
Can Epigenetics Guide the Production of Better Adapted Cultivars?




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Breeding Methods
2.1. Classical and Genomic Breeding
2.2. Transcriptomic Tools
3. Epigenetics and Its Potential Applications in Plant Breeding
3.1. Introduction to Epigenetics
3.2. Molecular Mechanisms
3.3. DNA Methylation
3.4. Quantifying DNA Methylation
3.5. Variation in DNA Methylation Patterns
3.5.1. Developmental Epigenetic Modifications
3.5.2. Acquired Epigenetic Modifications
3.6. Population Epigenetic Diversity
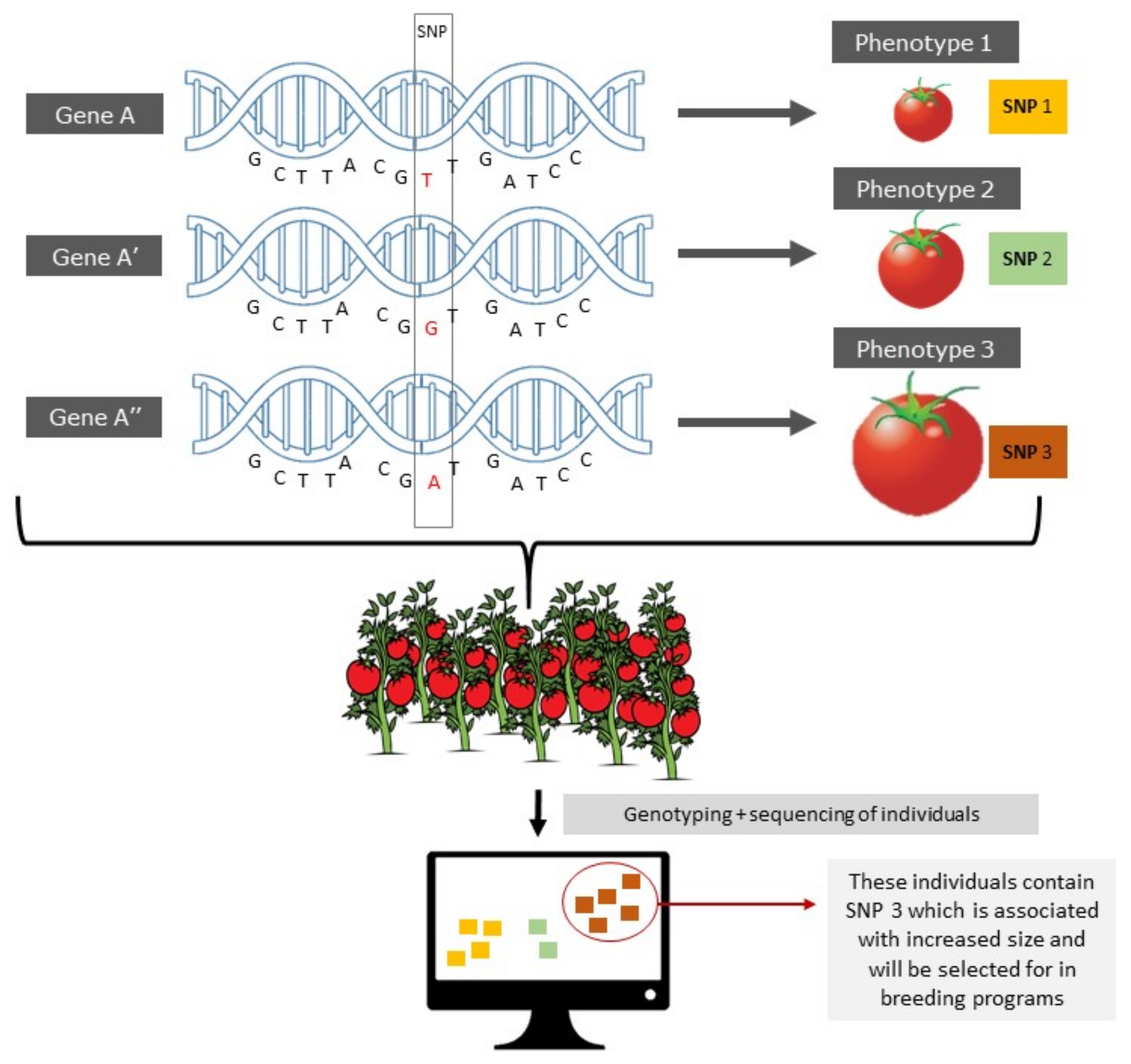
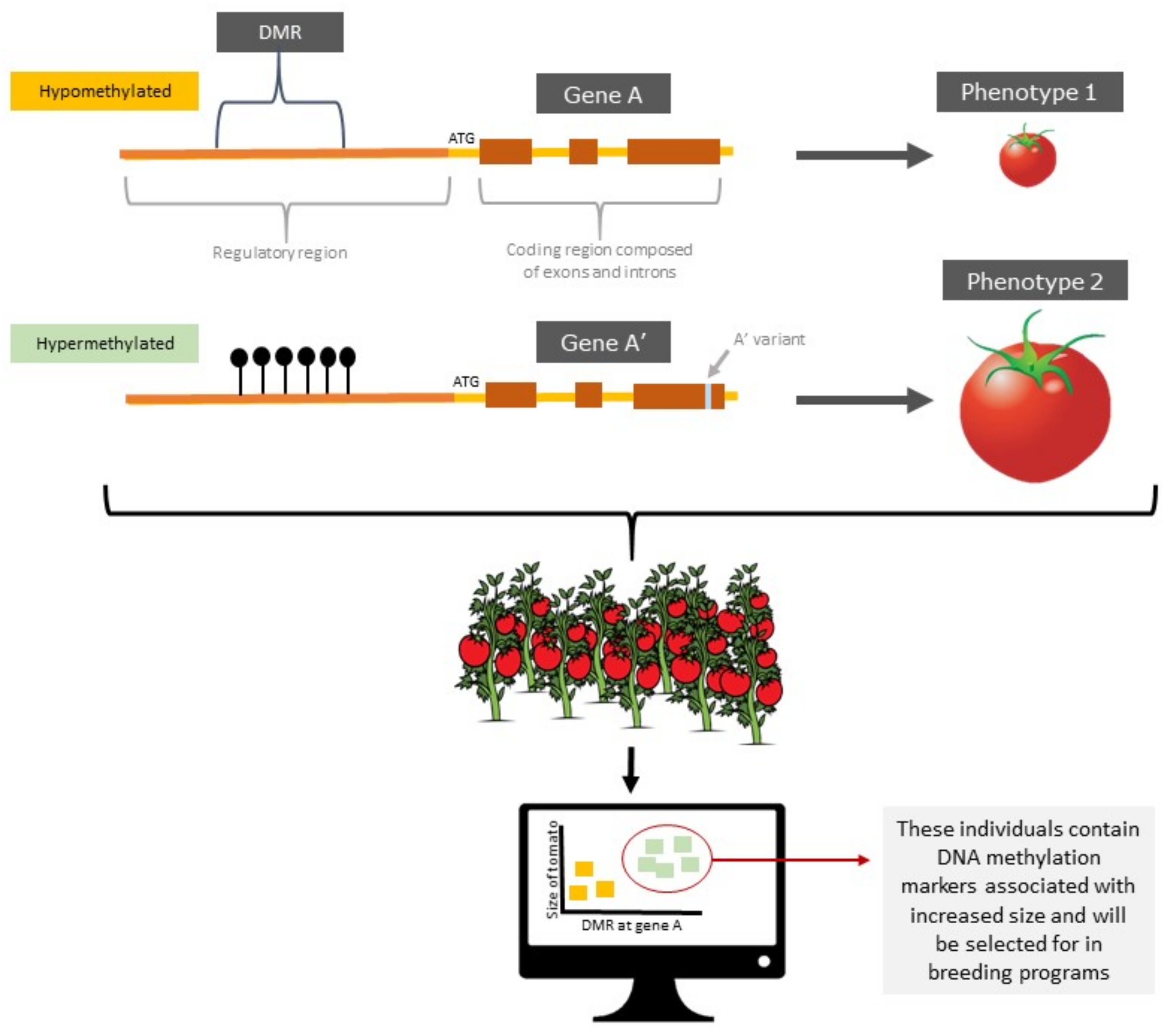
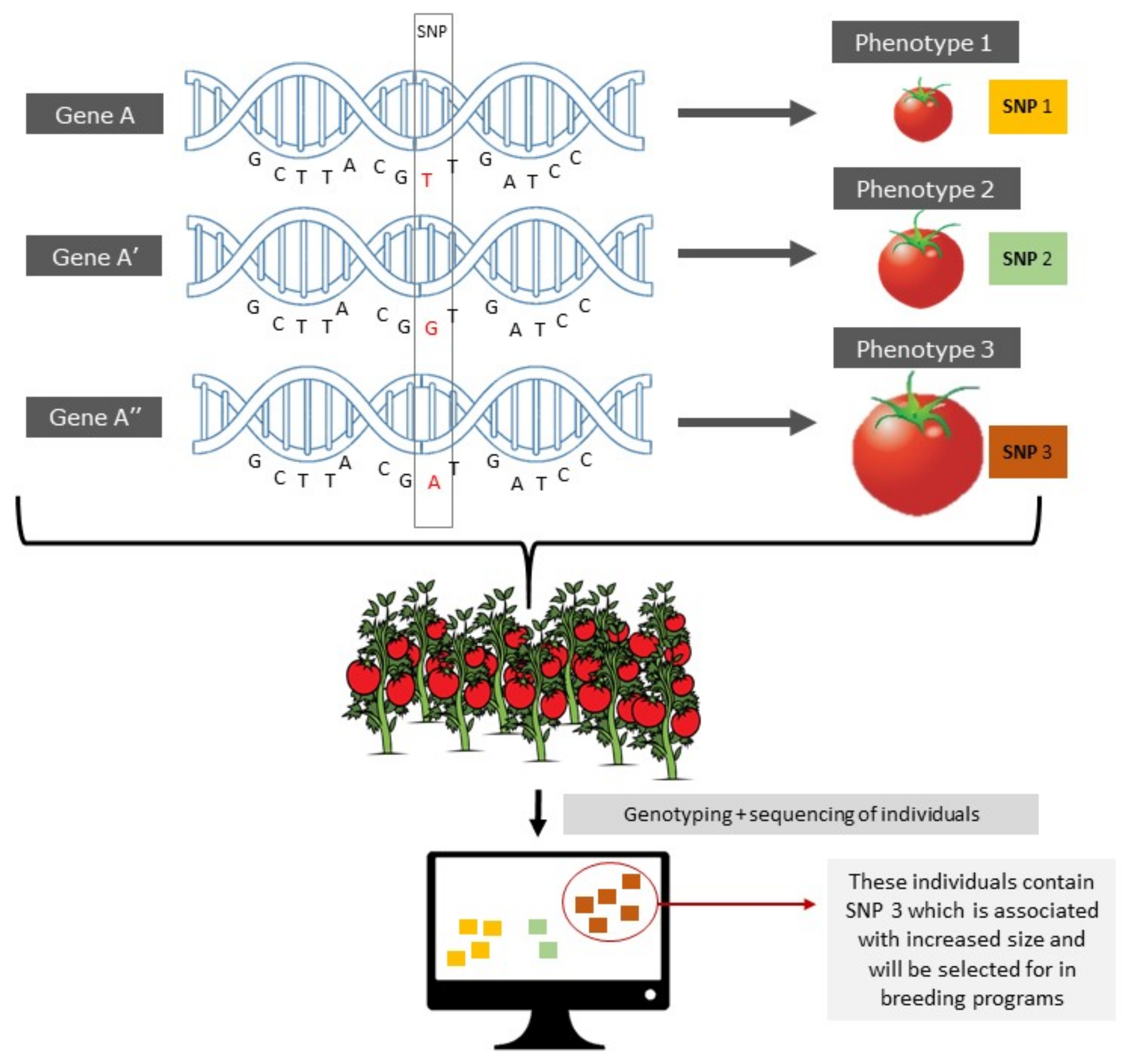
3.6.1. Epialleles and epiQTLs
3.6.2. Epigenome-Wide Association Mapping (EWAS)
3.7. Epigenetic Selection
3.8. Limitations of Epigenetic Markers
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, X.; Godwin, I.; Hayes, B.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperno, D.R.; Ranere, A.J.; Holst, I.; Iriarte, J.; Dickau, R. Starch grain and phytolith evidence for early ninth millennium B.P. maize from the Central Balsas River Valley, Mexico. Proc. Natl. Acad. Sci. USA 2009, 106, 5019–5024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamrick, J.L. Isozymes and the Analysis of Genetic Structure in Plant Populations. In Isozymes in Plant Biology; Springer: Dordrecht, The Netherlands, 1989; pp. 87–105. [Google Scholar]
- Nadeem, M.A.; Nawaz, M.A.; Shahid, M.Q.; Doğan, Y.; Comertpay, G.; Yıldız, M.; Hatipoğlu, R.; Ahmad, F.; Alsaleh, A.; Labhane, N.; et al. DNA molecular markers in plant breeding: Current status and recent advancements in genomic selection and genome editing. Biotechnol. Biotechnol. Equip. 2018, 32, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Gepts, P. A comparison between crop domestication, classical plant breeding, and genetic engineering. Crop Sci. 2002, 42, 1780–1790. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, M.A.; Frey, K.J. Yield-component Analysis of Oat Isolines that Produce Different Grain Yields. Crop Sci. 1977, 17, 165–168. [Google Scholar] [CrossRef]
- Brown, A.H.D.; Lawrence, G.J.; Jenkin, M.; Douglass, J.; Gregory, E. Linkage drag in backcross breeding in barley. J. Hered. 1989, 80, 234–239. [Google Scholar] [CrossRef]
- Zeven, A.C.; Knott, D.R.; Johnson, R. Investigation of linkage drag in near isogenic lines of wheat by testing for seedling reaction to races of stem rust, leaf rust and yellow rust. Euphytica 1983, 32, 319–327. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Jahufer, M.Z.Z.; Brouwer, J.B.; Pang, E.C.K. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: The basic concepts. Euphytica 2005, 142, 169–196. [Google Scholar] [CrossRef]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection: An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.-L. Molecular Markers and Marker-Assisted Breeding in Plants. In Plant Breeding from Laboratories to Fields; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Simmonds, N.W. Introgression and incorporation. Strategies for the use of crop genetic resources. Biol. Rev. 1993, 68, 539–562. [Google Scholar] [CrossRef]
