
Transcriptome and Metabolome Analyses of Salt Stress Response in Cotton (Gossypium hirsutum) Seed Pretreated with NaCl

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Material
2.2. RNA Isolation and Sequencing
2.3. Functional Annotation
2.4. Differentially Expressed Genes (DEGs)
2.5. Validation through qRT-PCR
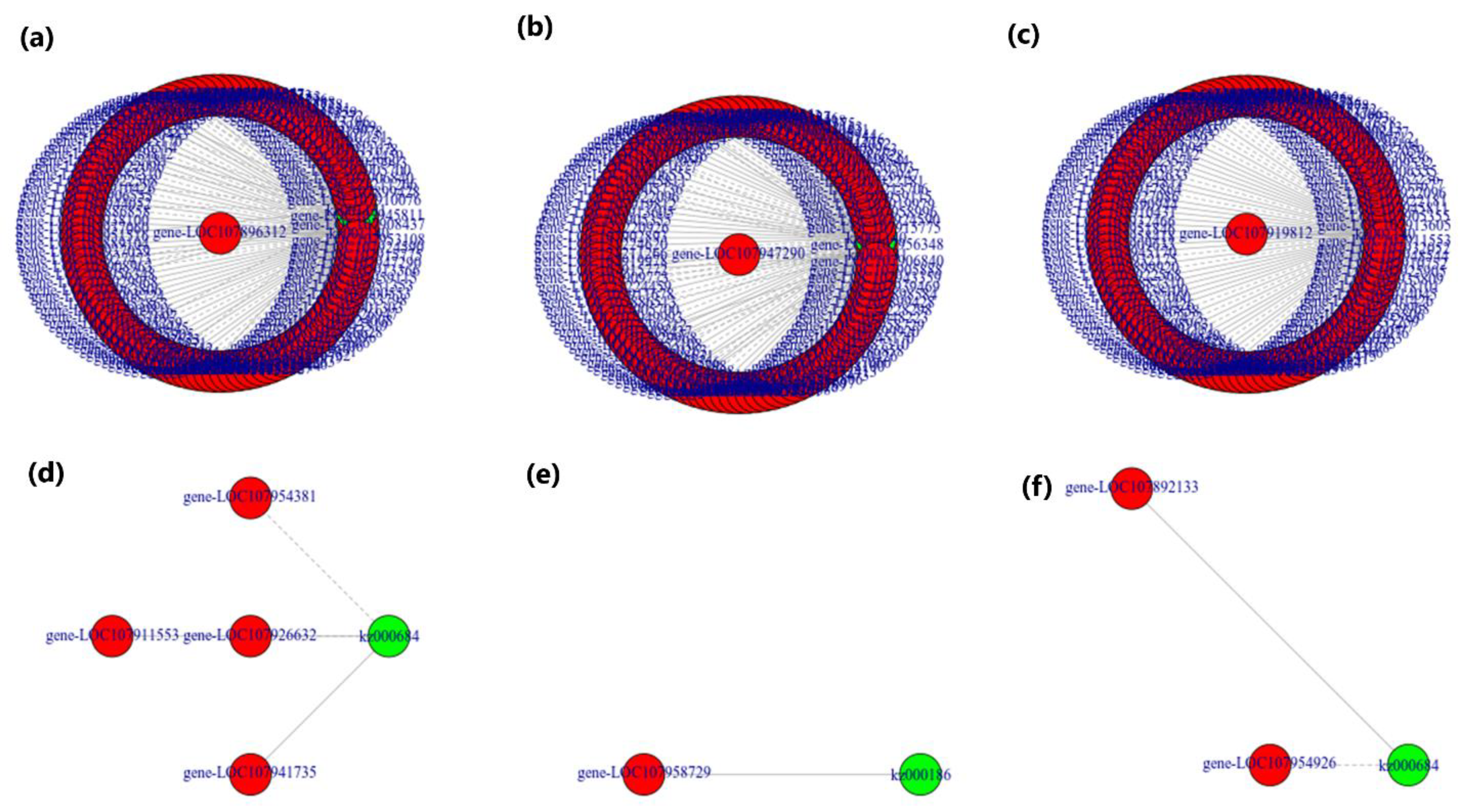
2.6. Network Analysis
2.7. Metabolite’s Profiling
2.8. Data Analyses
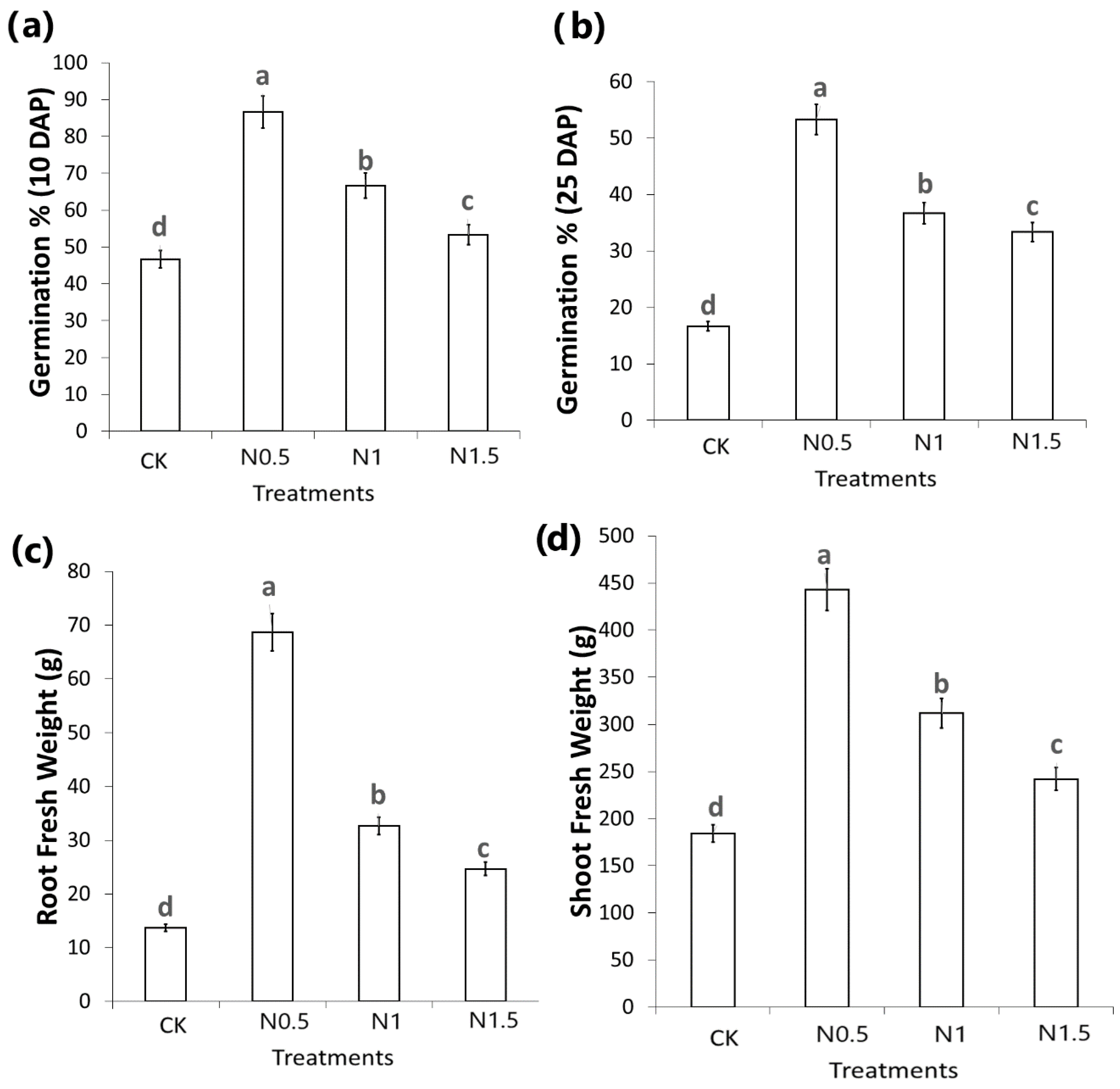
3. Results
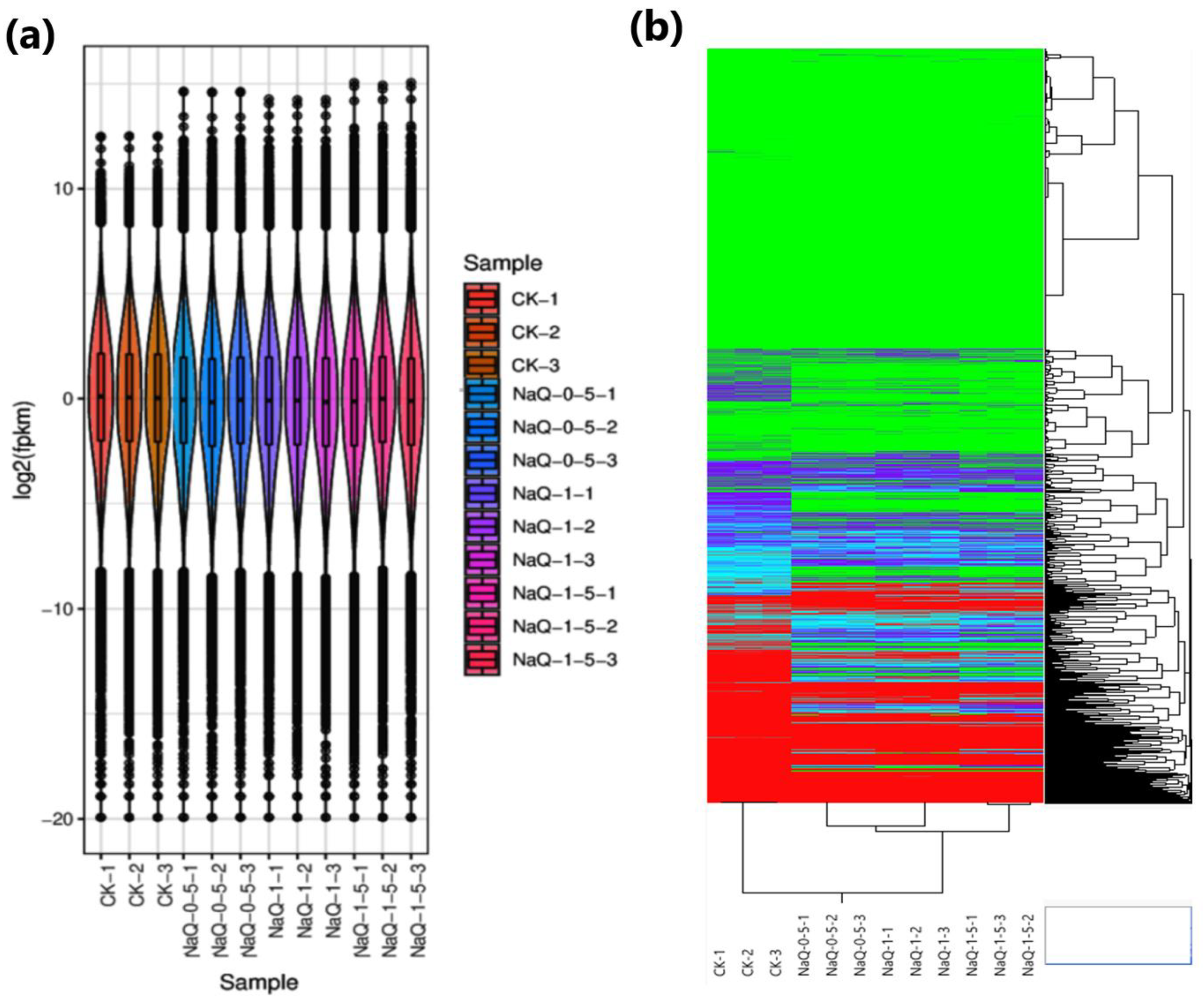
3.1. Transcriptome Profiling and Functional Annotations
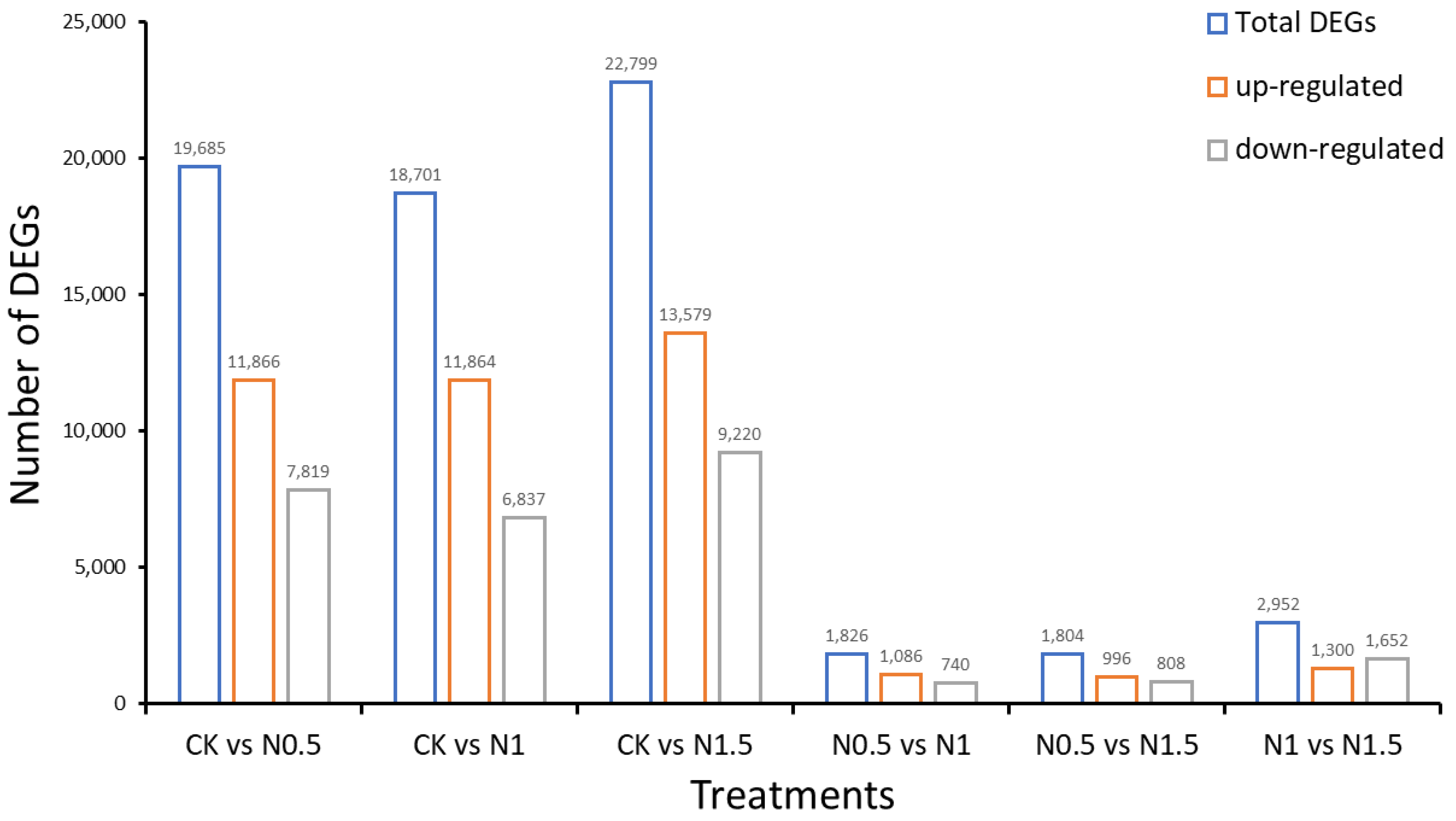
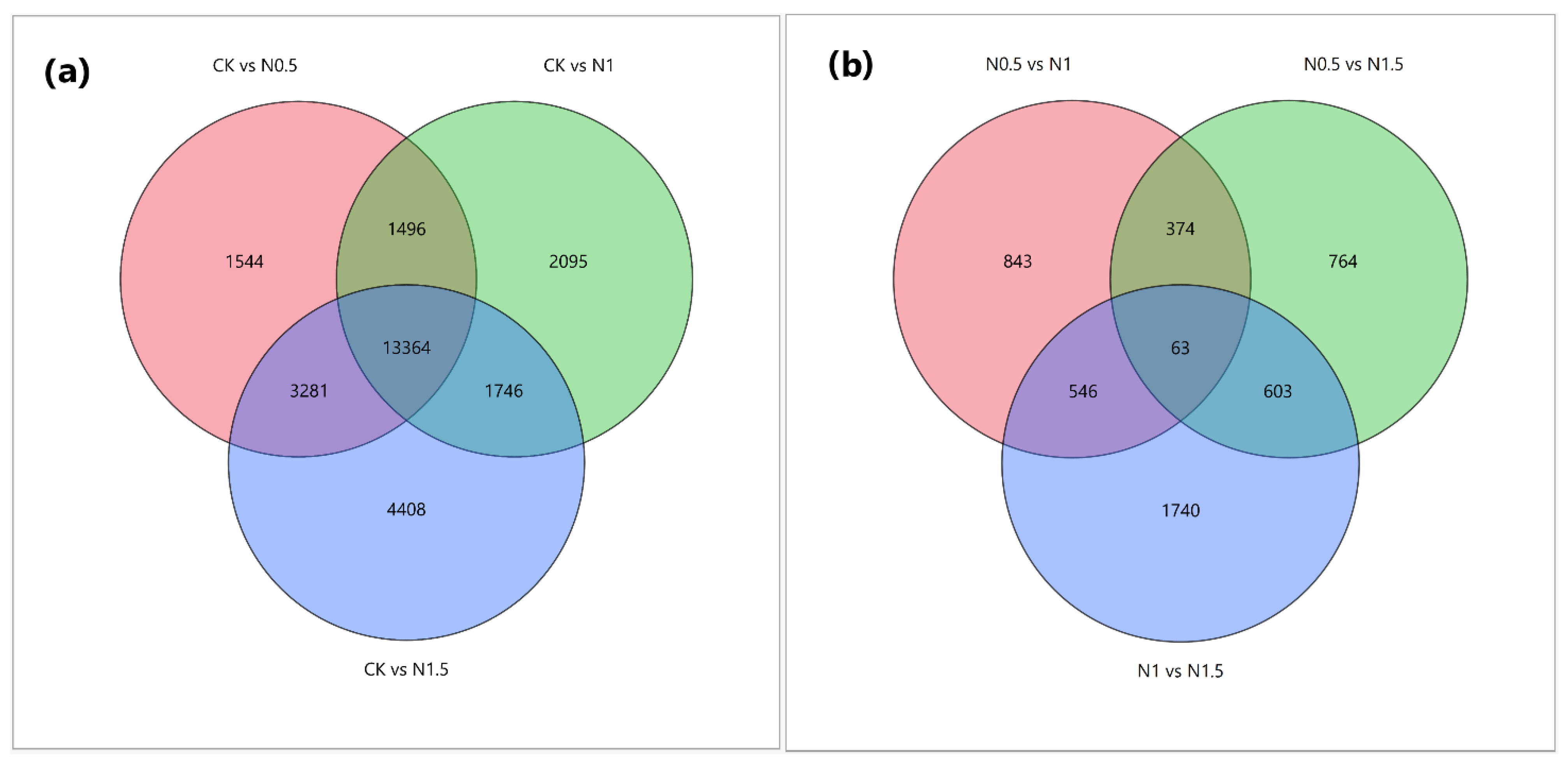
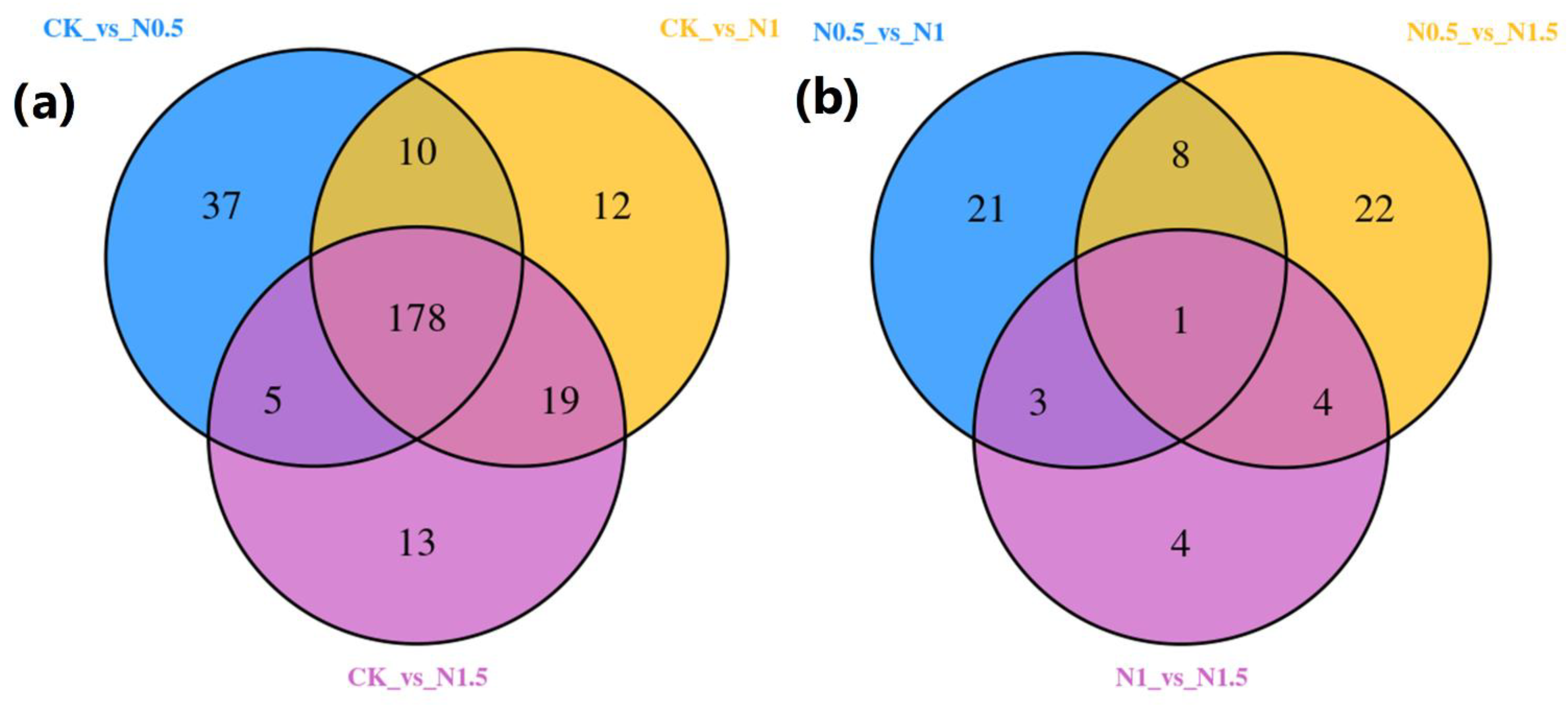
3.2. Identification of DEGs in CK and Treated Cotton Seedlings
3.3. Functional Annotation of DEGs
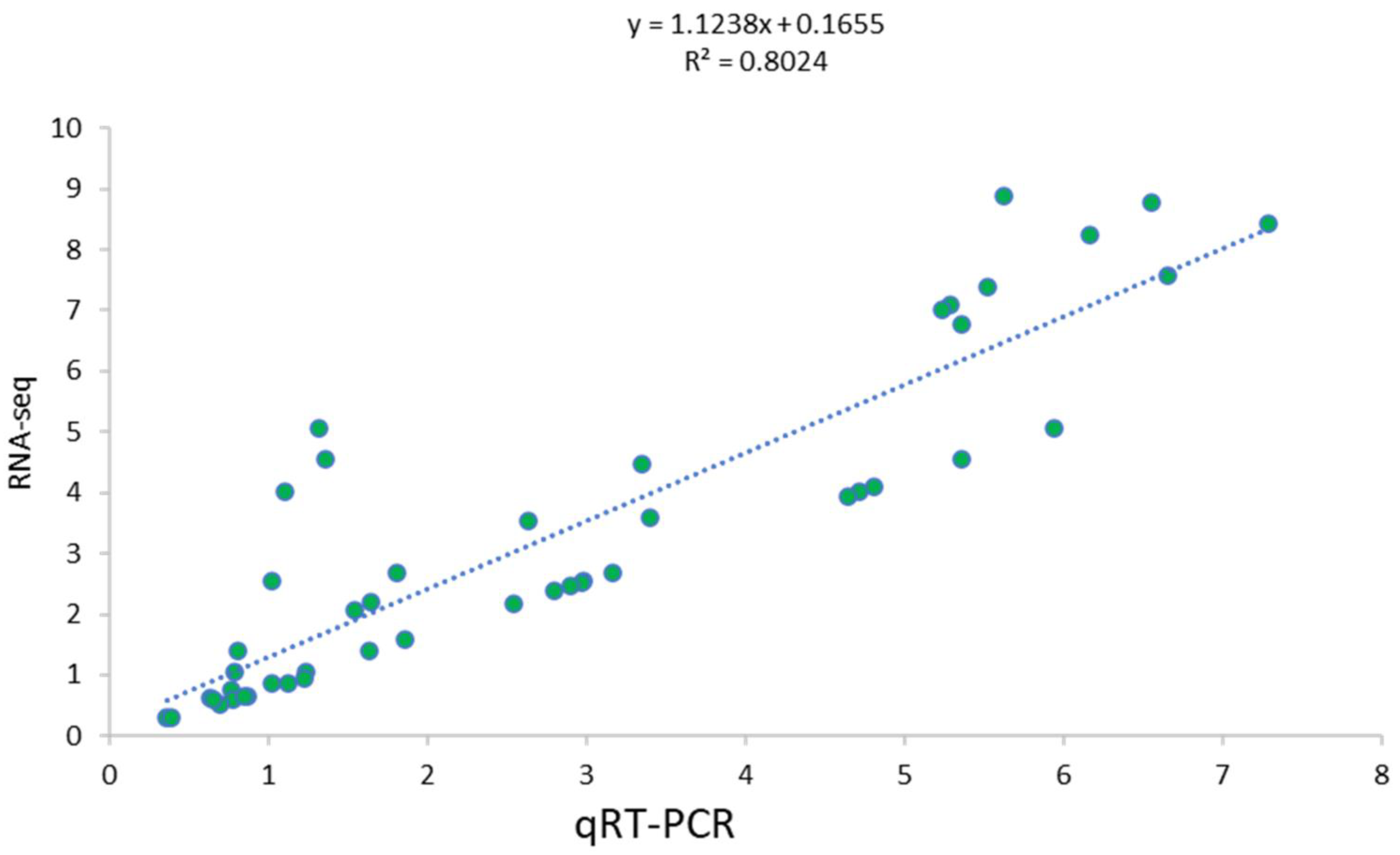
3.4. Validation Using qRT-PCR
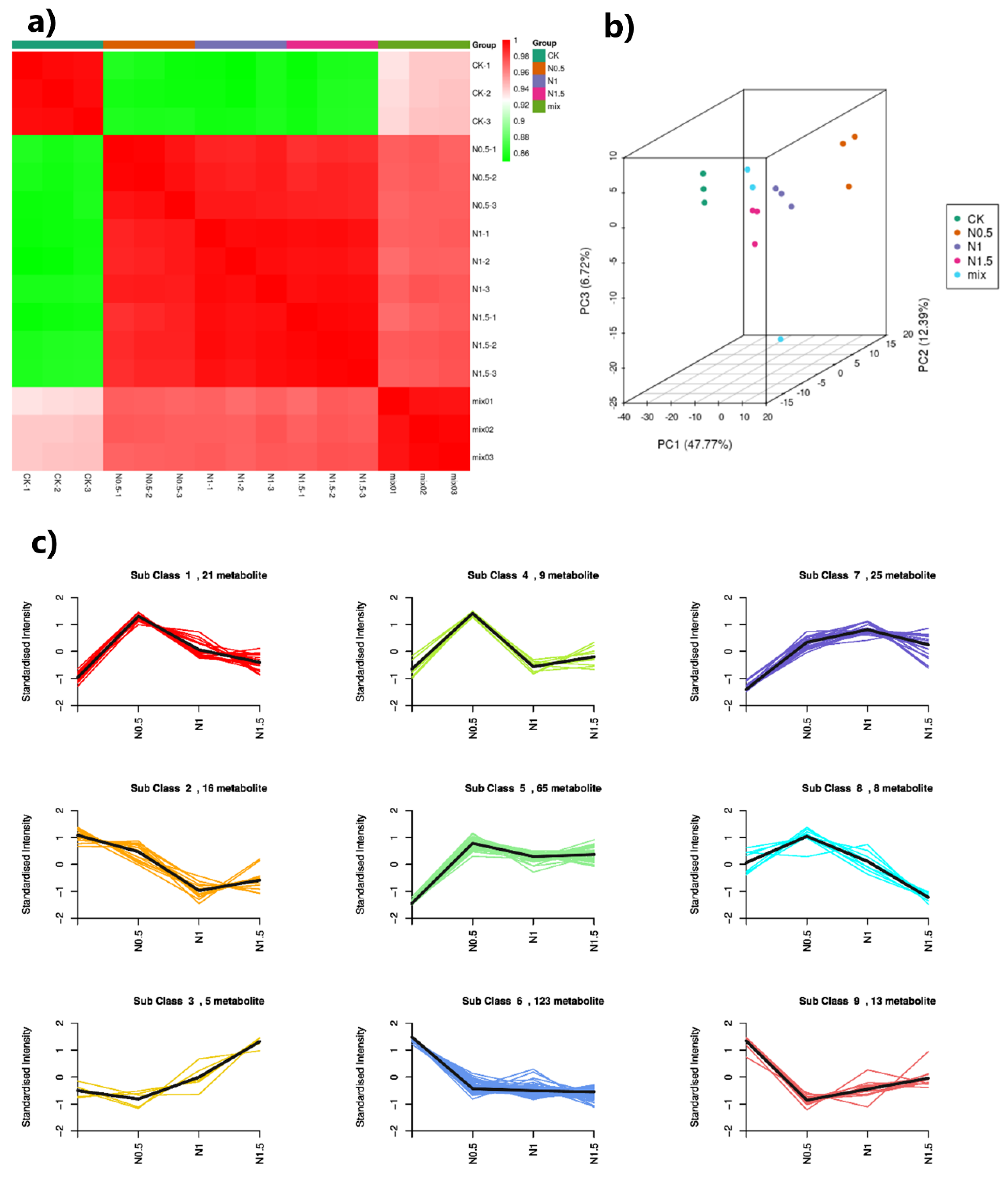
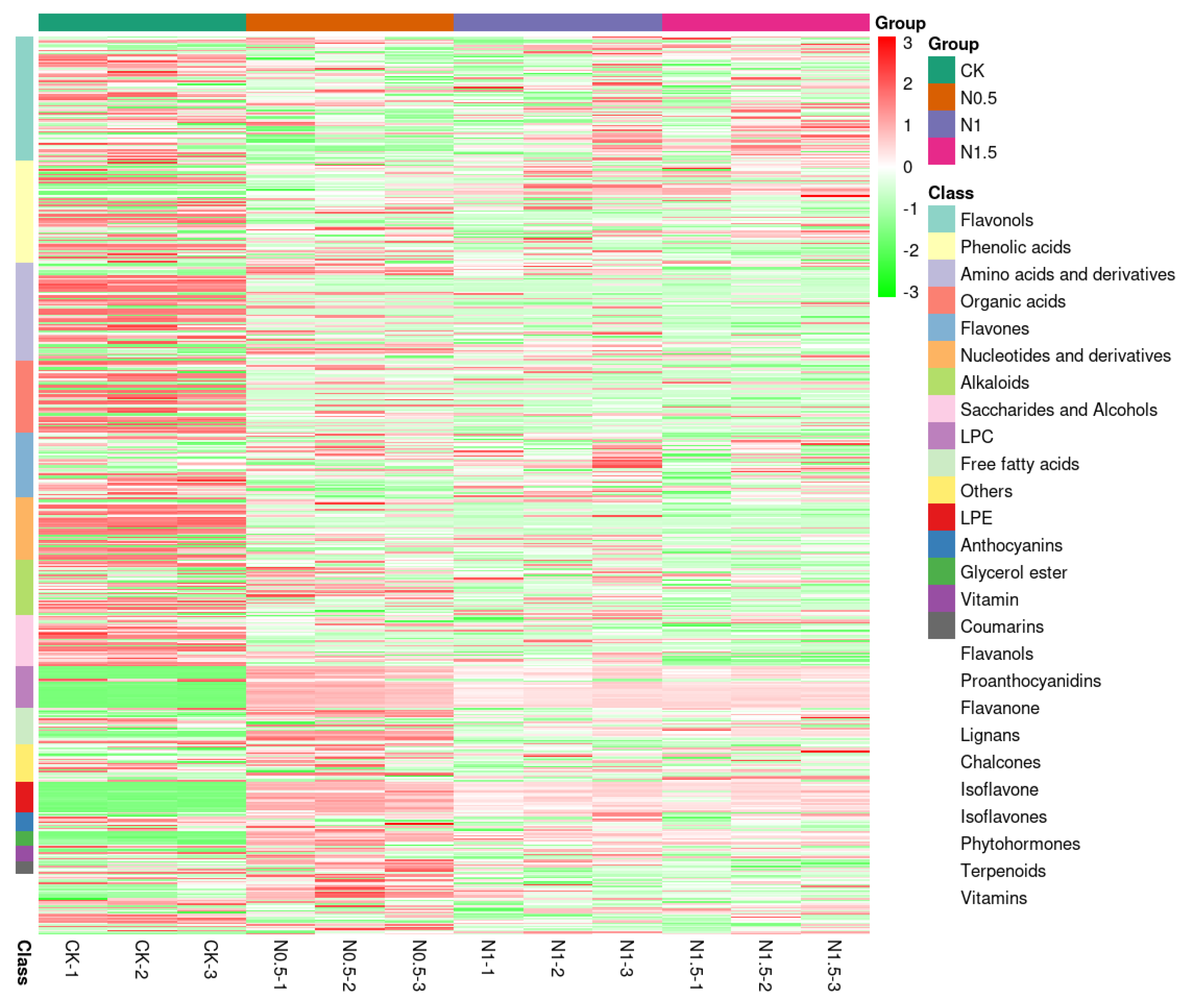
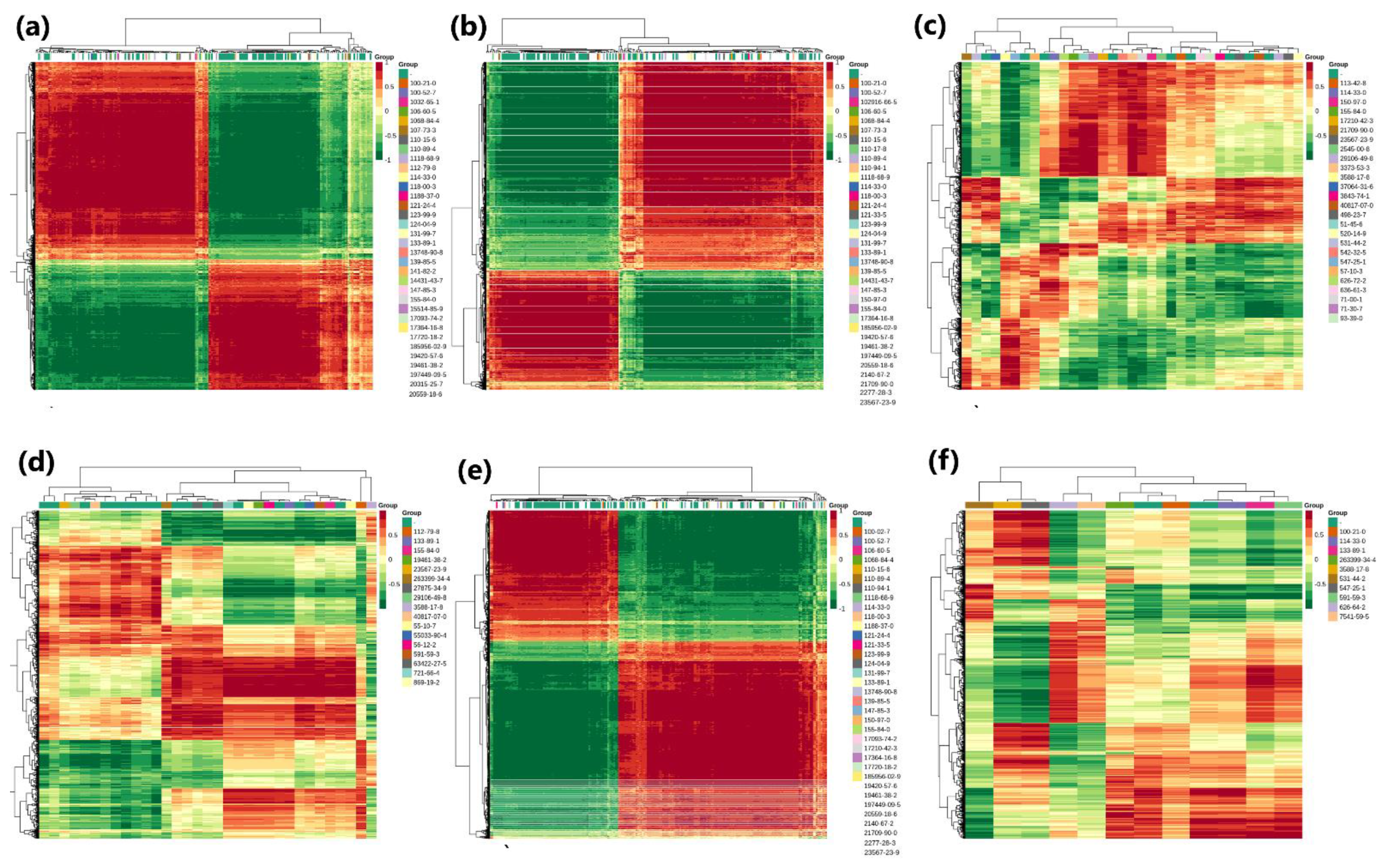
3.5. Metabolome Profiling
3.6. Combined Analysis of Transcriptome and Metabolome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhan, J.; Diao, Y.; Yin, G.; Sajjad, M.; Wei, X.; Lu, Z.; Wang, Y. Integration of mRNA and miRNA Analysis Reveals the Molecular Mechanism of Cotton Response to Salt Stress. Front. Plant Sci. 2021, 12, 767984. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Feng, J.; Ma, W.; Zhou, Y.; Ma, Z. GhCLCg-1, a vacuolar chloride channel, contributes to salt tolerance by regulating ion accumulation in upland cotton. Front. Plant Sci. 2021, 12, 765173. [Google Scholar] [CrossRef]
- Iqbal, M.M.; Khan, T.M.; Iqbal, M.S.; Khan, A.H. Estimation of genetic potential for salt tolerance in Gossypium hirsutum L. J. Agric. Res. 2013, 51, 03681157. [Google Scholar]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajihashemi, S.; Skalicky, M.; Brestic, M.; Pavla, V. Effect of sodium nitroprusside on physiological and anatomical features of salt-stressed Raphanus sativus. Plant Physiol. Biochem. 2021, 169, 160–170. [Google Scholar] [CrossRef]
- Wang, X.; An, M.; Wang, K.; Fan, H.; Shi, J.; Chen, K. Effects of Organic Polymer Compound Material on K+ and Na+ Distribution and Physiological Characteristics of Cotton under Saline and Alkaline Stresses. Front. Plant Sci. 2021, 12, 636536. [Google Scholar] [CrossRef]
- Ali, B.; Iqbal, M.S.; Shah, M.K.N.; Shabbir, G.; Cheema, N.M. Genetic analysis for various traits in Gossypium hirsutum L. Pak. J. Agric. Res. 2011, 24. [Google Scholar]
