Development of a Multipurpose Core Collection of Bread Wheat Based on High-Throughput Genotyping Data

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Genetic and Phenotypic Characterization
2.3. Creation of the Core Collections
2.4. Evaluation of the Core Collections
3. Results
3.1. Creation of the Core Collections
3.2. Evaluation of Core Collections
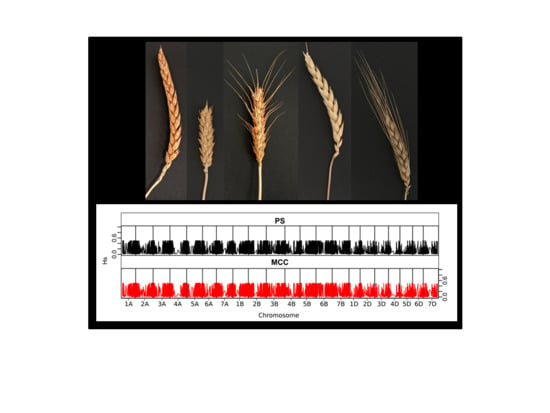
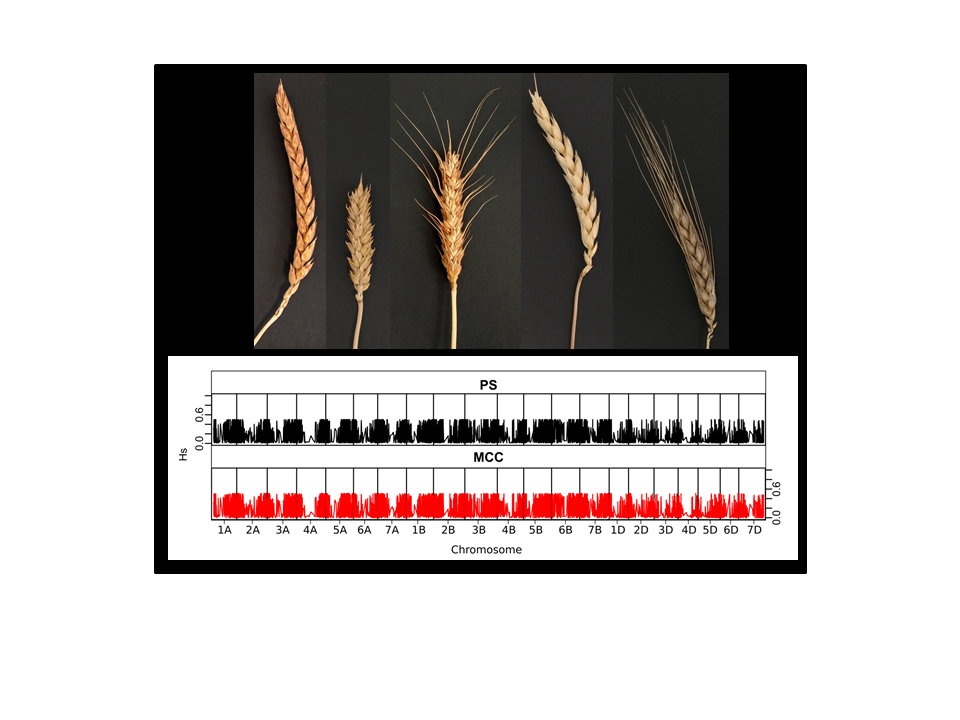
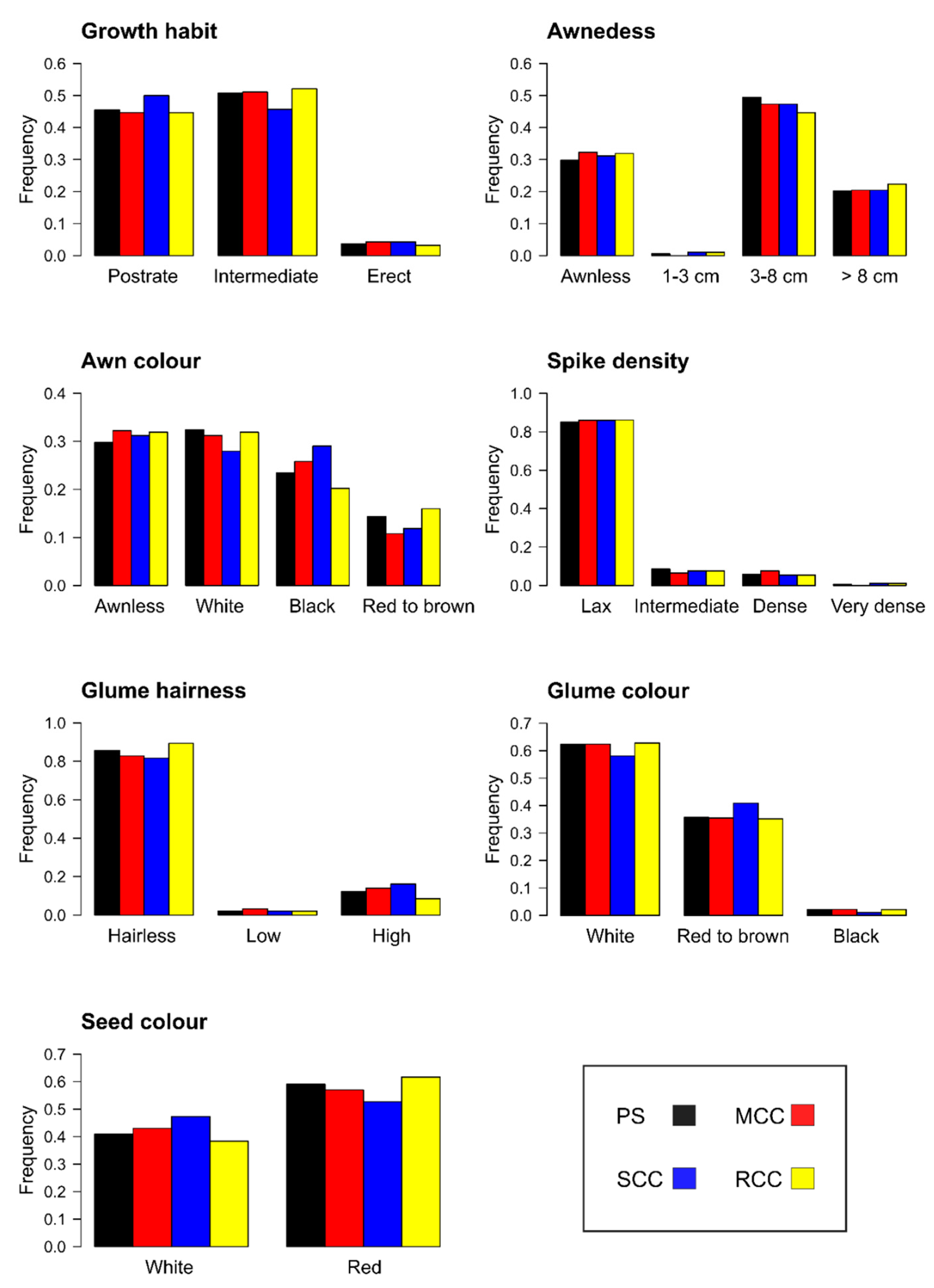
3.2.1. Representativeness of the Core Collections
3.2.2. Representativeness of the Core Collections
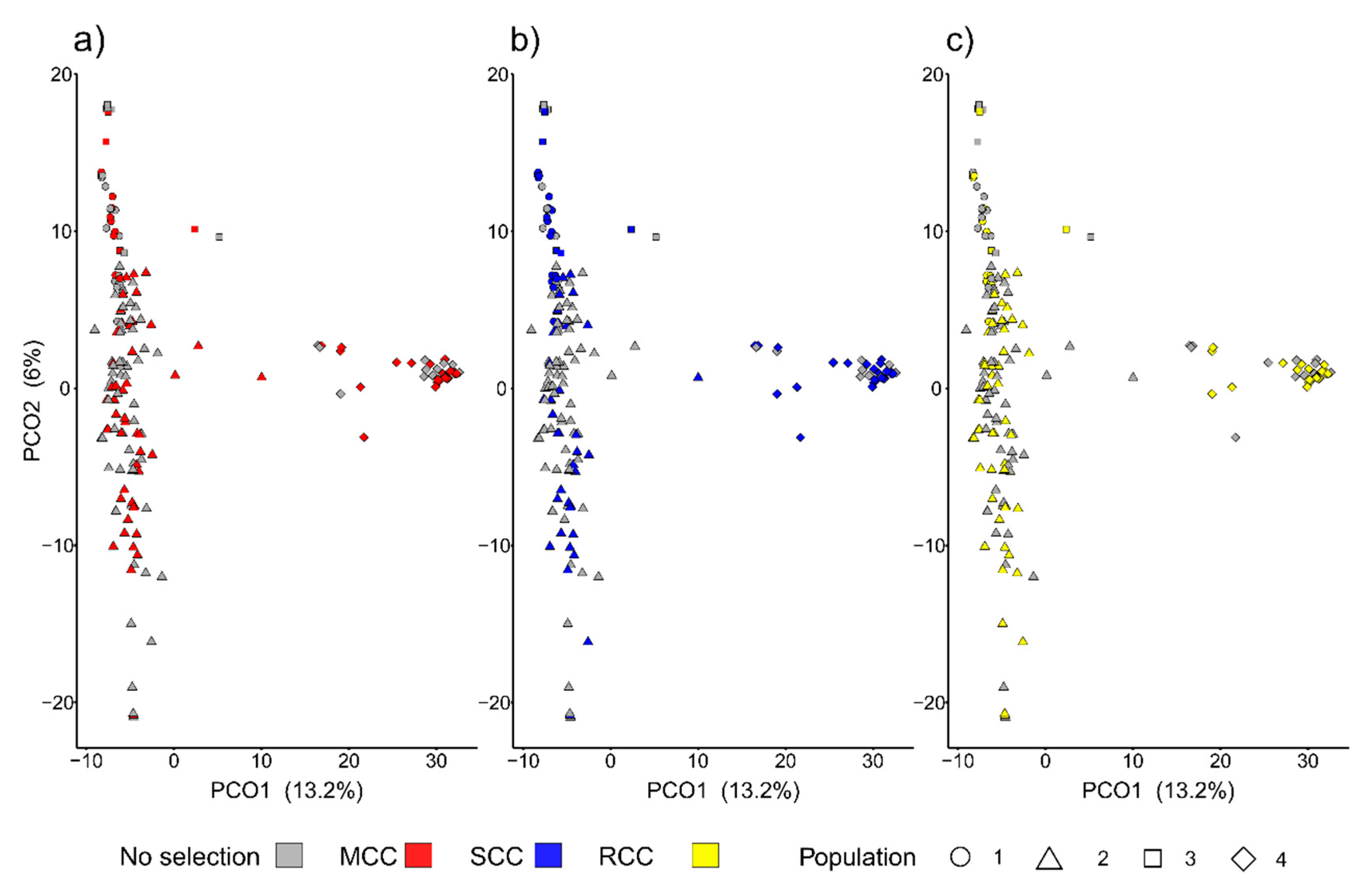
