The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis



Abstract
1. Introduction
2. Results
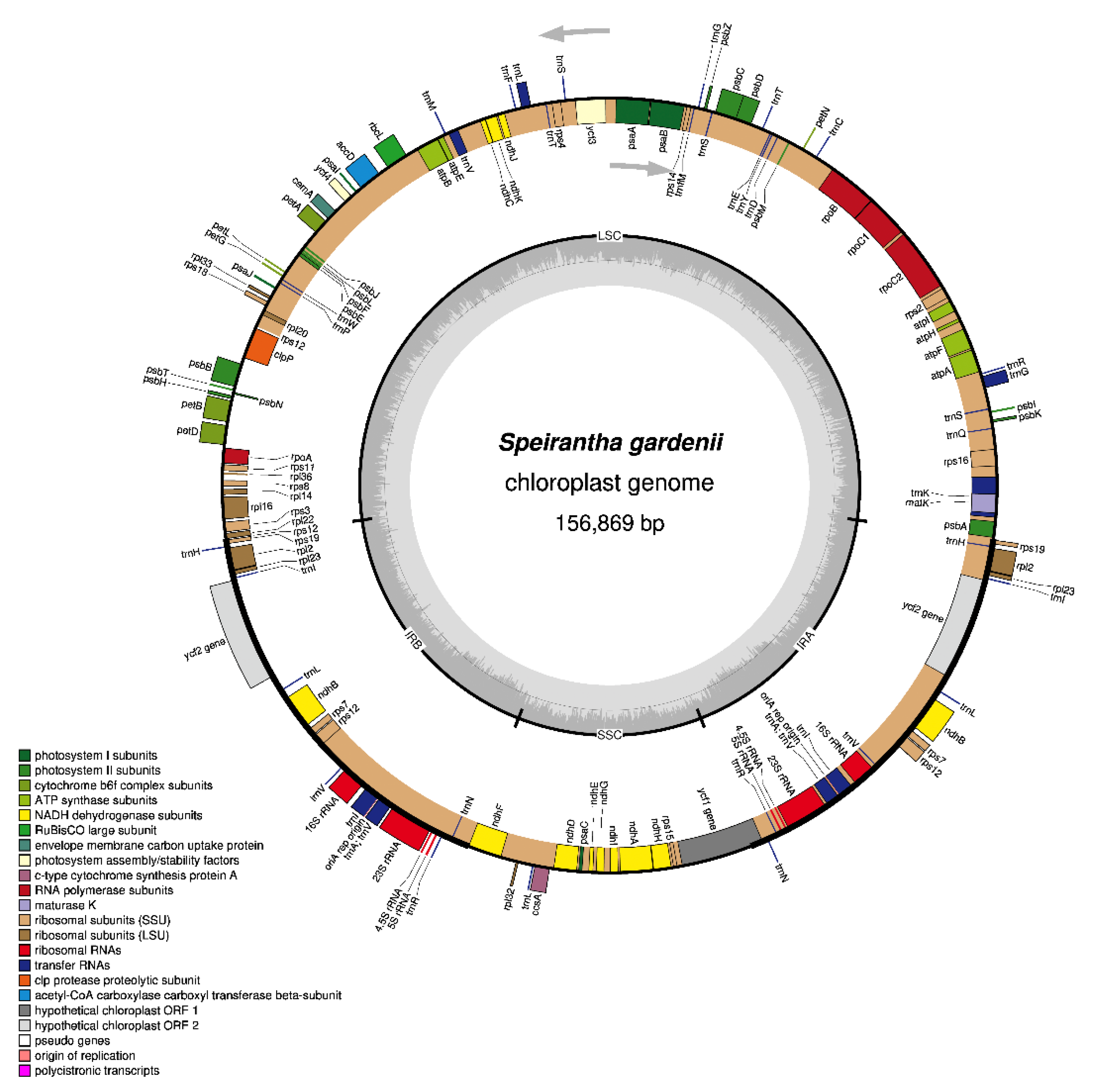
2.1. General Features of the Speirantha Gardenii Chloroplast Genome
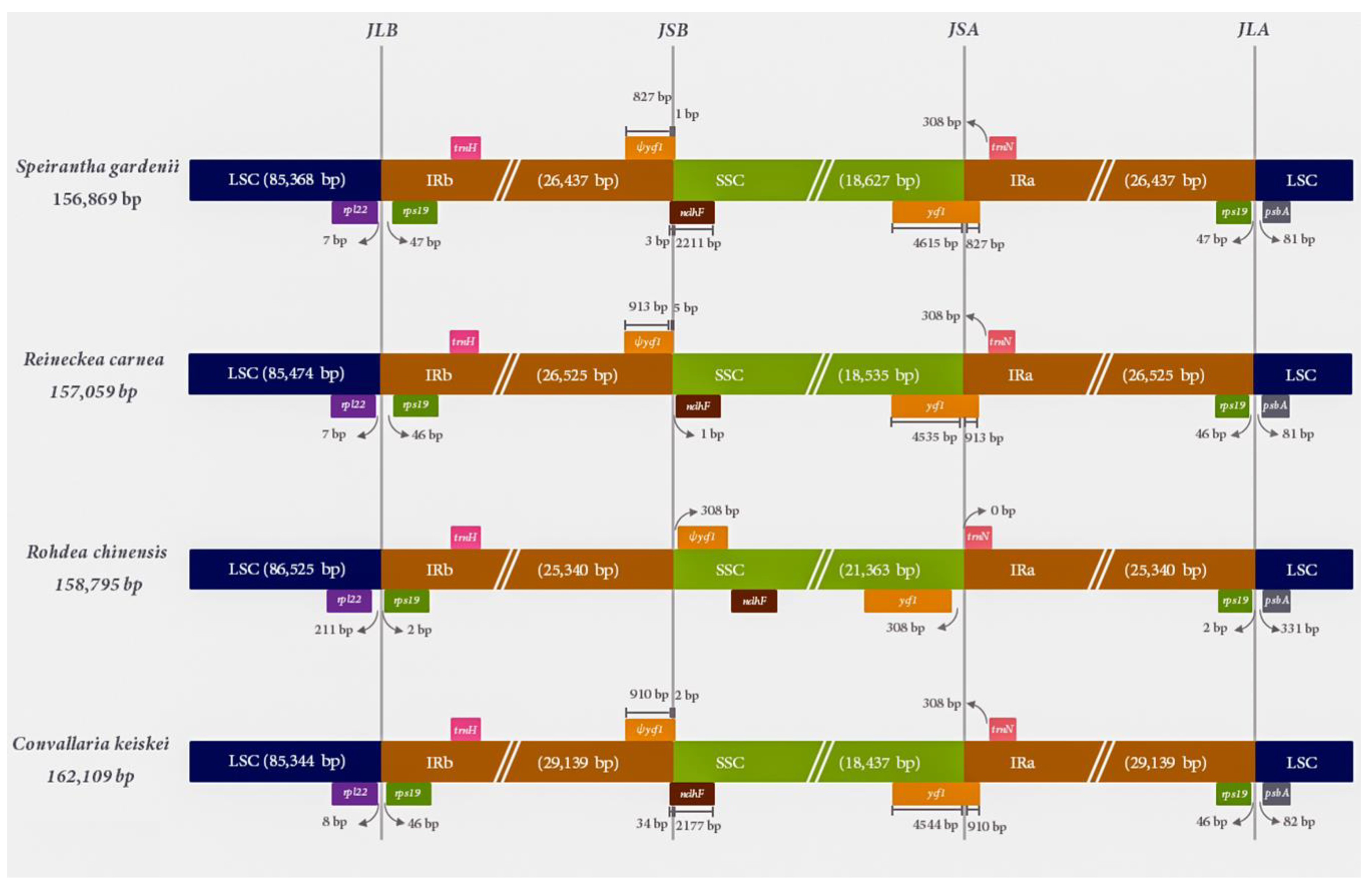
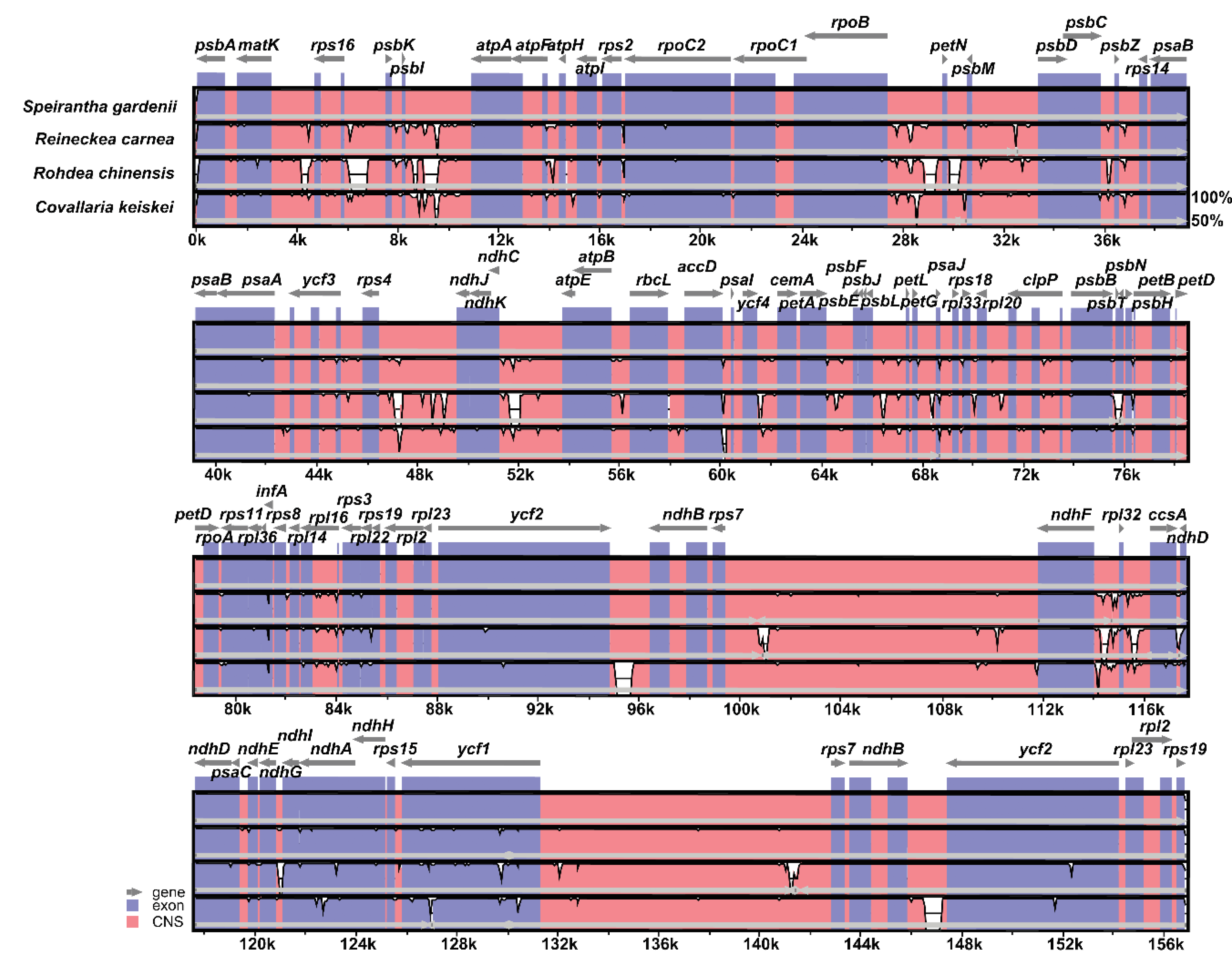
2.2. Comparative Analysis of the IR Contraction and Expansion in the Species of Nolinoideae
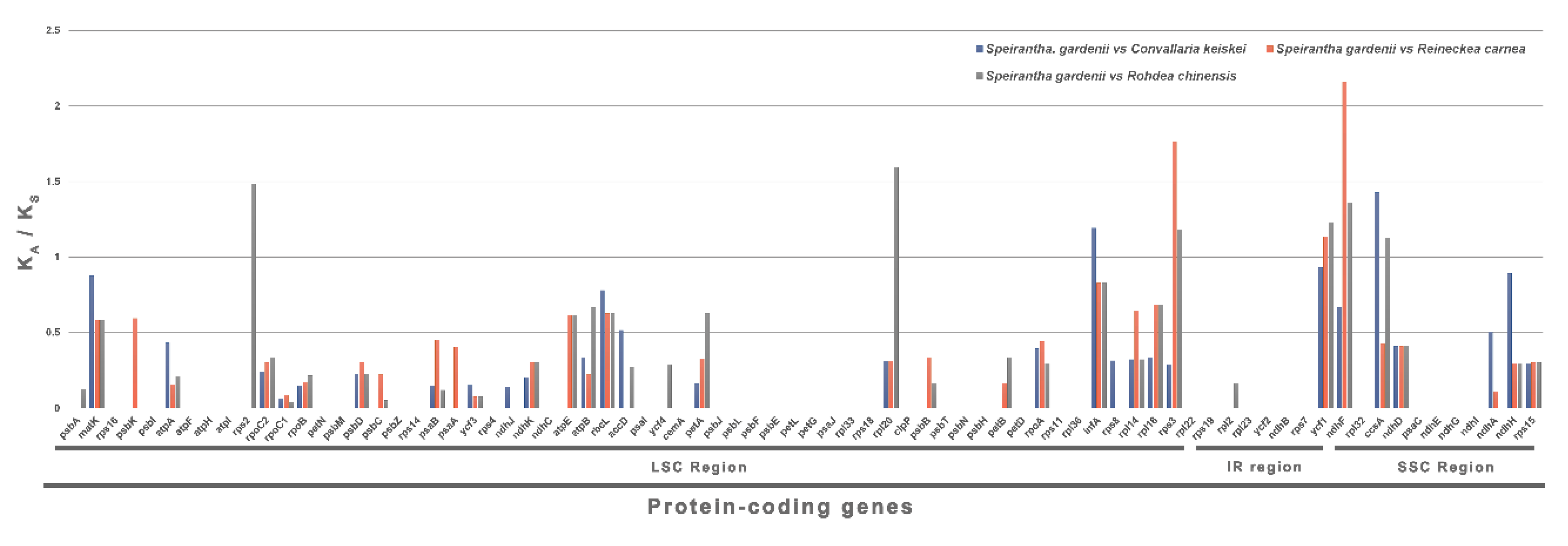
2.3. Synonymous (KS) and Nonsynonymous (KA) Substitution Rate Analysis
2.4. Selective Pressure Events
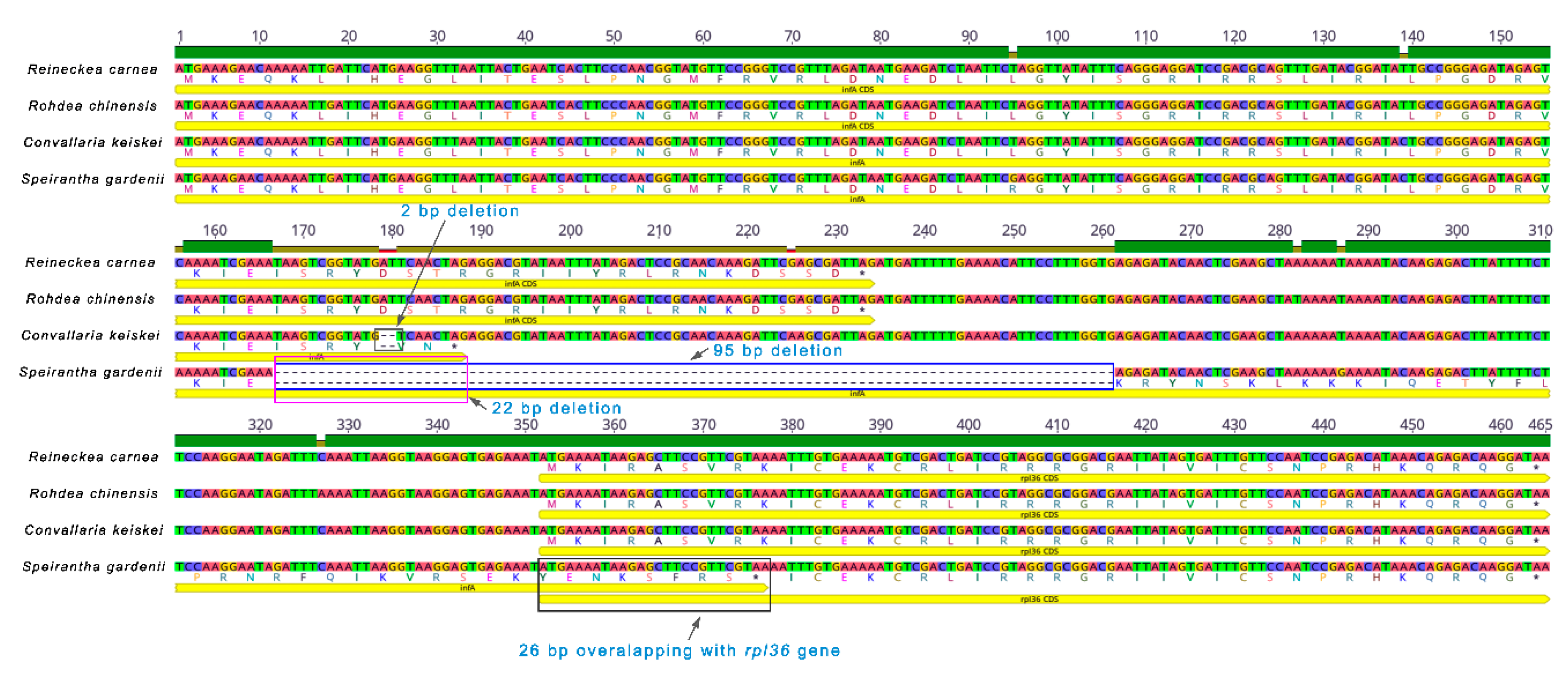
2.5. Analysis of infA Gene
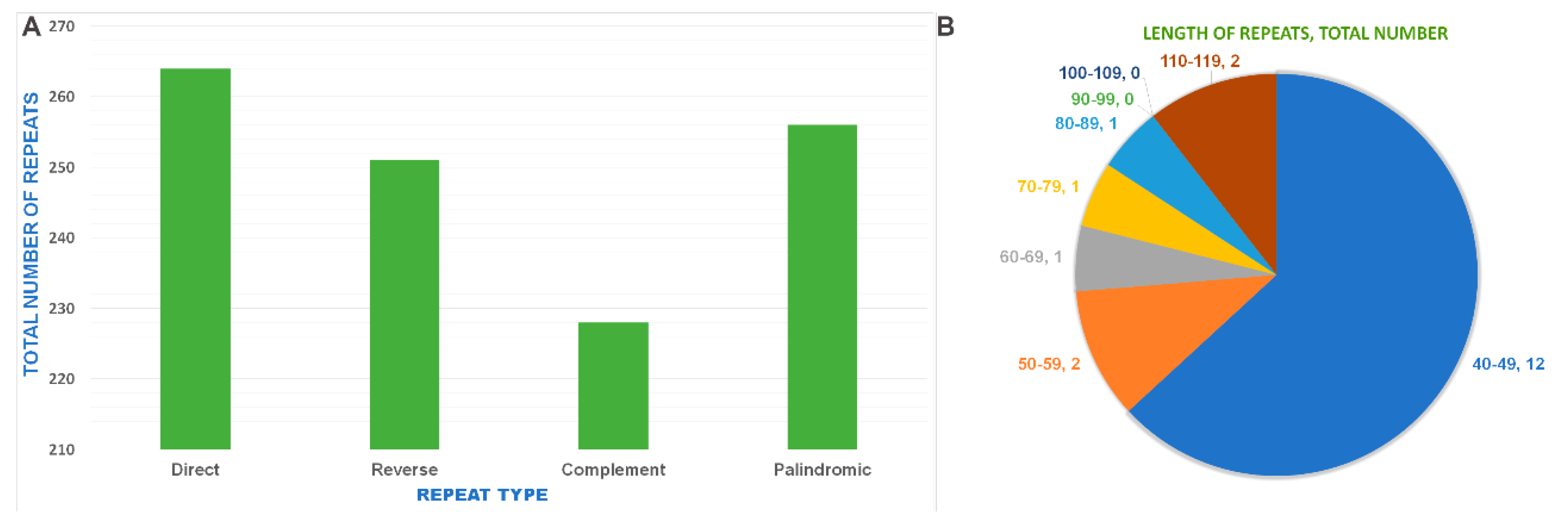
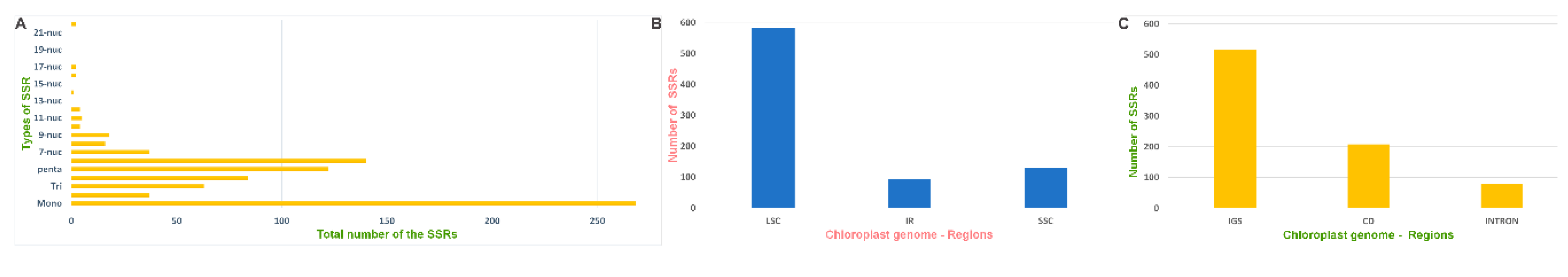
2.6. Repeat Sequence and Simple Sequence Repeat Analysis
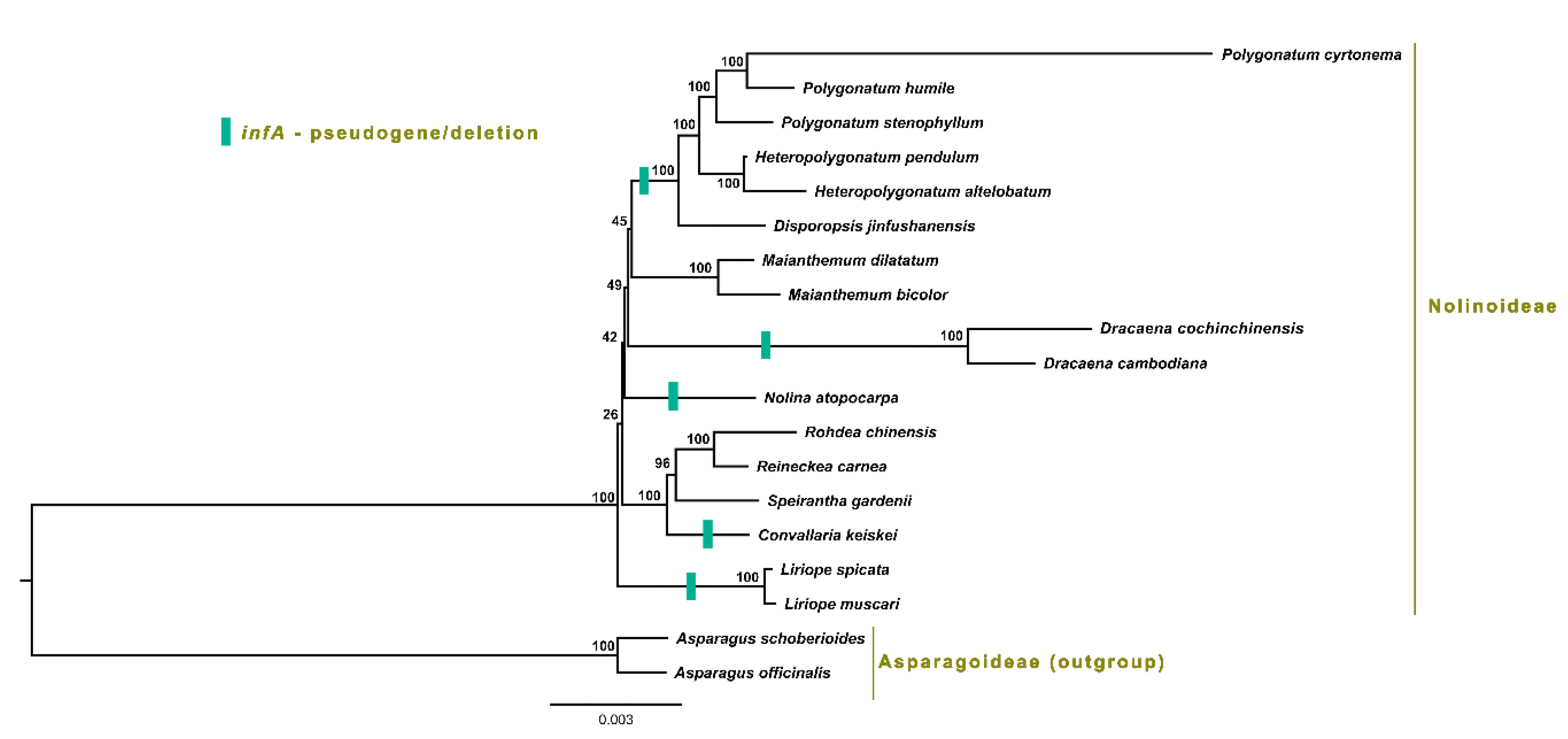
2.7. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. DNA Extraction and Sequencing
4.2. The Chloroplast Genome Annotation of the S. gardenii
4.3. Comparative Chloroplast Genome Analysis of the S. gardenii
4.4. Characterization of Substitution Rates
4.5. Positive Selection Analysis
4.6. Analysis of Repeat Sequences and Single Sequence Repeats (SSR)
4.7. Phylogenetic Tree Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LSC | Large single-copy |
| SSC | Small single-copy |
| IRs | Inverted repeats |
| CNVs | Copy number variations |
| DOGMA | Dual Organeller GenoMe Annotator |
| tRNA | Transfer RNA |
| rRNA | Ribosomal RNA |
| KS | Synonymous substitution |
| KA | Non-synonymous substitution |
| ω | Nonsynonymous vs. synonymous ratio |
| SSR | Simple sequence repeats |
| LRT | Likelihood ratio test |
References
- Ana, M.C.; Silvana, M.S.; Konstantin, R.; Paul, M.P.; Robert, J.S.; Fernando, O.Z.; Morrone, O. Phylogeny of Nassella (Stipeae, Pooideae, Poaceae) Based on Analyses of Chloroplast and Nuclear Ribosomal DNA and Morphology. Syst. Bot. 2014, 39, 814–828. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, W.; Xie, X.; Lu, Y.; Liu, Y.; Jin, X.; Suo, Z. Phylogenetic Resolution in Juglans Based on Complete Chloroplast Genomes and Nuclear DNA Sequences. Front. Plant Sci. 2017, 8, 1148. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, S.; Li, F.; Zhang, S.; Zhang, H.; Wang, X.; Sun, R.; Bonnema, G.; Borm, T.J. A Phylogenetic Analysis of Chloroplast Genomes Elucidates the Relationships of the Six Economically Important Brassica Species Comprising the Triangle of U. Front. Plant Sci. 2017, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Raman, G.; Park, S. Analysis of the Complete Chloroplast Genome of a Medicinal Plant, Dianthus superbus var. longicalyncinus, from a Comparative Genomics Perspective. PLoS ONE 2015, 10, e0141329. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Palmer, J.D. Comparative Organization of Chloroplast Genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef]
- Friis, E.M.; Pedersen, K.R.; Crane, P.R. Cretaceous angiosperm flowers: Innovation and evolution in plant reproduction. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2006, 232, 251–293. [Google Scholar] [CrossRef]
- Gurusamy, R.; Lee, E.M.; Nam, G.H.; Lee, B.; Park, S. The complete chloroplast genome of monocot plant, Maianthemum dilatatum. Mitochondrial DNA Part B 2018, 3, 1185–1186. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Y.; Shi, X.; Mao, S. The complete chloroplast genome sequence of Campylandra chinensis (Liliaceae). Mitochondrial DNA Part B 2018, 3, 780–781. [Google Scholar] [CrossRef]
- Lee, S.Y.; Zou, Y.; Liao, W.; Fan, Q. The complete chloroplast genome of a traditional medicinal and food plant, Polygonatum humile (Asparagaceae, Asparagales). Mitochondrial DNA Part B 2019, 4, 3184–3185. [Google Scholar] [CrossRef]
- Raman, G.; Park, S.; Lee, E.M.; Park, S. Evidence of mitochondrial DNA in the chloroplast genome of Convallaria keiskei and its subsequent evolution in the Asparagales. Sci. Rep. 2019, 9, 5028. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.-S.; Cao, P.-H.; Li, Y.-L.; Huang, S. Complete chloroplast genome of Reineckia carnea and its implications for the phylogenetic position within Nolinoideae (Asparagaceae). Mitochondrial DNA Part B 2019, 4, 2129–2130. [Google Scholar] [CrossRef]
