
Polarity-Based Sequential Extraction as a Simple Tool to Reveal the Structural Complexity of Humic Acids

 , ,
, ,  ,
, 
Abstract
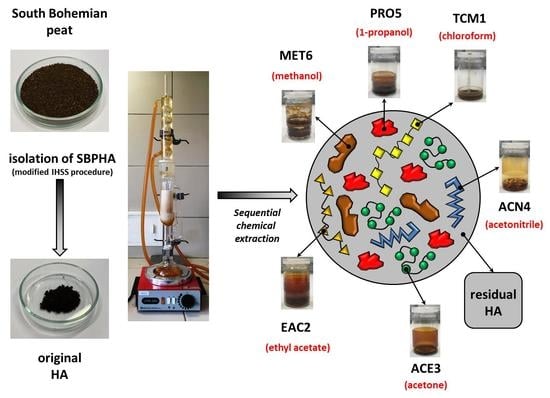
1. Introduction
2. Materials and Methods
2.1. Origin and Isolation of Humic Acids
2.2. Sequential Chemical Extraction
2.3. Organic Fractions Analysis
2.3.1. Elemental Composition
2.3.2. UV/Vis Spectrometry
2.3.3. ATR-FTIR Spectrometry
2.3.4. Fluorescence Spectrometry
2.3.5. Liquid-state 13C NMR Spectroscopy
2.3.6. Statistical Analysis
3. Results and Discussion
3.1. Yields, Ash Contents, and Elemental Analysis
3.2. UV/Vis Spectrometry
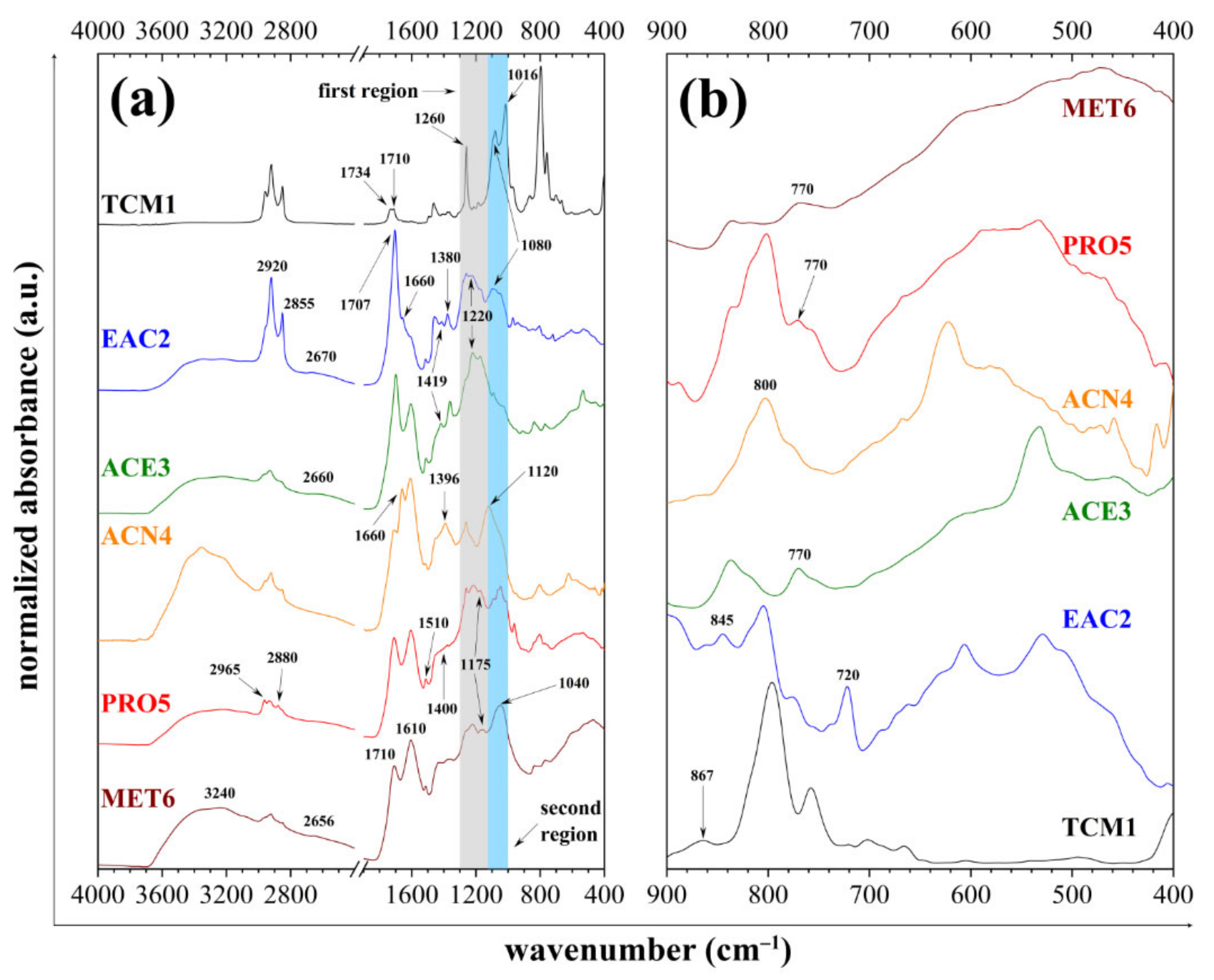
3.3. ATR-FTIR Spectrometry
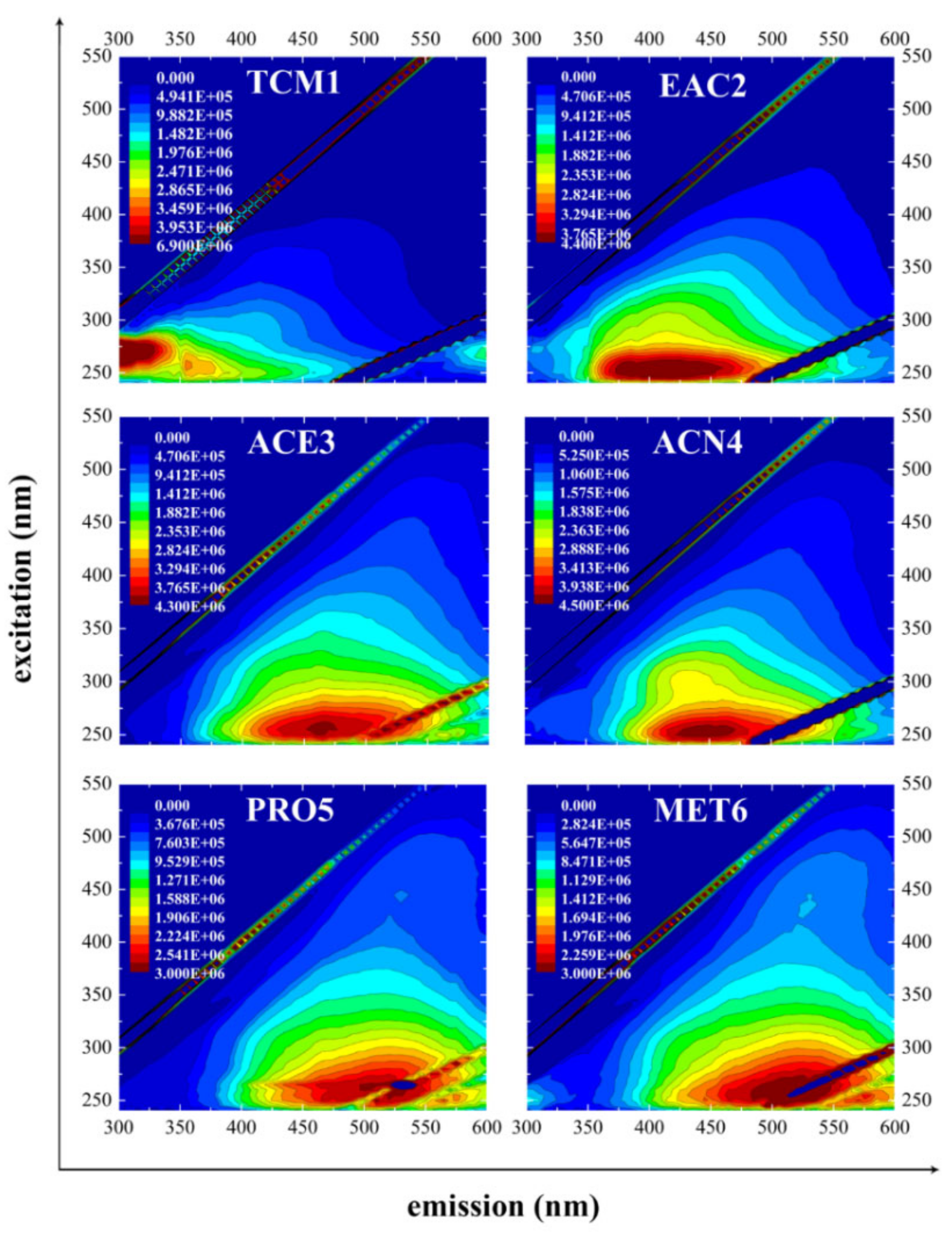
3.4. Fluorescence Spectrometry
3.5. Liquid-State 13C NMR Spectrometry
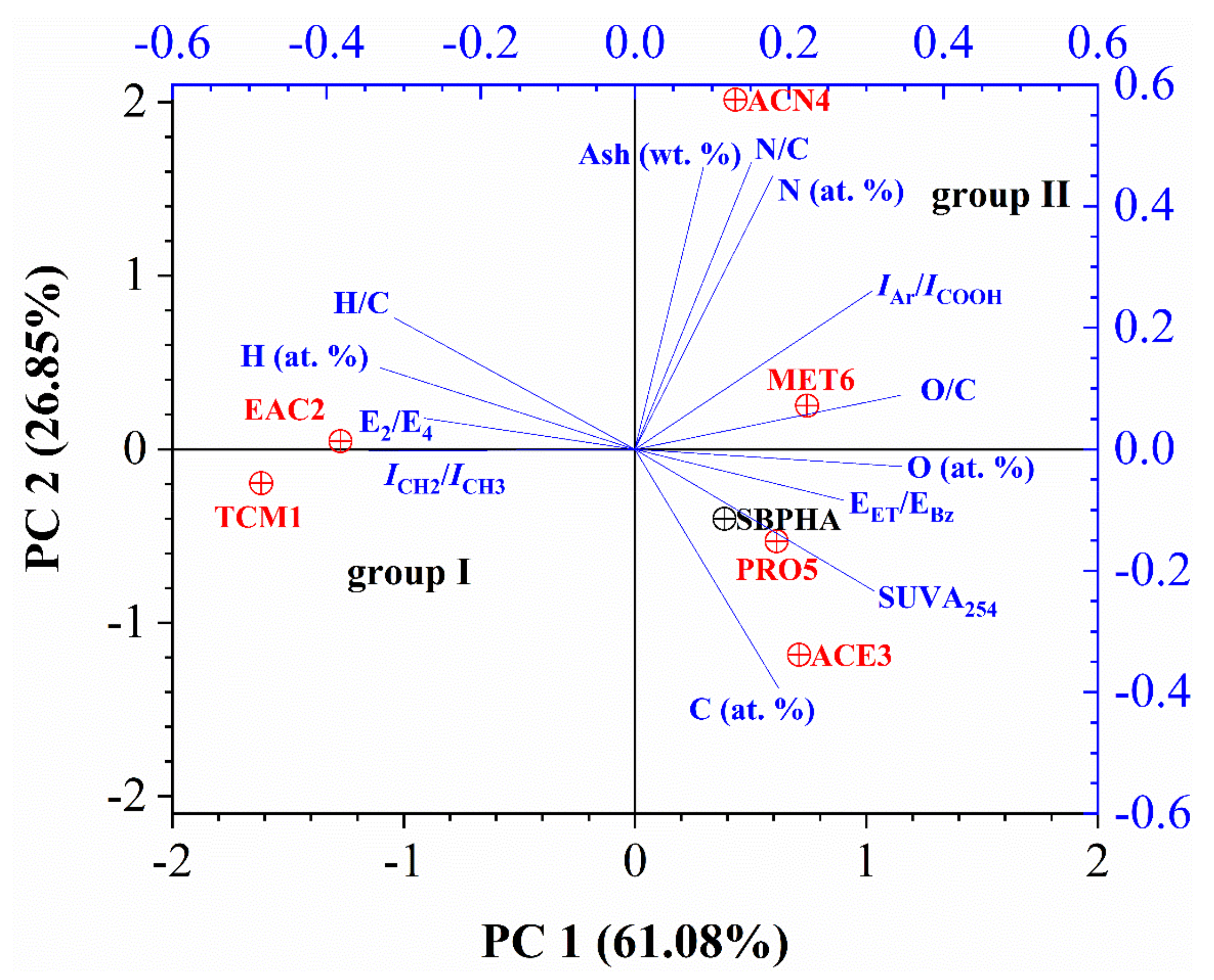
3.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kleber, M.; Lehmann, J. Humic substances extracted by alkali are invalid proxies for the dynamics and functions of organic matter in terrestrial and aquatic ecosystems. J. Environ. Qual. 2019, 48, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Olk, D.C.; Bloom, P.R.; Perdue, E.M.; McKnight, D.M.; Chen, Y.; Farenhorst, A.; Senesi, N.; Chin, Y.P.; Schmitt-Kopplin, P.; Hertkorn, N.; et al. Environmental and agricultural relevance of humic fractions extracted by alkali from soils and natural waters. J. Environ. Qual. 2019, 48, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Myneni, S.C.B. Chemistry of Natural Organic Matter—The Next Step: Commentary on a humic substances debate. J. Environ. Qual. 2019, 48, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zheng, Q.; Noll, L.; Zhang, S.; Wanek, W. Direct measurement of the in situ decomposition of microbial-derived soil organic matter. Soil Biol. Biochem. 2020, 141, 107660. [Google Scholar] [CrossRef]
- Hayes, M.H.B. Solvent systems for the isolation of organic components from soils. Soil Sci. Soc. Am. J. 2006, 70, 986–994. [Google Scholar] [CrossRef]
- Hildebrand, J.H.; Scott, R.L. Regular Solutions, 1st ed.; Prentice-Hall, Inc.: Englewood Cliffe, NJ, USA, 1962; p. 180. [Google Scholar]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007; p. 544. [Google Scholar]
- Senesi, N.; Testini, C.; Polemio, M. Chemical and spectroscopic characterization of soil organic matter fractions isolated by sequential extraction procedure. J. Soil Sci. 1983, 34, 801–813. [Google Scholar] [CrossRef]