- Naeem, M.; Ghouri, F.; Shahid, M.Q.; Iqbal, M.; Baloch, F.; Chen, L.; Ul-Allah, S.; Babar, M.; Rana, M. Genetic diversity in mutated and non-mutated rice varieties. Genet. Mol. Res. GMR 2015, 14, 17109–17123. [Google Scholar] [CrossRef]
- Tiwari, J.K.; Singh, B.P.; Gopal, J.; Patil, V.U. Molecular characterization of the Indian Andigena potato core collection using microsatellite markers. Afr. J. Biotechnol. 2013, 12, 1025–1033. [Google Scholar]
- Agarwal, P.; Parida, S.K.; Mahto, A.; Das, S.; Mathew, I.E.; Malik, N.; Tyagi, A.K. Agarwal 2014—Expanding frontiers in plant transcriptomics in aid of functional genomics and molecular breeding. Biotechnol. J. 2014, 9, 1480–1492. [Google Scholar] [CrossRef]
- Adams, M.D.; Kerlavage, A.R.; Fleischmann, R.D.; Fuldner, R.A. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature 1995, 377, 3–174. [Google Scholar]
- Velculescu, V.E.; Vogelsein, B.; Kinzler, K.W.; Zhang, L. Serial analysis of gene expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.L.; Ng, P.; Chiu, K.P.; Wong, C.H.; Ang, C.C.; Lipovich, L.; Liu, E.T.; Ruan, Y. 5′ Long serial analysis of gene expression (LongSAGE) and 3′ LongSAGE for transcriptome characterization and genome annotation. Proc. Natl. Acad. Sci. USA 2004, 101, 11701–11706. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, H.; Urasaki, N.; Yoshida, K.; Krüger, D.H.; Kahl, G.; Terauchi, R. SuperSAGE: Powerful Serial Analysis of Gene Expression. In RNA Abundance Analysis; Humana Press: Totowa, NJ, USA, 2012; Volume 883, pp. 1–17. [Google Scholar]
- Matsumura, H.; Yoshida, K.; Luo, S.; Kimura, E.; Fujibe, T.; Albertyn, Z.; Barrero, R.A.; Krüger, D.H.; Kahl, G.; Schrot, G.P.; et al. High-throughput superSAGE for digital gene expression analysis of multiple samples using next generation sequencing. PLoS ONE 2010, 5, e12010. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, K.L.; Høgh, A.L.; Emmersen, J. DeepSAGE—Digital transcriptomics with high sensitivity, simple experimental protocol and multiplexing of samples. Nucleic Acids Res. 2006, 34, e133. [Google Scholar] [CrossRef] [Green Version]
- Brenner, S.; Johnson, M.; Bridgham, J.; Golda, G.; Lloyd, D.H.; Johnson, D.; Luo, S.; McCurdy, S.; Foy, S.; Ewan, M.; et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat. Biotechnol. 2000, 18, 630–634. [Google Scholar] [CrossRef]
- Lashkari, D.A.; Derisi, J.L.; McCusker, J.H.; Namath, A.F.; Gentile, C.; Hwang, S.Y.; Brown, P.O.; Davis, R.W. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc. Natl. Acad. Sci. USA 1997, 94, 13057–13062. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Wang, X. Large-Scale Profiling of the Arabidopsis Transcriptome. Plant Physiol. 2000, 124, 1472–1476. [Google Scholar] [CrossRef] [Green Version]
