
Ensifer meliloti L6-AK89, an Effective Inoculant of Medicago lupulina Varieties: Phenotypic and Deep-Genome Screening

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Genome Sequences
2.3. Phage Typing
2.4. Plant Tests
2.5. Genome Sequencing and Assembly
2.6. Genome Analysis
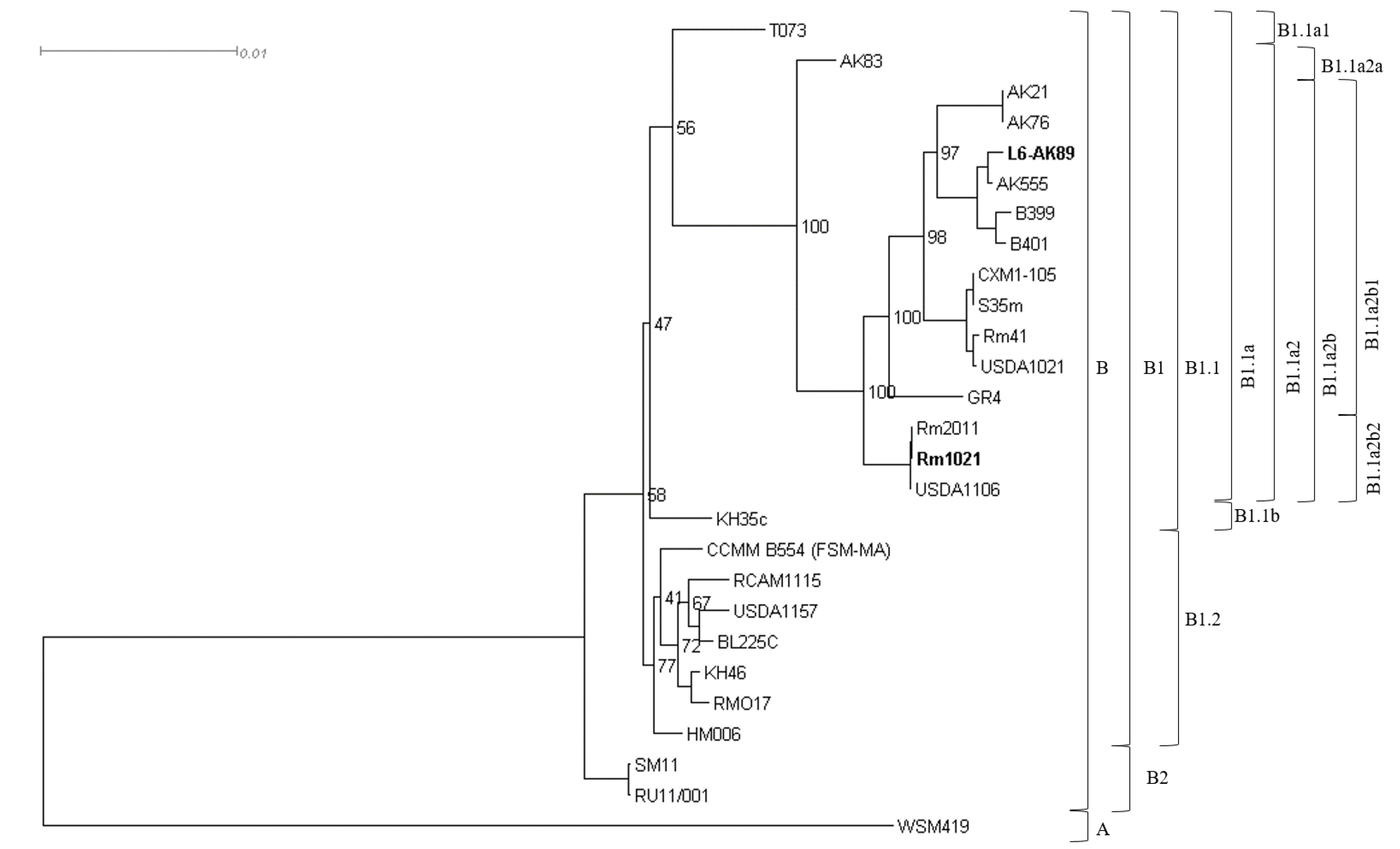
2.7. Phylogenetic Analysis
2.8. Microarray Analysis
3. Results and Discussion
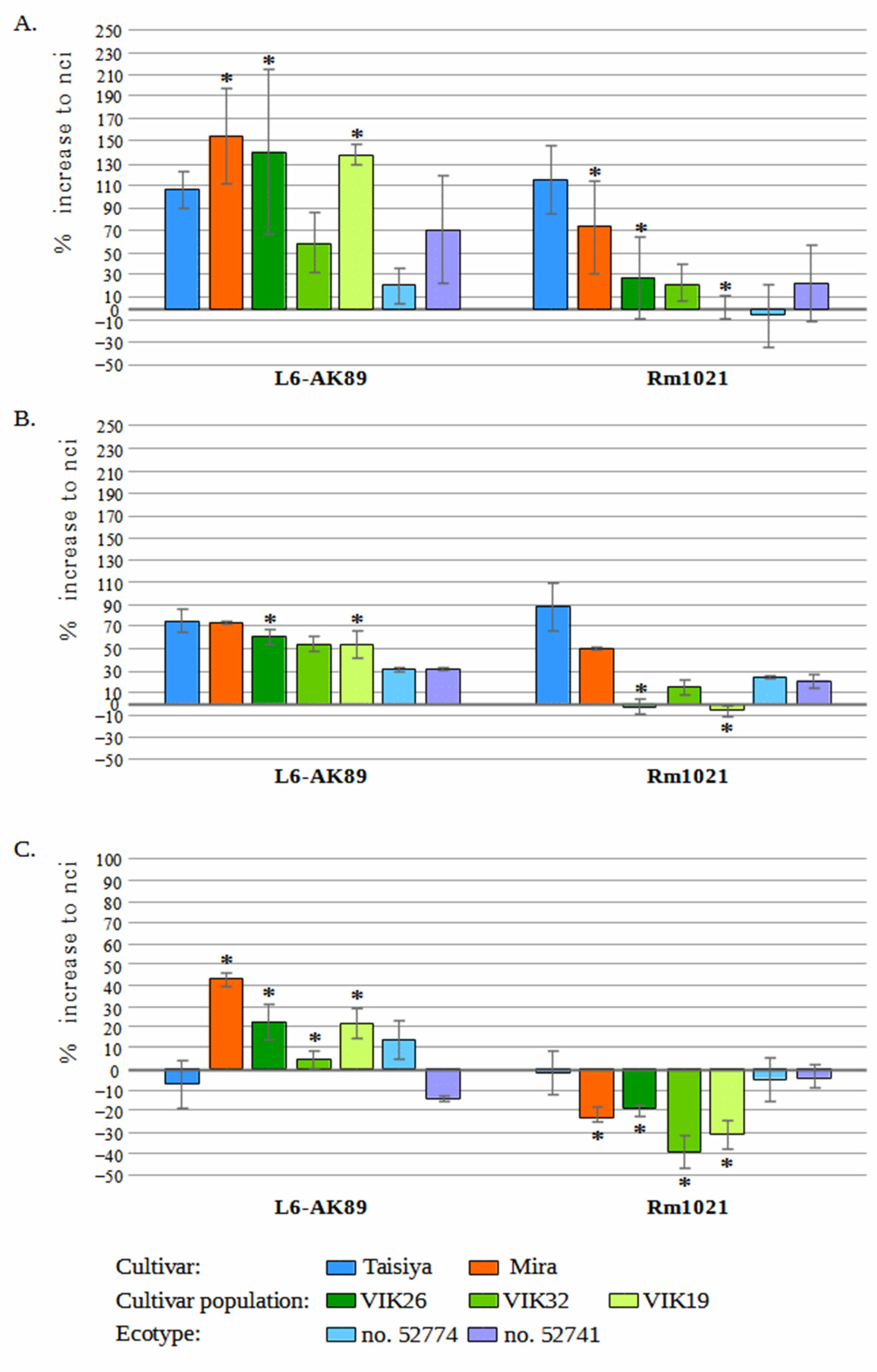
3.1. L6-AK89 in Symbiosis with Medicago spp.
3.2. L6-AK89 Genome Characteristics
3.3. Analysis of Coding Sequences
3.4. Mobilome of L6-AK89
3.4.1. Phage-Related Sequences Analysis
3.4.2. CRISPR/Cas Analysis
3.4.3. Abundance of IS Elements in Genome of L6-AK89
3.4.4. Potential Effect of IS Elements on Adjacent Genes in “Head-to-Tail” Position
3.5. Fitness Genes
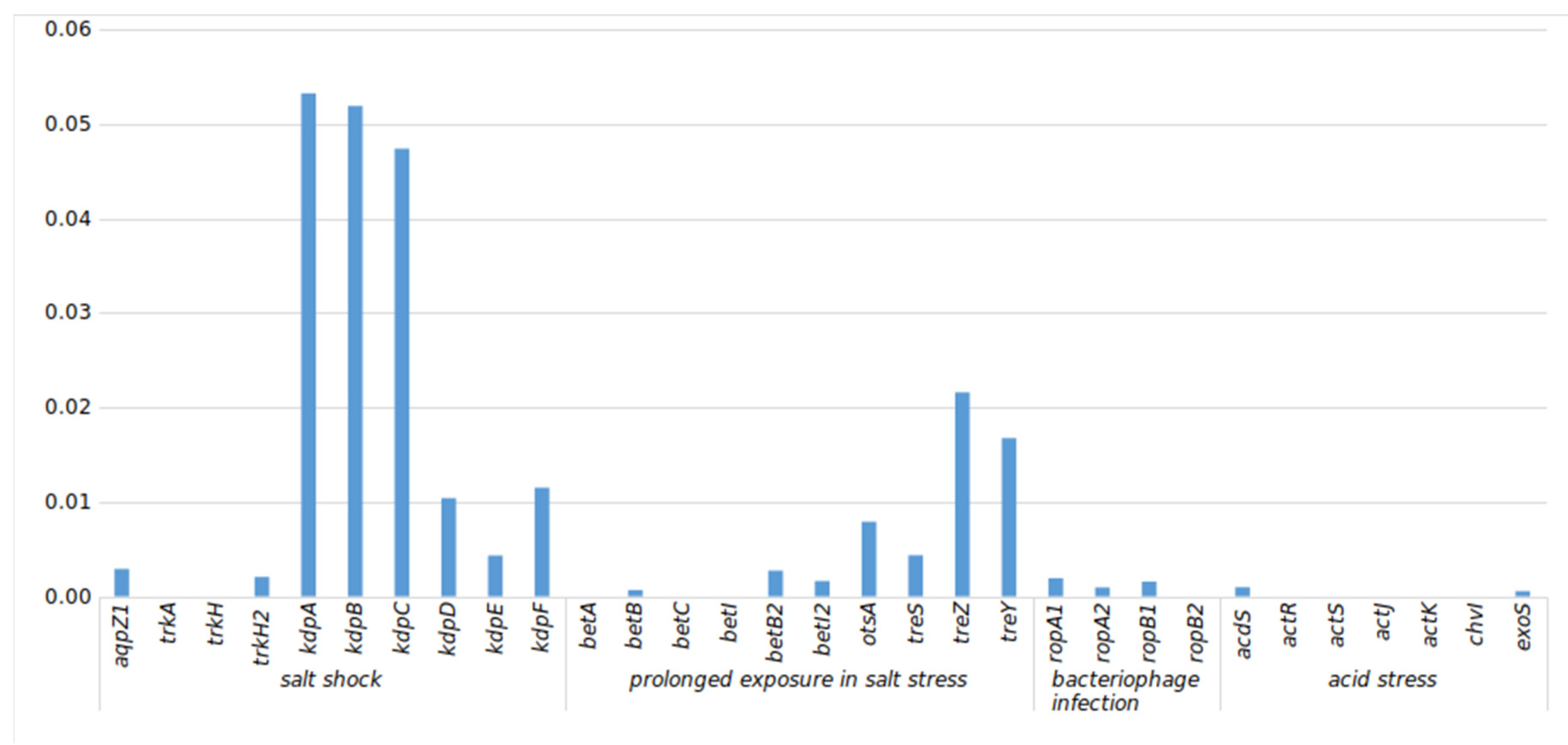
3.5.1. Stress-Related (str) Genes
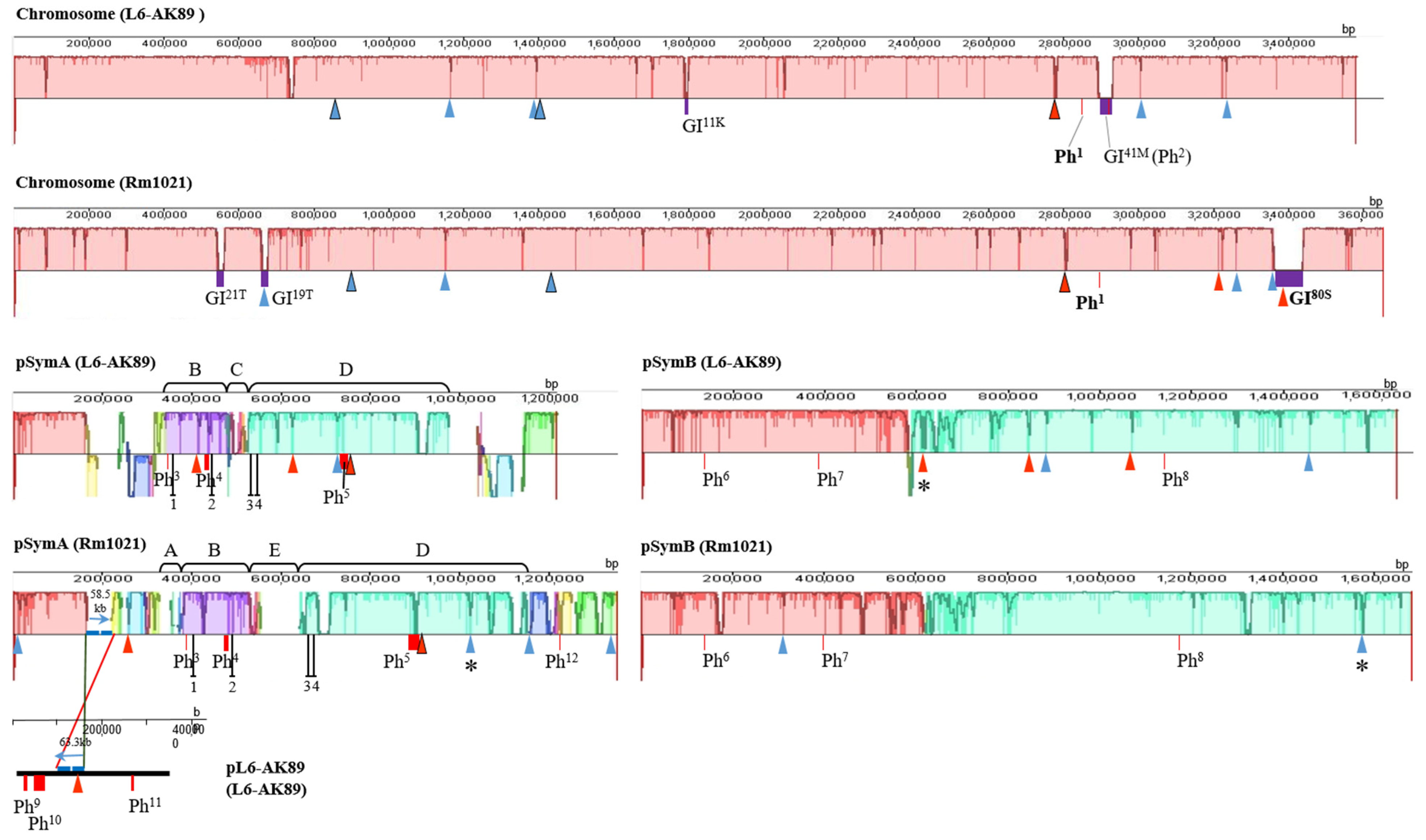
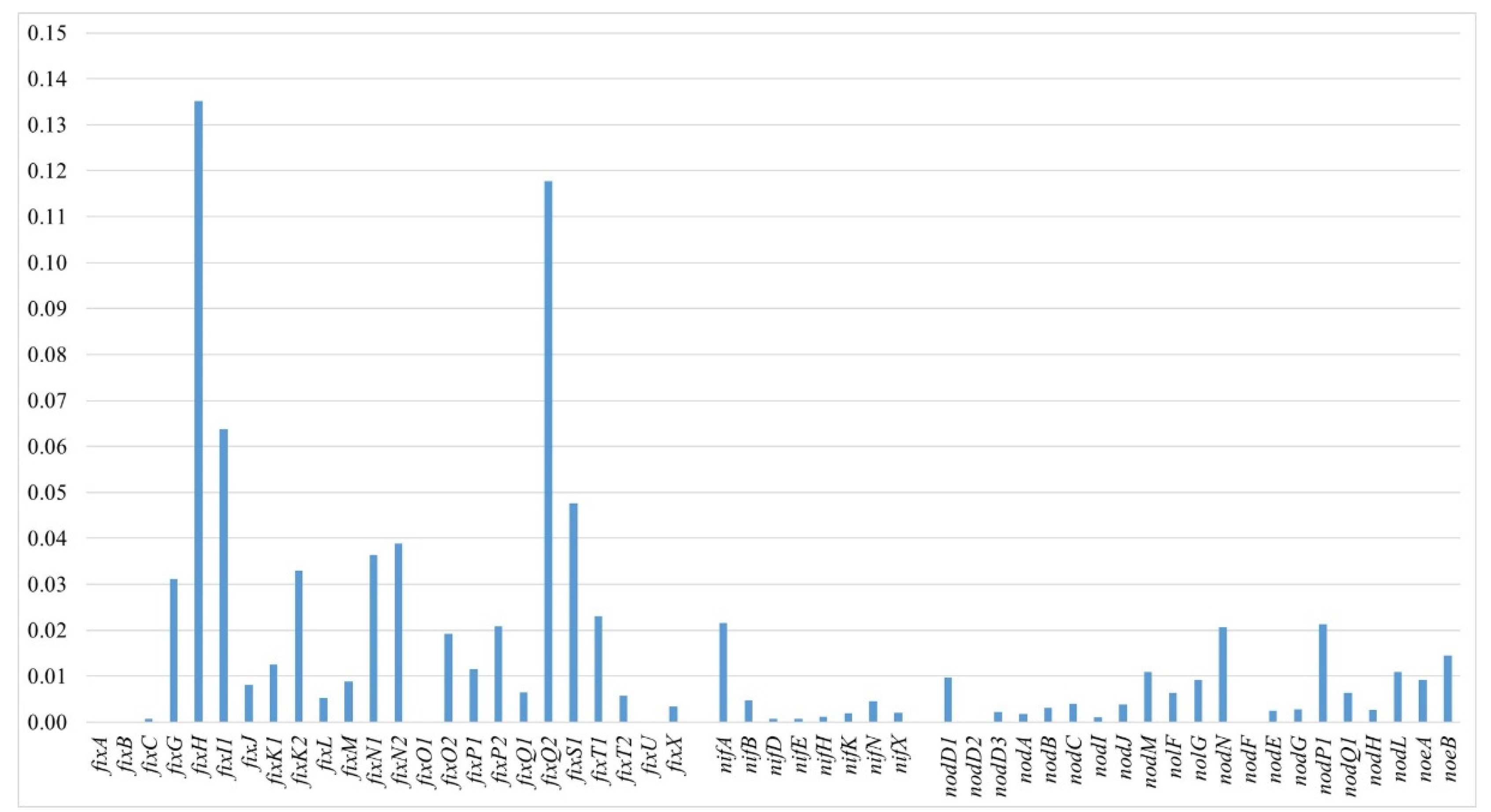
3.5.2. Sym Region Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Amer, N.; Al Chami, Z.; Al Bitar, L.; Mondelli, D.; Dumontet, S. Evaluation of Atriplex halimus, Medicago lupulina and Portulaca oleracea for phytoremediation of Ni, Pb, and Zn. Int. J. Phytoremediation 2013, 15, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Jakupović, L.; Kalvarešin, M.; Bukovina, K.; Poljak, V.; Vujić, L.; Zovko Končić, M. Optimization of two eco-friendly extractions of black medick (Medicago lupulina L.) phenols and their antioxidant, cosmeceutical, α-glucosidase and α-amylase inhibitory properties. Molecules 2021, 26, 1610. [Google Scholar] [CrossRef] [PubMed]
- Butkutė, B.; Padarauskas, A.; Cesevičienė, J.; Pavilonis, A.; Taujenis, L.; Lemežienė, N. Perennial legumes as a source of ingredients for healthy food: Proximate, mineral and phytoestrogen composition and antibacterial activity. J. Food Sci. Technol. 2017, 54, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Vorsheva, A.V.; Stepanova, G.V. Estimation of the productivity and nutritionality of forage mass of new varieties of Medicago lupulina. In Proceedings of the Plant Growing and Meadow Growing, Moscow, Russia, 18–19 November 2020; RSAU—MTAA: Moscow, Russia, 2020; pp. 628–631. (In Russian) [Google Scholar] [CrossRef]
- Stepanova, G.V.; Vorsheva, A.V. Formation of bicarp populations of hop medic. In Multifunctional Adaptive Fodder Production, 25th ed.; Cherniavskih, V.I., Kosolapov, V.M., Shpakov, A.S., Kutuzova, A.A., Pobednov, Y.A., Kostenko, S.I., Georgiadi, N.I., Svechnikova, G.N., Eds.; FSBOU DPO RAKO APK: Moscow, Russia, 2021; pp. 9–20. (In Russian) [Google Scholar]
- Bailly, X.; Giuntini, E.; Sexton, M.C.; Lower, R.P.; Harrison, P.W.; Kumar, N.; Young, J.P. Population genomics of Sinorhizobium medicae based on low-coverage sequencing of sympatric isolates. ISME J. 2011, 5, 1722–1734. [Google Scholar] [CrossRef]
- Harrison, T.L.; Wood, C.W.; Heath, K.D.; Stinchcombe, J.R. Geographically structured genetic variation in the Medicago lupulina-Ensifer mutualism. Evolution 2017, 71, 1787–1801. [Google Scholar] [CrossRef]
- Boyle, J.A.; Simonsen, A.K.; Frederickson, M.E.; Stinchcombe, J.R. Priority effects alter interaction outcomes in a legume-rhizobium mutualism. Proc. Biol. Sci. 2021, 288, 20202753. [Google Scholar] [CrossRef]
- Martens, M.; Delaere, M.; Coopman, R.; De Vos, P.; Gillis, M.; Willems, A. Multilocus sequence analysis of Ensifer and related taxa. Int. J. Syst. Evol. Microbiol. 2007, 57, 489–503. [Google Scholar] [CrossRef]
- Harrison, T.L.; Wood, C.W.; Borges, I.L.; Stinchcombe, J.R. No evidence for adaptation to local rhizobial mutualists in the legume Medicago lupulina. Ecol. Evol. 2017, 7, 4367–4376. [Google Scholar] [CrossRef]
- Biondi, E.G.; Pilli, E.; Giuntini, E.; Roumiantseva, M.L.; Andronov, E.E.; Onichtchouk, O.P.; Kurchak, O.N.; Simarov, B.V.; Dzyubenko, N.I.; Mengoni, A.; et al. Genetic relationship of Sinorhizobium meliloti and Sinorhizobium medicae strains isolated from Caucasian region. FEMS Microbiol. Lett. 2003, 220, 207–213. [Google Scholar] [CrossRef]
- Singh, R.; Interrante, S.M.; Butler, T.J.; Young, C.A. Characterization and effectiveness of co-inoculation of Sinorhizobium strains on annual medics. Crop Sci. 2012, 52, 932–942. [Google Scholar] [CrossRef]
- Roberts, R.; Jackson, R.W.; Mauchline, T.H.; Hirsch, P.R.; Shaw, L.J.; Döring, T.F.; Jones, H.E. Is there sufficient Ensifer and Rhizobium species diversity in UK farmland soils to support red clover (Trifolium pratense), white clover (T. repens), lucerne (Medicago sativa) and black medic (M. lupulina)? Appl. Soil Ecol. 2017, 120, 35–43. [Google Scholar] [CrossRef]
- Young, J.P.W.; Crossman, L.C.; Johnston, A.W.B.; Thomson, N.R.; Ghazoui, Z.F.; Hull, K.H.; Wexler, M.; Curson, A.R.J.; Todd, J.D.; Poole, P.S.; et al. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 2006, 7, R34. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Segerman, B. The genetic integrity of bacterial species: The core genome and the accessory genome, two different stories. Front. Cell. Infect. Microbiol. 2012, 2, 116. [Google Scholar] [CrossRef]
- Zhaxybayeva, O.; Lapierre, P.; Gogarten, J.P. Genome mosaicism and organismal lineages. Trends Genet. 2004, 20, 254–260. [Google Scholar] [CrossRef]
- Sim, S.H.; Yu, Y.; Lin, C.H.; Karuturi, R.K.; Wuthiekanun, V.; Tuanyok, A.; Chua, H.H.; Ong, C.; Paramalingam, S.S.; Tan, G.; et al. The core and accessory genomes of Burkholderia pseudomallei: Implications for human melioidosis. PLoS Pathog. 2008, 4, e1000178. [Google Scholar] [CrossRef]
- Mauchline, T.H.; Hayat, R.; Roberts, R.; Powers, S.J.; Hirsch, P.R. Assessment of core and accessory genetic variation in Rhizobium leguminosarum symbiovar trifolii strains from diverse locations and host plants using PCR-based methods. Lett. Appl. Microbiol. 2014, 59, 238–246. [Google Scholar] [CrossRef]
- Jiao, J.; Ni, M.; Zhang, B.; Young, J.P.W.; Chan, T.-F. Coordinated regulation of core and accessory genes in the multipartite genome of Sinorhizobium fredii. PLoS Genet. 2018, 14, e1007428. [Google Scholar] [CrossRef]
- Prozorov, A.A. Regularities of the location of genes having different functions and of some other nucleotide sequences in the bacterial chromosome. Microbiology 2007, 76, 383–392. (In Russian) [Google Scholar] [CrossRef]