- Du, Z.; Qian, J.; Jiang, Y.; Duan, B. The complete chloroplast genome of Polygonatum ordoratum (Mill.) Druce and its phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 1601–1602. [Google Scholar] [CrossRef]
- Jin, J.; Lao, J.; Zhong, C.; He, W.; Xie, J.; Hu, G.; Liu, H.; Yan, F.; Zhang, S. Complete chloroplast genome of a medicinal species Polygonatum kingianum in China (Asparagaceae, Asparagales). Mitochondrial DNA Part B 2020, 5, 959–960. [Google Scholar] [CrossRef]
- Wu, Z.; Raven, P. Flora of China (Flagellariaceae through Marantaceae); Missouri Botanical Garden Press: St. Louis, MO, USA, 2000. [Google Scholar]
- Curci, P.L.; De Paola, D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef]
- Raman, G.; Park, V.; Kwak, M.; Lee, B.; Park, S. Characterization of the complete chloroplast genome of Arabis stellari and comparisons with related species. PLoS ONE 2017, 12, e0183197. [Google Scholar] [CrossRef]
- Yang, J.B.; Yang, S.X.; Li, H.T.; Yang, J.; Li, D.Z. Comparative chloroplast genomes of Camellia species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef]
- Shi, H.; Yang, M.; Mo, C.; Xie, W.; Liu, C.; Wu, B.; Ma, X. Complete chloroplast genomes of two Siraitia Merrill species: Comparative analysis, positive selection and novel molecular marker development. PLoS ONE 2019, 14, e0226865. [Google Scholar] [CrossRef]
- Tomoko, O. Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J. Mol. Evol. 1995, 40, 56–63. [Google Scholar] [CrossRef]
- Hiratsuka, J.; Shimada, H.; Whittier, R.; Ishibashi, T.; Sakamoto, M.; Mori, M.; Kondo, C.; Honji, Y.; Sun, C.R.; Meng, B.Y. The complete sequence of the rice (Oryza sativa) chloroplast genome: Intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 1989, 217, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Maier, R.M.; Neckermann, K.; Igloi, G.L.; Kossel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995, 251, 614–628. [Google Scholar] [CrossRef] [PubMed]
