Nucleotide Diversity and Association Analysis of ZmMADS60 with Root Length in the Maize Seedling Stage

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Phenotypic Evaluation
2.2. ZmMADS60 Resequencing
2.3. Sequence Analysis, Genetic Diversity Analysis and Neutral Evolution Test
2.4. Natural Variation of the ZmMADS60 Gene Associated with Root Traits in Inbred Lines
3. Results
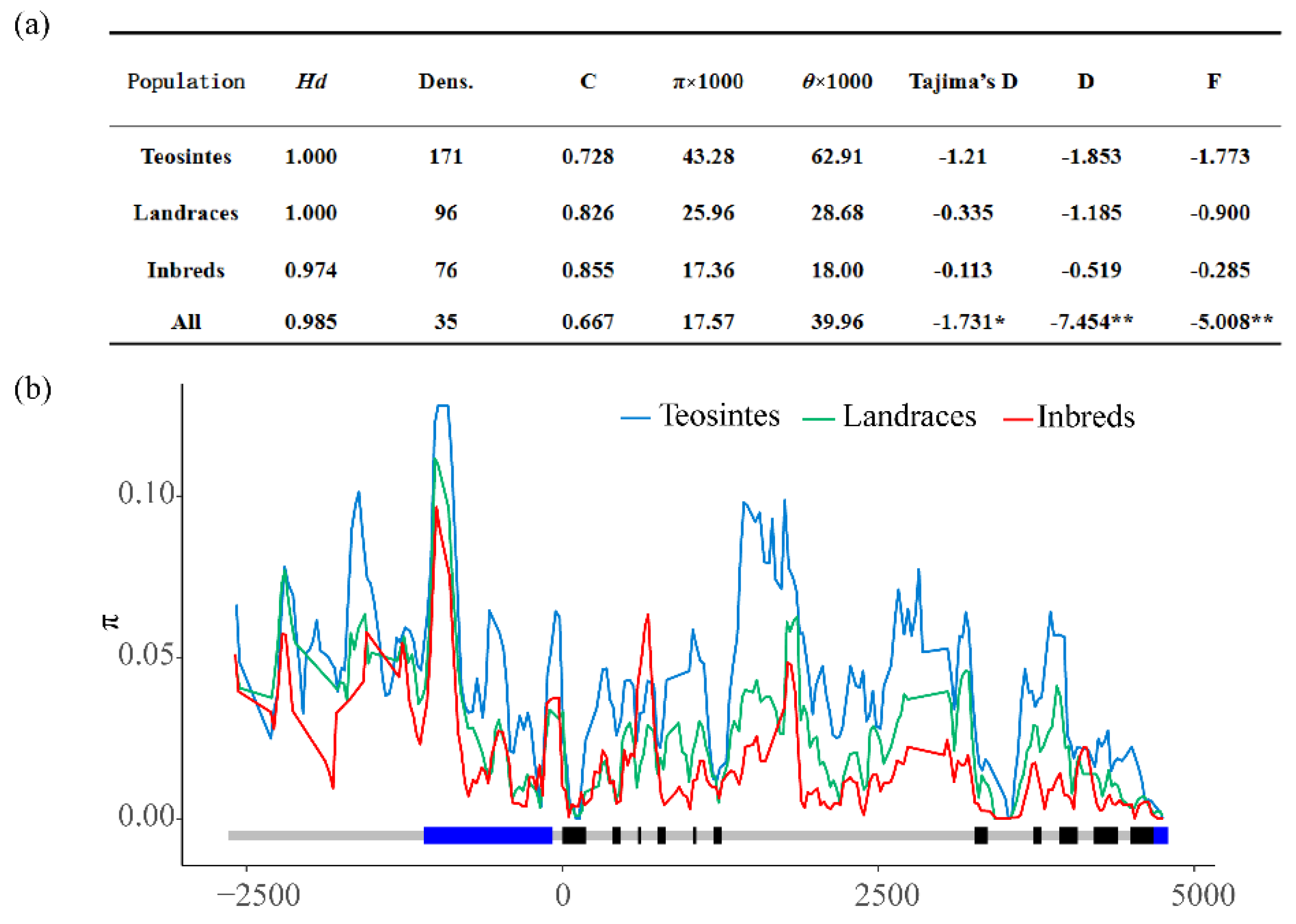
3.1. Nucleotide Diversity and Selection of ZmMADS60 in Inbred Lines, Landrace and Teosinte
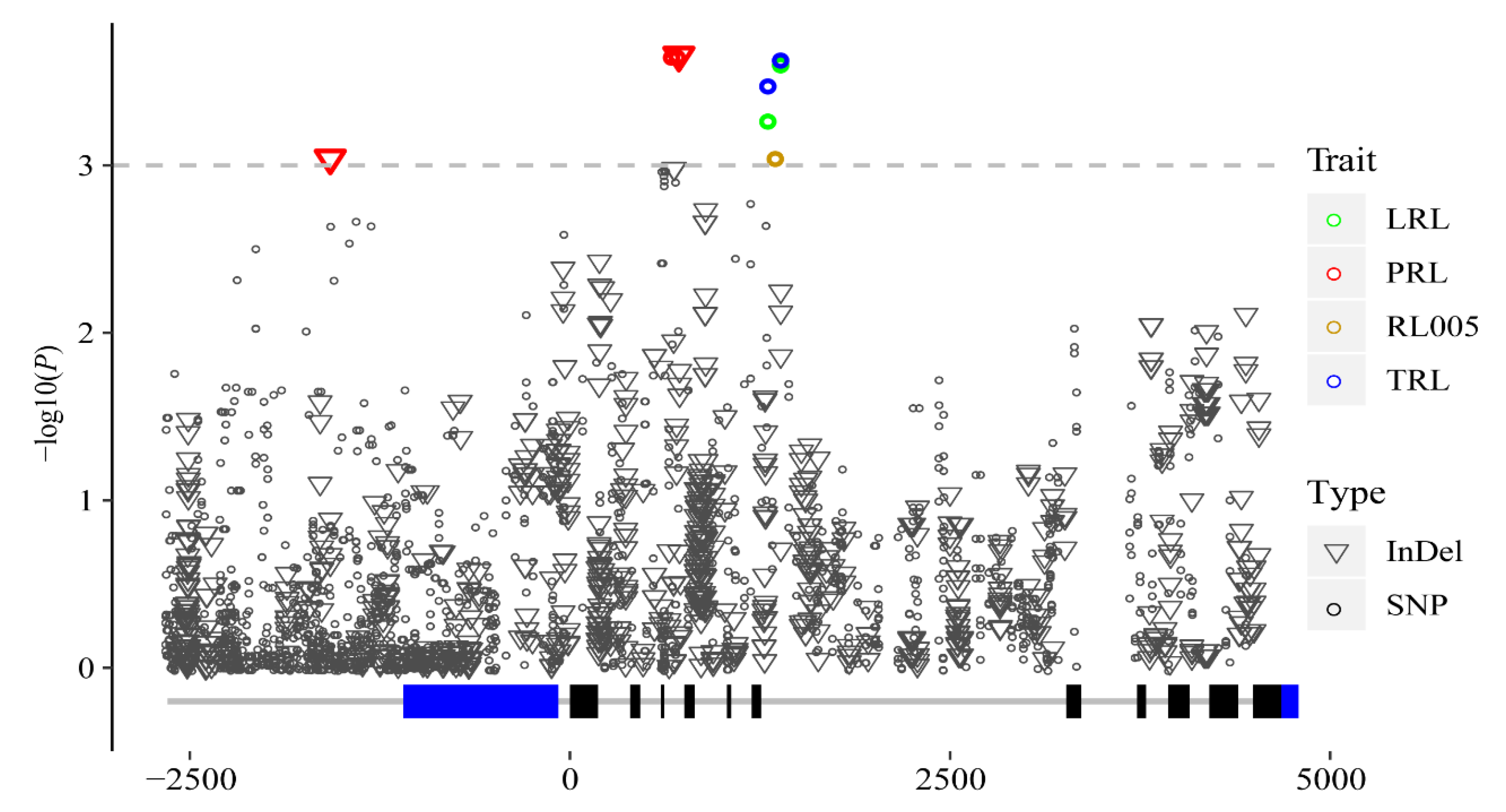
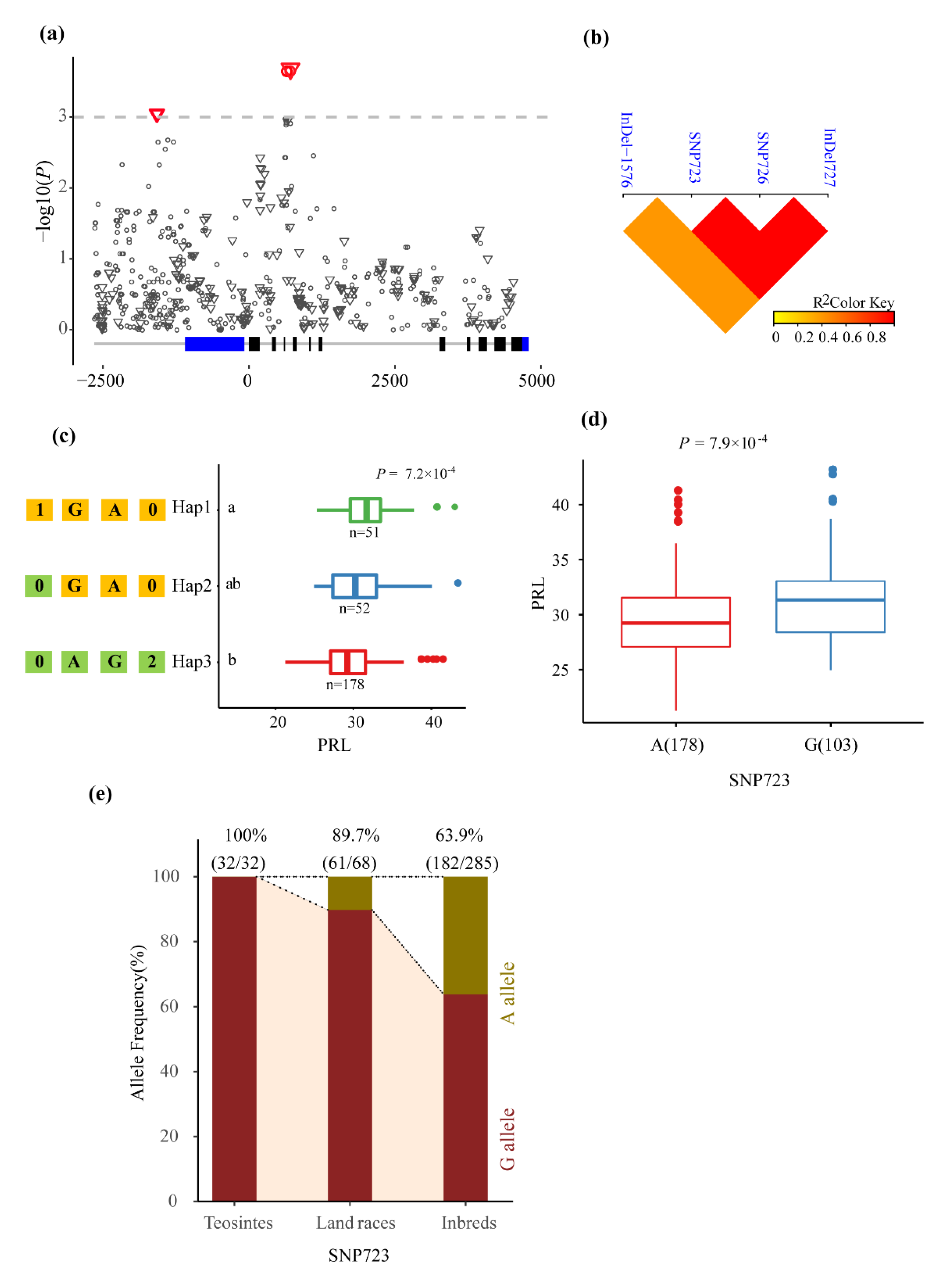
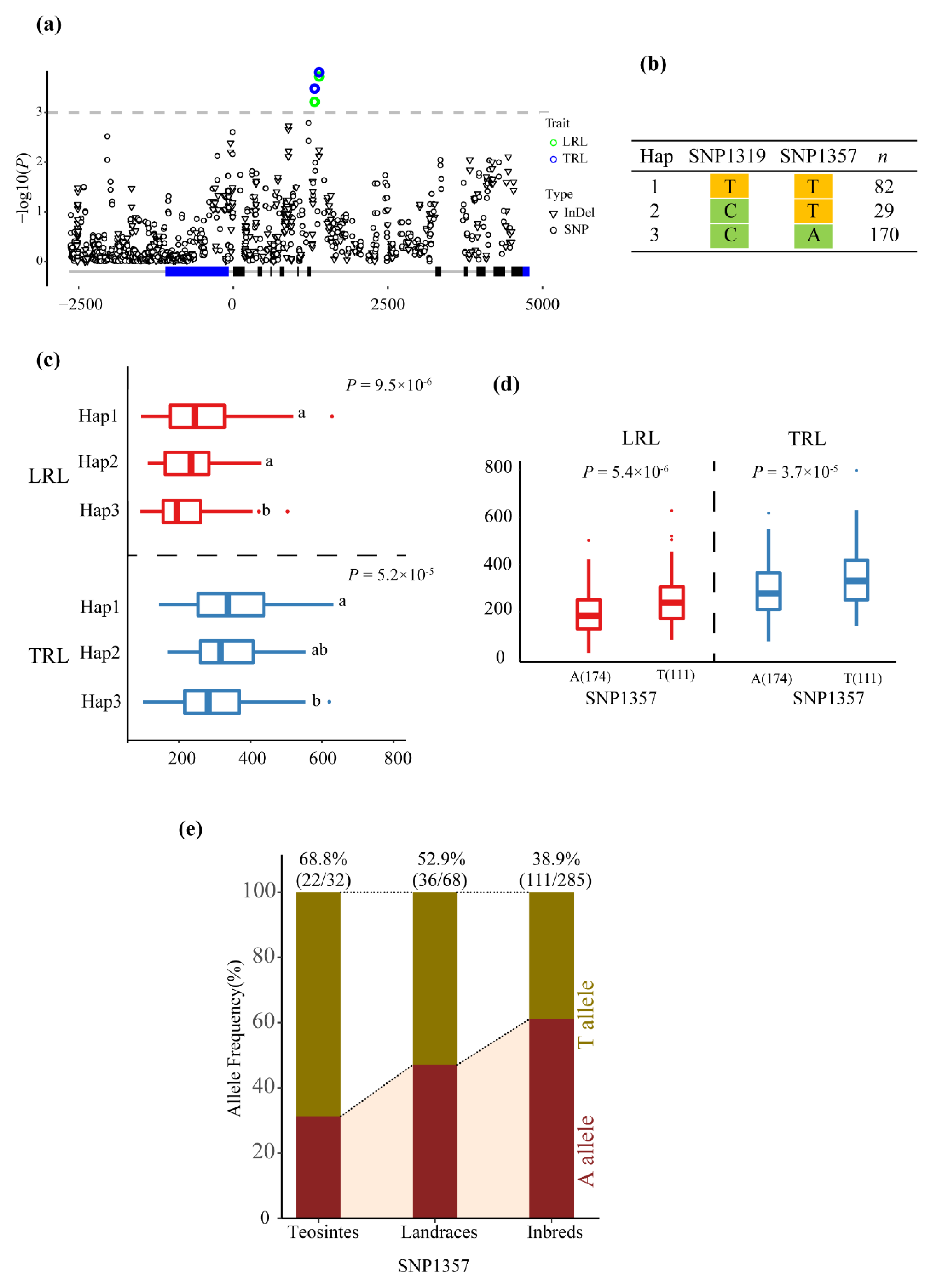
3.2. Association Analysis of Phenotypic Traits with ZmMADS60
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Osmont, K.S.; Sibout, R.; Hardtke, C.S. Hidden branches: Developments in root system architecture. Annu. Rev. Plant Biol. 2007, 58, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Hochholdinger, F.; Li, C.J. Plasticity of Lateral Root Branching in Maize. Front Plant Sci. 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Hochholdinger, F.; Yu, P.; Marcon, C. Genetic Control of Root System Development in Maize. Trends Plant Sci. 2018, 23, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Hochholdinger, F.; Park, W.J.; Sauer, M.; Woll, K. From weeds to crops: Genetic analysis of root development in cereals. Trends Plant Sci. 2004, 9, 42–48. [Google Scholar] [CrossRef]
- Stoeckle, D.; Thellmann, M.; Vermeer, J.E.M. Breakout-lateral root emergence in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2018, 41, 67–72. [Google Scholar] [CrossRef]
