
Genome-Wide Identification and Expression Analysis of Cytosine DNA Methyltransferase Genes Related to Somaclonal Variation in Pineapple (Ananas comosus L.)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Analysis of Methylation-Sensitive Amplification Polymorphism (MSAP)
2.3. Genome-Wide Identification of Cytosine DNA Methyltransferase Genes in Pineapple
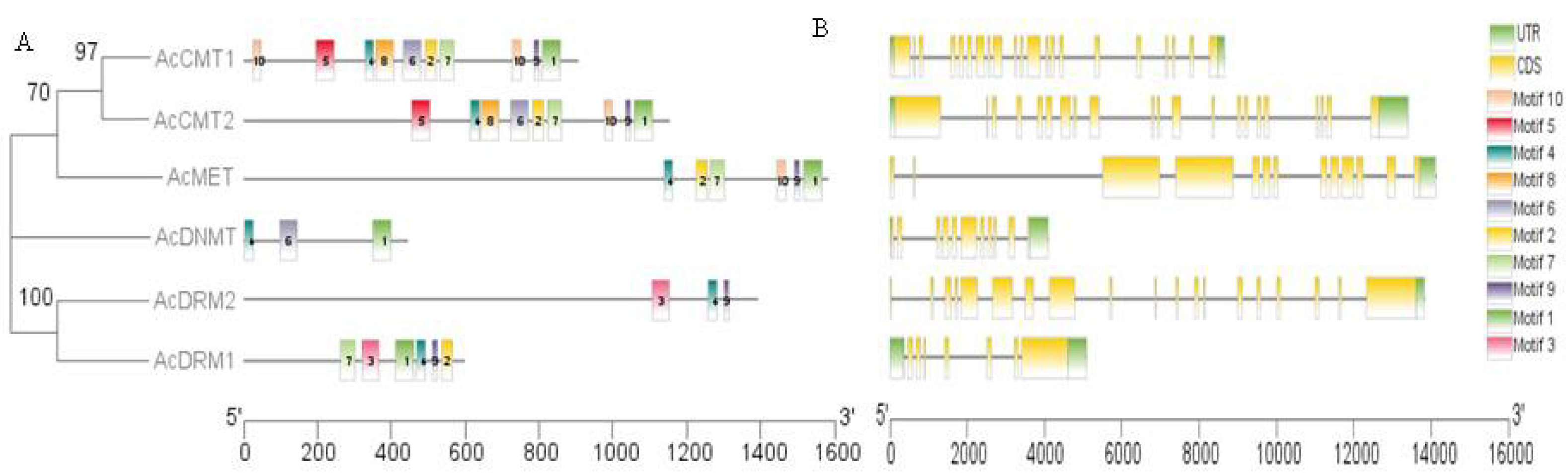
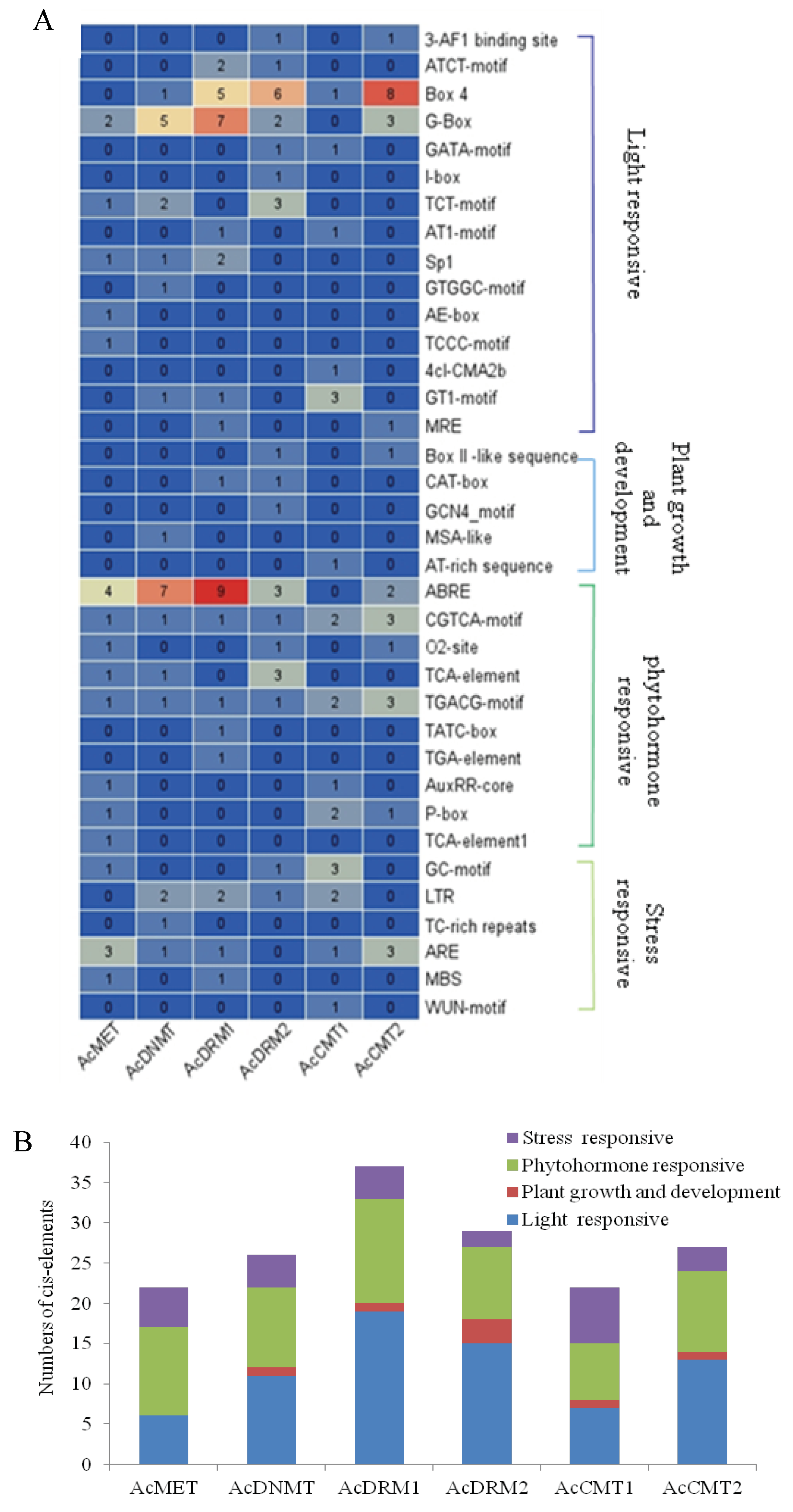
2.4. Analysis of Conserved Motif Gene Structure and Cis-Element
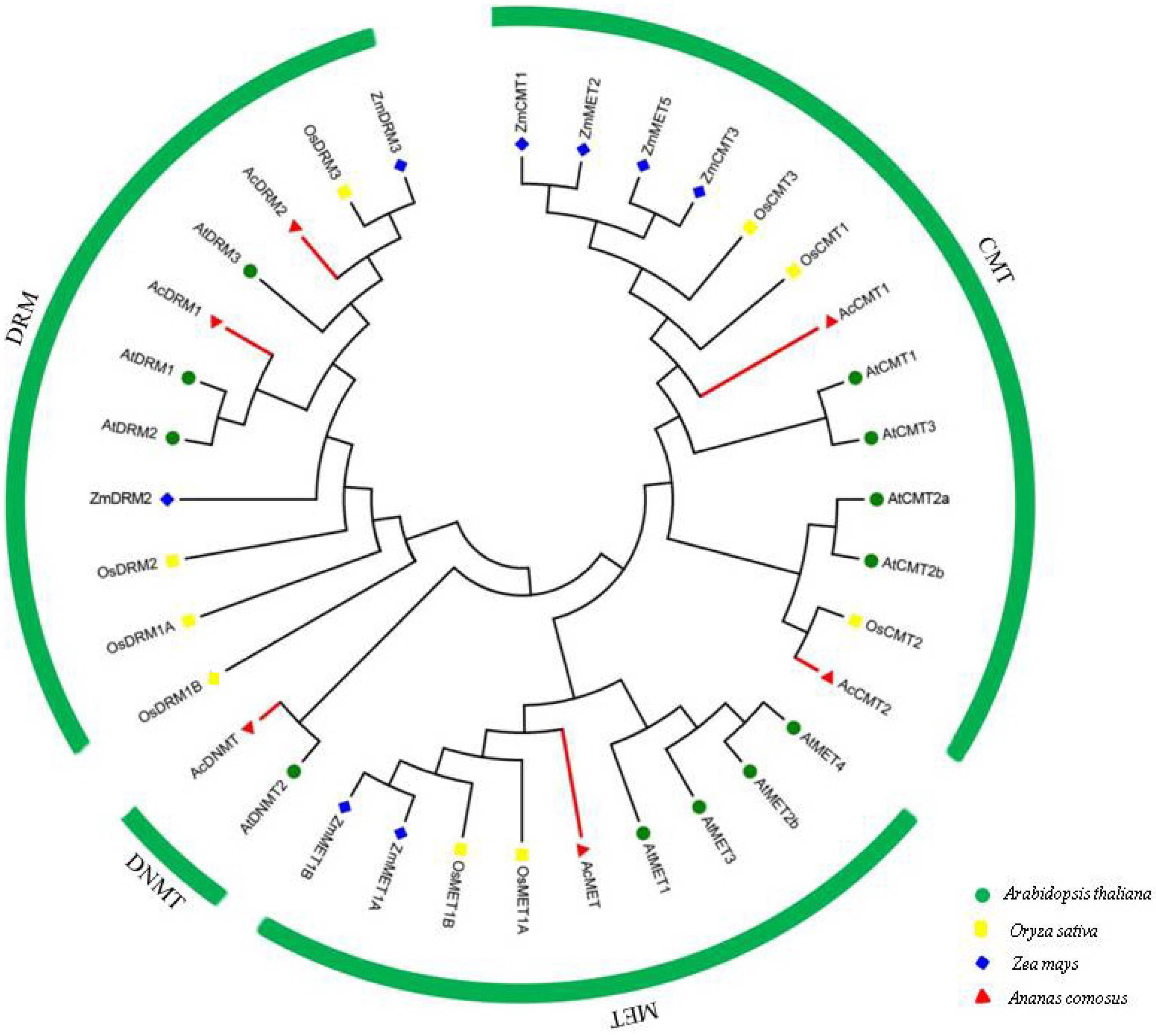
2.5. Construction of the Phylogenetic Tree
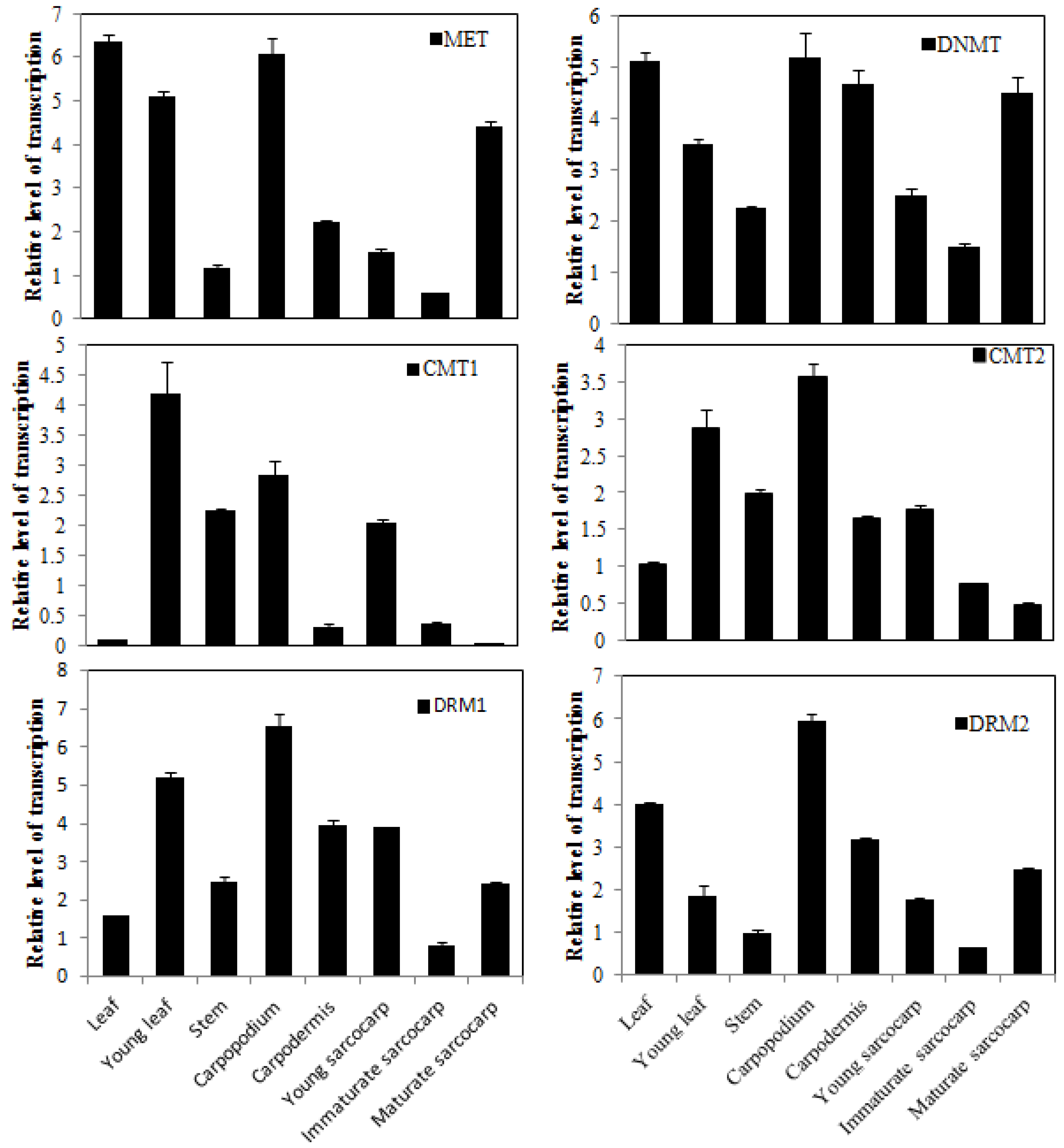
2.6. Analysis of Gene Expression
3. Results
3.1. Changes of DNA Methylation in Different Types of Somaclonal Variation
3.2. Classification and Sequence Characteristic Analysis of AcC5-MTase Genes in Pineapple
3.3. Analysis of Conserved Motifs and Structural Features in AcC5-MTase
3.4. Phylogenetic Analysis of C5-MTases in Pineapple and Other Plant Species
3.5. Cis-Element Analysis in the Promoter of AcC5-MTase Genes
3.6. Expression Patterns of AcC5-MTase Genes in Different Tissues during the Fruit Development of Pineapple
3.7. Expression Analysis of AcC5-MTase Genes in the Variant of Pineapple
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bednarek, P.T.; Orłowska, R. Plant tissue culture environment as a switch-key of (epi)genetic changes. Plant Cell Tissue Organ Cult. 2020, 140, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meilina, O.; Ordway, J.M.; Jiang, N.; Ooi, S.E.; Kok, S.; Sarpan, N.; Azimi, N.; TarmiziHashim, A.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Letter 2015, 525, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Igamberdiev, A.U.; Debnath, S.C. Tissue culture-induced DNA methylation in crop plants: A review. Mol. Biol. Rep. 2021, 48, 823–841. [Google Scholar] [CrossRef]
