
Effect of Hydroxyapatite Nanoparticles on the Degradability of Random Poly(butylene terephthalate-co-aliphatic dicarboxylate)s Having a High Content of Terephthalic Units

 , and
, and 
Abstract
:
1. Introduction
2. Experimental Section
- (a)
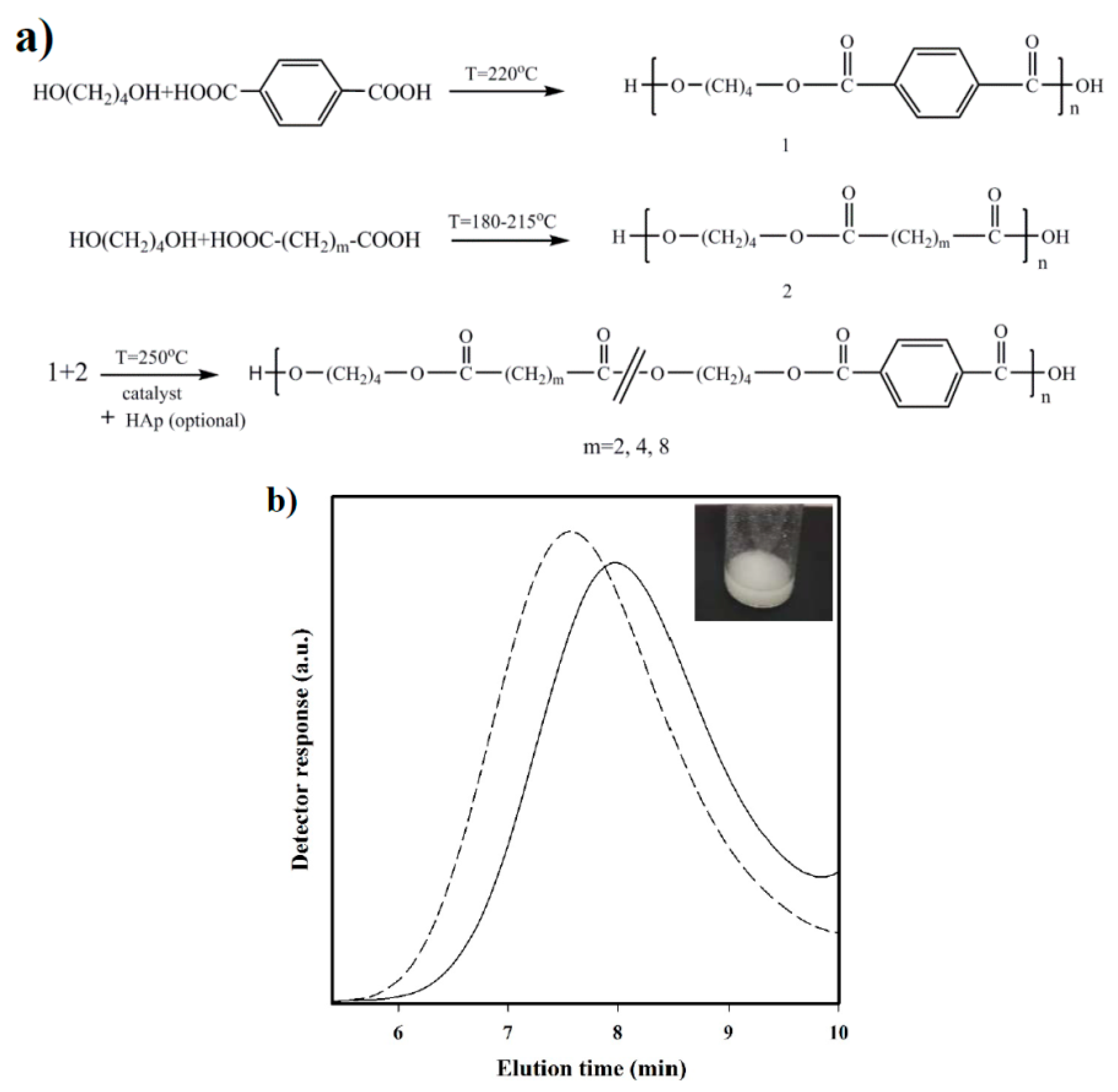
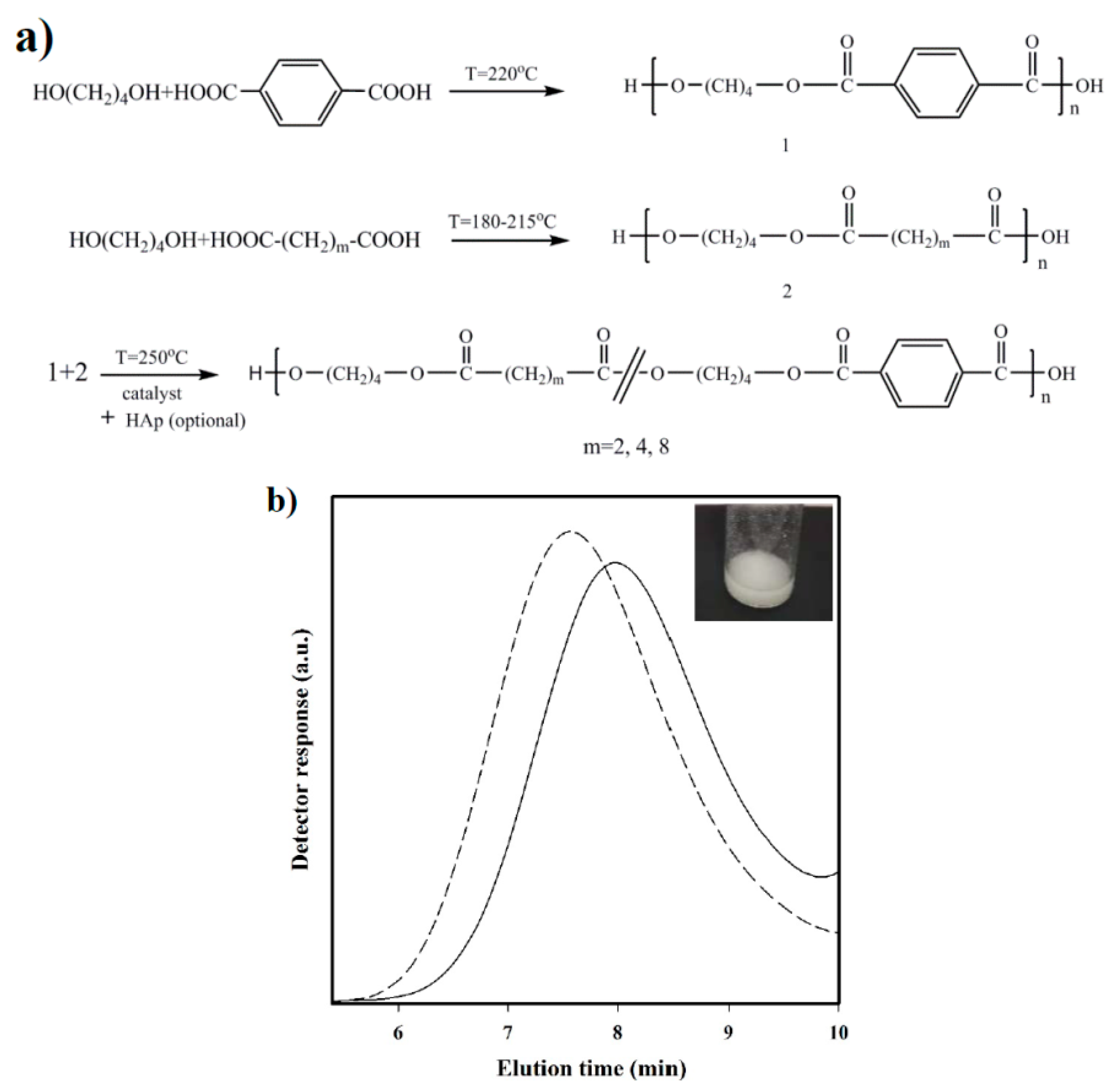
- Synthesis of prepolymers from 1,4-butanediol and the selected dicarboxylic unit. Polycondensation reactions were carried out using an excess of 1,4-butanediol (BDO) (i.e., 1.7:1 [OH]:[COOH]). After loading the reactor with 650 g of a mixture of BDO and the appropriate dicarboxylic acid, the reaction mixture was firstly stirred for 30 min at 140 °C under a pressure of 3–3.5 bars. A flow of N2 was provided to keep the required pressure while an electric condenser allowed separating the condensed water and the excess of alcohol. Reaction temperature was then increased to 215, 207, 200, and 220 °C, for sebacic, adipic, succinic, and terephthalic acid derivatives, respectively. The reaction was stopped when no more water could be recovered (approximately after 180 min). In fact, the reaction extent could be evaluated by weighing the water that was recovered at regular time intervals.
- (b)
- Vacuum polycondensation and thermal transesterification between aliphatic and aromatic prepolymers. The appropriate mixtures constituted by a 0.3:0.7 molar ratio of prepolymers derived from the aliphatic and aromatic acids and TBT as catalyst (1.4 mmol for 1 mol of dicarboxylic acid) were transferred to the reactor for 10 min at 200 °C and atmospheric pressure. Temperature was subsequently increased up to 250 °C while a vacuum (of 20 mbar) was applied. Once the mixer torque reached the desired value, the reaction was stopped (approximately after 150 min). Copolymers were dissolved in 1,1,1,3,3,3-hexafluroisopropanol (HFIP), precipitated in water, washed several times with water, methanol, and ether, and finally dried in a vacuum desiccator.
