
Copolymerization of Norbornene and Norbornadiene Using a cis-Selective Bimetallic W-Based Catalytic System

 ,
,  ,
, 

Abstract
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Catalytic Experiments
3. Results and Discussion
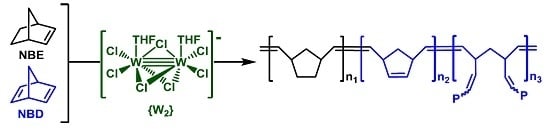
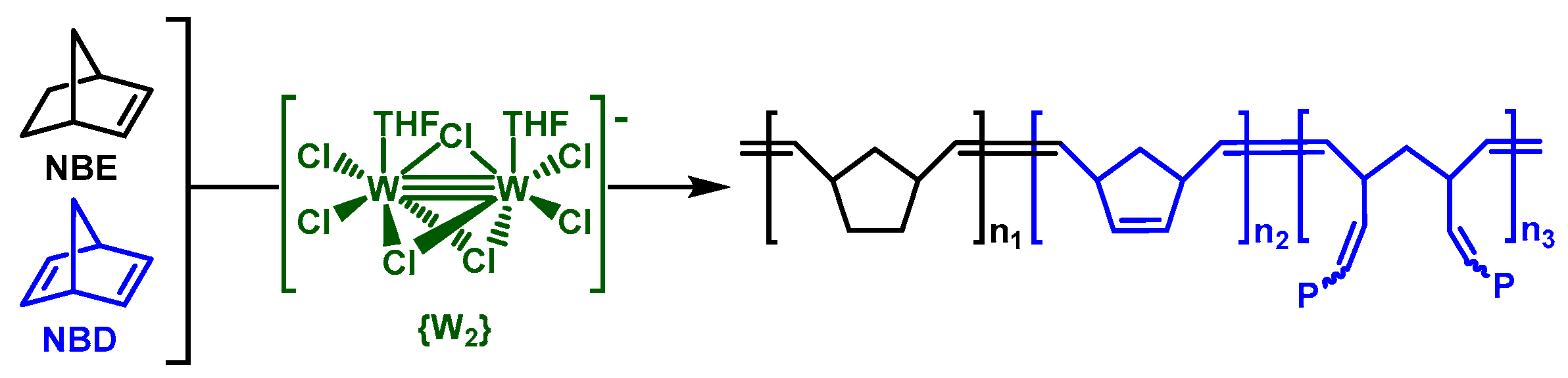
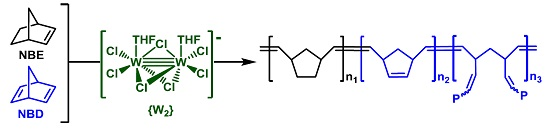
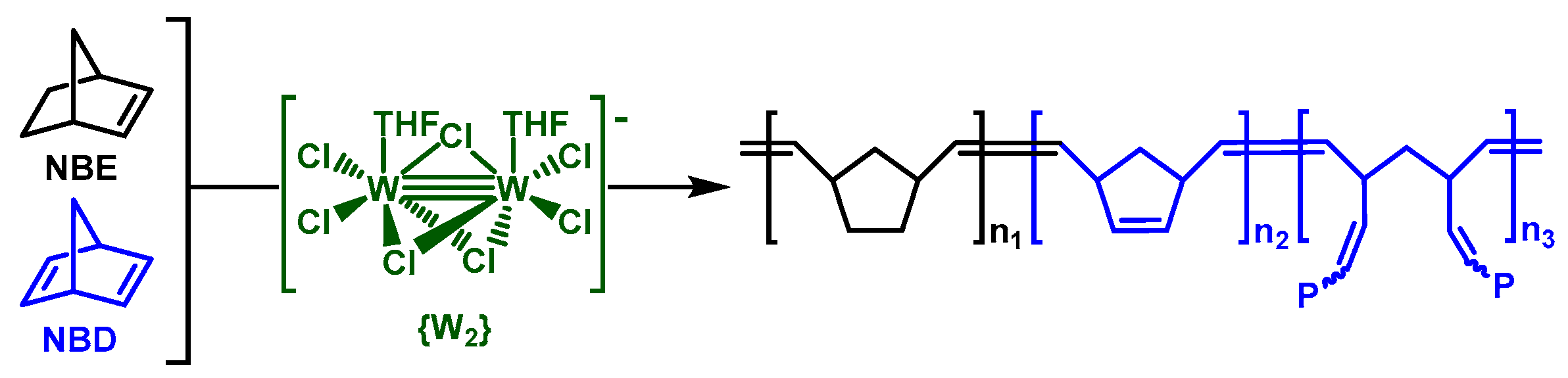
3.1. Catalyst and Catalytic Reactions
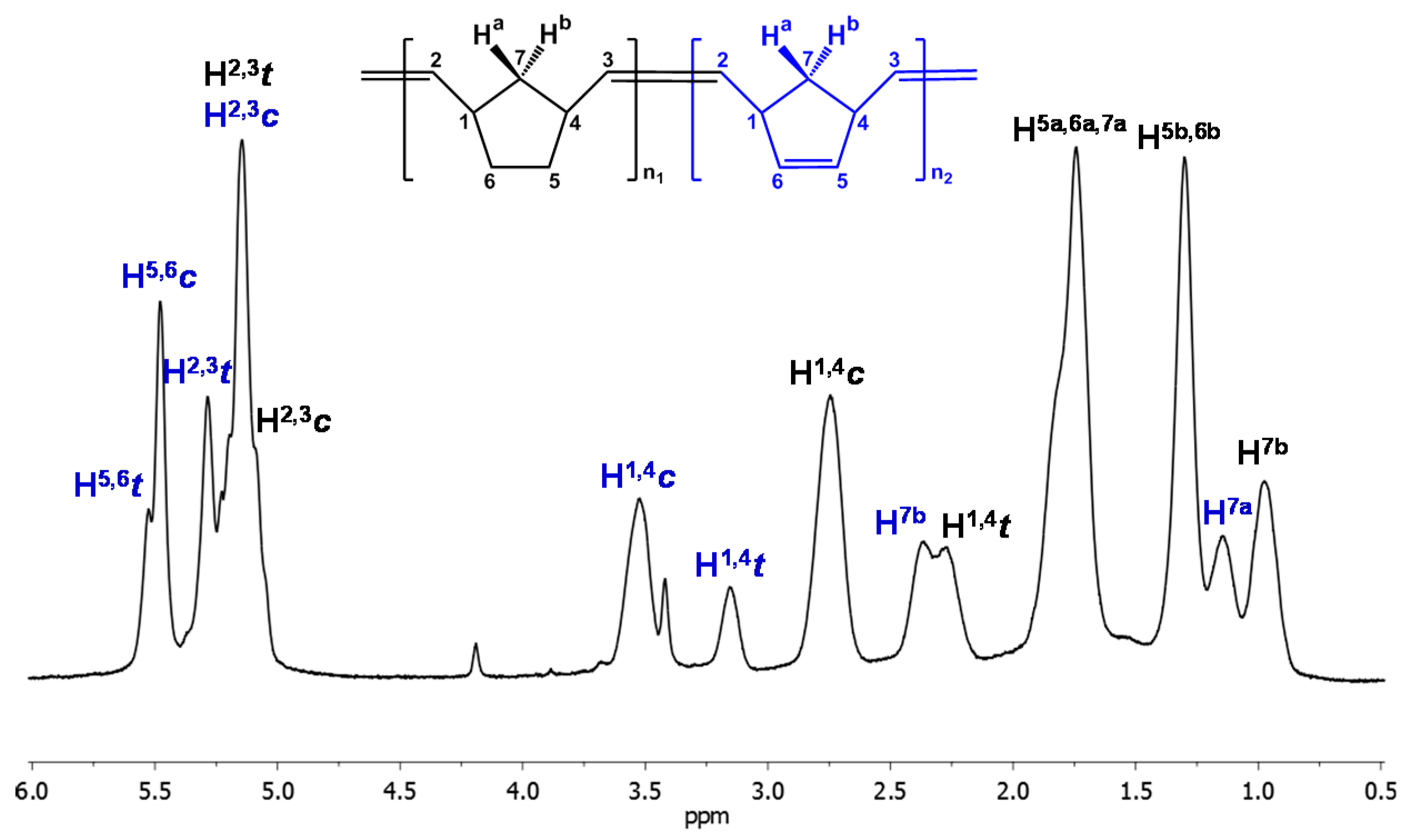
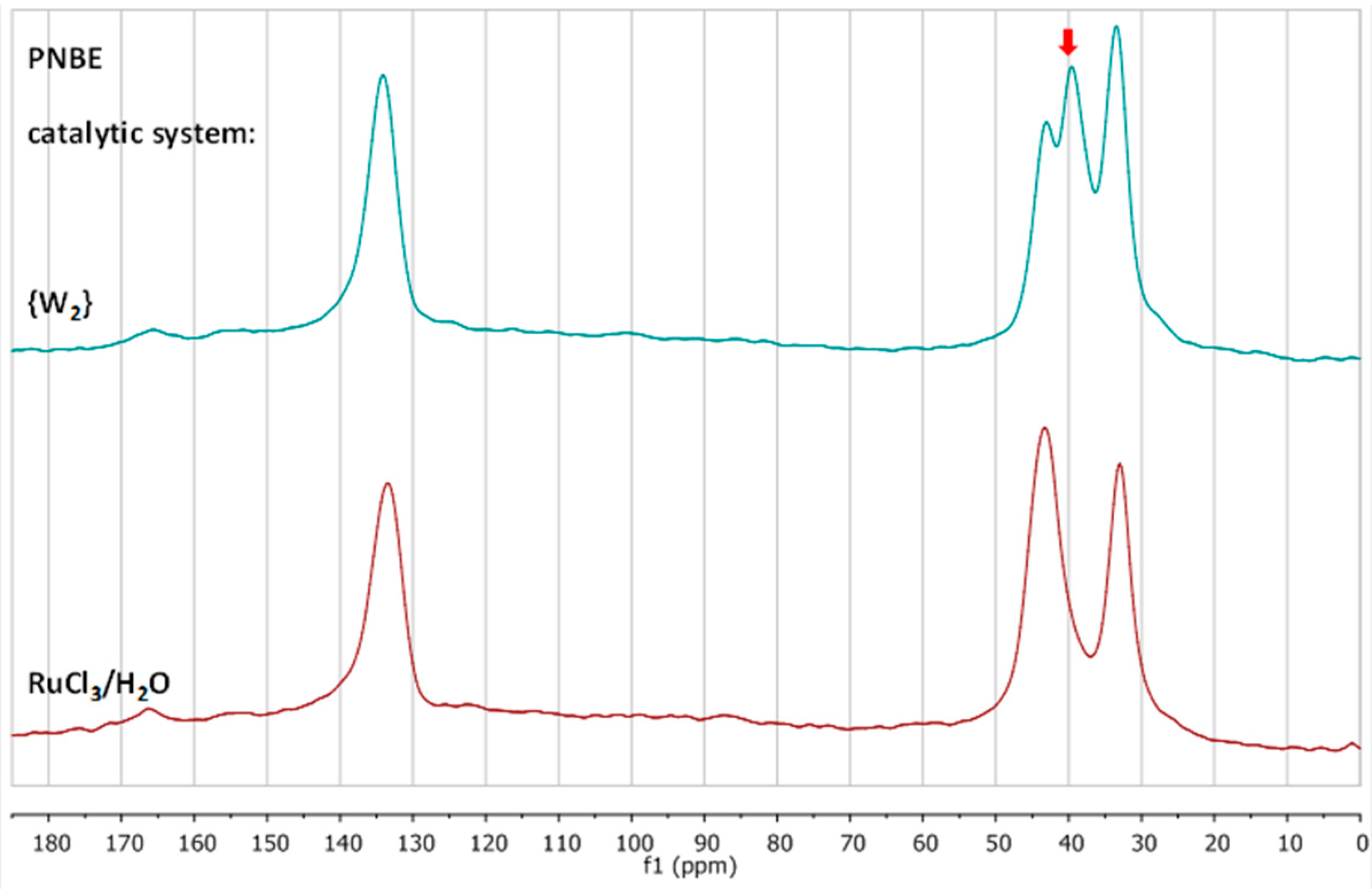
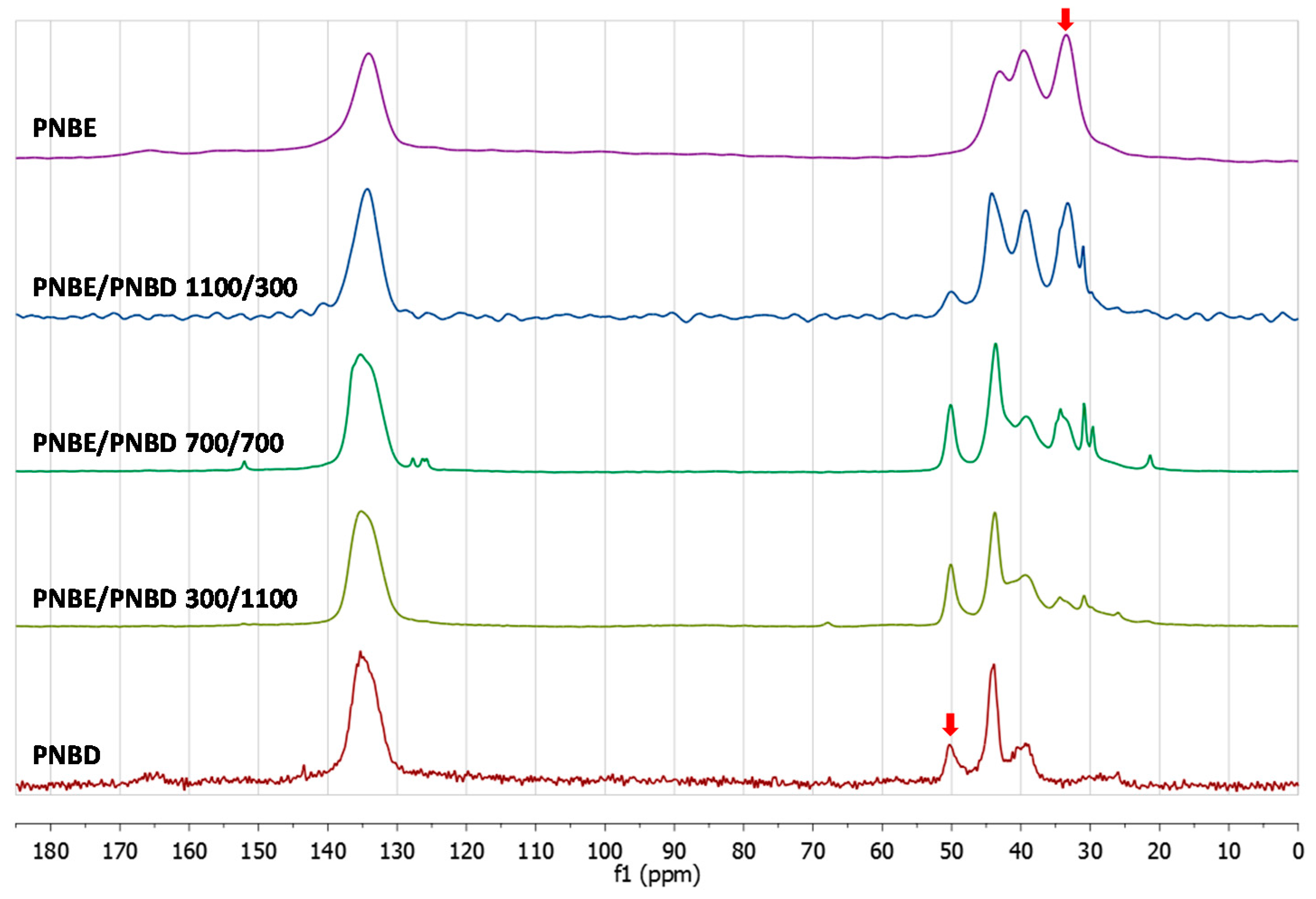
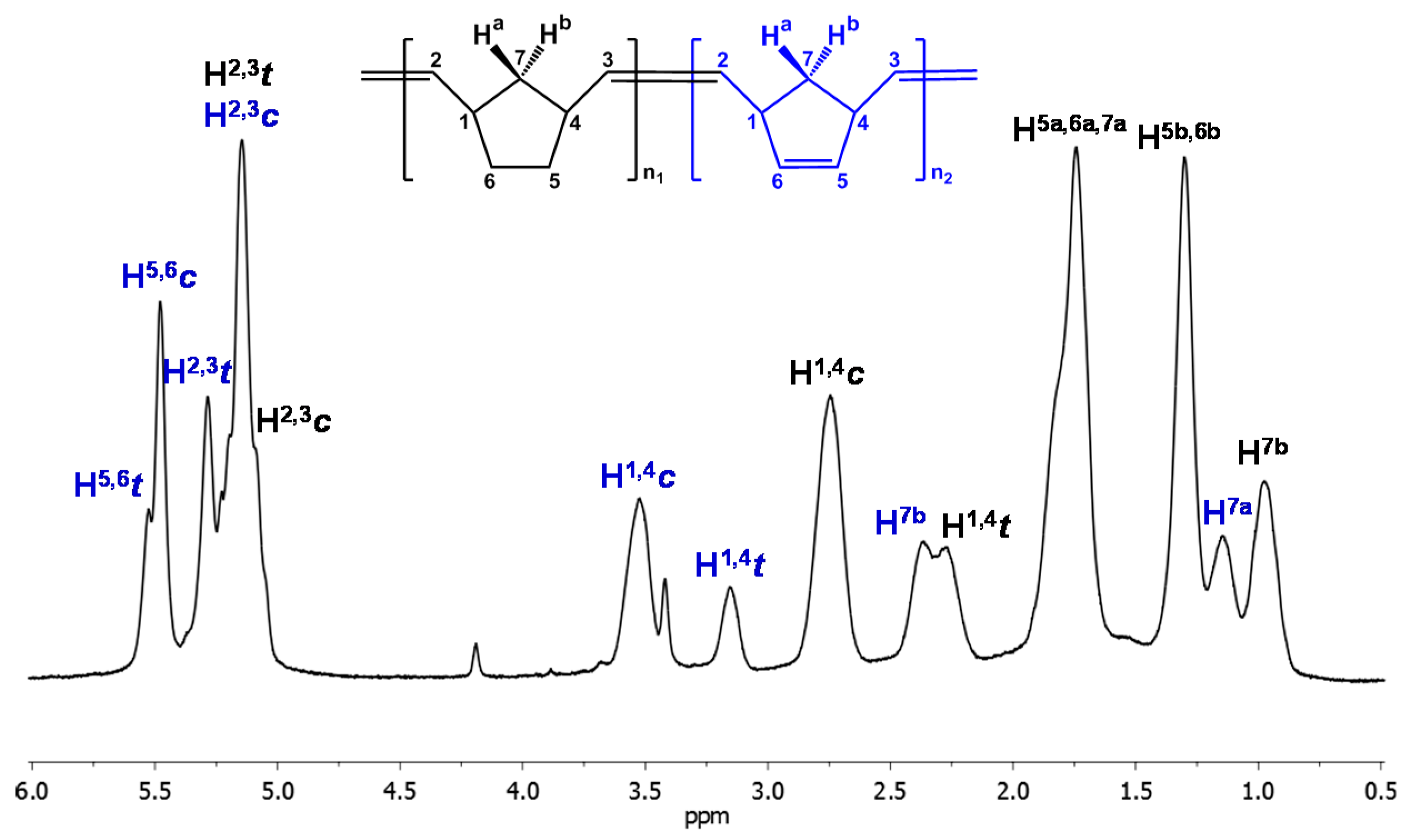
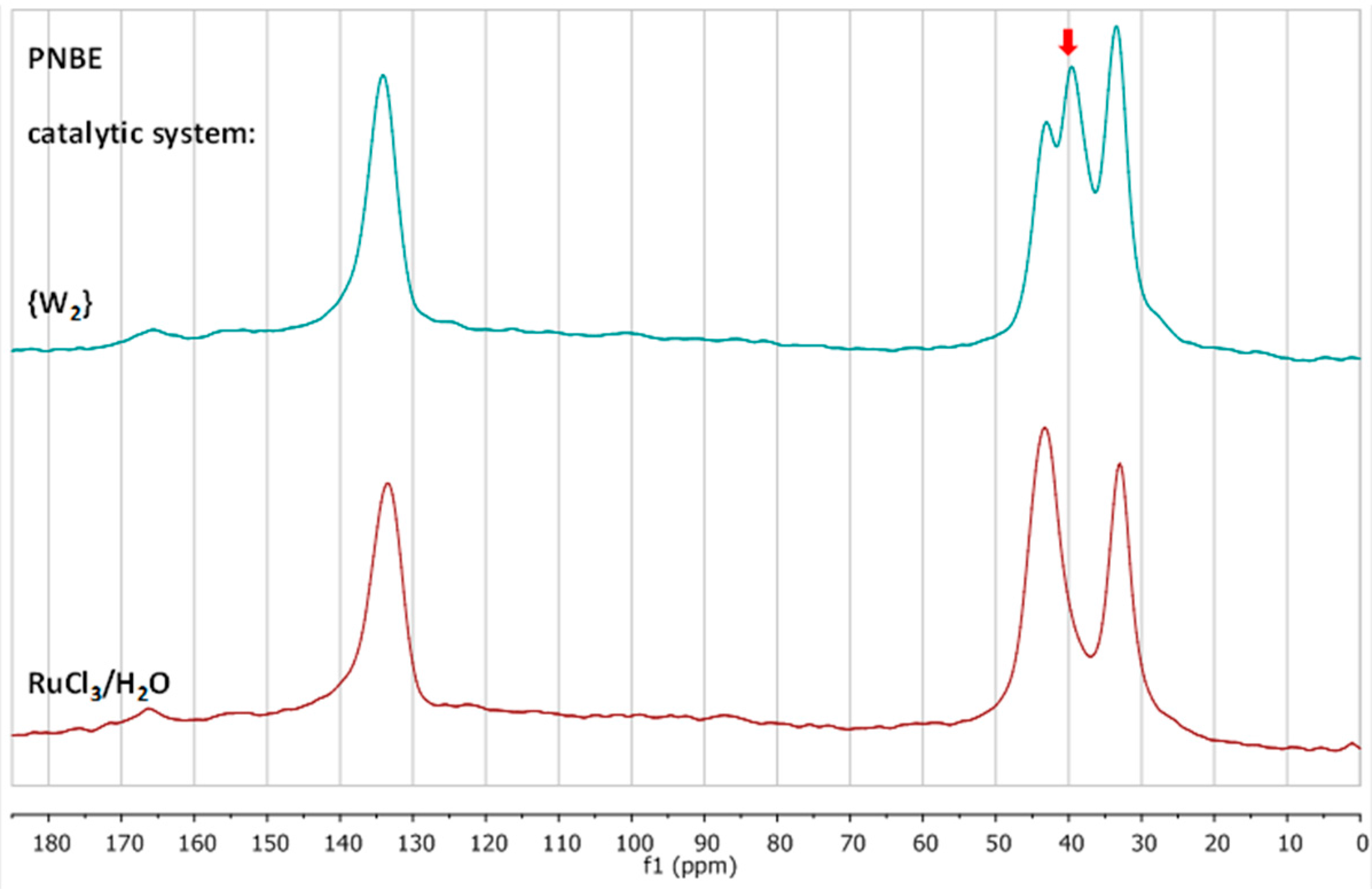
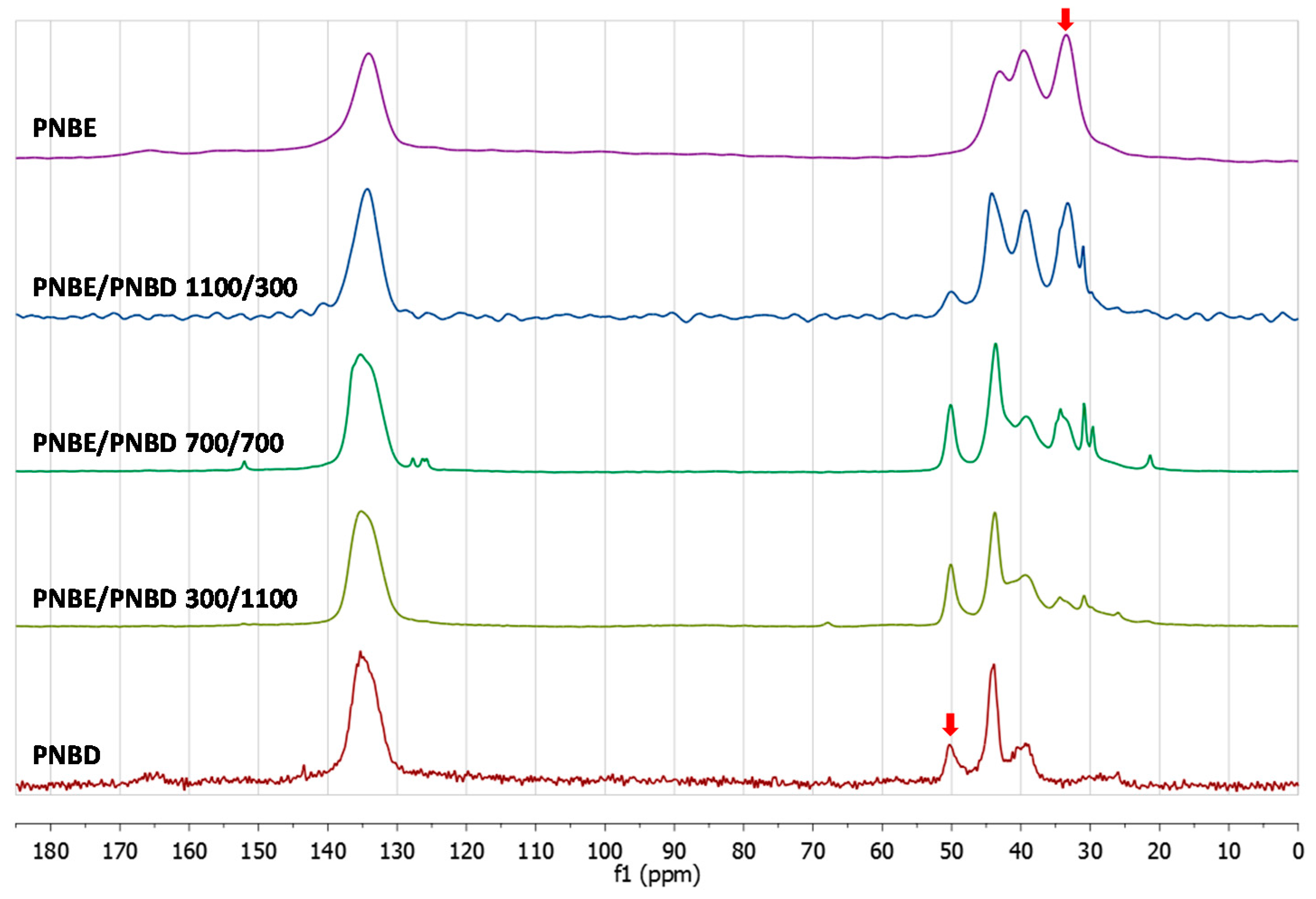
3.2. Chemical Characterization
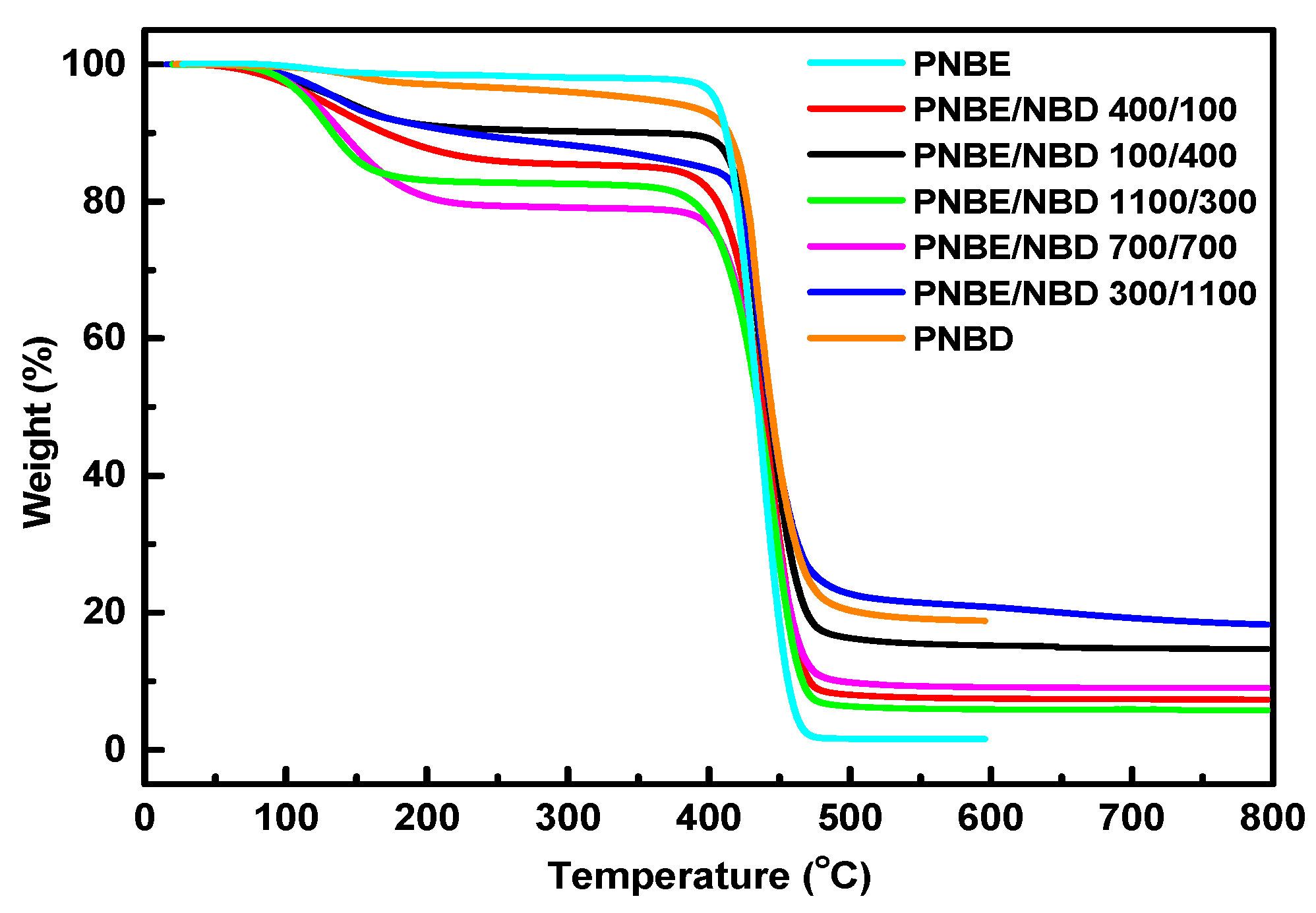
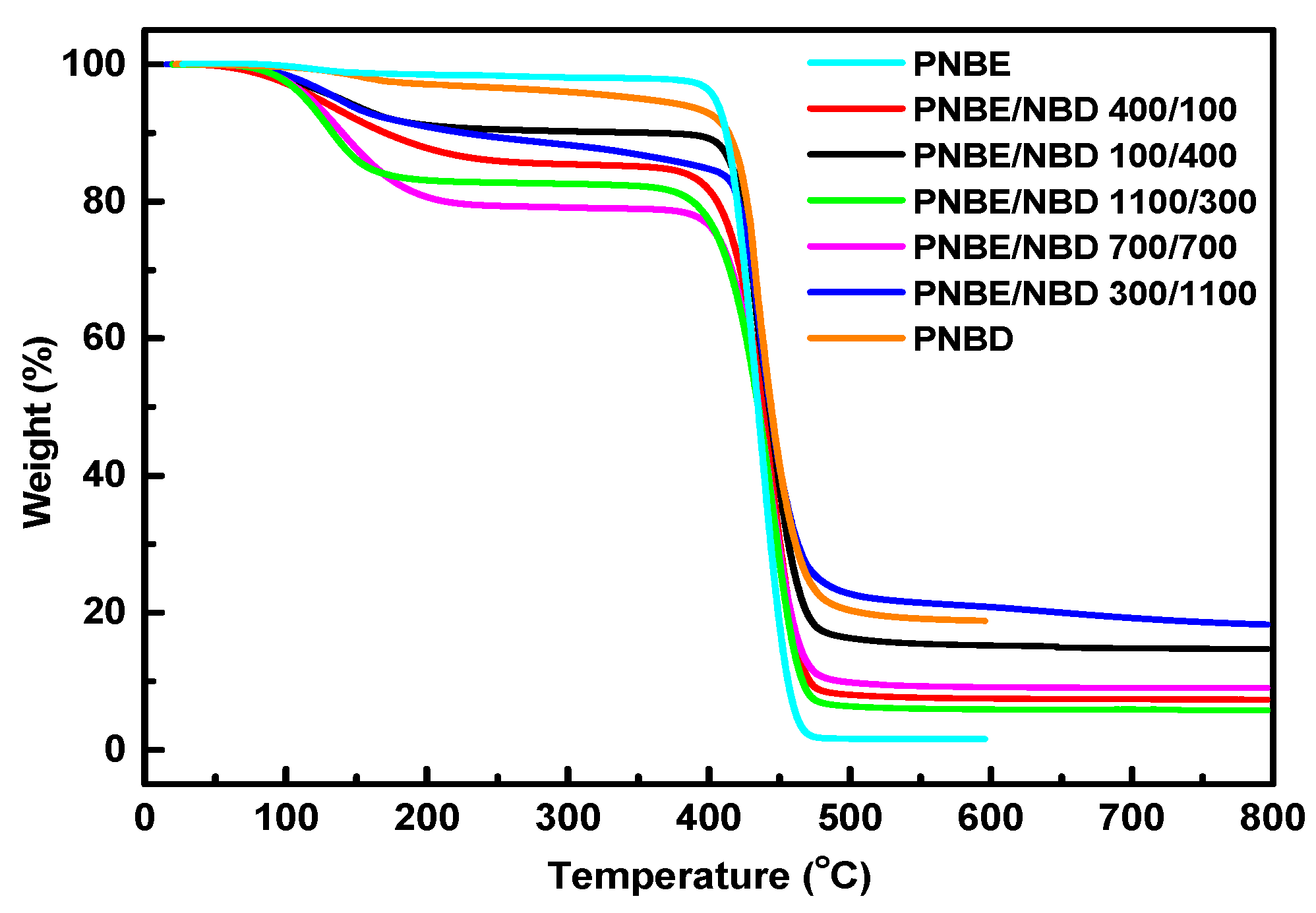
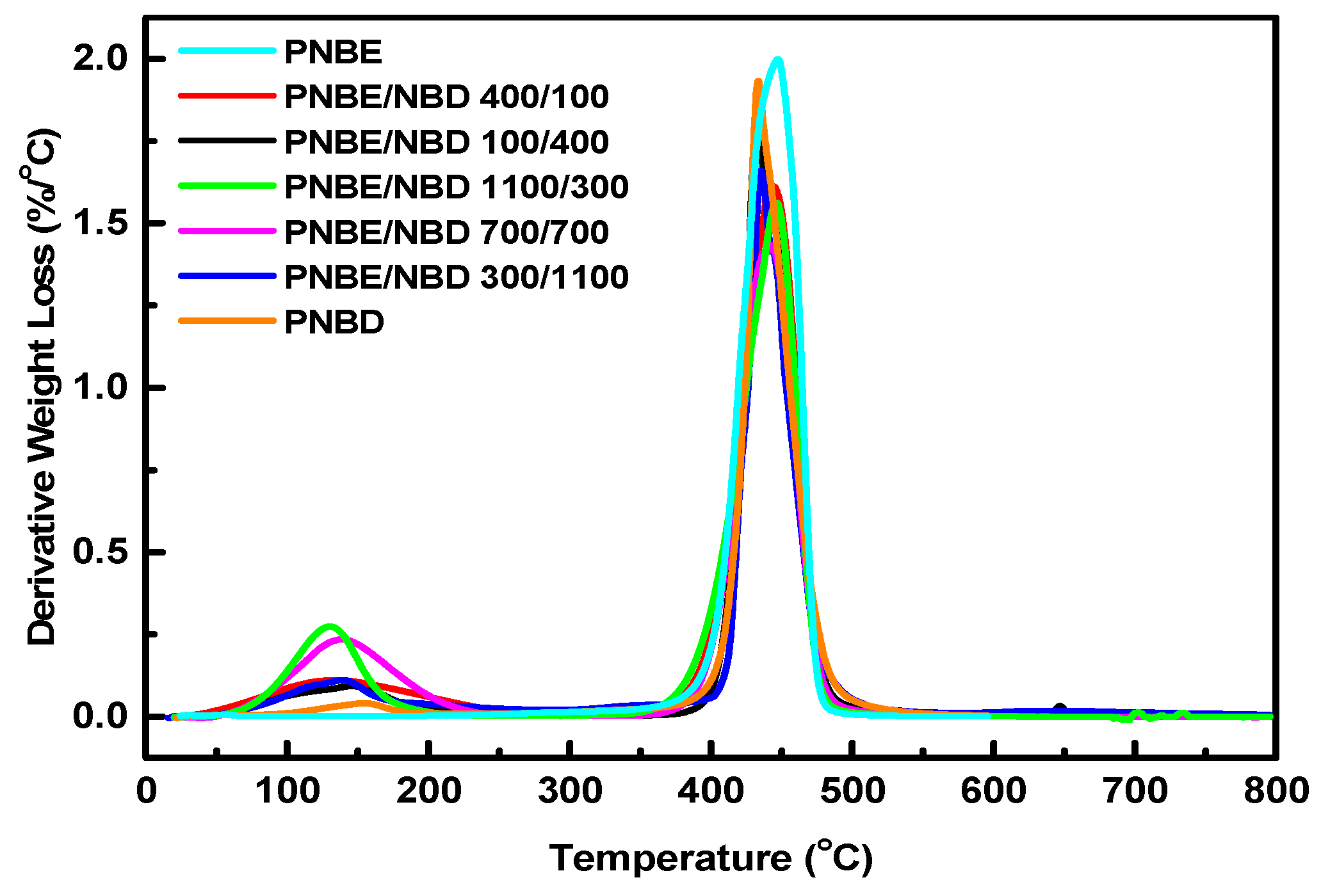
3.3. Thermal Properties
3.4. Morphology
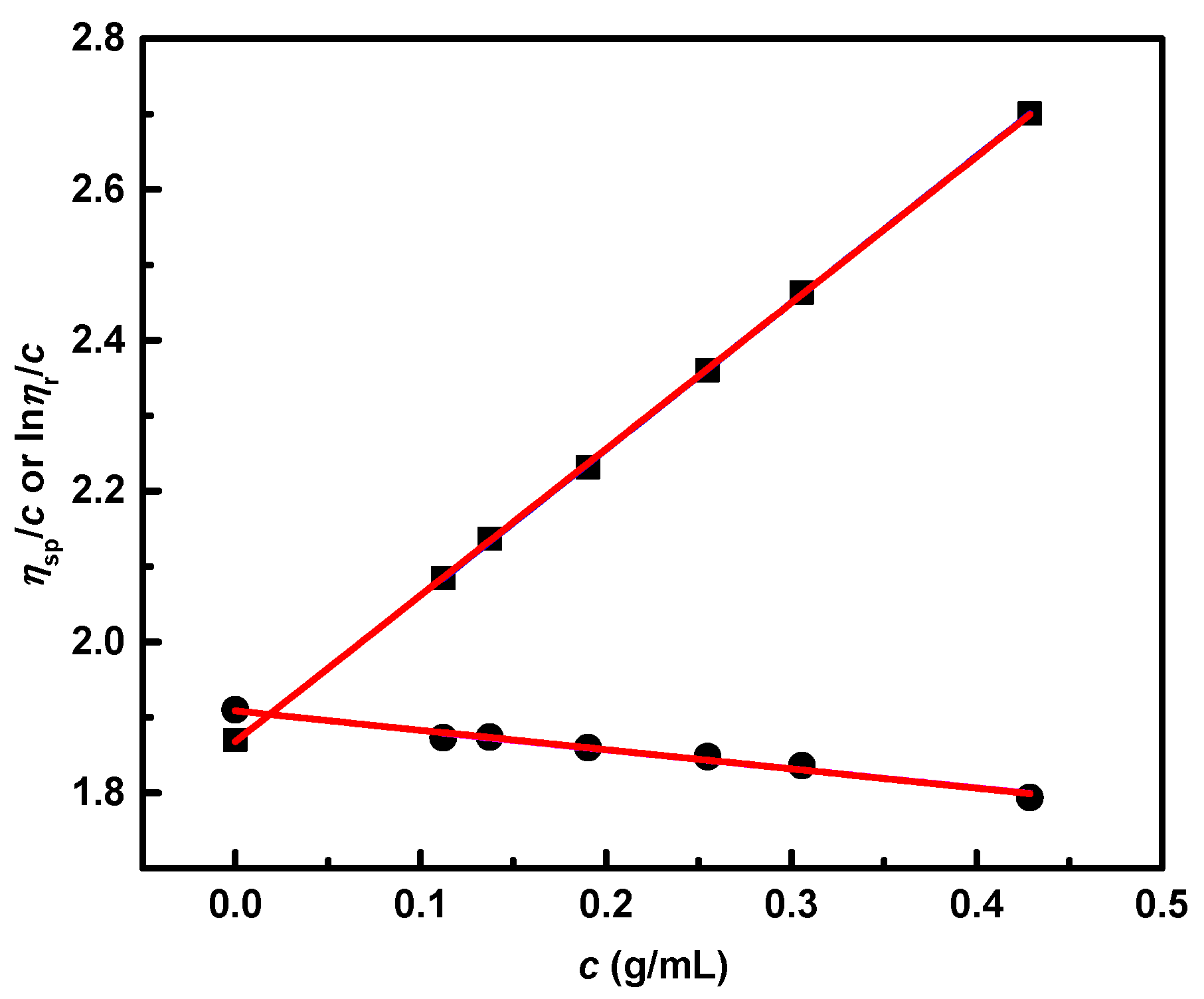
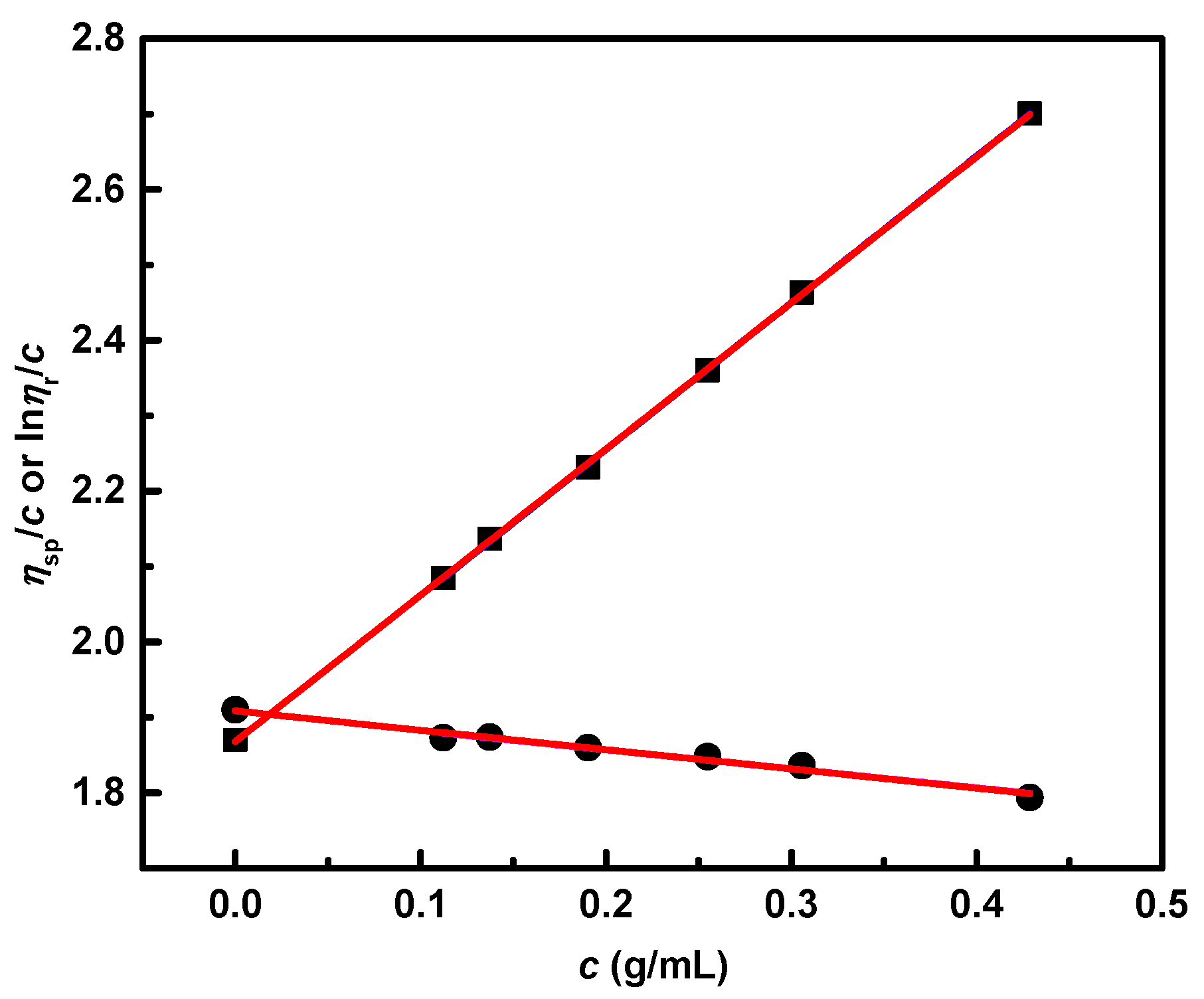
3.5. Dilute Solution Viscometry
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sutthasupa, S.; Shiotsuki, M.; Sanda, F. Recent advances in ring-opening metathesis polymerization, and application to synthesis of functional materials. Polym. J. 2010, 42, 905–915. [Google Scholar] [CrossRef]
- Leitgeb, A.; Wappel, J.; Slugovc, C. The ROMP toolbox upgraded. Polymer 2010, 51, 2927–2946. [Google Scholar] [CrossRef]
- Nomura, K.; Abdellatif, M.M. Precise synthesis of polymers containing functional end groups by living ring-opening metathesis polymerization (ROMP): Efficient tools for synthesis of block/graft copolymers. Polymer 2010, 51, 1861–1881. [Google Scholar] [CrossRef]
- Ivin, K.J.; Mol, J.C. Olefin Metathesis and Metathesis Polymerization; Academic Press: Cambridge, MA, USA, 1997. [Google Scholar]
- Dragutan, V.; Streck, R. Catalytic Polymerization of Cycloolefins: Ionic, Ziegler-Natta and Ring-Opening Metathesis Polymerization; Elsevier: Amsterdam, the Netherlands, 2000. [Google Scholar]
