
Enzymatic Synthesis of Biobased Polyesters and Polyamides



Abstract
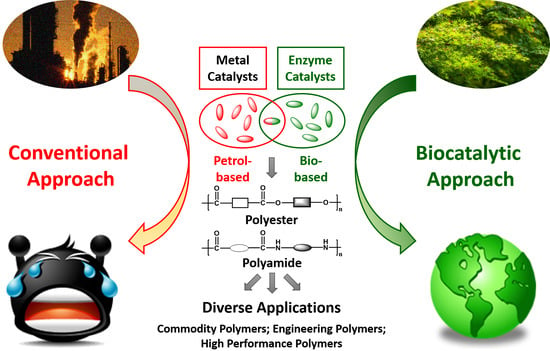
:
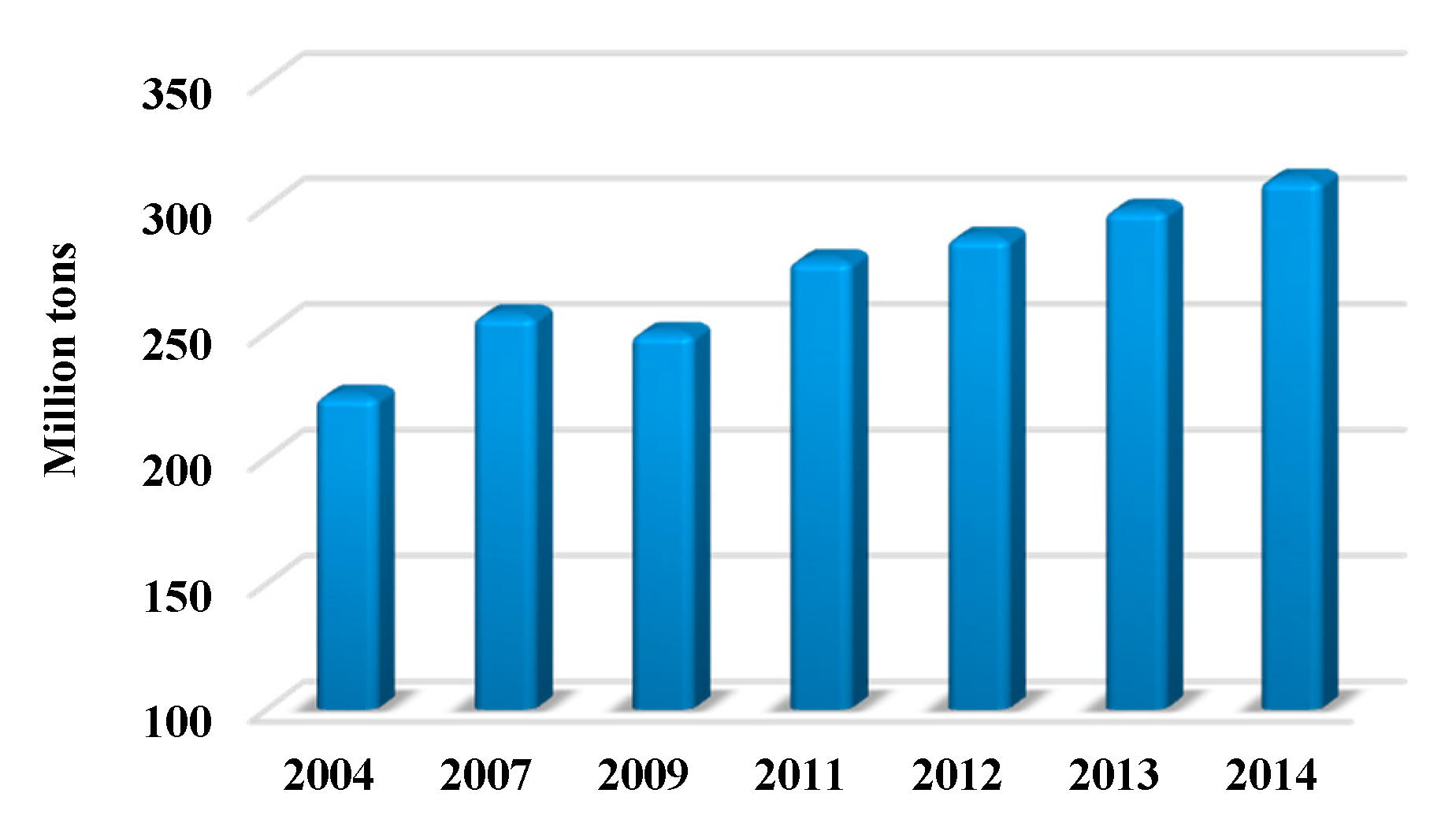
1. Polymers: From Petrol-Based to Biobased and Beyond
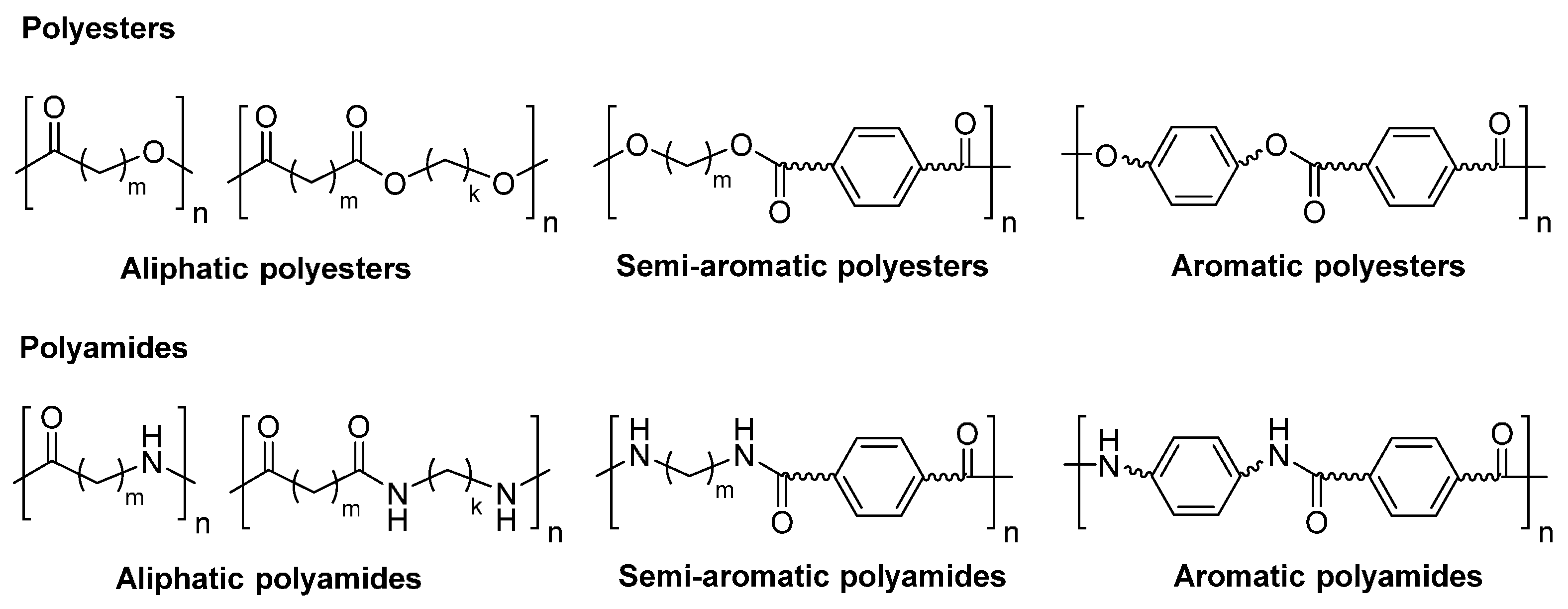