3. Results and Discussion
3.1. Synthesis and Characterization of Copolymers and Nanocomposites
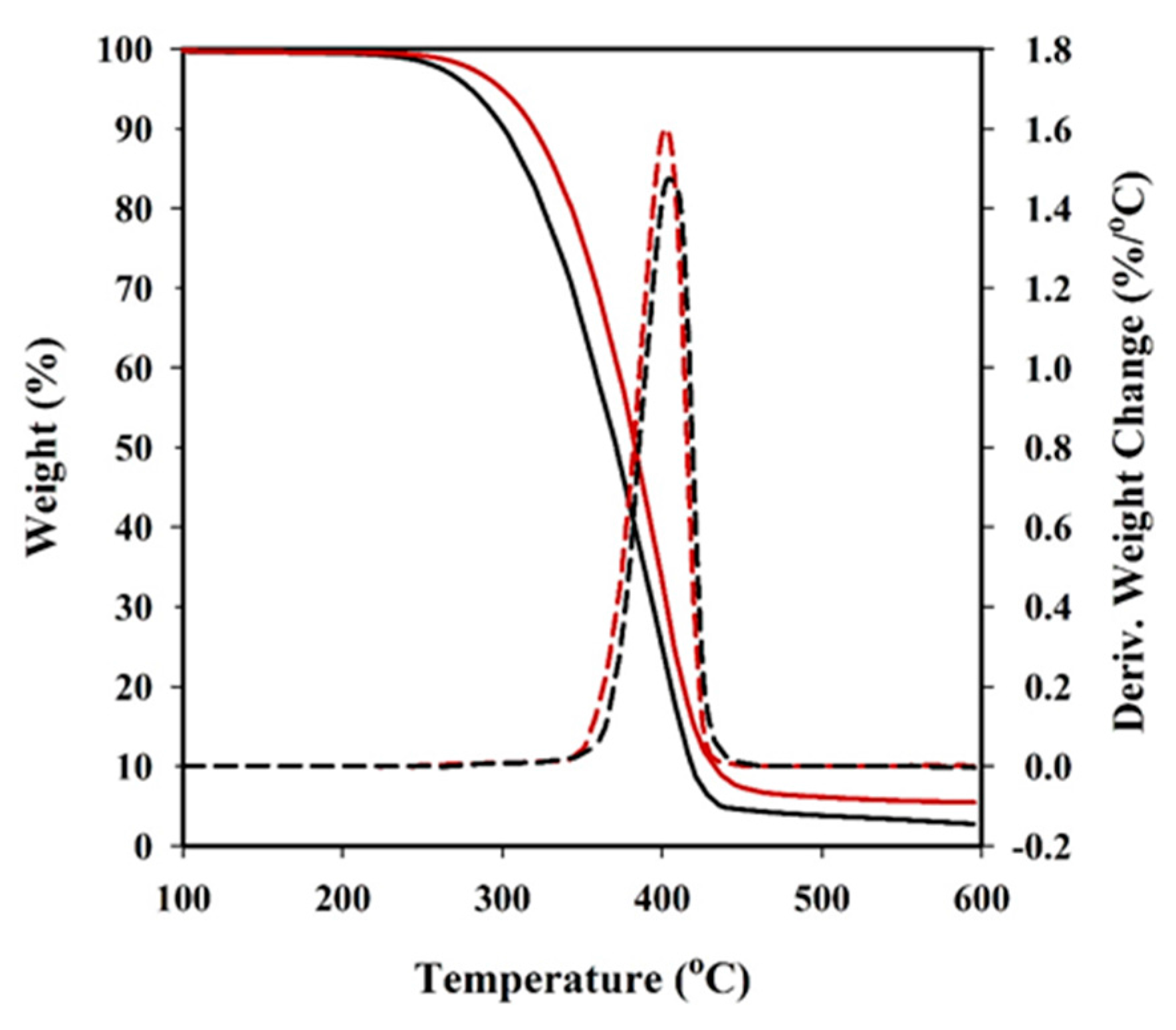
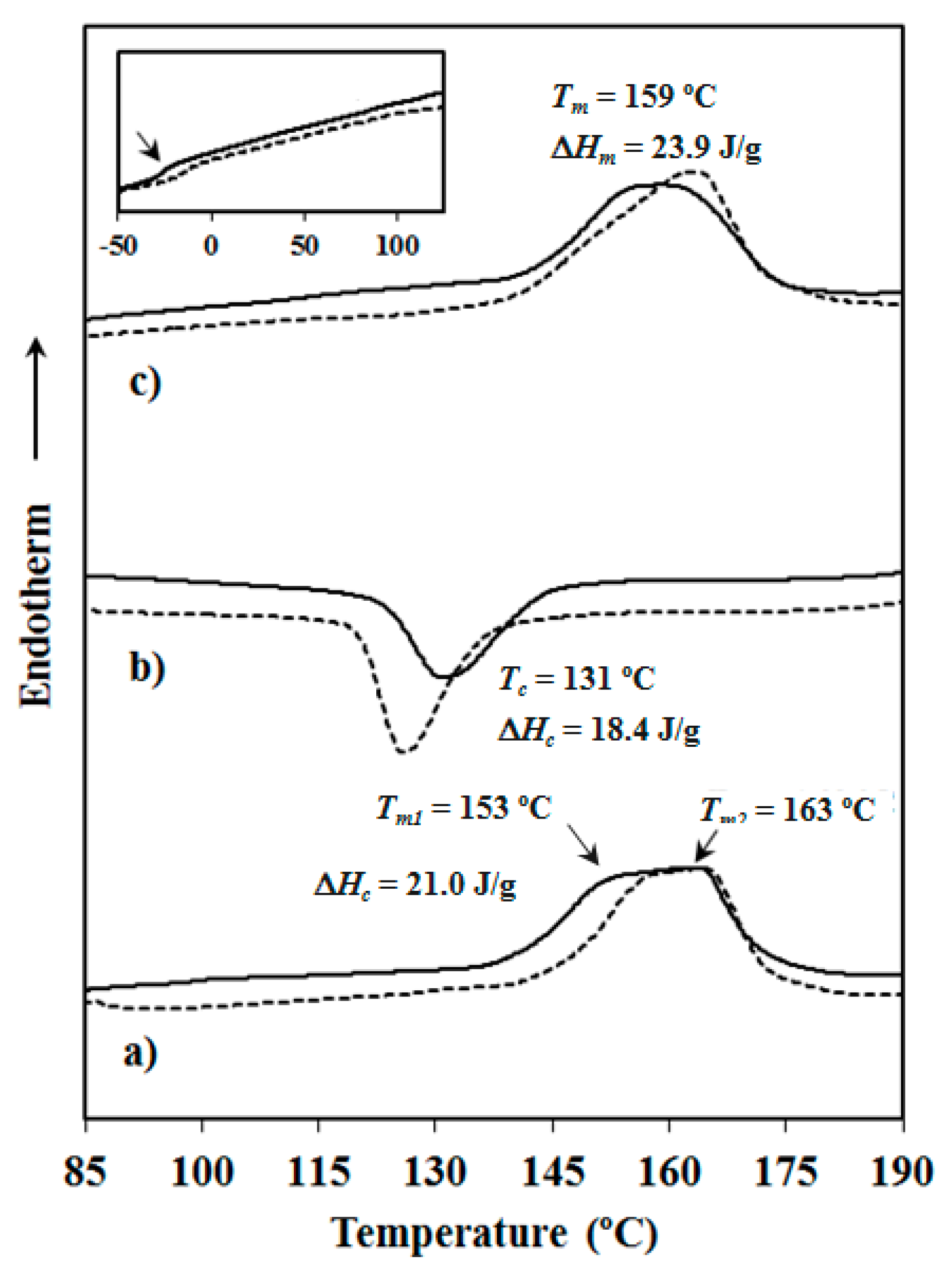
3.2. Thermal Properties of Copolyesters and Nanocomposites
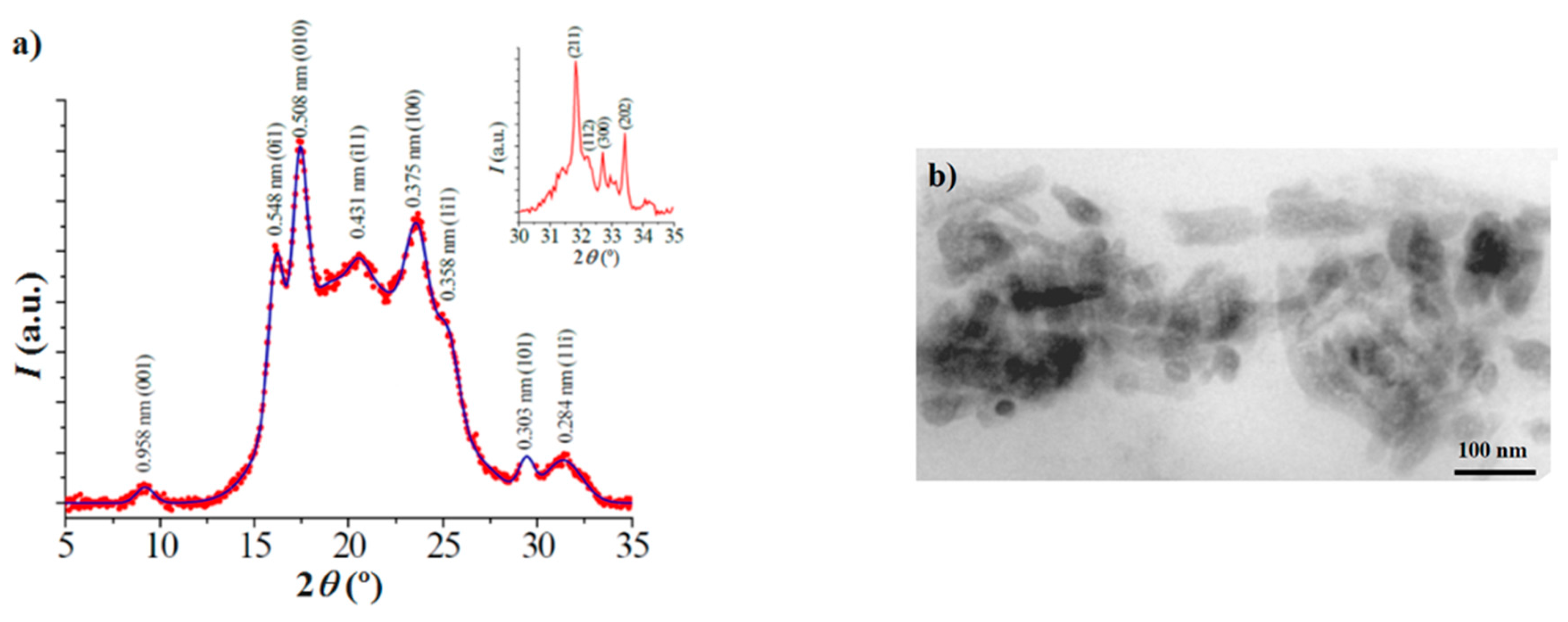
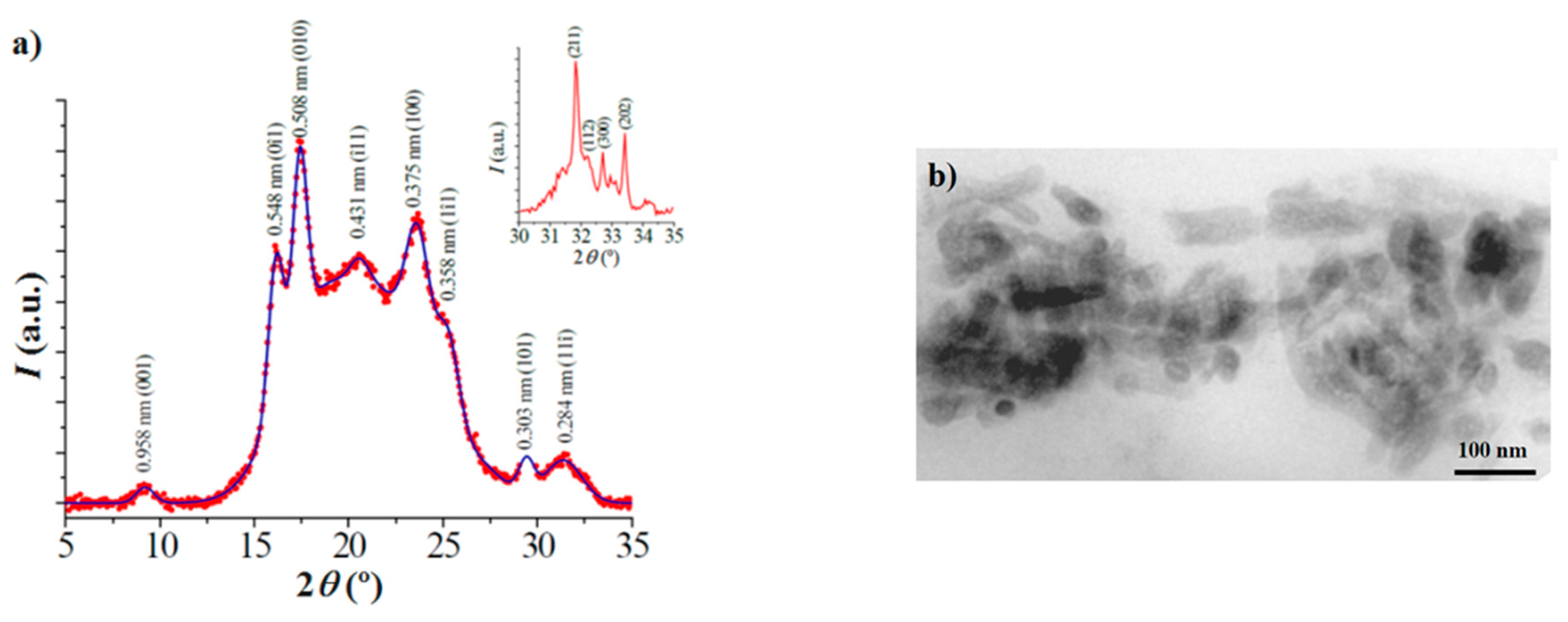
3.3. X-ray Diffraction Data
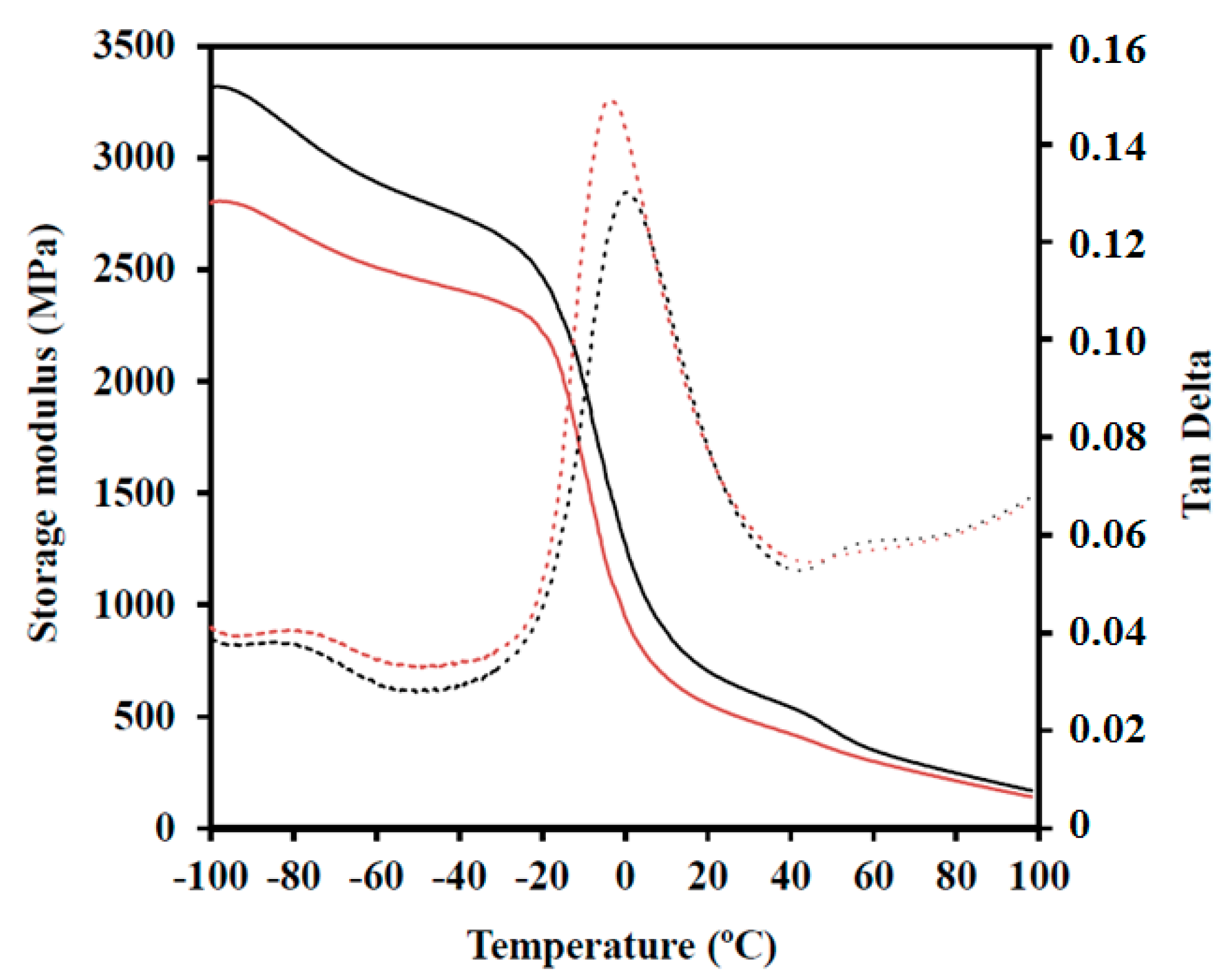
3.4. Mechanical Properties
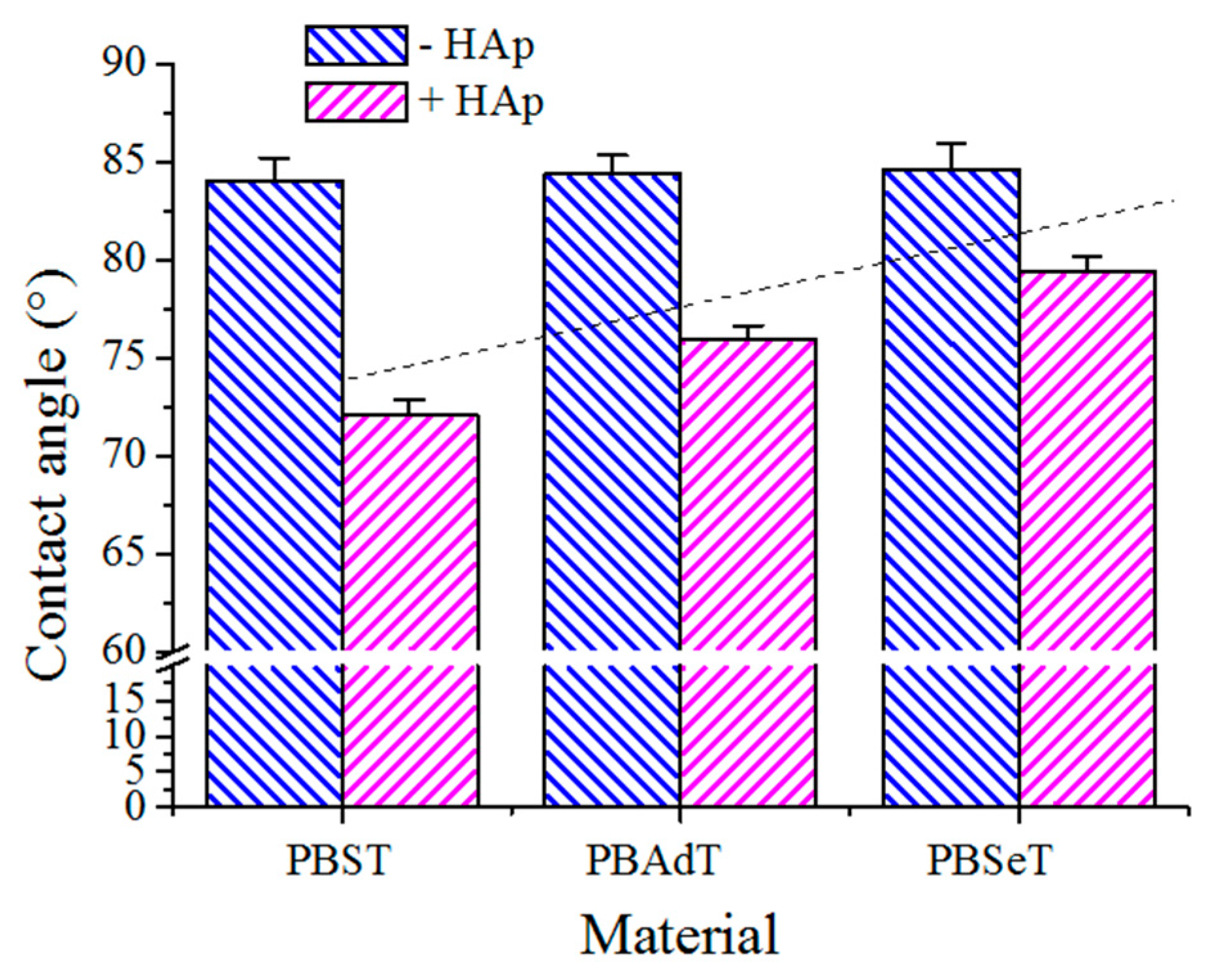
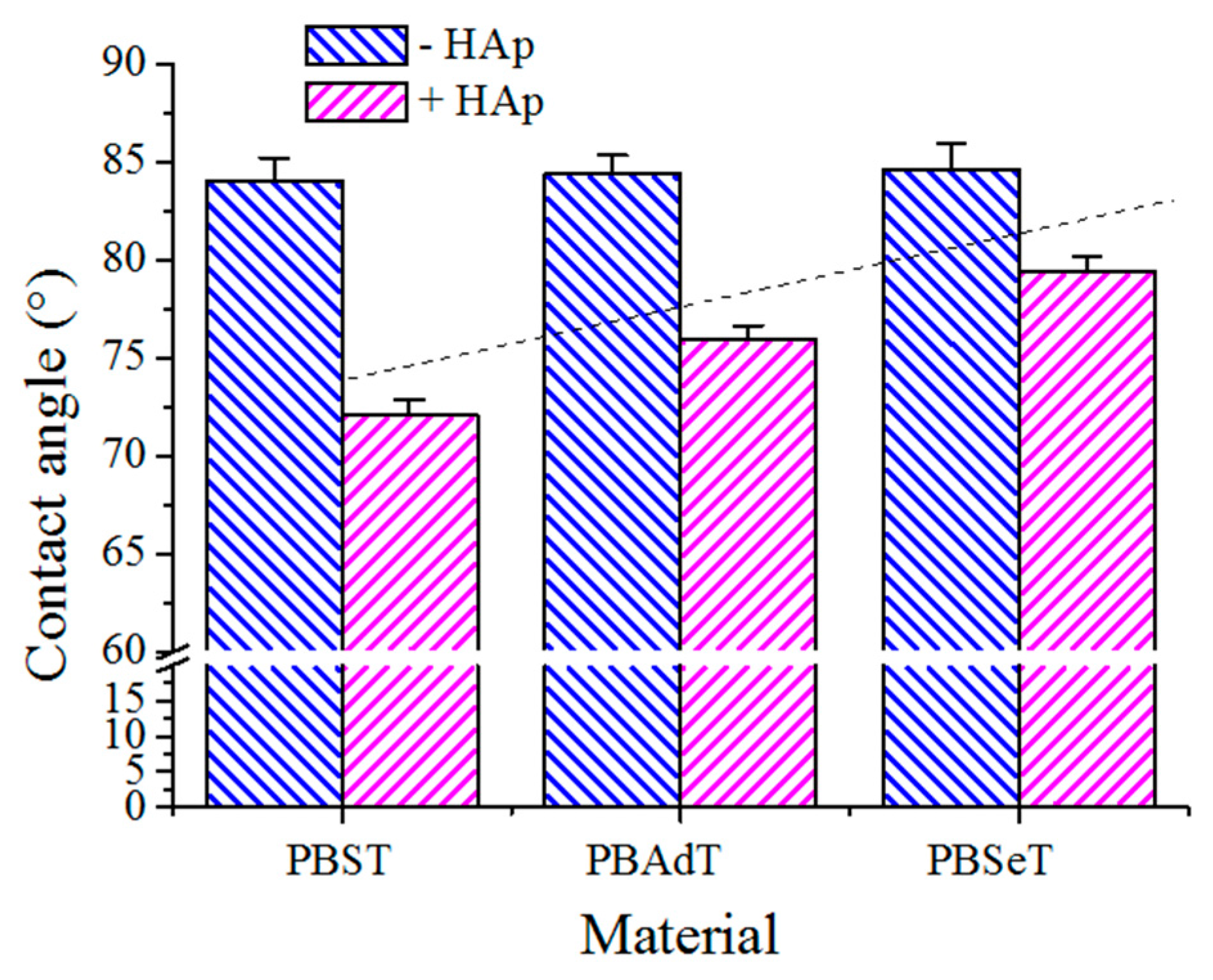
3.5. Contact Angle Measurements
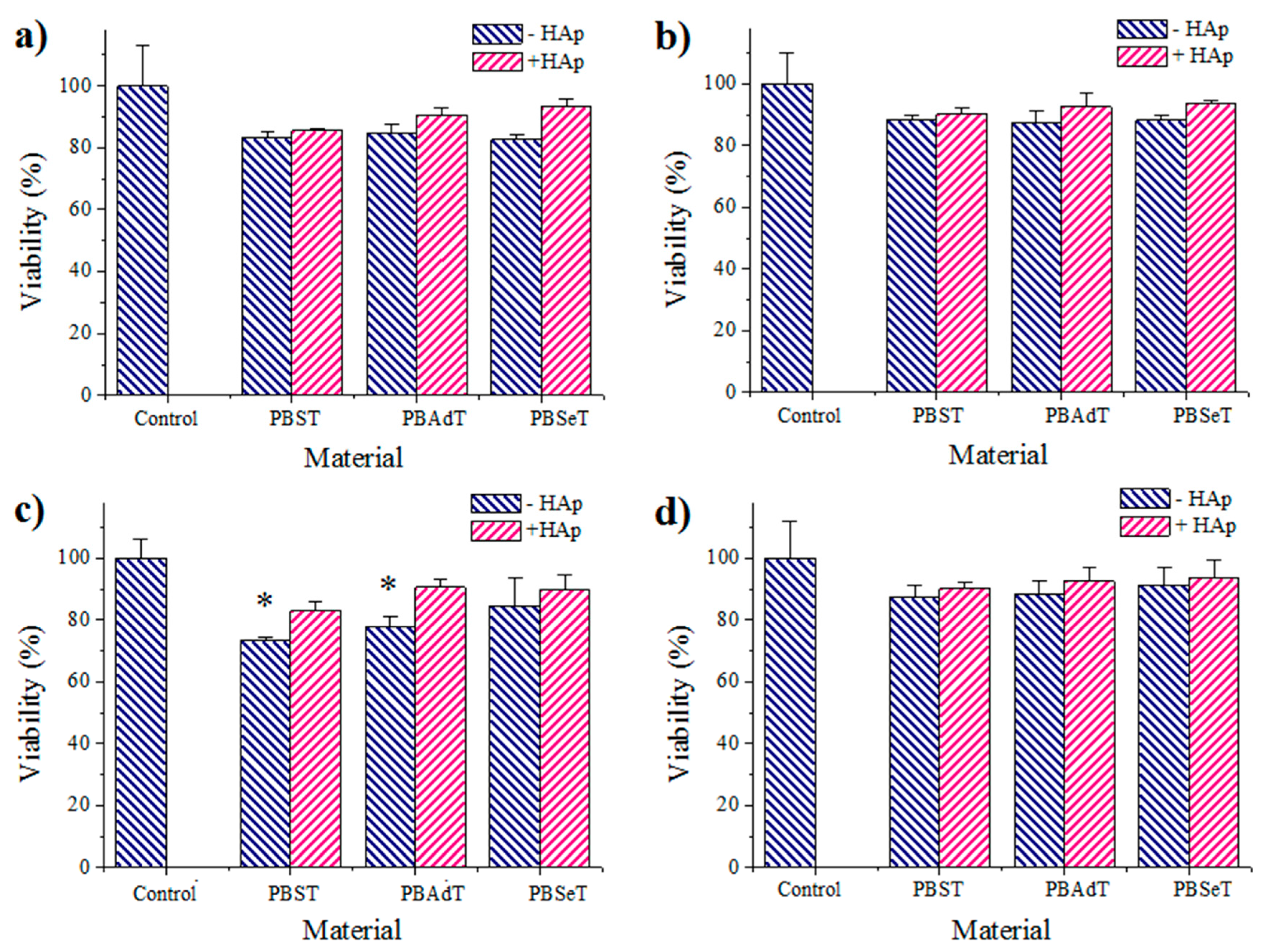
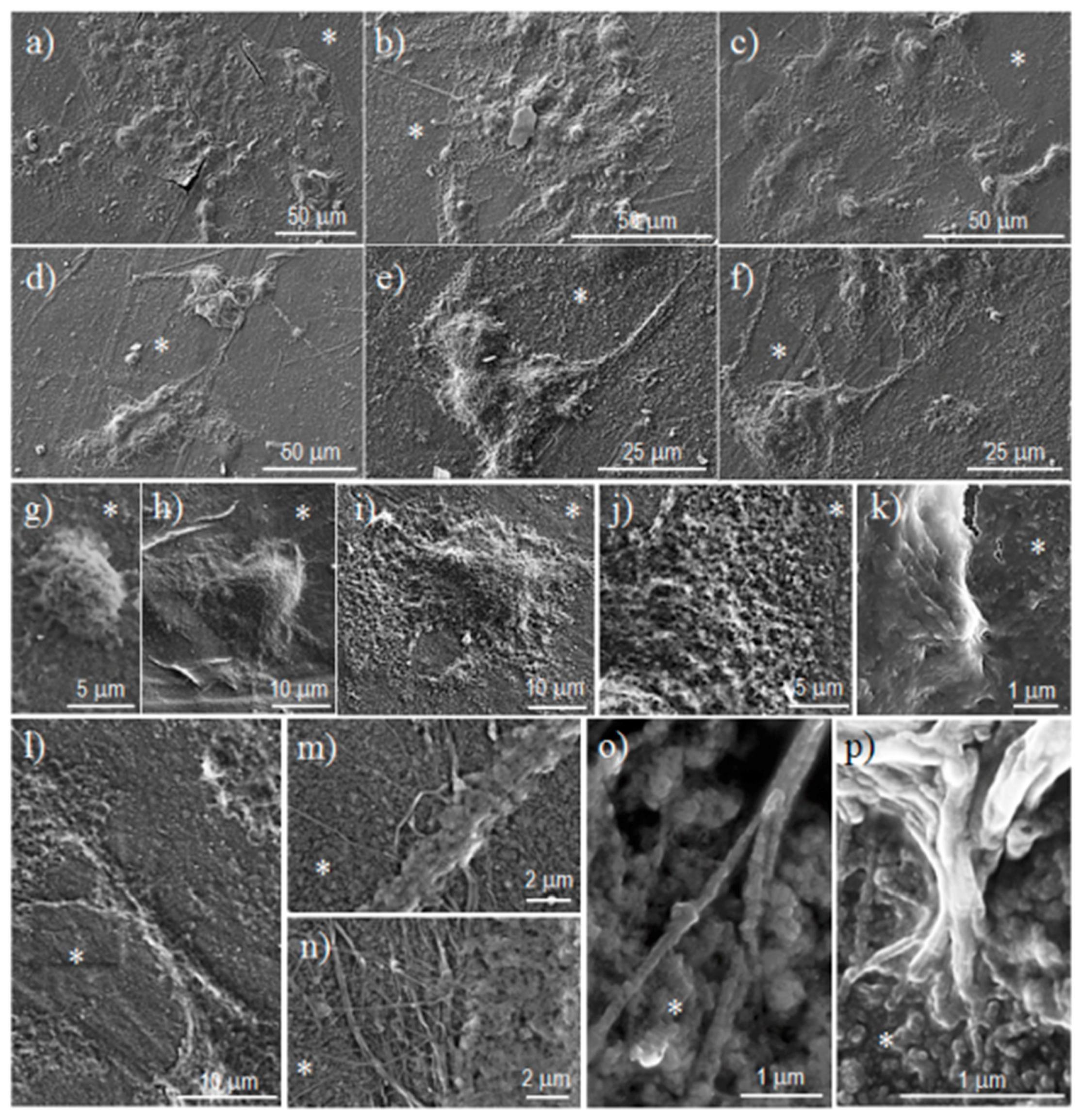
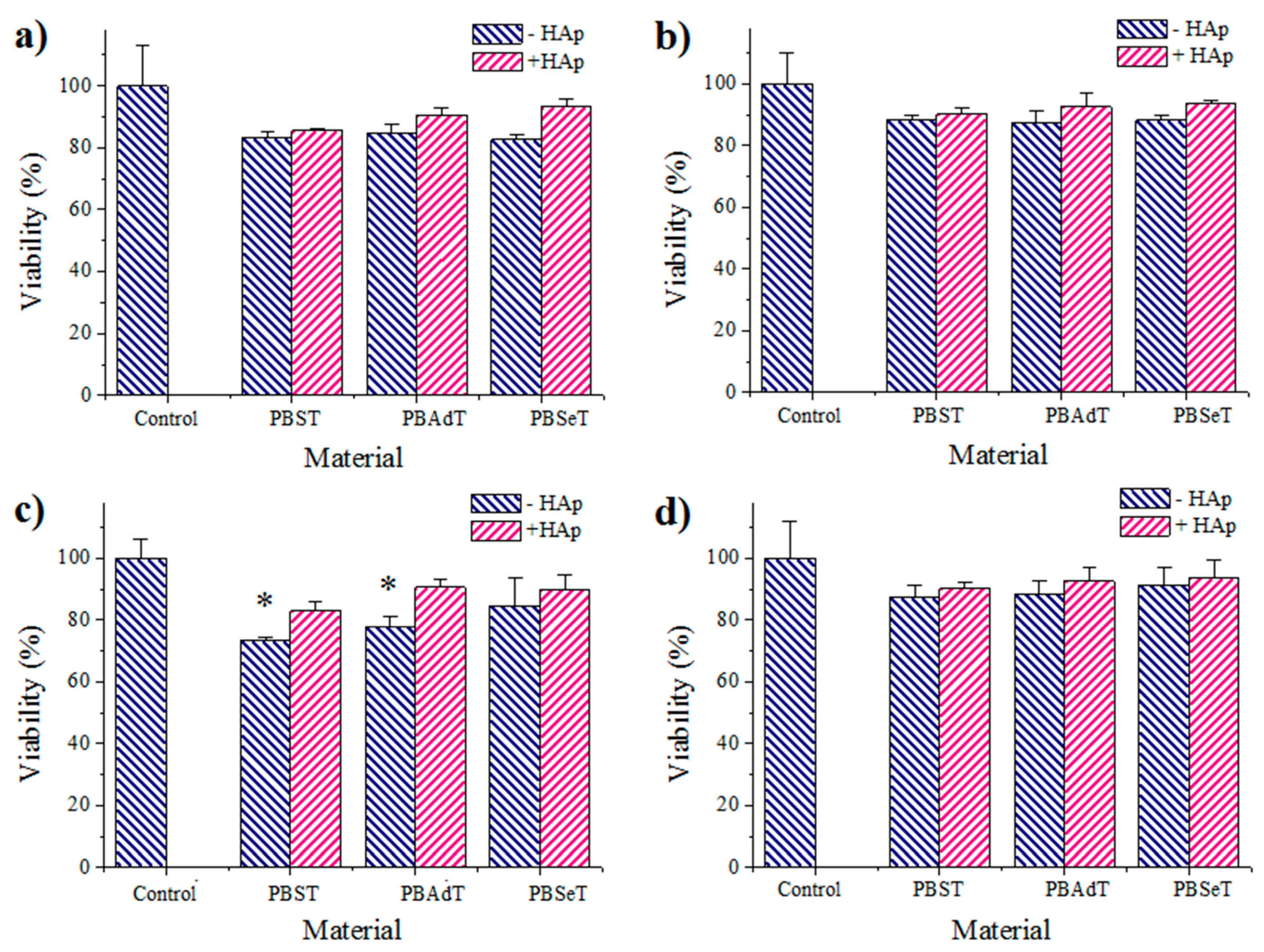
3.6. Cell Adhesion and Proliferation
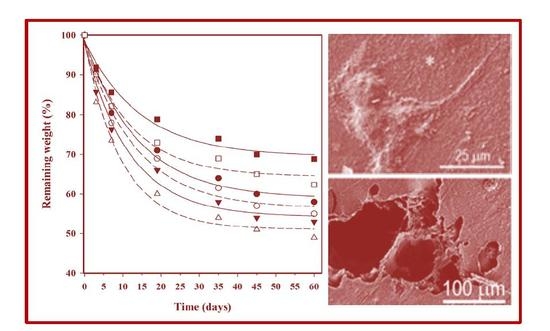
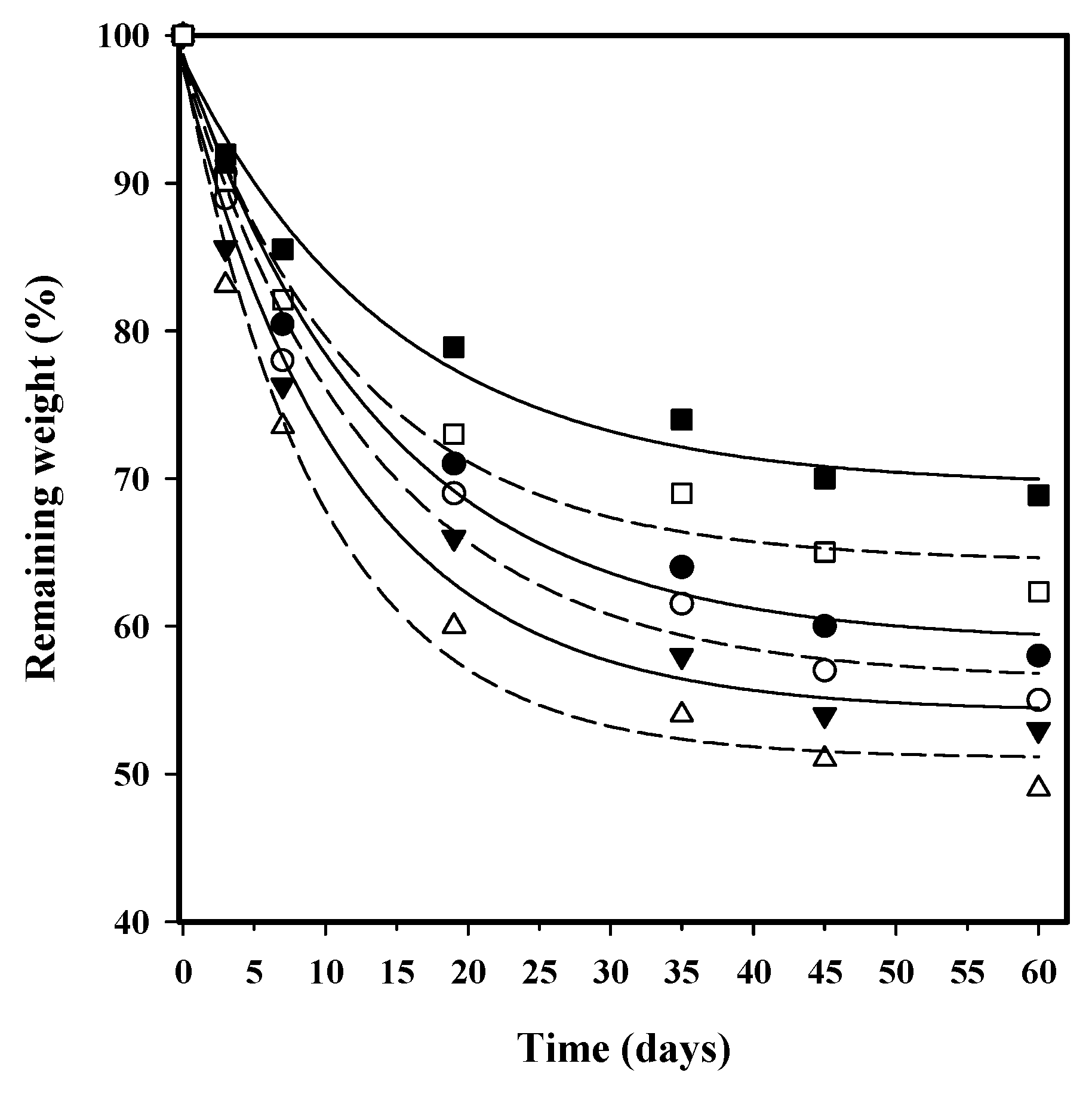
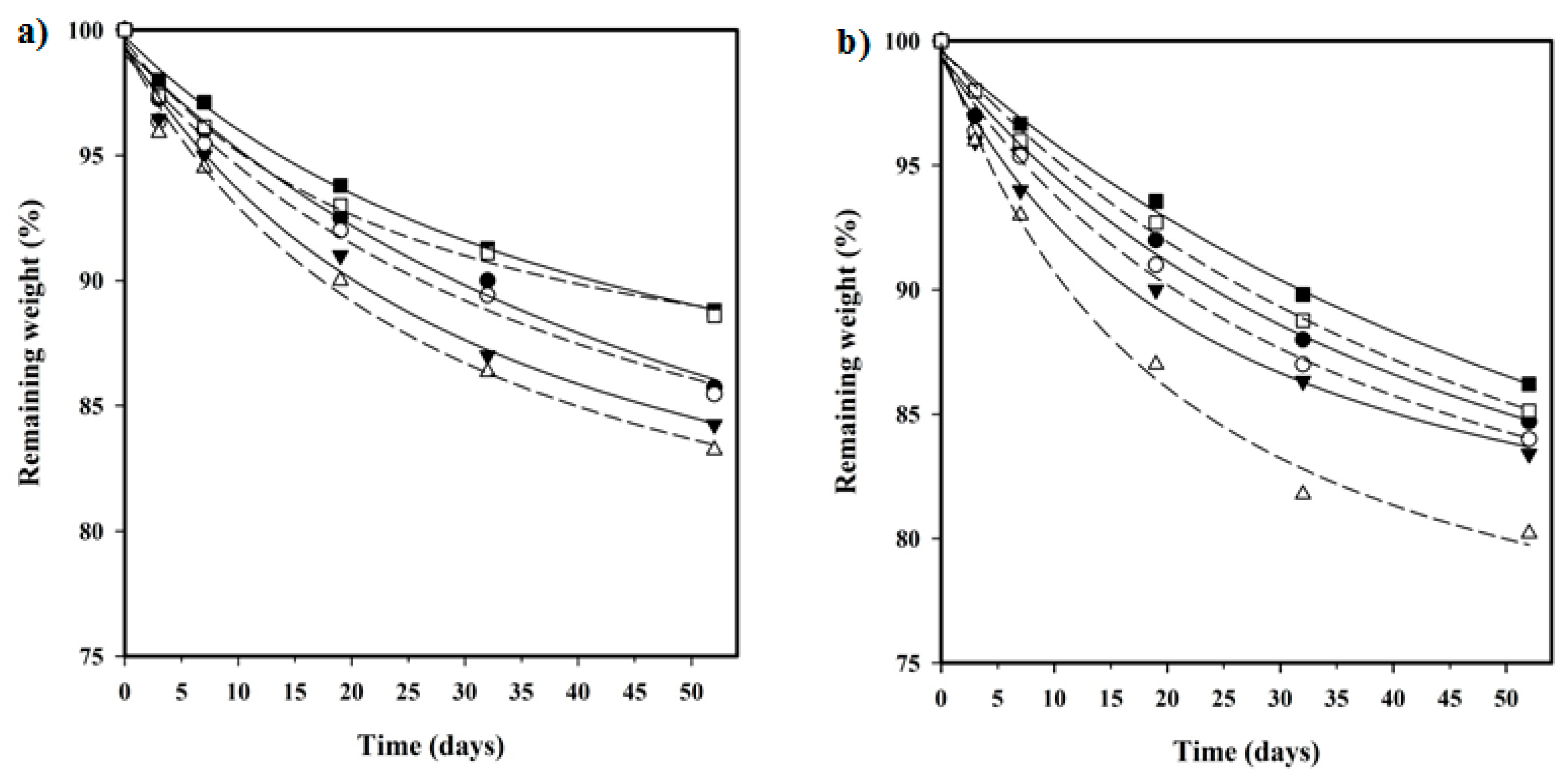
3.7. Hydrolytic Degradation
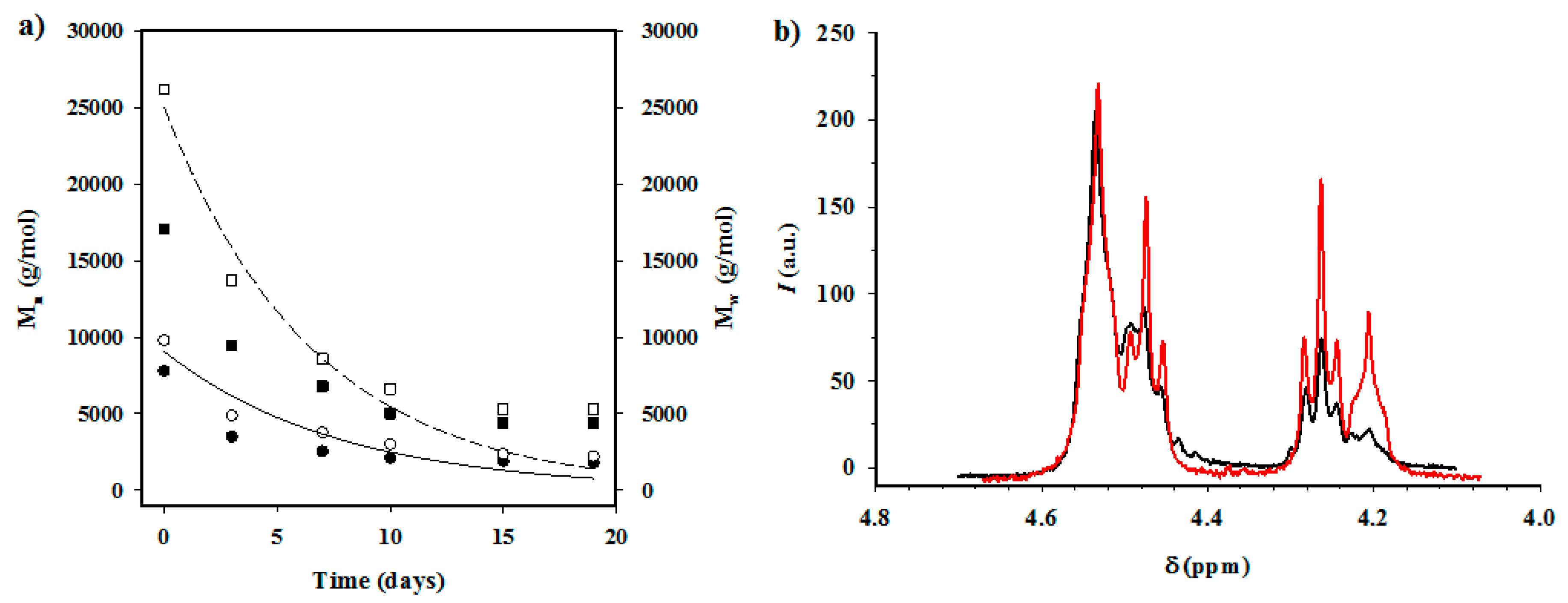
3.8. Enzymatic Degradation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical applications of biodegradable polymers. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef] [PubMed]
- Seyednejad, H.; Ghassemi, A.H.; van Nostrum, C.F.; Vermonden, T.; Hennink, W.E. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J. Control. Release 2011, 152, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Raquez, J.M.; Nabar, Y.; Narayan, R.; Dubois, P. Novel high-performance talc/poly[(butylene adipate)-co-terephthalate] hybrid materials. Macromol. Mater. Eng. 2008, 293, 310–320. [Google Scholar] [CrossRef]
- Tian, H.; Tang, Z.; Zhuang, X.; Chen, X.; Jing, X. Biodegradable synthetic polymers: Preparation, functionalization and biomedical application. Prog. Polym. Sci. 2012, 37, 237–280. [Google Scholar] [CrossRef]
- Musyanovych, A.; Landfester, K. Biodegradable polyester-based nanoparticle formation by miniemulsion technique. Mater. Matters 2012, 7, 30–32. [Google Scholar]
