
Multi-Species Transcriptome Assemblies of Cultivated and Wild Lentils (Lens sp.) Provide a First Glimpse at the Lentil Pangenome

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Transcriptome Assemblies and Qualities
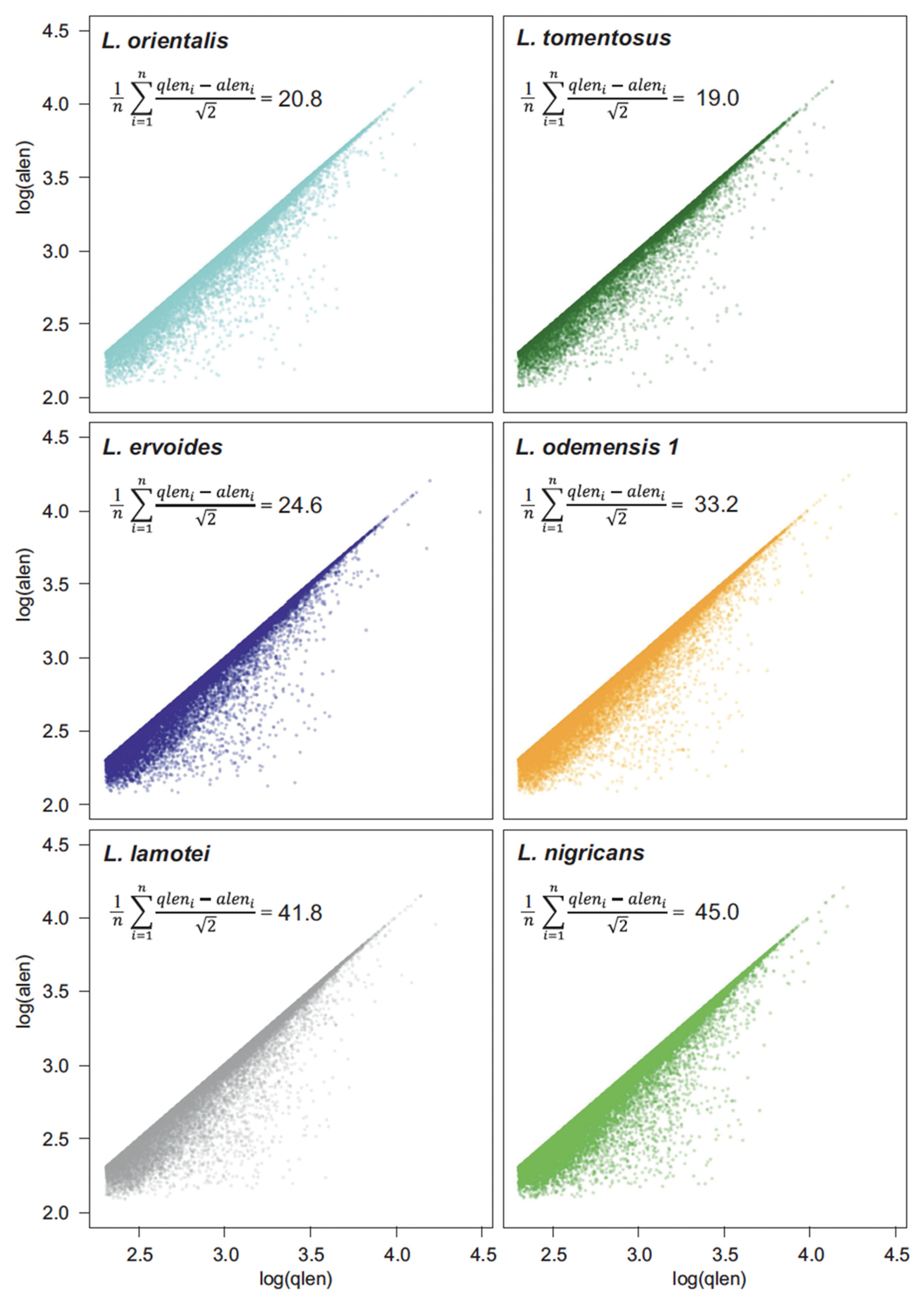
2.2. Comparison among Transcriptomes and with the Reference
2.3. Transcriptome Annotation and Multi-Species Pan-Transcriptome
3. Discussion
3.1. Transcriptome Quality Assessment
3.2. Divergency between Transcriptomes and the Lentil Reference Genome
3.3. First Glimpse at the Lentil Pangenome
4. Materials and Methods
4.1. Plant Material
4.2. RNA Extraction, Library Construction, and Sequencing
4.3. Transcript Assemblies and Quality Parameter Estimation
4.4. Transcript Abundance Estimation
4.5. Transcriptome Annotation and Multi-Species Pan-Transcriptome
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perez de la Vega, M.; Fratini, R.; Muehlbauer, F. Lentil. In Genetics, Genomics and Breeding of Cool Season Grain Legumes; Perez de la Vega, M., Torres, A.M., Cubero, J.I., Kole, C., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 98–150. [Google Scholar]
- Pérez de la Vega, M.; García García, P.; Gutierrez-Gonzalez, J.J.; Sáenz de Miera, L.E. Tackling Lentil Biotic Stresses in the Genomic Era. In Genomic Designing for Biotic Stress Resistant Pulse Crops; Kole, C., Ed.; Springer: Cham, Switzerland, 2022. [Google Scholar] [CrossRef]
- FAOSTAT 2019. Available online: https://www.fao.org/faostat (accessed on 15 March 2021).