- Katz, T.J. Metallcarbene mit niedriger oxidationsstufe als initiatoren für olefinmetathesen und verwandte reaktionen. Angew. Chem. 2005, 117, 3070–3079. [Google Scholar] [CrossRef]
- Katz, T.J. Olefin Metatheses and related reactions initiated by carbene derivatives of metals in low oxidation states. Angew. Chem. Int. Ed. 2005, 44, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Katz, T.J.; Lee, S.J. Initiation of acetylene polymerization by metal carbenes. J. Am. Chem. Soc. 1980, 102, 422–424. [Google Scholar] [CrossRef]
- Schrock, R.R. Multiple metal-carbon bonds for catalytic metathesis reactions (nobel lecture). Angew. Chem. Int. Ed. 2006, 45, 3748–3759. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, R.H. Olefin-metathesis catalysts for the preparation of molecules and materials (nobel lecture). Angew. Chem. Int. Ed. 2006, 45, 3760–3765. [Google Scholar] [CrossRef] [PubMed]
- Buchmeiser, M.R. Polymer-supported well-defined metathesis catalysts. Chem. Rev. 2009, 109, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Balcar, H.; Čejka, J. Mesoporous molecular sieves as advanced supports for olefin metathesis catalysts. Coord. Chem. Rev. 2013, 257, 3107–3124. [Google Scholar] [CrossRef]
- Raptopoulos, G.; Grigoropoulos, A.; Mertis, K.; Paraskevopoulou, P.; Pitsikalis, M. Multinuclear transition metal catalysts for metathesis polymerization. Current developments and future perspectives. Recent Res. Dev. Polym. Sci. 2014, 12, 83–106. [Google Scholar]
- Szymańska-Buzar, T. Structure and reactivity of tungsten(II) and molybdenum(II) compounds containing an MM′ bond. Coord. Chem. Rev. 2005, 249, 2195–2202. [Google Scholar] [CrossRef]
- Saragas, N.; Floros, G.; Paraskevopoulou, P.; Psaroudakis, N.; Koinis, S.; Pitsikalis, M.; Mertis, K. Polymerization of terminal alkynes with a triply bonded ditungsten halo-complex. J. Mol. Catal. Chem. 2009, 303, 124–131. [Google Scholar] [CrossRef]
- Chriti, D.; Grigoropoulos, A.; Raptopoulos, G.; Charalambidis, G.; Nikolaou, V.; Coutsolelos, A.G.; Pitsikalis, M.; Mertis, K.; Paraskevopoulou, P. Metathesis polymerization reactions induced by the bimetallic complex (Ph4P)2[W2(μ-Br)3Br6]. Polymers 2015, 7, 2611–2624. [Google Scholar] [CrossRef]
