Adv. Bot. Res. 2017, 82, 19–54. [Google Scholar]
- Molisch, H. Der Einfluss einer Pflanze auf die Andere-Allelopathie; Gustav Fisher Verlag: Jena, Germany, 1937. [Google Scholar]
- Rice, E.L. Allelopathy, 2nd ed.; Academic Press, Inc.: Orlando, FL, USA, 1984. [Google Scholar]
- Zhang, Z.; Liu, Y.; Yuan, L.; Weber, E.; van Kleunen, M. Effect of allelopathy on plant performance: A meta-analysis. Ecol. Lett. 2021, 24, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Bais, H.P. We are family: Kin recognition in crop plants. New Phytol. 2018, 220, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Biedrzycki, M.L.; Jalany, T.A.; Dudley, S.A.; Bais, H.P. Root exudates mediate kin recognition in plants. Commun. Integr. Biol. 2010, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Crepy, M.A.; Casal, J.J. Photoreceptor-mediated kin recognition in plants. New Phytol. 2015, 205, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Durret, R.; Levin, S. Allelopathy in Spatially Distributed Populations. J. Theor. Biol. 1997, 185, 165–171. [Google Scholar] [CrossRef]
- Inderjit, K.; Callaway, R.M. Experimental designs for the study of allelopathy. Plant Soil 2003, 256, 1–11. [Google Scholar]
- Inoye, B.; Stinchcombe, J.R. Relationships between ecological interaction modifications and diffuse coevolution: Similarities, differences, and causal links. Oikos 2001, 95, 353–360. [Google Scholar] [CrossRef]
- Guo, L.; Qiu, J.; Li, L.-F.; Lu, B.; Olsen, K.; Fan, L. Genomic clues for crop-weed interactions and evolution. Trends Plant Sci. 2018, 23, 1102–1115. [Google Scholar]
- Del Moral, R.; Willis, R.J.; Ashton, D.H. Suppression of coastal heath vegetation by Eucalyptus baxteri. Aust. J. Bot. 1978, 26, 203–219. [Google Scholar] [CrossRef]
- Fischer, N.H.; Williamson, G.B.; Weidenhamer, J.D.; Richardson, D.R. In search of allelopathy in the Florida scrub: The role of terpenoids. J. Chem. Ecol. 1994, 20, 1355–1380. [Google Scholar] [PubMed]
- Katz, D.A.; Sneh, B.; Friedman, J. The allelopathic potential of Coridothymus capitatus L. (Labiatae). Preliminary studies on the roles of the shrub in the inhibition of annuals germination and/or to promote allelopathically active actinomycetes. Plant Soil 1987, 98, 53–66. [Google Scholar]
- Weidenhamer, J.D.; Romeo, J.T. Allelopathic properties of Polygonella myriphylla: Field evidence and bioassay. J. Chem. Ecol. 1989, 15, 1957–1970. [Google Scholar]
- Bais, H.P.; Vepachedu, R.; Gilroy, S.; Callaway, R.M.; Vivanco, J.M. Allelopathy and exotic plant invasion: From molecules and genes to species interactions. Science 2003, 301, 1377–1380. [Google Scholar] [PubMed]
- Schenk, H.J.; Callaway, R.M.; Mahall, B.E. Spatial Root Segregation: Are Plants Territorial? Adv. Ecol. Res. 1999, 28, 145–180. [Google Scholar]
- Rizvi, S.G.H.; Rizvi, V. Allelopathy: Basic and Applied Aspects; Chapman and Hall: London, UK, 1992. [Google Scholar]
- Bais, H.P.; Walker, T.S.; Stermitz, F.R.; Hufbauer, R.A.; Vivanco, J.M. Enantiomeric-dependent phytotoxic and antimicrobial activity of (±)-catechin. A rhizosecreted racemic mixture from spotted knapweed. Plant Physiol. 2002, 128, 1173–1179. [Google Scholar]