- Piccolo, A. Characteristic of soil humic extracts obtained by some organic and inorganic solvents and purified by HCl-HF treatment. Soil Sci. 1988, 146, 418–426. [Google Scholar] [CrossRef]
- Fujitake, N.; Kusumato, A.; Tsukamato, M.; Kawahigashi, M.; Suzuki, T.; Otsuka, H. Properties of soil humic substances in fractions obtained by sequential extraction with pyrophosphate solutions at different pHs. I. Yield and particle size distribution. Soil Sci. Plant Nutr. 1998, 44, 253–260. [Google Scholar] [CrossRef]
- Li, L.; Huang, W.; Peng, P.; Sheng, G.; Fu, J. Chemical and molecular heterogeneity of humic acids repetitively extracted from a peat. Soil Sci. Soc. Am. J. 2003, 67, 740–746. [Google Scholar] [CrossRef]
- Olk, D.C. A Chemical fractionation for structure-function relations of soil organic matter in nutrient cycling. Soil Sci. Soc. Am. J. 2006, 70, 1013–1022. [Google Scholar] [CrossRef]
- Shirshova, L.T.; Ghabbour, E.A.; Davies, G. Spectroscopic characterization of humic acid fractions isolated from soil using different extraction procedures. Geoderma 2006, 133, 204–216. [Google Scholar] [CrossRef]
- Nebbioso, A.; Piccolo, A. Basis of a humeomics science: Chemical fractionation and molecular characterization of humic biosuprastructures. Biomacromolecules 2011, 12, 1187–1199. [Google Scholar] [CrossRef]
- Vinci, G.; Mazzei, P.; Drosos, M.; Zaccone, C.; Piccolo, A. Molecular characterization of ombrotrophic peats by humeomics. Chem. Biol. Technol. Agric. 2020, 7, 18. [Google Scholar] [CrossRef]
- Drosos, M.; Nebbioso, A.; Mazzei, P.; Vinci, G.; Spaccini, R.; Piccolo, A. A molecular zoom into soil Humeome by a direct sequential chemical fractionation of soil. Sci. Total Environ. 2017, 586, 807–816. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; WILEY-VCH: Weinheim, Germany, 2010; p. 718. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Baltimore, MD, USA, 2006; p. 954. [Google Scholar]
- Novák, F.; Šestauberová, M.; Hrabal, R. Structural features of lignohumic acids. J. Mol. Struct. 2015, 1093, 179–185. [Google Scholar] [CrossRef]
- Thomas, J.D.R. Chemistry of peat bitumen: Fractionation and infra-red studies. J. Appl. Chem. 2007, 12, 289–294. [Google Scholar] [CrossRef]
- Doskočil, L.; Enev, V.; Pekař, M.; Wasserbauer, J. The spectrometric characterization of lipids extracted from lignite samples from various coal basins. Org. Geochem. 2016, 95, 34–40. [Google Scholar] [CrossRef]