- Floros, G.; Saragas, N.; Paraskevopoulou, P.; Psaroudakis, N.; Koinis, S.; Pitsikalis, M.; Hadjichristidis, N.; Mertis, K. Ring opening metathesis polymerization of norbornene and derivatives by the triply bonded ditungsten complex Na[W2(µ-Cl)3Cl4(THF)2](THF)3. Polymers 2012, 4, 1657–1673. [Google Scholar] [CrossRef]
- Saragas, N.; Floros, G.; Raptopoulos, G.; Pitsikalis, M.; Paraskevopoulou, P.; Mertis, K. Exploring the Reactivity of Na[W2(μ-Cl)3Cl4(THF)2](THF)3 towards the polymerization of selected cycloolefins. Molecules 2015, 20, 21896–21908. [Google Scholar] [CrossRef] [PubMed]
- Raptopoulos, G.; Anyfantis, G.C.; Chriti, D.; Paraskevopoulou, P. Synthesis and structural characterization of poly(dicyclopentadiene) gels obtained with a novel ditungsten versus conventional W and Ru mononuclear catalysts. Inorg. Chim. Acta 2017, 460, 69–76. [Google Scholar] [CrossRef]
- Schrock, R.R.; Lee, J.-K.; O’Dell, R.; Oskam, J.H. Exploring factors that determine cis/trans structure and tacticity in polymers prepared by ring-opening metathesis polymerizations with initiators of the type syn- and anti-Mo(NAr)(CHCMe2Ph)(OR)2. observation of a temperature-dependent cis/trans ratio. Macromolecules 1995, 28, 5933–5940. [Google Scholar] [CrossRef]
- Schrock, R.R. Synthesis of stereoregular ROMP polymers using molybdenum and tungsten imido alkylidene initiators. Dalton Trans. 2011, 40, 7484–7495. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.R. Recent Advances in high oxidation state mo and w imido alkylidene chemistry. Chem. Rev. 2009, 109, 3211–3226. [Google Scholar] [CrossRef] [PubMed]
- Basset, J.-M.; Leconte, M.; Lefebvre, F.; Hamilton, J.G.; Rooney, J.J. Stereoselectivity in cyclic and acyclic metathesis reactions. Macromol. Chem. Phys. 1997, 198, 3499–3506. [Google Scholar] [CrossRef]