- Saski, C.; Lee, S.B.; Fjellheim, S.; Guda, C.; Jansen, R.K.; Luo, H.; Tomkins, J.; Rognli, O.A.; Daniell, H.; Clarke, J.L. Complete chloroplast genome sequences of Hordeum vulgare, Sorghum bicolor and Agrostis stolonifera, and comparative analyses with other grass genomes. Theor. Appl. Genet. 2007, 115, 591. [Google Scholar] [CrossRef][Green Version]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.C.; Chen, S.L.; Xiao, P.G. Molecular evolution and positive Darwinian selection of the chloroplast maturase matK. J. Plant Res. 2010, 123, 241–247. [Google Scholar] [CrossRef]
- Jiang, P.; Shi, F.X.; Li, M.R.; Liu, B.; Wen, J.; Xiao, H.X.; Li, L.F. Positive Selection Driving Cytoplasmic Genome Evolution of the Medicinally Important Ginseng Plant Genus Panax. Front. Plant Sci. 2018, 9, 359. [Google Scholar] [CrossRef]
- Zhang, Z.; An, M.; Miao, J.; Gu, Z.; Liu, C.; Zhong, B. The Antarctic sea ice alga Chlamydomonas sp. ICE-L provides insights into adaptive patterns of chloroplast evolution. BMC Plant Biol. 2018, 18, 53. [Google Scholar] [CrossRef]
- Heyduk, K.; Moreno-Villena, J.J.; Gilman, I.S.; Christin, P.A.; Edwards, E.J. The genetics of convergent evolution: Insights from plant photosynthesis. Nat. Rev. Genet. 2019, 20, 485–493. [Google Scholar] [CrossRef]
- Li, C.J.; Wang, R.N.; Li, D.Z. Comparative analysis of plastid genomes within the Campanulaceae and phylogenetic implications. PLoS ONE 2020, 15, e0233167. [Google Scholar] [CrossRef]
- Mrazek, J. Analysis of distribution indicates diverse functions of simple sequence repeats in Mycoplasma genomes. Mol. Biol. Evol. 2006, 23, 1370–1385. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pauwels, M.; Vekemans, X.; Gode, C.; Frerot, H.; Castric, V.; Saumitou-Laprade, P. Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytol. 2012, 193, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Cao, J.L.; Jiang, D.; Zhao, Z.Y.; Yuan, S.B.; Zhang, Y.J.; Zhang, T.; Zhong, W.H.; Yuan, Q.J.; Huang, L.Q. Development of Chloroplast Genomic Resources in Chinese Yam (Dioscorea polystachya). Biomed. Res. Int. 2018, 2018, 11. [Google Scholar] [CrossRef]
- Curci, P.L.; De Paola, D.; Sonnante, G. Development of chloroplast genomic resources for Cynara. Mol. Ecol. Resour. 2016, 16, 562–573. [Google Scholar] [CrossRef]