- Macías, F.A.; Mejías, F.J.R.; Molinillo, J.M.G. Recent advances in allelopathy for weed control: From knowledge to applications. Pest. Manag. Sci. 2019, 75, 2413–2436. [Google Scholar]
- Lupini, A.; Sorgonà, A.; Princi, M.P.; Sunseri, F.; Abenavoli, M.R. Morphological and physiological effects of trans-cinnamic acid and its hydroxylated derivatives on maize root types. Plant Growth Regul. 2016, 78, 263–273. [Google Scholar]
- Politycka, B. Free and glucosylated phenolics, phenol-beta-glucosyltransferase activity and membrane permeability in cucumber roots affected by derivatives of cinnamic and benzoic acid. Acta Physiol. Plantarum 1997, 19, 311–317. [Google Scholar]
- Cruz, O.R.; Anaya, A.L.; Hernandez-Bautista, B.E. Effects of allelochemical stress produced by sicyosdeppei on seedling root ultrastructure of Phaseolous vulgaris and Cucubita ficifolia. J. Chem. Ecol. 1998, 24, 2039–2057. [Google Scholar]
- Patterson, D.T. Effects of allelopathic chemicals on growth and physiological response of soybean (Glycine max). Weed Sci. 1981, 29, 53–58. [Google Scholar]
- Araniti, F.; Bruno, L.; Sunseri, F.; Pacenza, M.; Forgione, I.; Bitonti, M.B.; Abenavoli, M.R. The allelochemical farnesene affects Arabidopsis thaliana root meristem altering auxin distribution. Plant Physiol. Bioch. 2017, 121, 14–20. [Google Scholar]
- Yazaki, K.; Arimura, G.; Ohnishi, T. “Hidden” terpenoids in plants: Their biosynthesis, localization and ecological roles. Plant Cell Physiol. 2017, 58, 1615–1621. [Google Scholar]
- Gershenzon, J.; Dudareva, N. The function of terpene natural products in the natural world. Nat Chem Biol. 2007, 3, 408–414. [Google Scholar]
- Chou, C.C. Introduction to allelopathy. In Allelopathy: A Physiological Process with Ecological Implications; Reigosa, M.J., Pedrol, N., González, L., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 1–9. [Google Scholar]
- Zimdahl, R.L. Weed–Crop Competition: A Review, 2nd ed.; Blackwell: Hoboken, NJ, USA, 2004. [Google Scholar]
- MacLaren, C.; Storkey, J.; Menagat, A.; Metcalfe, H.; Dehnen-Schmutz, K. An ecological future for weed science to sustain crop production and the environment. A review. Agron. Sustain. Dev. 2020, 40, 24. [Google Scholar]
- Wink, M. Biochemistry, physiology and ecological functions of secondary metabolites. In Biochemistry of Plant Secondary Metabolism, 2nd ed.; Wink, M., Ed.; Wiley-Blackwell: West Sussex, UK, 2010; pp. 1–19. [Google Scholar]
- Boycheva, S.; Daviet, L.; Wolfender, J.; Fitzpatrick, T.B. The rise of operon-like gene clusters in plants. Trends Plant Sci. 2014, 19, 447–459. [Google Scholar]
- Cheng, F.; Cheng, Z.H.; Meng, H.W. Transcriptomic insights into the allelopathic effects of the garlic allelochemical diallyl disulfide on tomato roots. Sci. Rep. 2016, 6, 38902. [Google Scholar]
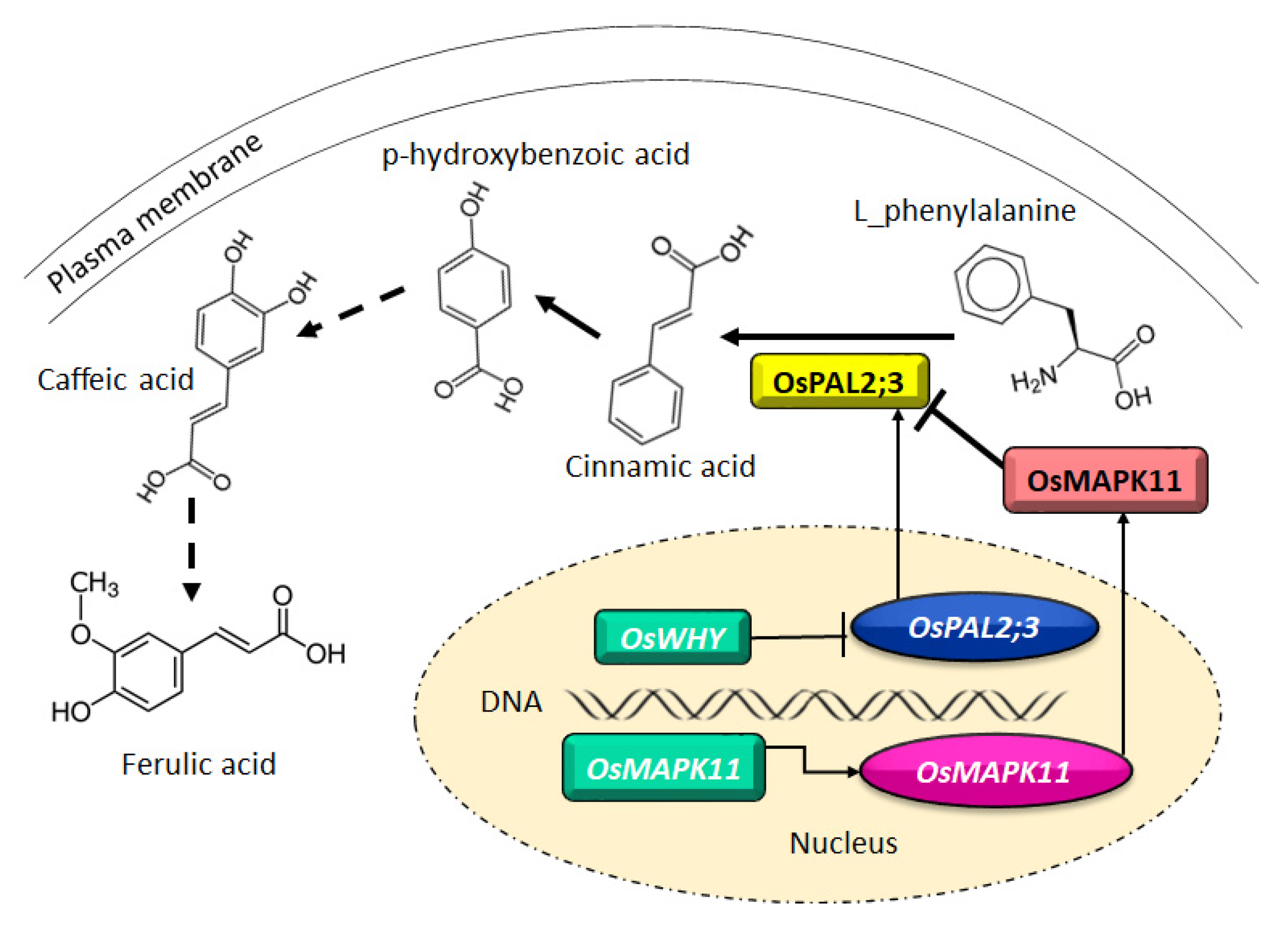
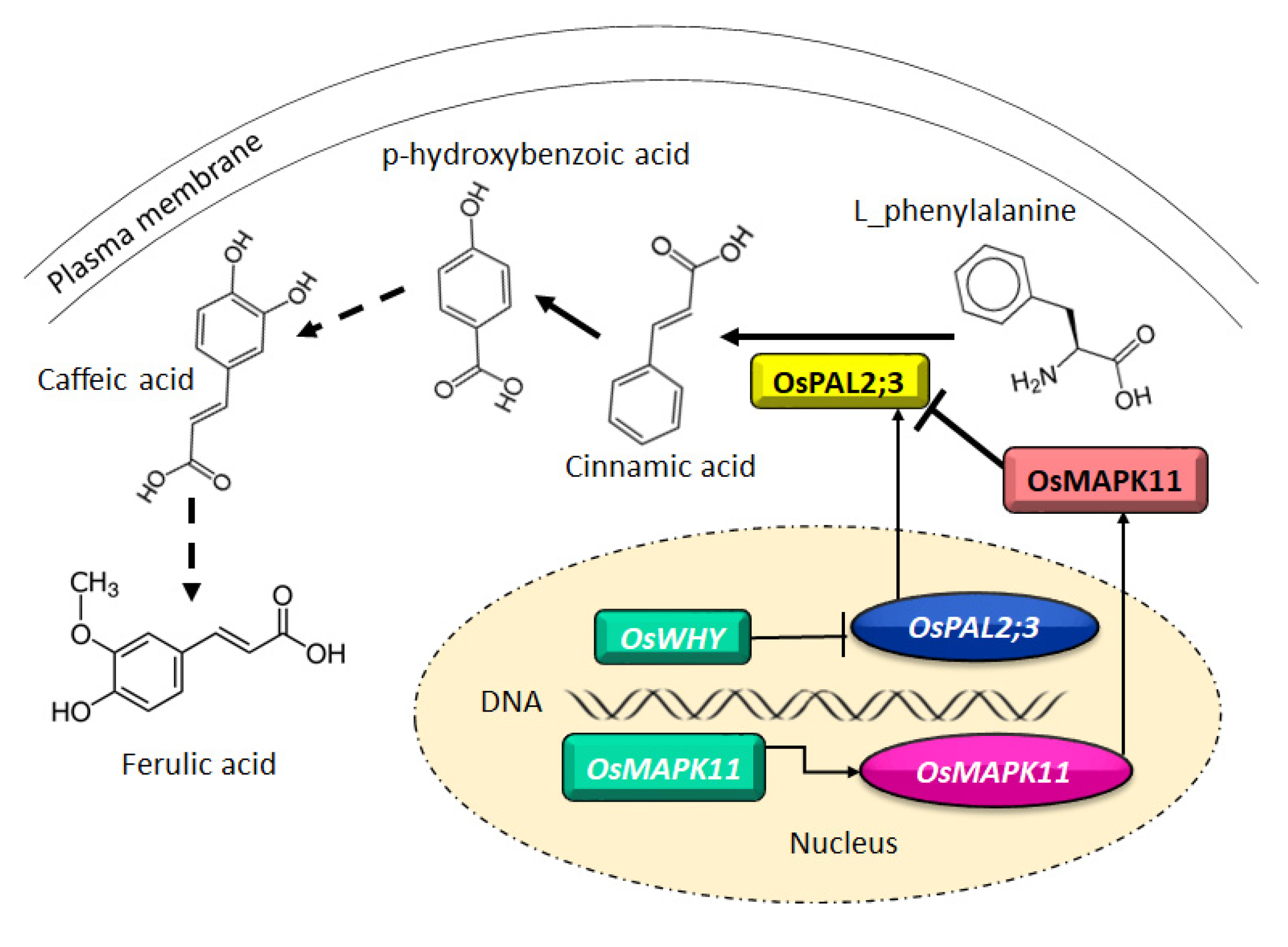
- Fang, C.; Yang, L.; Chen, W.; Li, L.; Zhang, P.; Li, Y.; He, H.; Lin, W. MYB57 transcriptionally regulates MAPK11 to interact with PAL2;3 and modulate rice allelopathy. J. Exp. Bot. 2020, 71, 2127–2142. [Google Scholar]
