
A Comprehensive Systematic Study on Thermoresponsive Gels: Beyond the Common Architectures of Linear Terpolymers

 ,
, 
Abstract
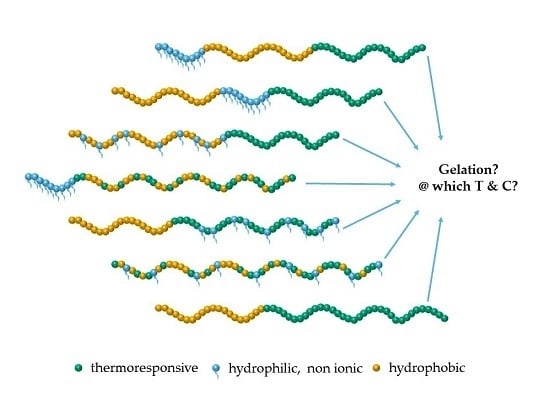
:
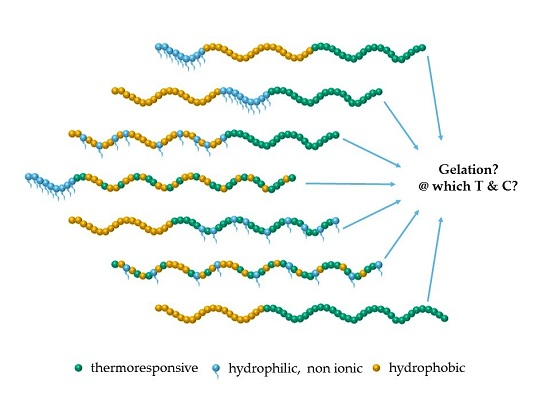
1. Introduction
2. Experimental
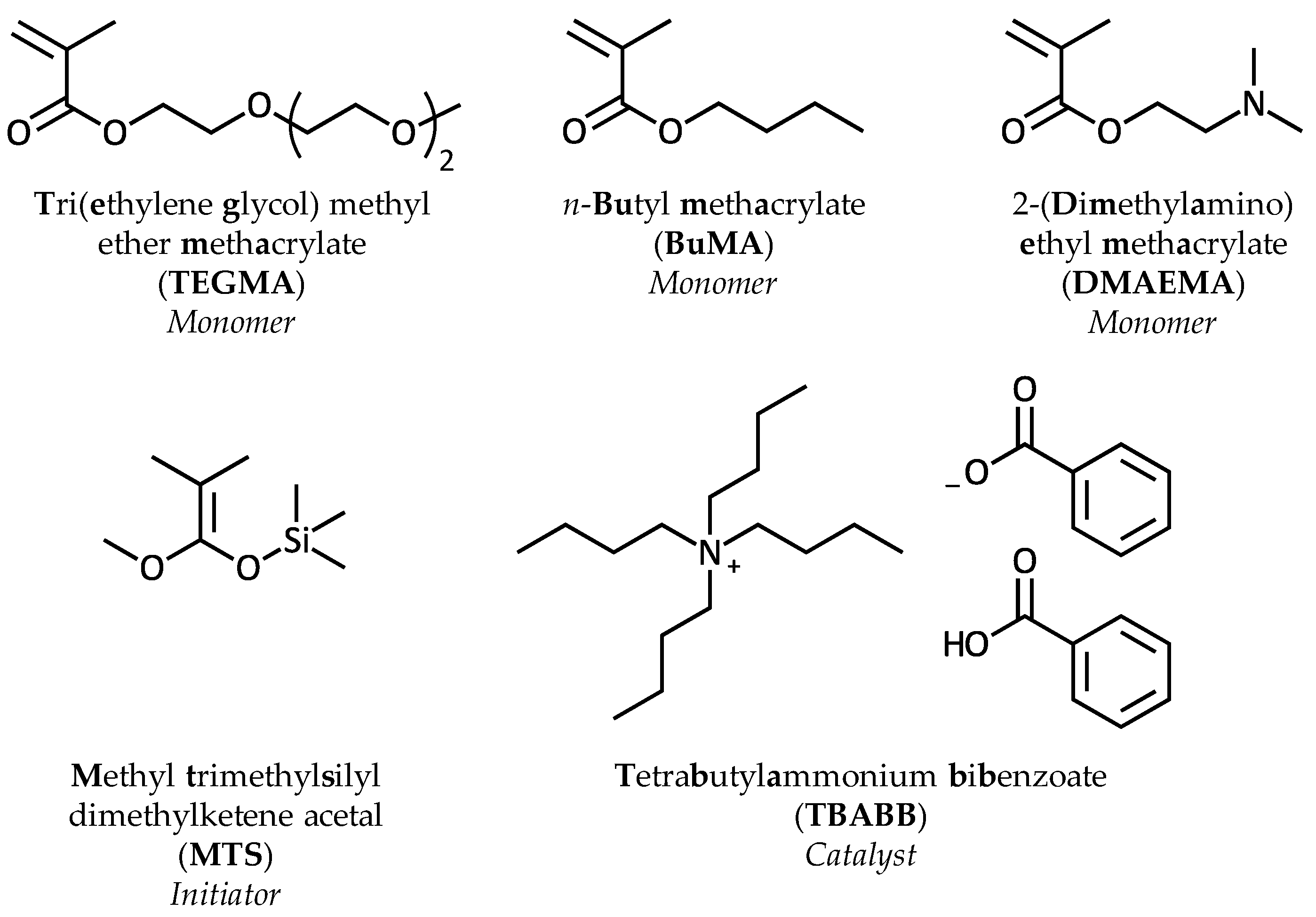
2.1. Materials
2.2. Purification of the Starting Materials
2.3. Copolymer Synthesis
2.4. Characterization in Organic Solvents
2.4.1. Gel Permeation Chromatography (GPC)
2.4.2. Proton Nuclear Magnetic Resonance Spectroscopy (1H-NMR)
2.5. Characterization in Aqueous Solution
2.5.1. Hydrogen Ion Titrations
2.5.2. Dynamic Light Scattering (DLS)
2.5.3. Transmission Electron Microscopy (TEM)
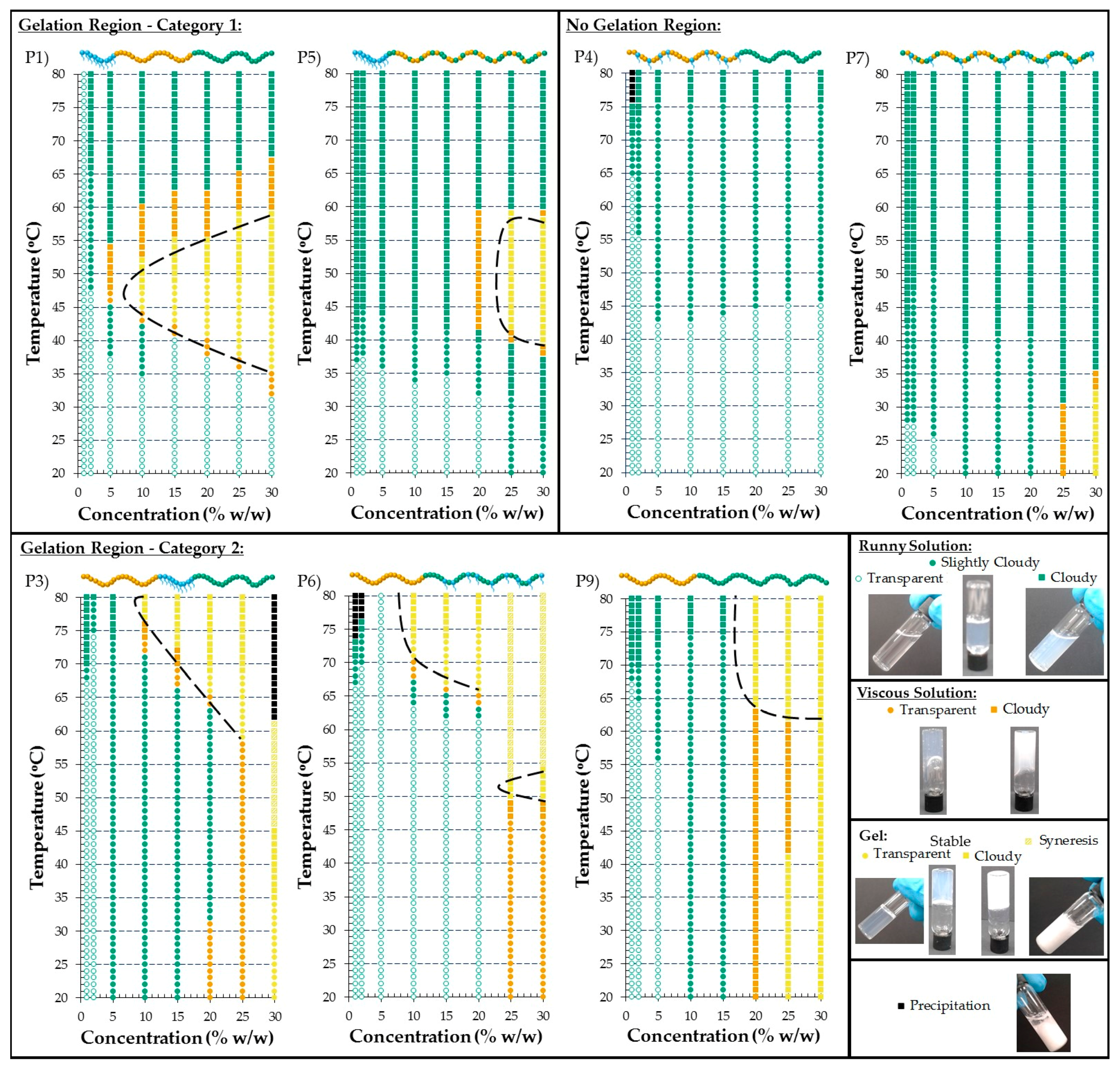
2.5.4. Visual Tests
3. Results and Discussion
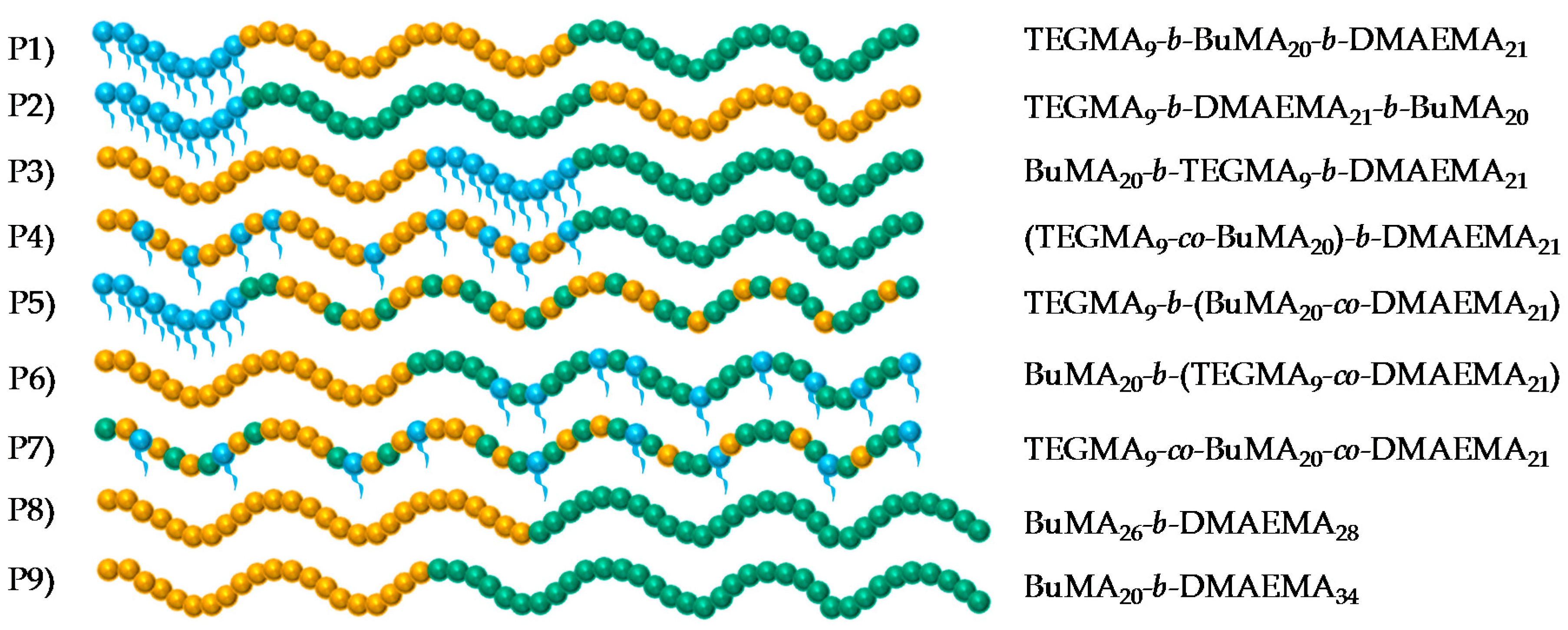
3.1. Structural Properties
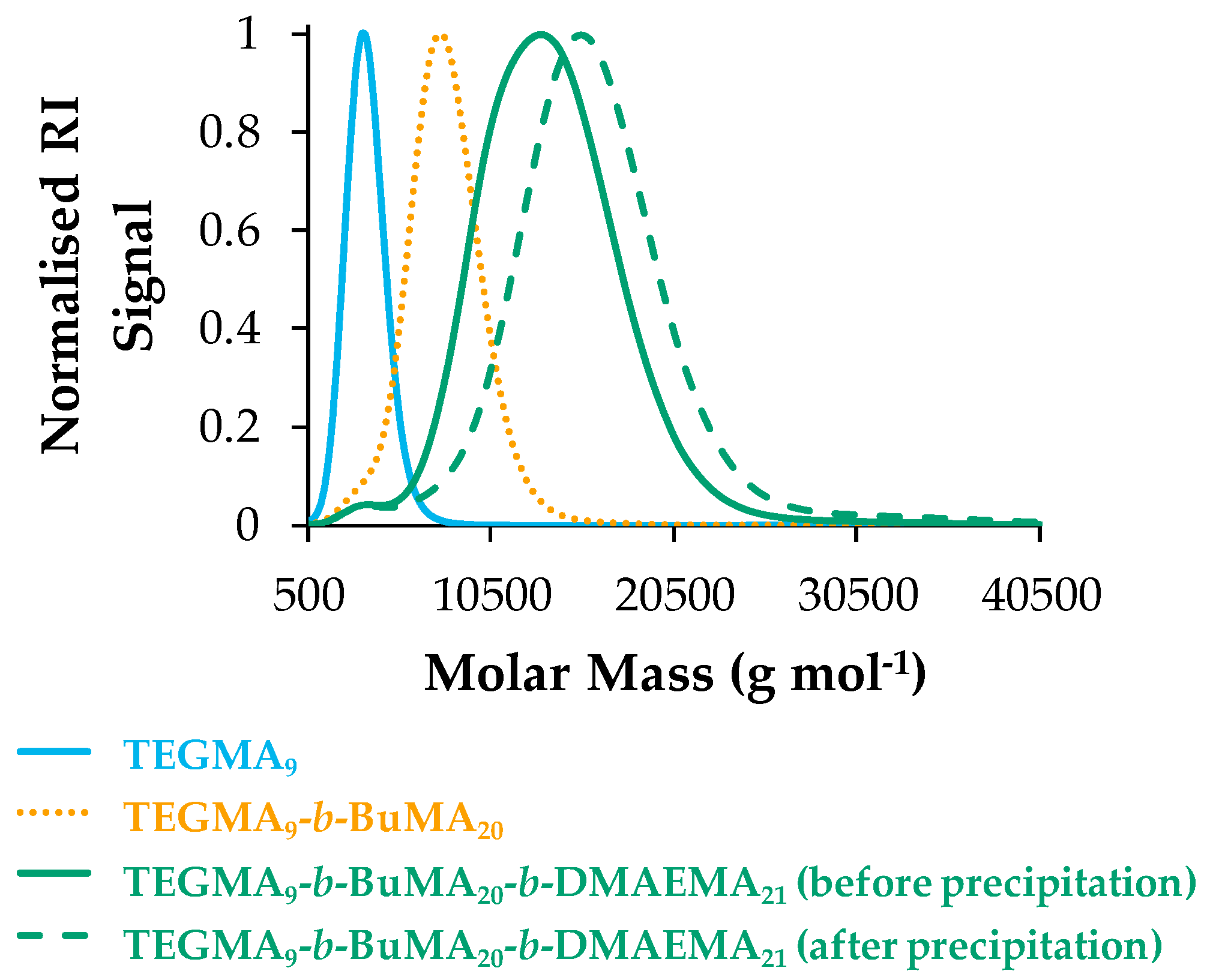
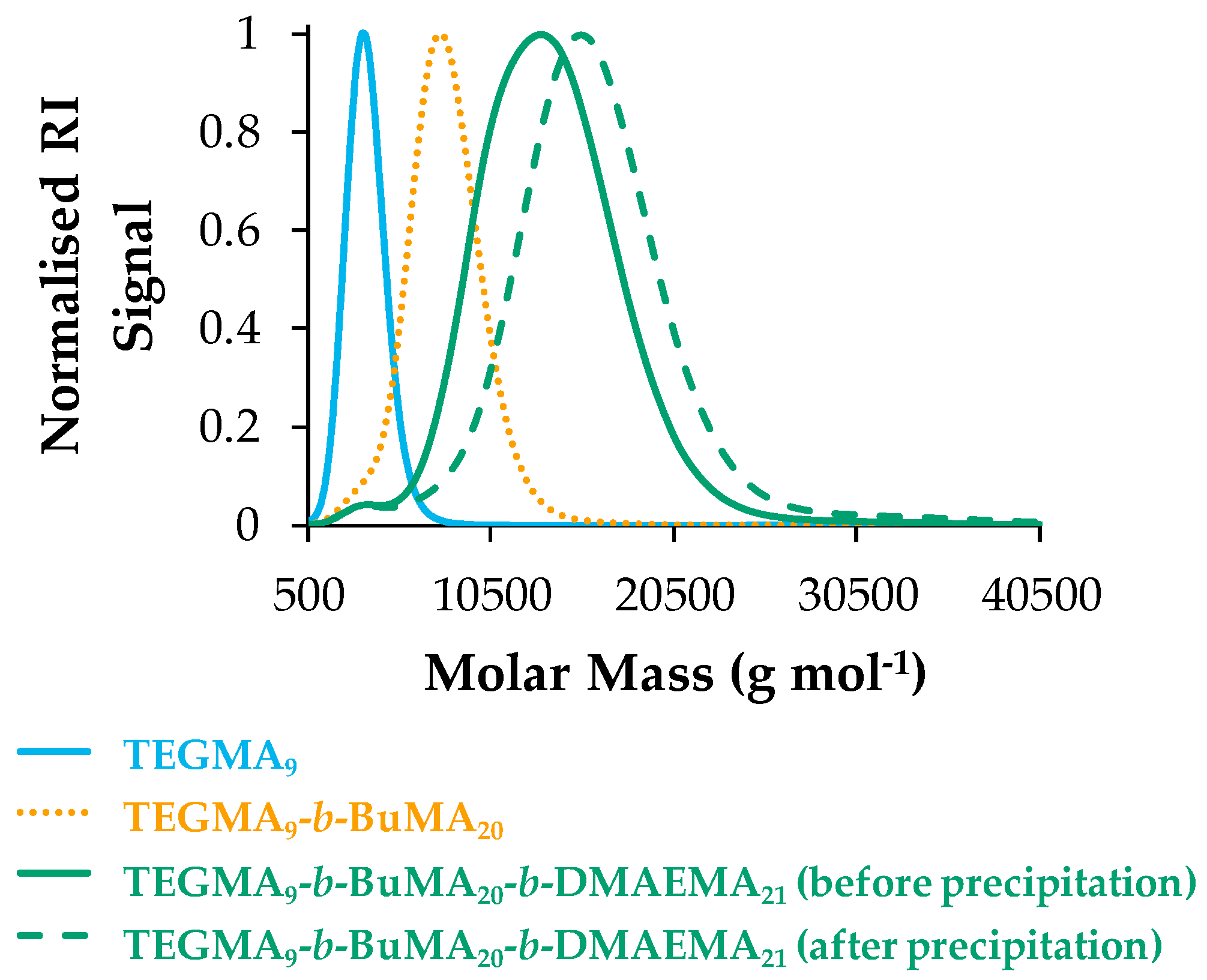
3.1.1. Molar Masses and Molar Mass Distributions
3.1.2. Compositions
3.2. Aqueous Solution Properties
3.2.1. Hydrodynamic Diameters
3.2.2. Transmission Electron Microscopy Images
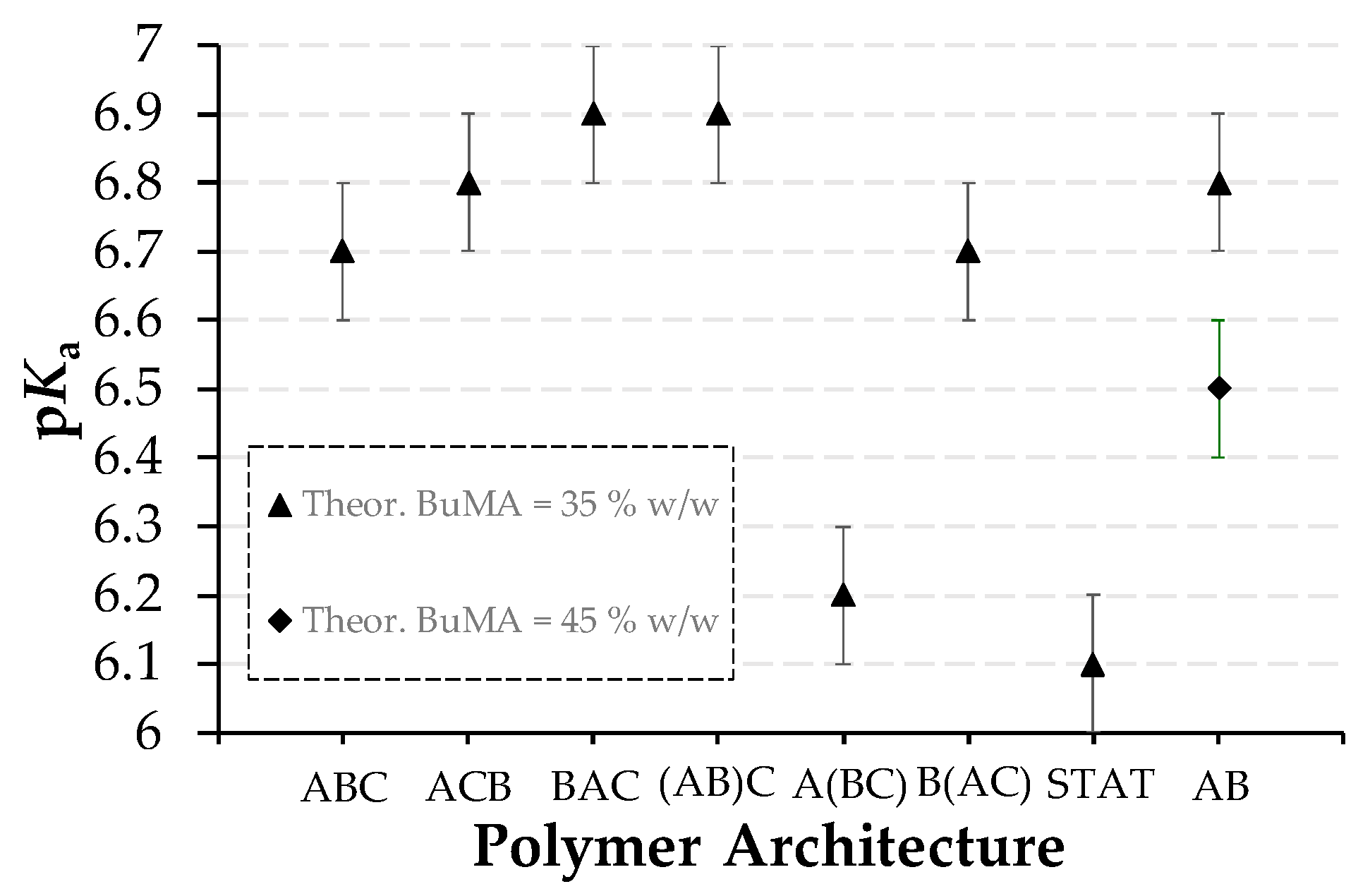
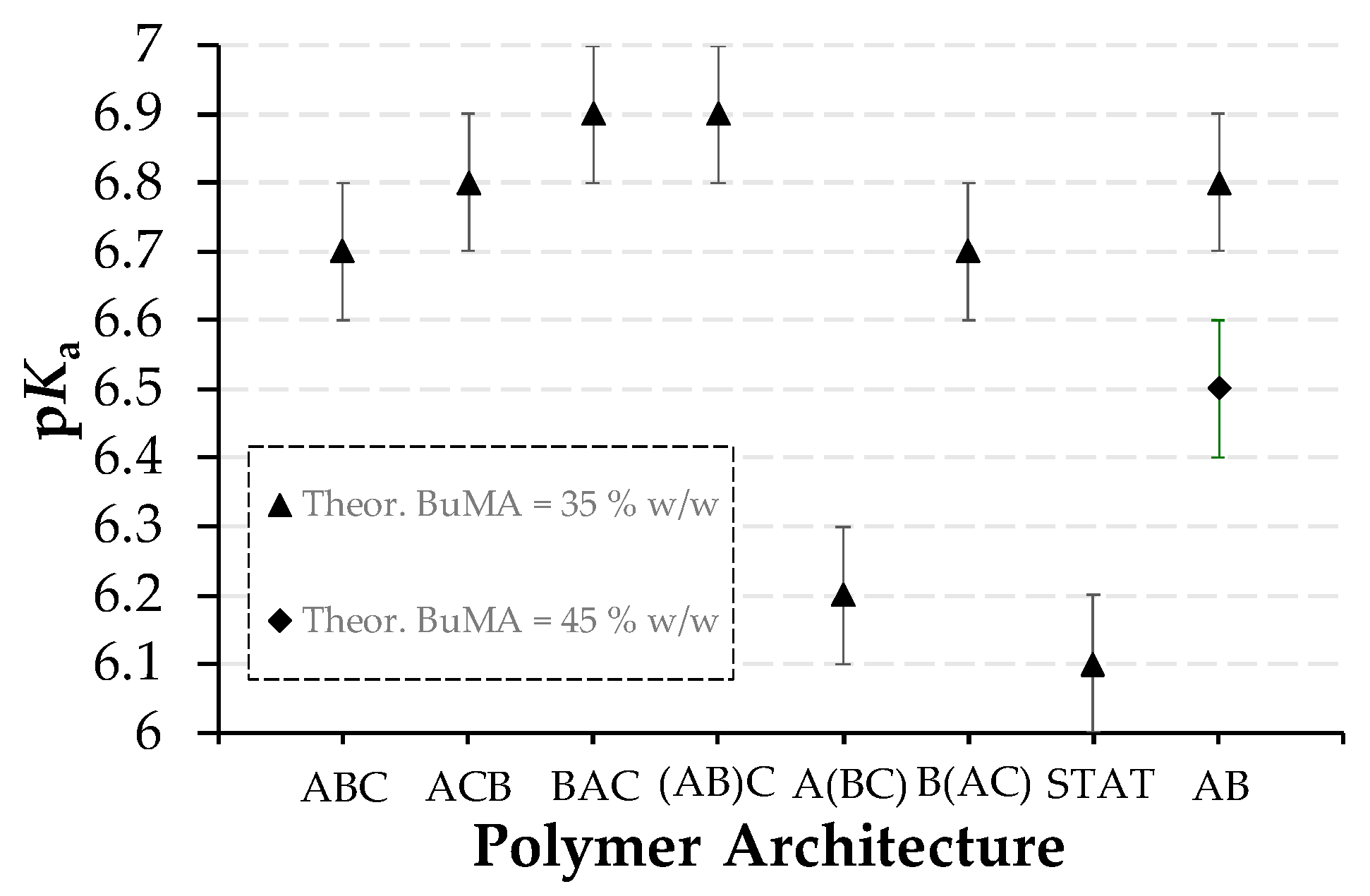
3.2.3. Effective pKas
3.2.4. Cloud Points
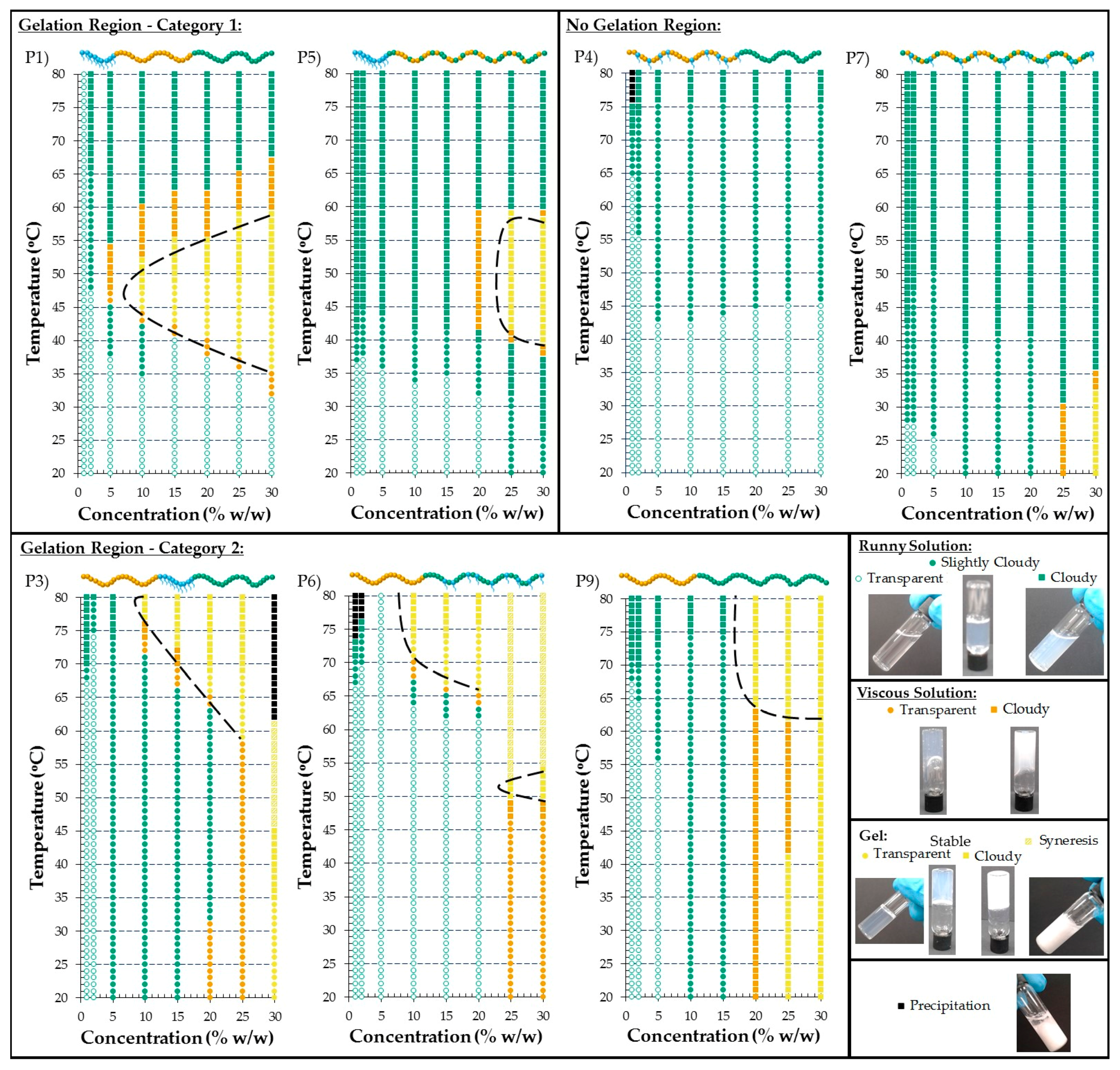
3.2.5. Visual Gel Points
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Constantinou, A.P.; Georgiou, T.K. Tuning the Gelation of Thermoresponsive Gels. Eur. Polym. J. 2016, 78, 366–375. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Polymers for Biomedical Applications. Polymers 2011, 3, 1215–1242. [Google Scholar] [CrossRef]