- Aharoni, A.; Keizer, L.C.P.; Bouwmeester, H.J.; Sun, Z.; Alvarez-Huerta, M.; Verhoeven, H.A.; Blaas, J.; van Houwelingen, A.M.; De Vos, R.C.; van der Voet, H.; et al. Identification of the SAAT gene involved in strawberry flavor biogenesis by use of DNA microarrays. Plant Cell 2000, 12, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Shoemaker, R.; Keim, P.; Vodkin, L.; Retzel, E.; Clifton, S.W.; Waterston, R.; Smoller, D.; Coryell, V.; Khanna, A.; Erpelding, J.; et al. A compilation of soybean ESTs: Generation and analysis. Genome 2002, 45, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Chen, C.; Liu, X.; Jiao, Y.; Su, N.; Li, L.; Wang, X.; Cao, M.; Sun, N.; Zhang, X.; et al. A microarray analysis of the rice transcriptome and its comparison to Arabidopsis. Genome Res. 2005, 15, 1274–1283. [Google Scholar] [CrossRef] [Green Version]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly Integrated Single-Base Resolution Maps of the Epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [Green Version]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 11, 951–969. [Google Scholar] [CrossRef] [Green Version]
- Cubillos, F.A.; Coustham, V.; Loudet, O. Lessons from eQTL mapping studies: Non-coding regions and their role behind natural phenotypic variation in plants. Curr. Opin. Plant Biol. 2012, 15, 192–198. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Samo, N.; Ebert, A.; Kopka, J.; Mozgová, I. Plant chromatin, metabolism and development—An intricate crosstalk. Curr. Opin. Plant Biol. 2021, 61, 102002. [Google Scholar] [CrossRef] [PubMed]
- Tirnaz, S.; Batley, J. Epigenetics: Potentials and Challenges in Crop Breeding. Mol. Plant 2019, 12, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Gallusci, P.; Dai, Z.; Génard, M.; Gauffretau, A.; Leblanc-Fournier, N.; Richard-Molard, C.; Vile, D.; Brunel-Muguet, S. Epigenetics for Plant Improvement: Current Knowledge and Modeling Avenues. Trends Plant Sci. 2017, 22, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Ishida, H. Nucleosome unwrapping and unstacking. Curr. Opin. Struct. Biol. 2020, 64, 119–125. [Google Scholar] [CrossRef]
- Agarwal, G.; Kudapa, H.; Ramalingam, A.; Choudhary, D.; Sinha, P.; Garg, V.; Singh, V.K.; Patil, G.B.; Pandey, M.K.; Nguyen, H.T.; et al. Epigenetics and epigenomics: Underlying mechanisms, relevance, and implications in crop improvement. Funct. Integr. Genom. 2020, 20, 739–761. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; Charles An, Y.Q.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, H.; Hirochika, H. Silencing of transposable elements in plants. Trends Plant Sci. 2001, 6, 527–534. [Google Scholar] [CrossRef]
- Ito, T.; Nishio, H.; Tarutani, Y.; Emura, N.; Honjo, M.N.; Toyoda, A.; Fujiyama, A.; Kakutani, T.; Kudoh, H. Seasonal stability and dynamics of dna methylation in plants in a natural environment. Genes 2019, 10, 544. [Google Scholar] [CrossRef] [Green Version]