- Jeltsch, A. Beyond Watson and Crick: DNA Methylation and Molecular Enzymology of DNA Methyltransferases. ChemBioChem 2002, 3, 274–293. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Hu, L.; Li, N.; Xu, C.; Zhong, S.; Lin, X.; Yang, J.; Zhou, T.; Liang, A.Y.; Wu, Y.; Chen, Y.R. Mutation of a major CG methylase in rice causes genome-wide hypomethylation, dysregulated genome expression, and seedling lethality. Proc. Natl. Acad. Sci. USA 2014, 111, 10642–10647. [Google Scholar] [CrossRef] [Green Version]
- Bewick, A.J.; Niederhuth, C.E.; Ji, L.; Rohr, N.A.; Griffin, R.T.; Mack, J.L.; Schmitz, R.J. The evolution of CHROMOMETHYLASES and gene body DNA methylation in plants. Genome Biol. 2017, 18, 65. [Google Scholar] [CrossRef] [Green Version]
- Ashapkin, V.V.; Kutueva, L.I.; Vanyushin, B.F. Plant DNA methyltransferase genes: Multiplicity, expression, methylation patterns. Biochem. Biokhimiia 2016, 81, 141–151. [Google Scholar] [CrossRef]
- Gao, Z.H.; Liu, H.L.; Liu, D.; Pontes, O.; He, X.J.; Qian, W.Q.; Lin, H.; Xie, M.; Lorkovic, Z.J.; Zhang, S.D.; et al. An RNA polymerase II-and AGO4-associated protein acts in RNA-directed DNA methylation. Nature 2010, 465, 106–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, A.; Ehrenhofer-Murray, A.; Jurkowski, T.P.; Lyko, F.; Reuter, G.; Ankri, S.; Nellen, W.; Schaefer, M.; Helm, M. Mechanism and biological role of Dnmt2 in nucleic acid methylation. RNA Biol. 2017, 14, 1108–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genger, R.K.; Kovac, K.A.; Dennis, E.S.; Peacock, W.J.; Finnegan, E.J. Multiple DNA methyltransferase genes in Arabidopsis thaliana. Plant Mol. Biol. 1999, 41, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Huang, X.; Lan, H.; Huma, T.; Bao, Y.; Huang, J.; Zhang, H. Comprehensive gene expression analysis of the DNA (cytosine-5) methyltransferase family in rice (Oryza sativa L.). Genet. Mol. Res. GMR 2014, 13, 5159–5172. [Google Scholar] [CrossRef]