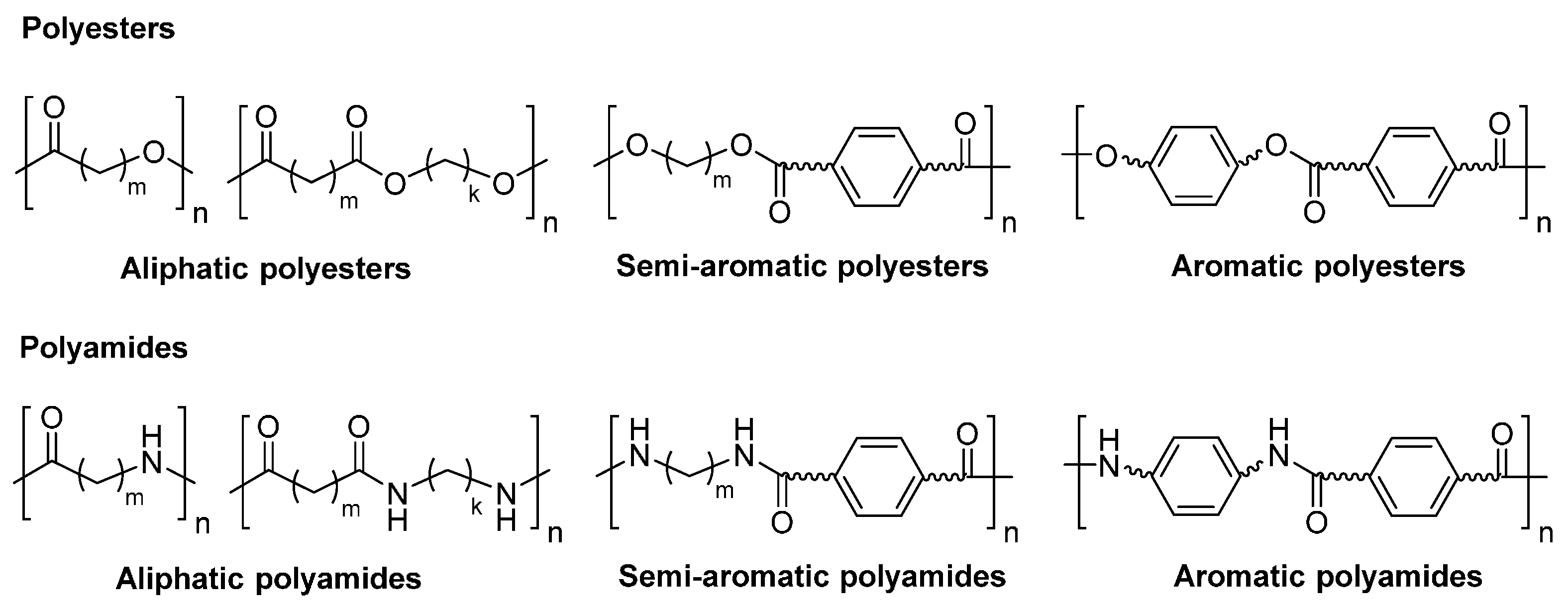
2. Polyesters
3. Polyamides
4. Biobased Monomers for Polyester and Polyamide Synthesis
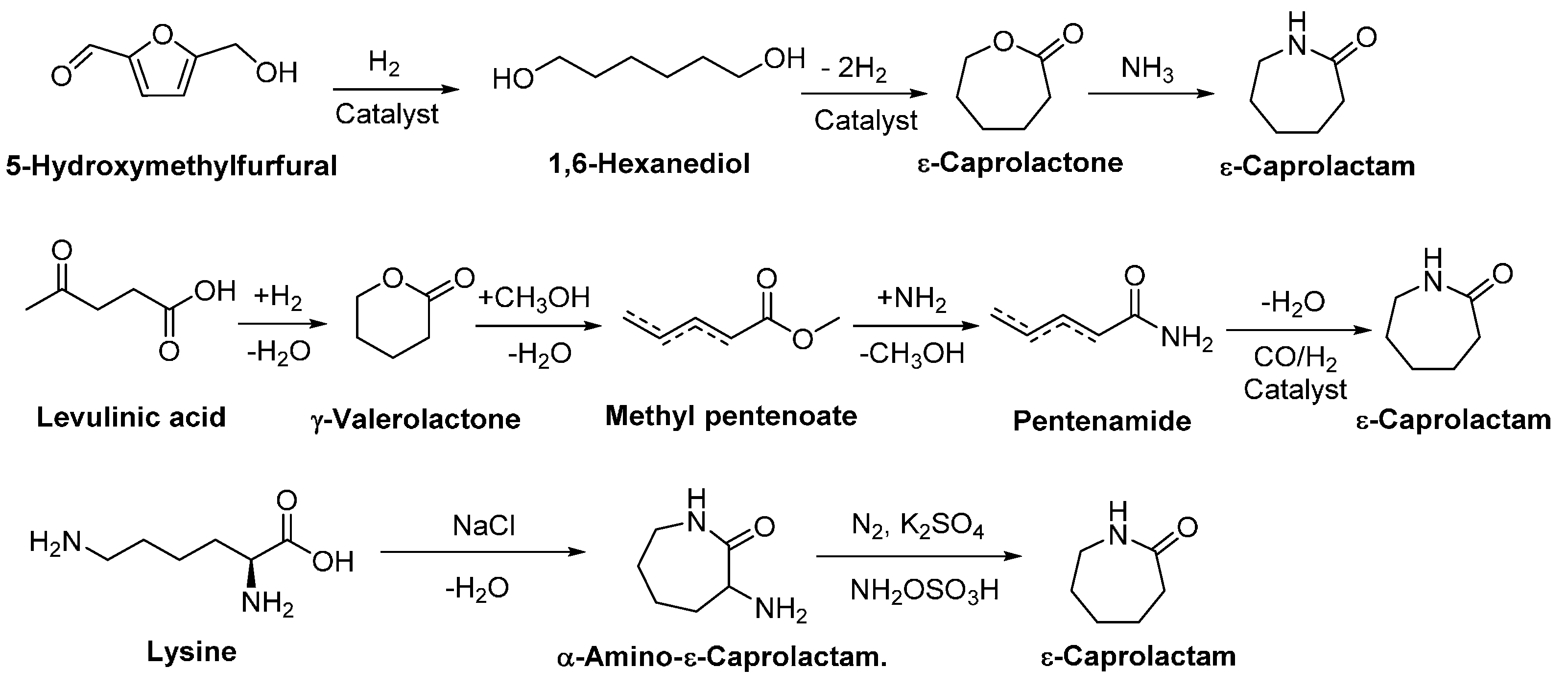
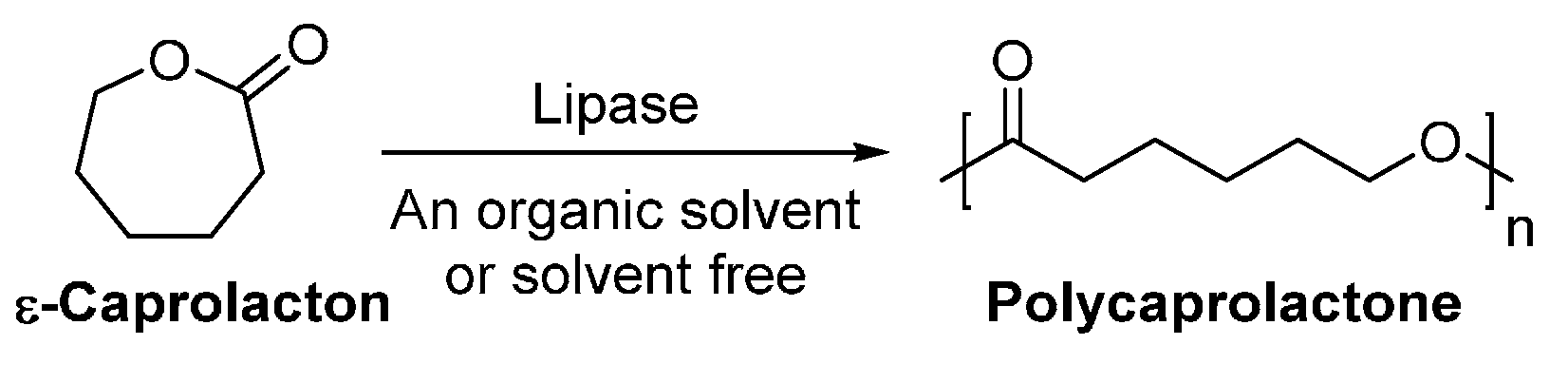