- Oliveira, P.H.; Touchon, M.; Cury, J.; Rocha, E.P.C. The chromosomal organization of horizontal gene transfer in bacteria. Nat. Commun. 2017, 8, 841. [Google Scholar] [CrossRef]
- Kivisaar, M. Mutation and recombination rates vary across bacterial chromosome. Microorganisms 2019, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; de Lorenzo, V.; Cebolla, A. Modulation of gene expression through chromosomal positioning in Escherichia coli. Microbiology 1997, 143, 2071–2078. [Google Scholar] [CrossRef] [PubMed]
- Block, D.H.; Hussein, R.; Liang, L.W.; Lim, H.N. Regulatory consequences of gene translocation in bacteria. Nucleic Acids Res. 2012, 40, 8979–8992. [Google Scholar] [CrossRef] [PubMed]
- Kopejtka, K.; Lin, Y.; Jakubovicova, M.; Koblizek, M.; Tomasch, J. Clustered core- and pan-genome content on Rhodobacteraceae chromosomes. Genome Biol. Evol. 2019, 11, 2208–2217. [Google Scholar] [CrossRef]
- Mira, A.; Ochman, H. Gene location and bacterial sequence divergence. Mol. Biol. Evol. 2002, 19, 1350–1358. [Google Scholar] [CrossRef]
- Juurik, T.; Ilves, H.; Teras, R.; Ilmjarv, T.; Tavita, K.; Ukkivi, K.; Kivisaar, M. Mutation frequency and spectrum of mutations vary at different chromosomal positions of Pseudomonas putida. PLoS ONE 2012, 7, e48511. [Google Scholar] [CrossRef]
- Lobry, J.R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996, 13, 660–665. [Google Scholar] [CrossRef]
- Salzberg, S.L.; Salzberg, A.J.; Kerlavage, A.R.; Tomb, J.F. Skewed oligomers and origins of replication. Gene 1998, 217, 57–67. [Google Scholar] [CrossRef]
- Lobry, J.R.; Louarn, J.M. Polarisation of prokaryotic chromosomes. Curr. Opin. Microbiol. 2003, 6, 101–108. [Google Scholar] [CrossRef]
- Song, J.; Ware, A.; Liu, S.L. Wavelet to predict bacterial ori and ter: A tendency towards a physical balance. BMC Genom. 2003, 4, 17. [Google Scholar] [CrossRef]
- Lesterlin, C.; Pages, C.; Dubarry, N.; Dasgupta, S.; Cornet, F. Asymmetry of chromosome replichores renders the DNA translocase activity of FtsK essential for cell division and cell shape maintenance in Escherichia coli. PLoS Genet. 2008, 4, e1000288. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K.; Sato, M.; Yoshikawa, H.; Tomita, M.; Itaya, M. Undesigned selection for replication termination of bacterial chromosomes. J. Mol. Biol. 2014, 426, 2918–2927. [Google Scholar] [CrossRef]
- Galardini, M.; Pini, F.; Bazzicalupo, M.; Biondi, E.G.; Mengoni, A. Replicon-dependent bacterial genome evolution: The case of Sinorhizobium meliloti. Genome Biol. Evol. 2013, 5, 542–558. [Google Scholar] [CrossRef]
- Lagares, A.; Sanjuán, J.; Pistorio, M. The plasmid mobilome of the model plant-symbiont Sinorhizobium meliloti: Coming up with new questions and answers. In Plasmids; Tolmasky, M.E., Alonso, J.C., Eds.; ASM Press: Washington, DC, USA, 2015; pp. 277–293. [Google Scholar] [CrossRef]
- Oresnik, I.J.; Liu, S.L.; Yost, C.K.; Hynes, M.F. Megaplasmid pRme2011a of Sinorhizobium meliloti is not required for viability. J. Bacteriol. 2000, 182, 3582–3586. [Google Scholar] [CrossRef]
- Guo, X.; Flores, M.; Mavingui, P.; Fuentes, S.I.; Hernández, G.; Dávila, G.; Palacios, R. Natural genomic design in Sinorhizobium meliloti: Novel genomic architectures. Genome Res. 2003, 13, 1810–1817. [Google Scholar] [CrossRef]
- Pistorio, M.; Giusti, M.A.; Del Papa, M.F.; Draghi, W.O.; Lozano, M.J.; Tejerizo, G.T.; Lagares, A. Conjugal properties of the Sinorhizobium meliloti plasmid mobilome. FEMS Microbiol. Ecol. 2008, 65, 372–382. [Google Scholar] [CrossRef]
- Nogales, J.; Blanca-Ordóñez, H.; Olivares, J.; Sanjuán, J. Conjugal transfer of the Sinorhizobium meliloti 1021 symbiotic plasmid is governed through the concerted action of one- and two-component signal transduction regulators. Environ. Microbiol. 2013, 15, 811–821. [Google Scholar] [CrossRef]
- Velázquez, E.; Mateos, P.F.; Pedrero, P.; Dazzo, F.B.; Martinez-Molina, E. Attenuation of symbiotic effectiveness by Rhizobium meliloti SAF22 related to the presence of a cryptic plasmid. Appl. Environ. Microbiol. 1995, 61, 2033–2036. [Google Scholar] [CrossRef]
- Barran, L.R.; Ritchot, N.; Bromfield, E.S. Sinorhizobium meliloti plasmid pRm1132f replicates by a rolling-circle mechanism. J. Bacteriol. 2001, 183, 2704–2708. [Google Scholar] [CrossRef]
- Roumiantseva, M.L.; Onishchuk, O.P.; Belova, V.S.; Kurchak, O.N.; Simarov, B.V. Polymorphism of Sinorhizobium meliloti strains isolated from diversity centers of alfalfa in various soil and climatic conditions, Russ. J. Genet. Appl. Res. 2011, 1, 97–102. [Google Scholar] [CrossRef]
- Saksaganskaia, A.; Vladimirova, M.; Afonin, A.; Simarov, B.; Roumiantseva, M. Cryptic plasmids essential for Sinorhizobium meliloti fitness. In Proceedings of the 20th International Multidisciplinary Scientific GeoConference SGEM 2020, Varna, Bulgaria, 18–24 August 2020; STEF92 Technology Ltd.: Sofia, Bulgaria, 2020; pp. 223–230. [Google Scholar] [CrossRef]
- Saksaganskaia, A.; Muntyan, V.; Muntyan, A.; Afonin, A.; Simarov, B.; Roumiantseva, M. Are cryptic plasmids of Sinorhizobium meliloti attractive within phage infection? FEBS Open Bio 2021, 11, 133. [Google Scholar] [CrossRef]
- Stiens, M.; Schneiker, S.; Keller, M.; Kuhn, S.; Pühler, A.; Schlüter, A. Sequence analysis of the 144-kilobase accessory plasmid pSmeSM11a, isolated from a dominant Sinorhizobium meliloti strain identified during a long-term field release experiment. Appl. Environ. Microbiol. 2006, 72, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Stiens, M.; Schneiker, S.; Puhler, A.; Schlüter, A. Sequence analysis of the 181-kb accessory plasmid pSmeSM11b, isolated from a dominant Sinorhizobium meliloti strain identified during a long-term field release experiment. FEMS Microbiol. Lett. 2007, 271, 297–309. [Google Scholar] [CrossRef] [PubMed]
- López, J.L.; Lozano, M.J.; Lagares, A., Jr.; Fabre, M.L.; Draghi, W.O.; Del Papa, M.F.; Pistorio, M.; Becker, A.; Wibberg, D.; Schlüter, A.; et al. Codon usage heterogeneity in the multipartite prokaryote genome: Selection-based coding bias associated with gene location, expression level, and ancestry. MBio 2019, 10, e00505-19. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hochhut, B.; Hentschel, U.; Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004, 2, 414–424. [Google Scholar] [CrossRef]