- Qian, Y.; Xi, Y.; Cheng, B.; Zhu, S. Genome-wide identification and expression profiling of DNA methyltransferase gene family in maize. Plant Cell Rep. 2014, 33, 1661–1672. [Google Scholar] [CrossRef]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; Mcquinn, R.; Gapper, N.E.; Liu, B.; Xiang, Z.Y.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; He, X.Q.; Zhao, H.C.; Xu, W.C.; Deng, H.; Wang, H.; Wang, S.Y.; Su, D.; Zheng, Z.L.; Yang, B.; et al. Genome-Wide identification of DNA methylases and demethylases in Kiwifruit (Actinidia chinensis). Front. Plant Sci. 2020, 11, 514993. [Google Scholar] [CrossRef]
- Montalvo, P.O.; Peña, C.D.; Oropeza, C.; Can, G.N.; Lara, I.C.; Castro, E.C.; Carbonell, L.S. A peak in global DNA methylation is a key step to initiate the somatic embryogenesis of coconut palm (Cocos nucifera L). Plant Cell Rep. 2020, 39, 1345–1357. [Google Scholar] [CrossRef]
- Rival, A.; Jaligot, E.; Beulé, T.; Finnegan, E.J. Isolation and expression analysis of genes encoding MET, CMT, and DRM methyltransferases in oil palm (Elaeis guineensis Jacq.) in relation to the ’mantled’ somaclonal variation. J. Exp. Bot. 2008, 59, 3271–3281. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.L.; Zhang, Z.H.; Han, B.M.; Li, H.; Dai, H.Y.; He, P.; Tian, H.Z. Isolation of DNA-methyltransferase genes from strawberry (Fragaria 3 ananassa Duch.) and their expression in relation to micropropagation. Plant Cell Rep. 2009, 28, 1373–1384. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.; Donoghue, M.T.; LeBlanc, C.; Wear, E.E.; Tanurdžić, M.; Berube, B.; Brooks, A.; Thompson, W.F.; Bowdoin, L.H.; Martienssen, R.A. Loss of small-RNA-directed DNA methylation in the plant cell cycle promotes germline reprogramming and somaclonal variation. Curr. Biol. 2021, 31, 591–600. [Google Scholar] [CrossRef]