3.2.3. Distances between Entries
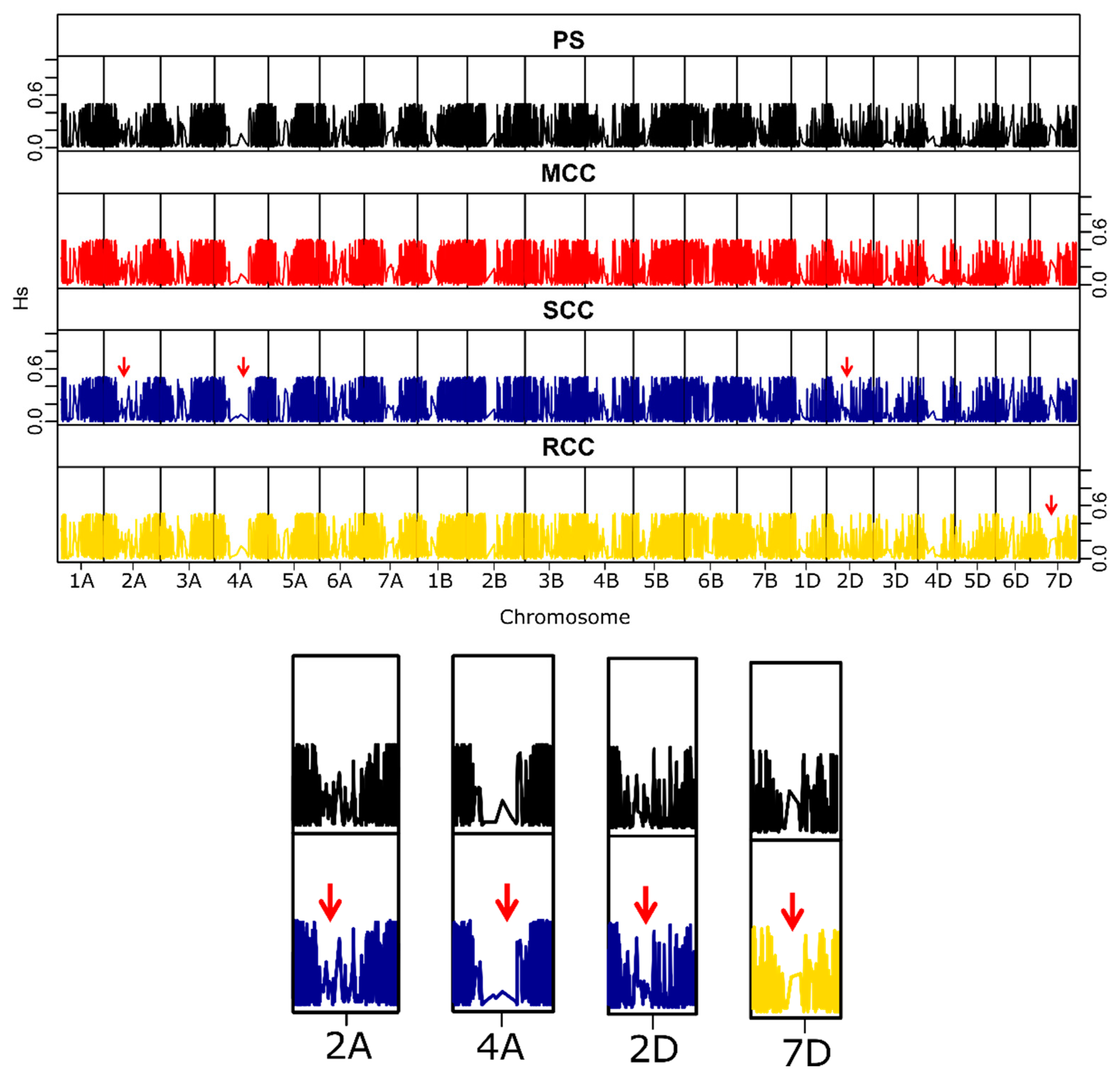
3.2.4. Distribution of Genetic Variability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAO. Harvesting Nature’s Diversity. Available online: http://www.fao.org/3/v1430e/V1430E04.htm (accessed on 2 March 2020).