- Frost, L.S.; Leplae, R.; Summers, A.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar] [CrossRef]
- Stokes, H.W.; Gillings, M.R. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 2011, 35, 790–819. [Google Scholar] [CrossRef]
- Jørgensen, T.S.; Kiil, A.S.; Hansen, M.A.; Sørensen, S.J.; Hansen, L.H. Current strategies for mobilome research. Front. Microbiol. 2015, 5, 750. [Google Scholar] [CrossRef]
- Rodríguez-Beltrán, J.; Sørum, V.; Toll-Riera, M.; de la Vega, C.; Peña-Miller, R.; San Millán, Á. Genetic dominance governs the evolution and spread of mobile genetic elements in bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 15755–15762. [Google Scholar] [CrossRef]
- Di Cenzo, G.C.; Checcucci, A.; Bazzicalupo, M.; Mengoni, A.; Viti, C.; Dziewit, L.; Finan, T.M.; Galardini, M.; Fondi, M. Metabolic modelling reveals the specialization of secondary replicons for niche adaptation in Sinorhizobium meliloti. Nat. Commun. 2016, 7, 12219. [Google Scholar] [CrossRef]
- Poole, P.; Ramachandran, V.; Terpolilli, J. Rhizobia: From saprophytes to endosymbionts. Nat. Rev. Microbiol. 2018, 16, 291–303. [Google Scholar] [CrossRef]
- Bernheim, A.; Sorek, R. The pan-immune system of bacteria: Antiviral defence as a community resource. Nat. Rev. Microbiol. 2020, 18, 113–119. [Google Scholar] [CrossRef]
- Vladimirova, M.; Muntyan, V.; Kozlova, A.; Grudinin, M.; Roumiantseva, M. CRISPR/Cas in genomes of Sinorhizobium meliloti. In Proceedings of the 20th International Multidisciplinary Scientific GeoConference SGEM 2020, Varna, Bulgaria, 18–24 August 2020; STEF92 Technology Ltd.: Sofia, Bulgaria, 2020; pp. 215–222. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, J.; Wang, B.; Han, J.; Hao, Y.; Wang, S.; Ma, X.; Yang, S.; Ma, L.; Yi, L.; et al. Endogenous type I CRISPR-Cas: From foreign DNA defense to prokaryotic engineering. Front. Bioeng. Biotechnol. 2020, 8, 62. [Google Scholar] [CrossRef]
- Pinilla-Redondo, R.; Russel, J.; Mayo-Muñoz, D.; Shah, S.A.; Garrett, R.A.; Nesme, J.; Madsen, J.S.; Fineran, P.C.; Sørensen, S.J. CRISPR-Cas systems are widespread accessory elements across bacterial and archaeal plasmids. Nucleic Acids Res. 2021, 1, gkab859. [Google Scholar] [CrossRef]
- Crook, M.B.; Draper, A.L.; Guillory, R.J.; Griffitts, J.S. The Sinorhizobium meliloti essential porin RopA1 is a target for numerous bacteriophages. J. Bacteriol. 2013, 95, 3663–3671. [Google Scholar] [CrossRef]
- Rüberg, S.; Tian, Z.X.; Krol, E.; Linke, B.; Meyer, F.; Wang, Y. Construction and validation of a Sinorhizobium meliloti whole genome DNA microarray: Genome-wide profiling of osmoadaptive gene expression. J. Biotechnol. 2003, 106, 255–268. [Google Scholar] [CrossRef]
- Domínguez-Ferreras, A.; Pérez-Arnedo, R.; Becker, A.; Olivares, J.; Soto, M.J.; Sanjuán, J. Transcriptome profiling reveals the importance of plasmid pSymB for osmoadaptation of Sinorhizobium meliloti. J. Bacteriol. 2006, 188, 7617–7625. [Google Scholar] [CrossRef]
- Hellweg, C.; Pühler, A.; Weidner, S. The time course of the transcriptomic response of Sinorhizobium meliloti 1021 following a shift to acidic pH. BMC Microbiol. 2009, 15, 37. [Google Scholar] [CrossRef]
- Tiwari, R.P.; Reeve, W.G.; Dilworth, M.J.; Glenn, A.R. Acid tolerance in Rhizobium meliloti strain WSM419 involves a two-component sensor-regulator system. Microbiology 1996, 142, 1693–1704. [Google Scholar] [CrossRef]
- Domínguez-Ferreras, A.; Muñoz, S.; Olivares, J.; Soto, M.J.; Sanjuán, J. Role of potassium uptake systems in Sinorhizobium meliloti osmoadaptation and symbiotic performance. J. Bacteriol. 2009, 191, 2133–2143. [Google Scholar] [CrossRef]
- Domínguez-Ferreras, A.; Soto, M.J.; Pérez-Arnedo, R.; Olivares, J.; Sanjuán, J. Importance of trehalose biosynthesis for Sinorhizobium meliloti osmotolerance and nodulation of alfalfa roots. J. Bacteriol. 2009, 191, 7490–7499. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kemp, J.; Da Fonseca, I.O.; Equi, R.C.; Sheng, X.; Charles, T.C.; Sobral, B.W.S. Sinorhizobium meliloti 1021 loss-of-function deletion mutation in chvI and its phenotypic characteristics. Mol. Plant Microbe Interact. 2010, 23, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Da-Silva, J.R.; Alexandre, A.; Brígido, C.; Oliveira, S. Can stress response genes be used to improve the symbiotic performance of rhizobia? AIMS Microbiol. 2017, 3, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Albicoro, F.J.; Draghi, W.; Martini, M.C.; Salas, M.E.; Torres Tejerizo, G.T.; Lozano, M.; Lopez, J.L.; Vacca, C.; Cafiero, J.H.; Pistorio, M.; et al. The two-component system ActJK is involved in acid stress tolerance and symbiosis in Sinorhizobium meliloti. J. Biotechnol. 2021, 329, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Rutten, P.J.; Poole, P.S. Oxygen regulatory mechanisms of nitrogen fixation in rhizobia. Adv. Microb. Physiol. 2019, 75, 325–389. [Google Scholar] [CrossRef] [PubMed]
- Lindström, K.; Mousavi, S.A. Effectiveness of nitrogen fixation in rhizobia. Microb. Biotechnol. 2020, 13, 1314–1335. [Google Scholar] [CrossRef]
- Barnett, M.J.; Long, S.R. The Sinorhizobium meliloti SyrM regulon: Effects on global gene expression are mediated by syrA and nodD3. J. Bacteriol. 2015, 197, 1792–1806. [Google Scholar] [CrossRef]
- Debellé, F.; Moulin, L.; Mangin, B.; Dénarié, J.; Boivin, C. Nod genes and Nod signals and the evolution of the Rhizobium legume symbiosis. Acta Biochim. Pol. 2001, 48, 359–365. [Google Scholar] [CrossRef]
- Aoki, S.; Ito, M.; Iwasaki, W. From β- to α-proteobacteria: The origin and evolution of rhizobial nodulation genes nodIJ. Mol. Biol. Evol. 2013, 30, 2494–2508. [Google Scholar] [CrossRef][Green Version]
- Geddes, B.A.; Kearsley, J.; Morton, R.; diCenzo, G.C.; Finan, T.M. The Genomes of Rhizobia. In Advances in Botanical Research; Elsevier: Amsterdam, The Netherlands, 2020; Volume 94, pp. 213–249. ISBN 978-0-08-102798-1. [Google Scholar]
- Ardourel, M.; Lortet, G.; Maillet, F.; Roche, P.; Truchet, G.; Promé, J.C.; Rosenberg, C. In Rhizobium meliloti, the operon associated with the nod box n5 comprises nodL, noeA and noeB, three host-range genes specifically required for the nodulation of particular Medicago species. Mol. Microbiol. 1995, 17, 687–699. [Google Scholar] [CrossRef]