- Gupta, B.; Huang, B. Mechanism of salinity tolerance in plants: Physiological, biochemical, and molecular characterization. Int. J. Genom. 2014, 2014, 701596. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Nahar, K.; Fujita, M. Plant Response to Salt Stress and Role of Exogenous Protectants to Mitigate Salt-Induced Damages. In Ecophysiology and Responses of Plants under Salt Stress; Ahmad, P., Azooz, M.M., Prasad, M.N.V., Eds.; Springer: New York, NY, USA, 2013; pp. 25–87. [Google Scholar]
- Nahar, K.; Hasanuzzaman, M.; Fujita, M. Roles of Osmolytes in Plant Adaptation to Drought and Salinity. In Osmolytes and Plants Acclimation to Changing Environment: Emerging Omics Technologies; Iqbal, N., Nazar, R., Khan, N.A., Eds.; Springer: New Delhi, India, 2016; pp. 37–68. [Google Scholar]
- Khajeh-Hosseini, M.; Powell, A.A.; Bingham, I.J. The interaction between salinity stress and seed vigour during germination of soyabean seeds. Seed Sci. Technol. 2003, 31, 715–725. [Google Scholar] [CrossRef]
- Adams, E.; Shin, R. Transport, signaling, and homeostasis of potassium and sodium in plants. J. Integr. Plant Biol. 2014, 56, 231–249. [Google Scholar] [CrossRef]
- Kronzucker, H.J.; Britto, D.T. Sodium transport in plants: A critical review. New Phytol. 2011, 189, 54–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, Z.; Li, S.; Qanmber, G.; Liu, L.; Guo, M.; Lu, L.; Ma, S.; Li, F.; Yang, Z. Genome-wide analysis of ZAT gene family revealed GhZAT6 regulates salt stress tolerance in G. hirsutum. Plant Sci. 2021, 312, 111055. [Google Scholar] [CrossRef] [PubMed]
- Sarfraz, Z.; Shah, M.M.; Iqbal, M.S. Genetic variability, heritability and genetic advance for agronomic traits among A-genome donor wheat genotypes. J. Agric. Res. 2016, 54, 15–20. [Google Scholar]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef]
- Xu, P.; Guo, Q.; Meng, S.; Zhang, X.; Xu, Z.; Guo, W.; Shen, X. Genome-wide association analysis reveals genetic variations and candidate genes associated with salt tolerance related traits in Gossypium hirsutum. BMC Genom. 2021, 22, 26. [Google Scholar] [CrossRef]
- Liu, B.; Wang, X.; Li, K.; Cai, Z. Spatially Resolved Metabolomics and Lipidomics Reveal Salinity and Drought-Tolerant Mechanisms of Cottonseeds. J. Agric. Food Chem. 2021, 69, 8028–8037. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.S.; Tang, S.; Sarfraz, Z.; Iqbal, M.S.; Li, H.; He, S.; Jia, Y.; Sun, G.; Pan, Z.; Xiaoli, G. Genetic Factors Underlying Single Fiber Quality in A-Genome Donor Asian Cotton (Gossypium arboreum). Front. Genet. 2021, 12, 758665. [Google Scholar] [CrossRef]