- Taramino, G.; Sauer, M.; Stauffer, J.L.; Multani, D.; Niu, X.M.; Sakai, H.; Hochholdinger, F. The maize (Zea mays L.) RTCS gene encodes a LOB domain protein that is a key regulator of embryonic seminal and post-embryonic shoot-borne root initiation. Plant J. 2007, 50, 649–659. [Google Scholar] [CrossRef]
- von Behrens, I.; Komatsu, M.; Zhang, Y.; Berendzen, K.W.; Niu, X.; Sakai, H.; Taramino, G.; Hochholdinger, F. Rootless with undetectable meristem 1 encodes a monocot-specific AUX/IAA protein that controls embryonic seminal and post-embryonic lateral root initiation in maize. Plant J. 2011, 66, 341–353. [Google Scholar] [CrossRef]
- Zhang, Y.; von Behrens, I.; Zimmermann, R.; Ludwig, Y.; Hey, S.; Hochholdinger, F. LATERAL ROOT PRIMORDIA 1 of maize acts as a transcriptional activator in auxin signalling downstream of the Aux/IAA gene rootless with undetectable meristem 1. J. Exp. Bot. 2015, 66, 3855–3863. [Google Scholar] [CrossRef]
- Abdel-Ghani, A.H.; Kumar, B.; Pace, J.; Jansen, C.; Gonzalez-Portilla, P.J.; Reyes-Matamoros, J.; San Martin, J.P.; Lee, M.; Lubberstedt, T. Association analysis of genes involved in maize (Zea mays L.) root development with seedling and agronomic traits under contrasting nitrogen levels. Plant Mol. Biol. 2015, 88, 133–147. [Google Scholar] [CrossRef]
- Kumar, B.; Abdel-Ghani, A.H.; Pace, J.; Reyes-Matamoros, J.; Hochholdinger, F.; Lubberstedt, T. Association analysis of single nucleotide polymorphisms in candidate genes with root traits in maize (Zea mays L.) seedlings. Plant Sci. 2014, 224, 9–19. [Google Scholar] [CrossRef]
- Liu, J.C.; Li, J.S.; Chen, F.J.; Zhang, F.S.; Ren, T.H.; Zhuang, Z.J.; Mi, G.H. Mapping QTLs for root traits under different nitrate levels at the seedling stage in maize (Zea mays L.). Plant Soil 2008, 305, 253–265. [Google Scholar] [CrossRef]