- Yu, Z.M.; Zhang, G.H.; Silva, J.T.; Li, M.Z.; Zhao, C.H.; He, C.M.; Si, C.; Zhang, M.Z. Genome-wide identification and analysis of DNA methyltransferase and demethylase gene families in Dendrobium officinale reveal their potential functions in polysaccharide accumulation. BMC Plant Biol. 2021, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Q.; Xiao, X.O.; Zhang, H.N.; Li, Y.H.; Liu, S.H.; Sun, W.S.; Zhang, X.M.; Wu, Q.S. Whole-genome bisulfite sequencing reveals a role for DNA methylation in variants from callus culture of pineapple (Ananas comosus L.). Genes 2019, 10, 877. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.Z.; Xu, C.G.; Saghai, M.A.; Zhang, Q.F. Patterns of cytosine methylation in an elite rice hybrid and its parental lines by a methylation–sensitive amplification polymorphism technique. Mol. Genet. 1999, 261, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Yang, ZPAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [CrossRef] [PubMed] [Green Version]
- Machczynska, J.; Orłowska, R.; Mankowski, D.R.; Zimny, J.; Bednarek, P.T. DNA methylation changes in triticale due to in vitro culture plant regeneration and consecutive reproduction. Plant Cell Tiss Organ Cult. 2014, 119, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Kubis, S.; Castilho, A.; Vershinin, A.; Heslop-Harrison, J. Retroelements, transposons and methylation status in the genome of oil palm (Elaeis guineensis) and the relationship to somaclonal variation. Plant Mol. Biol. 2003, 52, 69–79. [Google Scholar] [CrossRef]
- Orłowska, R.; Bednarek, P.T. Precise evaluation of tissue culture-induced variation during optimisation of in vitro regeneration regime in barley. Plant Mol. Biol. 2020, 103, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Coronel, C.J.; González, A.I.; Ruiz, M.L.; Polanco, C. Analysis of somaclonal variation in transgenic and regenerated plants of Arabidopsis thaliana using