Genome-Wide Association and Genomic Prediction for Fry Color in Potato



Abstract
1. Introduction
2. Results
2.1. Genotyping and Phenotyping Potato Lines
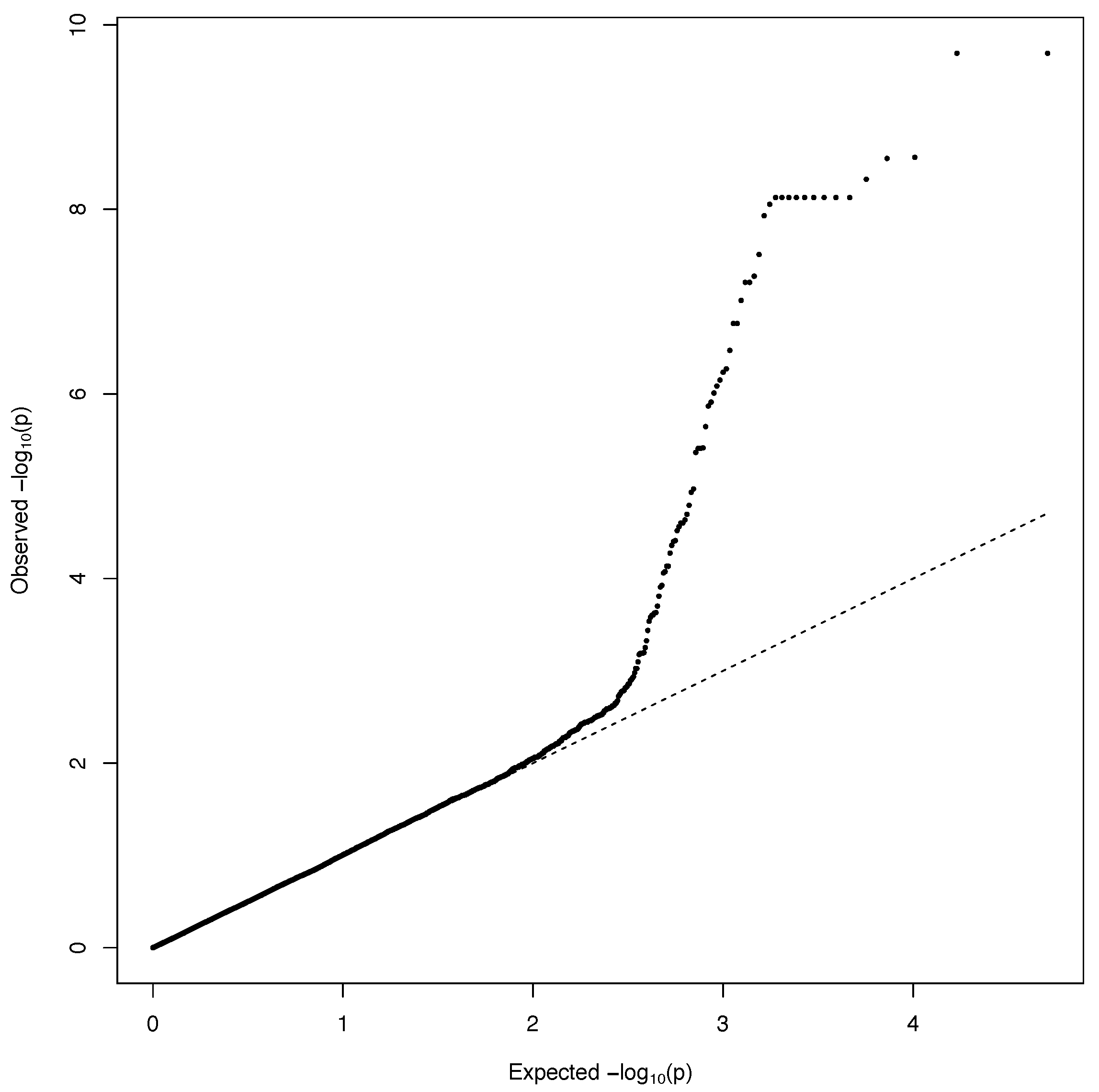
2.2. Genome-Wide Association Analysis
2.3. Using Genome-Wide Variants to Predict Fry Color
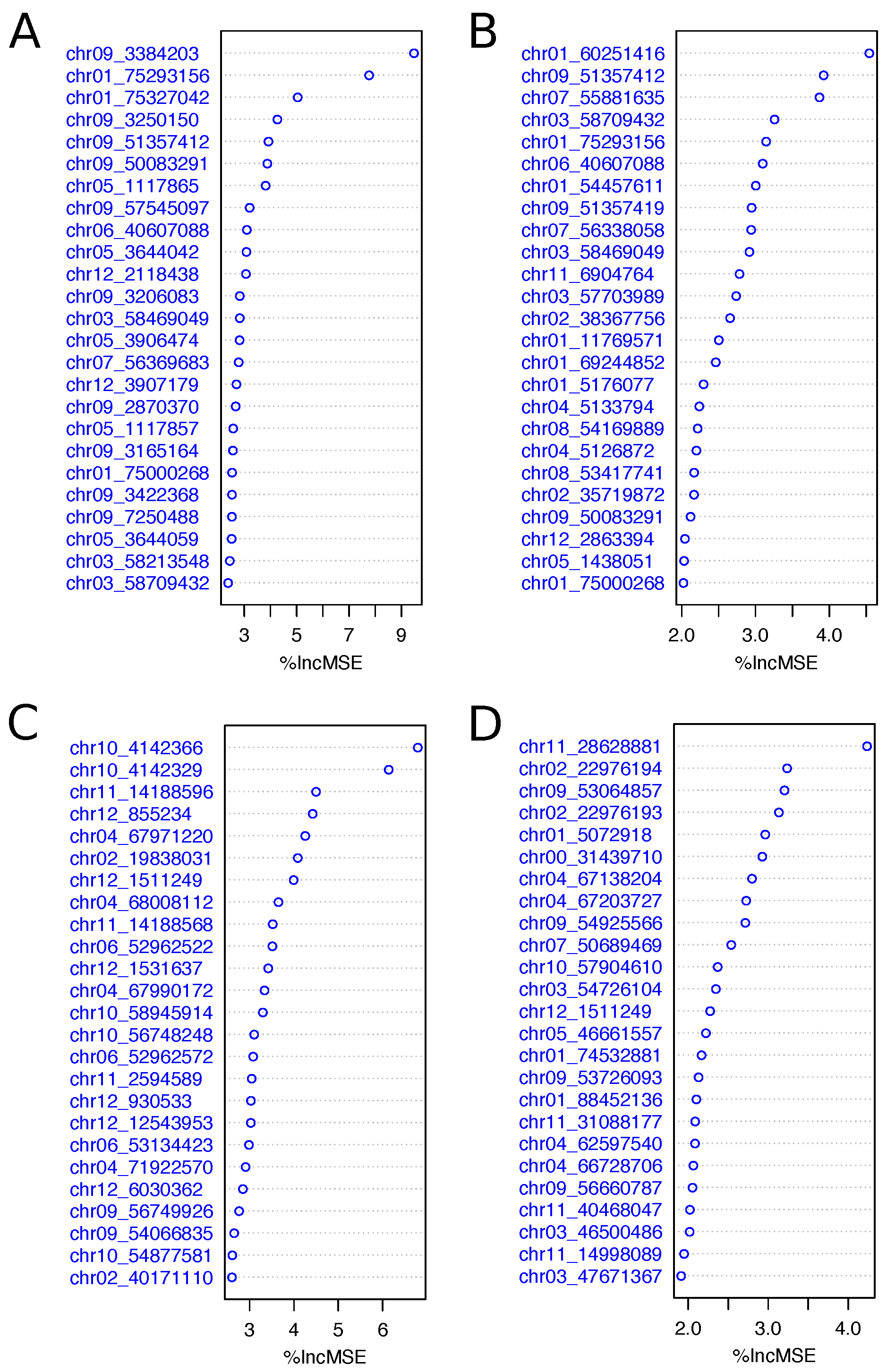
2.4. Using Selected Variants to Predict Fry Color
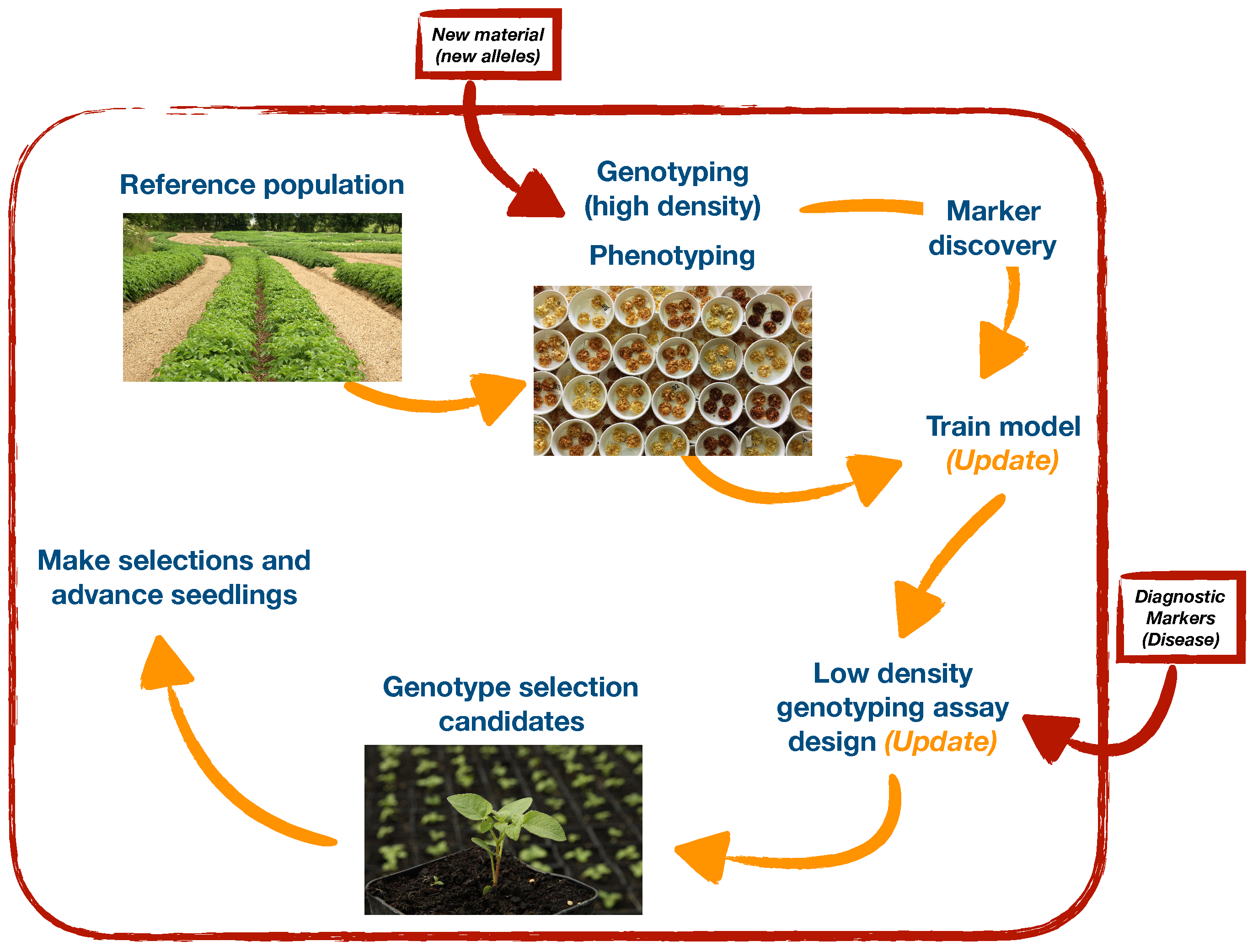
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Phenotyping Training and Test Panels
5.2. Genotyping Training and Testing Panels
5.3. GWAS to Identify QTL Associated with Fry Color
5.4. Genomic Prediction of Fry Color
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slater, A.T.; Cogan, N.O.; Forster, J.W.; Hayes, B.J.; Daetwyler, H.D. Improving genetic gain with genomic selection in autotetraploid potato. J. Abbr. 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Slater, A.T.; Wilson, G.M.; Cogan, N.O.; Forster, J.W.; Hayes, B.J. Improving the analysis of low heritability complex traits for enhanced genetic gain in potato. Theor. Appl. Genet. 2014, 127, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Annicchiarico, P.; Nazzicari, N.; Li, X.; Wei, Y.; Pecetti, L.; Brummer, E.C. Accuracy of genomic selection for alfalfa biomass yield in different reference populations. BMC Genom. 2015, 16, 1020. [Google Scholar] [CrossRef] [PubMed]
- F‘e, D.; Pedersen, M.G.; Jensen, C.S.; Jensen, J. Genomic prediction of seedling root length in maize (Zea mays L.). Plant J. 2015, 83, 903–912. [Google Scholar]