- Abadie, M.J.; Dimonie, M.; Couve, C.; Dragutan, V. New catalysts for linear polydicyclopentadiene synthesis. Eur. Polym. J. 2000, 36, 1213–1219. [Google Scholar] [CrossRef]
- Bokaris, E.P.; Kosmas, M.M. All cis-poly(NBE) derived by the ROMP catalysts based on WCl6. J. Mol. Catal. Chem. 2003, 192, 263–273. [Google Scholar] [CrossRef]
- Schrock, R.R. Synthesis of stereoregular polymers through ring-opening metathesis polymerization. Acc. Chem. Res. 2014, 47, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, A.; Schaaf, P.A.V.D.; Hafner, A.; Kolly, R.; Rime, F.; Kimer, H.-J. Ruthenium catalysts for ring-opening metathesis polymerization (ROMP) and related chemistry. In Ring Opening Metathesis Polymerisation and Related Chemistry; Khosravi, E., Szymanska-Buzar, T., Eds.; NATO Science Series; Springer: Amsterdam, The Netherlands, 2002; pp. 23–44. [Google Scholar]
- Amir-Ebrahimi, V.; Corry, D.A.; Hamilton, J.G.; Thompson, J.M.; Rooney, J.J. Characteristics of RuCl2(CHPh)(PCy3)2 as a catalyst for ring-opening metathesis polymerization. Macromolecules 2000, 33, 717–724. [Google Scholar] [CrossRef]
- Schaubroeck, D.; Brughmans, S.; Vercaemst, C.; Schaubroeck, J.; Verpoort, F. Qualitative FT-Raman investigation of the ring opening metathesis polymerization of dicyclopentadiene. J. Mol. Catal. Chem. 2006, 254, 180–185. [Google Scholar] [CrossRef]
- Ding, F.; Monsaert, S.; Drozdzak, R.; Dragutan, I.; Dragutan, V.; Sun, Y.; Gao, E.; Van Der Voort, P.; Verpoort, F. First FT-Raman and 1H-NMR comparative investigations in ring opening metathesis polymerization. Vib. Spectrosc. 2009, 51, 147–151. [Google Scholar] [CrossRef]
- Rosebrugh, L.E.; Marx, V.M.; Keitz, B.K.; Grubbs, R.H. Synthesis of highly cis, syndiotactic polymers via ring-opening metathesis polymerization using ruthenium metathesis catalysts. J. Am. Chem. Soc. 2013, 135, 10032–10035. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.G.; Ivin, K.J.; Rooney, J.J. Ring-opening polymerization of endo and exo-dicyclopentadiene and their 7,8-dihydro derivatives. J. Mol. Catal. 1986, 36, 115–125. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Eichhorn, B.W.; Folting, K.; Huffman, J.C.; Ontiveros, C.D.; Streib, W.E.; Van der Sluys, W.G. Preparation and characterization of sodium heptachloropentakis(THF)ditungstate(1-). A synthetically useful precursor for X3W.tplbond.WX3 compounds where X = CH2-tert-Bu, NMe2 and O-tert-Bu. Inorg. Chem. 1987, 26, 3182–3186. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. Multiple Bonds between Metal Atoms, 3rd ed.; Springer Science & Business Media, Inc.: New York, NY, USA, 2005. [Google Scholar]
- Düz, B.; Elbistan, C.K.; Ece, A.; Sevin, F. Application of carbon arc-generated Mo- and W-based catalyst systems to the ROMP of norbornene. Appl. Organomet. Chem. 2009, 23, 359–364. [Google Scholar] [CrossRef]
- Michelotti, F.W.; Keaveney, W.P. Coordinated polymerization of the bicyclo-[2.2.1]-heptene-2 ring system (norbornene) in polar media. J. Polym. Sci. A 1965, 3, 895–905. [Google Scholar] [CrossRef]
- Bell, B.; Hamilton, J.G.; Mackey, O.N.D.; Rooney, J.J. Microstructure of ring-opened polymers and copolymers of norbornadiene. J. Mol. Catal. 1992, 77, 61–73. [Google Scholar] [CrossRef]
- Carvalho, V.P., Jr.; Ferraz, C.P.; Lima-Neto, B.S. Tailored norbornene-based copolymer with systematic variation of norbornadiene as a crosslinker obtained via ROMP with alternative amine Ru catalysts. Eur. Polym. J. 2012, 48, 341–349. [Google Scholar] [CrossRef]
- Nemanich, R.J.; Solin, S.A. First- and second-order Raman scattering from finite-size crystals of graphite. Phys. Rev. B 1979, 20, 392–401. [Google Scholar] [CrossRef]
- Sperling, L.H. Introduction to Physical Polymer Science, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Ferraz, C.P.; Fonseca, L.R.; Tomazetti, V.; Silva, F.C.S.; Lima-Neto, B.S.; Carvalho, V.P. Copolymers from norbornene and norbornadiene with organized morphologies and high Tg values obtained via ROMP with a highly reactive [RuCl3(PCy3)2] complex. New J. Chem. 2016, 40, 9424–9431. [Google Scholar] [CrossRef]