- Chassenieux, C.; Tsitsilianis, C. Recent Trends in pH/Thermo-Responsive Self-Assembling Hydrogels: From Polyions to Peptide-Based Polymeric Gelators. Soft Matter 2016, 12, 1344–1359. [Google Scholar] [CrossRef] [PubMed]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Madsen, J.; Armes, S.P.; Bertal, K.; Lomas, H.; MacNeil, S.; Lewis, A.L. Biocompatible Wound Dressings Based on Chemically Degradable Triblock Copolymer Hydrogels. Biomacromolecules 2008, 9, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Place, E.S.; George, J.H.; Williams, C.K.; Stevens, M.M. Synthetic Polymer Scaffolds for Tissue Engineering. Chem. Soc. Rev. 2009, 38, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Elstad, N.L.; Fowers, K.D. OncoGel (ReGel/Paclitaxel)—Clinical Applications for a Novel Paclitaxel Delivery System. Adv. Drug Deliv. Rev. 2009, 61, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.M.; Rathi, R.; Shih, C.; McRea, J.C.; Seo, M.-H.; Oh, H.; Rhee, B.G.; Mestecky, J.; Moldoveanu, Z.; Morgan, M.; et al. Biodegradable Block Copolymers for Delivery of Proteins and Water-Insoluble Drugs. J. Control. Release 2001, 72, 203–215. [Google Scholar] [CrossRef]