- Liber, M.; Duarte, I.; Maia, A.T.; Oliveira, H.R. The History of Lentil (Lens culinaris subsp. Culinaris) Domestication and Spread as Revealed by Genotyping-by-Sequencing of Wild and Landrace Accessions. Front. Plant Sci. 2021, 12, 628439. [Google Scholar] [CrossRef]
- Koul, P.M.; Sharma, V.; Rana, M.; Chahota, R.K.; Kumar, S.; Sharma, T.R. Analysis of genetic structure and interrelationships in lentil species using morphological and SSR markers. 3 Biotech 2017, 7, 83. [Google Scholar] [CrossRef]
- Wong, M.M.L.; Gujaria-Verma, N.; Ramsay, L.; Yuan, H.Y.; Caron, C.; Diapari, M.; Vandenberg, A.; Bett, K.E. Classification and Characterization of Species within the Genus Lens Using Genotyping-by-Sequencing (GBS). PloS ONE 2015, 10, e0122025. [Google Scholar] [CrossRef]
- Kumar, H.; Singh, A.; Dikshit, H.K.; Mishra, G.P.; Aski, M.; Meena, M.C.; Kumar, S. Genetic dissection of grain iron and zinc concentrations in lentil (Lens culinaris Medik.). J. Genet. 2019, 98, 66. [Google Scholar] [CrossRef]
- Singh, D.; Singh, C.K.; Taunk, J.; Tomar, R.S.S.; Chaturvedi, A.K.; Gaikwad, K.; Pal, M. Transcriptome analysis of lentil (Lens culinaris Medikus) in response to seedling drought stress. BMC Genom. 2017, 18, 206. [Google Scholar] [CrossRef]
- Ramsay, L.; Koh, C.; Konkin, D.; Cook, D.; Penmetsa, V.; Dongying, G.; Coyne, C.; Humann, J.; Kaur, S.; Dolezel, J.; et al. Lens culinaris CDC Redberry Genome Assembly v2.0. 2019. Available online: https://knowpulse.usask.ca/genome-assembly/Lcu.2RBY (accessed on 7 April 2021).
- Ramsay, L.; Koh, C.S.; Kagale, S.; Gao, D.; Kaur, S.; Haile, T.; Gela, T.S.; Chen, L.-A.; Cao, Z.; Konkin, D.J.; et al. Genomic rearrangements have consequences for introgression breeding as revealed by genome assemblies of wild and cultivated lentil species. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gorim, L.Y.; Vandenberg, A. Evaluation of Wild Lentil Species as Genetic Resources to Improve Drought Tolerance in Cultivated Lentil. Front. Plant Sci. 2017, 8, 1129. [Google Scholar] [CrossRef]
- Dissanayake, R.; Braich, S.; Cogan, N.O.I.; Smith, K.; Kaur, S. Characterization of Genetic and Allelic Diversity Amongst Cultivated and Wild Lentil Accessions for Germplasm Enhancement. Front. Genet. 2020, 11, 546. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, J.J.; Garvin, D.F. Subgenome-specific assembly of vitamin E biosynthesis genes and expression patterns during seed development provide insight into the evolution of oat genome. Plant Biotechnol. J. 2016, 14, 2147–2157. [Google Scholar] [CrossRef]
- Hu, H.; Gutierrez-Gonzalez, J.J.; Liu, X.; Yeats, T.H.; Garvin, D.F.; Hoekenga, O.A.; Sorrells, M.E.; Gore, M.A.; Jannink, J.-L. Heritable temporal gene expression patterns correlate with metabolomic seed content in developing hexaploid oat seed. Plant Biotechnol. J. 2020, 18, 1211–1222. [Google Scholar] [CrossRef]
- Khorramdelazad, M.; Bar, I.; Whatmore, P.; Smetham, G.; Bhaskarla, V.; Yang, Y.; Bai, S.H.; Mantri, N.; Zhou, Y.; Ford, R. Transcriptome profiling of lentil (Lens culinaris) through the first 24 hours of Ascochyta lentis infection reveals key defence response genes. BMC Genom. 2018, 19, 108. [Google Scholar] [CrossRef]
- Mishra, G.P.; Aski, M.S.; Bosamia, T.; Chaurasia, S.; Mishra, D.C.; Bhati, J.; Kumar, A.; Javeria, S.; Tripathi, K.; Kohli, M.; et al. Insights into the Host-Pathogen Interaction Pathways through RNA-Seq Analysis of Lens culinaris Medik. in Response to Rhizoctonia bataticola Infection. Genes 2022, 13, 90. [Google Scholar] [CrossRef]