- Bochtler, M.; Kolano, A.; Xu, G.-L. DNA demethylation pathways: Additional players and regulators. Bioessays 2017, 39, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.N.; Zynda, G.J.; Song, J.; Han, Z.; Vaughn, M.W.; Li, Q.; Springer, N.M. Subtle perturbations of the maize methylome reveal genes and transposons silenced by chromomethylase or RNA-directed DNA methylation pathways. G3 Genes Genomes Genet. 2018, 8, 1921–1932. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Tarutani, Y.; Miyao, A.; Ito, T.; Yamazaki, M.; Sakai, H.; Fukai, E.; Hirochika, H. Loss of function mutations in the rice chromomethylase OsCMT3a cause a burst of transposition. Plant J. 2015, 83, 1069–1081. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.J.; Palanca-Ballester, C.; Urtasun, R.; Alemany-Cosme, E.; Lahoz, A.; Sandoval, J. Methods for analysis of specific DNA methylation status. Methods 2021, 187, 3–12. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA methylation analysis: Choosing the right method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Gravina, S.; Ganapathi, S.; Vijg, J. Single-cell, locus-specific bisulfite sequencing (SLBS) for direct detection of epimutations in DNA methylation patterns. Nucleic Acids Res. 2015, 43, e93. [Google Scholar] [CrossRef] [Green Version]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef]
- Suhua, F.; Zhenhui, Z.; Ming, W.; Steven, E.J. Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis with enzymatic methyl sequencing. Epigenetics Chromatin 2021, 13, 1–17. [Google Scholar]
- Shafi, A.; Mitrea, C.; Nguyen, T.; Draghici, S. A survey of the approaches for identifying differential methylation using bisulfite sequencing data. Brief. Bioinform. 2017, 19, 737–753. [Google Scholar] [CrossRef] [Green Version]
- Noshay, J.M.; Springer, N.M. Stories that can’t be told by SNPs; DNA methylation variation in plant populations. Curr. Opin. Plant Biol. 2021, 61, 101989. [Google Scholar] [CrossRef]
- Rajkumar, M.S.; Shankar, R.; Garg, R.; Jain, M. Bisulphite sequencing reveals dynamic DNA methylation under desiccation and salinity stresses in rice cultivars. Genomics 2020, 112, 3537–3548. [Google Scholar] [CrossRef]
- Ashapkin, V.V.; Kutueva, L.I.; Aleksandrushkina, N.I.; Vanyushin, B.F. Epigenetic mechanisms of plant adaptation to biotic and abiotic stresses. Int. J. Mol. Sci. 2020, 21, 7457. [Google Scholar] [CrossRef]
- Kong, L.; Liu, Y.; Wang, X.; Chang, C. Insight into the role of epigenetic processes in abiotic and biotic stress response in wheat and barley. Int. J. Mol. Sci. 2020, 21, 1480. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Wu, T.; Li, S.; He, Q.; Yang, Z.; Zhang, W.; Gan, Y.; Sun, P.; Xiang, G.; Zhang, H.; et al. The methylation patterns and transcriptional responses to chilling stress at the seedling stage in rice. Int. J. Mol. Sci. 2019, 20, 5089. [Google Scholar] [CrossRef] [Green Version]
- Grover, J.W.; Kendall, T.; Baten, A.; Burgess, D.; Freeling, M.; King, G.J.; Mosher, R.A. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 2018, 94, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef] [Green Version]
- Ikeuchi, M.; Iwase, A.; Sugimoto, K. Control of plant cell differentiation by histone modification and DNA methylation. Curr. Opin. Plant Biol. 2015, 28, 60–67. [Google Scholar] [CrossRef]