- Bena, G. Molecular phylogeny supports the morphologically based taxonomic transfer of the “medicagoid” Trigonella species to the genus Medicago L. Plant Syst. Evol. 2001, 229, 217–236. [Google Scholar] [CrossRef]
- Roumiantseva, M.L.; Vladimirova, M.E.; Muntyan, V.S.; Stepanova, G.V.; Saksaganskaya, A.S.; Kozhemyakov, A.P.; Orlova, A.G.; Becker, A.; Simarov, B.V. Highly effective root nodule inoculants of alfalfa (Medicago varia L.): Molecular-genetic analysis and practical usage in cultivar creation. Agric. Biol. 2019, 54, 1306–1323. [Google Scholar] [CrossRef]
- Reeve, W.; Ardley, J.; Tian, R.; Eshragi, L.; Yoon, J.W.; Ngamwisetkun, P.; Seshadri, R.; Ivanova, N.N.; Kyrpides, N.C. A Genomic Encyclopedia of the Root Nodule Bacteria: Assessing genetic diversity through a systematic biogeographic survey. Stand. Genom. Sci. 2015, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Ibragimova, M.V.; Rumyantseva, M.L.; Onishchuk, O.P.; Belova, V.S.; Kurchak, O.N.; Andronov, E.E.; Dziubenko, N.I.; Simarov, B.V. Symbiosis between the root-nodule bacterium Sinorhizobium meliloti and alfalfa (Medicago sativa) under salinization conditions. Microbiology 2006, 75, 77–81. [Google Scholar] [CrossRef]
- Greene, S.L.; Kisha, T.J.; Dzyubenko, N.I. Conserving alfalfa wild relatives: Is past introgression with Russian varieties evident today? Crop Sci. 2008, 48, 1853–1864. [Google Scholar] [CrossRef]
- Roumiantseva, M.L.; Muntyan, V.S. Root nodule bacteria Sinorhizobium meliloti: Tolerance to salinity and bacterial genetic determinants. Microbiology 2015, 84, 303–318. [Google Scholar] [CrossRef]
- Vincent, J.M. Serological studies of the root-nodule bacteria. I. Strains of Rhizobium meliloti. Proc. Linn. Soc. N.S.W. 1941, 66, 145–154. [Google Scholar]
- Beringer, J.E. R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 1974, 84, 188–198. [Google Scholar] [CrossRef]
- Clokie, M.R.J.; Kropinski, A.M. (Eds.) Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions, 1st ed.; Humana Press: New York, NY, USA, 2009; pp. 69–77. [Google Scholar] [CrossRef]
- Rumyantseva, M.L.; Simarov, B.V.; Onishchuk, O.P.; Andronov, E.E.; Chizhevskaya, E.P.; Belova, V.S.; Kurchak, O.N.; Muntyan, A.N.; Rumyantseva, T.B.; Zatovskaya, T.V. Biodiversity of Rhizobia in Ecosystems and Agrocenoses: Theoretical Bases and Methods, 1st ed.; ICZR: St. Petersburg, Russia, 2011; pp. 21–85. (In Russian) [Google Scholar]
- Smurygin, M.A.; Novoselova, A.S.; Konstantinova, A.M.; Rubcov, M.I.; Filimonov, M.A.; Bekhtin, N.S.; Voshchinin, P.A.; Ezhakova, O.F.; Katkov, V.A.; Kuleshov, G.F.; et al. Guidelines for the Selection of Perennial Grasses; FWRC FPA: Moscow, Russia, 1985; pp. 98–99. (In Russian) [Google Scholar]
- Roumiantseva, M.L.; Stepanova, G.V.; Kurchak, O.N.; Onishchuk, O.P.; Muntyan, V.S.; Dzyubenko, E.A.; Dzyubenko, N.I.; Simarov, B.V. Selection of salt tolerant alfalfa (Medicago L.) plants from different varieties and their morfo biological and symbiotic properties analysis. Agric. Biol. 2015, 50, 673–684. [Google Scholar] [CrossRef]
- Provorov, N.A.; Onishchuk, O.P.; Kurchak, O.N. Impacts of inoculation with Sinorhizobium meliloti strains differing in salt tolerance on the productivity and habitus of alfalfa (Medicago sativa L.). Agric. Biol. 2016, 51, 343–350. [Google Scholar] [CrossRef][Green Version]
- Lakin, G.F. Biometrics, 4th ed.; Vysshaya Shkola: Moscow, Russia, 1990; pp. 159–179. (In Russian) [Google Scholar]
- Green, M.R.; Sambrook, J. Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb. Protoc. 2017, 3, pdb-prot093450. [Google Scholar] [CrossRef]
- Afonin, A.M.; Gribchenko, E.S.; Sulima, A.S.; Zhukov, V.A. Complete genome sequence of an efficient Rhizobium leguminosarum bv. viciae strain, A1. Microbiol. Resour. Announc. 2020, 9, e00249-20. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Deepbinner: Demultiplexing barcoded Oxford Nanopore reads with deep convolutional neural networks. PLoS Comput. Biol. 2018, 14, e1006583. [Google Scholar] [CrossRef]
- Porechop. Available online: https://github.com/rrwick/Porechop (accessed on 20 October 2021).
- Filtlong. Available online: https://github.com/rrwick/Filtlong (accessed on 20 October 2021).
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies. Medaka. Available online: https://github.com/nanoporetech/medaka (accessed on 20 October 2021).
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Haft, D.H.; DiCuccio, M.; Badretdin, A.; Brover, V.; Chetvernin, V.; O’Neill, K.; Li, W.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; et al. RefSeq: An update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018, 46, D851–D860. [Google Scholar] [CrossRef]
- Li, W.; O’Neill, K.R.; Haft, D.H.; DiCuccio, M.; Chetvernin, V.; Badretdin, A.; Coulouris, G.; Chitsaz, F.; Derbyshire, M.K.; Durkin, A.S.; et al. RefSeq: Expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 2021, 49, D1020–D1028. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.M.; Lau, B.Y.; Williams, K.P. Islander: A database of precisely mapped genomic islands in tRNA and tmRNA genes. Nucleic Acids Res. 2015, 43, D48–D53. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Search and contextual analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef]
- Puterová, J.; Martínek, T. digIS: Towards detecting distant and putative novel insertion sequence elements in prokaryotic genomes. BMC Bioinform. 2021, 22, 258. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Li, X.; Xie, Y.; Liu, M.; Tai, C.; Sun, J.; Deng, Z.; Ou, H.Y. oriTfinder: A web-based tool for the identification of origin of transfers in DNA sequences of bacterial mobile genetic elements. Nucleic Acids Res. 2018, 46, W229–W234. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- De Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef]
- Bláha, L. Importance of root—shoot ratio for crops production. J. Agron. Agri. Sci. 2019, 2, 12. [Google Scholar] [CrossRef]
- Allito, B.B.; Ewusi-Mensah, N.; Logah, V.; Hunegnaw, D.K. Legume-rhizobium specificity effect on nodulation, biomass production and partitioning of faba bean (Vicia faba L.). Sci. Rep. 2021, 11, 3678. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.G.; Hendrickson, H. Lateral gene transfer: When will adolescence end? Mol. Microbiol. 2003, 50, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Young, J.P.W.; Wexler, M. Sym plasmid and chromosomal genotypes are correlated in field populations of Rhizobium leguminosarum. J. Gen. Microbiol. 1988, 134, 2731–2739. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.M.; Tian, C.F.; Sui, X.H. Robust markers reflecting phylogeny and taxonomy of rhizobia. PLoS ONE 2012, 7, e44936. [Google Scholar] [CrossRef]