- Pace, J.; Yu, X.; Lubberstedt, T. Genetic and environmental variation in a commercial breeding program of perennial ryegrass. Crop Sci. 2015, 55, 631–640. [Google Scholar]
- Ramstein, G.; Evans, J.; Kaeppler, S.; Mitchell, R.; Vogel, K.; Buell, C.; Casler, M.T. Accuracy of genomic prediction in switchgrass (Panicum virgatum L.) improved by accounting for linkage disequilibrium. G3 2016, 6, 1049–1062. [Google Scholar] [CrossRef]
- Sverrisdottie, E.; Byrne, S.; Sundmark, E.H.R.; Johnsen, H.O.; Kirk, H.G.; Asp, T.; Janss, L.; Nielsen, K.L. Genomic prediction of starch content and chipping quality in tetraploid potato using genotyping-by-sequencing. Theor. Appl. Genet. 2017, 130, 2091–2108. [Google Scholar] [CrossRef]
- Björn, B.; Keizer, P.L.; Paulo, M.J.; Visser, R.G.; van Eeuwijk, F.A.; van Eck, H.J. Identification of agronomically important QTL in tetraploid potato cultivars using a marker-trait association analysis. Theor. Appl. Genet. 2014, 127, 731–748. [Google Scholar]
- Bradshaw, J.E.; Hackett, C.A.; Pande, B.; Waugh, R.; Bryan, G.J. QTL mapping of yield, agronomic and quality traits in tetraploid potato (Solanum tuberosum subsp. tuberosum). Theor. Appl. Genet. 2008, 116, 193–211. [Google Scholar] [CrossRef]
- Björn, B.; Paulo, M.J.; Mank, R.A.; Van Eck, H.J.; Van Eeuwijk, F.A. Association mapping of quality traits in potato (Solanum tuberosum L.). Euphytica 2008, 161, 47–60. [Google Scholar]