- Sarfraz, Z.; Iqbal, M.S.; Geng, X.; Iqbal, M.S.; Nazir, M.F.; Ahmed, H.; He, S.; Jia, Y.; Pan, Z.; Sun, G. GWAS Mediated Elucidation of Heterosis for Metric Traits in Cotton (Gossypium hirsutum L.) Across Multiple Environments. Front. Plant Sci. 2021, 12, 566. [Google Scholar] [CrossRef]
- Uçarlı, C. Effects of salinity on seed germination and early seedling stage. Abiotic Stress Plants 2020, 211. [Google Scholar]
- Shen, J.; Chen, D.; Zhang, X.; Song, L.; Dong, J.; Xu, Q.; Hu, M.; Cheng, Y.; Shen, F.; Wang, W. Mitigation of salt stress response in upland cotton (Gossypium hirsutum) by exogenous melatonin. J. Plant Res. 2021, 134, 857–871. [Google Scholar] [CrossRef]
- Fan, R.; Su, X.; Guo, Y.; Sun, F.; Qu, Y.; Chen, Q. Cotton seedling drought tolerance is improved via salt preconditioning. Protoplasma 2021, 258, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Doğan, İ.; Özyiğit, İ.İ.; Demir, G. Mineral element distribution of cotton (Gossypium hirsutum L.) seedlings under different salinity levels. Pak. J. Bot. 2012, 44, 15–20. [Google Scholar]
- Liu, L.; Wang, B.; Liu, D.; Zou, C.; Wu, P.; Wang, Z.; Wang, Y.; Li, C. Transcriptomic and metabolomic analyses reveal mechanisms of adaptation to salinity in which carbon and nitrogen metabolism is altered in sugar beet roots. BMC Plant Biol. 2020, 20, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.A.; Iqbal, M.S.; Riaz, M.; Mahmood, A.; Shahid, M.R.; Abbas, G.; Farooq, J. Cause and effect estimates for yield contributing and morphological traits in upland cotton (Gossypium hirsutum L.). J. Agric. Res. 2013, 51, 393–398. [Google Scholar]
- Wang, W.; Li, W.; Cheng, Z.; Sun, J.; Gao, J.; Li, J.; Niu, X.; Amjid, M.W.; Yang, H.; Zhu, G.; et al. Transcriptome-wide N6-methyladenosine profiling of cotton root provides insights for salt stress tolerance. Environ. Exp. Bot. 2022, 194, 104729. [Google Scholar] [CrossRef]
- Xue, T.; Li, X.; Zhu, W.; Wu, C.; Yang, G.; Zheng, C. Cotton metallothionein GhMT3a, a reactive oxygen species scavenger, increased tolerance against abiotic stress in transgenic tobacco and yeast. J. Exp. Bot. 2009, 60, 339–349. [Google Scholar] [CrossRef]