- Ficiciyan, A.; Loos, J.; Sievers-Glotzbach, S.; Tscharntke, T. More than yield: Ecosystem services of traditional versus modern crop varieties revisited. Sustainability 2018, 10, 2834. [Google Scholar] [CrossRef]
- Lopes, M.S.; El-Basyoni, I.; Baenziger, P.S.; Singh, S.; Royo, C.; Ozbek, K.; Aktas, H.; Ozer, E.; Ozdemir, F.; Manickavelu, A.; et al. Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J. Exp. Bot. 2015, 66, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- CIMMYT. Seeds of Discovery. Available online: http://seedsofdiscovery.org/en/ (accessed on 5 March 2020).
- Brown, A. The case for core collections. In The Use of Plant Genetic Resources; Brown, A., Frankel, O., Marshall, D., Williams, J., Eds.; Cambridge University Press: Cambridge, UK, 1989; pp. 136–156. [Google Scholar]
- Brown, A. Core collections: A practical approach to genetic resources management. Genome 1989, 31, 818–824. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, D.; Wang, M.; Sun, J.; Qi, Y.; Li, J.; Han, L.; Qiu, Z.; Tang, S.; Li, Z. A core collection and mini core collection of Oryza sativa L in China. Appl. Genet. 2011, 122, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhu, J.; Xu, H. Methods of constructing core collections by stepwise clustering with three sampling strategies based on the genotypic values of crops. Appl. Genet. 2000, 101, 264–268. [Google Scholar] [CrossRef]
- Holtz, Y.; Ardisson, M.; Ranwez, V.; Besnard, A.; Leroy, P.; Poux, G.; Roumet, P.; Viader, V.; Santoni, S.; David, J. Genotyping by sequencing using specific allelic capture to build a high-density genetic map of durum wheat. PLoS ONE 2016, 11, e0154609. [Google Scholar] [CrossRef]
- Kilian, B.; Graner, A. NGS technologies for analyzing germplasm diversity in genebanks. Brief. Funct. Genom. 2012, 11, 38–50. [Google Scholar] [CrossRef]
- Heslot, N.; Rutkoski, J.; Poland, J.; Jannink, J.-L.; Sorrells, M.E. Impact of marker ascertainment bias on genomic selection accuracy and estimates of genetic diversity. PLoS ONE 2013, 8, e74612. [Google Scholar] [CrossRef]
- Manickavelu, A.; Jighly, A.; Ban, T. Molecular evaluation of orphan afghan common wheat (Triticum aestivum L.) landraces collected by Dr. Kihara using single nucleotide polymorphic markers. BMC Plant Biol. 2014, 14, 320. [Google Scholar] [CrossRef]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. Diversity arrays technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, 54. [Google Scholar] [CrossRef]
- Diversity Arrays Technology. Available online: https://www.diversityarrays.com (accessed on 5 March 2020).