- Lin, J.Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.F.; Chen, P.Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E9730–E9739. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.Q.; Zhang, Y.J.; Zhou, S.R.; Hu, W.Y.; Wu, X.T.; Ye, Y.J.; Wu, X.X.; Xiao, Y.P.; Li, X.; Xue, H.W. Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef] [Green Version]
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA methylation dynamics during early plant life. Genome Biol. 2017, 18, 179. [Google Scholar] [CrossRef]
- Rajkumar, M.S.; Gupta, K.; Khemka, N.K.; Garg, R.; Jain, M. DNA methylation reprogramming during seed development and its functional relevance in seed size/weight determination in chickpea. Commun. Biol. 2020, 3, 340. [Google Scholar] [CrossRef]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef]
- Daccord, N.; Celton, J.M.; Linsmith, G.; Becker, C.; Choisne, N.; Schijlen, E.; van de Geest, H.; Bianco, L.; Micheletti, D.; Velasco, R.; et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 2017, 49, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liu, R.; Niu, Q.; Tang, K.; Zhang, B.; Zhang, H.; Chen, K.; Zhu, J.K.; Lang, Z. Global increase in DNA methylation during orange fruit development and ripening. Proc. Natl. Acad. Sci. USA 2019, 116, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakatsu, T.; Stuart, T.; Valdes, M.; Breakfield, N.; Schmitz, R.J.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N.; et al. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 16058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.S.; Kawakatsu, T.; Kim, K.D.; Zhang, N.; Nguyen, C.T.; Khan, S.M.; Batek, J.M.; Joshi, T.; Schmutz, J.; Grimwood, J.; et al. Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. New Phytol. 2017, 214, 808–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widman, N.; Feng, S.; Jacobsen, S.E.; Pellegrini, M. Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 2014, 9, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, D.K.; Koenig, D.; Hagmann, J.; Becker, C.; Weigel, D. Evolution of DNA Methylation Patterns in the Brassicaceae is Driven by Differences in Genome Organization. PLoS Genetics 2014, 10, e1004785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Rong, T.; Cao, M. Analysis of DNA methylation in different maize tissues. J. Genet. Genom. 2008, 35, 41–48. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [Green Version]
- Gahlaut, V.; Zinta, G.; Jaiswal, V.; Kumar, S. Quantitative Epigenetics: A New Avenue for Crop Improvement. Epigenomes 2020, 4, 25. [Google Scholar] [CrossRef]
- Pan, Y.; Wane, W.; Zhao, X.; Zhu, L.; Fu, B.; Li, Z. DNA methylation alterations of rice in response to cold stress. Omics J. 2011, 4, 364–369. [Google Scholar]
- Zhang, W.; Wang, N.; Yang, J.; Guo, H.; Liu, Z.; Zheng, X.; Li, S.; Xiang, F. The salt-induced transcription factor GmMYB84 confers salinity tolerance in soybean. Plant Sci. 2020, 291, 110326. [Google Scholar] [CrossRef]