- Guo, J.; Chen, L.; Li, Y.X.; Shi, Y.S.; Song, Y.C.; Zhang, D.F.; Li, Y.; Wang, T.Y.; Yang, D.G.; Li, C.H. Meta-QTL analysis and identification of candidate genes related to root traits in maize. Euphytica 2018, 214, 223. [Google Scholar] [CrossRef]
- Pace, J.; Gardner, C.; Romay, C.; Ganapathysubramanian, B.; Lubberstedt, T. Genome-wide association analysis of seedling root development in maize (Zea mays L.). BMC Genom. 2015, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Drisch, R.C.; Stahl, Y. Function and regulation of transcription factors involved in root apical meristem and stem cell maintenance. Front. Plant Sci. 2015, 6, 505. [Google Scholar] [CrossRef]
- Zea mays Transcription Factors. Available online: http://planttfdb.cbi.pku.edu.cn/index.php?sp=Zma (accessed on 22 February 2020).
- Smaczniak, C.; Immink, R.G.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef]
- Wills, D.M.; Fang, Z.; York, A.M.; Holland, J.B.; Doebley, J.F. Defining the Role of the MADS-Box Gene, Zea Agamous-like1, a Target of Selection during Maize Domestication. J. Hered. 2018, 109, 333–338. [Google Scholar] [CrossRef]
- Alter, P.; Bircheneder, S.; Zhou, L.Z.; Schluter, U.; Gahrtz, M.U.; Sonnewald, U.; Dresselhaus, T. Flowering Time-Regulated Genes in Maize Include the Transcription Factor ZmMADS1. Plant Physiol. 2016, 172, 389–404. [Google Scholar] [CrossRef]