- Hsieh, F.-Y.; Lin, H.-Y.; Hsu, S.-H. 3D Bioprinting of Neural Stem Cell-Laden Thermoresponsive Biodegradable Polyurethane Hydrogel and Potential in Central Nervous System Repair. Biomaterials 2015, 71, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-C.; Li, S.; Hu, S.-G.; Chang, W.-C.; Jeng, U.-S.; Hsu, S.-H. Synthesis of Thermoresponsive Amphiphilic Polyurethane Gel as a New Cell Printing Material near Body Temperature. ACS Appl. Mater. Interfaces 2015, 7, 27613–27623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Vora, A.; Han, W.; Wojtecki, R.J.; Maune, H.; Le, A.B.A.; Thompson, L.E.; McClelland, G.M.; Ribet, F.; Engler, A.C.; et al. Dual-Responsive Hydrogels for Direct-Write 3D Printing. Macromolecules 2015, 48, 6482–6488. [Google Scholar] [CrossRef]
- Kolesky, D.B.; Truby, R.L.; Gladman, A.S.; Busbee, T.A.; Homan, K.A.; Lewis, J.A. 3D Bioprinting of Vascularized, Heterogeneous Cell-Laden Tissue Constructs. Adv. Mater. 2014, 26, 3124–3130. [Google Scholar] [CrossRef] [PubMed]
- Buwalda, S.J.; Boere, K.W.M.; Dijkstra, P.J.; Feijen, J.; Vermonden, T.; Hennink, W.E. Hydrogels in a Historical Perspective: From Simple Networks to Smart Materials. J. Control. Release 2014, 190, 254–273. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Atala, A. Biomaterials for Integration with 3-D Bioprinting. Ann. Biomed. Eng. 2015, 43, 730–746. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.J. Polymer Chemistry: A Practical Approach; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Webster, O.W.; Hertler, W.R.; Sogah, D.Y.; Farnham, W.B.; RajanBabu, T.V. Group-Transfer Polymerization. 1. A New Concept for Addition Polymerization with Organosilicon Initiators. J. Am. Chem. Soc. 1983, 105, 5706–5708. [Google Scholar] [CrossRef]