- Dong, W.P.; Liu, H.; Xu, C.; Zuo, Y.J.; Chen, Z.J.; Zhou, S.L. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 1–8. [Google Scholar] [CrossRef]
- Li, B.; Lin, F.R.; Huang, P.; Guo, W.Y.; Zheng, Y.Q. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biol. Res. 2020, 53, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tangphatsornruang, S.; Uthaipaisanwong, P.; Sangsrakru, D.; Chanprasert, J.; Yoocha, T.; Jomchai, N.; Tragoonrung, S. Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 2011, 475, 104–112. [Google Scholar] [CrossRef]
- Wang, L.; Wuyun, T.-N.; Du, H.; Wang, D.; Cao, D. Complete chloroplast genome sequences of Eucommia ulmoides: Genome structure and evolution. Tree Genet. Genomes 2016, 12, 12. [Google Scholar] [CrossRef]
- Carbonell-Caballero, J.; Alonso, R.; Ibañez, V.; Terol, J.; Talon, M.; Dopazo, J. A Phylogenetic Analysis of 34 Chloroplast Genomes Elucidates the Relationships between Wild and Domestic Species within the Genus Citrus. Mol. Biol. Evol. 2015, 32, 2015–2035. [Google Scholar] [CrossRef]
- Henriquez, C.L.; Arias, T.; Pires, J.C.; Croat, T.B.; Schaal, B.A. Phylogenomics of the plant family Araceae. Mol. Phylogenet. Evol. 2014, 75, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Shen, X.F.; Liao, B.S.; Xu, J.; Hou, D.Y. Comparing and phylogenetic analysis chloroplast genome of three Achyranthes species. Sci. Rep. 2020, 10, 1–13. [Google Scholar]
- Satish, K.V.; Pasha, S.V.; Krishna, P.H.; Reddy, C.S. Achyranthes coynei Santapau (Amaranthaceae): An Endemic and Threatened Species from Kachchh Desert, India. Natl. Acad. Sci. Lett. 2015, 38, 281–282. [Google Scholar]
- Schwartz, L.M.; Gibson, D.J.; Young, B.G. Life history of Achyranthes japonica (Amaranthaceae): An invasive species in southern Illinois. J. Torrey Bot. Soc. 2016, 143, 93–102. [Google Scholar]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. USA 2007, 104, 19363–19368. [Google Scholar]
- Floden, A.; Schilling, E.E. Using phylogenomics to reconstruct phylogenetic relationships within tribe Polygonateae (Asparagaceae), with a special focus on Polygonatum. Mol. Phylogenet. Evol. 2018, 129, 202–213. [Google Scholar]