- Martins, T.; Saab, S.D.C.; Milori, D.M.B.P.; Brinatti, A.M.; Rosa, J.A.; Cassaro, F.A.M.; Pires, L.F. Soil organic matter humification under different tillage managements evaluated by Laser Induced Fluorescence (LIF) and C/N ratio. Soil Tillage Res. 2011, 111, 231–235. [Google Scholar] [CrossRef]
- Ma, X.; Green, S.A. Fractionation and spectroscopic properties of fulvic acid and its extract. Chemosphere 2008, 72, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Korshin, G.V.; Li, C.-W.; Benjamin, M.M. Monitoring the properties of natural organic matter through UV spectroscopy: A consistent theory. Water Res. 1997, 31, 1787–1795. [Google Scholar] [CrossRef]
- del Vecchio, R.; Blough, N.V. On the origin of the optical properties of humic substances. Environ. Sci. Technol. 2004, 38, 3885–3891. [Google Scholar] [CrossRef] [PubMed]
- Baigorri, R.; Fuentes, M.; González-Gaitano, G.; García-Mina, J.M. Analysis of molecular aggregation in humic substances in solution. Colloids Surf. A Physicochem. Eng. Asp. 2007, 302, 301–306. [Google Scholar] [CrossRef]
- Song, J.; Jin, X.; Wang, X.C.; Jin, P. Preferential binding properties of carboxyl and hydroxyl groups with aluminium salts for humic acid removal. Chemosphere 2019, 234, 478–487. [Google Scholar] [CrossRef]
- Boguta, P.; Sokołowska, Z. Zinc binding to fulvic acids: Assessing the impact of pH, metal concentrations and chemical properties of fulvic acids on the mechanism and stability of formed soluble complexes. Molecules 2020, 25, 1297. [Google Scholar] [CrossRef]
- Chen, Y.; Senesi, N.; Schnitzer, M. Information provided on humic substances by E4/E6 ratios. Soil Sci. Soc. Am. J. 1977, 41, 352–358. [Google Scholar] [CrossRef]
- Li, P.; Hur, J. Utilization of UV-Vis spectroscopy and related data analyses for dissolved organic matter (DOM) studies: A review. Crit. Rev. Environ. Sci. Technol. 2017, 47, 131–154. [Google Scholar] [CrossRef]
- Zykova, M.V.; Schepetkin, I.A.; Belousov, M.V.; Krivoshchekov, S.V.; Logvinova, L.A.; Bratishko, K.A.; Yusubov, M.S.; Romanenko, S.V.; Quinn, M.T. Physicochemical characterization and antioxidant activity of humic acids isolated from peat of various origins. Molecules 2018, 23, 753. [Google Scholar] [CrossRef]
- Rodríguez, F.J.; Schlenger, P.; García-Valverde, M. Monitoring changes in the structure and properties of humic substances following ozonation using UV-Vis, FTIR and 1H NMR techniques. Sci. Total Environ. 2016, 541, 623–637. [Google Scholar] [CrossRef]