- Menéndez, C.M.; Ritter, E.; Schäfer-Pregl, R.; Walkemeier, B.; Kalde, A.; Salamini, F.; Gebhardt, C. Cold sweetening in diploid potato: Mapping quantitative trait loci and candidate genes. Genetics 2002, 162, 1423–1434. [Google Scholar] [PubMed]
- Li, L.; Tacke, E.; Hofferbert, H.R.; Lübeck, J.; Strahwald, J.; Draffehn, A.M.; Walkemeier, B.; Gebhardt, C. Validation of candidate gene markers for marker-assisted selection of potato cultivars with improved tuber quality. Theor. Appl. Genet. 2013, 126, 1039–1052. [Google Scholar] [CrossRef]
- Fischer, M.; Schreiber, L.; Colby, T.; Kuckenberg, M.; Tacke, E.; Hofferbert, H.R.; Schmidt, J.; Gebhardt, C. Novel candidate genes influencing natural variation in potato tuber cold sweetening identified by comparative proteomics and association mapping. BMC Plant Biol. 2013, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, L.; Nader-Nieto, A.C.; Schönhals, E.M.; Walkemeier, B.; Gebhardt, C. SNPs in genes functional in starch-sugar interconversion associate with natural variation of tuber starch and sugar content of potato (Solanum tuberosum L.). G3 Genes Genom. Genet. 2014, 4, 1797–1811. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Paulo, M.J.; Strahwald, J.; Lübeck, J.; Hofferbert, H.R.; Tacke, E.; Junghans, H.; Wunder, J.; Draffehn, A.; van Eeuwijk, F.; et al. Natural DNA variation at candidate loci is associated with potato chip color, tuber starch content, yield and starch yield. Theor. Appl. Genet. 2008, 116, 1167–1181. [Google Scholar] [CrossRef][Green Version]
- Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef]
- Uitdewilligen, J.G.; Wolters, A.M.A.; Bjorn, B.; Borm, T.J.; Visser, R.G.; van Eck, H.J. A next-generation sequencing method for genotyping-by-sequencing of highly heterozygous autotetraploid potato. PLoS ONE 2013, 8, e62355. [Google Scholar] [CrossRef]
- Li, X.; Wei, Y.; Acharya, A.; Jiang, Q.; Kang, J.; Brummer, E.C. A saturated genetic linkage map of autotetraploid alfalfa (Medicago sativa L.) developed using genotyping-by-sequencing is highly syntenous with the Medicago truncatula genome. G3 Genes Genom. Genet. 2014, 4, 1971–1979. [Google Scholar]
- Tan, B.; Grattapaglia, D.; Martins, G.S.; Ferreira, K.Z.; Sundberg, B.; Ingvarsson, P.K. Evaluating the accuracy of genomic prediction of growth and wood traits in two eucalyptus species and their f 1 hybrids. BMC Plant Biol. 2017, 17, 110. [Google Scholar] [CrossRef]
- Wang, Y.; Mette, M.F.; Miedaner, T.; Gottwald, M.; Wilde, P.; Reif, J.C.; Zhao, Y. The accuracy of prediction of genomic selection in elite hybrid rye populations surpasses the accuracy of marker-assisted selection and is equally augmented by multiple field evaluation locations and test years. BMC Genom. 2014, 15, 556. [Google Scholar] [CrossRef]
- Nielsen, N.H.; Jahoor, A.; Jensen, J.D.; Orabi, J.; Cericola, F.; Edriss, V.; Jensen, J. Genomic prediction of seed quality traits using advanced barley breeding lines. PLoS ONE 2016, 11, 0164494. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Tetens, J.; Seefried, F.-R.; Lichtner, P.; Thaller, G. The impact of genetic relationship information on genomic breeding values in german holstein cattle. Genet. Sel. Evol. 2010, 42, 5. [Google Scholar] [CrossRef] [PubMed]