- Qian, Y.; Hu, W.; Liao, J.; Zhang, J.; Ren, Q. The Dynamics of DNA methylation in the maize (Zea mays L.) inbred line B73 response to heat stress at the seedling stage. Biochem. Biophys. Res. Commun. 2019, 512, 742–749. [Google Scholar] [CrossRef]
- Gardiner, L.J.; Joynson, R.; Omony, J.; Rusholme-Pilcher, R.; Olohan, L.; Lang, D.; Bai, C.; Hawkesford, M.; Salt, D.; Spannagl, M.; et al. Hidden variation in polyploid wheat drives local adaptation. Genome Res. 2018, 28, 1319–1332. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Bhushan, B.; Gaikwad, K.; Yadav, O.P.; Kumar, S.; Rai, R.D. Induced defence responses of contrasting bread wheat genotypes under differential salt stress imposition. Indian J. Biochem. Biophys. 2015, 52, 75–85. [Google Scholar]
- Lämke, J.; Bäurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef]
- Kumar, G.; Rattan, U.K.; Singh, A.K. Chilling-mediated DNA methylation changes during dormancy and its release reveal the importance of epigenetic regulation during winter dormancy in Apple (Malus x domestica Borkh.). PLoS ONE 2016, 11, e0149934. [Google Scholar] [CrossRef] [Green Version]
- Bäurle, I. Plant Heat Adaptation: Priming in response to heat stress. F1000Research 2016, 5, 694. [Google Scholar] [CrossRef]
- Liu, T.; Li, Y.; Duan, W.; Huang, F.; Hou, X. Cold acclimation alters DNA methylation patterns and confers tolerance to heat and increases growth rate in Brassica rapa. J. Exp. Bot. 2017, 68, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Kawakatsu, T.; Huang, S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic diversity in global collection of Arabidopsis thaliana accession. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef] [Green Version]
- Eichten, S.R.; Briskine, R.; Song, J.; Li, Q.; Swanson-Wagner, R.; Hermanson, P.J.; Waters, A.J.; Starr, E.; West, P.T.; Tiffin, P.; et al. Epigenetic and Genetic Influences on DNA Methylation Variation in Maize Populations. Plant Cell 2013, 25, 2783–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Zhang, J.; Liu, Y.; Liu, S.; Liu, Z.; Duan, Z.; Wang, Z.; Zhu, B.; Guo, Y.L.; Tian, Z. DNA methylation footprints during soybean domestication and improvement. Genome Biol. 2018, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xie, L.; Zhang, Q.; Ouyang, W.; Deng, L.; Guan, P.; Ma, M.; Li, Y.; Zhang, Y.; Xiao, Q.; et al. Integrative analysis of reference epigenomes in 20 rice varieties. Nat. Commun. 2020, 11, 2658. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.J.; He, Y.; Valdés-López, O.; Khan, S.M.; Joshi, T.; Urich, M.A.; Nery, J.R.; Diers, B.; Xu, D.; Stacey, G.; et al. Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 2013, 23, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Takata, M.; Kishima, Y.; Sano, Y. DNA Methylation Polymorphisms in Rice and Wild Rice Strains: Detection of Epigenetic Markers. Breed. Sci. 2005, 55, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.J.; Ecker, J.R. Epigenetic and epigenomic variation in Arabidopsis thaliana. Trends Plant Sci. 2012, 17, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Ong-Abdullah, M.; Ordway, J.M.; Jiang, N.; Ooi, S.E.; Kok, S.Y.; Sarpan, N.; Azimi, N.; Hashim, A.T.; Ishak, Z.; Rosli, S.K.; et al. Loss of Karma transposon methylation underlies the mantled somaclonal variant of oil palm. Nature 2015, 525, 533–537. [Google Scholar] [CrossRef] [Green Version]