- Chen, J.; Gu, B.; LeBooeuf, E.; Pan, H.; Dai, S. Spectroscopic characterization of the structural and functional properties of natural organic matter fractions. Chemosphere 2002, 48, 59–68. [Google Scholar] [CrossRef]
- Maeng, S.K.; Timmes, T.C.; Kim, H.-C. Characterization of EfOM fraction responsible for short-term fouling in ultrafiltration. Sep. Sci. Technol. 2015, 50, 2697–2707. [Google Scholar] [CrossRef]
- Tinoco, P.; Almendros, G.; González-Vila, F.J.; Sanz, J.; González-Pérez, J.A. Revisiting molecular characteristics responsive for the aromaticity of soil humic acids. J. Soils Sediments 2015, 15, 781–791. [Google Scholar] [CrossRef]
- Yakimenko, O.; Khundzhua, D.; Izosimov, A.; Yuzhakov, V.; Patsaeva, S. Source indicator of commercial humic products: UV-Vis and fluorescence proxies. J. Soils Sediments 2018, 18, 1279–1291. [Google Scholar] [CrossRef]
- Kumada, K.; Sato, O. Characteristics of the green fraction of P type humic acid. J. Soil Sci. Plant Nutr. 1980, 26, 309–316. [Google Scholar] [CrossRef]
- Davis, W.M.; Erickson, C.L.; Johnston, C.T.; Delfino, J.J.; Porter, J.E. Quantitative fourier transform infrared spectroscopic investigation humic substance functional group composition. Chemosphere 1999, 38, 2913–2928. [Google Scholar] [CrossRef]
- Senesi, N.; Sipos, S. Molecular weight distribution, analytical and spectroscopic characterization of humic fractions sequentially isolated by organic solvents from a brown coal humic acid. Org. Geochem. 1985, 8, 157–162. [Google Scholar] [CrossRef]
- Stuart, B.H. Infrared Spectroscopy: Fundamentals and Applications, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004; p. 244. [Google Scholar]
- Baes, A.U.; Bloom, P.R. Diffuse reflectance and transmission fourier transform infrared (DRIFT) spectroscopy of humic and fulvic acids. Soil Sci. Soc. Am. J. 1989, 53, 695–700. [Google Scholar] [CrossRef]
- Terkhi, M.C.; Taleb, F.; Gossart, P.; Semmoud, A.; Addou, A. Fourier transform infrared study of mercury interaction with carboxyl groups in humic acids. J. Photochem. Photobiol. A 2008, 198, 205–214. [Google Scholar] [CrossRef]
- Tatzber, M.; Stemmer, M.; Spiegel, H.; Katzlberger, C.; Haberhauer, G.; Mentler, A.; Gerzabek, M.H. FTIR-spectroscopic characterization of humic acids and humin fractions obtained by advanced NaOH, Na4P2O7, and Na2CO3 extraction procedures. J. Plant Nutr. Soil Sci. 2007, 170, 522–529. [Google Scholar] [CrossRef]