- Guo, Y.H.; Yu, Y.P.; Wang, D.; Wu, C.A.; Yang, G.D.; Huang, J.G.; Zheng, C.C. GhZFP1, a novel CCCH-type zinc finger protein from cotton, enhances salt stress tolerance and fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5. New Phytol. 2009, 183, 62–75. [Google Scholar] [CrossRef]
- Zhao, J.; Gao, Y.; Zhang, Z.; Chen, T.; Guo, W.; Zhang, T. A receptor-like kinase gene (GbRLK) from Gossypium barbadense enhances salinity and drought-stress tolerance in Arabidopsis. BMC Plant Biol. 2013, 13, 110. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.-Q.; Chen, M.; Xia, L.-Q.; Xiu, H.-J.; Xu, Z.-S.; Li, L.-C.; Zhao, C.-P.; Cheng, X.-G.; Ma, Y.-Z. A cotton (Gossypium hirsutum) DRE-binding transcription factor gene, GhDREB, confers enhanced tolerance to drought, high salt, and freezing stresses in transgenic wheat. Plant Cell Rep. 2009, 28, 301–311. [Google Scholar] [CrossRef]
- Chu, X.; Wang, C.; Chen, X.; Lu, W.; Li, H.; Wang, X.; Hao, L.; Guo, X. The Cotton WRKY Gene GhWRKY41 Positively Regulates Salt and Drought Stress Tolerance in Transgenic Nicotiana benthamiana. PLoS ONE 2015, 10, e0143022. [Google Scholar] [CrossRef]
- Yan, H.; Jia, H.; Chen, X.; Hao, L.; An, H.; Guo, X. The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production. Plant Cell Physiol. 2014, 55, 2060–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.-Q.; Li, W.; Zhou, W.; Zhang, J.-M.; Li, D.-D.; Gong, S.-Y.; Li, X.-B. Seven cotton genes encoding putative NAC domain proteins are preferentially expressed in roots and in responses to abiotic stress during root development. Plant Growth Regul. 2013, 71, 101–112. [Google Scholar] [CrossRef]
- Meng, C.; Cai, C.; Zhang, T.; Guo, W. Characterization of six novel NAC genes and their responses to abiotic stresses in Gossypium hirsutum L. Plant Sci. 2009, 176, 352–359. [Google Scholar] [CrossRef]
- Yasir, M.; He, S.; Sun, G.; Geng, X.; Pan, Z.; Gong, W.; Jia, Y.; Du, X. A Genome-Wide Association Study Revealed Key SNPs/Genes Associated with Salinity Stress Tolerance In Upland Cotton. Genes 2019, 10, 829. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Xing, H.; Zeng, W.; Xu, J.; Mao, L.; Wang, L.; Feng, W.; Tao, J.; Wang, H.; Zhang, H.; et al. Genome-wide association and differential expression analysis of salt tolerance in Gossypium hirsutum L. at the germination stage. BMC Plant Biol. 2019, 19, 394. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Magwanga, R.O.; Yang, X.; Jin, D.; Cai, X.; Hou, Y.; Wei, Y.; Zhou, Z.; Wang, K.; Liu, F. Genetic regulatory networks for salt-alkali stress in Gossypium hirsutum with differing morphological characteristics. BMC Genom. 