- Cocciolone, S.M.; Chopra, S.; Flint-Garcia, S.A.; McMullen, M.D.; Peterson, T. Tissue-specific patterns of a maize Myb transcription factor are epigenetically regulated. Plant J. 2001, 27, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Quadrana, L.; Almeida, J.; Asís, R.; Duffy, T.; Dominguez, P.G.; Bermúdez, L.; Conti, G.; Corrêa da Silva, J.V.; Peralta, I.E.; Colot, V.; et al. Natural occurring epialleles determine vitamin e accumulation in tomato fruits. Nat. Commun. 2014, 5, 4027. [Google Scholar] [CrossRef] [Green Version]
- Hofmeister, B.T.; Lee, K.; Rohr, N.A.; Hall, D.W.; Schmitz, R.J. Stable inheritance of DNA methylation allows creation of epigenotype maps and the study of epiallele inheritance patterns in the absence of genetic variation. Genome Biol. 2017, 18, 155. [Google Scholar] [CrossRef]
- Cortijo, S.; Wardenaar, R.; Colomé-Tatché, M.; Gilly, A.; Etcheverry, M.; Labadie, K.; Caillieux, E.; Hospital, F.; Aury, J.M.; Wincker, P.; et al. Mapping the Epigenetic Basis of Complex Traits. Science 2014, 343, 1145–1148. [Google Scholar] [CrossRef]
- Furci, L.; Jain, R.; Stassen, J.; Berkowitz, O.; Whelan, J.; Roquis, D.; Baillet, V.; Colot, V.; Johannes, F.; Ton, J. Identification and characterisation of hypomethylated DNA loci controlling quantitative resistance in Arabidopsis. eLife 2019, 8, e40655. [Google Scholar] [CrossRef]
- Can, S.N.; Nunn, A.; Galanti, D.; Langenberger, D.; Becker, C.; Volmer, K.; Heer, K.; Opgenoorth, L.; Fernandez-Pozo, N.; Rensing, S.A. The EpiDiverse Plant Epigenome-Wide Association Studies (EWAS) Pipeline. Epigenomes 2021, 5, 12. [Google Scholar] [CrossRef]
- McCartney, D.L.; Hillary, R.F.; Stevenson, A.J.; Ritchie, S.J.; Walker, R.M.; Zhang, Q.; Morris, S.W.; Bermingham, M.L.; Campbell, A.; Murray, A.D.; et al. Epigenetic prediction of complex traits and death. Genome Biol. 2018, 19, 136. [Google Scholar] [CrossRef] [Green Version]
- Johannes, F.; Porcher, E.; Teixeira, F.K.; Saliba-Colombani, V.; Simon, M.; Agier, N.; Bulski, A.; Albuisson, J.; Heredia, F.; Audigier, P.; et al. Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet. 2009, 5, e1000530. [Google Scholar] [CrossRef]
- Hu, Y.; Morota, G.; Rosa, G.J.; Gianola, D. Prediction of plant height in arabidopsis thaliana using DNA methylation data. Genetics 2015, 201, 779–793. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chen, G.; Hermanson, P.J.; Xu, Q.; Sun, C.; Chen, W.; Kan, Q.; Li, M.; Crisp, P.A.; Yan, J.; et al. Population-level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biol. 2019, 20, 243. [Google Scholar] [CrossRef]
- López, C.M.R.; Wilkinson, M.J. Epi-fingerprinting and epi-interventions for improved crop production and food quality. Front. Plant Sci. 2015, 6, 397. [Google Scholar]