- Díaz, A.; Katsarava, R.; Puiggalí, J. Synthesis, properties and applications of biodegradable polymers derived from diols and dicarboxylic acids: From polyesters to poly(ester amide)s. Int. J. Mol. Sci. 2014, 15, 7064–7123. [Google Scholar] [CrossRef] [PubMed]
- Fujimaki, T. Processability and properties of aliphatic polyesters, “BIONOLLE”, synthesized by polycondensation reaction. Polym. Degrad. Stab. 1998, 59, 209–214. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Mizukoshi, T. Bionolle (polybutylenesuccinate). In Synthetic Biodegradable Polymers; Rieger, B., Künkel, A., Coates, W.G., Reichardt, R., Dinjus, E., Zevaco, A.T., Eds.; Springer: Berlin & Heidelberg, Germany, 2012; pp. 285–313. [Google Scholar]
- Witt, U.; Müller, R.-J.; Deckwer, W.-D. Biodegradation behavior and material properties of aliphatic/aromatic polyesters of commercial importance. J. Environ. Polym. Degrad. 1997, 5, 81–89. [Google Scholar] [CrossRef]
- Müller, R.J.; Witt, U.; Rantze, E.; Deckwer, W.D. Architecture of biodegradable copolyesters containing aromatic constituents. Polym. Degrad. Stab. 1998, 59, 203–208. [Google Scholar] [CrossRef]
- Witt, U.; Müller, R.J.; Deckwer, W.D. Studies on sequence distribution on aliphatic/aromatic copolyesters by high-resolution 13C nuclear magnetic resonance spectroscopy for evaluation of biodegradability. Macromol. Chem. Phys. 1996, 197, 1525–1535. [Google Scholar] [CrossRef]
- Kleeberg, I.; Hetz, C.; Kroppenstedt, R.M.; Müller, R.J.; Deckwer, W.D. Biodegradation of aliphatic/aromatic copolyesters by thermophilic actinomycetes. Appl. Environ. Microbiol. 1998, 64, 1731–1735. [Google Scholar] [PubMed]
