
16S rRNA Gene-Based Metagenomic Analysis of Rhizosphere Soil Bacteria in Arkansas Rice Crop Fields


 ,
,
Abstract
:1. Introduction
1.1. Rice as a Major Crop in Arkansas and the World
1.2. Soil Microbiome
2. Materials and Methods
2.1. Soil Sampling and Processing
2.2. DNA Extraction and DNA Sequence Analysis
2.3. Bioinformatics Analysis
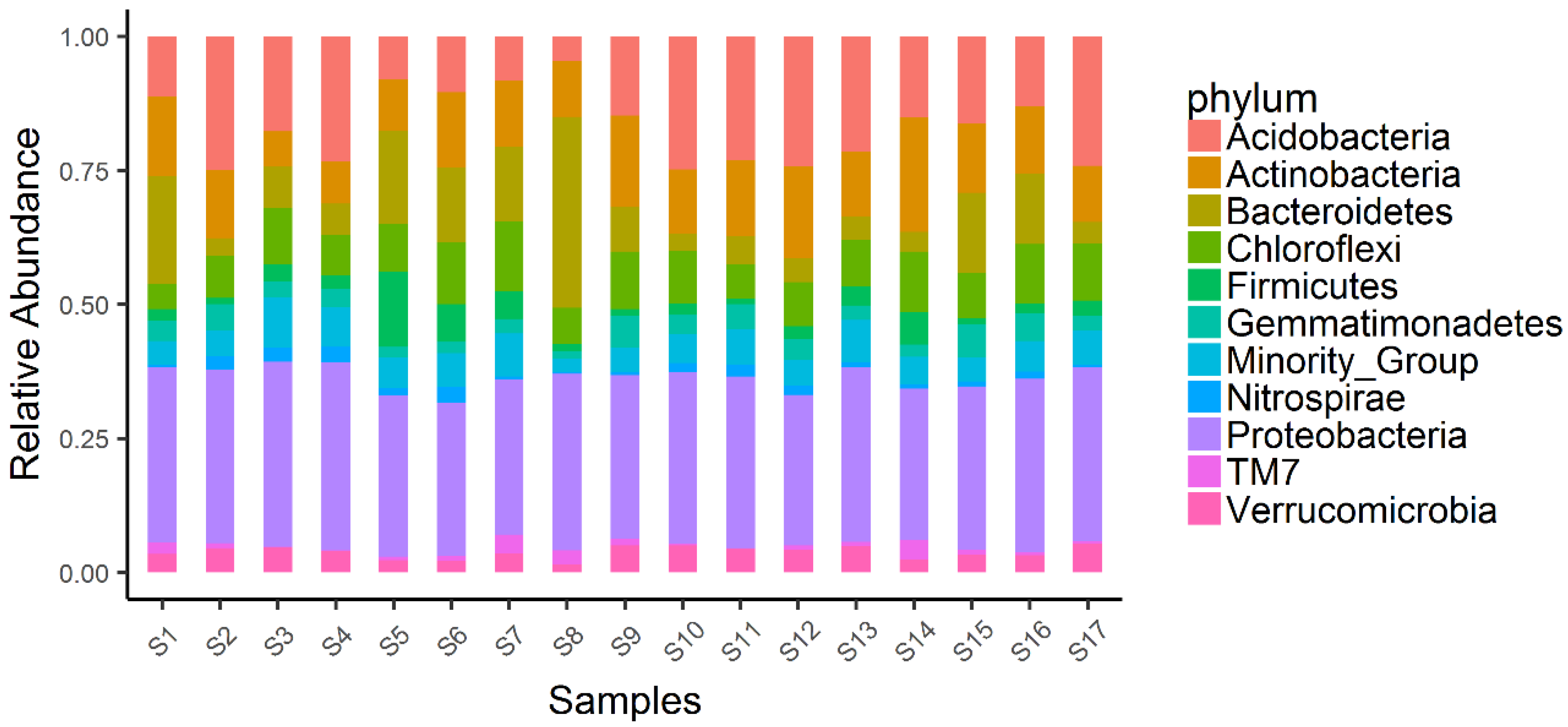
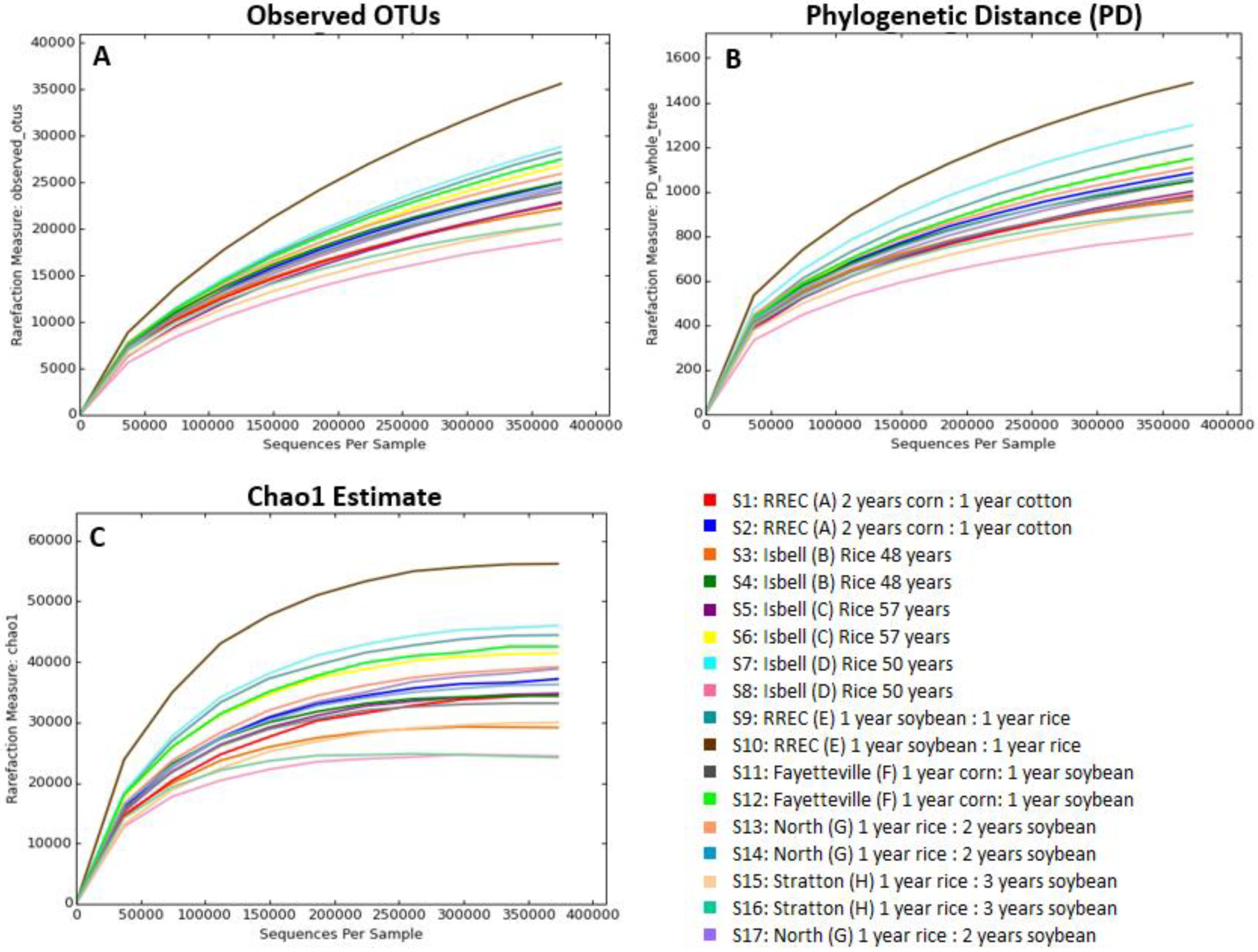
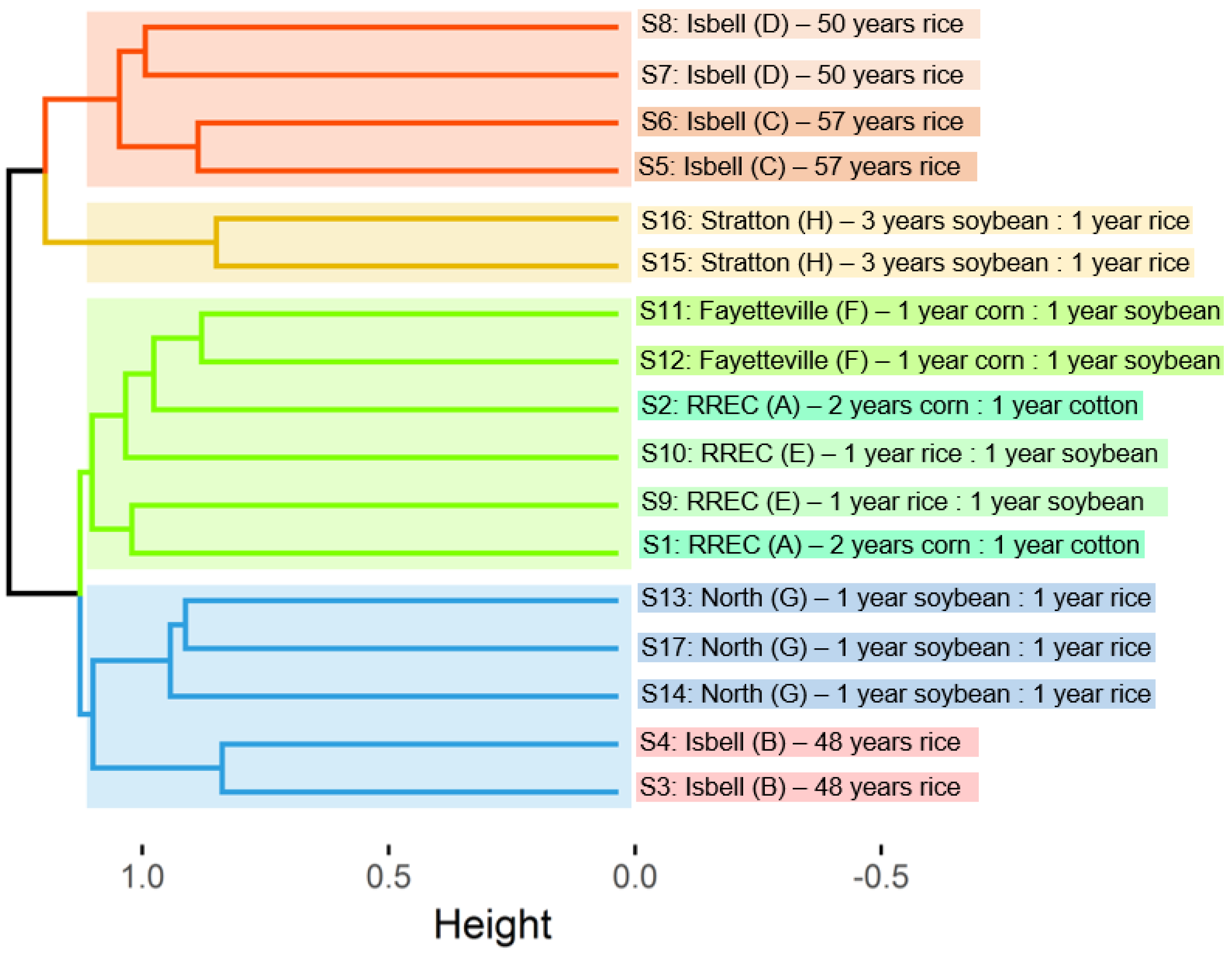
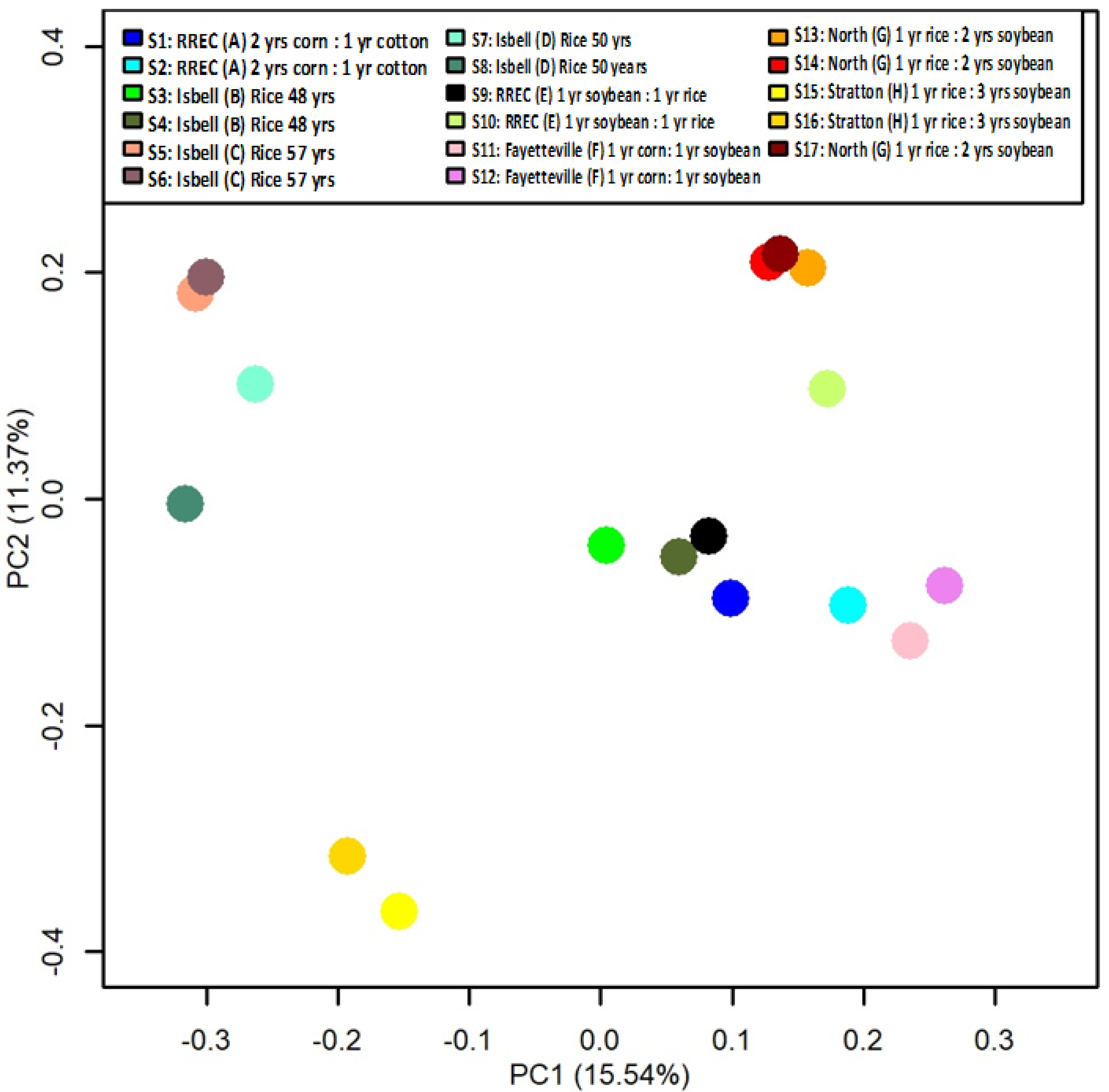
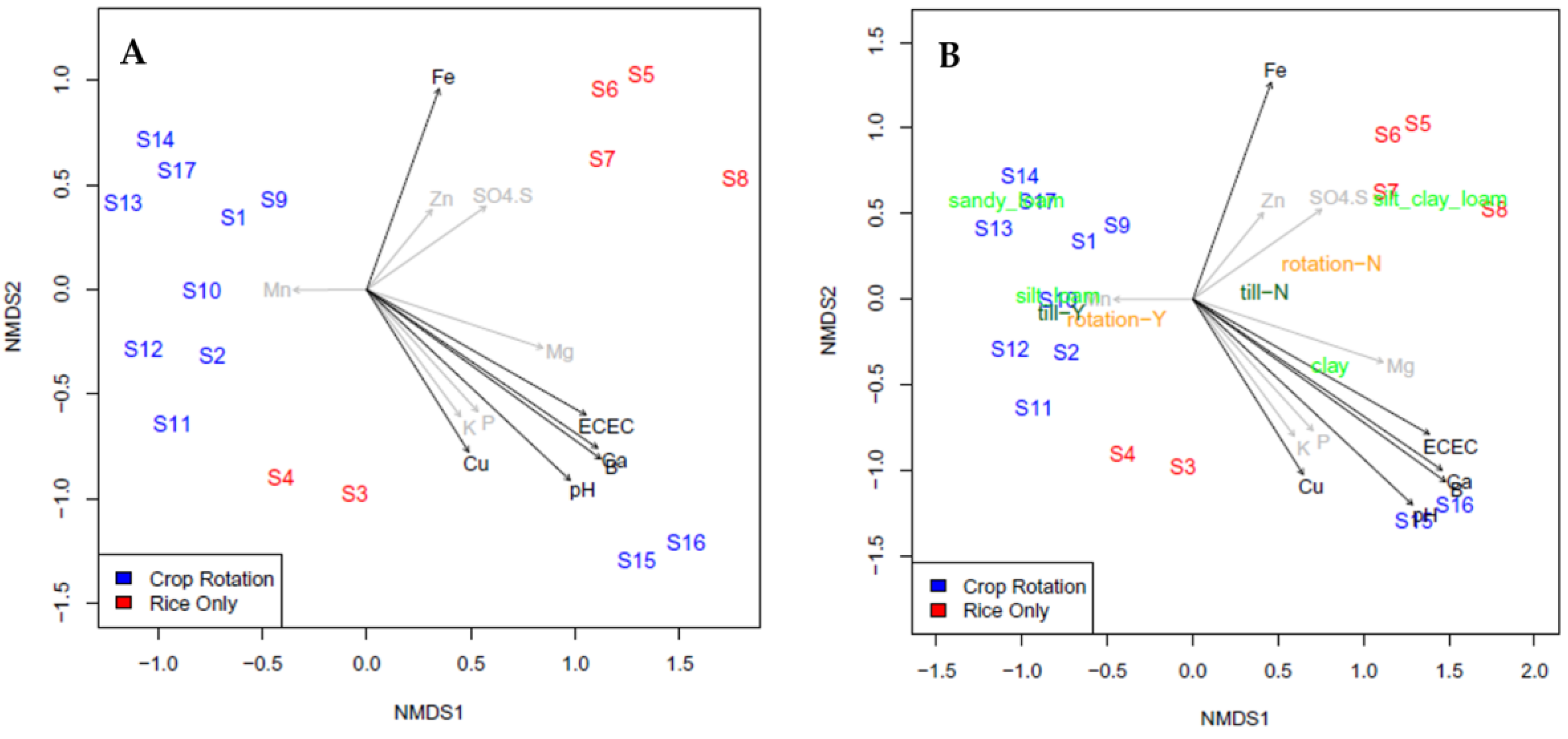
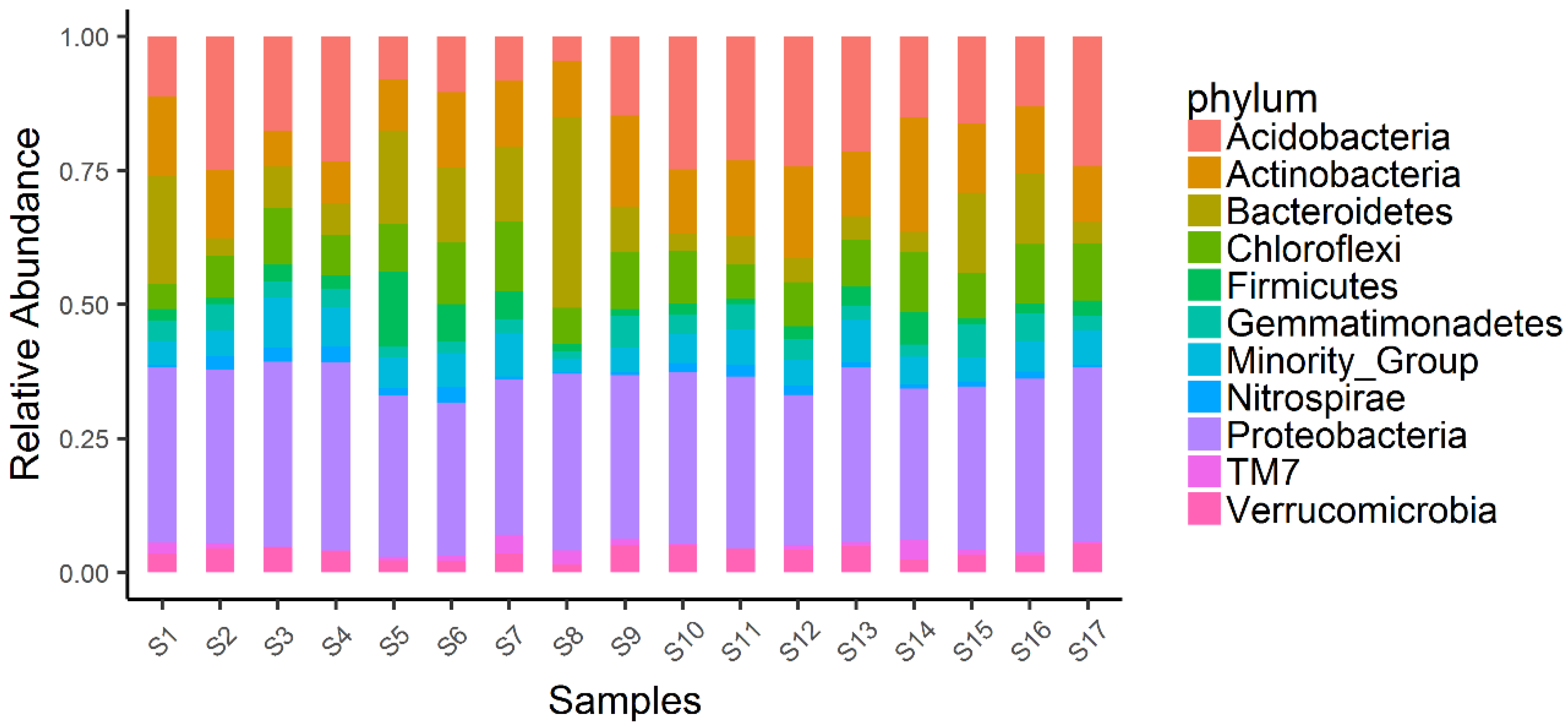
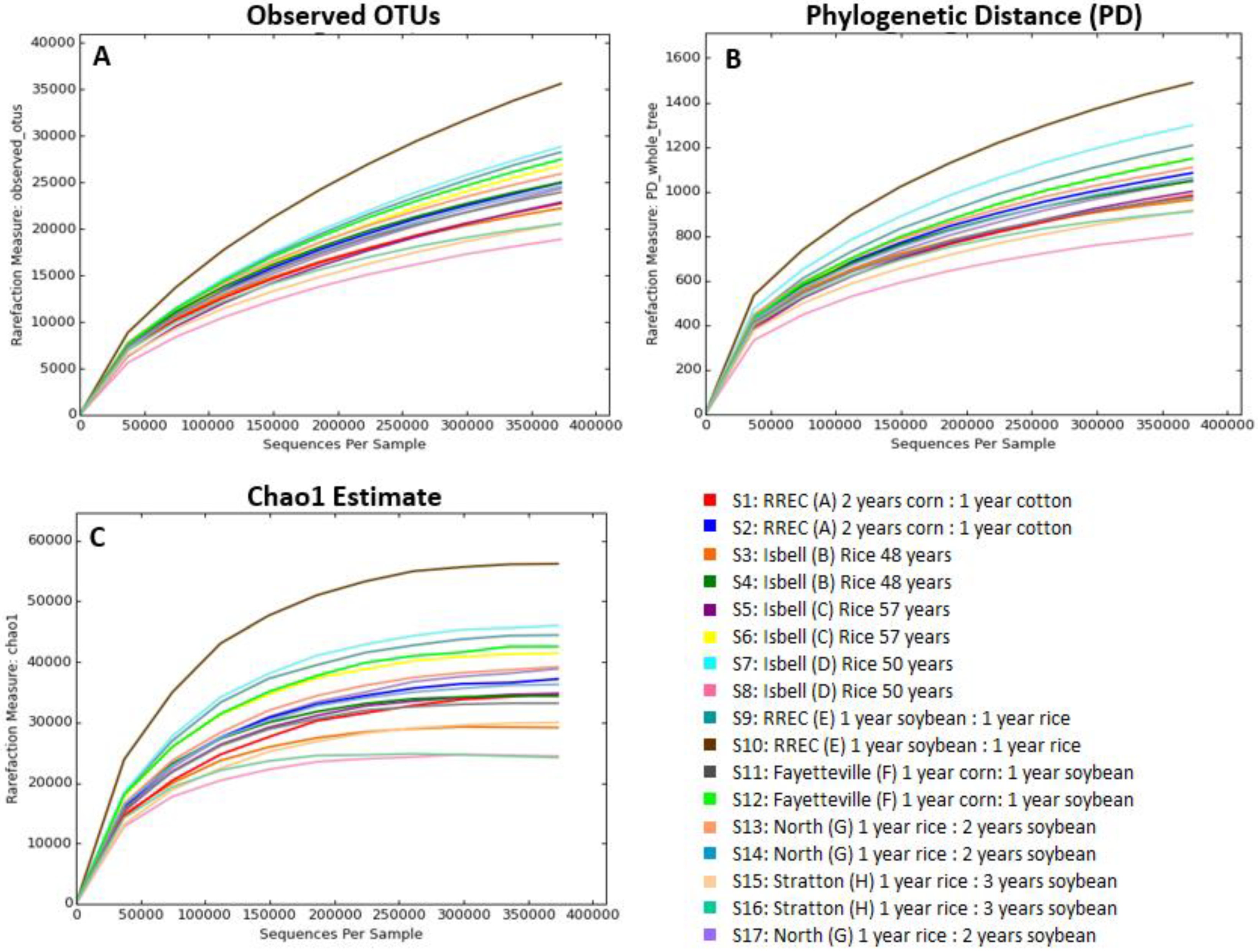
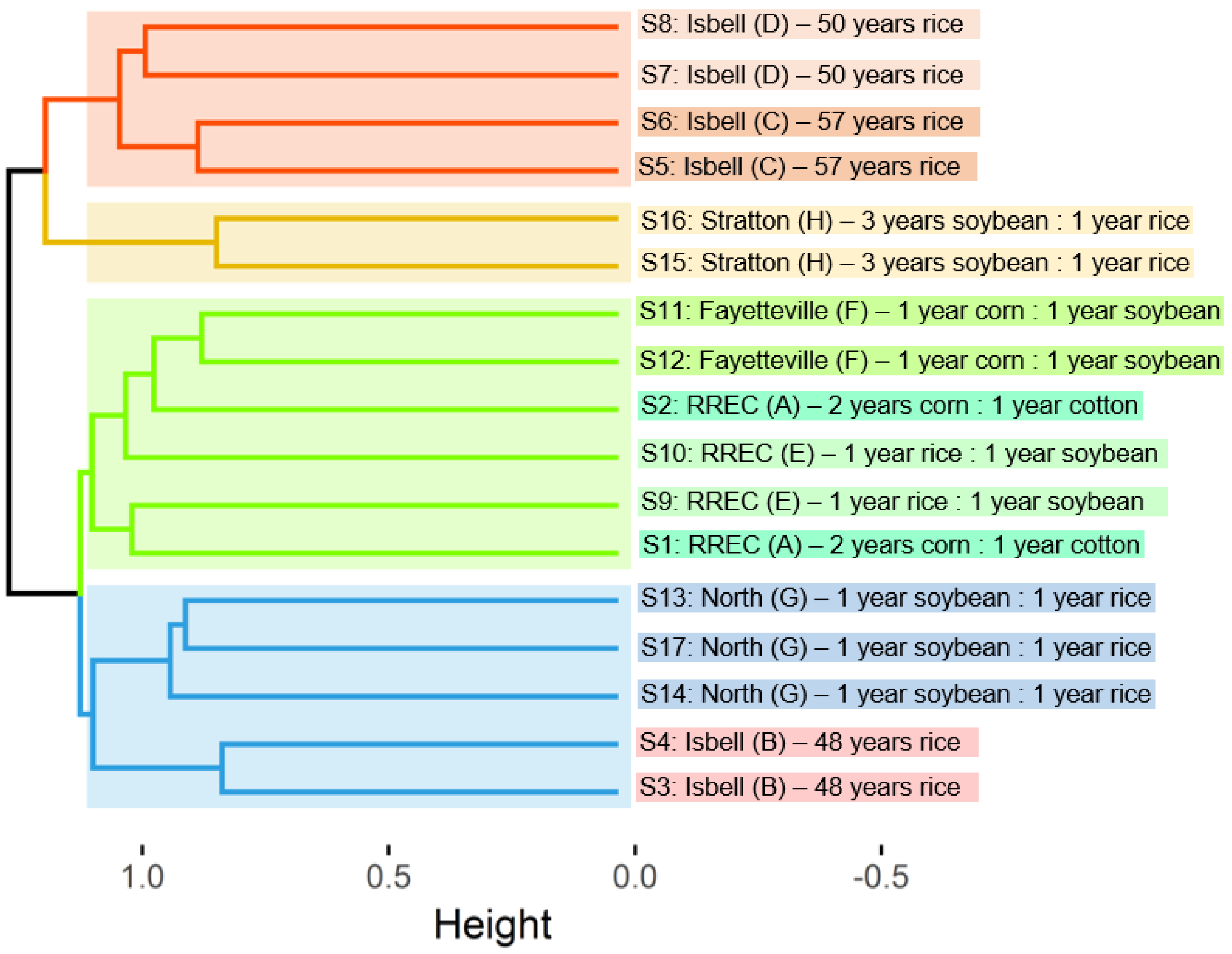
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Philippot, L.; Spor, A.; Hénault, C.; Bru, D.; Bizouard, F.; Jones, C.M.; Sarr, A.; Maron, P.-A. Loss in microbial diversity affects nitrogen cycling in soil. ISME J. 2013, 7, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Wagg, C.; Bender, S.F.; Widmer, F.; van der Heijden, M.G. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. USA 2014, 111, 5266–5270. [Google Scholar] [CrossRef] [Green Version]