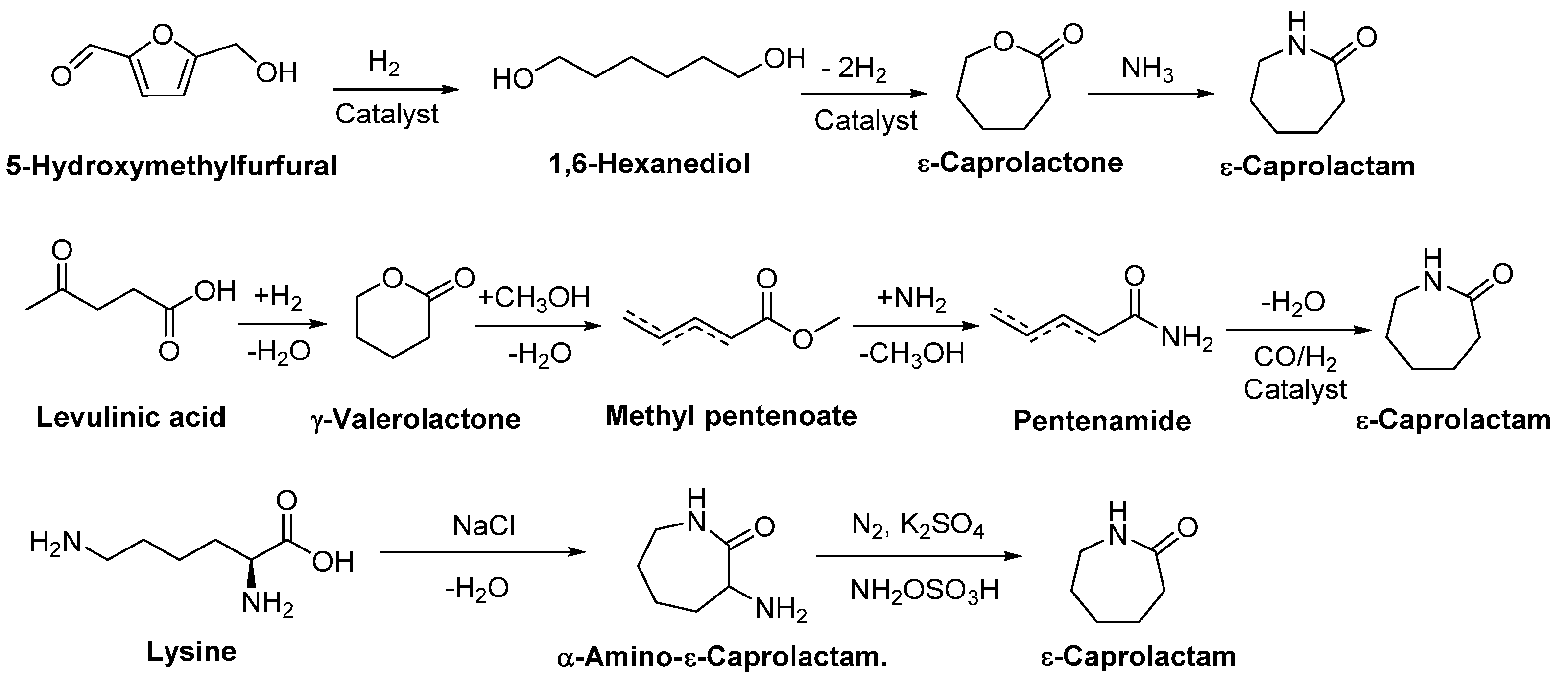
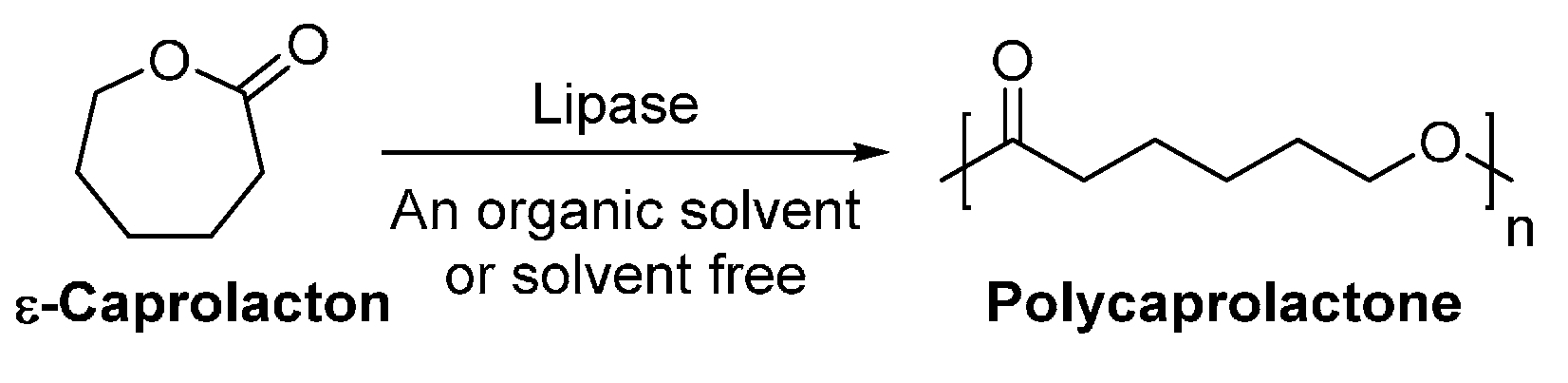
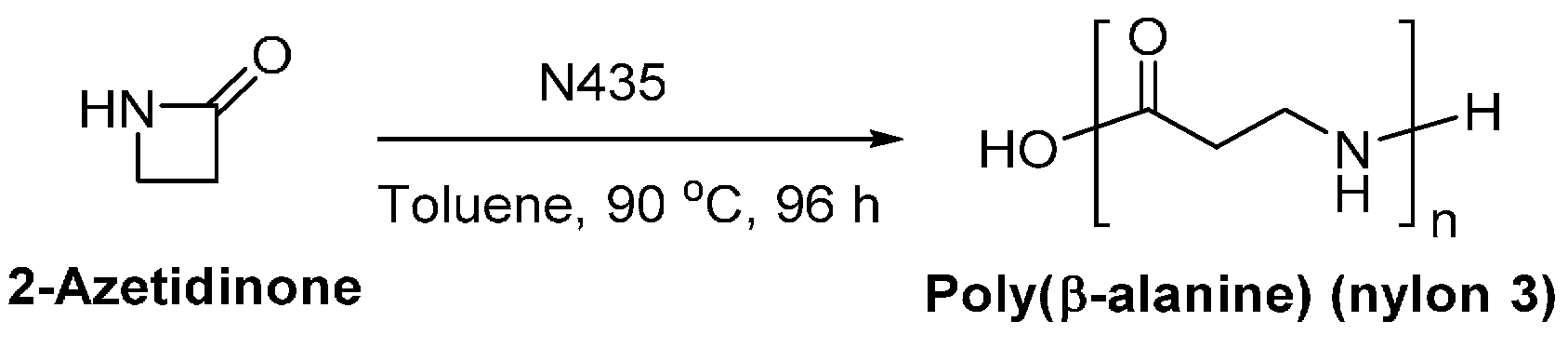
4.1. Biobased Lactones and Lactams
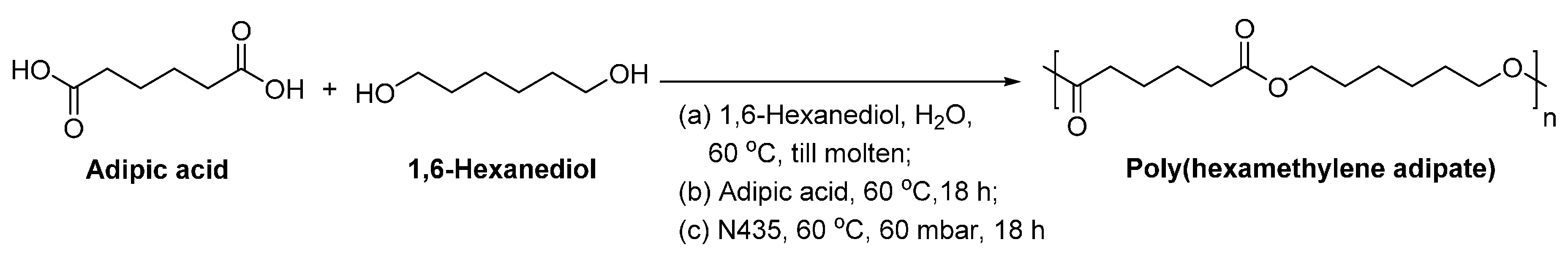
4.2. Biobased Aliphatic Diacids