- Hauben, M.; Haesendonckx, B.; Standaert, E.; Van Der Kelen, K.; Azmi, A.; Akpo, H.; Breusegem, F.V.; Guisez, Y.; Bots, M.; Lambert, B.; et al. Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. USA 2009, 106, 20109–20114. [Google Scholar] [CrossRef] [Green Version]
- Tricker, P.J.; López, C.M.R.; Gibbings, G.; Hadley, P.; Wilkinson, M.J. Transgenerational, dynamic methylation of stomata genes in response to low relative humidity. Int. J. Mol. Sci. 2013, 14, 6674–6689. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Guo, Z.; Gao, L.; Zhao, G.; Zhang, W.; Zhou, R.; Wu, Y.; Wang, H.; An, H.; Jia, J. DNA methylation pattern of Photoperiod-B1 is associated with photoperiod insensitivity in wheat (Triticum aestivum). New Phytol. 2014, 204, 682–692. [Google Scholar] [CrossRef]
- Kakoulidou, I.; Avramidou, E.V.; Baránek, M.; Brunel-Muguet, S.; Farrona, S.; Johannes, F.; Kaiserli, E.; Liberman-Lazarovich, M.; Martinelli, F.; Mladenov, V.; et al. Epigenetics for crop improvement in times of global change. Biology 2021, 10, 766. [Google Scholar] [CrossRef]
- Tricker, P.J. Transgenerational inheritance or resetting of stress-induced epigenetic modifications: Two sides of the same coin. Front. Plant Sci. 2015, 6, 699. [Google Scholar] [CrossRef] [Green Version]
- Varotto, S.; Tani, E.; Abraham, E.; Krugman, T.; Kapazoglou, A.; Melzer, R.; Radanović, A.; Miladinović, D. Epigenetics: Possible applications in climate-smart crop breeding. J. Exp. Bot. 2020, 71, 5223–5236. [Google Scholar] [CrossRef]
- Latutrie, M.; Gourcilleau, D.; Pujol, B. Epigenetic variation for agronomic improvement: An opportunity for vegetatively propagated crops. Am. J. Bot. 2019, 106, 1281–1284. [Google Scholar] [CrossRef]
- Sarkar, D.; Kar, S.K.; Chattopadhyay, A.; Shikha Rakshit, A.; Tripathi, V.K.; Dubey, P.; Abhilash, P. Low input sustainable agriculture: A viable climate-smart option for boosting food production in a warming world. Ecol. Indic. 2020, 115, 106412. [Google Scholar] [CrossRef]
- Xu, G.; Lyu, J.; Li, Q.; Liu, H.; Wang, D.; Zhang, M.; Springer, N.M.; Ross-Ibarra, J.; Yang, J. Evolutionary and functional genomics of DNA methylation in maize domestication and improvement. Nat. Commun. 2020, 11, 5539. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turcotte, H.; Hooker, J.; Samanfar, B.; Parent, J.-S. Can Epigenetics Guide the Production of Better Adapted Cultivars? Agronomy 2022, 12, 838. https://doi.org/10.3390/agronomy12040838
Turcotte H, Hooker J, Samanfar B, Parent J-S. Can Epigenetics Guide the Production of Better Adapted Cultivars? Agronomy. 2022; 12(4):838. https://doi.org/10.3390/agronomy12040838
Chicago/Turabian StyleTurcotte, Haley, Julia Hooker, Bahram Samanfar, and Jean-Sébastien Parent. 2022. "Can Epigenetics Guide the Production of Better Adapted Cultivars?" Agronomy 12, no. 4: 838. https://doi.org/10.3390/agronomy12040838
APA StyleTurcotte, H., Hooker, J., Samanfar, B., & Parent, J.-S. (2022). Can Epigenetics Guide the Production of Better Adapted Cultivars? Agronomy, 12(4), 838. https://doi.org/10.3390/agronomy12040838