2020, 21, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Shi, G.; Guo, X.; Zhang, L.; Xu, W.; Wang, Y.; Su, Z.; Hua, J. Transcriptome analysis reveals that distinct metabolic pathways operate in salt-tolerant and salt-sensitive upland cotton varieties subjected to salinity stress. Plant Sci. 2015, 238, 33–45. [Google Scholar] [CrossRef]
- Zhang, L.; Xi, D.; Li, S.; Gao, Z.; Zhao, S.; Shi, J.; Wu, C.; Guo, X. A cotton group C MAP kinase gene, GhMPK2, positively regulates salt and drought tolerance in tobacco. Plant Mol. Biol. 2011, 77, 17–31. [Google Scholar] [CrossRef]
- Slonim, D.K.; Yanai, I. Getting started in gene expression microarray analysis. PLoS Comput. Biol. 2009, 5, e1000543. [Google Scholar] [CrossRef]
- Ari, Ş.; Arikan, M. Next-Generation Sequencing: Advantages, Disadvantages, and Future. In Plant Omics: Trends and Applications; Hakeem, K.R., Tombuloğlu, H., Tombuloğlu, G., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 109–135. [Google Scholar]
- Peng, W.; Song, N.; Li, W.; Yan, M.; Huang, C.; Yang, Y.; Duan, K.; Dai, L.; Wang, B. Integrated Analysis of MicroRNA and Target Genes in Brachypodium distachyon Infected by Magnaporthe oryzae by Small RNA and Degradome Sequencing. Front. Plant Sci. 2021, 12, 742347. [Google Scholar] [CrossRef]
- Singh, R.; Kumar, K.; Bharadwaj, C.; Verma, P.K. Broadening the horizon of crop research: A decade of advancements in plant molecular genetics to divulge phenotype governing genes. Planta 2022, 255, 46. [Google Scholar] [CrossRef] [PubMed]
- Kowal, E.; Llamas, B. Race in a genome: Long read sequencing, ethnicity-specific reference genomes and the shifting horizon of race. J. Anthropol. Sci. 2019, 97, 91–106. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. STRING v10: Protein—Protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.; Zhang, H.; Liu, X.; Yu, S.; Xiong, L.; Luo, J. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Xia, J. MetaboAnalystR: An R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 2018, 34, 4313–4314. [Google Scholar] [CrossRef] [Green Version]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Hejnák, V.; Tatar, Ö.; Atasoy, G.D.; Martinková, J.; Çelen, A.E.; Hnilička, F.; Skalický, M. Growth and photosynthesis of Upland and Pima cotton: Response to drought and heat stress. Plant Soil Environ. 