
Targeted Sequencing of the Short Arm of Chromosome 6V of a Wheat Relative Haynaldia villosa for Marker Development and Gene Mining


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Chromosome Sorting and DNA Sequencing
2.3. Identification of Repetitive Sequences
2.4. Transcriptome Data
2.5. Identification of Coding Sequences
2.6. Development of Intron Target Markers
3. Results
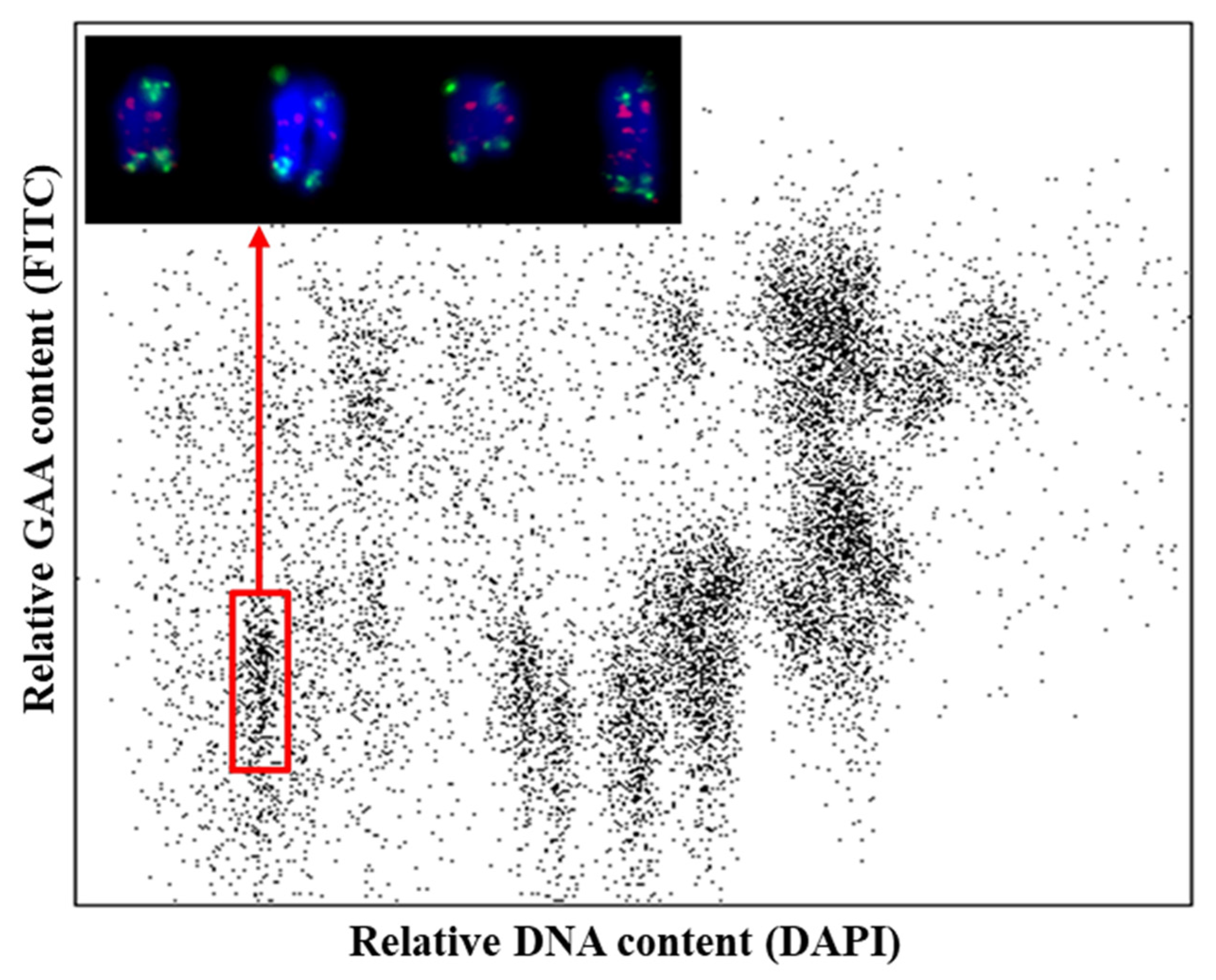
3.1. Flow Sorting and Sequencing of Chromosome Arm 6VS of H. villosa
3.2. The Repetitive DNA Elements in the 6VS Sequence
3.3. Gene Content of Chromosome Arm 6VS
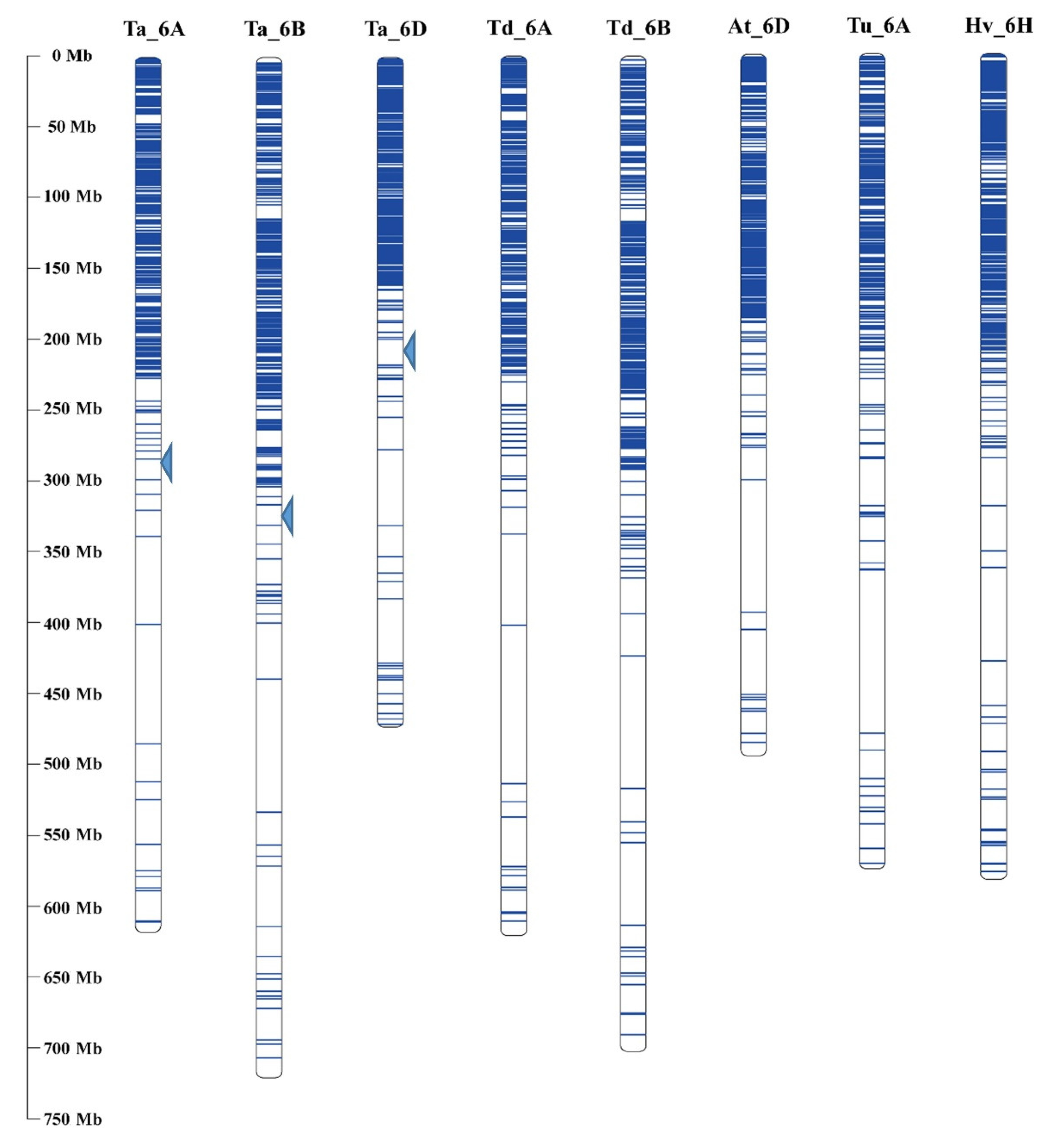
3.4. Comparative Genome Analysis of 6VS Sequence Composition
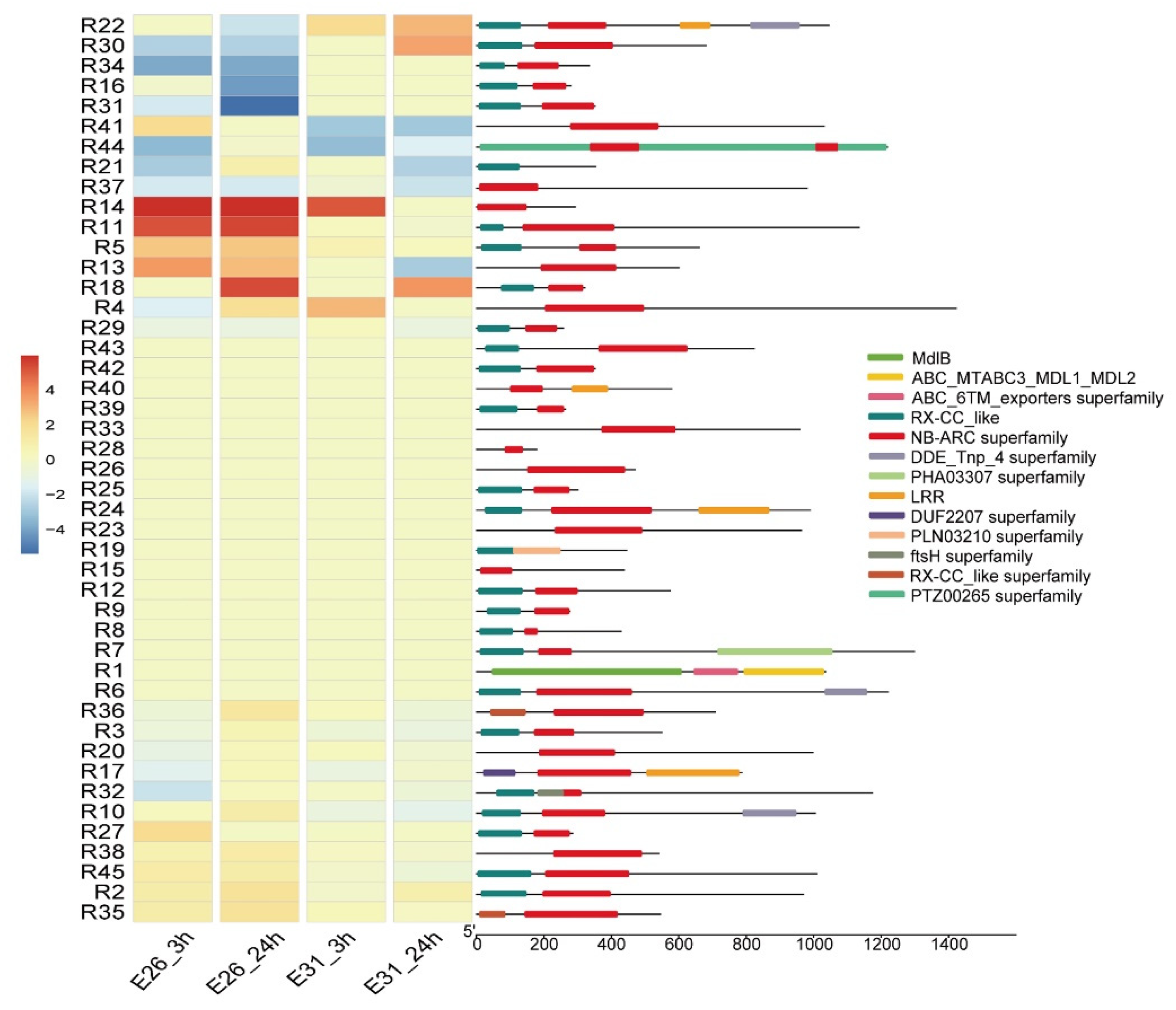
3.5. NB-ARC Domain Proteins Enrichment and their Expression Profiling after Bgt Infection
3.6. Haplotype Analysis of GW2-6V
3.7. Development of 6VS Specific Intron Targeted (IT) Markers
4. Discussion
4.1. Aneuploid Germplasm Facilitates Flow-Sorting Target Chromosomes or Chromosome Arms
4.2. The Available 6VS Sequences Would Facilitate the Introduction of Interest Genes with Minimized Linkage Drag by Chromosome Engineering
4.3. The High TGW for 6VS/6AL Translocation Line Was Not Attributable to GW2-6V
4.4. Development of Specific Molecular Markers Using Chromosome Sorting Strategy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grądzielewska, A. The genus Dasypyrum—Part 2. Dasypyrum villosum—A wild species used in wheat improvement. Euphytica 2006, 152, 441–454. [Google Scholar]
- Mohammad, P.; Hossain, M.; Khodaker, N.; Shiraishi, M. Study for morphological characters of species alien to wheat in Bangladesh. Sarhad J. Agric. 1997, 13, 541–550. [Google Scholar]
- Okocha, P. Peculiarities of nucleo-cytoplasmic interactions in allocytoplasmic forms of wheat. Glob. J. Pure Appl. Sci. 1999, 5, 431–436. [Google Scholar]