- Chaves, H.K.; Ferraz, C.P.; Carvalho, V.P., Jr.; Lima-Neto, B.S. Tuning the activity of alternative Ru-based initiators for ring-opening metathesis polymerization of norbornene and norbornadiene by the substituent in 4-CH2R-piperidine. J. Mol. Catal. Chem. 2014, 385, 46–53. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Driva, P.; Sakellariou, G.; Chatzichristidi, M. 6.03—Polymers with star-related structures: Synthesis, properties, and applications. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 29–111. [Google Scholar]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Mays, J. Macromolecular architectures by living and controlled/living polymerizations. Prog. Polym. Sci. 2006, 31, 1068–1132. [Google Scholar] [CrossRef]
- Kyriakou, K. Metathesis (Co)Polymerization Reactions of Cycloolefins with Dimetallic Tungsten Complexes. Master’s Thesis, National and Kapodistrian University of Athens, Athens, Greece, 2015. [Google Scholar]
- Roovers, J. Branched Polymers. In Encyclopedia of Polymer Science and Technology, 2nd ed.; Mark, H.F., Bikales, N.M., Overberger, C.G., Menges, G., Kroschwitz, J.I., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1985. [Google Scholar]
- Zimm, B.M.; Stockmayer, W.M. The Dimensions of Chain Molecules Containing Branches and Rings. J. Chem. Phys. 1949, 17, 1301–1314. [Google Scholar] [CrossRef]
- Bazan, G.C.; Schrock, R.R. Synthesis of star block copolymers by controlled ring-opening metathesis polymerization. Macromolecules 1991, 24, 817–823. [Google Scholar] [CrossRef]
- Saunders, R.S.; Cohen, R.E.; Wong, S.J.; Schrock, R.R. Synthesis of amphiphilic star block copolymers using ring-opening metathesis polymerization. Macromolecules 1992, 25, 2055–2057. [Google Scholar] [CrossRef]
- Dounis, P.; James Feast, W. A route to low polydispersity linear and star polyethylenes via ring-opening metathesis polymerization. Polymer 1996, 37, 2547–2554. [Google Scholar] [CrossRef]
- Beerens, H.; Wang, W.; Verdonck, L.; Verpoort, F. Multi-nuclear dendritic Ru-complexes as catalysts for ROMP; synthesis and characterization of starpolymers. J. Mol. Catal. Chem. 2002, 190, 1–7. [Google Scholar] [CrossRef]