- Hanc, A.; Enev, V.; Hrebeckova, T.; Klucakova, M.; Pekar, M. Characterization of humic acids in a continuous-feeding vermicomposting system with horse manure. Waste Manag. 2019, 99. [Google Scholar] [CrossRef] [PubMed]
- Senesi, N.; Miano, T.M.; Provenzano, M.R.; Brunetti, G. Characterization, differentiation, and classification of humic substances by fluorescence spectroscopy. Soil Sci. 1991, 152, 259–271. [Google Scholar] [CrossRef]
- Enev, V.; Pospíšilová, Ľ.; Klučáková, M.; Liptaj, T.; Doskočil, L. Spectral characterization of selected humic substances. Soil Water Res. 2014, 9, 9–17. [Google Scholar] [CrossRef]
- Stylianou, S.K.; Katsoyiannis, I.A.; Ernst, M.; Zouboulis, A.I. Impact of O3 or O3/H2O2 treatment via a membrane contacting system on the composition and characteristics of the natural organic matter of surface waters. Environ. Sci. Pollut. Res. 2018, 25, 12246–12255. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-F.; Dou, S.; Wang, Z.-G. Structural analysis of humic acid in soil at different corn straw returning modes through fluorescence spectroscopy and infrared spectroscopy. Int. J. Anal. Chem. 2019, 2019. [Google Scholar] [CrossRef]
- Sierra, M.M.D.; Giovanela, M.; Parlanti, E.; Soriano-Sierra, E.J. Fluorescence fingerprint of fulvic and humic acids from varied origins as viewed by single-scan and excitation/emission matrix techniques. Chemosphere 2005, 58, 715–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Benedetti, M.F.; Han, W.; Korshin, G.V. Comparison of the properties of standard soil and aquatic fulvic and humic acids based on the data of differential absorbance and fluorescence spectroscopy. Chemosphere 2020, 261, 128189. [Google Scholar] [CrossRef]
- Alberts, J.J.; Takács, M. Total luminescence spectra of IHSS standard and reference fulvic acids, humic acids and natural organic matter: Comparison of aquatic and terrestrial source terms. Org. Geochem. 2004, 35, 243–256. [Google Scholar] [CrossRef]
- Rodríguez, F.J.; Schlenger, P.; García-Valverde, M. A comprehensive structural evaluation of humic substances using several fluorescence techniques before and after ozonation. Part I: Structural characterization of humic substances. Sci. Total Environ. 2014, 476–477, 718–730. [Google Scholar] [CrossRef]
- Doskočil, L.; Burdíková-Szewieczková, J.; Enev, V.; Kalina, L.; Wasserbauer, J. Spectral characterization and comparison of humic acids isolated from some European lignites. Fuel 2018, 213, 123–132. [Google Scholar] [CrossRef]