- Zhang, H.; Rutherford, S.; Qi, S.; Huang, P.; Dai, Z. Transcriptome profiling of Arabidopsis thaliana roots in response to allelopathic effects of Conyza canadensis. Ecotoxicology 2022, 31, 53–63. [Google Scholar]
- Duke, S.O.; Baerson, S.R.; Pan, Z.; Kagan, I.A.; Sanchez-Moreiras, A.; Reigosa, M.J.; Pedrol, N.; Schulz, M. Genomic Approaches to Understanding Allelochemical Effects on Plants. In Allelopathy in Sustainable Agriculture and Forestry; Zeng, R.S., Mallik, A.U., Luo, S.M., Eds.; Springer: New York, NY, USA, 2008. [Google Scholar]
- Yuan, Y.; Lee, H.T.; Hu, H.; Scheben, A.; Edwards, D. Single-Cell genomics analysis in plants. Genes 2018, 9, 50. [Google Scholar]
- He, H.Q.; Lin, W.X.; Liang, Y.Y.; Song, B.Q.; Ke, Y.Q.; Guo, Y.C.; Liang, K.J. Analyzing the molecular mechanism of crop allelopathy by using differential proteomics. Acta Ecol. Sin. 2005, 25, 3141–3146. [Google Scholar]
- Chi, W.-C.; Chen, Y.-A.; Hsiung, Y.-C.; Fu, S.-F.; Chou, C.-H.; Trinh, N.N.; Chen, Y.-C.; Huang, H.-J. Autotoxicity mechanism of Oryza sativa: Transcriptome response in rice roots exposed to ferulic acid. BMC Genom. 2013, 14, 351. [Google Scholar]
- Zhang, Q.; Zheng, X.-Y.; Lin, S.-X.; Gu, C.-Z.; Li, L.; Li, J.-Y.; Fang, C.-X.; He, H.-B. Transcriptome analysis reveals that barnyard grass exudates increase the allelopathic potential of allelopathic and non-allelopathic rice (Oryza sativa) accessions. Rice 2019, 12, 30. [Google Scholar] [CrossRef]
- Li, L.-L.; Zhao, H.-H.; Kong, C.-H. (-)-Loliolide, the most ubiquitous lactone, is involved in barnyard grass-induced rice allelopathy. J. Exp. Bot. 2020, 71, 1540–1550. [Google Scholar]
- Yang, Y.; Zhang, Z.; Li, R.; Yi, Y.; Yang, H.; Wang, C.; Wang, Z.; Liu, Y. RgC3H involves in the biosynthesis of allelopathic phenolic acids and alters their release amount in Rehmannia glutinosa roots. Plants 2020, 9, 567. [Google Scholar] [CrossRef] [PubMed]
- Horvath, D.P.; Hansen, S.A.; Moriles-Miller, J.P.; Pierik, R.; Yan, C.; Clay, D.E.; Scheffler, B.; Clay, S.A. RNAseq reveals weed-induced PIF3-like as candidate target to manipulate weed stress response in soybean. New Phytol. 2015, 207, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.-M.; Ham, T.-H.; Cho, G.-W.; Kwon, S.-W.; Lee, Y.; Seo, J.; An, Y.-J.; Kim, S.-Y.; Lee, J. Study of quantitative trait loci (QTLs) associated with allelopathic trait in rice. Genes 2020, 11, 479. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Q.; Lin, S.; Wang, P.; Li, J.; Wang, H.; He, H. Dynamic analysis on weed inhibition and phenolic acids of allelopathic rice in field test. Arch. Agron. Soil Sci. 2020, 67, 1809–1821. [Google Scholar] [CrossRef]
- Wu, H.; Pratley, J.; Ma, W.; Haig, T. Quantitative trait loci and molecular markers associated with wheat allelopathy. Theor. Appl. Genet. 2003, 107, 1477–1481. [Google Scholar] [CrossRef]
- Zeng, D.; Qian, Q.; Teng, S.; Dong, G.; Fujimoto, H.; Yasufumi, K.; Zhu, L. Genetic analysis of rice allelopathy. Chin. Sci. Bull. 2003, 48, 265–268. [Google Scholar] [CrossRef]