- Guo, H.J.; Wang, E.T.; Zhang, X.X.; Li, Q.Q.; Zhang, Y.M.; Tian, C.F.; Chen, W.X. Replicon-dependent differentiation of symbiosis-related genes in Sinorhizobium strains nodulating Glycine max. Appl. Environ. Microbiol. 2014, 80, 1245–1255. [Google Scholar] [CrossRef]
- Stefan, A.; Van Cauwenberghe, J.; Rosu, C.M.; Stedel, C.; Labrou, N.E.; Flemetakis, E.; Efrose, R.C. Genetic diversity and structure of Rhizobium leguminosarum populations associated with clover plants are influenced by local environmental variables. Syst. Appl. Microbiol. 2018, 41, 251–259. [Google Scholar] [CrossRef]
- Cherkasova, M.E.; Muntyan, V.S.; Saksaganskaia, A.S.; Simarov, B.V.; Roumiantseva, M.L. Sinorhizobium meliloti: Chromosomal types and genomic islands. Ecol. Genet. 2019, 17, 23–38. [Google Scholar] [CrossRef]
- Roumiantseva, M.L.; Muntyan, V.S.; Cherkasova, M.E.; Andronov, E.E.; Saksaganskaya, A.S.; Dzyubenko, E.A.; Dzyubenko, N.I.; Simarov, B.V. A comparative analysis of genomic characters of reference Sinorhizobium meliloti strains, the alfalfa symbionts (review). Agric. Biol. 2017, 52, 928–939. [Google Scholar] [CrossRef]
- Wang, B.; Guo, F.; Huang, C.; Zhao, H. Unraveling the iterative type I polyketide synthases hidden in Streptomyces. Proc. Natl. Acad. Sci. USA 2020, 117, 8449–8454. [Google Scholar] [CrossRef]
- Strieker, M.; Tanović, A.; Marahiel, M.A. Nonribosomal peptide synthetases: Structures and dynamics. Curr. Opin. Struct. Biol. 2010, 20, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.; O’Brien, J.; Welch, T.; Clarke, P.; Cuív, P.O.; Crosa, J.H.; O’Connell, M. Genetic organization of the region encoding regulation, biosynthesis, and transport of rhizobactin 1021, a siderophore produced by Sinorhizobium meliloti. J. Bacteriol. 2001, 8, 2576–2585. [Google Scholar] [CrossRef] [PubMed]
- Sagot, B.; Gaysinski, M.; Mehiri, M.; Guigonis, J.M.; Le Rudulier, D.; Alloing, G. Osmotically induced synthesis of the dipeptide N-acetylglutaminylglutamine amide is mediated by a new pathway conserved among bacteria. Proc. Natl. Acad. Sci. USA 2010, 107, 12652–12657. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Araya, J.; Pollender, A.; Huynh, D.; Schlömann, M.; Chávez, R.; Levicán, G. Osmotic imbalance, cytoplasm acidification and oxidative stress induction support the high toxicity of chloride in acidophilic bacteria. Front. Microbiol 2019, 10, 2455. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiang, S.; Deng, Z.; Dedon, P.C.; Chen, S. DNA phosphorothioate modification-a new multi-functional epigenetic system in bacteria. FEMS Microbiol. Rev. 2019, 43, 109–122. [Google Scholar] [CrossRef]
- Bittner, A.N.; Foltz, A.; Oke, V. Only one of five groEL genes is required for viability and successful symbiosis in Sinorhizobium meliloti. J. Bacteriol. 2007, 189, 1884–1889. [Google Scholar] [CrossRef]
- Schneiker-Bekel, S. The complete genome sequence of the dominant Sinorhizobium meliloti field isolate SM11 extends the S. meliloti pan-genome. J. Biotechnol. 2011, 155, 20–33. [Google Scholar] [CrossRef]
- Kuhn, S.; Stiens, M.; Pühler, A.; Schlüter, A. Prevalence of pSmeSM11a-like plasmids in indigenous Sinorhizobium meliloti strains isolated in the course of a field release experiment with genetically modified S. meliloti strains. FEMS Microbiol. Ecol. 2008, 63, 118–131. [Google Scholar] [CrossRef]
- Garcia-Vallve, S.; Guzman, E.; Montero, M.A.; Romeu, A. HGT-DB: A database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 2003, 31, 187–189. [Google Scholar] [CrossRef]
- Tuller, T. Codon bias, tRNA pools, and horizontal gene transfer. Mob. Genet. Elements 2011, 1, 75–77. [Google Scholar] [CrossRef]
- Bedhomme, S.; Amorós-Moya, D.; Valero, L.M.; Bonifaci, N.; Pujana, M.À.; Bravo, I.G. Evolutionary changes after translational challenges imposed by horizontal gene transfer. Genome Biol. Evol. 2019, 11, 814–831. [Google Scholar] [CrossRef]
- Selbitschka, W.; Arnold, W.; Jording, D.; Kosier, B.; Toro, N.; Pühler, A. The insertion sequence element ISRm2011-2 belongs to the IS630-Tc1 family of transposable elements and is abundant in Rhizobium meliloti. Gene 1995, 163, 59–64. [Google Scholar] [CrossRef]
- Roumiantseva, M.L.; Andronov, E.E.; Sharypova, L.A.; Dammann-Kalinowski, T.; Keller, M.; Young, J.P.; Simarov, B.V. Diversity of Sinorhizobium meliloti from the Central Asian alfalfa gene center. Appl. Environ. Microbiol. 2002, 68, 4694–4697. [Google Scholar] [CrossRef]
- Biondi, E.G.; Toro, N.; Bazzicalupo, M.; Martínez-Abarca, F. Spread of the group II intron RmInt1 and its insertion sequence target sites in the plant endosymbiont Sinorhizobium meliloti. Mob. Genet. Elements 2011, 1, 2–7. [Google Scholar] [CrossRef]
- Toro, N.; Martínez-Abarca, F.; Molina-Sánchez, M.D.; García-Rodríguez, F.M.; Nisa-Martínez, R. Contribution of mobile group II introns to Sinorhizobium meliloti genome evolution. Front. Microbiol. 2018, 9, 627. [Google Scholar] [CrossRef]
- Schlüter, J.P.; Reinkensmeier, J.; Daschkey, S.; Evguenieva-Hackenberg, E.; Janssen, S.; Jänicke, S.; Becker, J.D.; Giegerich, R.; Becker, A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genom. 2010, 11, 245. [Google Scholar] [CrossRef]
- Hernandez-Lucas, I.; Ramirez-Trujillo, J.A.; Gaitan, M.A.; Guo, X.; Flores, M.; Martinez-Romero, E.; Perez-Rueda, E.; Mavingui, P. Isolation and characterization of functional insertion sequences of rhizobia. FEMS Microbiol. Lett. 2006, 261, 25–31. [Google Scholar] [CrossRef][Green Version]
- Mandon, K.; Osterås, M.; Boncompagni, E.; Trinchant, J.C.; Spennato, G.; Poggi, M.C.; Le Rudulier, D. The Sinorhizobium meliloti glycine betaine biosynthetic genes (betlCBA) are induced by choline and highly expressed in bacteroids. Mol. Plant Microbe Interact. 2003, 16, 709–719. [Google Scholar] [CrossRef]
- Wang, D.; Xue, H.; Wang, Y.; Yin, R.; Xie, F.; Luo, L. The Sinorhizobium meliloti ntrX gene is involved in succinoglycan production, motility, and symbiotic nodulation on alfalfa. Appl. Environ. Microbiol. 2013, 79, 7150–7159. [Google Scholar] [CrossRef]