- Liang, Y.M.; Liu, Q.; Wang, X.F.; Huang, C.; Xu, G.H.; Hey, S.; Lin, H.Y.; Li, C.; Xu, D.Y.; Wu, L.S.; et al. ZmMADS69 functions as a flowering activator through the ZmRap2.7-ZCN8 regulatory module and contributes to maize flowering time adaptation. New Phytol. 2019, 221, 2335–2347. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Garcia-Ponce, B.; Sanchez, M.P.; Espinosa-Soto, C.; Garcia-Gomez, M.L.; Pineyro-Nelson, A.; Garay-Arroyo, A. MADS-box genes underground becoming mainstream: Plant root developmental mechanisms. New Phytol. 2019, 223, 1143–1158. [Google Scholar] [CrossRef]
- Zhang, H.; Forde, B.G. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science 1998, 279, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.P.; Xu, N.; Chen, H.L.; Wang, G.X.; Huang, J.L. OsMADS25 regulates root system development via auxin signalling in rice. Plant J. 2018, 95, 1004–1022. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Zhou, H.Z.; Wu, Y.; Zhang, H.; Lin, J.; Jiang, X.; He, Q.; Zhu, J.; Li, Y.; Yu, H.; et al. OsSPL3, an SBP-Domain Protein, Regulates Crown Root Development in Rice. Plant Cell 2019, 31, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, J.; Li, P.; Wang, Y.; Ge, Z.; Qian, J.; Fan, Y.; Ni, J.; Xu, Y.; Yang, Z. Integrating GWAS and Gene Expression Analysis Identifies Candidate Genes for Root Morphology Traits in Maize at the Seedling Stage. Genes 2019, 10, 773. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloglu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Hall, T.; Biosciences, I.; Carlsbad, C. BioEdit: An important software for molecular biology. GERF Bull Biosci. 2011, 2, 60–61. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [PubMed]
- Rogers, E.D.; Benfey, P.N. Regulation of plant root system architecture: Implications for crop advancement. Curr. Opin. Biotechnol. 2015, 32, 93–98. [Google Scholar] [CrossRef]