- Van Der Heijden, M.G.A.; Bardgett, R.D.; Van Straalen, N.M. The unseen majority: Soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 2008, 11, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [Green Version]
- Riesenfeld, C.S.; Schloss, P.D.; Handelsman, J. Metagenomics: Genomic Analysis of Microbial Communities. Annu. Rev. Genet. 2004, 38, 525–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handelsman, J. Metagenomics: Application of Genomics to Uncultured Microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S. Fundamentals of Rice Crop Science; International Rice Research Institute: Los Baños, Philippines, 1981. [Google Scholar]
- FAO. Food and Agriculture Organization of the United Nations. 2018. Available online: http://www.fao.org/faostat/en/#data/QC/Vis (accessed on 12 December 2021).
- Hardke, J.T. Arkansas Rice Production Handbook; University of Arkansas Division of Agriculture Cooperative Extension Service—MP192: Little Rock, AR, USA, 2013. [Google Scholar]
- Frink, C.R.; Waggoner, P.E.; Ausubel, J.H. Nitrogen fertilizer: Retrospect and prospect. Proc. Natl. Acad. Sci. USA 1999, 96, 1175–1180. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.; Lu, C.; Yu, Z. Historical nitrogen fertilizer use in agricultural ecosystems of the contiguous United States during 1850–2015: Application rate, timing, and fertilizer types. Earth Syst. Sci. Data 2018, 10, 969–984. [Google Scholar] [CrossRef] [Green Version]
- Souza, R.C.; Hungria, M.; Cantão, M.E.; Vasconcelos, A.T.R.; Nogueira, M.A.; Vicente, V.A. Metagenomic analysis reveals microbial functional redundancies and specificities in a soil under different tillage and crop-management regimes. Appl. Soil Ecol. 2015, 86, 106–112. [Google Scholar] [CrossRef]
- Bullock, D.G. Crop rotation. Crit. Rev. Plant Sci. 1992, 11, 309–326. [Google Scholar] [CrossRef]
- Peters, R.D.; Sturz, A.V.; Carter, M.R.; Sanderson, J.B. Developing disease-suppressive soils through crop rotation and tillage management practices. Soil Tillage Res. 2003, 72, 181–192. [Google Scholar] [CrossRef]
