
Highly Branched poly(5-amino-1-pentanol-co-1,4-butanediol diacrylate) for High Performance Gene Transfection


 ,
, 
Abstract
: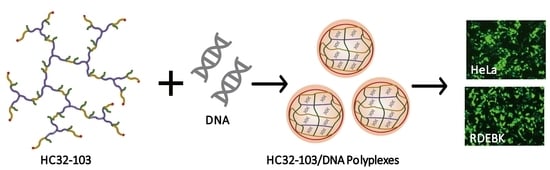
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
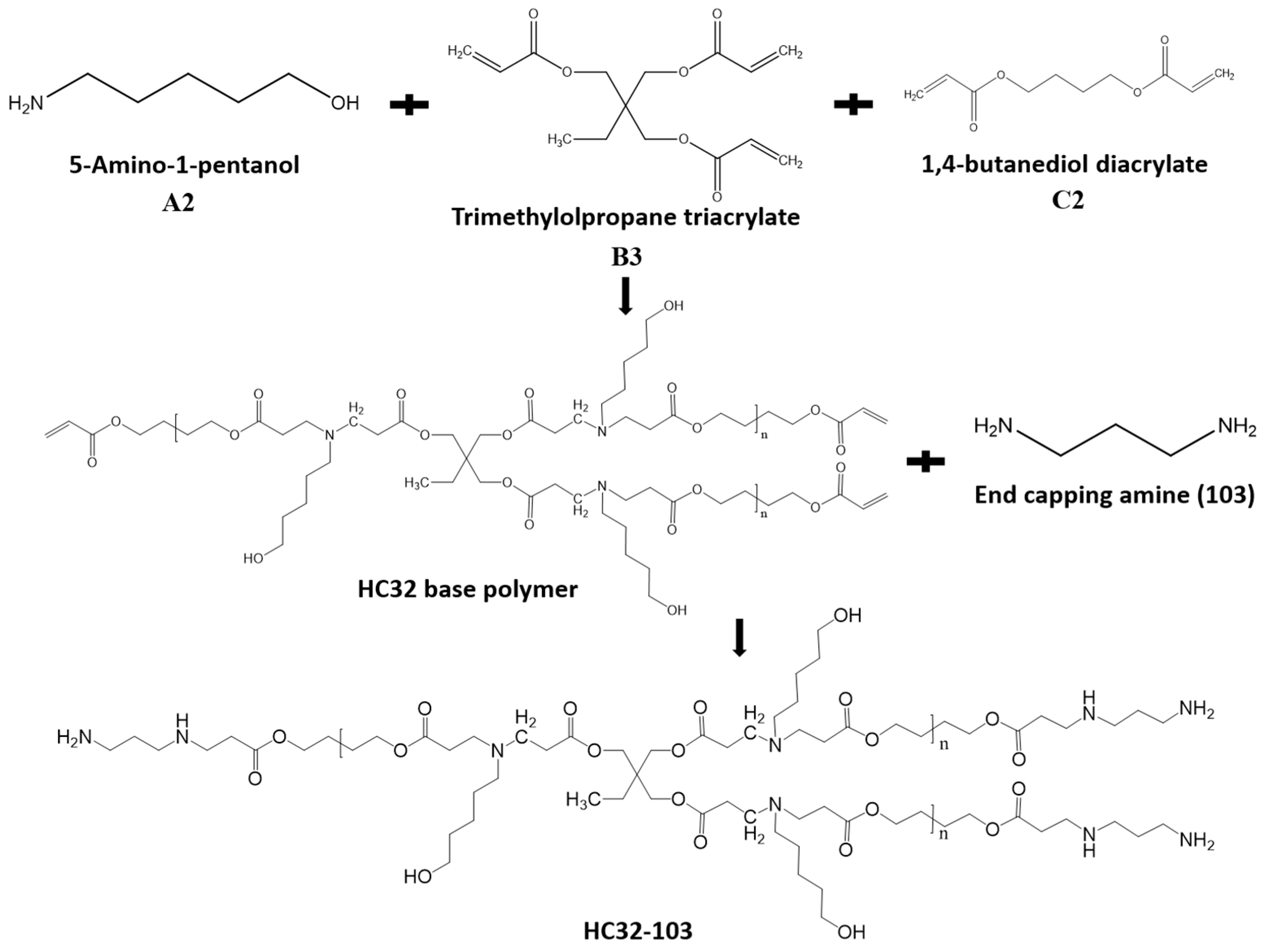
2.2. Polymer Synthesis
2.3. Molecular Weight Measurements
2.4. Proton Nuclear Magnetic Resonance (1H NMR)
2.5. Acid-Base Titration
2.6. Picogreen Assays
2.7. Size and Zeta Potential of Polyplexes
2.8. Polyplex Stability Measurements
2.9. Polymer Degradation
2.10. DNA Release
2.11. Cell Culture
2.12. Transfection Experiments
2.13. Evaluation of Gene Transfection Efficiency Using Gluciferase Assays and GFP Expression
2.14. Cytotoxicity Assessment after Transfection Using Alamarblue Assay
2.15. Statistics
3. Results and Discussion
3.1. Polymers Synthesis and Characterization
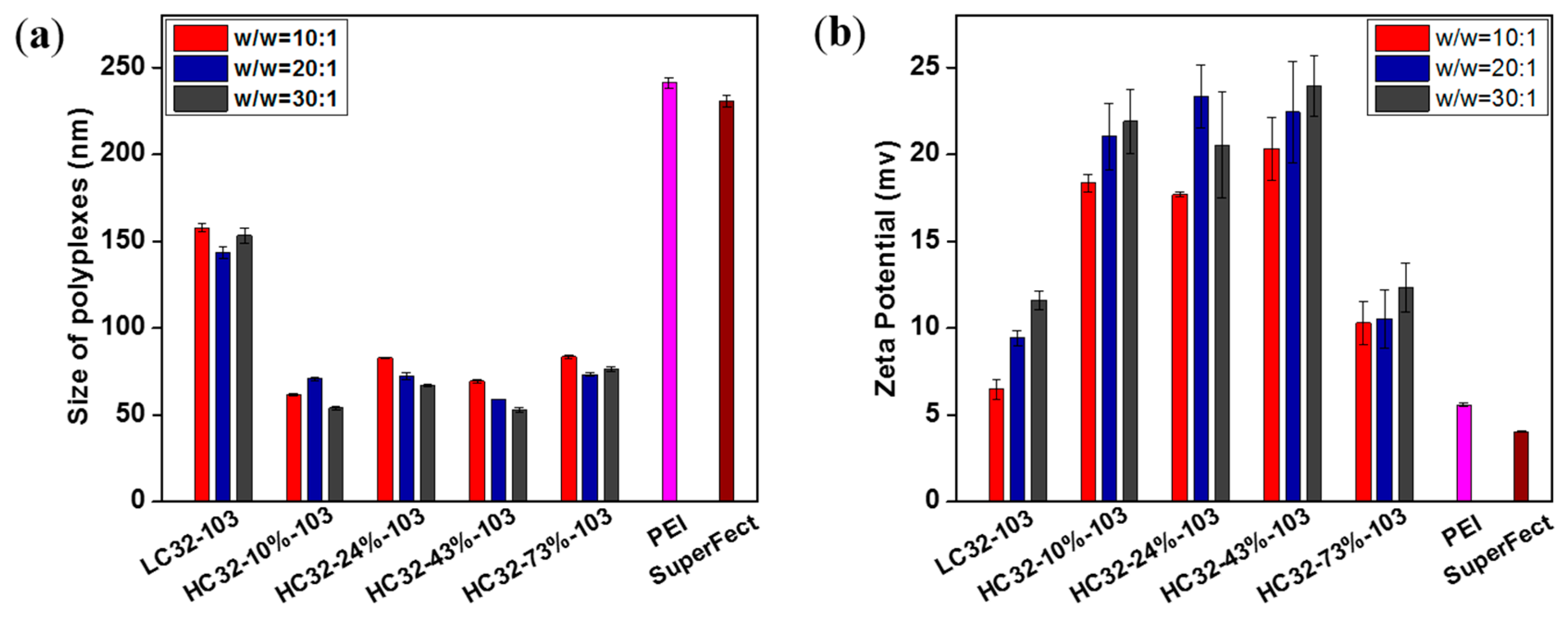
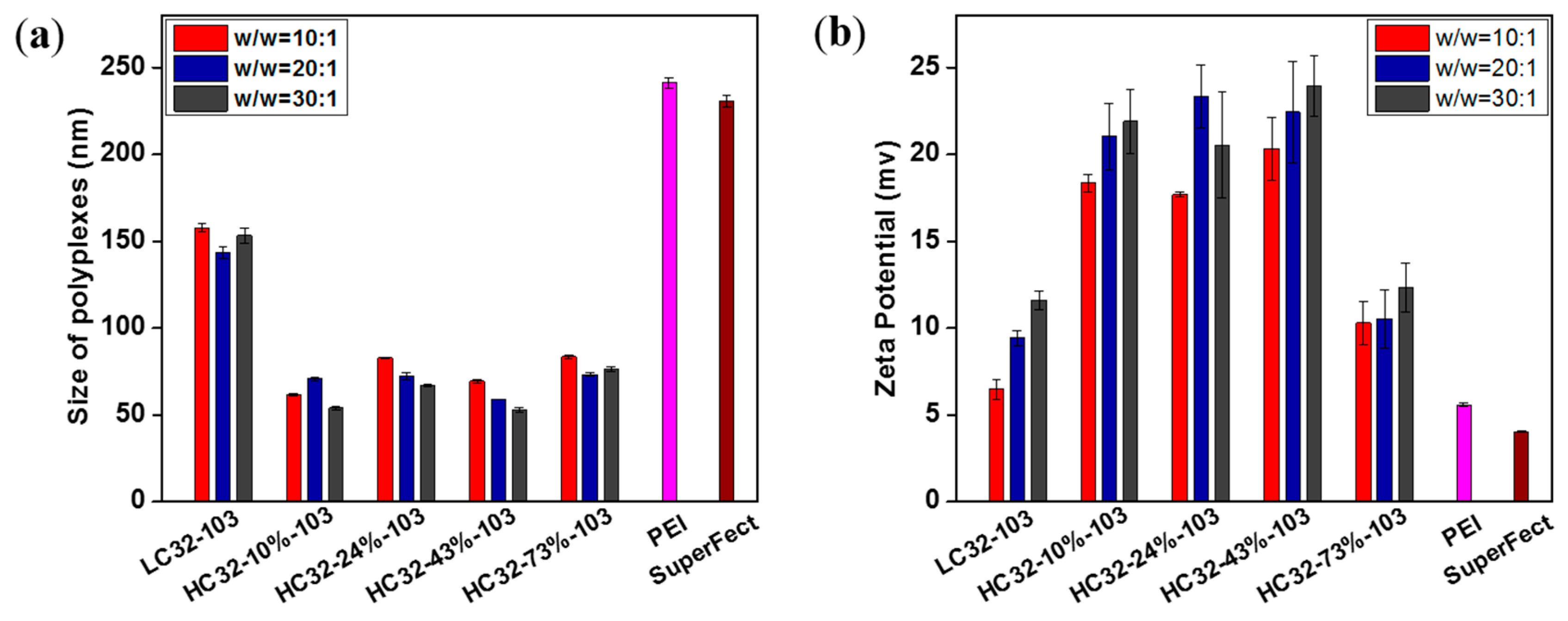
3.2. Biophysical Properties of LC32/DNA and HC32/DNA Polyplexes
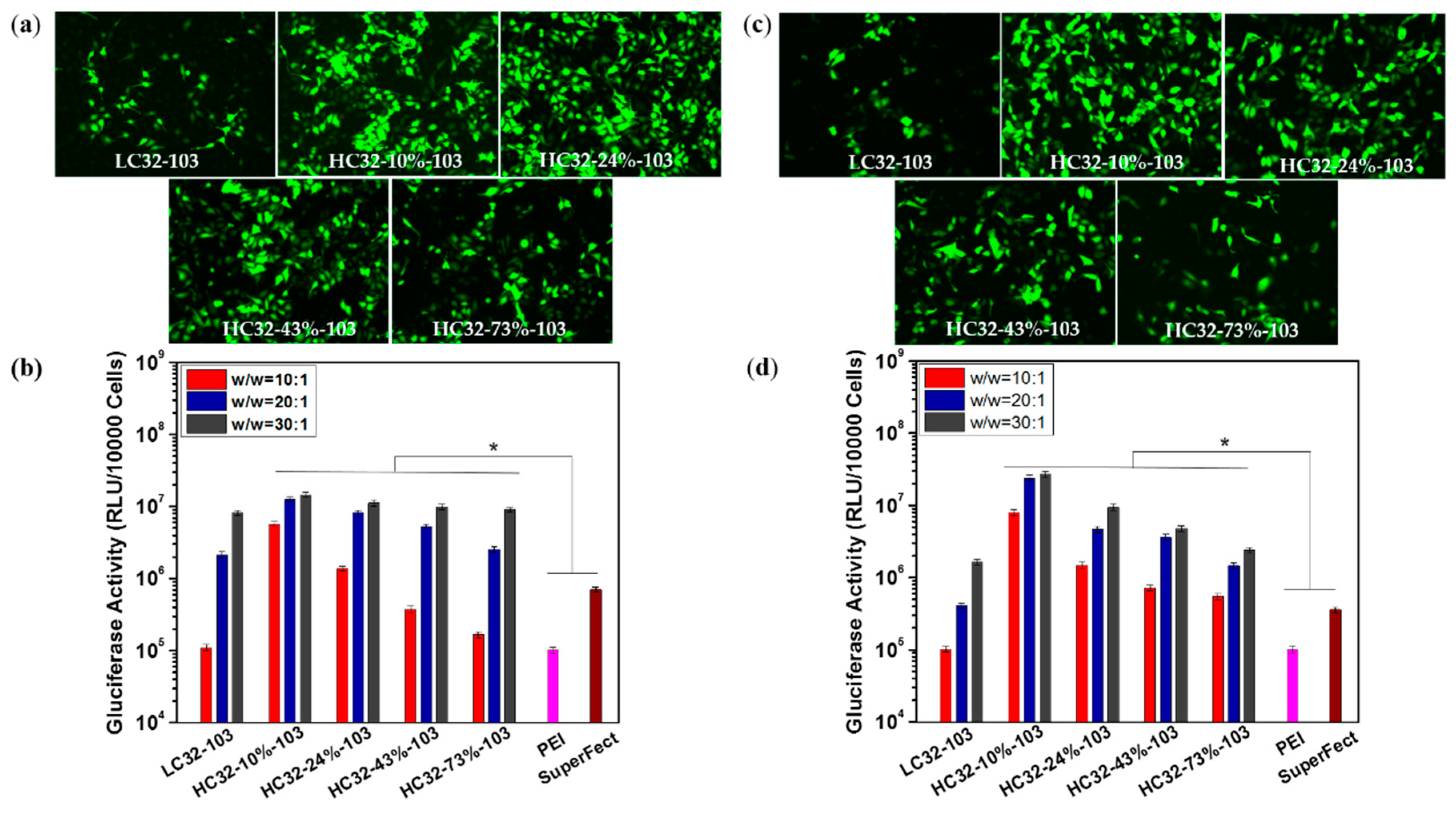
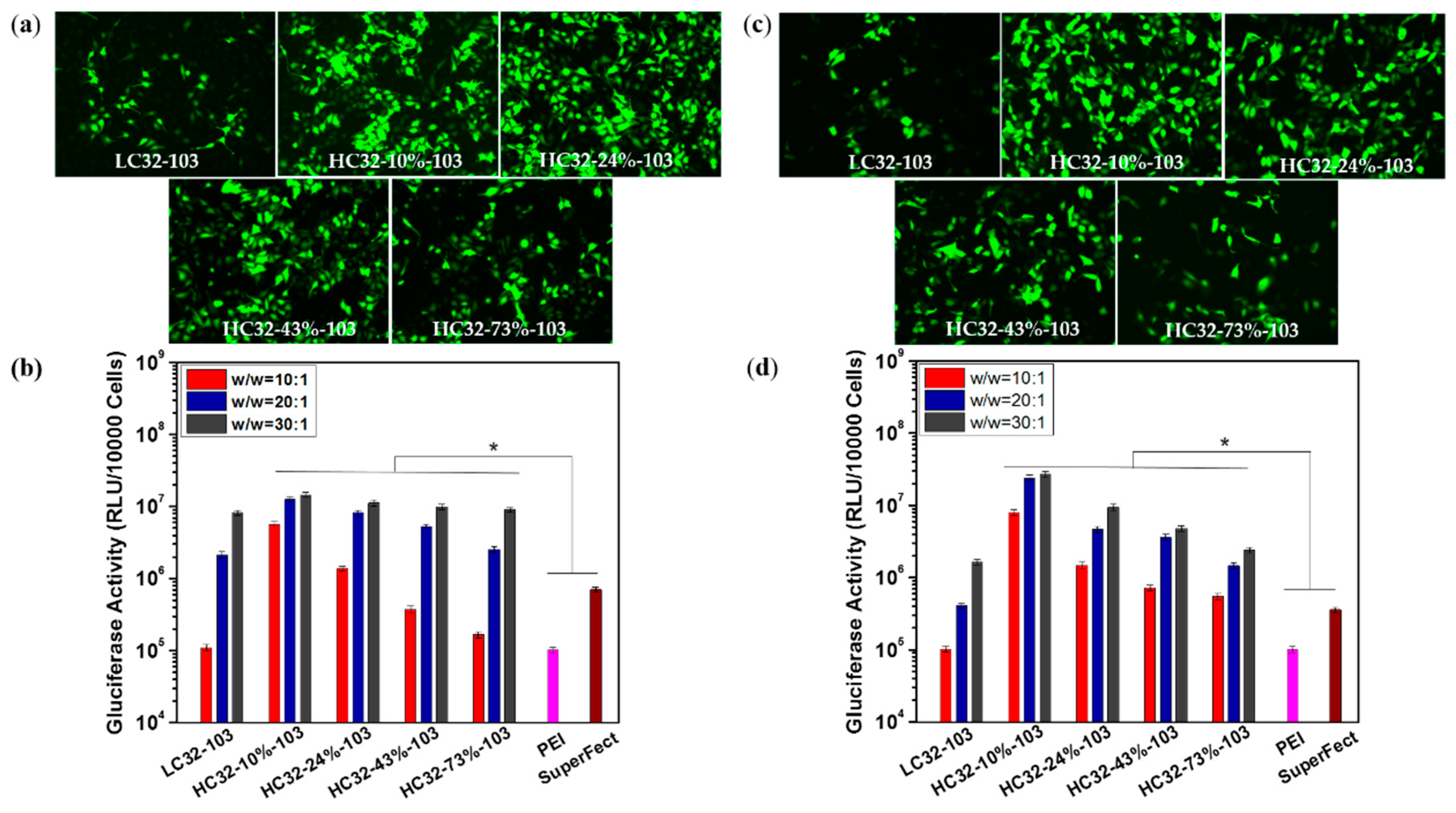
3.3. Transfection Efficiency and Cytotoxicity Measurement