- Witt, U.; Einig, T.; Yamamoto, M.; Kleeberg, I.; Deckwer, W.-D.; Müller, R.-J. Biodegradation of aliphatic–aromatic copolyesters: Evaluation of the final biodegradability and ecotoxicological impact of degradation intermediates. Chemosphere 2001, 44, 289–299. [Google Scholar] [CrossRef]
- Kleeberg, I.; Welzel, K.; VandenHeuvel, J.; Müller, R.J.; Deckwer, W.-D. Characterization of a new extracellular hydrolase from thermobifida fusca degrading aliphatic-aromatic copolyesters. Biomacromolecules 2005, 6, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, J.; Guo, B.; Xie, X. Crystallization kinetics and morphology of biodegradable poly(butylene succinate-co-propylene succinate)s. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 420–428. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.N. Synthesis cocrystallization enzymatic degradation of novel poly(butylene-co-propylene succinate) copolymers. Biomacromolecules 2007, 8, 2437–2449. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.F.; Chen, M.; Shih, Y.C.; Chen, C.H. Nonisothermal crystallization kinetics of biodegradable poly(butylene succinate-co-propylene succinate)s. J. Polym. Sci. Part B Polym. Phys. 2010, 48, 1299–1308. [Google Scholar] [CrossRef]
- Herrera, R.; Franco, L.; Rodríguez-Galán, A.; Puiggalí, J. Characterization and degradation behavior of poly(butylene adipate-co-terephthalate)s. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 4141–4157. [Google Scholar] [CrossRef]
- Marten, E.; Müller, R.-J.; Deckwer, W.-D. Studies on the enzymatic hydrolysis of polyesters. II. aliphatic-aromatic copolyesters. Polym. Degrad. Stab. 2005, 88, 371–381. [Google Scholar] [CrossRef]
- Jaisankar, V.; Nanthini, R.; Ravi, A.; Karunanidhi, M. A study on biodegradation of aliphatic-aromatic random copolyesters. J. Polym. Mater. 2009, 26, 157–166. [Google Scholar]