methylation related metAFLP and TMD markers. Plant Cell Rep. 2018, 37, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Jaligot, E.; Rival, A.; Beulé, T.; Dussert, S.; Verdeil, J. Somaclonal variation in oil palm (Elaeis guineensis Jacq.): The DNA methylation hypothesis. Plant Cell Rep. 2000, 19, 684–690. [Google Scholar] [CrossRef]
- Azizi p Hanaf, M.; Sahebi, M.; Harikrishna, J.; Taheri, S.; Yassoralipour, A.; Nasehi, A. Epigenetic changes and their relationship to somaclonal variation: A need to monitor the micropropagation of plantation crops. Funct. Plant Biol. 2020, 47, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Aké, F.; Castillo-Castro, E.; Pool, F.B.; Espadas, F.; Santamaría, J.; Robert, M.; De-la-Peña, C. Physiological differences and changes in global DNA methylation levels in Agave angustifolia Haw. albino variant somaclones during the micropropagation process. Plant Cell Rep. 2016, 35, 2489–2502. [Google Scholar] [CrossRef] [PubMed]
- Ming, R.; Vanburen, R.; Wai, C.M.; Tang, H.B.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.L.; Chen, J.; Bigger, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Hu, S.N.; Wang, J.; Wong, G.K.; Li, S.G.; Liu, B.; Deng, Y.J.; Dai, L.; Zhou, Y.; Zhang, X.Q.; et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 2002, 5565, 79–92. [Google Scholar] [CrossRef]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [Green Version]
- Miguel, C.; Marum, L. An epigenetic view of plant cells cultured in vitro: Somaclonal variation and beyond. J. Exp. Bot. 2011, 62, 3713–3725. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.H.; Xie, Q.; Li, B.Y.; Su, H.Z. Molecular characterization and transcription analysis of DNA methyltransferase genes in tomato (Solanum lycopersicum). Genet. Mol. Biol. 2020, 43, e20180295. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access 35. H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Stroud, H.; Do, T.; Du, J.M.; Stroud, H.; Do, T.; Du, J.; Zhong, X.H.; Feng, S.H.; Johnson, L.; Patel, D.J.; et al. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Ohr, H.; Lee, J.W.; Hyun, Y.; Fischer, R.L.; Choi, Y. Temporal and spatial downregulation of Arabidopsis MET1 activity results in global DNA hypomethylation and developmental defects. Mol. Cells 2008, 26, 611–615. [Google Scholar] [PubMed]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Han, S.S.; Wang, Y.; Zhang, X.Z.; Han, Z.H. Variations in leaf morphology and DNA methylation following in vitro culture of Malus xiaojinensis. Plant Cell Tissue Organ Cult. 2012, 111, 153–161. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CK | Somaclonal Variation | Number of Sites | ||||||
|---|---|---|---|---|---|---|---|---|
| Types of Band | MspI | HpaII | MspI | HpaII | Narrow Leaves | Yellow-Striped Leaves | Green-Striped Leaves | Spiny Leaves |
| A1 | 1 | 1 | 1 | 1 | 83 | 98 | 116 | 121 |
| A2 | 0 | 1 | 0 | 1 | 66 | 51 | 63 | 53 |
| A3 | 1 | 0 | 1 | 0 | 22 | 24 | 23 | 32 |
| Total bands | 171 | 173 | 202 | 206 | ||||
| No change (%) | 61.96 | 64.07 | 53.30 | 63.19 | ||||
| B1 | 1 | 1 | 0 | 1 | 16 | 13 | 9 | 14 |
| B2 | 1 | 1 | 1 | 0 | 18 | 19 | 8 | 6 |
| B3 | 1 | 0 | 0 | 1 | 8 | 15 | 12 | 8 |
| Total bands | 42 | 47 | 29 | 28 | ||||
| Methylation (%) | 15.22 | 17.41 | 7.65 | 8.59 | ||||
| C1 | 1 | 0 | 1 | 1 | 26 | 20 | 92 | 32 |
| C2 | 0 | 1 | 1 | 1 | 29 | 21 | 44 | 38 |
| C3 | 0 | 1 | 1 | 0 | 8 | 9 | 12 | 22 |
| Total bands | 63 | 50 | 148 | 92 | ||||
| Demethylation (%) | 22.83 | 18.52 | 39.05 | 28.22 | ||||
| Gene ID | Gene Name | Chromosome Location | CDS Length (bp) | Exon NO. | Amino Acid (aa) | Molecular Weight (kDa) | GRAVY Value | Theoretical pI | Predicted Subcellular Localization |
|---|---|---|---|---|---|---|---|---|---|