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.C. Immune responses to gene therapy vectors: Influence on vector function and effector mechanisms. Gene Ther. 2004, 11, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Baum, C.; Kustikova, O.; Modlich, U.; Li, Z.; Fehse, B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum. Gene Ther. 2006, 17, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Waehler, R.; Russell, S.J.; Curiel, D.T. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 2007, 8, 573–587. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Bouard, D.; Alazard-Dany, D.; Cosset, F.L. Viral vectors: From virology to transgene expression. Br. J. Pharmacol. 2009, 157, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy—An overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef] [PubMed]
- Pack, D.W.; Hoffman, A.S.; Pun, S.; Stayton, P.S. Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 2005, 4, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, M.A.; Simanek, E.E. Nonviral vectors for gene delivery. Chem. Rev. 2009, 109, 259–302. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.M.; Langer, R. Degradable poly(β-amino esters): Synthesis, characterization, and self-assembly with plasmid DNA. J. Am. Chem. Soc. 2000, 122, 10761–10768. [Google Scholar] [CrossRef]
- Green, J.J.; Langer, R.; Anderson, D.G. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc. Chem. Res. 2008, 41, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.G.; Lynn, D.M.; Langer, R. Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angew. Chem. Int. Ed. 2003, 42, 3153–3158. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.Y.; Green, J.J. Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for sirna and DNA delivery to human brain cancer. Adv. Healthc. Mater. 2013, 2, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.J.; Ketola, T.M.; Tzeng, S.Y.; Sunshine, J.C.; Urtti, A.; Lemmetyinen, H.; Vuorimaa-Laukkanen, E.; Yliperttula, M.; Green, J.J. The effect and role of carbon atoms in poly(β-amino ester)s for DNA binding and gene delivery. J. Am. Chem. Soc. 2013, 135, 6951–6957. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Akanda, M.I.; Li, D.; Kozielski, K.L.; Green, J.J. Effects of base polymer hydrophobicity and end-group modification on polymeric gene delivery. Biomacromolecules 2011, 12, 3592–3600. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, J.C.; Peng, D.Y.; Green, J.J. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol. Pharm. 2012, 9, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Cazares, H.; Tzeng, S.Y.; Young, N.P.; Abutaleb, A.O.; Quinones-Hinojosa, A.; Green, J.J. Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo. ACS Nano 2014, 8, 5141–5153. [Google Scholar] [CrossRef] [PubMed]