- Kijchavengkul, T.; Auras, R.; Rubino, M.; Selke, S.; Ngouajio, M.; Fernandez, R.T. Biodegradation and hydrolysis rate of aliphatic aromatic polyester. Polym. Degrad. Stab. 2010, 95, 2641–2647. [Google Scholar] [CrossRef]
- Armentano, I.; Dottori, M.; Fortunati, E.; Mattioli, S.; Kenny, J.M. Biodegradable polymer matrix nanocomposites for tissue engineering: A review. Polym. Degrad. Stab. 2010, 95, 2126–2146. [Google Scholar] [CrossRef]
- Furuichi, K.; Oaki, Y.; Imai, H. Preparation of nanotextured and nanofibrous hydroxyapatite through dicalcium phosphate with gelatin. Chem. Mater. 2006, 18, 229–234. [Google Scholar] [CrossRef]
- Llorens, E.; del Valle, L.J.; Díaz, A.; Casas, M.T.; Puiggalí, J. polylactide nanofibers loaded with vitamin B6 and polyphenols as bioactive platform for tissue engineering. Macromol. Res. 2013, 21, 775–787. [Google Scholar] [CrossRef]
- Ou, C.-F.; Chao, M.-S.; Huang, S.-L. The crystallization behaviors of poly(butylene terephthalate) blended with co[poly(butylene terephthalate-p-oxybenzoate)] copolyesters. Eur. Polym. J. 2000, 36, 2665–2670. [Google Scholar] [CrossRef]
- Illers, K.-H. Heat of fusión and specific volumen of poly(ethylene terephthalate) and poly(butylene terephthalate). Colloid Polym. Sci. 1980, 258, 117–124. [Google Scholar] [CrossRef]
- Lum, R.M. Thermal decomposition of poly(butylene terephthalate). J. Polym Sci. Polym. Chem. Ed. 1979, 17, 203–213. [Google Scholar] [CrossRef]
- Li, F.; Xu, X.; Li, Q.; Li, Y.; Zhang, H.; Yu, J.; Cao, A. Thermal degradation and their kinetics of biodegradable poly(butylene succinate-co-butylene terephthate)s undernitrogen and air atmospheres. Polym. Degrad. Stab. 2006, 91, 1685–1693. [Google Scholar] [CrossRef]
- Song, K. Formation of polymorphic structure and its influences on properties in uniaxially stretched polybutylene terephthalate films. J. Appl. Polym. Sci. 2000, 78, 412–423. [Google Scholar] [CrossRef]