- Fellman, J.B.; Hood, E.; Spencer, R.G.M. Fluorescence spectroscopy opens new windows into dissolved organic matter dynamics in freshwater ecosystems: A review. Limnol. Oceanogr. 2010, 55, 2452–2462. [Google Scholar] [CrossRef]
- Yang, L.; Hur, J.; Zhuang, W. Occurrence and behaviors of fluorescence EEM-PARAFAC components in drinking water and wastewater treatment systems and their applications: A review. Environ. Sci. Pollut. Res. 2015, 22, 6500–6510. [Google Scholar] [CrossRef]
- Yu, M.-D.; Xi, B.-D.; Zhu, Z.-Q.; Zhang, L.; Yang, C.; Geng, C.-M.; He, X.-S. Fate and removal of aromatic organic matter upon a combined leachate treatment process. Chem. Eng. J. 2020, 401, 126157. [Google Scholar] [CrossRef]
- Zhou, Y.; Martin, P.; Müller, M. Composition and cycling of dissolved organic matter from tropical peatlands of coastal Sarawak, Borneo, revealed by fluorescence spectroscopy and parallel factor analysis. Biogeosciences 2019, 16, 2733–2749. [Google Scholar] [CrossRef]
- Chen, W.; Westerhoff, P.; Leenheer, J.A.; Booksh, K. Fluorescence excitation-emission matrix regional integration to quantify spectra for dissolved organic matter. Environ. Sci. Technol. 2003, 37, 5701–5710. [Google Scholar] [CrossRef]
- Sun, J.; Guo, L.; Li, Q.; Zhao, Y.; Gao, M.; She, Z.; Jin, C. Three-dimensional fluorescence excitation-emission matrix (EEM) spectroscopy with regional integration analysis for assessing waste sludge hydrolysis at different pretreated temperatures. Environ. Sci. Pollut. Res. 2016, 23, 24061–24067. [Google Scholar] [CrossRef] [PubMed]
- Birdwell, J.E.; Engel, A.S. Characterization of dissolved organic matter in cave and spring waters using UV-Vis absorbance and fluorescence spectroscopy. Org. Geochem. 2010, 41, 270–280. [Google Scholar] [CrossRef]
- Cao, J.; Jiang, J. Reducing capacities in continuously released low molecular weight fractions from bulk humic acids. J. Environ. Manag. 2019, 244, 172–179. [Google Scholar] [CrossRef]
- Coble, P.G.; Lead, J.; Baker, A.; Reynolds, D.M.; Spencer, R.G.M. Aquatic Organic Matter Fluorescence, 1st ed.; Cambridge University Press: New York, NY, USA, 2014; p. 418. [Google Scholar]
- McKnight, D.M.; Boyer, E.W.; Westerhoff, P.K.; Doran, P.T.; Kulbe, T.; Andersen, D.T. Spectrofluorometric characterization of dissolved organic matter for indication of precursor organic material and aromaticity. Limnol. Oceanogr. 2001, 46, 38–48. [Google Scholar] [CrossRef]