- Quader, M.; Daggard, G.; Barrow, R.; Walker, S.; Sutherland, M.W. Allelopathy, Dimboa production and genetic variability in accessions of Triticum speltoides. J. Chem. Ecol. 2001, 27, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhuang, Y.; Xu, T.; Li, Y.; Li, Y.; Lin, W. Changes in rice allelopathy and rhizosphere microflora by inhibiting rice phenylalanine ammonialyase gene expression. J. Chem. Ecol. 2013, 39, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Galhano, R.; Wiemann, P.; Bueno, E.; Tiernan, M.; Wu, W.; Peters, R.J. Genetic evidence for natural product-mediated plant-plant allelopathy in rice (Oryza sativa). New Phytol. 2012, 193, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lin, S.; Zhang, Q.; Wang, P.; Wang, H.; He, H. Comparative analysis on metabolites and gene expression difference of various allelopathic potential rice under weed stress. Weed Res. 2022; in press. [Google Scholar] [CrossRef]
- Shehzad, T.; Okuno, K. Genetic analysis of QTLs controlling allelopathic characteristics in sorghum. PLoS ONE 2020, 15, e0235896. [Google Scholar] [CrossRef]
- Sultana, M.H.; Liu, F.; Alamin, M.; Mao, L.; Jia, L.; Chen, H.; Wu, D.; Wang, Y.; Fu, F.; Wu, S.; et al. Gene modules co-regulated with biosynthetic gene custers for allelopathy between rice and bernyargrass. Int. J. Mol. Sci. 2019, 20, 3846. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.; Chen, Q.; Miao, Y.; Peng, Z.; Huang, B.; Guo, L.; Liu, D.; Du, H. Allelopathic effect of Artemisia argyi on the germination and growth of various weeds. Sci. Rep. 2021, 11, 4303. [Google Scholar] [CrossRef]
- Guo, L.; Qiu, J.; Ye, C.; Jin, G.; Mao, L.; Zhang, H.; Yang, X.; Peng, Q.; Wang, Y.; Jia, L.; et al. Echinochloa crus-galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nat. Commun. 2017, 8, 1031. [Google Scholar] [CrossRef]
- Gierl, A.; Frey, M. Evolution of benzoxazinone biosynthesis and indole production in maize. Planta 2001, 213, 493–498. [Google Scholar] [CrossRef]
- Frey, M.; Chomet, P.; Glawischnig, E.; Stettner, C.; Grün, S.; Winklmair, A.; Eisenreich, W.; Bacher, A.; Meerley, R.B.; Briggs, S.P.; et al. Analysis of a chemical plant defense mechanism in grasses. Science 1997, 277, 696–699. [Google Scholar] [CrossRef]
- Song, B.; Xiong, J.; Fang, C.; Qiu, L.; Lin, R.; Liang, Y.; Lin, W. Allelopathic enhancement and differential gene expression in rice under low nitrogen treatment. J. Chem. Ecol. 2008, 34, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.X.; Xiong, J.; Qiu, L.; Wang, H.B.; Song, B.Q.; He, H.B.; Lin, R.Y.; Lin, W.X. Analysis of gene expressions associated with increased allelopathy in rice (Oryza sativa L.) induced by exogenous salicylic acid. Plant Growth Regul. 2009, 57, 163–172. [Google Scholar] [CrossRef]
- Fang, C.; Li, Y.; Li, C.; Li, B.; Ren, Y.; Zheng, H.; Zeng, X.; Shen, L.; Lin, W. Identification and comparative analysis of microRNAs in barnyard grass (Echinochloa crus-galli) in response to rice allelopathy. Plant Cell Environ. 2015, 38, 1368–1381. [Google Scholar] [CrossRef]
- Dixon, R.A.; Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell 1995, 7, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- He, H.B.; Wang, H.B.; Fang, C.X.; Wu, H.; Guo, X.K.; Lin, Z.H.; Lin, W.X. Barnyard grass stress up regulates the biosynthesis of phenolic compounds in allelopathic rice. J. Plant Physiol. 2012, 169, 1747–1753. [Google Scholar] [CrossRef] [PubMed]