- Green, J.J.; Zugates, G.T.; Tedford, N.C.; Huang, Y.H.; Griffith, L.G.; Lauffenburger, D.A.; Sawicki, J.A.; Langer, R.; Anderson, D.G. Combinatorial modification of degradable polymers enables transfection of human cells comparable to adenovirus. Adv. Mater. 2007, 19, 2836–2842. [Google Scholar] [CrossRef]
- Eltoukhy, A.A.; Chen, D.L.; Alabi, C.A.; Langer, R.; Anderson, D.G. Degradable terpolymers with alkyl side chains demonstrate enhanced gene delivery potency and nanoparticle stability. Adv. Mater. 2013, 25, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Green, J.J.; Dinio, T.; Keung, L.; Cho, S.W.; Park, H.; Langer, R.; Anderson, D.G. Gene delivery to human adult and embryonic cell-derived stem cells using biodegradable nanoparticulate polymeric vectors. Gene Ther. 2009, 16, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Cho, S.W.; Son, S.M.; Bogatyrev, S.R.; Singh, D.; Green, J.J.; Mei, Y.; Park, S.; Bhang, S.H.; Kim, B.S.; et al. Genetic engineering of human stem cells for enhanced angiogenesis using biodegradable polymeric nanoparticles. Proc. Natl. Acad. Sci. USA 2010, 107, 3317–3322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.F.; Huang, W.; Liu, J.Y.; Zhu, X.Y.; Yan, D.Y. Self-assembly of hyperbranched polymers and its biomedical applications. Adv. Mater. 2010, 22, 4567–4590. [Google Scholar] [CrossRef] [PubMed]