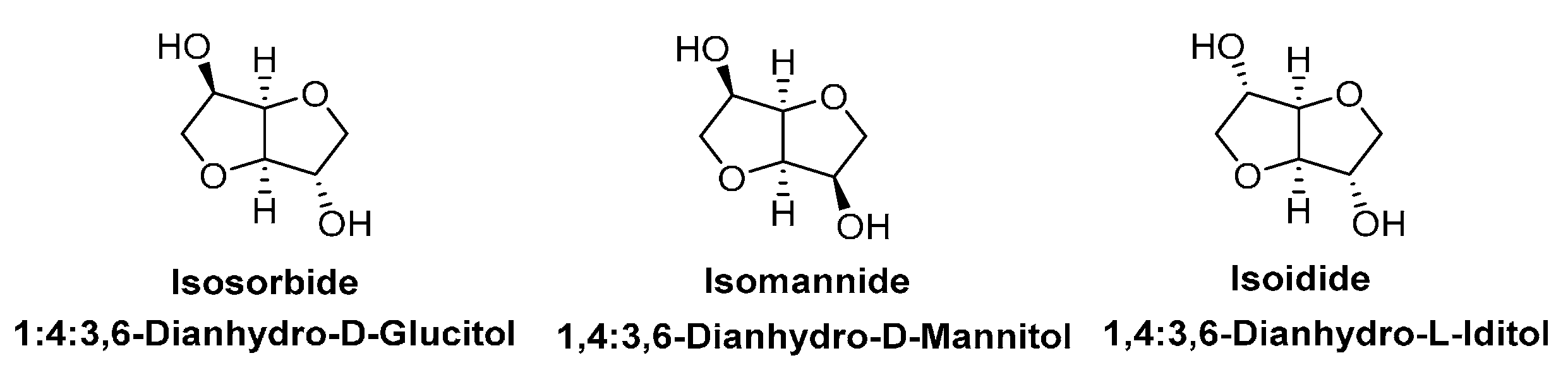
4.3. Biobased Aliphatic Diols and Polyols
4.4. Biobased Aliphatic Diamines
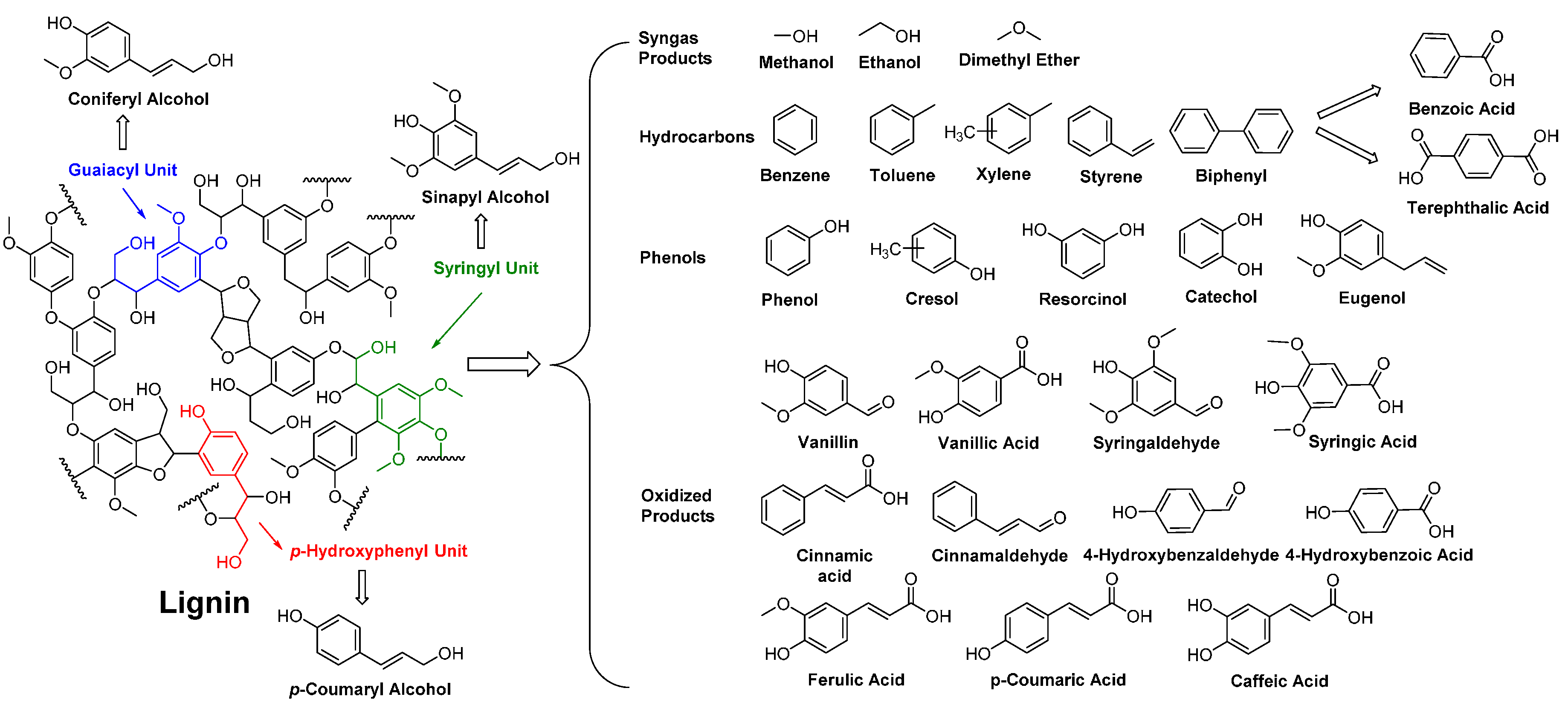
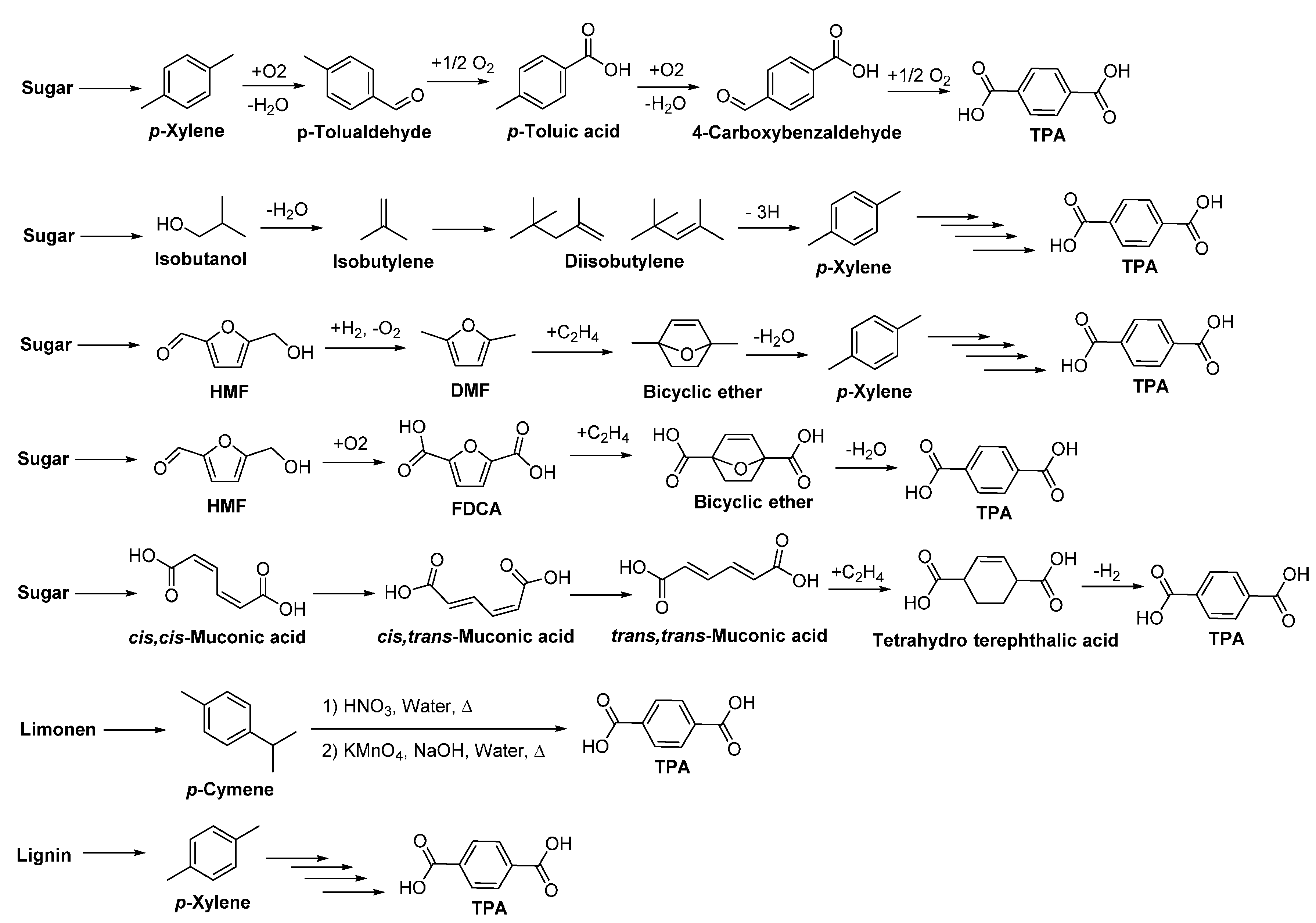
4.5. Biobased Aromatic Monomers