- Field, B.; Jordan, F.; Osbourn, A. First encounters—Deployment of defence related natural products by plants. New Phytol. 2006, 172, 193–207. [Google Scholar] [CrossRef]
- Kato, T.; Kabuto, C.; Sasaki, N.; Tsunagawa, M.; Aizawa, H.; Fujita, K.; Kato, Y.; Kitahara, Y.; Takahashi, N. Momilactones, growth inhibitors from rice, Oryza sativa L. Tetrahedron Lett. 1973, 14, 3861–3864. [Google Scholar] [CrossRef]
- Takahashi, N.; Kato, T.; Tsunagawa, M.; Sasaki, N.; Kitahara, Y. Mechanisms of dormancy in rice seeds: II. New growth inhibitors, momilactone-A and-B isolated from the hulls of rice seeds. Jpn. J. Breed. 1976, 26, 91–98. [Google Scholar] [CrossRef]
- Kato-Noguchi, H.; Peters, R.J. The role of momilactones in rice allelopathy. J. Chem. Ecol. 2013, 39, 175–185. [Google Scholar] [CrossRef]
- Serra Serra, N.; Shanmuganathan, R.; Becker, C. Allelopathy in rice: A story of momilactones, kin recognition, and weed management. J. Exp. Bot. 2021, 72, 4022–4037. [Google Scholar] [CrossRef]
- Peters, R.J. Uncovering the complex metabolic network underlying diterpenoid phytoalexin biosynthesis in rice and other cereal crop plants. Phytochemistry 2006, 67, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Toyomasu, T. Recent advances regarding diterpene cyclase genes in higher plants and fungi. Biosci. Biotechnol. Biochem. 2008, 72, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, C.; Wu, C.; Xiong, L.; Chen, G.; Zhang, Q.; Wang, S. RMD: A rice mutant database for functional analysis of the rice genome. Nucl. Acids Res. 2006, 34, D745–D748. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.S.; Lee, S.; Jung, K.H.; Jun, S.H.; Jeong, D.H.; Lee, J.; Kim, C.; Jang, S.; Yang, K.; Nam, J.; et al. T-DNA insertional mutagenesis for functional genomics in rice. Plant J. 2000, 22, 561–570. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Afzal, F.; Gul, A.; Kazi, A.M. Next-generation sequencing technologies and plant improvement. In Plant Omics: Trends and Applications, 1st ed.; Hakeem, K.R., Tombuloğlu, H., Tombuloğlu, G., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 271–294. [Google Scholar]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2006, 437, 376–380. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, L.; Li, J.; Wang, H.; Fang, C.; Yang, X.; He, H. Increasing rice allelopathy by induction of barnyard grass (Echinochloa crus-galli) root exudates. J. Plant Growth Regul. 2018, 37, 745–754. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Zhu, Y.; Guo, L.; Ji, R.; Miao, Y.; Guo, L.; Du, H.; Liu, D. Caffeic Acid, an Allelochemical in Artemisia argyi, Inhibits Weed Growth via Suppression of Mitogen-Activated Protein Kinase Signaling Pathway and the Biosynthesis of Gibberellin and Phytoalexin. Front. Plant Sci. 2021, 12, 802198. [Google Scholar] [CrossRef] [PubMed]