2015, 61, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Guo, Y. Unraveling salt stress signaling in plants. J. Integr. Plant Biol. 2018, 60, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.; Ma, X.; Wan, P.; Liu, L. Plant salt-tolerance mechanism: A review. Biochem. Biophys. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.T.; Ma, S.L.; Bai, L.P.; Zhang, L.; Ma, H.; Jia, P.; Liu, J.; Zhong, M.; Guo, Z.F. Signal transduction during cold, salt, and drought stresses in plants. Mol. Biol. Rep. 2012, 39, 969–987. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Bressan, R.A.; Hasegawa, P.M.; Pardo, J.M. Ion Homeostasis in NaCl Stress Environments. Plant Physiol. 1995, 109, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef]
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055. [Google Scholar] [CrossRef] [Green Version]
- Koornneef, M.; Bentsink, L.; Hilhorst, H. Seed dormancy and germination. Curr. Opin. Plant Biol. 2002, 5, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Nonogaki, H.; Bassel, G.W.; Bewley, J.D. Germination—Still a mystery. Plant Sci. 2010, 179, 574–581. [Google Scholar] [CrossRef]
- An, Y.-Q.; Lin, L. Transcriptional regulatory programs underlying barley germination and regulatory functions of gibberellin and abscisic acid. BMC Plant Biol. 2011, 11, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Lu, S. Biosynthesis and regulation of phenylpropanoids in plants. Crit. Rev. Plant Sci. 2017, 36, 257–290. [Google Scholar] [CrossRef]
- Khan, M.S.; Khan, M.N. Understanding Plant Responses to Drought and Salt Stresses: Advances and Challenges in “Omics” Approaches. In Transgenic Crops-Emerging Trends and Future Perspectives; IntechOpen: London, UK, 2019. [Google Scholar]
- Wang, Y.; Huang, L.; Du, F.; Wang, J.; Zhao, X.; Li, Z.; Wang, W.; Xu, J.; Fu, B. Comparative transcriptome and metabolome profiling reveal molecular mechanisms underlying OsDRAP1-mediated salt tolerance in rice. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Tu, W.; Mu, L.; Sun, Z.; Hu, Q.; Yang, Y. Saline alkali water desalination project in Southern Xinjiang of China: A review of desalination planning, desalination schemes and economic analysis. Renew. Sustain. Energy Rev. 2019, 113, 109268. [Google Scholar] [CrossRef]