- Della Gatta, C.; Tanzarella, O.; Resta, P.; Blanco, A. Protein content in a population of Haynaldia villosa and electrophoretic pattern of the amphiploid T. durum × H. villosa. In Breeding Methodologies in Durum Wheat and Triticale; Tuscia University: Viterbo, Italy, 1984; pp. 39–43. [Google Scholar]
- Pace, C.; Paolini, R.; Scarascia, M.; Qualset, C.; Delre, V. Evaluation and utilization of Dasypyrum villosum as a genetic resource for wheat improvement. In Wheat Genetic Resources: Meeting Diverse Needs; John Wiley & Sons: Hoboken, NJ, USA, 1990; pp. 279–379. [Google Scholar]
- De Pace, C.; Snidaro, D.; Ciaffi, M.; Vittori, D.; Ciofo, A.; Cenci, A.; Tanzarella, O.; Qualset, C.; Mugnozza, G.S. Introgression of Dasypyrum villosum chromatin into common wheat improves grain protein quality. Euphytica 2001, 117, 67–75. [Google Scholar] [CrossRef]
- Qualset, C.; De Pace, C.; Jan, C.; Mugnozza, G.S.; Tanzarella, O.; Greco, B. Haynaldia villosa (L.) Schur: A species with potential use in wheat breeding. Am. Soc. Agron. Abstr. 1981, 36, 191. [Google Scholar]
- Zhong, G.Y.; Dvořák, J. Evidence for common genetic mechanisms controlling the tolerance of sudden salt stress in the tribe Triticeae. Plant Breed. 1995, 114, 297–302. [Google Scholar] [CrossRef]
- Zhao, C.; Lv, X.; Li, Y.; Li, F.; Geng, M.; Mi, Y.; Ni, Z.; Wang, X.; Xie, C.; Sun, Q. Haynaldia villosa NAM-V1 is linked with the powdery mildew resistance gene Pm21 and contributes to increasing grain protein content in wheat. BMC Genet. 2016, 17, 82. [Google Scholar]
- Xing, L.; Hu, P.; Liu, J.; Witek, K.; Zhou, S.; Xu, J.; Zhou, W.; Gao, L.; Huang, Z.; Zhang, R.; et al. Pm21 from Haynaldia villosa Encodes a CC-NBS-LRR that Confers Powdery Mildew Resistance in Wheat. Mol. Plant 2018, 11, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Bauer, E.; Schmutzer, T.; Barilar, I.; Mascher, M.; Gundlach, H.; Martis, M.M.; Twardziok, S.O.; Hackauf, B.; Gordillo, A.; Wilde, P. Towards a whole-genome sequence for rye (Secale cereale L.). Plant J. 2017, 89, 853–869. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.Q.; Zhao, S.; Liu, D.; Wang, J.; Sun, H.; Zhang, C.; Fan, H.; Li, D.; Dong, L.; Tao, Y.; et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 2013, 496, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhao, S.; Kong, X.; Li, Y.; Zhao, G.; He, W.; Appels, R.; Pfeifer, M.; Tao, Y.; Zhang, X.; et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 2013, 496, 91–95. [Google Scholar] [CrossRef]
- Doležel, J.; Kubaláková, M.; Paux, E.; Bartoš, J.; Feuillet, C. Chromosome-based genomics in the cereals. Chromosome Res. 2007, 15, 51–66. [Google Scholar] [CrossRef]
- Berkman, P.J.; Skarshewski, A.; Lorenc, M.T.; Lai, K.; Duran, C.; Ling, E.Y.; Stiller, J.; Smits, L.; Imelfort, M.; Manoli, S.; et al. Sequencing and assembly of low copy and genic regions of isolated Triticum aestivum chromosome arm 7DS. Plant Biotechnol. J. 2011, 9, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Vitulo, N.; Albiero, A.; Forcato, C.; Campagna, D.; Dal Pero, F.; Bagnaresi, P.; Colaiacovo, M.; Faccioli, P.; Lamontanara, A.; Simkova, H.; et al. First survey of the wheat chromosome 5A composition through a next generation sequencing approach. PLoS ONE 2011, 6, e26421. [Google Scholar] [CrossRef] [PubMed]
- Berkman, P.J.; Skarshewski, A.; Manoli, S.; Lorenc, M.T.; Stiller, J.; Smits, L.; Lai, K.; Campbell, E.; Kubalakova, M.; Simkova, H.; et al. Sequencing wheat chromosome arm 7BS delimits the 7BS/4AL translocation and reveals homoeologous gene conservation. TAG Theor. Appl. Genet. 2012, 124, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.J.; Akpinar, B.A.; Šimková, H.; Kubaláková, M.; Doležel, J.; Budak, H. Next-generation sequencing of flow-sorted wheat chromosome 5D reveals lineage-specific translocations and widespread gene duplications. BMC Genom. 2014, 15, 1080. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kobayashi, F.; Joshi, G.P.; Onuki, R.; Sakai, H.; Kanamori, H.; Wu, J.; Simkova, H.; Nasuda, S.; Endo, T.R.; et al. Next-generation survey sequencing and the molecular organization of wheat chromosome 6B. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2014, 21, 103–114. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Helguera, M.; Rivarola, M.; Clavijo, B.; Martis, M.M.; Vanzetti, L.S.; Gonzalez, S.; Garbus, I.; Leroy, P.; Simkova, H.; Valarik, M.; et al. New insights into the wheat chromosome 4D structure and virtual gene order, revealed by survey pyrosequencing. Plant Sci. Int. J. Exp. Plant Biol. 2015, 233, 200–212. [Google Scholar] [CrossRef]
- Tiwari, V.K.; Wang, S.; Danilova, T.; Koo, D.H.; Vrána, J.; Kubaláková, M.; Hřibová, E.; Rawat, N.; Kalia, B.; Singh, N.; et al. Exploring the tertiary gene pool of bread wheat: Sequence assembly and analysis of chromosome 5M(g) of Aegilops geniculata. Plant J. Cell Mol. Biol. 2015, 84, 733–746. [Google Scholar] [CrossRef]
- Xiao, J.; Dai, K.; Fu, L.; Vrána, J.; Kubaláková, M.; Wan, W.; Sun, H.; Zhao, J.; Yu, C.; Wu, Y.; et al. Sequencing flow-sorted short arm of Haynaldia villosa chromosome 4V provides insights into its molecular structure and virtual gene order. BMC Genom. 2017, 18, 791. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, X.; Xiao, J.; Yuan, C.; Wu, Y.; Cao, A.; Xing, L.; Chen, P.; Zhang, S.; Wang, X.; et al. Whole genome development of intron targeting (IT) markers specific for Dasypyrum villosum chromosomes based on next-generation sequencing technology. Mol. Breed. 2017, 37, 115. [Google Scholar] [CrossRef]
- Wang, H.; Dai, K.; Xiao, J.; Yuan, C.; Zhao, R.; Dolezel, J.; Wu, Y.; Cao, A.; Chen, P.; Zhang, S.; et al. Development of intron targeting (IT) markers specific for chromosome arm 4VS of Haynaldia villosa by chromosome sorting and next-generation sequencing. BMC Genom. 