- Yamashita, J.; Tamura, M.N. Phylogenetic analyses and chromosome evolution in Convallarieae (Ruscaceae sensu lato), with some taxonomic treatments. J. Plant Res. 2004, 117, 363–370. [Google Scholar] [CrossRef]
- Meng, Y.; Nie, Z.L.; Deng, T.; Wen, J.; Yang, Y.P. Phylogenetics and evolution of phyllotaxy in the Solomon’s seal genus Polygonatum (Asparagaceae: Polygonateae). Bot. J. Linn. Soc. 2014, 176, 435–451. [Google Scholar]
- Chen, S.C.; Kim, D.K.; Chase, M.W.; Kim, J.H. Networks in a Large-Scale Phylogenetic Analysis: Reconstructing Evolutionary History of Asparagales (Lilianae) Based on Four Plastid Genes. PLoS ONE 2013, 8, e59472. [Google Scholar]
- Andrews, S. FASTQC: A Quality Control Tool for High Throughput Sequence Data; Elsevier: Amsterdam, The Netherland, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Mayer, C.; Leese, F.; Tollrian, R. Genome-wide analysis of tandem repeats in Daphnia pulex—A comparative approach. BMC Genom. 2010, 11, 277. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Region | S. gardenii | |
|---|---|---|
| Total chloroplast genome size (bp) | 156,869 | |
| LSC length (bp) | 85,368 | |
| SSC length (bp) | 18,627 | |
| IR length (bp) | 26,437 | |
| Total number of genes | 136 | |
| Protein-coding genes | 90 | |
| tRNA genes | 38 | |
| rRNA genes | 8 | |
| Genes duplicated by IR | 21 | |
| Genes with introns | 17 | |
| GC content | Total (%) | 37.6 |
| LSC (%) | 35.6 | |
| SSC (%) | 31.5 | |
| IR (%) | 43.0 | |
| CDS (%) | 38.1 | |
| rRNA (%) | 55.3 | |
| tRNA (%) | 53.2 | |
| All genes (%) | 39.8 | |
| Protein-coding genes (%bp) | 50.13 | |
| All genes (%bp) | 71.42 | |
| Non-coding regions (%) | 28.58 |
| Protein-Coding Genes | Comparison between Models | 2ΔLnL | d.f. | p-Value | Positive Sites (M7 vs. M8) |
|---|---|---|---|---|---|
| ccsA | M0 vs. M3 | 0 | 4 | 1 | 81 N 0.642, 125 S 0.642, 223 S 0.642, 280 S 0.642, 292 I 0.636 |
| M1 vs. M2A | 0 | 2 | 1 | ||
| M7 vs. M8 | 0.000002 | 2 | 0.999999 | ||
| M8a vs. M8 | 0.001524 | 1 | 0.968859739 | ||
| infA | M0 vs. M3 | 15.845366 | 4 | 0.003233836 | 32 L 0.759, 56 I 0.881, 57 G 0.966 *, 58 M 0.965 *, 59 Q 0.737, 60 L 0.961 * |