4.6. Other Biobased Monomers
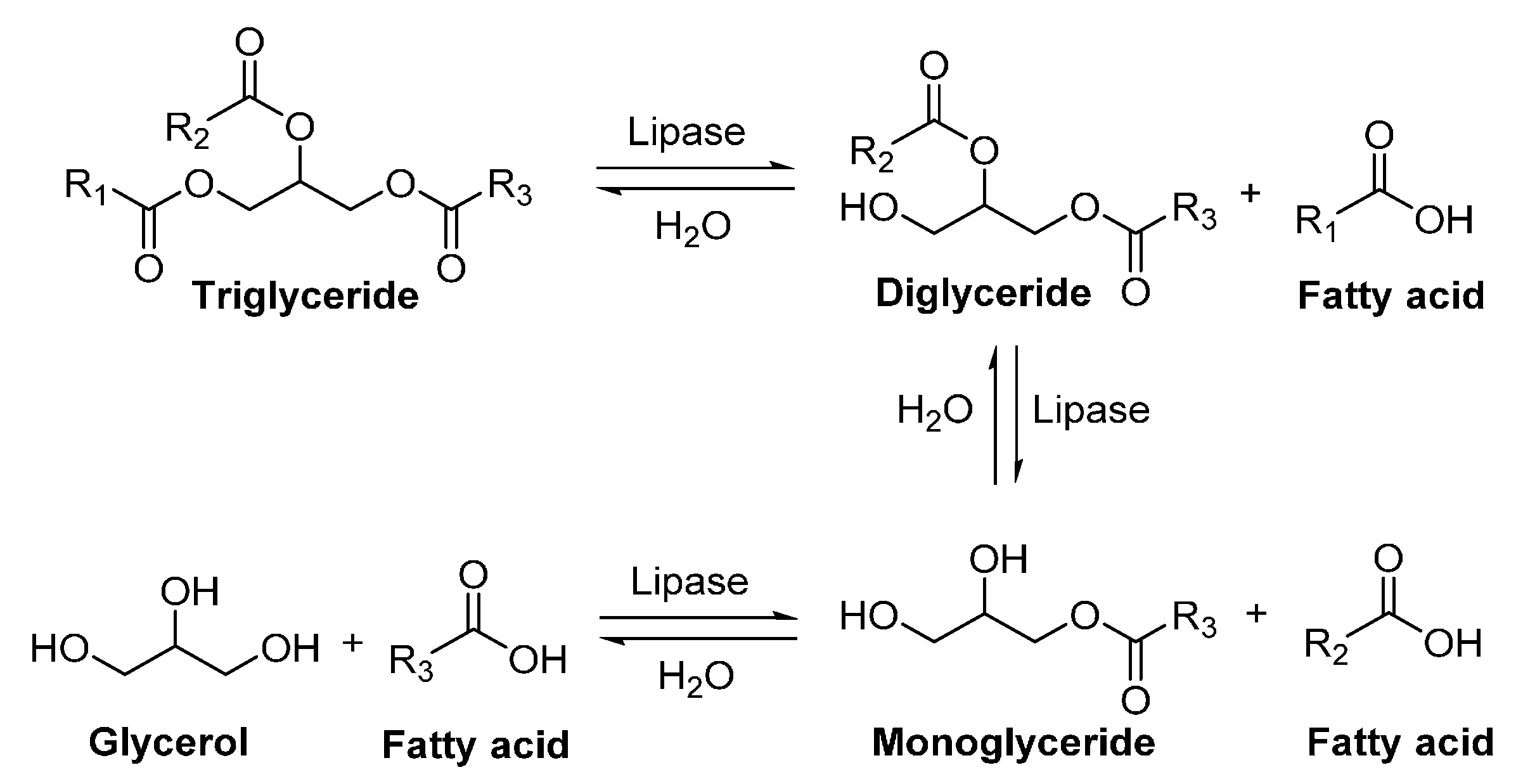
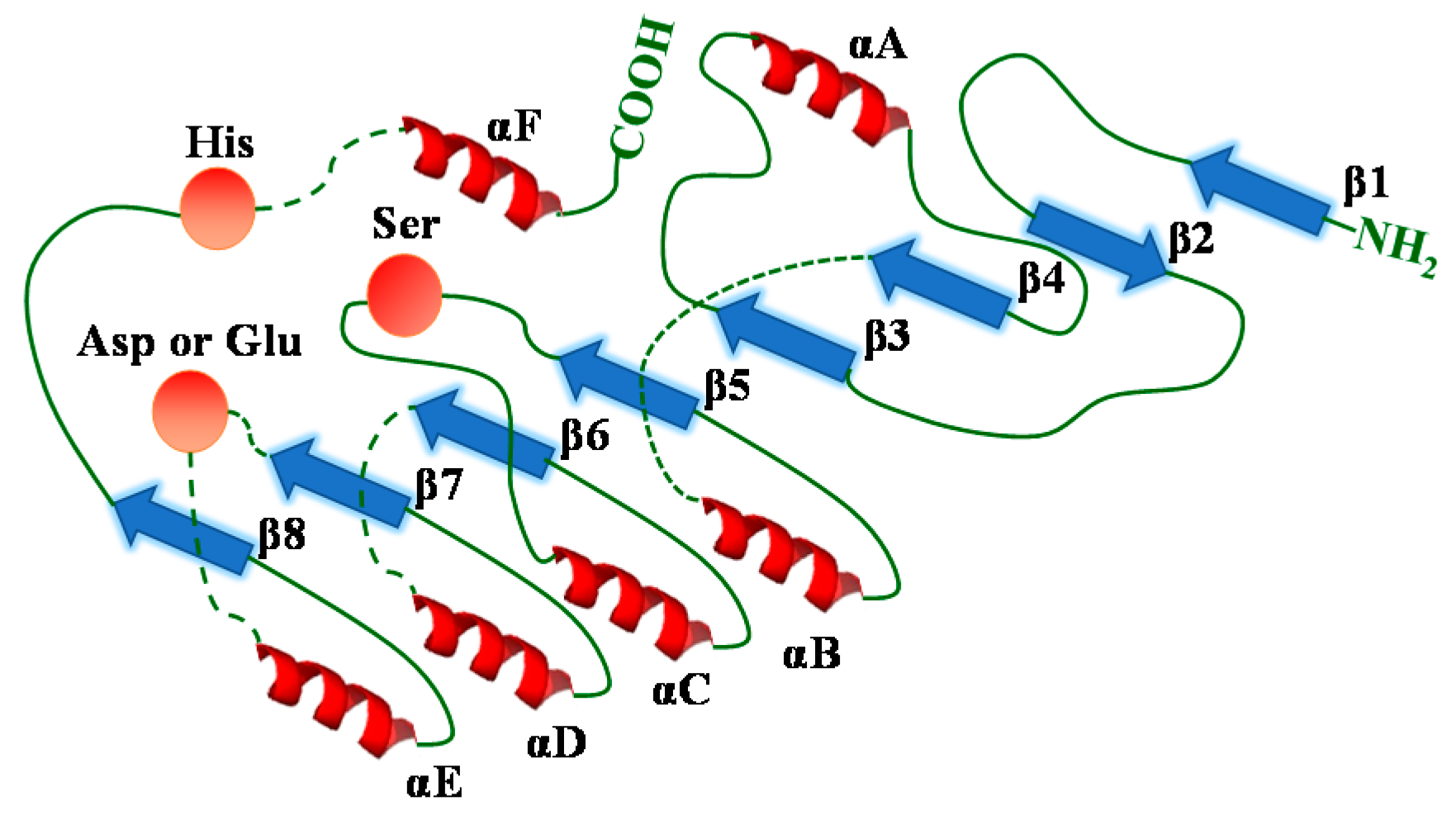
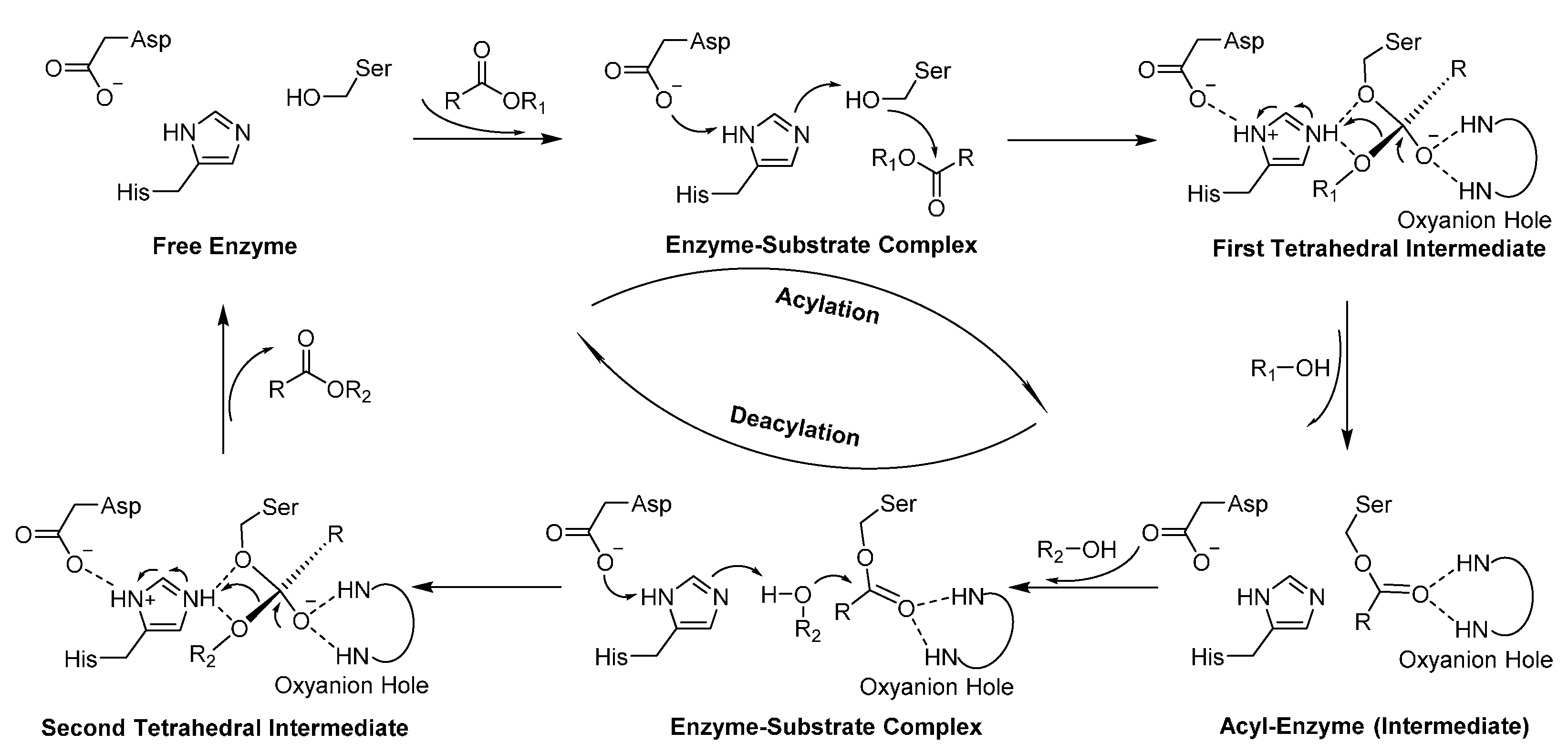
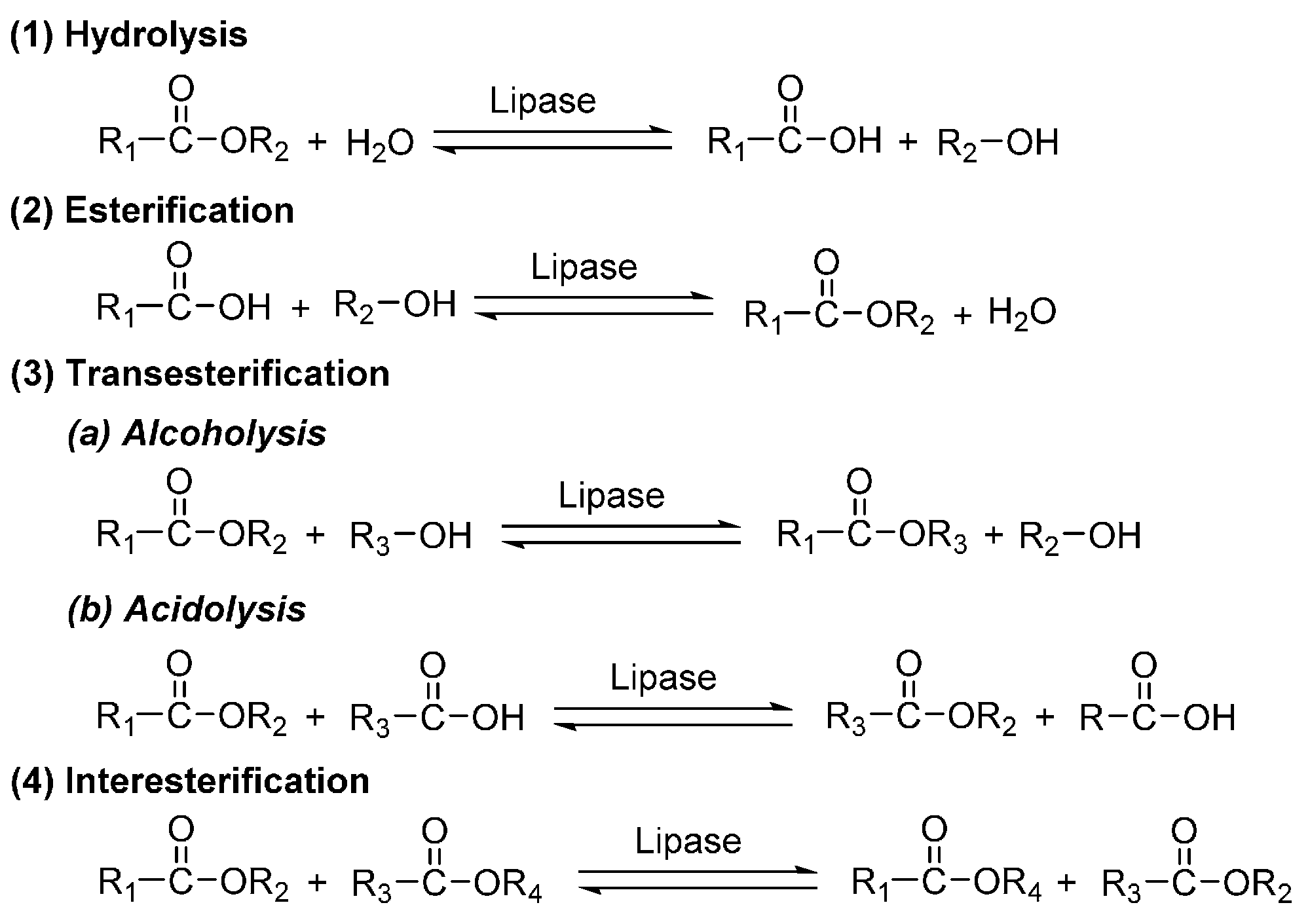
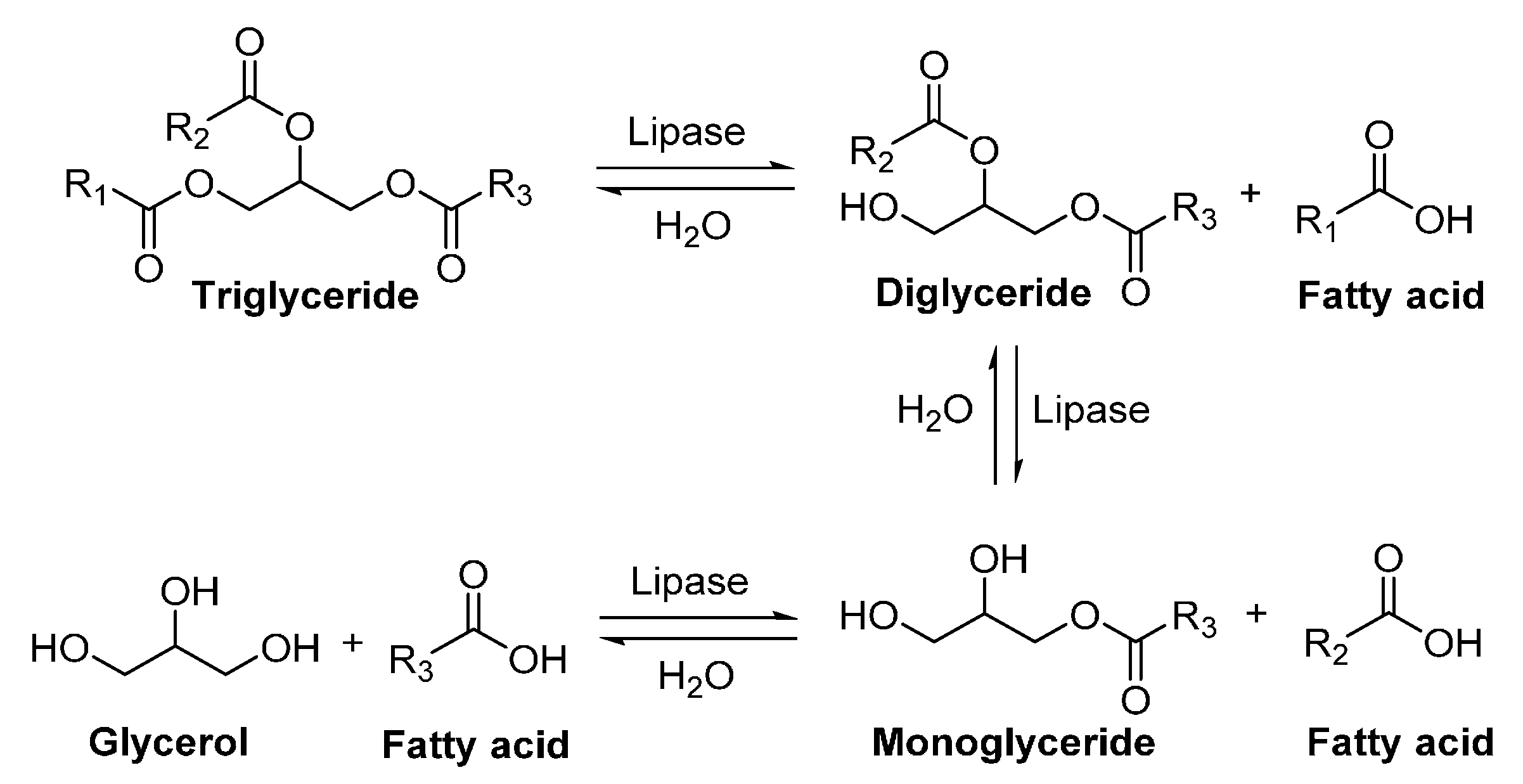