- Hatada, K.; Kitayama, T.; Vogl, O. Macromolecular Desing of Polymeric Materials; Marcel Dekker, Inc.: New York, NY, USA, 1997; p. 878. [Google Scholar]
- Ward, M.A.; Georgiou, T.K. Multicompartment Thermoresponsive Gels: Does the Length of the Hydrophobic Side Group Matter? Polym. Chem. 2013, 4, 1893–1902. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Gels Based on ABA Triblock Copolymers: Does the Asymmetry Matter? J. Polym. Sci. A 2013, 51, 2850–2859. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Terpolymers Based on Methacrylate Monomers: Effect of Architecture and Composition. J. Polym. Sci. A 2010, 48, 775–783. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Triblock Copolymers Based on Methacrylate Monomers: Effect of Molecular Weight and Composition. Soft Matter 2012, 8, 2737–2745. [Google Scholar] [CrossRef]
- Constantinou, A.P.; Georgiou, T.K. Thermoresponsive Gels Based on ABC Triblock Copolymers: Effect of the Length of the PEG Side Group. Polym. Chem. 2016, 7, 2045–2056. [Google Scholar] [CrossRef]
- Dicker, I.B.; Cohen, G.M.; Farnham, W.B.; Hertler, W.R.; Laganis, E.D.; Sogah, D.Y. Oxyanions Catalyze Group-Transfer Polymerization to Give Living Polymers. Macromolecules 1990, 23, 4034–4041. [Google Scholar] [CrossRef]