- Siarot, L.; Toyazaki, H.; Hidaka, M.; Kurumisawa, K.; Hirakawa, T.; Morohashi, K.; Aono, T. A Novel regulatory pathway for K+ uptake in the legume symbiont Azorhizobium caulinodans in which TrkJ represses the kdpFABC operon at high extracellular K+ concentrations. Appl. Environ. Microbiol. 2017, 83, e01197-17. [Google Scholar] [CrossRef]
- van Loo, B.; Schober, M.; Valkov, E.; Heberlein, M.; Bornberg-Bauer, E.; Faber, K.; Hyvönen, M.; Hollfelder, F. Structural and mechanistic analysis of the choline sulfatase from Sinorhizobium melliloti: A class I sulfatase specific for an alkyl sulfate ester. J. Mol. Biol. 2018, 430, 1004–1023. [Google Scholar] [CrossRef]
- Sharma, M.P.; Grover, M.; Chourasiya, D.; Bharti, A.; Agnihotri, R.; Maheshwari, H.S.; Pareek, A.; Buyer, J.S.; Sharma, S.K.; Schütz, L.; et al. Deciphering the role of trehalose in tripartite symbiosis among rhizobia, arbuscular mycorrhizal fungi, and legumes for enhancing abiotic stress tolerance in crop plants. Front. Microbiol. 2020, 11, 509919. [Google Scholar] [CrossRef] [PubMed]
- Vanaporn, M.; Titball, R.W. Trehalose and bacterial virulence. Virulence 2020, 11, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Mom, R.; Muries, B.; Benoit, P.; Robert-Paganin, J.; Réty, S.; Venisse, J.S.; Padua, A.; Label, P.; Auguin, D. Voltage-gating of aquaporins, a putative conserved safety mechanism during ionic stresses. FEBS Lett. 2021, 595, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Kosolapova, A.O.; Belousov, M.V.; Sulatskaya, A.I.; Belousova, M.E.; Sulatsky, M.I.; Antonets, K.S.; Volkov, K.V.; Lykholay, A.N.; Shtark, O.Y.; Vasilieva, E.N.; et al. Two novel amyloid proteins, RopA and RopB, from the root nodule bacterium Rhizobium leguminosarum. Biomolecules 2019, 9, 694. [Google Scholar] [CrossRef]
- Boussau, B.; Karlberg, E.O.; Frank, A.C.; Legault, B.A.; Andersson, S.G. Computational inference of scenarios for alpha-proteobacterial genome evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 9722–9727. [Google Scholar] [CrossRef]
- Paço, A.; Brígido, C.; Alexandre, A.; Mateos, P.F.; Oliveira, S. The symbiotic performance of chickpea rhizobia can be improved by additional copies of the clpB chaperone gene. PLoS ONE 2016, 11, e0148221. [Google Scholar] [CrossRef]
- Kopat, V.V.; Chirak, E.R.; Kimeklis, A.K.; Safronova, V.I.; Belimov, A.A.; Kabilov, M.R.; Andronov, E.E.; Provorov, N.A. Evolution of fixNOQP genes encoding cytochrome oxidase with high affinity to oxygen in rhizobia and related bacteria. Russ. J. Genet. 2017, 53, 766–774. [Google Scholar] [CrossRef]
- Cren, M.; Kondorosi, A.; Kondorosi, E. NolR controls expression of the Rhizobium meliloti nodulation genes involved in the core Nod factor synthesis. Mol. Microbiol. 1995, 15, 733–747. [Google Scholar] [CrossRef]
- Burrus, V.; Waldor, M.K. Formation of SXT tandem arrays and SXT-R391 hybrids. J. Bacteriol. 2004, 186, 2636–2645. [Google Scholar] [CrossRef]
- Kung, V.L.; Ozer, E.A.; Hauser, A.R. The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2010, 74, 621–641. [Google Scholar] [CrossRef]
- Guerillot, R.; Da Cunha, V.; Sauvage, E.; Bouchier, C.; Glaser, P. Modular evolution of TnGBSs, a new family of ICEs associating IS transposition, plasmid replication and conjugation for their spreading. J. Bacteriol. 2013, 195, 1979–1990. [Google Scholar] [CrossRef]
- Berger, C.; Rückert, C.; Blom, J.; Rabaey, K.; Kalinowski, J.; Rosenbaum, M.A. Estimation of pathogenic potential of an environmental Pseudomonas aeruginosa isolate using comparative genomics. Sci. Rep. 2021, 11, 1370. [Google Scholar] [CrossRef]
- Bakaev, M.; Vladimirova, M.; Kozlova, A.; Muntyan, V.; Afonin, A.; Grudinin, M.; Roumiantseva, M. Abundant of CRISPR/Cas elements in genomes of nitrogen-fixing bacteria from the genus Sinorhizobium. FEBS Open Bio 2021, 11, 134. [Google Scholar] [CrossRef]
- Muntyan, V.S.; Afonin, A.M.; Vladimirova, M.E.; Saksaganskaya, A.S.; Gribchenko, E.S.; Baturina, O.; Roumiantseva, M.L. Complete genome sequence of Sinorhizobium meliloti S35m, a salt-tolerant isolate from alfalfa rhizosphere in soil native to the Caucasus region. Microbiol. Resour. Announc. 2021, 10, e01417-20. [Google Scholar] [CrossRef]
- Charbonneau, D.M.; Aubé, A.; Rachel, N.M.; Guerrero, V.; Delorme, K.; Breault-Turcot, J.; Masson, J.F.; Pelletier, J.N. Development of Escherichia coli asparaginase II for immunosensing: A trade-off between receptor density and sensing efficiency. ACS Omega 2017, 2, 2114–2125. [Google Scholar] [CrossRef]
- Zhouravleva, G.A.; Rovinski, N.S. Adaptive role of tmRNA. Ecol. Genet. 2005, 3, 12–18. [Google Scholar] [CrossRef][Green Version]
- Gillet, R.; Felden, B. Emerging views on tmRNA mediated protein tagging and ribosome rescue. Mol. Microbiol. 2001, 42, 879–885. [Google Scholar] [CrossRef]
- Withey, J.H.; Friedman, D.I. The biological roles of trans-translation. Curr. Opin. Microbiol. 2002, 5, 154–159. [Google Scholar] [CrossRef]
- Marketon, M.M.; González, J.E. Identification of two quorum-sensing systems in Sinorhizobium meliloti. J. Bacteriol. 2002, 184, 3466–3475. [Google Scholar] [CrossRef]
- Gurich, N.; González, J.E. Role of quorum sensing in Sinorhizobium meliloti-alfalfa symbiosis. J. Bacteriol. 2009, 191, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Coggin, A.; Yagnik, K.; Teplitski, M. Role of specific quorum-sensing signals in the regulation of exopolysaccharide II production within Sinorhizobium meliloti spreading colonies. PLoS ONE 2012, 7, e42611. [Google Scholar] [CrossRef] [PubMed]

 I-XI, ISRm17;
I-XI, ISRm17;  I-XIX, ISRm2011–2; /. Figures framed in black have identical localization of ISRm17 and ISRm2011–2, respectively, on replicons of strains L6-AK89 and Rm1021. * Disrupted IS element sequences (2 ORFs identified). Genomic islands (GIs):
I-XIX, ISRm2011–2; /. Figures framed in black have identical localization of ISRm17 and ISRm2011–2, respectively, on replicons of strains L6-AK89 and Rm1021. * Disrupted IS element sequences (2 ORFs identified). Genomic islands (GIs):  Width of rectangle corresponds to GI coordinates in chromosome: GI11K, GI41M, GI19T, GI21T, GI80S, where superscript indicates length of GI in kb and capital letter corresponds to tRNA gene of a certain amino acid (site of integration). Incomplete prophages (Phs):