- Riggio, V.; Abdel-Aziz, M.; Matika, O.; Moreno, C.; Carta, A.; Bishop, S. Accuracy of genomic prediction within and across populations for nematode resistance and body weight traits in sheep. Animal 2014, 8, 520–528. [Google Scholar] [CrossRef]
- Rosyara, U.R.; De Jong, W.S.; Douches, D.S.; Endelman, J.B. Software for genome-wide association studies in autopolyploids and its application to potato. Plant Genome 2016, 9. [Google Scholar] [CrossRef]
- Campbell, N.R.; Harmon, S.A.; Narum, S.R. Genotyping-in-thousands by sequencing (gt-seq): A cost effective snp genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour. 2015, 15, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (gbs) approach for high diversity species. PLoS ONE 2011, 6, 19379. [Google Scholar] [CrossRef]
- Buffalo, V. Scythe—A Bayesian Adapter Trimmer Version 0.994 Beta. Available online: https://github.com/vsbuffalo/scythe (accessed on 7 November 2015).
- Joshi, F. Sickle—A Windowed Adaptive Trimming Tool for Fastq Files Using Quality. 2011. Available online: https://github.com/ucdavis-bioinformatics/sickle (accessed on 7 November 2015).
- Joshi, F. Sabre—A Barcode Demultiplexing and Trimming Tool for Fastq Files. 2011. Available online: https://github.com/najoshi/sabre (accessed on 7 November 2015).