- Patrickios, C.S.; Lowe, A.B.; Armes, S.P.; Billingham, N.C. ABC Triblock Polymethacrylates: Group Transfer Polymerization Synthesis of the ABC, ACB, and BAC Topological Isomers and Solution Characterization. J. Polym. Sci. A 1998, 36, 617–631. [Google Scholar] [CrossRef]
- Raduan, N.H.; Horozov, T.S.; Georgiou, T.K. “Comb-Like” Non-Ionic Polymeric Macrosurfactants. Soft Matter 2010, 6, 2321–2329. [Google Scholar] [CrossRef]
- Hadjiyannakou, S.C.; Vamvakaki, M.; Patrickios, C.S. Synthesis, Characterization and Evaluation of Amphiphilic Diblock Copolymer Emulsifiers Based on Methoxy Hexa(Ethylene Glycol) Methacrylate and Benzyl Methacrylate. Polymer 2004, 45, 3681–3692. [Google Scholar] [CrossRef]
- Wu, C.; Ying, A.; Ren, S. Fabrication of Polymeric Micelles with Core-Shell-Corona Structure for Applications in Controlled Drug Release. Colloid Polym. Sci. 2013, 291, 827–834. [Google Scholar] [CrossRef]
- Georgiou, T.K.; Vamvakaki, M.; Patrickios, C.S.; Yamasaki, E.N.; Phylactou, L.A. Nanoscopic Cationic Methacrylate Star Homopolymers: Synthesis by Group Transfer Polymerization, Characterization and Evaluation as Transfection Reagents. Biomacromolecules 2004, 5, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, T.K.; Phylactou, L.A.; Patrickios, C.S. Synthesis, Characterization, and Evaluation as Transfection Reagents of Ampholytic Star Copolymers: Effect of Star Architecture. Biomacromolecules 2006, 7, 3505–3512. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.R.; Patrickios, C.S. Synthesis and Aqueous Solution Characterization of Catalytically Active Block Copolymers Containing Imidazole. Macromolecules 1998, 31, 9075–9077. [Google Scholar] [CrossRef]