2017, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.K.; Wang, S.; Sehgal, S.; Vrána, J.; Friebe, B.; Kubaláková, M.; Chhuneja, P.; Doležel, J.; Akhunov, E.; Kalia, B.; et al. SNP Discovery for mapping alien introgressions in wheat. BMC Genom. 2014, 15, 273. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.; Martis, M.; Hedley, P.E.; Šimková, H.; Liu, H.; Morris, J.A.; Steuernagel, B.; Taudien, S.; Roessner, S.; Gundlach, H.; et al. Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 2011, 23, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Martis, M.M.; Zhou, R.; Haseneyer, G.; Schmutzer, T.; Vrána, J.; Kubaláková, M.; Konig, S.; Kugler, K.G.; Scholz, U.; Hackauf, B.; et al. Reticulate evolution of the rye genome. Plant Cell 2013, 25, 3685–3698. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Sanchez-Martin, J.; Steuernagel, B.; Ghosh, S.; Herren, G.; Hurni, S.; Adamski, N.; Vrána, J.; Kubaláková, M.; Krattinger, S.G.; Wicker, T.; et al. Rapid gene isolation in barley and wheat by mutant chromosome sequencing. Genome Biol. 2016, 17, 221. [Google Scholar] [CrossRef]
- Thind, A.K.; Wicker, T.; Šimková, H.; Fossati, D.; Moullet, O.; Brabant, C.; Vrána, J.; Doležel, J.; Krattinger, S.G. Rapid cloning of genes in hexaploid wheat using cultivar-specific long-range chromosome assembly. Nat. Biotechnol. 2017, 35, 793. [Google Scholar] [CrossRef]
- Cao, A.; Xing, L.; Wang, X.; Yang, X.; Wang, W.; Sun, Y.; Qian, C.; Ni, J.; Chen, Y.; Liu, D.; et al. Serine/threonine kinase gene Stpk-V, a key member of powdery mildew resistance gene Pm21, confers powdery mildew resistance in wheat. Proc. Natl. Acad. Sci. USA 2011, 108, 7727–7732. [Google Scholar] [CrossRef]
- Xing, L.; Yuan, L.; Lv, Z.; Wang, Q.; Yin, C.; Huang, Z.; Liu, J.; Cao, S.; Zhang, R.; Peidu, C.; et al. Long range assembly of sequences helps to unravel the genome structure and small variation of the wheat-Haynaldia villosa translocated chromosome 6VS 6AL. Plant Biotechnol. J. 2021, 19, 1567–1578. [Google Scholar] [CrossRef]