- Aschi, A.; Aubert, M.; Riah-Anglet, W.; Nélieu, S.; Dubois, C.; Akpa-Vinceslas, M.; Trinsoutrot-Gattin, I. Introduction of Faba bean in crop rotation: Impacts on soil chemical and biological characteristics. Appl. Soil Ecol. 2017, 120, 219–228. [Google Scholar] [CrossRef]
- Weller, D.M.; Raaijmakers, J.M.; Gardener, B.B.; Thomashow, L.S. Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu. Rev. Phytopathol. 2002, 40, 309–348. [Google Scholar] [CrossRef] [Green Version]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef]
- Nielsen, M.N.; Winding, A.; Binnerup, S.J.; Hansen, B.M.; Hendriksen, N.B.; Kroer, N. Microorganisms as Indicators of Soil Health; Ministry of the Environment, National Environmental Research Institute: Copenhagen, Denmark, 2002. [Google Scholar]
- Anderson, N.A. The Genetics and Pathology of Rhizoctonia Solani. Annu. Rev. Phytopathol. 1982, 20, 329–347. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.; Kim, H.R.; Rahmatallah, Y.; Wiggins, G.; Yang, Q.; Singh, R.; Glazko, G.; Mukherjee, A. RNA-seq reveals differentially expressed genes in rice (Oryza sativa) roots during interactions with plant-growth promoting bacteria, Azospirillum brasilense. PLoS ONE 2019, 14, e0217309. [Google Scholar] [CrossRef]
- Okubo, T.; Ikeda, S.; Sasaki, K.; Ohshima, K.; Hattori, M.; Sato, T.; Minamisawa, K. Phylogeny and functions of bacterial communities associated with field-grown rice shoots. Microbes Environ. 2014, 29, 329–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessitsch, A.; Hardoim, P.; Döring, J.; Weilharter, A.; Krause, A.; Woyke, T.; Mitter, B.; Hauberg-Lotte, L.; Friedrich, F.; Rahalkar, M.; et al. Functional Characteristics of an Endophyte Community Colonizing Rice Roots as Revealed by Metagenomic Analysis. Mol. Plant Microbe Interact. 2012, 25, 28–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Meth. 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Hamady, M.; Lozupone, C.; Knight, R. Fast UniFrac: Facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010, 4, 17–27. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Package “Vegan”. Community Ecology Package, Version 2. 2013. Available online: https://cran.ism.ac.jp/web/packages/vegan/vegan.pdf (accessed on 12 December 2021).