- Emileh, A.; Vasheghani-Farahani, E.; Imani, M. Swelling Behavior, Mechanical Properties and Network Parameters of pH- and Temperature-Sensitive Hydrogels of Poly((2-Dimethyl Amino) Ethyl Methacrylate-co-Butyl Methacrylate). Eur. Polym. J. 2007, 43, 1986–1995. [Google Scholar] [CrossRef]
- Philippova, O.E.; Hourdet, D.; Audebert, R.; Khokhlov, A.R. pH-Responsive Gels of Hydrophobically Modified Poly(Acrylic Acid). Macromolecules 1997, 30, 8278–8285. [Google Scholar] [CrossRef]
- De Souza, J.C.P.; Naves, A.F.; Florenzano, F.H. Specific Thermoresponsiveness of PMMA-Block-PDMAEMA to Selected Ions and Other Factors in Aqueous Solution. Colloid Polym. Sci. 2012, 290, 1285–1291. [Google Scholar] [CrossRef]
- Fournier, D.; Hoogenboom, R.; Thijs, H.M.L.; Paulus, R.M.; Schubert, U.S. Tunable pH- and Temperature-Sensitive Copolymer Libraries by Reversible Addition-Fragmentation Chain Transfer Copolymerizations of Methacrylates. Macromolecules 2007, 40, 915–920. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer No. | Theoretical polymer structure a | % w/w TEGMA-BuMA-DMAEMA | MM Theor. b (g·mol−1) | Mn c (g·mol−1) | Ð c | |
|---|---|---|---|---|---|---|
| Theoretical | 1H-NMR | |||||
| 1 | TEGMA9 | 100-00-00 | 100-00-00 | 2125 | 3020 | 1.12 |
| TEGMA9-b-BuMA20 | 42-58-00 | 42-58-00 | 4960 | 6600 | 1.12 | |
| TEGMA9-b-BuMA20-b-DMAEMA21 | 25-35-40 | 25-35-40 | 8200 | 10,700 | 1.17 | |