- Asins, M.J.; Bernet, G.P.; Villalta, I.; Carbonell, E.A. QTL Analysis in Plant Breeding. In Molecular Techniques in Crop Improvement; Jain, S., Brar, D., Eds.; Springer: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Olofsdotter, M.; Jensen, L.B.; Courtois, B. Improving crop competitive ability using allelopathy—An example from rice. Plant Breed. 2002, 121, 1–9. [Google Scholar] [CrossRef]
- Niemeyer, H.M.; Jerez, J.M. Chromosomal location of genes for hydroxamic acid accumulation in wheat using wheat aneuploids and wheat substitution lines. Heredity 1997, 79, 10–14. [Google Scholar] [CrossRef]
- Ebana, K.; Yan, W.; Dilday, R.H.; Namai, H.; Okuno, K. Analysis of QTL Associated with the Allelopathic Effect of Rice Using Water-soluble Extracts. Breed. Sci. 2001, 51, 47–51. [Google Scholar] [CrossRef] [Green Version]
- Bharamappanavara, M.; Siddaiah, A.M.; Ponnuvel, S.; Ramappa, L.; Patil, B.; Appaiah, M.; Maganti, S.M.; Sundaram, R.M.; Kadadanamari, S.; Tuti, M.D.; et al. Mapping QTL hotspots associated with weed competitive traits in backcross population derived from Oryza sativa L. and O. glaberrima Steud. Sci. Rep. 2020, 10, 22103. [Google Scholar] [CrossRef]
- Fierer, N. Embracing the unknown: Disentangling the complexities of the soil microbiome. Nat. Rev. Microbiol. 2017, 15, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.; Berg, C.; Landa, B.B.; Auerbach, A.; Moissl-Eichinger, C.; Berg, G. Plant genotype-specific archaeal and bacterial endophytes but similar Bacillus antagonists colonize mediterranean olive trees. Front. Microbiol. 2015, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Frąc, M.; Hannula, S.E.; Bełka, M.; Jędryczka, M. Fungal Biodiversity and Their Role in Soil Health. Front. Microbiol. 2018, 9, 707. [Google Scholar] [CrossRef]
- Barrera, S.E.; Sarango-Flóres, S.W.; Montenegro-Gómez, S.P. The phyllosphere microbiome and its potential application in horticultural crops. A review. Rev. Colomb. Cienc. Hortic. 2019, 13, 384–396. [Google Scholar] [CrossRef]
- Ding, L.-J.; Cui, H.-L.; Nie, S.-A.; Long, X.-E.; Duan, G.-L.; Zhu, Y.-G. Microbiomes inhabiting rice roots and rhizosphere. FEMS Microbiol. Ecol. 2019, 95, fiz040. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Teixeira, P.J. Root-exuded coumarin shapes the root microbiome. Proc. Natl. Acad. Sci. USA 2018, 115, 5629–5631. [Google Scholar] [CrossRef] [PubMed]
- Stringlis, I.A.; Yu, K.; Feussner, K.; de Jonge, R.; Van Bentum, S.; Van Verk, M.C.; Berendsen, R.; Bakker, P.A.H.M.; Feussner, I.; Pieterse, C.M.J. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jian, X.; Li, Y.; Zeng, X.; Xu, L.; Khan, M.U.; Lin, W. OsPAL2-1 Mediates Allelopathic Interactions Between Rice and Specific Microorganisms in the Rhizosphere Ecosystem. Front. Microbiol. 2020, 23, 1411. [Google Scholar] [CrossRef]
- Escudero-Martinez, C.; Coulter, M.; Alegria Terrazas, R.; Foito, A.; Kapadia, R.; Pietrangelo, L.; Maver, M.; Sharma, R.; Aprile, A.; Morris, J.; et al. Identifying plant genes shaping microbiota composition in the barley rhizosphere. Nat. Commun. 2022, 13, 3443. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.A.; Obermeier, M.M.; Berg, G. Bioprospecting plant-associated microbiomes. J. Biotechnol. 2016, 235, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Egamberdieva, D.; Wirth, S.; Alqarawi, A.A.; Elsayed, F.A.A.; Hashem, A. Phytohormones and Beneficial Microbes: Essential Components for Plants to Balance Stress and Fitness. Front. Microbiol. 2017, 8, 2104. [Google Scholar] [CrossRef]