- Bishopp, A.J.P.; Lynch, J.P. The hidden half of crop yields. Nat. Plants 2015, 1, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Bray, A.L.; Topp, C.N. The Quantitative Genetic Control of Root Architecture in Maize. Plant Cell Physiol. 2018, 59, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Uga, Y.; Sugimoto, K.; Ogawa, S.; Rane, J.; Ishitani, M.; Hara, N.; Kitomi, Y.; Inukai, Y.; Ono, K.; Kanno, N.; et al. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 2013, 45, 1097–1102. [Google Scholar] [CrossRef]
- Mu, X.H.; Chen, F.J.; Wu, Q.P.; Chen, Q.W.; Wang, J.F.; Yuan, L.X.; Mi, G.H. Genetic improvement of root growth increases maize yield via enhanced post-silking nitrogen uptake. Eur. J. Agron. 2015, 63, 55–61. [Google Scholar] [CrossRef]
- Yu, L.H.; Miao, Z.Q.; Qi, G.F.; Wu, J.; Cai, X.T.; Mao, J.L.; Xiang, C.B. MADS-Box Transcription Factor AGL21 Regulates Lateral Root Development and Responds to Multiple External and Physiological Signals. Mol. Plant 2014, 7, 1653–1669. [Google Scholar] [CrossRef]
- Gamuyao, R.; Chin, J.H.; Pariasca-Tanaka, J.; Pesaresi, P.; Catausan, S.; Dalid, C.; Slamet-Loedin, I.; Tecson-Mendoza, E.M.; Wissuwa, M.; Heuer, S. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 2012, 488, 535–539. [Google Scholar] [CrossRef]
- Li, P.C.; Chen, F.J.; Cai, H.G.; Liu, J.C.; Pan, Q.C.; Liu, Z.G.; Gu, R.L.; Mi, G.H.; Zhang, F.S.; Yuan, L.X. A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. J. Exp. Bot. 2015, 66, 3175–3188. [Google Scholar] [CrossRef]