| 2 | TEGMA9 | 100-00-00 | 100-00-00 | 2125 | 3030 | 1.11 |
| TEGMA9-b-DMAEMA21 | 38-00-62 | 38-00-62 | 5365 | 7650 | 1.11 | |
| TEGMA9-b-DMAEMA21-b-BuMA20 | 25-35-40 | 26-34-40 | 8200 | 10,800 | 1.16 | |
| 3 | BuMA20 | 00-100-00 | 00-100-00 | 2935 | 2980 | 1.12 |
| BuMA20-b-TEGMA9 | 42-58-00 | 41-59-00 | 4960 | 6700 | 1.08 | |
| BuMA20-b-TEGMA9-b-DMAEMA21 | 25-35-40 | 26-35-39 | 8200 | 10,100 | 1.13 | |
| 4 | TEGMA9-co-BuMA20 | 42-58-00 | 42-58-00 | 4960 | 5940 | 1.09 |
| (TEGMA9-co-BuMA20)-b-DMAEMA21 | 25-35-40 | 26-35-39 | 8200 | 8780 | 1.10 | |
| 5 | TEGMA9 | 100-00-00 | 100-00-00 | 2125 | 2810 | 1.12 |
| TEGMA9-b-(BuMA20-co-DMAEMA21) | 25-35-40 | 26-34-40 | 8200 | 9950 | 1.14 | |
| 6 | BuMA20 | 00-100-00 | 00-100-00 | 2935 | 4180 | 1.10 |
| BuMA20-b-(TEGMA9-co-DMAEMA21) | 25-35-40 | 26-37-37 | 8200 | 8580 | 1.13 | |
| 7 | TEGMA9-co-BuMA20-co-DMAEMA21 | 25-35-40 | 26-35-39 | 8200 | 8760 | 1.10 |
| 8 | BuMA26 | 00-100-00 | 00-100-00 | 3826 | 6240 | 1.07 |
| BuMA26-b-DMAEMA28 | 00-46-54 | 00-47-53 | 8200 | 11,000 | 1.11 | |
| 9 | BuMA20 | 00-100-00 | 00-100-00 | 2935 | 4040 | 1.10 |
| BuMA20-b-DMAEMA34 | 00-35-65 | 00-37-63 | 8200 | 9380 | 1.10 | |
| Polymer No. | Theoretical polymer structure a | Hydrodynamic diameter (nm) | Effective pKas ± 0.1 | Cloud Points ± 2 °C | |||
|---|---|---|---|---|---|---|---|
| Theoretical b | Experimental ± 0.5 | 0% H+ | 10% H+ | ||||
| pH = 7 | pH = 6 | ||||||
| 1 | TEGMA9-b-BuMA20-b-DMAEMA21 | 20.5 c | 8.7 | 6.5 | 6.7 | 29 | No CP |