- Gatard, S.; Kahlal, S.; Méry, D.; Nlate, S.; Cloutet, E.; Saillard, J.-Y.; Astruc, D. Synthesis, chemistry, DFT calculations, and ROMP activity of monomeric benzylidene complexes containing a chelating diphosphine and of four generations of metallodendritic analogues: Positive and negative dendritic effects and formation of dendritic ruthenium-polynorbornene stars. Organometallics 2004, 23, 1313–1324. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | {W2}/ΝΒΕ/NBD (Molar Ratio) | t (min) | Yield (%) | Mw × 10−3 b | Mw/Mn b |
|---|---|---|---|---|---|
| PNBE c | 1/500/0 | 60 | 96 | 529 | 1.2 |
| PNBE/PNBD 400/100 | 1/400/100 | 10 | 61 | 129 | 2.0 |
| PNBE/PNBD 100/400 | 1/100/400 | 3 | 99 | d | d |
| PNBE/PNBD 1100/300 | 1/1100/300 | 12 | 11 | 252 | 2.5 |
| PNBE/PNBD 700/700 | 1/700/700 | 7 | 18 | d | d |
| PNBE/PNBD 300/1100 | 1/300/1100 | 5 | 66 | d | d |
| PNBD c | 1/0/500 | 6 | >99 | d | d |
| Sample | Start (°C) | Finish (°C) | Peak (°C) | Residue (%) |
|---|---|---|---|---|
| PNBE | 376.11 | 490.49 | 445.96 | 1.60 |
| PNBE/PNBD 400/100 | 353.54 | 495.96 | 443.78 | 7.35 |
| PNBE/PNBD 100/400 | 377.78 | 509.09 | 432.86 | 14.75 |
| PNBE/PNBD 1100/300 | 348.48 | 491.92 | 446.59 | 5.78 |
| PNBE/PNBD 700/700 | 363.64 | 508.08 | 442.67 | 9.06 |
| PNBE/PNBD 300/1100 | 389.90 | 539.39 | 435.51 | 18.46 |
| PNBD | 377.69 | 561.96 | 433.42 | 18.89 |
| Sample | Tg (°C) |
|---|---|
| PNBE | 57.9 |
| PNBE/PNBD 400/100 | 37.9 |
| PNBE/PNBD 100/400 | 39.5 |
| PNBE/PNBD 1100/300 | 39.4 |
| PNBE/PNBD 700/700 | 29.1 |
| PNBE/PNBD 300/1100 | 47.5 |
| PNBD | 50.1 |
| Entry | CH2Cl2 (mL) | NBD (μL) | t1 b (min) | t2 c (h) | Yield (%) | Mw × 10−3 d | Mw/Mn d |
|---|---|---|---|---|---|---|---|
| 1 | 15 | 10 | 10 | 1 | 9 | - | - |
| 2 | 15 | 20 | 5 | 1 | 22 | 320 | 1.41 |
| 3 | 25 | 10 | 15 | 21 | 32 | 429 | 1.42 |
| 4 | 25 | 20 | 5 | 21 | 16 | 771 | 1.29 |
| 5 | 25 | 10 | 40 | 6 | 8 | 764 | 1.30 |