- Singh, D.; Singh, C.K.; Taunk, J.; Jadon, V.; Pal, M.; Gaikwad, K. Genome wide transcriptome analysis reveals vital role of heat responsive genes in regulatory mechanisms of lentil (Lens culinaris Medikus). Sci. Rep. 2019, 9, 12976. [Google Scholar] [CrossRef]
- Sohrabi, S.S.; Ismaili, A.; Nazarian-Firouzabadi, F.; Fallahi, H.; Hosseini, S.Z. Identification of key genes and molecular mechanisms associated with temperature stress in lentil. Gene 2022, 807, 145952. [Google Scholar] [CrossRef]
- Morgil, H.; Tardu, M.; Cevahir, G.; Kavakli, I.H. Comparative RNA-seq analysis of the drought-sensitive lentil (Lens culinaris) root and leaf under short- and long-term water deficits. Funct. Integr. Genom. 2019, 19, 715–727. [Google Scholar] [CrossRef]
- Singh, C.K.; Singh, D.; Taunk, J.; Chaudhary, P.; Tomar, R.S.S.; Chandra, S.; Singh, D.; Pal, M.; Konjengbam, N.S.; Singh, M.P.; et al. Comparative Inter- and IntraSpecies Transcriptomics Revealed Key Differential Pathways Associated with Aluminium Stress Tolerance in Lentil. Front. Plant Sci. 2021, 12, 693630. [Google Scholar] [CrossRef]
- García-García, P.; Vaquero, F.; Vences, F.J.; De Miera, L.E.S.; Polanco, C.; González, A.I.; Horres, R.; Krezdorn, N.; Rotter, B.; Winter, P.; et al. Transcriptome profiling of lentil in response to Ascochyta lentis infection. Span. J. Agric. Res. 2019, 17, e0703. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Cantalapiedra, C.P.; García-Pereira, M.J.; Gordon, S.P.; Vogel, J.P.; Igartua, E.; Casas, A.M.; Vinuesa, P. Analysis of Plant Pan-Genomes and Transcriptomes with GET_HOMOLOGUES-EST, a Clustering Solution for Sequences of the Same Species. Front. Plant Sci. 2017, 8, 184. [Google Scholar] [CrossRef]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-Pangenome by Integrating the Wild Side of a Species for Accelerated Crop Improvement. Trends Plant Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Multiple wheat genomes reveal global variation in modern breeding. Nature 2020, 588, 277–283. [Google Scholar] [CrossRef]
- Gao, L.; Gonda, I.; Sun, H.; Ma, Q.; Bao, K.; Tieman, D.M.; Burzynski-Chang, E.A.; Fish, T.L.; Stromberg, K.A.; Sacks, G.L.; et al. The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 2019, 51, 1044–1051. [Google Scholar] [CrossRef]
- Li, Y.-H.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.-G.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.; et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef]
- Golicz, A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef]
- Gusev, A.; Ko, A.; Shi, H.; Bhatia, G.; Chung, W.; Penninx, B.W.J.H.; Jansen, R.; de Geus, E.J.C.; I Boomsma, D.; Wright, F.A.; et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 2016, 48, 245–252. [Google Scholar] [CrossRef]
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Peñagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the Maize Pan-Genome and Pan-Transcriptome. Plant Cell 2014, 26, 121–135. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; A Simão, F.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, J.J.; Tu, Z.J.; Garvin, D.F. Analysis and annotation of the hexaploid oat seed transcriptome. BMC Genom. 2013, 14, 471. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef]
- Davidson, N.M.; Hawkins, A.D.K.; Oshlack, A. SuperTranscripts: A data driven reference for analysis and visualisation of transcriptomes. Genome Biol. 2017, 18, 148. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, J.J.; Garvin, D.F. Reference Genome-Directed Resolution of Homologous and Homeologous Relationships within and between Different Oat Linkage Maps. Plant Genome 2011, 4, 178–190. [Google Scholar] [CrossRef]