| Aco013381.1 | AcCMT1 | LG15:10716493–10725146 | 2709 | 20 | 903 | 101,168.17 | −0.616 | 5.43 | Nucleus |
| Aco015994.1 | AcCMT2 | LG22:399865–413271 | 3456 | 20 | 1152 | 126,757.86 | −0.545 | 8.64 | Nucleus |
| Aco005236.1 | AcMET | LG07:3182723–3196828 | 4662 | 12 | 1554 | 175,261.41 | −0.499 | 5.94 | Cytoplasm |
| Aco006129.1 | AcDNMT | LG16:8638388–8642481 | 1206 | 10 | 402 | 45,882.35 | −0.321 | 5.9 | Nucleus |
| Aco007653.1 | AcDRM1 | LG08:988482–993571 | 1788 | 18 | 596 | 66,733.66 | −0.518 | 5.05 | Nucleus |
| Aco001739.1 | AcDRM2 | LG18:8333848–8347665 | 4167 | 7 | 1389 | 152,876.07 | −0.545 | 6.03 | Cytoplasm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, W.; Xiao, X.; Sun, W.; Liu, S.; Wu, Q.; Yao, Y.; Zhang, H.; Zhang, X. Genome-Wide Identification and Expression Analysis of Cytosine DNA Methyltransferase Genes Related to Somaclonal Variation in Pineapple (Ananas comosus L.). Agronomy 2022, 12, 1039. https://doi.org/10.3390/agronomy12051039
Lin W, Xiao X, Sun W, Liu S, Wu Q, Yao Y, Zhang H, Zhang X. Genome-Wide Identification and Expression Analysis of Cytosine DNA Methyltransferase Genes Related to Somaclonal Variation in Pineapple (Ananas comosus L.). Agronomy. 2022; 12(5):1039. https://doi.org/10.3390/agronomy12051039
Chicago/Turabian StyleLin, Wenqiu, Xi’ou Xiao, Weisheng Sun, Shenghui Liu, Qingsong Wu, Yanli Yao, Hongna Zhang, and Xiumei Zhang. 2022. "Genome-Wide Identification and Expression Analysis of Cytosine DNA Methyltransferase Genes Related to Somaclonal Variation in Pineapple (Ananas comosus L.)" Agronomy 12, no. 5: 1039. https://doi.org/10.3390/agronomy12051039
APA StyleLin, W., Xiao, X., Sun, W., Liu, S., Wu, Q., Yao, Y., Zhang, H., & Zhang, X. (2022). Genome-Wide Identification and Expression Analysis of Cytosine DNA Methyltransferase Genes Related to Somaclonal Variation in Pineapple (Ananas comosus L.). Agronomy, 12(5), 1039. https://doi.org/10.3390/agronomy12051039