- Boyle, E.S.; Guerriero, N.; Thiallet, A.; del Vecchio, R.; Blough, N.V. Optical properties of humic substances and CDOM: Relation to structure. Environ. Sci. Technol. 2009, 43, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Boguta, P.; D’Orazio, V.; Sokołowska, Z.; Senesi, N. Effects of selected chemical and physicochemical properties of humic acids from peat soils on their interaction mechanisms with copper ions at various pHs. J. Geochem. Explor. 2016, 168, 119–126. [Google Scholar] [CrossRef]
- Conte, P.; Piccolo, A.; van Lagen, B.; Buurman, P.; de Jager, P.A. Quantitative differences in evaluating soil humic substances by liquid- and solid-state 13C-NMR spectroscopy. Geoderma 1997, 80, 339–352. [Google Scholar] [CrossRef]
- Khatami, S.; Deng, Y.; Tien, M.; Hatcher, P.G. Formation of water-soluble organic matter through fungal degradation of lignin. Org. Geochem. 2019, 135, 64–70. [Google Scholar] [CrossRef]
- Hatcher, P.G.; Waggoner, D.; Chen, H. Evidence for the existence of humic acids in peat soils based on solid-state 13C NMR. J. Environ. Qual. 2019, 48, 1571–1577. [Google Scholar] [CrossRef]
- Schnitzer, M. Soil organic matter—The next 75 years. Soil Sci. 1991, 151, 41–58. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Yield | Ash | C | H | N | O | H/C | O/C | N/C | |
|---|---|---|---|---|---|---|---|---|---|
| (wt.%) 1 | (wt.%) 1 | (at.%) 2 | |||||||
| SBPHA | - | 0.7 | 35.00 ± 0.24 | 49.84 ± 0.16 | 1.12 ± 0.09 | 14.03 | 1.42 | 0.40 | 0.032 |
| TCM1 | 3.76 | 0.07 ± 0.02 | 30.83 ± 0.11 | 67.10 ± 0.32 | 0.15 ± 0.02 | 1.92 | 2.18 | 0.06 | 0.005 |
| EAC2 | 1.10 | 1.81 ± 0.09 | 33.30 ± 0.16 | 65.65 ± 0.28 | 0.63 ± 0.05 | 0.42 | 1.97 | 0.01 | 0.019 |
| ACE3 | 13.00 | 0.34 ± 0.04 | 43.78 ± 0.20 | 34.62 ± 0.13 | 0.82 ± 0.05 | 20.77 | 0.79 | 0.47 | 0.019 |
| ACN4 | 7.83 | 4.10 ± 0.08 | 30.52 ± 0.34 | 49.87 ± 0.19 | 3.30 ± 0.10 | 16.31 | 1.63 | 0.53 | 0.108 |
| PRO5 | 19.74 | 0.83 ± 0.17 | 37.22 ± 0.21 | 44.23 ± 0.36 | 0.94 ± 0.07 | 17.61 | 1.19 | 0.47 | 0.025 |
| MET6 | 11.91 | 2.99 ± 0.12 | 34.62 ± 0.37 | 46.18 ± 0.22 | 1.43 ± 0.12 | 17.77 | 1.33 | 0.51 | 0.041 |
| UV/Vis Ratios | UV Parameter | FTIR Intensity Ratios | |||
|---|---|---|---|---|---|
| EET/EBz | E2/E4 | SUVA254 1 | IAr/ICOOH2 | ICH2/ICH33 | |
| SBPHA | 0.74 ± 0.06 | 5.53 ± 0.12 | 5.68 | 1.26 | 1.06 |
| TCM1 | 0.51 ± 0.02 | 11.87 ± 0.15 | 0.24 | 0.22 | 1.81 |