- Youssef, N.H.; Elshahed, M.S. Diversity rankings among bacterial lineages in soil. ISME J. 2008, 3, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bent, S.J.; Forney, L.J. The tragedy of the uncommon: Understanding limitations in the analysis of microbial diversity. ISME J. 2008, 2, 689–695. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Chellemi, D.O.; Graham, J.H.; Martin, K.J.; Rosskopf, E.N. Comparison of soil bacterial communities under diverse agricultural land management and crop production practices. Microb. Ecol. 2008, 55, 293–310. [Google Scholar] [CrossRef]
- Rousk, J.; Baath, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Knight, R. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. USA 2007, 104, 11436–11440. [Google Scholar] [CrossRef] [Green Version]
- Bossio, D.A.; Scow, K. Impacts of carbon and flooding on soil microbial communities: Phospholipid fatty acid profiles and substrate utilization patterns. Microb. Ecol. 1998, 35, 265–278. [Google Scholar] [CrossRef]
- Jangid, K.; Williams, M.A.; Franzluebbers, A.J.; Schmidt, T.M.; Coleman, D.C.; Whitman, W.B. Land-use history has a stronger impact on soil microbial community composition than aboveground vegetation and soil properties. Soil Biol. Biochem. 2011, 43, 2184–2193. [Google Scholar] [CrossRef]
- Upchurch, R.; Chiu, C.-Y.; Everett, K.; Dyszynski, G.; Coleman, D.C.; Whitman, W.B. Differences in the composition and diversity of bacterial communities from agricultural and forest soils. Soil Biol. Biochem. 2008, 40, 1294–1305. [Google Scholar] [CrossRef]
- Navarro-Noya, Y.E.; Gómez-Acata, S.; Montoya-Ciriaco, N.; Rojas-Valdez, A.; Suárez-Arriaga, M.C.; Valenzuela-Encinas, C.; Jiménez-Bueno, N.; Verhulst, N.; Govaerts, B.; Dendooven, L. Relative impacts of tillage, residue management and crop-rotation on soil bacterial communities in a semi-arid agroecosystem. Soil Biol. Biochem. 2013, 65, 86–95. [Google Scholar] [CrossRef]
- Venter, Z.S.; Jacobs, K.; Hawkins, H.-J. The impact of crop rotation on soil microbial diversity: A meta-analysis. Pedobiologia 2016, 59, 215–223. [Google Scholar] [CrossRef]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.R.; Blair, P.L.; Boyd, C.; Cody, B.; Hazel, A.; Hedrick, A.; Kathuria, H.; Khurana, P.; Kramer, B.; Muterspaw, K.; et al. Microbial community responses to soil tillage and crop rotation in a corn/soybean agroecosystem. Ecol. Evol. 2016, 6, 8075–8084. [Google Scholar] [CrossRef] [PubMed]
- De Moura, M.S.; Silva, B.M.; Mota, P.K.; Borghi, E.; Resende, A.V.d.; Acuña-Guzman, S.F.; Araújo, G.S.S.; da Silva, L.d.C.M.; de Oliveira, G.C.; Curi, N. Soil management and diverse crop rotation can mitigate early-stage no-till compaction and improve least limiting water range in a Ferralsol. Agric. Water Manag. 2021, 243, 106523. [Google Scholar] [CrossRef]
- Mathew, R.P.; Feng, Y.; Githinji, L.; Ankumah, R.; Balkcom, K.S. Impact of No-Tillage and Conventional Tillage Systems on Soil Microbial Communities. Appl. Environ. Soil Sci. 2012, 2012, 548620. [Google Scholar] [CrossRef] [Green Version]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food Security: The Challenge of Feeding 9 Billion People. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Bargaz, A.; Lyamlouli, K.; Chtouki, M.; Zeroual, Y.; Dhiba, D. Soil Microbial Resources for Improving Fertilizers Efficiency in an Integrated Plant Nutrient Management System. Front. Microbiol. 2018, 9, 1606. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