- Rasheed, A.; Mujeeb-Kazi, A.; Ogbonnaya, F.C.; He, Z.; Rajaram, S. Wheat genetic resources in the post-genomics era: Promise and challenges. Ann. Bot. 2018, 121, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Schoen, D.J.; Brown, A. Conservation of allelic richness in wild crop relatives is aided by assessment of genetic markers. Proc. Natl. Acad. Sci. USA 1993, 90, 10623–10627. [Google Scholar] [CrossRef] [PubMed]
- Erskine, W.; Muehlbauer, F. Allozyme and morphological variability, outcrossing rate and core collection formation in lentil germplasm. Appl. Genet. 1991, 83, 119–125. [Google Scholar] [CrossRef]
- Bataillon, T.M.; David, J.L.; Schoen, D.J. Neutral genetic markers and conservation genetics: Simulated germplasm collections. Genetics 1996, 144, 409–417. [Google Scholar]
- Franco, J.; Crossa, J.; Taba, S.; Shands, H. A sampling strategy for conserving genetic diversity when forming core subsets. Crop Sci. 2005, 45, 1035–1044. [Google Scholar] [CrossRef]
- Franco, J.; Crossa, J.; Warburton, M.L.; Taba, S. Sampling strategies for conserving maize diversity when forming core subsets using genetic markers. Crop Sci. 2006, 46, 854–864. [Google Scholar] [CrossRef]
- Thachuk, C.; Crossa, J.; Franco, J.; Dreisigacker, S.; Warburton, M.; Davenport, G.F. Core hunter: An algorithm for sampling genetic resources based on multiple genetic measures. BMC Bioinform. 2009, 10, 243. [Google Scholar] [CrossRef]
- Díez, C.M.; Imperato, A.; Rallo, L.; Barranco, D.; Trujillo, I. Worldwide core collection of olive cultivars based on simple sequence repeat and morphological markers. Crop Sci. 2012, 52, 211–221. [Google Scholar] [CrossRef]
- Krishnan, R.R.; Sumathy, R.; Ramesh, S.; Bindroo, B.; Naik, G.V. SimEli: Similarity elimination method for sampling distant entries in development of core collections. Crop Sci. 2014, 54, 1070–1078. [Google Scholar] [CrossRef]
- INIA. National Inventory of Plant Genetic Resources. Available online: http://webx.inia.es/web_inventario_nacional/Introduccioneng.asp (accessed on 5 March 2020).
- Aparicio, N.; Alvaro, F.; Sillero, J.C.; Ruiz, M.; López, P.; Cátedra, M.; Codesal, P. Bread Wheat (Triticum aestivum, L.) Core Collection based in Spanish landraces. In Proceedings of the 8th International Wheat Conference, St. Petersburg, Russia, 1–4 June 2010; Dzyubenko, N.I., Ed.; NI Vavilov Research Institute of Plant Industry (VIR): Saint Petersburg, Russia, 2005; p. 85. [Google Scholar]
- Odong, T.; Jansen, J.; Van Eeuwijk, F.; van Hintum, T.J. Quality of core collections for effective utilisation of genetic resources review, discussion and interpretation. Appl. Genet. 2013, 126, 289–305. [Google Scholar] [CrossRef]
- Pascual, L.; Ruiz, M.; López-Fernández, M.; Pérez-Peña, H.; Benavente, E.; Vázquez, J.F.; Sansaloni, C.; Giraldo, P. Genomic analysis of Spanish wheat landraces reveals their variability and potential for breeding. BMC Genom. 2020, 21, 122. [Google Scholar] [CrossRef]
- Payne, P.I.; Nightingale, M.A.; Krattiger, A.F.; Holt, L.M. The relationship between HMW glutenin subunit composition and the bread-making quality of British-grown wheat varieties. J. Sci. Food Agric. 1987, 40, 51–65. [Google Scholar] [CrossRef]
- IBPGR. Revised Descriptor List for Wheat (Triticum ssp); International Board for Plant Genetic Resources: Rome, Italy, 1985; p. 12. [Google Scholar]
- Pacheco, A.; Alvarado, G.; Rodríguez, F.; Burgueño, J. BIO-R (Biodiversity Analysis with R for Windows) Version 2.0; Cimmyt: Sonora, Mexico, 2016. [Google Scholar]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef]
- R Core Team R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Fisher, R. Statistical Methods for Research Workers; Oliver and Boyd: Edinburgh, Scotland, 1954. [Google Scholar]
- Gower, J.C. A general coefficient of similarity and some of its properties. Biometrics 1971, 857–871. [Google Scholar] [CrossRef]