- Li, H.; Durbin, R. Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- De Pristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M. A framework for variation discovery and genotyping using next-generation dna sequencing data. Nat Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Park, T.; Casella, G. The bayesian lasso. J. Am. Stat. Assoc. 2008, 103, 681–686. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- De los Campos, G.; Pérez-Rodríguez, P. Bayesian Generalized Linear Regression. Available online: https://rdrr.io/cran/BGLR/ (accessed on 12 December 2019).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OTF | C + 104d | C + 237d | C + 111d | C + 183d | C + 230d | |
|---|---|---|---|---|---|---|
| OTF | 1 | |||||
| C + 104d | 0.92 | 1 | ||||
| C + 237d | 0.84 | 0.88 | 1 | |||
| C + 111d | 0.84 | 0.91 | 0.86 | 1 | ||
| C + 183d | 0.78 | 0.87 | 0.83 | 0.91 | 1 | |
| C + 230d | 0.77 | 0.86 | 0.83 | 0.90 | 0.91 | 1 |
| Population | OTF | LTS |
|---|---|---|
| 2015 | 192 | 192 |
| 2016 | 45 | 88 |
| 2017 | 219 | 219 |
| test panel | 56 | - |
| Chrom | bp | −log10(p) |
|---|---|---|
| chr04 | 67971220 | 6.23 |
| chr04 | 68008112 | 5.91 |
| chr10 | 49770199 | 6.15 |
| chr10 | 53208176 | 5.87 |
| chr10 | 54783863 | 8.33 |
| chr10 | 54800561 | 7.93 |
| chr10 | 54966754 | 9.69 |
| chr10 | 55285966 | 9.69 |
| chr10 | 55358563 | 6.09 |
| chr10 | 55639153 | 8.06 |
| chr10 | 55889244 | 8.55 |
| chr10 | 55921128 | 6.47 |
| chr10 | 56255214 | 8.13 |
| chr10 | 56255215 | 8.13 |
| chr10 | 56372149 | 8.13 |
| chr10 | 56514796 | 7.21 |
| chr10 | 56514804 | 7.21 |
| chr10 | 56748248 | 8.13 |
| chr10 | 56903243 | 8.13 |
| chr10 | 57498778 | 7.01 |
| chr10 | 57627246 | 8.56 |
| chr10 | 57699003 | 7.28 |
| chr10 | 57778018 | 7.51 |
| chr10 | 57780687 | 6.01 |
| chr10 | 57837337 | 6.27 |
| chr10 | 58032412 | 8.13 |
| chr10 | 58082084 | 8.13 |
| chr10 | 58263956 | 6.76 |
| chr10 | 58263973 | 6.76 |
| chr10 | 58305552 | 8.13 |
| chr10 | 58403467 | 8.13 |
| Train Set | Test Set | Markers | rrBLUP | BayesA | Bayesian Lasso | Random Forest |
|---|---|---|---|---|---|---|
| off-the-field | ||||||
| 2015 | 2016 | 26,045 | 0.26 (0.43) | 0.25 (0.45) | 0.26 (0.49) | 0.11 (0.30) |
| 2015 | 2017 | 38,041 | 0.75 (1.05) | 0.75 (1.13) | 0.75 (1.17) | 0.68 (1.38) |
| 2017 | 2015 | 38,041 | 0.77 (1.29) | 0.77 (1.38) | 0.77 (1.40) | 0.72 (1.73) |
| 2017 | 2016 | 28,655 | 0.48 (1.05) | 0.44 (1.03) | 0.46 (1.06) | 0.45 (1.24) |
| 2016 | 2017 | 28,655 | 0.56 (3.26) | 0.55 (3.16) | 0.48 (2.94) | 0.32 (1.54) |
| 2016 | 2015 | 26,045 | 0.49 (2.59) | 0.49 (2.44) | 0.50 (2.88) | 0.43 (2.10) |
| 2015 | Test panel | 35,242 | 0.67 (0.77) | 0.67 (0.86) | 0.67 (0.82) | 0.60 (1.10) |