- Li, G.; Serba, D.D.; Saha, M.C.; Bouton, J.H.; Lanzatella, C.L.; Tobias, C.M. Genetic Linkage Mapping and Transmission Ratio Distortion in a Three-Generation Four-Founder Population of Panicum virgatum (L.). G3 Genes|Genomes|Genet. 2014, 4, 913–923. [Google Scholar] [CrossRef][Green Version]
- Bhadauria, V.; Ramsay, L.; Bett, K.E.; Banniza, S. QTL mapping reveals genetic determinants of fungal disease resistance in the wild lentil species Lens ervoides. Sci. Rep. 2017, 7, 3231. [Google Scholar] [CrossRef]
- Polanco, C.; de Miera, L.E.S.; González, A.I.; García, P.G.; Fratini, R.; Vaquero, F.; Vences, F.J.; De La Vega, M.P. Construction of a high-density interspecific (Lens culinaris × L. odemensis) genetic map based on functional markers for mapping morphological and agronomical traits, and QTLs affecting resistance to Ascochyta in lentil. PLoS ONE 2019, 14, e0214409. [Google Scholar] [CrossRef]
- Vinuesa, P.; Contreras-Moreira, B. Robust Identification of Orthologues and Paralogues for Microbial Pan-Genomics Using GET_HOMOLOGUES: A Case Study of pIncA/C Plasmids. In Bacterial Pangenomics; Mengoni, A., Galardini, M., Fondi, M., Eds.; Humana Press: New York, NY, USA, 2015; Volume 1231, pp. 203–232. [Google Scholar] [CrossRef]
- Marroni, F.; Pinosio, S.; Morgante, M. Structural variation and genome complexity: Is dispensable really dispensable? Curr. Opin. Plant Biol. 2014, 18, 31–36. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010; Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 15 March 2021).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Thierry-Mieg, J.; Thierry-Mieg, D.; Busby, B.; Madden, T.L. Magic-BLAST, an accurate RNA-seq aligner for long and short reads. BMC Bioinform. 2019, 20, 405. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 15 April 2021).
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome Analysis of Multiple Pathogenic Isolates of Streptococcus agalactiae: Implications for the Microbial ‘pan-genome’. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Vinuesa, P.; Ochoa-Sánchez, L.E.; Contreras-Moreira, B. GET_PHYLOMARKERS, a Software Package to Select Optimal Orthologous Clusters for Phylogenomics and Inferring Pan-Genome Phylogenies, Used for a Critical Geno-Taxonomic Revision of the Genus Stenotrophomonas. Front. Microbiol. 2018, 9, 771. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutierrez-Gonzalez, J.J.; García, P.; Polanco, C.; González, A.I.; Vaquero, F.; Vences, F.J.; Pérez de la Vega, M.; Sáenz de Miera, L.E. Multi-Species Transcriptome Assemblies of Cultivated and Wild Lentils (Lens sp.) Provide a First Glimpse at the Lentil Pangenome. Agronomy 2022, 12, 1619. https://doi.org/10.3390/agronomy12071619
Gutierrez-Gonzalez JJ, García P, Polanco C, González AI, Vaquero F, Vences FJ, Pérez de la Vega M, Sáenz de Miera LE. Multi-Species Transcriptome Assemblies of Cultivated and Wild Lentils (Lens sp.) Provide a First Glimpse at the Lentil Pangenome. Agronomy. 2022; 12(7):1619. https://doi.org/10.3390/agronomy12071619
Chicago/Turabian StyleGutierrez-Gonzalez, Juan J., Pedro García, Carlos Polanco, Ana Isabel González, Francisca Vaquero, Francisco Javier Vences, Marcelino Pérez de la Vega, and Luis E. Sáenz de Miera. 2022. "Multi-Species Transcriptome Assemblies of Cultivated and Wild Lentils (Lens sp.) Provide a First Glimpse at the Lentil Pangenome" Agronomy 12, no. 7: 1619. https://doi.org/10.3390/agronomy12071619
APA StyleGutierrez-Gonzalez, J. J., García, P., Polanco, C., González, A. I., Vaquero, F., Vences, F. J., Pérez de la Vega, M., & Sáenz de Miera, L. E. (2022). Multi-Species Transcriptome Assemblies of Cultivated and Wild Lentils (Lens sp.) Provide a First Glimpse at the Lentil Pangenome. Agronomy, 12(7), 1619. https://doi.org/10.3390/agronomy12071619