- Ho, N.-G.; Mathew, D.C.; Huang, C.-C. Plant-Microbe Ecology: Interactions of Plants and Symbiotic Microbial Communities. In Plant Ecology-Traditional Approaches to Recent Trends; IntechOpen: London, UK, 2017. [Google Scholar]
- Yi, H.-S.; Yang, J.W.; Ryu, C.-M. ISR meets SAR outside: Additive action of the endophyte Bacillus pumilus INR7 and the chemical inducer, benzothiadiazole, on induced resistance against bacterial spot in field-grown pepper. Front. Plant Sci. 2013, 4, 122. [Google Scholar] [CrossRef]
- Abbas, T.; Zahir, Z.A.; Naveed, M.; Abbas, S.; Alwahibi, M.S.; Elshikh, M.S.; Adnan, M. Large Scale Screening of Rhizospheric Allelopathic Bacteria and Their Potential for the Biocontrol of Wheat-Associated Weeds. Agronomy 2020, 10, 1469. [Google Scholar] [CrossRef]
- Lee, S.; Behringer, G.; Hung, R.; Bennett, J. Effects of fungal volatile organic compounds on Arabidopsis thaliana growth and gene expression. Fungal Ecol. 2019, 37, 1–9. [Google Scholar] [CrossRef]
- Gao, Y.; Feng, J.; Wu, J.; Wang, K.; Wu, S.; Liu, H.; Jiang, M. Transcriptome analysis of the growth-promoting effect of volatile organic compounds produced by Microbacterium aurantiacum GX14001 on tobacco (Nicotiana benthamiana). BMC Plant Biol. 2022, 22, 208. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Rout, M.E.; Southworth, D. The root microbiome influences scales from molecules to ecosystems: The unseen majority. Am. J. Bot. 2013, 100, 1689–1691. [Google Scholar] [CrossRef]
- Bever, J.D. Feedback between plants and their soil communities in an old field community. Ecology 1994, 75, 1965–1977. [Google Scholar] [CrossRef]
- Baldrich, P.; Rutter, B.D.; Karimi, H.Z.; Podicheti, R.; Meyers, B.C.; Innes, R.W. Plant extracellular vesicles contain diverse small RNA species and are enriched in 10- to 17-nucleotide “tiny” RNAs. Plant Cell 2019, 31, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, H.; Yergeau, E.; Monard, C.; Combier, J.P.; El Amrani, A. Rhizospheric plant-microbe interactions: miRNAs as a key mediator. Trends Plant Sci. 2021, 26, 132–141. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.-L.; Zhao, J.-H.; Wang, S.; Jin, Y.; Chen, Z.-Q.; Fang, Y.-Y.; Hua, C.-L.; Ding, S.-W.; Guo, H.-S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef]
- Wang, M.; Weiberg, A.; Dellota, E., Jr.; Yamane, D.; Jin, H. Botrytis small RNA Bc -siR37 suppresses plant defense genes by cross-kingdom RNAi. RNA Biol. 2017, 14, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Qiao, L.; Wang, M.; He, B.; Lin, F.-M.; Palmquist, J.; Huang, S.-D.; Jin, H. Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 2018, 360, 1126–1129. [Google Scholar] [CrossRef]
- He, B.; Cai, Q.; Qiao, L.; Huang, C.-Y.; Wang, S.; Miao, W.; Ha, T.; Wang, Y.; Jin, H. RNA-binding proteins contribute to small RNA loading in plant extracellular vesicles. Nat. Plants 2021, 7, 342–352. [Google Scholar] [CrossRef]