| 2016 | Test panel | 26,869 | 0.48 (2.31) | 0.47 (2.38) | 0.46 (2.30) | 0.39 (1.72) |
| 2017 | Test panel | 38,582 | 0.66 (0.70) | 0.68 (0.79) | 0.68 (0.87) | 0.64 (1.11) |
| low-temperature-storage | ||||||
| 2015 | 2016 | 29,421 | 0.36 (0.70) | 0.34 (0.75) | 0.34 (0.76) | 0.26 (0.82) |
| 2015 | 2017 | 38,041 | 0.65 (1.03) | 0.65 (1.12) | 0.66 (1.22) | 0.61 (1.55) |
| 2017 | 2015 | 38,041 | 0.62 (1.14) | 0.65 (1.33) | 0.66 (1.39) | 0.64 (2.02) |
| 2017 | 2016 | 32,315 | 0.29 (0.64) | 0.29 (0.71) | 0.29 (0.76) | 0.24 (0.91) |
| 2016 | 2017 | 32,315 | 0.50 (5.49) | 0.47 (1.45) | 0.47 (1.45) | 0.44 (2.39) |
| 2016 | 2015 | 29,421 | 0.61 (8.96) | 0.52 (2.22) | 0.52 (2.08) | 0.46 (3.42) |
| 10 | 25 | 50 | 100 | 500 | 5000 | ||
|---|---|---|---|---|---|---|---|
| off-the-field | |||||||
| 2015 to 2017 | Selected | 0.59 (0.96) | 0.62 (1.05) | 0.62 (0.96) | 0.65 (0.99) | 0.68 (1.11) | 0.72 (1.18) |
| Random | 0.27 (0.69) | 0.38 (0.74) | 0.46 (0.77) | 0.55 (0.85) | 0.67 (0.94) | 0.74 (1.03) | |
| 2017 to 2015 | Selected | 0.50 (0.69) | 0.59 (0.81) | 0.60 (0.79) | 0.67 (0.84) | 0.69 (0.90) | 0.74 (1.03) |
| Random | 0.32 (1.06) | 0.43 (1.06) | 0.50 (1.03) | 0.57 (1.04) | 0.67 (1.09) | 0.76 (1.24) | |
| low-temperature-storage | |||||||
| 2015 to 2017 | Selected | 0.50 (0.85) | 0.49 (0.77) | 0.51 (0.76) | 0.50 (0.79) | 0.59 (0.83) | 0.66 (0.99) |
| Random | 0.24 (0.70) | 0.31 (0.70) | 0.38 (0.74) | 0.45 (0.83) | 0.57 (0.93) | 0.62 (0.98) | |
| 2017 to 2015 | Selected | 0.51 (1.20) | 0.54 (1.12) | 0.49 (1.02) | 0.53 (1.17) | 0.55 (1.06) | 0.61 (1.05) |
| Random | 0.26 (1.15) | 0.36 (1.06) | 0.45 (1.11) | 0.50 (1.10) | 0.58 (1.13) | 0.62 (1.13) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byrne, S.; Meade, F.; Mesiti, F.; Griffin, D.; Kennedy, C.; Milbourne, D. Genome-Wide Association and Genomic Prediction for Fry Color in Potato. Agronomy 2020, 10, 90. https://doi.org/10.3390/agronomy10010090
Byrne S, Meade F, Mesiti F, Griffin D, Kennedy C, Milbourne D. Genome-Wide Association and Genomic Prediction for Fry Color in Potato. Agronomy. 2020; 10(1):90. https://doi.org/10.3390/agronomy10010090
Chicago/Turabian StyleByrne, Stephen, Fergus Meade, Francesca Mesiti, Denis Griffin, Colum Kennedy, and Dan Milbourne. 2020. "Genome-Wide Association and Genomic Prediction for Fry Color in Potato" Agronomy 10, no. 1: 90. https://doi.org/10.3390/agronomy10010090
APA StyleByrne, S., Meade, F., Mesiti, F., Griffin, D., Kennedy, C., & Milbourne, D. (2020). Genome-Wide Association and Genomic Prediction for Fry Color in Potato. Agronomy, 10(1), 90. https://doi.org/10.3390/agronomy10010090