- Yasuniwa, M.; Tsubakihara, S.; Ohoshita, K.; Tokudome, S. X-ray studies on the double melting behavior of poly(butylene terephthalate). J. Polym. Sci. Part B Polym. Phys. 2001, 39, 2005–2015. [Google Scholar] [CrossRef]
- Gigli, M.; Fabbri, M.; Lotti, N.; Gamberini, R.; Rimini, B.; Munari, A. Poly(butylene succinate)-based polyesters for biomedical applications: A review. Eur. Polym. J. 2016, 75, 431–460. [Google Scholar] [CrossRef]
- Hakkarainen, M.; Albertsson, A.-C. Degradation products of aliphatic and aliphatic–aromatic polyesters. In Chromatography for Sustainable Polymeric Materials; Hakkarainen, M., Albertsson, A.-C., Eds.; Springer: Berlin & Heidelberg, Germany, 2008; pp. 85–116. [Google Scholar]
- Anderson, J.; Hollinger, M. Perspectives on the in vivo responses of biodegradable polymers. In Biomedical Applications of Synthetic Biodegradable Polymers; Hollinger, J.O., Ed.; CRC Press: Boca Raton, FL, USA, 1995; pp. 223–233. [Google Scholar]
- Li, S.; Vert, M. Biodegradation of aliphatic polyesters. In Degradable Polymers: Principles and Applications; Scott, G., Gilead, D., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 1995; pp. 43–87. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Yield (%) | COOH (meq/g) | Mn (g/mol) | Mw (g/mol) | PDI |
|---|---|---|---|---|---|
| PBST | 89% | 17 | 9540 | 20,200 | 2.12 |
| PBAdT | 85% | 31 | 7800 | 17,100 | 2.19 |
| PBSeT | 90% | 25 | 7000 | 17,100 | 2.44 |
| PBST-HAp | 91% | 15 | 9500 | 23,100 | 2.43 |
| PBAdT-HAp | 88% | 23 | 9800 | 26,200 | 2.67 |
| PBSeT-HAp | 90% | 23 | 9000 | 22,000 | 2.44 |
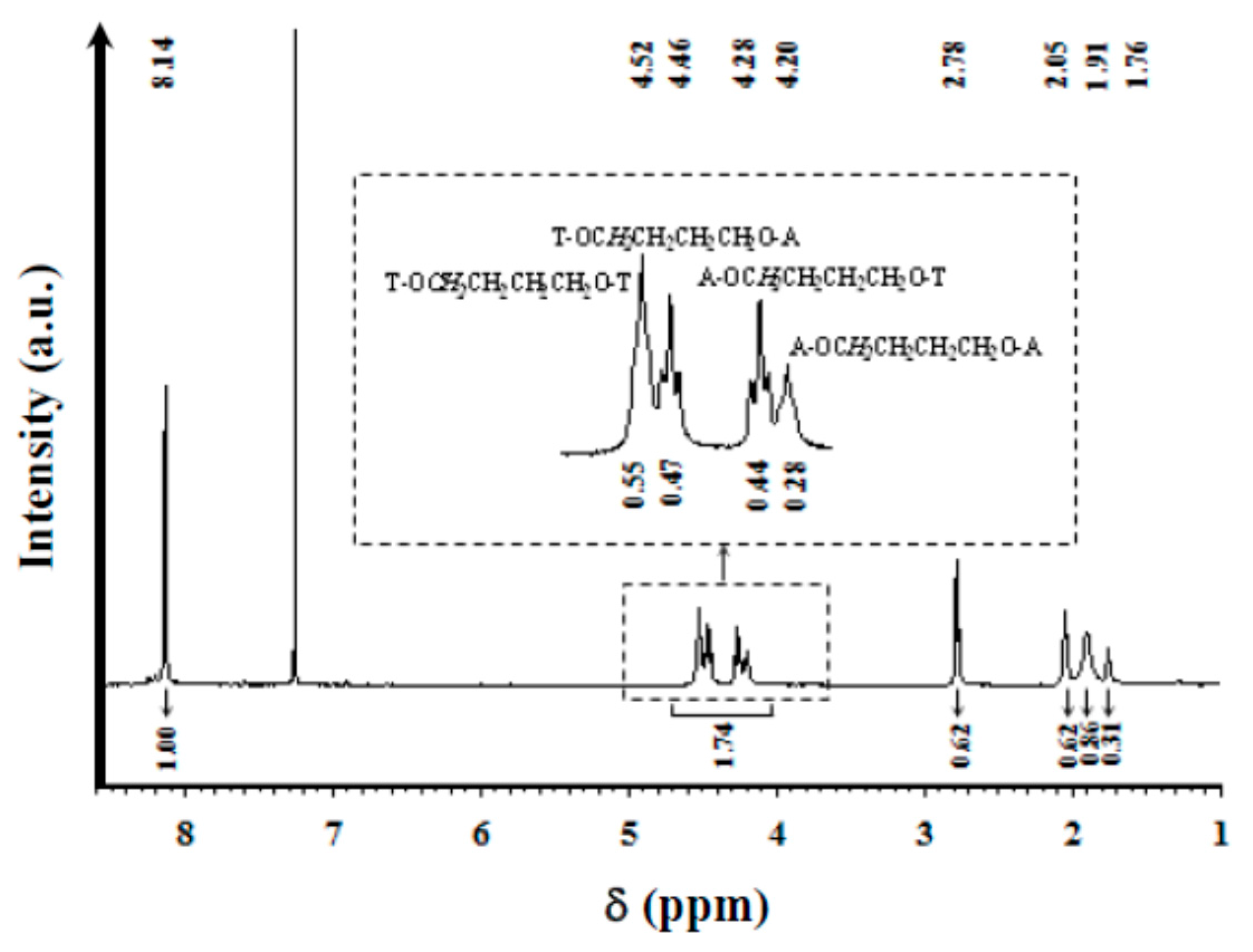
| Sequence | Chemical Shift (ppm) | ||
|---|---|---|---|
| PBST | PBAdT | PBSeT | |