- Ruiz, M.; Giraldo, P.; Royo, C.; Villegas, D.; Jose Aranzana, M.; Carrillo, J.M. Diversity and genetic structure of a collection of spanish durum wheat landraces. Crop Sci. 2012, 52, 2262–2275. [Google Scholar] [CrossRef]
- Ruiz, M.; Rodriguez-Quijano, M.; Metakovsky, E.V.; Vazquez, J.F.; Carrillo, J.M. Polymorphism, variation and genetic identity of Spanish common wheat germplasm based on gliadin alleles. Field Crop. Res. 2002, 79, 185–196. [Google Scholar] [CrossRef]
- Giraldo, P.; Rodriguez-Quijano, M.; Simon, C.; Vazquez, J.F.; Carrillo, J.M. Allelic variation in HMW glutenins in Spanish wheat landraces and their relationship with bread quality. Span. J. Agric. Res. 2010, 8, 1012–1023. [Google Scholar] [CrossRef]
- Martinez, F.; Niks, R.E.; Moral, A.; Urbano, J.M.; Rubiales, D. Search for partial resistance to leaf rust in a collection of ancient Spanish wheats. Hereditas 2001, 135, 193–197. [Google Scholar] [CrossRef][Green Version]
- Vázquez, J.F.; Chacón, E.A.; Carrillo, J.M.; Benavente, E. Grain mineral density of bread and durum wheat landraces from geochemically diverse native soils. Crop Pasture Sci. 2018, 69, 335–346. [Google Scholar] [CrossRef]
- Ruiz, M.; Giraldo, P.; Royo, C.; Carrillo, J.M. Creation and validation of the spanish durum wheat core collection. Crop Sci. 2013, 53, 2530–2537. [Google Scholar] [CrossRef]
- Igartua, E.; Gracia, M.; Lasa, J.; Medina, B.; Molina-Cano, J.; Montoya, J.; Romagosa, I. The Spanish barley core collection. Genet. Resour. Crop Evol. 1998, 45, 475–481. [Google Scholar] [CrossRef]
- Silvar, C.; Flath, K.; Kopahnke, D.; Gracia, M.; Lasa, J.; Casas, A.; Igartua, E.; Ordon, F. Analysis of powdery mildew resistance in the Spanish barley core collection. Plant Breed. 2011, 130, 195–202. [Google Scholar] [CrossRef]
- Yahiaoui, S.; Cuesta-Marcos, A.; Gracia, M.P.; Medina, B.; Lasa, J.M.; Casas, A.M.; Ciudad, F.J.; Montoya, J.L.; Moralejo, M.; Molina-Cano, J.L. Spanish barley landraces outperform modern cultivars at low-productivity sites. Plant Breed. 2014, 133, 218–226. [Google Scholar] [CrossRef]
- Ruiz, M.; Giraldo, P.; González, J.M. Phenotypic variation in root architecture traits and their relationship with eco-geographical and agronomic features in a core collection of tetraploid wheat landraces (Triticum turgidum L.). Euphytica 2018, 214, 54. [Google Scholar] [CrossRef]
- Ruiz, M.; Bernal, G.; Giraldo, P. An update of low molecular weight glutenin subunits in durum wheat relevant to breeding for quality. J. Cereal Sci. 2018, 83, 236–244. [Google Scholar] [CrossRef]
- Martínez-Moreno, F.; Ruiz, M.; Blandón, M.; Giraldo, P. Resistance to leaf rust in a core collection of ancient Spanish tetraploid wheats. In Proceedings of the I Spanish Symposium on Cereal Physiology and Breeding, Zaragoza, Spain, 9–10 April 2018; p. 34. [Google Scholar]
- Acuña-Matamoros, C.L.; Reyes-Valdés, M.H. Comparison of optimization methods for core subset selection from a large collection of Mexican wheat landraces characterized by SNP markers. Plant Genet. Resour. 2018, 16, 228–236. [Google Scholar] [CrossRef]
- Kobayashi, F.; Tanaka, T.; Kanamori, H.; Wu, J.; Katayose, Y.; Handa, H. Characterization of a mini core collection of Japanese wheat varieties using single-nucleotide polymorphisms generated by genotyping-by-sequencing. Breed. Sci. 2016, 66, 213–225. [Google Scholar] [CrossRef]
- Wingen, L.U.; Orford, S.; Goram, R.; Leverington-Waite, M.; Bilham, L.; Patsiou, T.S.; Ambrose, M.; Dicks, J.; Griffiths, S. Establishing the AE Watkins landrace cultivar collection as a resource for systematic gene discovery in bread wheat. Appl. Genet. 2014, 127, 1831–1842. [Google Scholar] [CrossRef]