- Sun, S.; Zhou, Y.; Chen, J.; Shi, J.P.; Zhao, H.M.; Zhao, H.N.; Song, W.B.; Zhang, M.; Cui, Y.; Dong, X.M.; et al. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nat. Genet. 2018, 50, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Poorter, H.; Fiorani, F.; Pieruschka, R.; Wojciechowski, T.; van der Putten, W.H.; Kleyer, M.; Schurr, U.; Postma, J. Pampered inside, pestered outside? Differences and similarities between plants growing in controlled conditions and in the field. New Phytol. 2016, 212, 838–855. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.A.; Pound, M.P.; Bennett, M.J.; Wells, D.M. Uncovering the hidden half of plants using new advances in root phenotyping. Curr. Opin. Biotechnol. 2019, 55, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Vigouroux, Y.; Goodman, M.M.; Sanchez, G.J.; Buckler, E.; Doebley, J. A single domestication for maize shown by multilocus microsatellite genotyping. Proc. Natl. Acad. Sci. USA 2002, 99, 6080–6084. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, M.; Tenaillon, M.I.; Bi, I.V.; Schroeder, S.G.; Sanchez-Villeda, H.; Doebley, J.F.; Gaut, B.S.; McMullen, M.D. A large-scale screen for artificial selection in maize identifies candidate agronomic loci for domestication and crop improvement. Plant Cell 2005, 17, 2859–2872. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Pan, S.; Kennedy, A.; Melzer, R. MADS-box genes and crop domestication: The jack of all traits. J. Exp. Bot. 2018, 69, 1447–1469. [Google Scholar] [CrossRef]
- Zhao, Q.; Weber, A.L.; McMullen, M.D.; Guill, K.; Doebley, J. MADS-box genes of maize: Frequent targets of selection during domestication. Genet. Res. 2011, 93, 65–75. [Google Scholar] [CrossRef]
- Singh, J.; Gezan, S.A.; Vallejos, C.E. Developmental Pleiotropy Shaped the Roots of the Domesticated Common Bean (Phaseolus vulgaris). Plant Physiol. 2019, 180, 1467–1479. [Google Scholar] [CrossRef]