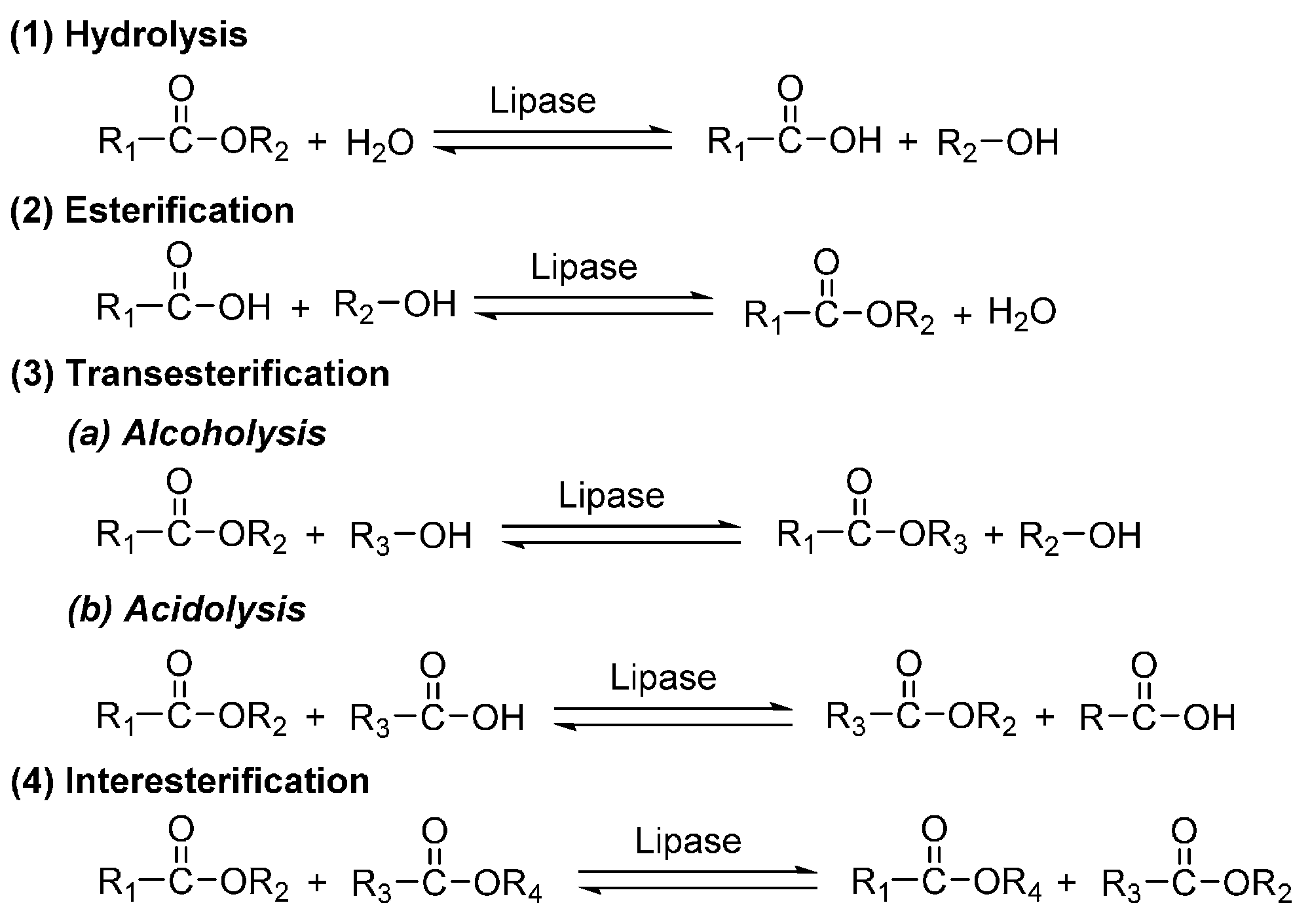
5. Lipases
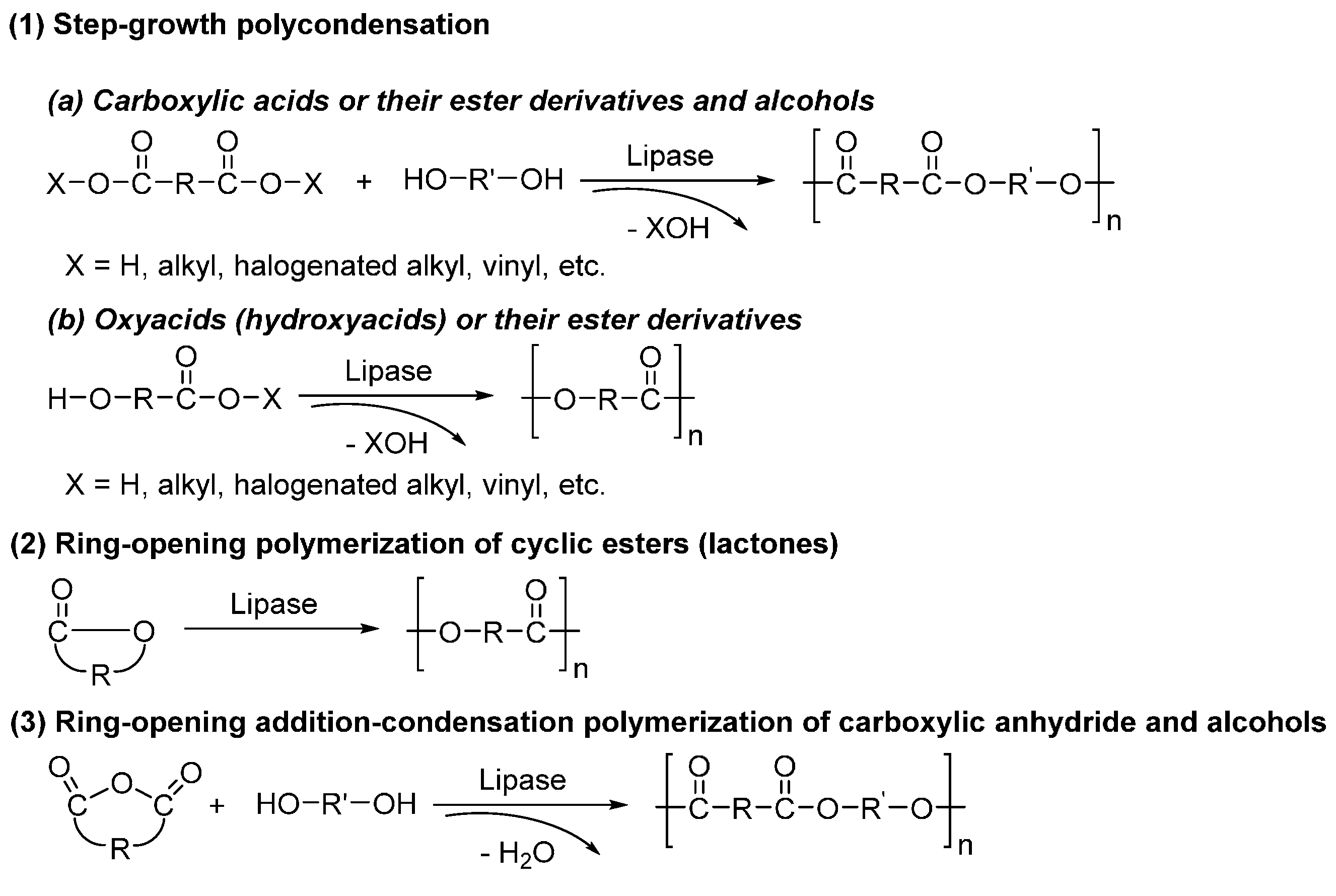
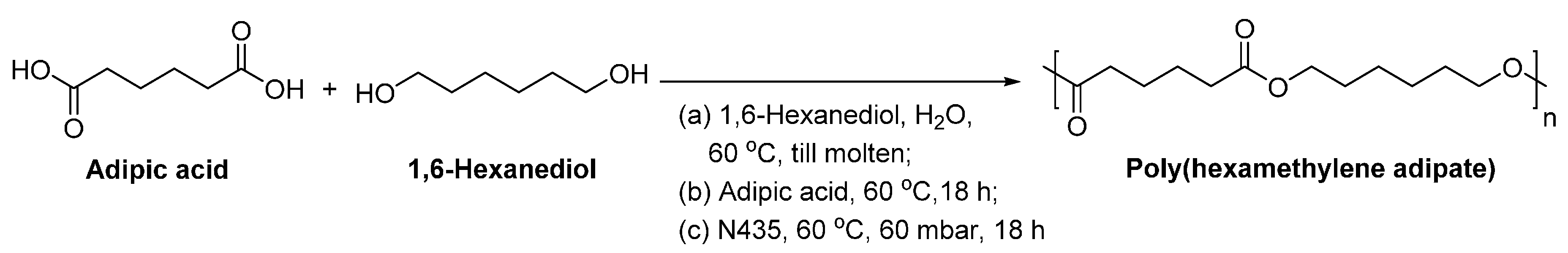
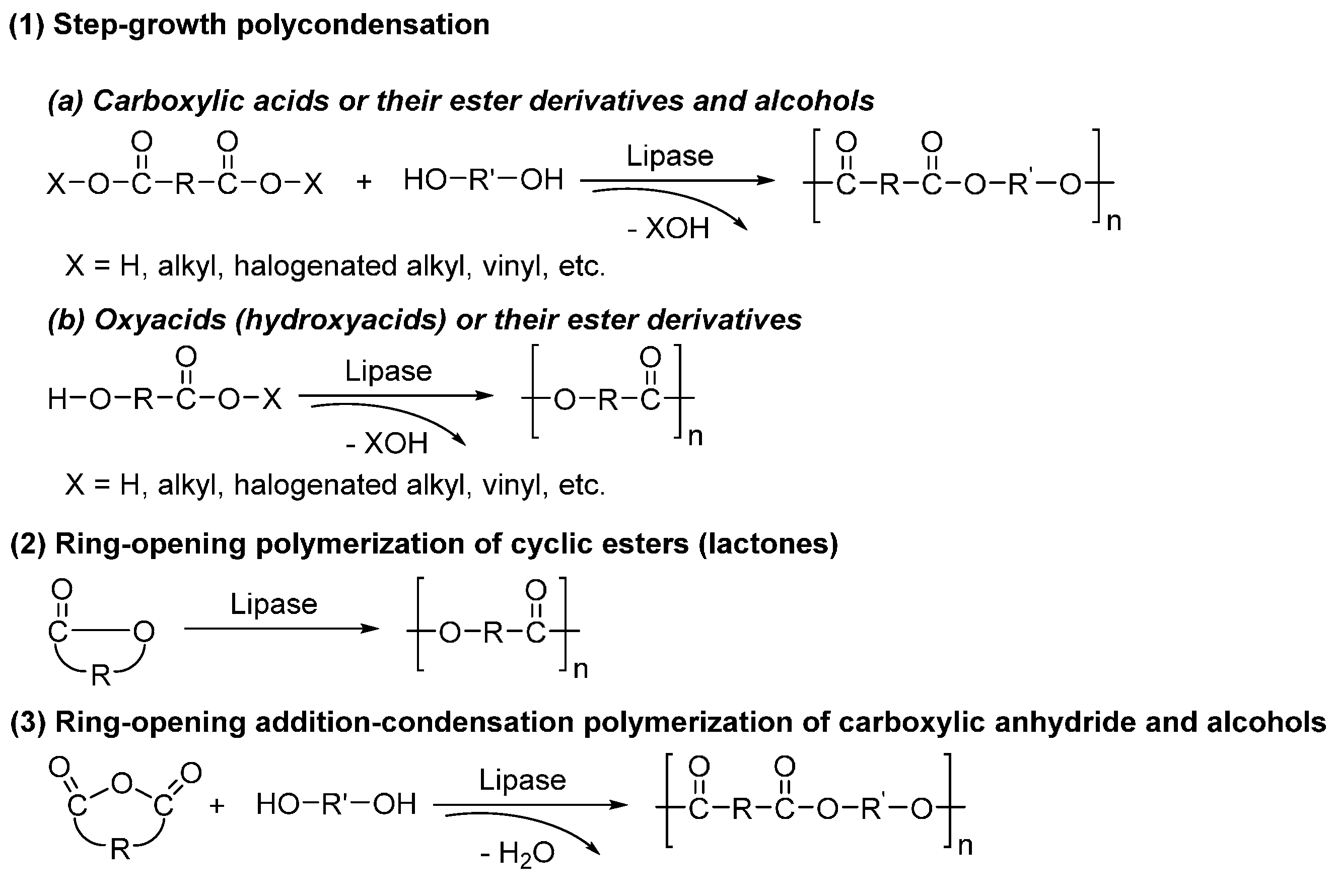
6. Enzyme-Catalyzed Synthesis of Polyesters
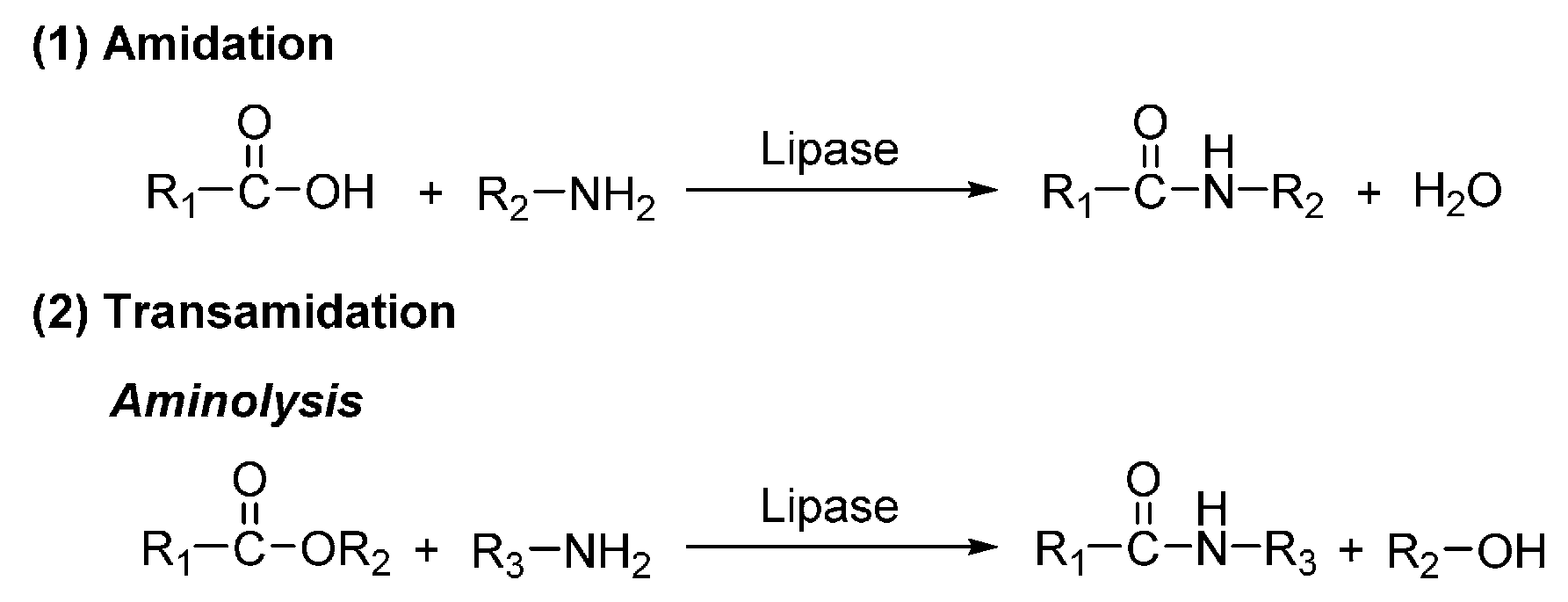