| 2 | TEGMA9-b-DMAEMA21-b-BuMA20 | 26.6 d | 32.7 | 32.7 | 6.8 | NA | No CP |
| 3 | BuMA20-b-TEGMA9-b-DMAEMA21 | 24.8 d | 18.2 | 13.5 | 6.9 | NA | No CP |
| 4 | (TEGMA9-co-BuMA20)-b-DMAEMA21 | 19.1 e | 21.0 | 24.4 | 6.9 | No CP | No CP |
| 5 | TEGMA9-b-(BuMA20-co-DMAEMA21) | 18.1 f | 25.5 | 21.0 | 6.2 | 32 | No CP |
| 6 | BuMA20-b-(TEGMA9-co-DMAEMA21) | 20.8 d | 28.2 | 18.2 | 6.7 | NA | No CP |
| 7 | TEGMA9-co-BuMA20-co-DMAEMA21 | 2.7 g | NA | 15.7 | 6.1 | NA | NA |
| 8 | BuMA26-b-DMAEMA28 | 28.1 c | 32.7 | 15.7 | 6.5 | NA | No CP |
| 9 | BuMA20-b-DMAEMA34 | 25.3 c | 21.0 | 15.7 | 6.8 | NA | No CP |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Constantinou, A.P.; Zhao, H.; McGilvery, C.M.; Porter, A.E.; Georgiou, T.K. A Comprehensive Systematic Study on Thermoresponsive Gels: Beyond the Common Architectures of Linear Terpolymers. Polymers 2017, 9, 31. https://doi.org/10.3390/polym9010031
Constantinou AP, Zhao H, McGilvery CM, Porter AE, Georgiou TK. A Comprehensive Systematic Study on Thermoresponsive Gels: Beyond the Common Architectures of Linear Terpolymers. Polymers. 2017; 9(1):31. https://doi.org/10.3390/polym9010031
Chicago/Turabian StyleConstantinou, Anna P., Hanyi Zhao, Catriona M. McGilvery, Alexandra E. Porter, and Theoni K. Georgiou. 2017. "A Comprehensive Systematic Study on Thermoresponsive Gels: Beyond the Common Architectures of Linear Terpolymers" Polymers 9, no. 1: 31. https://doi.org/10.3390/polym9010031