- Newland, B.; Aied, A.; Pinoncely, A.V.; Zheng, Y.; Zhao, T.; Zhang, H.; Niemeier, R.; Dowd, E.; Pandit, A.; Wang, W. Untying a nanoscale knotted polymer structure to linear chains for efficient gene delivery in vitro and to the brain. Nanoscale 2014, 6, 7526–7533. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.Y.; Xiong, W.; Wei, J.Z.; Wang, Y.; Chen, X.S.; Jing, X.B.; Zhu, Q.Y. Gene transfection of hyperbranched PEI grafted by hydrophobic amino acid segment PBLG. Biomaterials 2007, 28, 2899–2907. [Google Scholar] [CrossRef] [PubMed]
- Kadlecova, Z.; Baldi, L.; Hacker, D.; Wurm, F.M.; Klok, H.A. Comparative study on the in vitro cytotoxicity of linear, dendritic, and hyperbranched polylysine analogues. Biomacromolecules 2012, 13, 3127–3137. [Google Scholar] [CrossRef] [PubMed]
- Kadlecova, Z.; Rajendra, Y.; Matasci, M.; Baldi, L.; Hacker, D.L.; Wurm, F.M.; Klok, H.A. DNA delivery with hyperbranched polylysine: A comparative study with linear and dendritic polylysine. J. Control. Release 2013, 169, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.Y.; Zhang, H.; Newland, B.; Aied, A.; Zhou, D.Z.; Wang, W.X. Significance of branching for transfection: Synthesis of highly branched degradable functional poly(dimethylaminoethyl methacrylate) by vinyl oligomer combination. Angew. Chem. Int. Ed. 2014, 53, 6095–6100. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Narain, R. The effect of molecular weight, compositions and lectin type on the properties of hyperbranched glycopolymers as non-viral gene delivery systems. Biomaterials 2012, 33, 3990–4001. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.B.; Kim, S.M.; Lee, Y.; Lee, W.K.; Yang, T.G.; Lee, M.J.; Suh, H.; Park, J.S. Cationic hyperbranched poly(amino ester): A novel class of DNA condensing molecule with cationic surface, biodegradable three-dimensional structure, and tertiary amine groups in the interior. J. Am. Chem. Soc. 2001, 123, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, D.C.; Ma, Y.X.; Tang, G.P.; Wang, S.; He, C.B.; Chung, T.S.; Goh, S. Novel poly(amino ester)s obtained from michael addition polymerizations of trifunctional amine monomers with diacrylates: Safe and efficient DNA carriers. Chem. Commun. 2003, 2630–2631. [Google Scholar] [CrossRef]
- Zhou, D.Z.; Cutlar, L.; Gao, Y.S.; Wang, W.; O’Keeffe-Ahern, J.; McMahon, S.; Duarte, B.; Larcher, F.; Rodriguez, B.J.; Greiser, U. The transition from linear to highly branched poly(β-amino ester)s: Branching matters for gene delivery. Sci. Adv. 2016, 2, e1600102. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, M.; Zhou, D.; Ng, S.; Ahern, J.O.; Alshehri, F.; Gao, Y.; Pierucci, L.; Greiser, U.; Wang, W. Highly Branched poly(5-amino-1-pentanol-co-1,4-butanediol diacrylate) for High Performance Gene Transfection. Polymers 2017, 9, 161. https://doi.org/10.3390/polym9050161
Zeng M, Zhou D, Ng S, Ahern JO, Alshehri F, Gao Y, Pierucci L, Greiser U, Wang W. Highly Branched poly(5-amino-1-pentanol-co-1,4-butanediol diacrylate) for High Performance Gene Transfection. Polymers. 2017; 9(5):161. https://doi.org/10.3390/polym9050161
Chicago/Turabian StyleZeng, Ming, Dezhong Zhou, Singwei Ng, Jonathan O′Keeffe Ahern, Fatma Alshehri, Yongsheng Gao, Luca Pierucci, Udo Greiser, and Wenxin Wang. 2017. "Highly Branched poly(5-amino-1-pentanol-co-1,4-butanediol diacrylate) for High Performance Gene Transfection" Polymers 9, no. 5: 161. https://doi.org/10.3390/polym9050161
APA StyleZeng, M., Zhou, D., Ng, S., Ahern, J. O., Alshehri, F., Gao, Y., Pierucci, L., Greiser, U., & Wang, W. (2017). Highly Branched poly(5-amino-1-pentanol-co-1,4-butanediol diacrylate) for High Performance Gene Transfection. Polymers, 9(5), 161. https://doi.org/10.3390/polym9050161