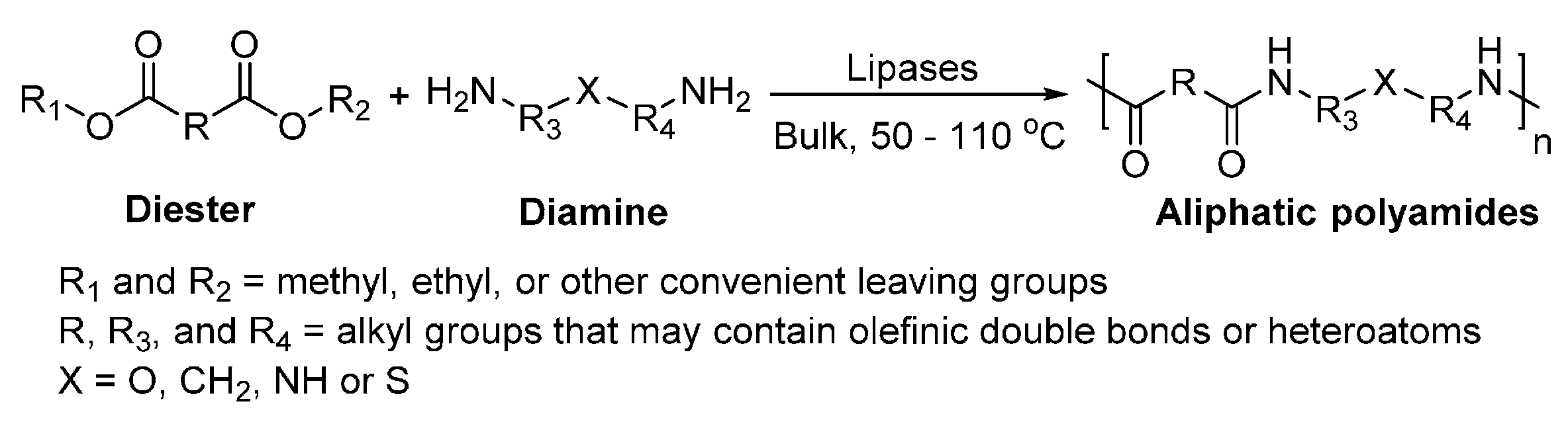
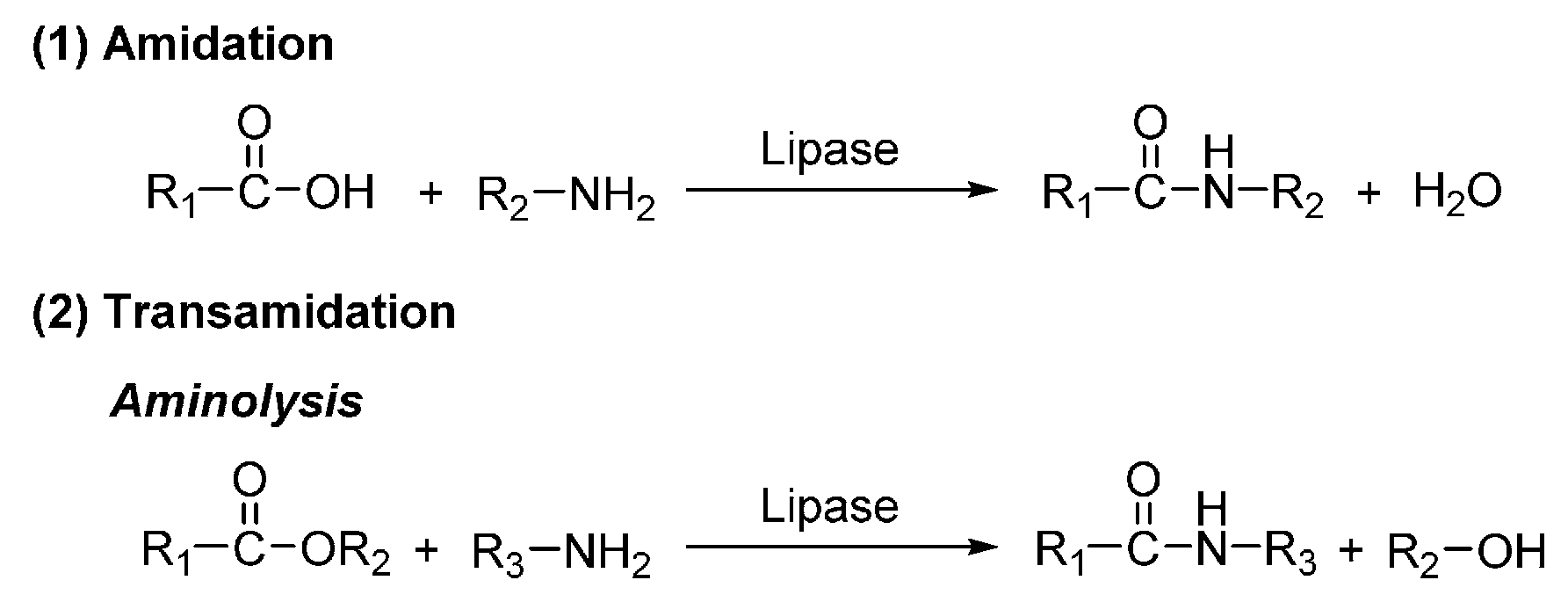
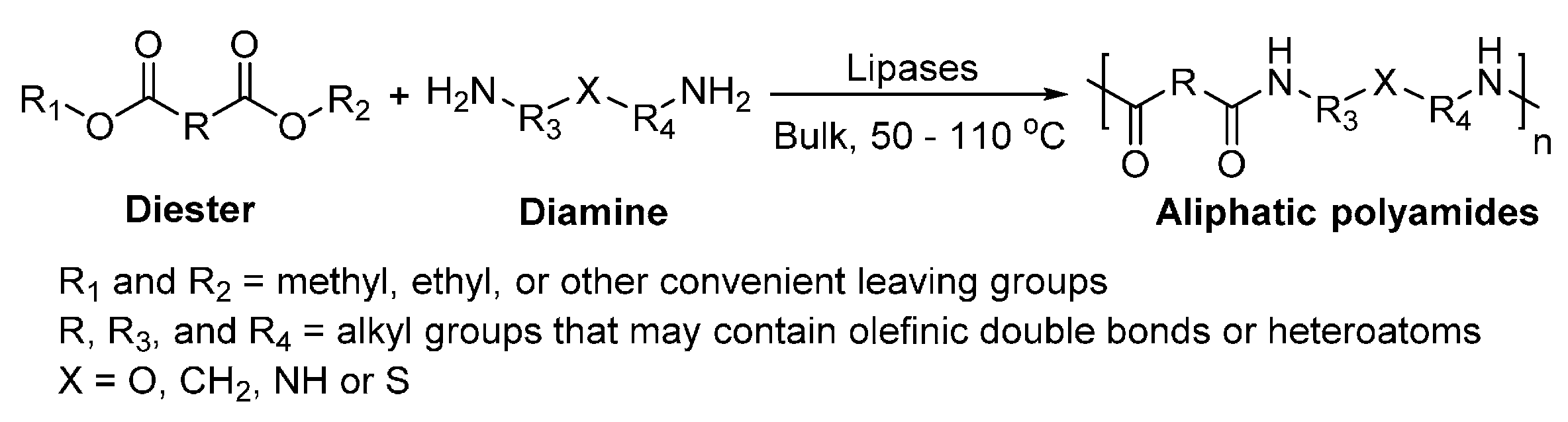
7. Enzyme-Catalyzed Synthesis of Polyamides
8. Lipase-Catalyzed Synthesis of Sustainable Polyesters