- Liang, J.; Li, Y.; Si, B.; Wang, Y.; Chen, X.; Wang, X.; Chen, H.; Wang, H.; Zhang, F.; Bai, Y. Optimizing biochar application to improve soil physical and hydraulic properties in saline-alkali soils. Sci. Total Environ. 2021, 771, 144802. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Kutueva, L.I.; Aleksandrushkina, N.I.; Vanyushin, B.F. Epigenetic Mechanisms of Plant Adaptation to Biotic and Abiotic Stresses. Int. J. Mol. Sci. 2020, 21, 7457. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, Q.; Wang, X.; Mei, J.; Sharma, A.; Tripathi, D.K.; Yuan, H.; Zheng, B. Genome-wide identification and expression profiles of ABCB gene family in Chinese hickory (Carya cathayensis Sarg.) during grafting. Plant Physiol. Biochem. 2021, 168, 477–487. [Google Scholar] [CrossRef]
- Rushton, P.J.; Somssich, I.E.; Ringler, P.; Shen, Q.J. WRKY transcription factors. Trends Plant Sci. 2010, 15, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, Y.; Li, S.; Zhang, L.; Zou, C.; Yu, D. The role of WRKY transcription factors in plant abiotic stresses. Biochim. Biophys. Acta 2012, 1819, 120–128. [Google Scholar] [CrossRef]
- Yokoi, S.; Quintero, F.J.; Cubero, B.; Ruiz, M.T.; Bressan, R.A.; Hasegawa, P.M.; Pardo, J.M. Differential expression and function of Arabidopsis thaliana NHX Na+/H+ antiporters in the salt stress response. Plant J. 2002, 30, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Apse, M.P.; Aharon, G.S.; Snedden, W.A.; Blumwald, E. Salt tolerance conferred by overexpression of a vacuolar Na+/H+ antiport in Arabidopsis. Science 1999, 285, 1256–1258. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [Green Version]
- Zirek, N.S.; Ozlem, U. The developmental and metabolic effects of different magnesium dozes in pepper plants under salt stress. Not. Bot. Horti Agrobot. Cluj-Napoca 2020, 48, 967–977. [Google Scholar] [CrossRef]
- Chini, A.; Fonseca, S.; Fernández, G.; Adie, B.; Chico, J.M.; Lorenzo, O.; García-Casado, G.; López-Vidriero, I.; Lozano, F.M.; Ponce, M.R.; et al. The JAZ family of repressors is the missing link in jasmonate signalling. Nature 2007, 448, 666–671. [Google Scholar] [CrossRef]
- Figueroa, P.; Browse, J. The Arabidopsis JAZ2 promoter contains a G-Box and thymidine-rich module that are necessary and sufficient for jasmonate-dependent activation by MYC transcription factors and repression by JAZ proteins. Plant Cell Physiol. 2012, 53, 330–343. [Google Scholar] [CrossRef] [Green Version]
- Shu, J.; Ma, X.; Ma, H.; Huang, Q.; Zhang, Y.; Guan, M.; Guan, C. Transcriptomic, proteomic, metabolomic, and functional genomic approaches of Brassica napus L. during salt stress. PLoS ONE 2022, 17, e0262587. [Google Scholar] [CrossRef]