- York, L.M.; Galindo-Castañeda, T.; Schussler, J.R.; Lynch, J.P. Evolution of US maize (Zea mays L.) root architectural and anatomical phenes over the past 100 years corresponds to increased tolerance of nitrogen stress. J. Exp. Bot. 2015, 66, 2347–2358. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Lin, Z.; Wang, J.; Xu, M.; Lai, J.; Yu, J.; Lin, Z. The genetic architecture of nodal root number in maize. Plant J. 2018, 93, 1032–1044. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Upstream | 5′UTR | Coding Region | 3′UTR | Entire Region |
|---|---|---|---|---|---|
| Total length of amplicons(bp) | 1543 | 1019 | 4755 | 112 | 7429 |
| Number of all of the sequence vanants | 171 | 198 | 823 | 7 | 1199 |
| Frequency of all of the sequence variants | 0.111 | 0.194 | 0.173 | 0.063 | 0.161 |
| Number of nucleotide substitutions (bp) | 141 | 164 | 706 | 7 | 1018 |
| Frequency of polymorphic sites per bp | 0.091 | 0.161 | 0.148 | 0.063 | 0.137 |
| Number of InDels | 30 | 34 | 117 | 0 | 181 |
| Number of InDel sites | 77 | 155 | 331 | 0 | 563 |
| Average InDel length | 3 | 6 | 3.504 | 0 | 3.89 |
| Frequency of indels per bp | 0.019 | 0.033 | 0.025 | 0 | 0.024 |
| π × 1000 | 37.59 | 17.83 | 15.27 | 0.34 | 17.57 |
| θ × 1000 | 56.75 | 40.68 | 38.45 | 10.02 | 39.96 |
| Tajima’s D | −1.016 | −1.696 | −1.858 * | −1.899 * | −1.731 * |
| Fu and Li’s D * | −3.849 ** | −6.084 ** | −7.9 ** | −6.006 ** | −7.454 ** |
| Fu and Li’s F * | −2.831 * | −4.503 ** | −5.355 ** | −5.459 ** | −5.008 ** |
| Trait | Marker | Alleles | p Value | −lg(P) | R2 | Type |