- Qi, L.L.; Chen, P.D.; Liu, D.J.; Gill, B.S. Homoeologous relationships of Haynaldia villosa chromosomes with those of Triticum aestivum as revealed by RFLP analysis. Genes Genet. Syst. 1999, 74, 77–82. [Google Scholar] [CrossRef][Green Version]
- Kubaláková, M.; Vrána, J.; Číhaliková, J.; Šimková, H.; Doležel, J. Flow karyotyping and chromosome sorting in bread wheat (Triticum aestivum L.). TAG Theor. Appl. Genet. 2002, 104, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Vrána, J.; Kubaláková, M.; Šimková, H.; Číhaliková, J.; Lysák, M.A.; Doležel, J. Flow sorting of mitotic chromosomes in common wheat (Triticum aestivum L.). Genetics 2000, 156, 2033–2041. [Google Scholar] [CrossRef]
- Giorgi, D.; Farina, A.; Grosso, V.; Gennaro, A.; Ceoloni, C.; Lucretti, S. FISHIS: Fluorescence In Situ Hybridization in Suspension and Chromosome Flow Sorting Made Easy. PLoS ONE 2013, 8, e57994. [Google Scholar] [CrossRef] [PubMed]
- Kubaláková, M.; Valárik, M.; Bartoš, J.; Vrána, J.; Číhaliková, J.; Molnár-Lang, M.; Doležel, J. Analysis and sorting of rye (Secale cereale L.) chromosomes using flow cytometry. Genome 2003, 46, 893–905. [Google Scholar] [CrossRef]
- Sun, H.; Song, J.; Lei, J.; Song, X.; Dai, K.; Xiao, J.; Yuan, C.; An, S.; Wang, H.; Wang, X. Construction and application of oligo-based FISH karyotype of Haynaldia villosa. J. Genet. Genom. 2018, 45, 463–466. [Google Scholar] [CrossRef]
- Šimková, H.; Svensson, J.T.; Condamine, P.; Hřibová, E.; Suchánková, P.; Bhat, P.R.; Bartoš, J.; Šafař, J.; Close, T.J.; Doležel, J. Coupling amplified DNA from flow-sorted chromosomes to high-density SNP mapping in barley. BMC Genom. 2008, 9, 294. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; The International Wheat Genome Sequencing Consortium (IWGSC); et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, V.; Gahlaut, V.; Mathur, S.; Agarwal, P.; Khandelwal, M.K.; Khurana, J.P.; Tyagi, A.K.; Balyan, H.S.; Gupta, P.K. Identification of Novel SNP in Promoter Sequence of TaGW2-6A Associated with Grain Weight and Other Agronomic Traits in Wheat (Triticum aestivum L.). PLoS ONE 2015, 10, e0129400. [Google Scholar] [CrossRef]
- Li, H.; Chen, X.; Xin, Z.Y.; Ma, Y.Z.; Xu, H.J.; Chen, X.Y.; Jia, X. Development and identification of wheat—Haynaldia villosa T6DL.6VS chromosome translocation lines conferring resistance to powdery mildew. Plant Breed. 2005, 124, 203–205. [Google Scholar] [CrossRef]
- Khan, S.J.; Muafia, S.; Nasreen, Z.; Salariya, A.M. Genetically Modified Organisms (GMOs): Food Security or Threat to Food Safety. Pak. J. Sci. 2012, 64, 6–12. [Google Scholar]
- Abrouk, M.; Balcárková, B.; Šimková, H.; Komínková, E.; Martis, M.M.; Jakobson, I.; Timofejeva, L.; Rey, E.; Vrána, J.; Kilian, A.; et al. The in silico identification and characterization of a bread wheat/Triticum militinae introgression line. Plant Biotechnol. J. 2017, 15, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, P.; Zhang, S.; Wang, X.; He, Z.; Zhang, Y.; Zhao, H.; Huang, H.; Zhou, X. Effects of the 6VS.6AL translocation on agronomic traits and dough properties of wheat. Euphytica 2006, 155, 305–313. [Google Scholar] [CrossRef]
- Su, Z.; Hao, C.; Wang, L.; Dong, Y.; Zhang, X. Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor. Appl. Genet. 2011, 122, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bai, Z.; Li, X.; Wang, P.; Wu, Q.; Yang, L.; Li, L.; Li, X. SNP identification and allelic-specific PCR markers development for TaGW2, a gene linked to wheat kernel weight. Theor. Appl. Genet. 2012, 125, 1057–1068. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
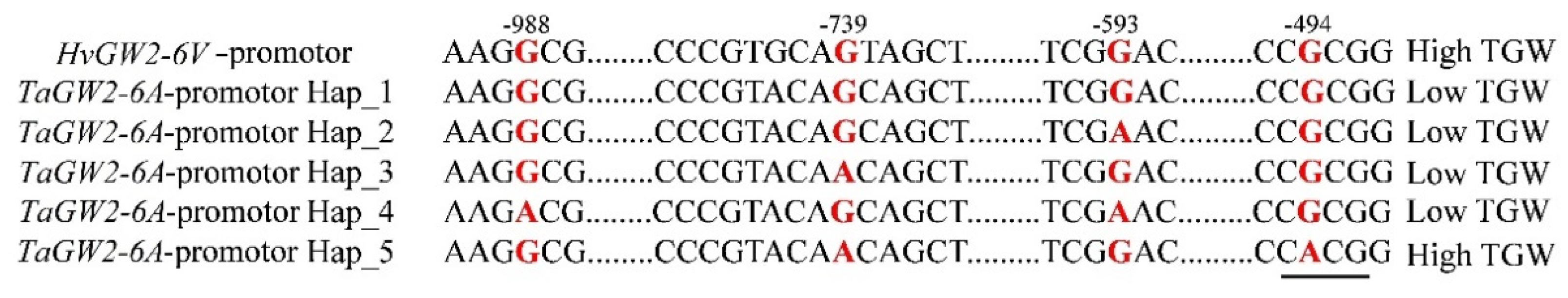{kind=link}
{kind=link}
| Total bases (Gbp) | 47.7 |
| Number of assembly scaffolds | 153,177 |
| Total assembly bases (bp) | 230,388,792 |