- Balfourier, F.; Roussel, V.; Strelchenko, P.; Exbrayat-Vinson, F.; Sourdille, P.; Boutet, G.; Koenig, J.; Ravel, C.; Mitrofanova, O.; Beckert, M. A worldwide bread wheat core collection arrayed in a 384-well plate. Appl. Genet. 2007, 114, 1265–1275. [Google Scholar] [CrossRef]
- Van Hintum, T.J.; Brown, A.; Spillane, C. Core Collections of Plant Genetic Resources; Bioversity International: Rome, Italy, 2000; p. 48. [Google Scholar]
- Varshney, R.K.; Nayak, S.N.; May, G.D.; Jackson, S.A. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 2009, 27, 522–530. [Google Scholar] [CrossRef]
- Gale, M.; Marshall, G.A. The chromosomal location of Gai 1 and Rht 1, genes for gibberellin insensitivity and semi-dwarfism, in a derivative of Norin 10 wheat. Heredity 1976, 37, 283–289. [Google Scholar] [CrossRef]
- Murai, M.; Takamure, I.; Sato, S.; Tokutome, T.; Sato, Y. Effects of the dwarfing gene originating from ‘Dee-geo-woo-gen’on yield and its related traits in rice. Breed. Sci. 2002, 52, 95–100. [Google Scholar] [CrossRef]
- Vikram, P.; Franco, J.; Burgueño-Ferreira, J.; Li, H.; Sehgal, D.; Saint Pierre, C.; Ortiz, C.; Sneller, C.; Tattaris, M.; Guzman, C. Unlocking the genetic diversity of Creole wheats. Sci. Rep. 2016, 6, 23092. [Google Scholar] [CrossRef]
- Marshall, D.; Brown, A. Optimum sampling strategies in genetic conservation. In Crop Genetic Resources for Today and Tomorrow; Frankel, O.H., Hawkes, J.G., Eds.; Cambridge University Press: Cambridge, UK, 1975; pp. 53–70. [Google Scholar]
- Odong, T.; van Heerwaarden, J.; Jansen, J.; van Hintum, T.J.; van Eeuwijk, F. Statistical techniques for defining reference sets of accessions and microsatellite markers. Crop Sci. 2011, 51, 2401–2411. [Google Scholar] [CrossRef]
- Reyes-Valdés, M.H.; Burgueño, J.; Singh, S.; Martínez, O.; Sansaloni, C.P. An informational view of accession rarity and allele specificity in germplasm banks for management and conservation. PLoS ONE 2018, 13, e0193346. [Google Scholar] [CrossRef]
- Wen, W.; Franco, J.; Chavez-Tovar, V.H.; Yan, J.; Taba, S. Genetic characterization of a core set of a tropical maize race Tuxpeño for further use in maize improvement. PLoS ONE 2012, 7, e32626. [Google Scholar] [CrossRef]
- Corrado, G.; Caramante, M.; Piffanelli, P.; Rao, R. Genetic diversity in Italian tomato landraces: Implications for the development of a core collection. Sci. Hortic. 2014, 168, 138–144. [Google Scholar] [CrossRef]
- Informe Cereal de Invierno GENVCE 2018. Available online: www.genvce.org/repositorio/informe-cereal-de-invierno-genvce-2018 (accessed on 23 February 2020).
- Map of Soil pH in Europe. Available online: https://esdac.jrc.ec.europa.eu/content/soil-ph-europe (accessed on 23 February 2020).
- Zhang, X.K.; Xiao, Y.G.; Zhang, Y.; Xia, X.C.; Dubcovsky, J.; He, Z.H. Allelic variation at the vernalization genes Vrn-A1, Vrn-B1, Vrn-D1, and Vrn-B3 in Chinese wheat cultivars and their association with growth habit. Crop Sci. 2008, 48, 458–470. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; García-Pereira, M.J.; Gracia, M.P.; Igartua, E.; Casas, A.M.; Contreras-Moreira, B. Large differences in gene expression responses to drought and heat stress between elite barley cultivar scarlett and a spanish landrace. Front. Plant Sci. 2017, 8, 647. [Google Scholar] [CrossRef]
- Monteagudo Gálvez, A.; Casas Cendoya, A.M.; Pérez Cantalapiedra, C.; Contreras-Moreira, B.; Gracia Gimeno, M.P.; Igartua Arregui, E. Harnessing novel diversity from landraces to improve an elite barley variety. Front. Plant Sci. 2019, 10, 434. [Google Scholar] [CrossRef]
- Ruiz, M.; Zambrana, E.; Fite, R.; Sole, A.; Tenorio, J.L.; Benavente, E. Yield and quality performance of traditional and improved bread and durum wheat varieties under two conservation tillage systems. Sustainability 2019, 11, 4522. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop 1 | Pop 2 | Pop 3 | Pop 4 | |
|---|---|---|---|---|