| EAC2 | 0.70 ± 0.02 | 12.00 ± 0.06 | 0.66 | 0.34 | 1.87 |
| ACE3 | 0.74 ± 0.01 | 9.77 ± 0.11 | 5.89 | 0.80 | 1.11 |
| ACN4 | 0.66 ± 0.00 | 9.53 ± 0.10 | 2.03 | 1.44 | 1.11 |
| PRO5 | 0.74 ± 0.04 | 7.64 ± 0.14 | 6.40 | 1.06 | 0.98 |
| MET6 | 0.83 ± 0.02 | 7.18 ± 0.22 | 5.79 | 1.21 | 1.04 |
| Fluorescence Peak Region | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | C | V | B | H | ||||||
| Ex/Em (nm) | IF (CPS) 1 | Ex/Em (nm) | IF (CPS) 1 | Ex/Em (nm) | IF (CPS) 1 | Ex/Em (nm) | IF (CPS) 1 | Ex/Em (nm) | IF (CPS) 1 | |
| SBPHA | 255/495 | 1.77 | ||||||||
| TCM1 | 270/305 | 6.86 | 255/355 | 3.23 | ||||||
| EAC2 2 | 250/415 | 4.36 | 270/305 | 0.99 | ||||||
| ACE3 | 255/465 | 4.22 | ||||||||
| ACN4 | 255/445 | 4.49 | 300/435 | 2.74 | 270/315 | 0.98 | ||||
| PRO5 | 265/500 | 2.76 | 445/530 | 0.80 | ||||||
| MET6 | 260/505 | 2.57 | 435/525 | 0.72 | 250/300 | 0.99 | ||||
| Organic Fractions | Average Distribution of Individual Carbon Types (%) | (fa) 1 | |||||
|---|---|---|---|---|---|---|---|
| 0–45 ppm | 45–106 ppm | 106–145 ppm | 145–165 ppm | 165–190 ppm | 190–220 ppm | ||
| TCM1 | 78 | 8 | 8 | 2 | 3 | 1 | 0.10 |
| ACE3 | 23 | 19 | 36 | 11 | 9 | 2 | 0.53 |
| PRO5 | 24 | 22 | 33 | 9 | 10 | 2 | 0.48 |
| MET6 | 16 | 30 | 31 | 8 | 13 | 2 | 0.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enev, V.; Sedláček, P.; Kubíková, L.; Sovová, Š.; Doskočil, L.; Klučáková, M.; Pekař, M. Polarity-Based Sequential Extraction as a Simple Tool to Reveal the Structural Complexity of Humic Acids. Agronomy 2021, 11, 587. https://doi.org/10.3390/agronomy11030587
Enev V, Sedláček P, Kubíková L, Sovová Š, Doskočil L, Klučáková M, Pekař M. Polarity-Based Sequential Extraction as a Simple Tool to Reveal the Structural Complexity of Humic Acids. Agronomy. 2021; 11(3):587. https://doi.org/10.3390/agronomy11030587
Chicago/Turabian StyleEnev, Vojtěch, Petr Sedláček, Leona Kubíková, Šárka Sovová, Leoš Doskočil, Martina Klučáková, and Miloslav Pekař. 2021. "Polarity-Based Sequential Extraction as a Simple Tool to Reveal the Structural Complexity of Humic Acids" Agronomy 11, no. 3: 587. https://doi.org/10.3390/agronomy11030587
APA StyleEnev, V., Sedláček, P., Kubíková, L., Sovová, Š., Doskočil, L., Klučáková, M., & Pekař, M. (2021). Polarity-Based Sequential Extraction as a Simple Tool to Reveal the Structural Complexity of Humic Acids. Agronomy, 11(3), 587. https://doi.org/10.3390/agronomy11030587