|---|---|---|---|---|---|---|
| LRL | SNP1319 | C/T | 0.000536 | 3.271 | 0.044 | Intron7 |
| LRL | SNP1357 | A/T | 0.000226 | 3.645 | 0.05 | Intron7 |
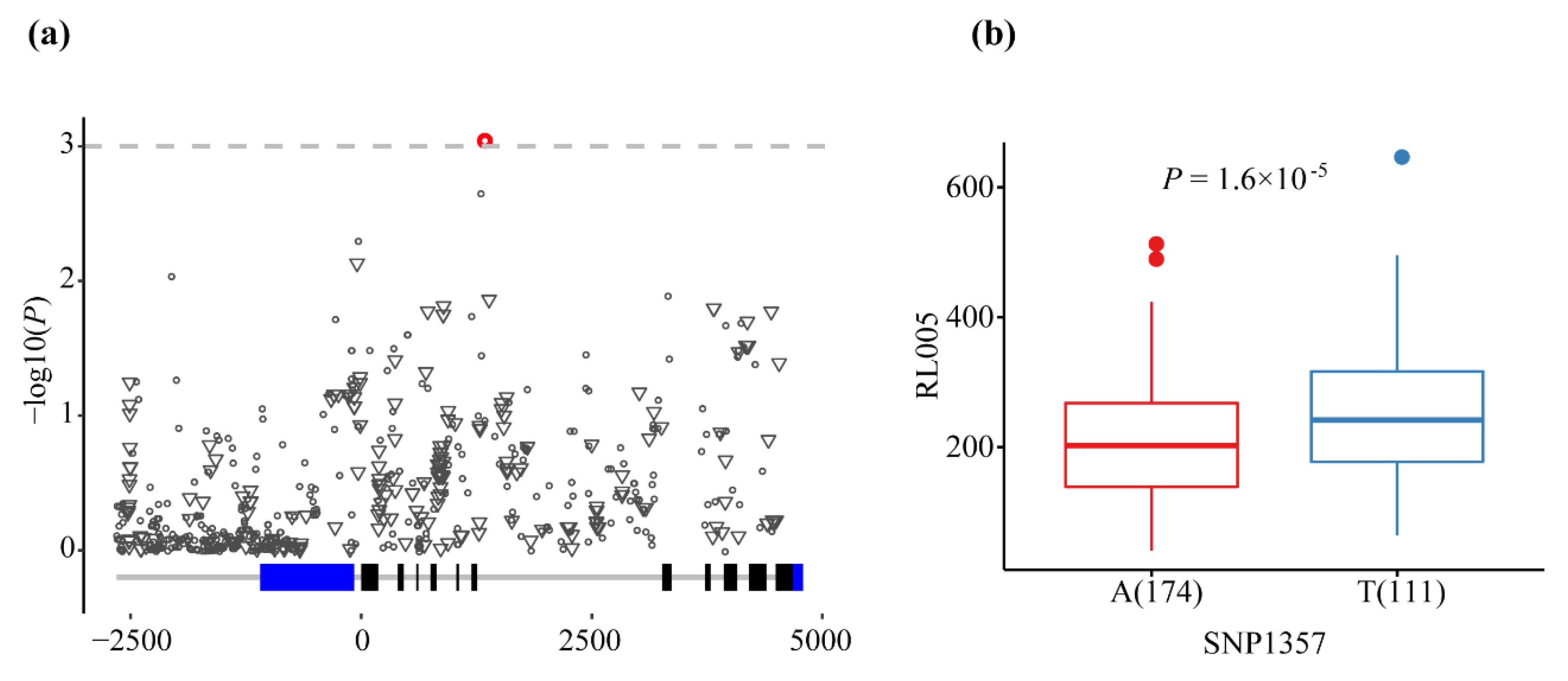
| RL005 | SNP1357 | A/T | 0.000896 | 3.048 | 0.039 | Intron7 |
| TRL | SNP1319 | C/T | 0.00033 | 3.481 | 0.046 | Intron7 |
| TRL | SNP1357 | A/T | 0.000232 | 3.635 | 0.048 | Intron7 |
| PRL | InDel-1576 | -/A | 0.000907 | 3.042 | 0.041 | Upstream |
| PRL | SNP723 | A/G | 0.000223 | 3.653 | 0.051 | Intron4 |
| PRL | SNP726 | G/A | 0.000223 | 3.653 | 0.051 | Intron4 |
| PRL | InDel727 | TT/-- | 0.000234 | 3.631 | 0.05 | Intron4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Ge, Z.; Wang, H.; Wei, J.; Wang, Y.; Xu, Y.; Yang, Z.; Xu, C. Nucleotide Diversity and Association Analysis of ZmMADS60 with Root Length in the Maize Seedling Stage. Agronomy 2020, 10, 342. https://doi.org/10.3390/agronomy10030342
Li P, Ge Z, Wang H, Wei J, Wang Y, Xu Y, Yang Z, Xu C. Nucleotide Diversity and Association Analysis of ZmMADS60 with Root Length in the Maize Seedling Stage. Agronomy. 2020; 10(3):342. https://doi.org/10.3390/agronomy10030342
Chicago/Turabian StyleLi, Pengcheng, Zhenzhen Ge, Houmiao Wang, Jie Wei, Yunyun Wang, Yang Xu, Zefeng Yang, and Chenwu Xu. 2020. "Nucleotide Diversity and Association Analysis of ZmMADS60 with Root Length in the Maize Seedling Stage" Agronomy 10, no. 3: 342. https://doi.org/10.3390/agronomy10030342
APA StyleLi, P., Ge, Z., Wang, H., Wei, J., Wang, Y., Xu, Y., Yang, Z., & Xu, C. (2020). Nucleotide Diversity and Association Analysis of ZmMADS60 with Root Length in the Maize Seedling Stage. Agronomy, 10(3), 342. https://doi.org/10.3390/agronomy10030342