Width of rectangle corresponds to GI coordinates in chromosome: GI11K, GI41M, GI19T, GI21T, GI80S, where superscript indicates length of GI in kb and capital letter corresponds to tRNA gene of a certain amino acid (site of integration). Incomplete prophages (Phs):  Width of rectangle corresponds to Ph coordinates in replicon: Ph1−12, where superscript indicates number of phages in list: 1 Pseudomonas phage nickie(NC_042091); 2 Sulfitobacter phage NYA-2014a(NC_027299); 3 Salmonella phage vB_SosS_Oslo (NC_018279); 4 Xanthomonas phage Carpasina (NC_047962); 5,10 Stx2-converting phage Stx2a_F451(NC_049924); 6 Escherichia phage ArgO145(NC_049918); 7 Synechococcus phage S-CBWM1(NC_048106); 8 Streptococcus phage Dp-1(NC_015274); 9 Sinorhizobium phage phiLM21(NC_029046); 11 Escherichia phage SH2026Stx1(NC_049919); 12 Stx2-converting phage 1717(NC_011357). On pSymA only: B and D collinear homologous regions; A, E, and C unique regions for Rm1021 or L6-AK89; ┴ sym genes: 1 (nodD2), 2 (nodM), 3 (fixS1), 4 (fixL).
I-XI, ISRm17; I-XIX, ISRm2011–2; /. Figures framed in black have identical localization of ISRm17 and ISRm2011–2, respectively, on replicons of strains L6-AK89 and Rm1021. * Disrupted IS element sequences (2 ORFs identified). Genomic islands (GIs): Width of rectangle corresponds to GI coordinates in chromosome: GI11K, GI41M, GI19T, GI21T, GI80S, where superscript indicates length of GI in kb and capital letter corresponds to tRNA gene of a certain amino acid (site of integration). Incomplete prophages (Phs): Width of rectangle corresponds to Ph coordinates in replicon: Ph1−12, where superscript indicates number of phages in list: 1 Pseudomonas phage nickie(NC_042091); 2 Sulfitobacter phage NYA-2014a(NC_027299); 3 Salmonella phage vB_SosS_Oslo (NC_018279); 4 Xanthomonas phage Carpasina (NC_047962); 5,10 Stx2-converting phage Stx2a_F451(NC_049924); 6 Escherichia phage ArgO145(NC_049918); 7 Synechococcus phage S-CBWM1(NC_048106); 8 Streptococcus phage Dp-1(NC_015274); 9 Sinorhizobium phage phiLM21(NC_029046); 11 Escherichia phage SH2026Stx1(NC_049919); 12 Stx2-converting phage 1717(NC_011357). On pSymA only: B and D collinear homologous regions; A, E, and C unique regions for Rm1021 or L6-AK89; ┴ sym genes: 1 (nodD2), 2 (nodM), 3 (fixS1), 4 (fixL).
Width of rectangle corresponds to Ph coordinates in replicon: Ph1−12, where superscript indicates number of phages in list: 1 Pseudomonas phage nickie(NC_042091); 2 Sulfitobacter phage NYA-2014a(NC_027299); 3 Salmonella phage vB_SosS_Oslo (NC_018279); 4 Xanthomonas phage Carpasina (NC_047962); 5,10 Stx2-converting phage Stx2a_F451(NC_049924); 6 Escherichia phage ArgO145(NC_049918); 7 Synechococcus phage S-CBWM1(NC_048106); 8 Streptococcus phage Dp-1(NC_015274); 9 Sinorhizobium phage phiLM21(NC_029046); 11 Escherichia phage SH2026Stx1(NC_049919); 12 Stx2-converting phage 1717(NC_011357). On pSymA only: B and D collinear homologous regions; A, E, and C unique regions for Rm1021 or L6-AK89; ┴ sym genes: 1 (nodD2), 2 (nodM), 3 (fixS1), 4 (fixL).
I-XI, ISRm17; I-XIX, ISRm2011–2; /. Figures framed in black have identical localization of ISRm17 and ISRm2011–2, respectively, on replicons of strains L6-AK89 and Rm1021. * Disrupted IS element sequences (2 ORFs identified). Genomic islands (GIs): Width of rectangle corresponds to GI coordinates in chromosome: GI11K, GI41M, GI19T, GI21T, GI80S, where superscript indicates length of GI in kb and capital letter corresponds to tRNA gene of a certain amino acid (site of integration). Incomplete prophages (Phs): Width of rectangle corresponds to Ph coordinates in replicon: Ph1−12, where superscript indicates number of phages in list: 1 Pseudomonas phage nickie(NC_042091); 2 Sulfitobacter phage NYA-2014a(NC_027299); 3 Salmonella phage vB_SosS_Oslo (NC_018279); 4 Xanthomonas phage Carpasina (NC_047962); 5,10 Stx2-converting phage Stx2a_F451(NC_049924); 6 Escherichia phage ArgO145(NC_049918); 7 Synechococcus phage S-CBWM1(NC_048106); 8 Streptococcus phage Dp-1(NC_015274); 9 Sinorhizobium phage phiLM21(NC_029046); 11 Escherichia phage SH2026Stx1(NC_049919); 12 Stx2-converting phage 1717(NC_011357). On pSymA only: B and D collinear homologous regions; A, E, and C unique regions for Rm1021 or L6-AK89; ┴ sym genes: 1 (nodD2), 2 (nodM), 3 (fixS1), 4 (fixL).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain | Assembly | Species | Strain | Assembly |
|---|---|---|---|---|---|
| E. meliloti | Rm1021 | GCA_000006965 | E. meliloti | KH35c | GCA_002197105 |
| Rm2011 | GCA_000346065 | KH46 | GCA_002197465 | ||
| AK21 | GCA_009664245 | RCAM1115 | GCA_014189595 | ||
| AK555 | GCA_003044175 | Rm41 | GCA_002197045 | ||
| AK76 | GCA_016406285 | RMO17 | GCA_000747295 | ||
| AK83 | GCA_000147795 | RU11/001 | GCA_001050915 | ||
| B399 | GCA_002302375 | S35m | GCA_015689095 | ||
| B401 | GCA_002302355 | SM11 | GCA_000218265 | ||
| BL225C | GCA_000147775 | T073 | GCA_002197145 | ||
| CCMM B554(FSM-MA) | GCA_002215195 | USDA1021 | GCA_002197445 | ||
| CXM1–105 | GCA_003044215 | USDA1106 | GCA_002197065 | ||
| GR4 | GCA_000320385 | USDA1157 | GCA_002197025 | ||
| HM006 | GCA_002197165 | E. medicae | WSM419 | GCA_000017145 |
| Replicon | Size (bp) * | Number of | GC Content (%) * | |||
|---|---|---|---|---|---|---|
| tRNA * | rRNA * | tmRNA * | ORFs * | |||
| Chromosome | 3,580,141/3,654,135 | 53 **/52 | 9/9 | 1/1 | 3333/3315 | 62.9/63.7 |
| pSymA | 1,217,792/1,354,226 | 1/2 | 0/0 | 0/0 | 1156/1176 | 60.4/60.4 |
| pSymB | 1,645,902/1,683,333 | 2/1 | 0/0 | 0/0 | 1486/1463 | 62.5/62.4 |
| pL6-AK89 | 349,886 | 1 | 0 | 0 | 353 | 59.1 |
| Total: | 6,793,721/6,691,694 | 57/55 | 9/9 | 1/1 | 6328/5954 | 62.1/62.2 |
| Replicon | Cluster Coordinates | Cover/Identity (%) *** | Cluster Name | Putative Cluster Function | |
|---|---|---|---|---|---|
| L6-AК89 | Rm1021 | ||||
| Chromosome | 593,047–640,555 * | 620,572–668,086 * [620,572–658,665] ** | 100/99.54 | T1PKS | polyketide synthesis [133] |
| 1,210,650–1,253,307 |
1,244,088–1,286,745 [1,244,088–1,286,734] | 100/99.94 | NRPS-like | nonribosomal peptide synthesis [134] | |
| 1,619,597–1,640,439 |
1,653,352–1,674,194 [1,653,352–1,674,194] | 100/99.98 | terpene | terpene biosynthesis | |
| 1,963,942–1,984,586 |