- Holden, H.M.; Rayment, I.; Thoden, J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem. 2003, 278, 43885–43888. [Google Scholar] [CrossRef] [Green Version]
- Verbert, A. Biosynthesis 2b. from Glc3Man9GlcNAc2-protein to Man5GlcNAc2-protein: Transfer ‘en bloc’and processing. In New Comprehensive Biochemistry; Elsevier: Amsterdam, The Netherlands, 1995; Volume 29, pp. 145–152. [Google Scholar]
- Sandhoff, K.; Van Echten, G.; Schröder, M.; Schnabel, D.; Suzuki, K. Metabolism of glycolipids: The role of glycolipid-binding proteins in the function and pathobiochemistry of lysosomes. Biochem. Soc. Trans. 1992, 20, 695–699. [Google Scholar] [CrossRef]
- Mollerach, M.; García, E. The galU gene of Streptococcus pneumoniae that codes for a UDP–glucose pyrophosphorylase is highly polymorphic and suitable for molecular typing and phylogenetic studies. Gene 2000, 260, 77–86. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, Y.; Liu, J.; Lv, F.; Chen, J.; Zhou, Z. The effects of fruiting positions on cellulose synthesis and sucrose metabolism during cotton (Gossypium hirsutum L.) fiber development. PLoS ONE 2014, 9, e89476. [Google Scholar] [CrossRef]
- Delmer, D.P.; Haigler, C.H. The regulation of metabolic flux to cellulose, a major sink for carbon in plants. Metab. Eng. 2002, 4, 22–28. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, L.; Liu, J.; Luo, J.; Zhao, X.; Dong, H.; Ma, Y.; Sui, N.; Zhou, Z.; Meng, Y. Effects of Soil Salinity on Sucrose Metabolism in Cotton Fiber. PLoS ONE 2016, 11, e0156398. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, W.; Wang, Q.; Chen, L.; Ren, Y. Transcriptome and Metabolome Analyses of Salt Stress Response in Cotton (Gossypium hirsutum) Seed Pretreated with NaCl. Agronomy 2022, 12, 1849. https://doi.org/10.3390/agronomy12081849
Ren W, Wang Q, Chen L, Ren Y. Transcriptome and Metabolome Analyses of Salt Stress Response in Cotton (Gossypium hirsutum) Seed Pretreated with NaCl. Agronomy. 2022; 12(8):1849. https://doi.org/10.3390/agronomy12081849
Chicago/Turabian StyleRen, Wei, Qian Wang, Li Chen, and Yanping Ren. 2022. "Transcriptome and Metabolome Analyses of Salt Stress Response in Cotton (Gossypium hirsutum) Seed Pretreated with NaCl" Agronomy 12, no. 8: 1849. https://doi.org/10.3390/agronomy12081849
APA StyleRen, W., Wang, Q., Chen, L., & Ren, Y. (2022). Transcriptome and Metabolome Analyses of Salt Stress Response in Cotton (Gossypium hirsutum) Seed Pretreated with NaCl. Agronomy, 12(8), 1849. https://doi.org/10.3390/agronomy12081849