| M1 vs. M2A | 11.350176 | 2 | 0.003430367 | ||
| M7 vs. M8 | 11.352554 | 2 | 0.003426291 | ||
| M8a vs. M8 | 11.350166 | 1 | 0.000754412 | ||
| ndhF | M0 vs. M3 | 12.435874 | 4 | 0.014387898 | 67 F 0.543, 281 F 0.568, 472 Y 0.581, 486 E 0.606, 505 N 0.571, 560 H 0.583, 574 L 0.617, 596 L 0.665, 614 S 0.616, 636 G 0.632, 675 L 0.580, 680 Q 0.628, 728 F 0.568, 732 L 0.963 *, 734 F 0.931 |
| M1 vs. M2A | 9.771726 | 2 | 0.007552603 | ||
| M7 vs. M8 | 8.535662 | 2 | 0.014012143 | ||
| M8a vs. M8 | 8.529956 | 1 | 0.003493481 | ||
| rpl20 | M0 vs. M3 | 0 | 4 | 1 | 43 L 0.620, 80 R 0.612, 116 M 0.612, 117 K 0.603 |
| M1 vs. M2A | 0 | 2 | 1 | ||
| M7 vs. M8 | 0.00013 | 2 | 0.999935002 | ||
| M8a vs. M8 | 0.00004 | 1 | 0.994953769 | ||
| rps2 | M0 vs. M3 | 24.069072 | 4 | 0.000077368 | 48 T 0.838, 59 D 0.839, 91 A 0.838, 160 E 0.840, 199 L 0.850, 237—0.998 ** |
| M1 vs. M2A | 23.735088 | 2 | 0.000007014 | ||
| M7 vs. M8 | 15.316822 | 2 | 0.000472057 | ||
| M8a vs. M8 | 13.715290 | 1 | 0.000212716 | ||
| rps3 | M0 vs. M3 | 17.514528 | 4 | 0.001534958 | 28 N 0.937, 30 S 0.599, 68 Q 0.584, 105 F 0.608, 106 H 0.580, 221—0.988 * |
| M1 vs. M2A | 12.631522 | 2 | 0.00180759 | ||
| M7 vs. M8 | 14.593672 | 2 | 0.00067768 | ||
| M8a vs. M8 | 13.505938 | 1 | 0.00023781 | ||
| ycf1 | M0 vs. M3 | 26.084252 | 4 | 0.000030431 | 267 Y 0.939, 271 Y 0.517, 290 D 0.520, 296 Y 0.995 **, 407 L 0.523, 438 R 0.536, 447 S 0.503, 491 T 0.545, 507 Q 0.503, 558 K 0.920, 601 I 0.539, 804 L 0.523, 864 I 0.503, 921 F 0.535, 953 R 0.936, 1068 S 0.558, 1082 Q 0.927, 1096 S 0.516, 1108 R 0.935, 1162 R 0.510, 1255 L 0.559, 1340 L 0.926, 1370 Q 0.524, 1373 Q 0.503, 1375 F 0.515, 1484 Q 0.509, 1499 I 0.523, 1526 F 0.541, 1543 L 0.505, 1639 Q 0.530, 1671 H 0.925, 1672 F 0.515 |
| M1 vs. M2A | 19.806836 | 2 | 0.000050003 | ||
| M7 vs. M8 | 19.228316 | 2 | 0.000066777 | ||
| M8a vs. M8 | 18.528045 | 1 | 0.000016742 |
| Protein-Coding Genes | Comparison between Models | 2ΔLnL | d.f. | p-Value | Positive Sites (M7 vs. M8) |
|---|---|---|---|---|---|