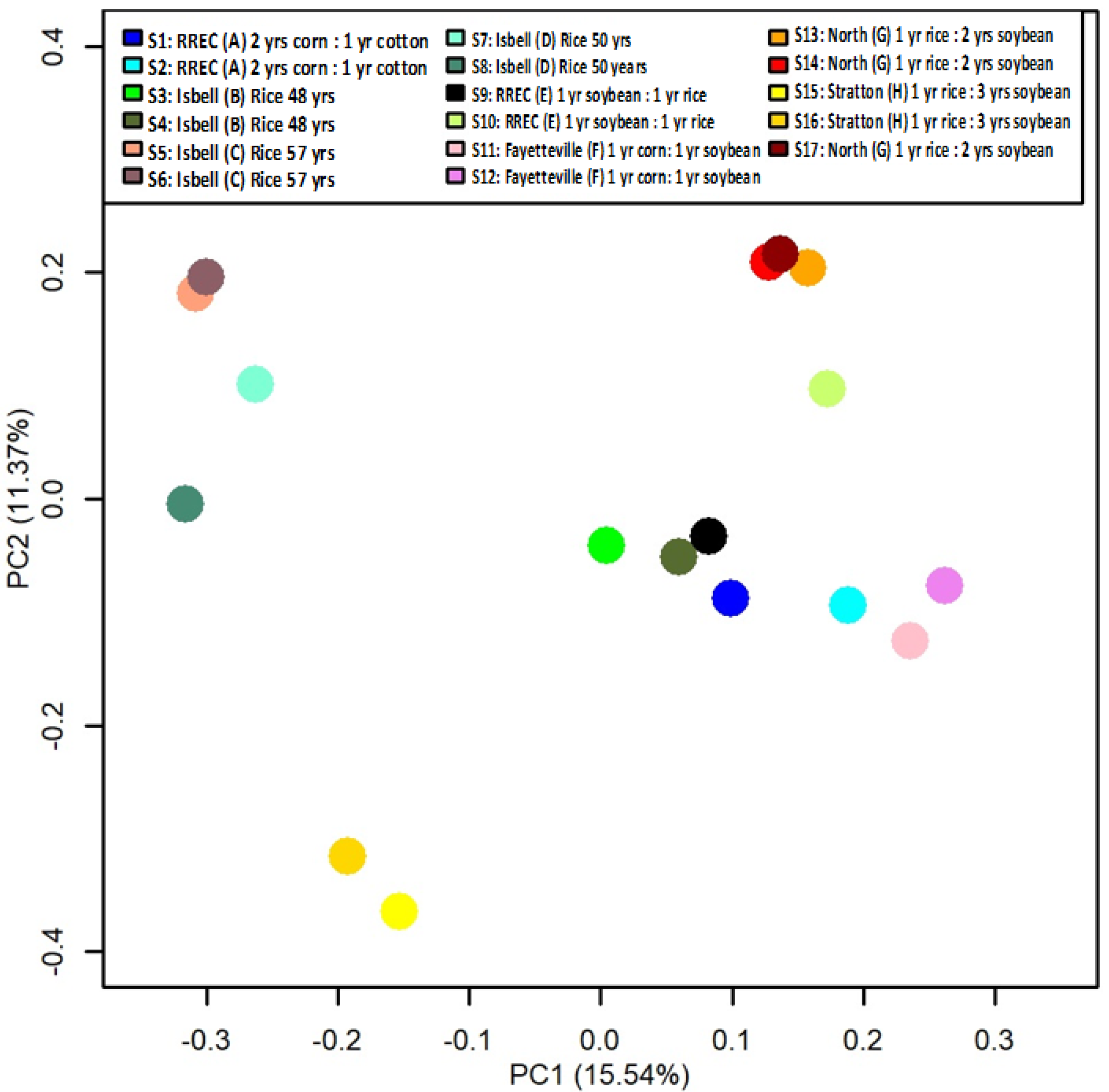
| Location | Field | Coordinates | Last Crop | Current Crop (Nov. 2017) | Rotation |
|---|---|---|---|---|---|
| A | RREC | 34°27′44″ N 91°24′08″ W | Corn | Cotton | 2 years corn: 1 year cotton |
| B | Isbell | 34°34′56″ N 91°45′47″ W | Rice | Rice | Rice 48 years |
| C | Isbell | 34°35′46″ N 91°45′28″ W | Rice | Rice | Rice 57 years |
| D | Isbell | 34°37′32″ N 91°45′12″ W | Rice | Rice | Rice 50 years |
| E | RREC | 34°27′48″ N 91°25′10″ W | soybean | Rice | 1 year soybean: 1 year rice |
| F | Fayetteville | 36°05′56″ N 94°10′23″ W | Corn | Soybean | 1 year corn: 1 year soybean |
| G | North | 34°17′19″ N 91°34′32″ W | Rice | Soybean | 1 year rice: 2 years soybean |
| H | Stratton | 34°29′42″ N 91°34′42″ W | Rice | Soybean | 1 year rice: 3 years soybean |
| Location (Field) | P | K | Ca | Mg | SO4-S | Zn | Fe | Mn | Cu | B | Soil pH (1:2 Soil-Water) | Soil ECEC (cmolc/kg) | Estimated Soil Texture | Crop Rotation Yes/No | Till Yes/No |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A (RREC) | 109 | 1033 | 69 | 13 | 3.0 | 141 | 190 | 0.8 | 0.3 | 6.3 | 9 | Silt loam | Yes 2 years corn: 1 year cotton | Yes | |
| 19 | 131 | 1097 | 75 | 11 | 6.2 | 136 | 223 | 1.5 | 0.3 | 6.3 | 9 | ||||
| B (Isbell) | 12 | 279 | 3289 | 850 | 65 | 3.2 | 263 | 53 | 7.1 | 0.7 | 6.4 | 29 | Clay | No Rice 48 years | No |
| 20 | 376 | 2715 | 631 | 84 | 4.3 | 337 | 63 | 4.0 | 0.8 | 5.8 | 25 | ||||
| C (Isbell) | 11 | 237 | 3753 | 728 | 29 | 3.6 | 369 | 116 | 2.0 | 0.7 | 6.8 | 29 | Clay | No Rice 57 years | No |
| 10 | 196 | 3142 | 674 | 26 | 3.0 | 406 | 166 | 1.9 | 0.7 | 6.9 | 25 | ||||
| D (Isbell) | 49 | 161 | 1700 | 266 | 119 | 7.1 | 434 | 28 | 3.2 | 0.7 | 6.5 | 15 | Silt loam—Silty clay loam | No Rice 50 years | No |
| 57 | 158 | 1869 | 276 | 259 | 6.1 | 388 | 35 | 3.3 | 0.7 | 6.5 | 16 | ||||
| E (RREC) | 26 | 71 | 600 | 89 | 12 | 5.5 | 382 | 189 | 1.0 | 0.4 | 5.7 | 8 | Silt loam | Yes 1 year soybean: 1 year rice | Yes |
| 16 | 69 | 649 | 101 | 9 | 6.7 | 225 | 202 | 1.4 | 0.3 | 5.9 | 7 | ||||
| F (Fayetteville) | 21 | 61 | 659 | 44 | 7 | 1.2 | 61 | 149 | 1.3 | 0.1 | 6.6 | 6 | Silt loam | Yes 1 year corn: 1 year soybean | Yes |
| 30 | 80 | 618 | 42 | 7 | 1.4 | 74 | 165 | 1.4 | 0.2 | 6.3 | 6 | ||||
| G (North) | 44 | 272 | 367 | 72 | 21 | 3.7 | 262 | 12 | 1.1 | 0.2 | 5.3 | 8 | Sandy loam | Yes 1 year rice: 2 years soybean | No |
| 67 | 220 | 483 | 101 | 18 | 4.7 | 343 | 20 | 1.0 | 0.3 | 5.2 | 9 | ||||
| 25 | 174 | 534 | 85 | 148 | 4.3 | 523 | 11 | 0.3 | 0.4 | 4.6 | 10 | Silt Loam | |||
| H (Stratton) | 81 | 236 | 3619 | 338 | 19 | 3.5 | 174 | 67 | 2.1 | 1.1 | 8 | 24 | Clay | Yes 1 year rice: 3 years soybean | No |
| 101 | 375 | 4895 | 390 | 33 | 4.9 | 201 | 66 | 3.1 | 1.5 | 8.1 | 31 |
| Location | Field | Metagenomics Sample ID | Total Reads | Reads Passing Quality Filtering | % Reads Passing Quality Filtering | Aligned Merged Reads per Sample |