1,986,863–2,007,507 [1,986,863–2,007,507] | 100/99.97 | hserlactone | quorum sensing | |
| 2,224,345–2,246,364 |
2,245,746–2,267,765 [2,245,742–2,267,771] | 100/99.91 | TfuA-related | trifolitoxin production | |
| pSymA | 493,358–513,978 | - | - | hserlactone | quorum sensing |
| - | 1,304,062–1,319,558 | - | siderophore | rhizobactin synthesis [135] | |
| pSymB | 459,736–481,971 |
- [487,573–503,480] | 99/96.93 | NAGGN | osmotic shock response [136] |
| - | 548,445–559,374 | - | bacteriocin | antibiotic | |
| pL6-AK89 | 201,714–279,196 | - | - | phosphonate, ectoine | osmotic stress protection [137] |
| L6-AK89 Replicon | Type of Insertion Sequence (GII/PhII) | Integration Site of GI/attP of Ph | Ph | GI/Ph Coordinates | GI/Ph Length (bp) | GI/Ph GC% |
|---|---|---|---|---|---|---|
| Chromosome | GI | tRNA-Lys(TTT) | - | 1,787,380–1,797,997 | 10618 | 60.4 |
| inc-Ph1 | TGCAGCGTTCATA | Pseudomonas phage nickie (NC_042091) III−1 | 2,847,359–2,859,574 | 12216 | 62.5 | |
| GI (inc-Ph2) | tRNA-Met(CAT) | Sulfitobacter phage III−2 | GI: 2,889,780–2,930,558 (Ph: 2,903,909–2,920,668) | GI: 40,779 Ph: 16,760 | GI: 59.3 Ph: 61.4 | |
| pSymA | inc-Ph3 * | - | Salmonella phage vB_SosS_Oslo (NC_018279) III−3 | 347,835–353,703 | 5869 | 58.8 |
| inc-Ph4 * | - | Xanthomonas phage Carpasina (NC_047962) III−4 | 423,074–432,147 | 9074 | 59.6 | |
| inc-Ph5 *, ** | TGGCGGGCGCTC | Stx2-converting phage Stx2a_F451 (NC_049924) III−5 | 728,573–746,657, 746,650–753,387 | 18,085 6738 | 58.9 58.2 | |
| pSymB | inc-Ph6 | - | Escherichia phage ArgO145 (NC_049918) | 133,663–143,079 | 9459.6 | 62.1 |
| inc-Ph7 | - | Synechococcus phage S-CBWM1 (NC_048106) | 387,843–396,892 | 9050 | 62.3 | |
| inc-Ph8 | - | Streptococcus phage Dp-1 (NC_015274) III−6 | 1,142,006–1,148,376 | 6371 | 62.5 | |
| pL6-AK89 | inc-Ph9 | - | Sinorhizobium phage phiLM21 (NC_029046) III−7 | 17,254–27,049 | 9796 | 58.7 |
| inc-Ph10 | CCGTCGCCTTCC | Stx2-converting phage Stx2a_F451 (NC_049924) | 43,164–66,618 | 23,455 | 58.1 | |
| inc-Ph11 | - | Escherichia phages SH2026Stx1(NC_049919) III −8 | 264,406–272,442 | 8037 | 57.8 |
| IS Element | Number of IS Elements (Full-Sized/Fragments, n.c.) per Replicon II/III | Origin IV | Identity V(%) | Total Number: (Full-Sized/ Fragments, n.c.) | |||||
|---|---|---|---|---|---|---|---|---|---|
| IS Family I | Group I/II | IS Element II | Chromosome | pSymA | pSymB | pL6-AK89 | |||
| IS256 | - | ISRm5 | 2/0 | - | 1/2 (≤79%, ≤14%) | 0/1 (≤45%) | E. mel. | 98–99 | 3/3, 0 |
| - | ISRm3 | - | 1/0 | 2/0 | 2/0 | E. mel. | 99 | 5/0, 0 | |
| IS4 | IS4Sa | ISRm16 (ISRm22) VI | 5/0 | - | - | - | E. mel. | 99–100 | 5/0, 0 |
| IS630 | - | ISRm2011–2 | 6/0 | 0/1 (≤91%) | 2/0 | - | E. mel. | 99–100 | 8/1, 0 |
| - | n.c. III | - | - | 1714, 1696 | 1669 | - | - | 0/0, 3 | |
| ISAs1 | - | ISRm21 | 1/0 | - | - | - | E. mel. | 98 | 1/0, 0 |
| IS1595 | ISNwi1 | ISRm32 | - | 0/1 (≤66%) | - | 0/2 (≤61%, ≤18%) | E. mel. | 92–96 | 0/3, 0 |
| IS481 | - | ISRm20 | 2/2 (≤84%, ≤17%) | - | - | - | E. mel. | 99–100 | 2/2, 0 |
| - | n.c. | - | - | 11143 | - | - | - | 0/0, 1 | |
| IS3 | IS407 | ISRm7 | - | 1/0 | - | - | E. mel. | 99 | 1/0, 0 |
| IS2 | ISRm1 | - | - | - | 1/2 (≤97%, ≤3%) | E. mel. | 95–99 | 1/2, 0 | |
| IS407 | ISRle5 | - | - | - | 1/0 | R. leg. | 89 | 1/0, 0 | |
| ISNCY | ISDol1 | ISRm17 | 1/0 | 3/0 | 2/2 (≤96%, ≤4%) | 1/0 | E. mel. | 99–100 | 7/2, 0 |
| IS1202 | ISRel26 | 0/1 (≤87%) | 0/1 (≤88%) | - | 0/1 (≤87%) | R. etli | 83–85 | 0/3, 0 | |
| IS1202 | ISRel10 | - | 0/1 (≤26%) | - | - | R. etli | 83 | 0/1, 0 | |
| IS5 | IS903 | ISRm33 | - | - | - | 0/1 (≤72%) | E. mel. | 98 | 0/1, 0 |
| IS1031 | ISRm4–1 | - | - | - | *1/0 | E. mel. | 98 | 1/0, 0 | |
| None model1 | ISNisp7 | - | 0/1 (≤70%) | - | - | Nitr. sp. | 79 | 0/1, 0 | |
| IS427 | ISRel13 | - | - | - | *0/1 (≤93%) | R. etli | 91 | 0/1, 0 | |
| IS66 | - | ISRm14 | - | - | - | *1/0 | E. mel. | 97 | 1/0, 0 |
| - | ISSme3 | - | 1/0 | - | *1/0 | E. med. | 95 | 2/0, 0 | |
| - | ISRle3 | - | - | - | 0/1 (≤84%) | R. leg. | 82 | 0/1, 0 | |
| - | n.c. | - | - | - | 1732 | - | - | 0/0, 1 | |
| - | n.c. | - | 11533 | 11533 | - | - | - | 0/0, 2 | |
| - | n.c. | 11596 | - | - | - | - | - | 0/0, 1 | |
| IS21 | - | ISPve1 | 0/1 (≤44%) | p. ver. | 81 | 0/1, 0 | |||
| - | ISRel16 | - | - | 0/1 (≤95%) | 1/0 | R. etli | 79–80 | 1/1, 0 | |
| - | ISSme2 | - | - | - | 1/0 | E. med. | 97 | 1/0, 0 | |
| - | n.c. | - | - | - | 1999 | - | - | 0/0, 1 | |
| IS6 | - | ISEc59 | - | - | - | * 0/1 (≤85%) | E. coli | 82 | 0/1, 0 |
| IS30 | - | n.c. | - | 1905 | - | - | - | - | 0/0, 1 |
| IS110 | - | n.c. | 1453 | - | 1990 | - | - | - | 0/0, 2 |
| - | n.c. | - | - | - | 11068 | - | - | 0/0, 1 | |
| - | n.c. | - | - | - | 1657 | - | - | 0/0, 1 | |
| - | n.c. | - | - | - | 1864 | - | - | 0/0, 1 | |
| ISL3 | - | n.c. | - | - | - | 11740 | - | - | 0/0, 1 |
| In total: 15 IS-families | Total number: full-sized/fragments, n.c. IS | 17/3, 2 | 6/5, 2 | 7/5, 5 | 10/11, 7 | - | - | 40/24, 16 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roumiantseva, M.L.; Vladimirova, M.E.; Saksaganskaia, A.S.; Muntyan, V.S.; Kozlova, A.P.; Afonin, A.M.; Baturina, O.A.; Simarov, B.V. Ensifer meliloti L6-AK89, an Effective Inoculant of Medicago lupulina Varieties: Phenotypic and Deep-Genome Screening. Agronomy 2022, 12, 766. https://doi.org/10.3390/agronomy12040766
Roumiantseva ML, Vladimirova ME, Saksaganskaia AS, Muntyan VS, Kozlova AP, Afonin AM, Baturina OA, Simarov BV. Ensifer meliloti L6-AK89, an Effective Inoculant of Medicago lupulina Varieties: Phenotypic and Deep-Genome Screening. Agronomy. 2022; 12(4):766. https://doi.org/10.3390/agronomy12040766
Chicago/Turabian StyleRoumiantseva, Marina L., Maria E. Vladimirova, Alla S. Saksaganskaia, Victoria S. Muntyan, Alexandra P. Kozlova, Alexey M. Afonin, Olga A. Baturina, and Boris V. Simarov. 2022. "Ensifer meliloti L6-AK89, an Effective Inoculant of Medicago lupulina Varieties: Phenotypic and Deep-Genome Screening" Agronomy 12, no. 4: 766. https://doi.org/10.3390/agronomy12040766
APA StyleRoumiantseva, M. L., Vladimirova, M. E., Saksaganskaia, A. S., Muntyan, V. S., Kozlova, A. P., Afonin, A. M., Baturina, O. A., & Simarov, B. V. (2022). Ensifer meliloti L6-AK89, an Effective Inoculant of Medicago lupulina Varieties: Phenotypic and Deep-Genome Screening. Agronomy, 12(4), 766. https://doi.org/10.3390/agronomy12040766