| 6 | 25 | 20 | 40 | 6 | 3 | 661 | 1.24 |
| 7 | 25 | 10 | 90 | 30 | 10 | 423 | 1.37 |
| 8 | 25 | 20 | 90 | 30 | 35 | 566 | 1.31 |
| Entry | NBD (μL) | t1 b (min) | t2 c (h) | Yield (%) | Mw × 10−3 d | Mw/Mn d |
|---|---|---|---|---|---|---|
| 1 | 10 | 40 | 30 | 10 | 430 | 1.41 |
| 2 | 20 | 40 | 30 | 5 | 436 | 1.60 |
| 3 | 10 | 40 | 30 | 5 | 352 | 1.53 |
| 4 | 20 | 40 | 30 | 10 | 564 | 1.52 |
| 5 | 10 | 30 | 30 | 15 | 463 | 1.56 |
| 6 | 100 | 30 | 30 | 30 | 390 | 1.42 |
| Sample (Table 4) | Mw × 10−3 a | [η] (mL/g) | [η]lin (mL/g) | g’ | Functionality |
|---|---|---|---|---|---|
| 2 | 320 | 186.629 | 233.47 | 0.799 | 4.9 |
| 3 | 429 | 529.04 | 282.22 | 1.874 | |
| 8 | 566 | 65.321 | 336.83 | 0.194 | 59.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raptopoulos, G.; Kyriakou, K.; Mali, G.; Scarpellini, A.; Anyfantis, G.C.; Mavromoustakos, T.; Pitsikalis, M.; Paraskevopoulou, P. Copolymerization of Norbornene and Norbornadiene Using a cis-Selective Bimetallic W-Based Catalytic System. Polymers 2017, 9, 141. https://doi.org/10.3390/polym9040141
Raptopoulos G, Kyriakou K, Mali G, Scarpellini A, Anyfantis GC, Mavromoustakos T, Pitsikalis M, Paraskevopoulou P. Copolymerization of Norbornene and Norbornadiene Using a cis-Selective Bimetallic W-Based Catalytic System. Polymers. 2017; 9(4):141. https://doi.org/10.3390/polym9040141
Chicago/Turabian StyleRaptopoulos, Grigorios, Katerina Kyriakou, Gregor Mali, Alice Scarpellini, George C. Anyfantis, Thomas Mavromoustakos, Marinos Pitsikalis, and Patrina Paraskevopoulou. 2017. "Copolymerization of Norbornene and Norbornadiene Using a cis-Selective Bimetallic W-Based Catalytic System" Polymers 9, no. 4: 141. https://doi.org/10.3390/polym9040141
APA StyleRaptopoulos, G., Kyriakou, K., Mali, G., Scarpellini, A., Anyfantis, G. C., Mavromoustakos, T., Pitsikalis, M., & Paraskevopoulou, P. (2017). Copolymerization of Norbornene and Norbornadiene Using a cis-Selective Bimetallic W-Based Catalytic System. Polymers, 9(4), 141. https://doi.org/10.3390/polym9040141