| HS | 0.21 | 0.32 | 0.13 | 0.23 |
| PS | 25 | 112 | 16 | 36 |
| MCC | 13 | 50 | 8 | 23 |
| SCC | 21 | 37 | 10 | 26 |
| RCC | 10 | 55 | 8 | 21 |
| Latitude | Longitude | Elevation (m) | |
|---|---|---|---|
| PS | 433310N–281820N | 0174928W–0041559E | 10–1610 |
| MCC | 433310N–281820N | 0174928W–0041559E | 22–1540 |
| SCC | 433310N–281820N | 0174928W–0041559E | 35–1540 |
| RCC | 433310N–281820N | 0162421W–0031238E | 63–1540 |
| Days to Heading (Days) | Days to Maturity (Days) | Plant Height (cm) | Spike Length (mm) | Spikelets Per Spike (Number) | Evaluation Parameter | |||
|---|---|---|---|---|---|---|---|---|
| Mean | PS | 171.23 | 206.86 | 88.27 | 117.03 | 19.11 | MD | - |
| MCC | 171.48 | 206.81 | 87.89 | 118.36 | 19.31 | 0 | ||
| SCC | 172.08 | 206.9 | 85.97 | 115.98 | 19.02 | 0 | ||
| RCC | 171.19 | 207 | 88.19 | 119 | 19.28 | 0 | ||
| Variance | PS | 49.16 | 11.34 | 137.87 | 365.54 | 4.16 | VD | - |
| MCC | 47.48 | 10.65 | 160.14 | 403.72 | 4.52 | 0 | ||
| SCC | 48.83 | 9.59 | 137.73 | 340.78 | 4.22 | 0 | ||
| RCC | 48.97 | 10.54 | 159.7 | 385.03 | 3.51 | 0 | ||
| Coefficient of Variation | PS | 4.09 | 1.63 | 13.3 | 16.34 | 10.68 | VR | - |
| MCC | 4.02 | 1.58 | 14.4 | 16.98 | 11.01 | 102.06 | ||
| SCC | 4.04 | 1.49 | 13.58 | 15.83 | 10.74 | 97.92 | ||
| RCC | 4.09 | 1.57 | 14.33 | 16.49 | 9.72 | 99.18 | ||
| Range | PS | 155–188 | 199–216 | 53–119 | 59–168 | 14–24 | CR | - |
| MCC | 157–188 | 199–216 | 53–115 | 67–168 | 14–24 | 91.53 | ||
| SCC | 157–188 | 199–215 | 53–114 | 59–168 | 14–24 | 91.64 | ||
| RCC | 155–188 | 200–216 | 53–119 | 59–153 | 14–23 | 89.46 | ||
| Glu-1 Homoeoloci | Vrn-A1 Alleles (%) | |||||
|---|---|---|---|---|---|---|
| Glu-A1 | Glu-B1 | Glu-D1 | Vrn-A1 | Vrn-A1a | Vrn-A1b | |
| PS | a,b,c,y | a,al,am,aq,d,e,f,h,i,u,n2,n3,n4,n5,n6 | a,c,d,h,j,l,n6 | 18.52 | 52.38 | 29.10 |
| MCC | a,b,c,y | a,al,am,aq,d,e,f,h,i,u,n2,n3,n4,n5,n6 | a,c,d,h,j,l,n6 | 18.09 | 48.94 | 32.98 |
| SCC | a,b,c,y | a,al,am,aq,d,e,f,h,i,u,n2,n3,n4,n5,n6 | a,c,d,h,j,l,n6 | 12.77 | 55.32 | 31.91 |
| RCC | a,b,c,y | a,al,am,aq,d,e,f,h,i,u,n3,n4,n5,n6 | a,c,d,h,j,l | 15.96 | 50.00 | 34.04 |
| SNP Markers (Number) | Fixed SNP Markers (Number) | |||
|---|---|---|---|---|
| MAF | PS | MCC | SCC | RCC |
| >0.1 | 6376 | 0 | 0 | 0 |
| ≥0.05 | 8394 | 0 | 6 | 2 |
| ≥0.03 | 10,092 | 3 | 43 | 8 |
| >0.01 | 14,002 | 534 | 685 | 586 |
| ≤0.01 | 828 | 387 | 419 | 453 |
| Total | 14,830 | 921 | 1104 | 1049 |
| HS | 0.20 | 0.20 | 0.19 | 0.19 |
| E-E | A-NE | E-NE | Min E-NE | |
|---|---|---|---|---|
| PS | 0.189 | - | 0.073 | 0.0061 |
| MCC | 0.192 | 0.043 | 0.090 | 0.0067 |
| SCC | 0.190 | 0.051 | 0.081 | 0.0065 |
| RCC | 0.191 | 0.043 | 0.081 | 0.0067 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascual, L.; Fernández, M.; Aparicio, N.; López-Fernández, M.; Fité, R.; Giraldo, P.; Ruiz, M. Development of a Multipurpose Core Collection of Bread Wheat Based on High-Throughput Genotyping Data. Agronomy 2020, 10, 534. https://doi.org/10.3390/agronomy10040534
Pascual L, Fernández M, Aparicio N, López-Fernández M, Fité R, Giraldo P, Ruiz M. Development of a Multipurpose Core Collection of Bread Wheat Based on High-Throughput Genotyping Data. Agronomy. 2020; 10(4):534. https://doi.org/10.3390/agronomy10040534
Chicago/Turabian StylePascual, Laura, Mario Fernández, Nieves Aparicio, Matilde López-Fernández, Rosario Fité, Patricia Giraldo, and Magdalena Ruiz. 2020. "Development of a Multipurpose Core Collection of Bread Wheat Based on High-Throughput Genotyping Data" Agronomy 10, no. 4: 534. https://doi.org/10.3390/agronomy10040534
APA StylePascual, L., Fernández, M., Aparicio, N., López-Fernández, M., Fité, R., Giraldo, P., & Ruiz, M. (2020). Development of a Multipurpose Core Collection of Bread Wheat Based on High-Throughput Genotyping Data. Agronomy, 10(4), 534. https://doi.org/10.3390/agronomy10040534