| –C6H4 | 8.14 | 8.17 | 8.14 |
| T–OCH2CH2CH2CH2O–T | 4.52 | 4.57 | 4.64 |
| T–OCH2CH2CH2CH2O–A | 4.46 | 4.52 | 4.59 |
| A–OCH2CH2CH2CH2O-T | 4.28 | 4.31 | 4.37 |
| A–OCH2CH2CH2CH2O–A | 4.20 | 4.25 | 4.31 |
| COCH2–… | 2.78 | 2.52 | 2.42 |
| T–OCH2CH2CH2CH2O–T | 2.05 | 2.08 | 2.06 |
| T–OCH2CH2CH2CH2O–A | 1.91 | 1.92 | 1.92 |
| A–OCH2CH2CH2CH2O–A | 1.76 | 1.80 | 1.79 |
| COCH2CH2–… | - | 1.72 | 1.64, 1.32 |
| Coplymer | Composition molar fraction | Fraction of diads centered in the butylene units | Probability of finding units | Block lengths | Degree of randomness | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| fT | fA | fTT | fTA | fAT | fAA | PTA | PTA | LnTB | LnAB | r | |
| PBST | 0.65 | 0.35 | 0.40 | 0.24 | 0.24 | 0.12 | 0.38 | 0.65 | 2.63 | 1.53 | 1.03 |
| PBAdT | 0.64 | 0.36 | 0.38 | 0.25 | 0.25 | 0.12 | 0.39 | 0.61 | 2.56 | 1.64 | 1.00 |
| PBSeT | 0.65 | 0.35 | 0.45 | 0.22 | 0.22 | 0.11 | 0.34 | 0.70 | 2.94 | 1.43 | 1.04 |
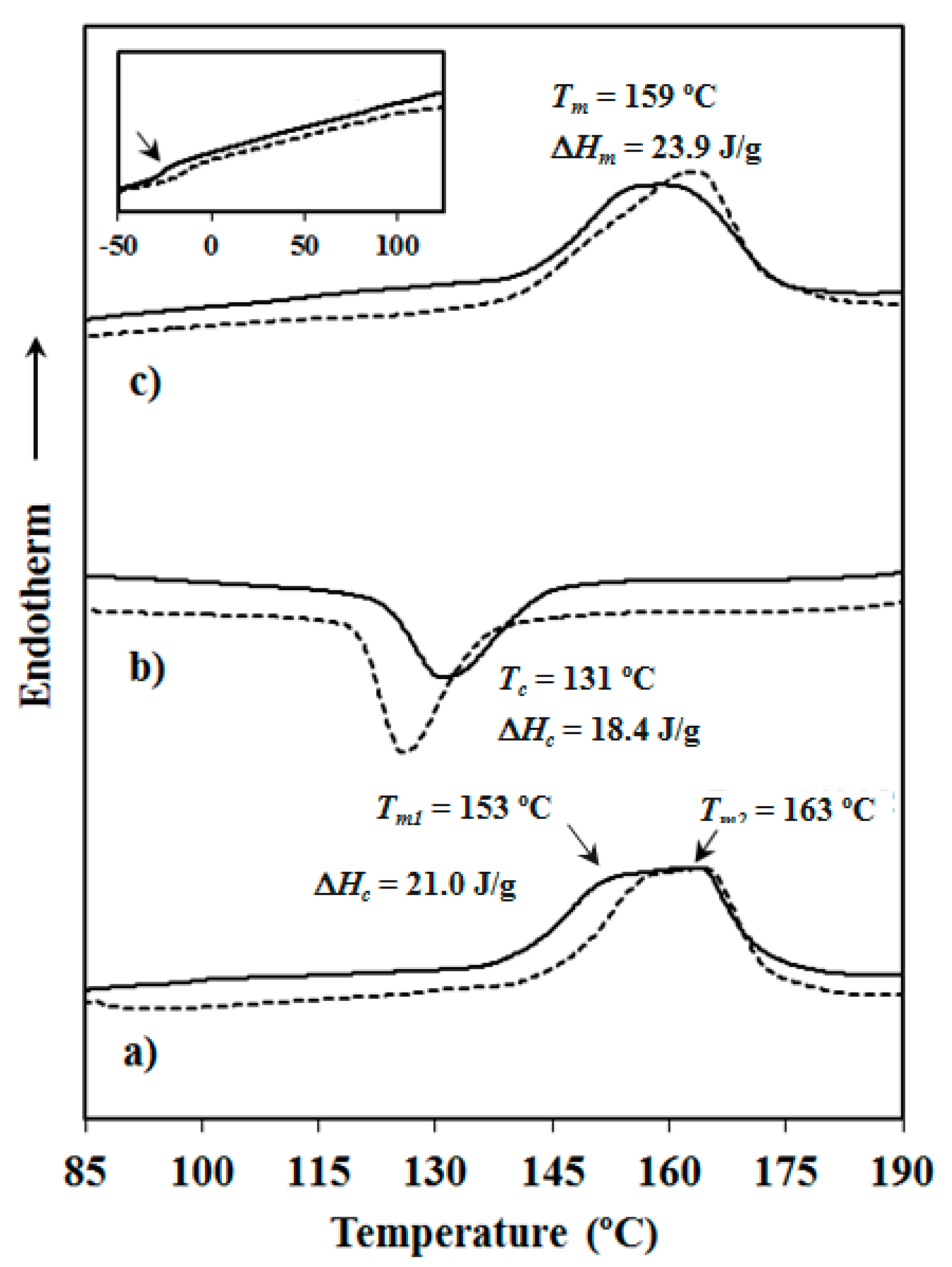
| Sample | 1st Heating run | Cooling run | 2nd Heating run | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Tm (°C) | ΔHf (J/g) | χα (%) | Tc (°C) | ΔHc (J/g) | Tg (°C) | Tm (°C) | ΔHf (J/g) | χα (%) | |
| PBST | 153, 159 | 19.6 | 21 | 119 | 15.9 | −7 | 155, 159 | 20.8 | 22 |
| PBAdT | 153, 163 | 21.0 | 23 | 131 | 18.4 | −27 | 159 | 23.9 | 26 |
| PBSeT | 159, 163 | 23.7 | 26 | 128 | 19.1 | −27 | 164 | 20.6 | 22 |
| PBST-HAp | 149, 159 | 19.1 | 20 | 119 | 17.4 | −7 | 158 | 21.9 | 23 |
| PBAdT-HAp | 158, 164 | 19.6 | 22 | 126 | 19.5 | −14 | 163 | 23.8 | 26 |
| PBSeT-HAp | 160, 166 | 22.7 | 26 | 129 | 22.3 | −27 | 162 | 23.4 | 27 |
| Sample | Tonset (°C) | Tpeak (°C) | Char yield (%) |
|---|---|---|---|
| PBST | 315 | 402 | 2.8 |
| PBAdT | 318 | 405 | 5.6 |
| PBSeT | 322 | 405 | 5.2 |