|---|---|---|---|---|---|---|
| A | RREC | S1 | 936,364 | 899,156 | 96.0 | 749,518 |
| S2 | 771,282 | 743,354 | 96.4 | 617,111 | ||
| B | Isbell | S3 | 483,099 | 464,663 | 96.2 | 372,886 |
| S4 | 623,931 | 594,092 | 95.2 | 460,853 | ||
| C | Isbell | S5 | 892,264 | 860,061 | 96.4 | 725,334 |
| S6 | 909,293 | 875,900 | 96.3 | 713,702 | ||
| D | Isbell | S7 | 1,028,520 | 980,191 | 95.3 | 794,318 |
| S8 | 597,720 | 576,338 | 96.4 | 478,296 | ||
| E | RREC | S9 | 906,681 | 873,317 | 96.3 | 730,662 |
| S10 | 904,034 | 869,247 | 96.2 | 705,984 | ||
| F | Fayetteville | S11 | 495,227 | 476,026 | 96.1 | 376,829 |
| S12 | 860,221 | 829,259 | 96.4 | 677,953 | ||
| G | North | S13 | 886,695 | 848,726 | 95.7 | 692,452 |
| S14 | 803,096 | 775,715 | 96.6 | 642,329 | ||
| S17 | 913,691 | 881,743 | 96.5 | 746,262 | ||
| H | Stratton | S15 | 948,914 | 908,703 | 95.8 | 741,907 |
| S16 | 476,703 | 459,319 | 96.4 | 378,918 |
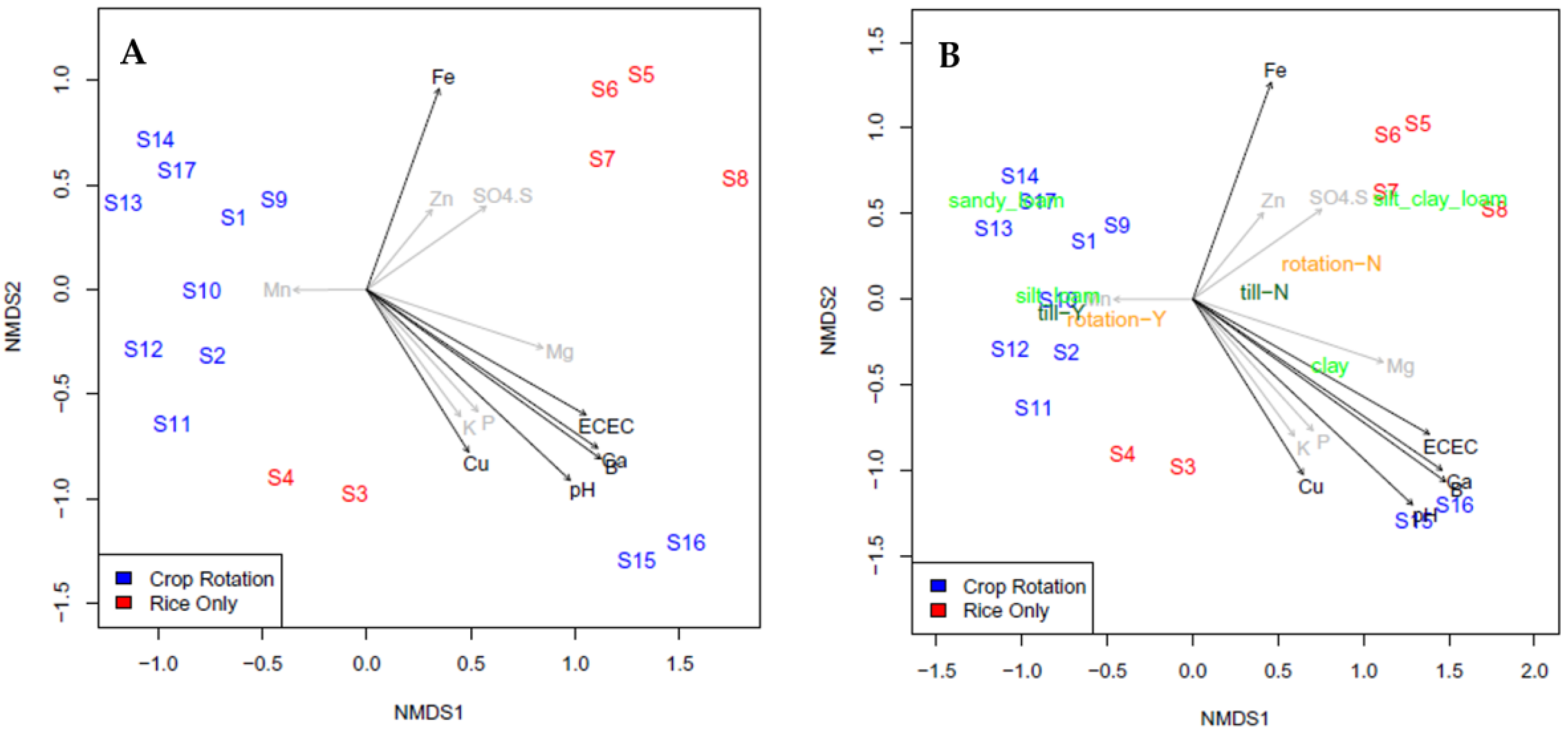
| Pearson’s Correlation with Shannon Diversity | Sum Sq | Df | F-Value | Pr (>F) | ||
|---|---|---|---|---|---|---|
| SO4-S | −0.657 | 0.248172 | 1 | 446.0492 | 0.002234 | ** |
| Fe | −0.347 | 0.012271 | 1 | 22.05459 | 0.042474 | * |
| Mg | −0.294 | 0.000161 | 1 | 0.290031 | 0.644121 | |
| ECEC | −0.274 | 0.035436 | 1 | 63.69047 | 0.015341 | * |
| Ca | −0.241 | 0.036469 | 1 | 65.54779 | 0.014916 | * |
| B | −0.195 | 0.058488 | 1 | 105.1231 | 0.009379 | ** |
| Mn | 0.187 | 0.016118 | 1 | 28.97011 | 0.032828 | * |
| pH | −0.135 | 0.007749 | 1 | 13.92685 | 0.064893 | . |
| Cu | −0.121 | 0.120177 | 1 | 215.9979 | 0.004598 | ** |
| Zn | −0.086 | 0.012036 | 1 | 21.63242 | 0.04325 | * |
| K | −0.063 | 0.001383 | 1 | 2.485264 | 0.255624 | |
| P | −0.043 | 0.249766 | 1 | 448.9144 | 0.00222 | ** |
| Rotation | 0.001886 | 1 | 3.390498 | 0.206919 | ||
| Till | 0.007491 | 1 | 13.46463 | 0.066902 | . | |
| Residuals | 0.001113 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, C.; Shakiba, E.; North, D.; McGraw, M.; Ballard, E.; Barrett-D’Amico, M.; Glazko, G.; Rahmatallah, Y. 16S rRNA Gene-Based Metagenomic Analysis of Rhizosphere Soil Bacteria in Arkansas Rice Crop Fields. Agronomy 2022, 12, 222. https://doi.org/10.3390/agronomy12010222
Oliveira C, Shakiba E, North D, McGraw M, Ballard E, Barrett-D’Amico M, Glazko G, Rahmatallah Y. 16S rRNA Gene-Based Metagenomic Analysis of Rhizosphere Soil Bacteria in Arkansas Rice Crop Fields. Agronomy. 2022; 12(1):222. https://doi.org/10.3390/agronomy12010222
Chicago/Turabian StyleOliveira, Cássia, Ehsan Shakiba, Dustin North, Madison McGraw, Ethan Ballard, Marissa Barrett-D’Amico, Galina Glazko, and Yasir Rahmatallah. 2022. "16S rRNA Gene-Based Metagenomic Analysis of Rhizosphere Soil Bacteria in Arkansas Rice Crop Fields" Agronomy 12, no. 1: 222. https://doi.org/10.3390/agronomy12010222
APA StyleOliveira, C., Shakiba, E., North, D., McGraw, M., Ballard, E., Barrett-D’Amico, M., Glazko, G., & Rahmatallah, Y. (2022). 16S rRNA Gene-Based Metagenomic Analysis of Rhizosphere Soil Bacteria in Arkansas Rice Crop Fields. Agronomy, 12(1), 222. https://doi.org/10.3390/agronomy12010222