- Knief, C. Analysis of plant microbe interactions in the era of next generation sequencing technologies. Front. Plant Sci. 2014, 5, 216. [Google Scholar] [CrossRef]
- Bringel, F.; Couée, I. Pivotal roles of phyllosphere microorganisms at the interface between plant functioning and atmospheric trace gas dynamics. Front. Microbiol. 2015, 6, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Plant Material | Methods | Targets | References |
|---|---|---|---|
| Common reed | RNA-seq | Phytohormones | He et al. [39] |
| Rice/barnyard grass | Microarray | Phytohormones | Chi et al. [40] |
| Tomato | RNA-seq | Antioxidants and Hormones | Cheng et al. [34] |
| Rice/barnyard grass | RNA-seq | Shikimic acid and acetic acid pathways | Zhang et al. [41] |
| Rice/barnyard grass | RNA-seq | Diterpenoid and flavonoid biosynthesis pathway | Li et al. [42] |
| Rehmannia glutinosa | Cloning, qRT-PCR | Phenolic biosynthesis: C3H gene | Yang et al. [43] |
| Soybeans | RNA-seq | Oxidative stress and jasmonic acid signaling (PIF3) | Horvath et al. [44] |
| Rice | SNPs genotyping | QTL regions | Chung et al. [45] |
| Rice | qRT-PCR | Biosynthesis of phenolic acids | Zhang et al. [46] |
| Wheat/ryegrass | AFLP, RFLP and SSR genotyping | QTL regions | Wu et al. [47] |
| Lettuce/rice | RFLP genotyping | QTL regions | Zeng et al. [48] |
| Lettuce/Triticum Speltoides | RAPD genotyping | Genetic diversity in allelopathic potential | Quader et al. [49] |
| Rice | RNA interference | PAL gene expression | Fang et al. [50] |
| Rice | T-DNA insertion | OsCPS4, OsKSL4 | Xu et al. [51] |
| Rice/barnyard grass | qRT-PCR | PAL, C4H, F5H, and COMT genes | Zhang et al. [52] |
| Sorghum | SSR genotyping | QTL regions | Shehzad et al. [53] |
| Rice/barnyard grass | qRT-PCR, ChIP-seq, ChIP-qPCR | MYB transcription factor | Fang et al. [35] |
| Rice | qRT-PCR, RNA-seq | Biosynthetic gene clusters | Sultana et al. [54] |
| Arabidopsis | RNA-seq | Signal transduction, nutrient transporter, detoxification genes | Zhang et al. [36] |
| Rice | RNA-seq | Chlorophyll and nitrogen metabolisms | Li et al. [55] |
| Rice | Genome sequencing | Detoxification-related genes (CYP450, GST) DIMBOA gene cluster. | Guo et al. [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aci, M.M.; Sidari, R.; Araniti, F.; Lupini, A. Emerging Trends in Allelopathy: A Genetic Perspective for Sustainable Agriculture. Agronomy 2022, 12, 2043. https://doi.org/10.3390/agronomy12092043
Aci MM, Sidari R, Araniti F, Lupini A. Emerging Trends in Allelopathy: A Genetic Perspective for Sustainable Agriculture. Agronomy. 2022; 12(9):2043. https://doi.org/10.3390/agronomy12092043
Chicago/Turabian StyleAci, Meriem Miyassa, Rossana Sidari, Fabrizio Araniti, and Antonio Lupini. 2022. "Emerging Trends in Allelopathy: A Genetic Perspective for Sustainable Agriculture" Agronomy 12, no. 9: 2043. https://doi.org/10.3390/agronomy12092043
APA StyleAci, M. M., Sidari, R., Araniti, F., & Lupini, A. (2022). Emerging Trends in Allelopathy: A Genetic Perspective for Sustainable Agriculture. Agronomy, 12(9), 2043. https://doi.org/10.3390/agronomy12092043