| ccsA | M0 vs. M3 | 25.070924 | 4 | 0.000048685 | 170 D 0.989 *, 175 R 0.969 *, 178 F 0.764, 184 F 0.978 *, 186 D 0.871, 206 R 0.868, 278 S 0.582 |
| M1 vs. M2A | 13.324234 | 2 | 0.001278437 | ||
| M7 vs. M8 | 13.687792 | 2 | 0.001065942 | ||
| M8a vs. M8 | 13.319022 | 1 | 0.000262727 | ||
| ndhF | M0 vs. M3 | 74.589864 | 4 | 0 | 77 V 0.814, 463 K 0.804, 486 A 0.610, 513 G 0.702, 514 R 0.638, 523 H 0.828, 531 T 0.942, 546 V 0.979 *, 560 N 0.998 **, 584 P 0.726, 586 F 0.947, 588 G 0.550, 590 P 0.961 *, 596 L 0.698, 636 G 0.975 *, 638 P 0.850, 675 L 0.637, 680 Q 0.871, 728 F 0.830, 729 F 0.720, 732 L 0.973 *, 733 F 0.627 |
| M1 vs. M2A | 17.810044 | 2 | 0.000135706 | ||
| M7 vs. M8 | 18.041236 | 2 | 0.000120891 | ||
| M8a vs. M8 | 17.827478 | 1 | 0.000024187 | ||
| rpl20 | M0 vs. M3 | 0.407316 | 4 | 0.98187355 | 76 Y 0.698 |
| M1 vs. M2A | 0.016874 | 2 | 0.991598492 | ||
| M7 vs. M8 | 0.067732 | 2 | 0.966701034 | ||
| M8a vs. M8 | 0.01735 | 1 | 0.895206241 | ||
| rps2 | M0 vs. M3 | 108.22369 | 4 | 0 | 32 A 0.524, 131 N 0.504, 199 L 0.612, 237—1.000 ** |
| M1 vs. M2A | 108.22376 | 2 | 0 | ||
| M7 vs. M8 | 108.237424 | 2 | 0 | ||
| M8a vs. M8 | 108.245618 | 1 | 0 | ||
| rps3 | M0 vs. M3 | 2.187614 | 4 | 0.70129752 | 30 S 0.571, 86 E 0.568 |
| M1 vs. M2A | 0 | 2 | 1 | ||
| M7 vs. M8 | 0.000004 | 2 | 0.999998 | ||
| M8a vs. M8 | 0.025852 | 1 | 0.87226229 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raman, G.; Park, S. The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis. Agronomy 2020, 10, 1405. https://doi.org/10.3390/agronomy10091405
Raman G, Park S. The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis. Agronomy. 2020; 10(9):1405. https://doi.org/10.3390/agronomy10091405
Chicago/Turabian StyleRaman, Gurusamy, and SeonJoo Park. 2020. "The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis" Agronomy 10, no. 9: 1405. https://doi.org/10.3390/agronomy10091405
APA StyleRaman, G., & Park, S. (2020). The Complete Chloroplast Genome Sequence of the Speirantha gardenii: Comparative and Adaptive Evolutionary Analysis. Agronomy, 10(9), 1405. https://doi.org/10.3390/agronomy10091405