| PBST-HAp | 352 | 405 | 5.6 |
| PBAdT-HAp | 342 | 404 | 7.9 |
| PBSeT-HAp | 348 | 407 | 6.3 |
| Sample | Tg (°C) | E’−20 °C (MPa) | E’20 °C (MPa) | E’70 (MPa) |
|---|---|---|---|---|
| PBSTS | 10.5 | 1830 | 480 | 180 |
| PBAdT | −2.5 | 1840 | 550 | 250 |
| PBSeT | −6.5 | 2030 | 370 | 80 |
| PBST-HAp | 11.0 | 2210 | 590 | 220 |
| PBAdT-HAp | 2.3 | 2470 | 700 | 290 |
| PBSeT-HAp | −3.5 | 2200 | 400 | 100 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heidarzadeh, N.; Rafizadeh, M.; Taromi, F.A.; Del Valle, L.J.; Franco, L.; Puiggalí, J. Effect of Hydroxyapatite Nanoparticles on the Degradability of Random Poly(butylene terephthalate-co-aliphatic dicarboxylate)s Having a High Content of Terephthalic Units. Polymers 2016, 8, 253. https://doi.org/10.3390/polym8070253
Heidarzadeh N, Rafizadeh M, Taromi FA, Del Valle LJ, Franco L, Puiggalí J. Effect of Hydroxyapatite Nanoparticles on the Degradability of Random Poly(butylene terephthalate-co-aliphatic dicarboxylate)s Having a High Content of Terephthalic Units. Polymers. 2016; 8(7):253. https://doi.org/10.3390/polym8070253
Chicago/Turabian StyleHeidarzadeh, Nina, Mehdi Rafizadeh, Faramarz Afshar Taromi, Luís Javier Del Valle, Lourdes Franco, and Jordi Puiggalí. 2016. "Effect of Hydroxyapatite Nanoparticles on the Degradability of Random Poly(butylene terephthalate-co-aliphatic dicarboxylate)s Having a High Content of Terephthalic Units" Polymers 8, no. 7: 253. https://doi.org/10.3390/polym8070253
APA StyleHeidarzadeh, N., Rafizadeh, M., Taromi, F. A., Del Valle, L. J., Franco, L., & Puiggalí, J. (2016). Effect of Hydroxyapatite Nanoparticles on the Degradability of Random Poly(butylene terephthalate-co-aliphatic dicarboxylate)s Having a High Content of Terephthalic Units. Polymers, 8(7), 253. https://doi.org/10.3390/polym8070253