| Max. length of assembly scaffolds (bp) | 138,620 |
| Min. length of assembly scaffolds (bp) | 100 |
| N50 (bp) | 9.788 |
| Mean length (bp) | 1.464 |
| GC-content (%) | 45.68 |
| Type | Subtype | Total Length (bp) | % Genome |
|---|---|---|---|
| DNA transposon | |||
| TIR | 11,269,751 | 6.53 | |
| Helitron | 189,843 | 0.11 | |
| retrotransposon | |||
| LTR_Copia | 18,656,357 | 10.81 | |
| LTR_Gypsy | 89,588,481 | 51.91 | |
| LTR_Unknown | 3,831,370 | 2.22 | |
| SINE | 1,691,326 | 0.98 | |
| Unknown | 4,694,291 | 2.72 | |
| tandem repeat | 535,011 | 0.31 | |
| unknown | 4,694,291 | 2.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wan, W.; Li, M.; Yu, Z.; Liu, J.; Holušová, K.; Vrána, J.; Doležel, J.; Wu, Y.; Wang, H.; et al. Targeted Sequencing of the Short Arm of Chromosome 6V of a Wheat Relative Haynaldia villosa for Marker Development and Gene Mining. Agronomy 2021, 11, 1695. https://doi.org/10.3390/agronomy11091695
Zhang X, Wan W, Li M, Yu Z, Liu J, Holušová K, Vrána J, Doležel J, Wu Y, Wang H, et al. Targeted Sequencing of the Short Arm of Chromosome 6V of a Wheat Relative Haynaldia villosa for Marker Development and Gene Mining. Agronomy. 2021; 11(9):1695. https://doi.org/10.3390/agronomy11091695
Chicago/Turabian StyleZhang, Xu, Wentao Wan, Mengli Li, Zhongyu Yu, Jia Liu, Kateřina Holušová, Jan Vrána, Jaroslav Doležel, Yufeng Wu, Haiyan Wang, and et al. 2021. "Targeted Sequencing of the Short Arm of Chromosome 6V of a Wheat Relative Haynaldia villosa for Marker Development and Gene Mining" Agronomy 11, no. 9: 1695. https://doi.org/10.3390/agronomy11091695
APA StyleZhang, X., Wan, W., Li, M., Yu, Z., Liu, J., Holušová, K., Vrána, J., Doležel, J., Wu, Y., Wang, H., Xiao, J., & Wang, X. (2021). Targeted Sequencing of the Short Arm of Chromosome 6V of a Wheat Relative Haynaldia villosa for Marker Development and Gene Mining. Agronomy, 11(9), 1695. https://doi.org/10.3390/agronomy11091695