and Polyamides from Biobased Monomers
8.1. Biobased Saturated Aliphatic Polyesters
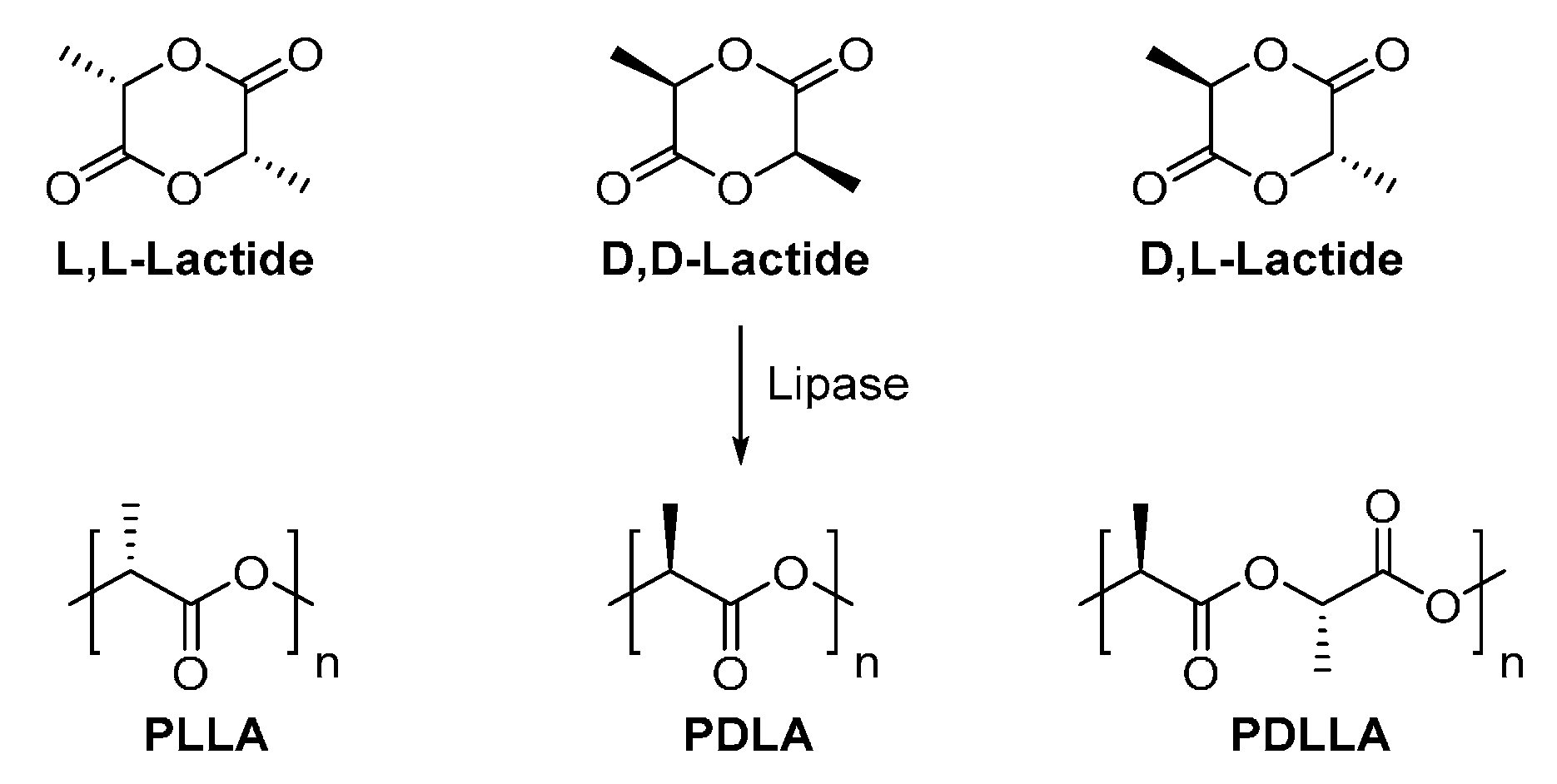
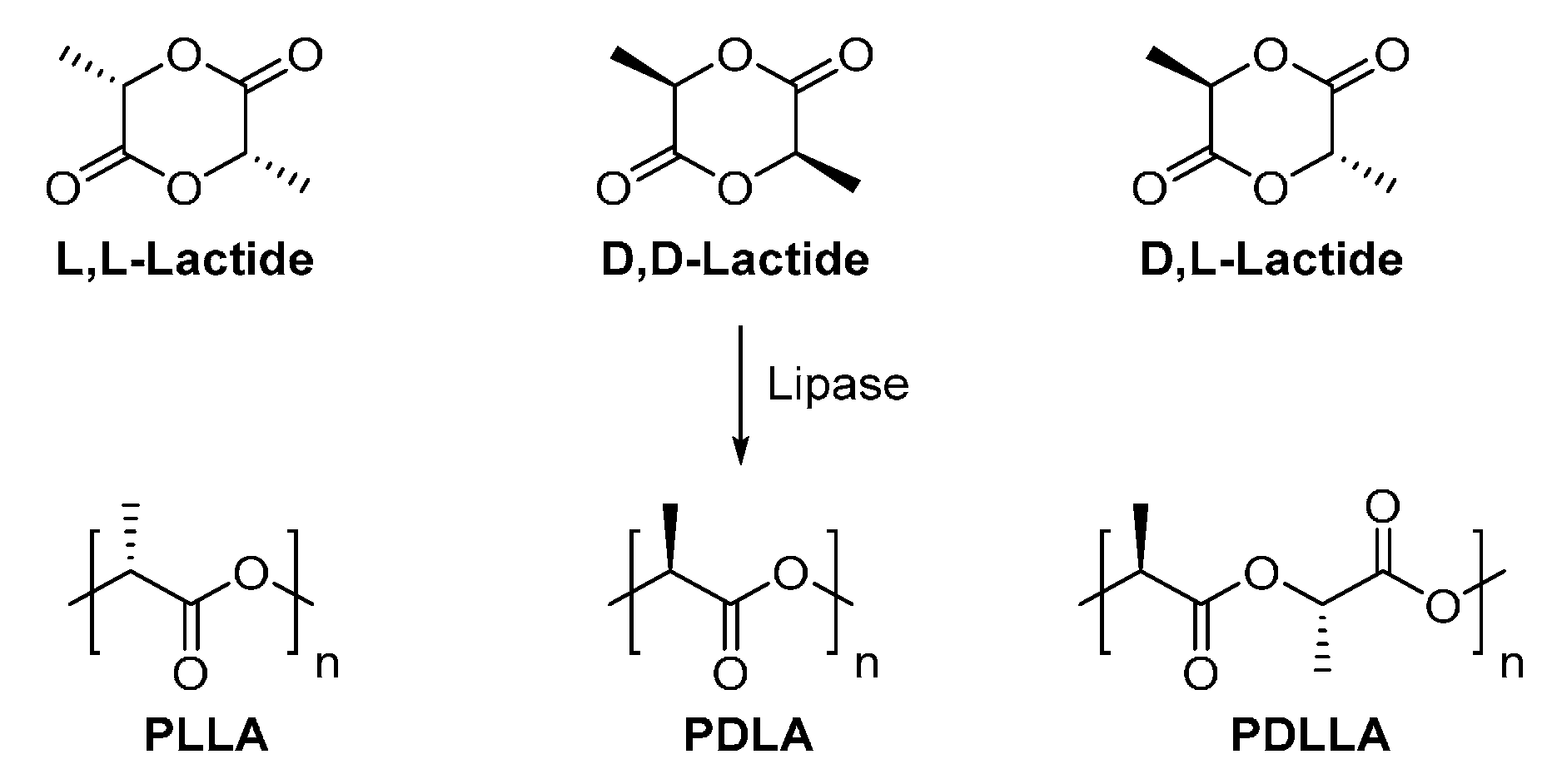
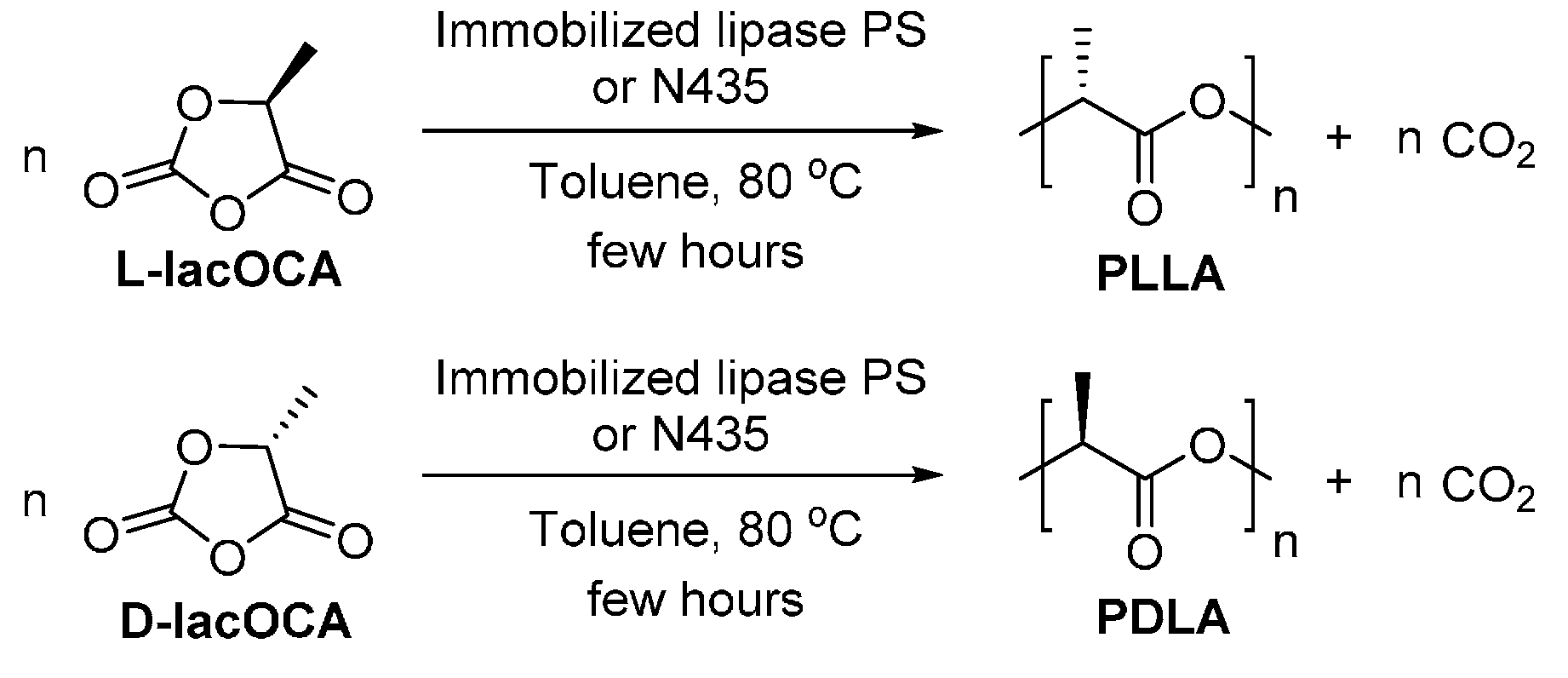
8.1.1. Poly(lactic acid)
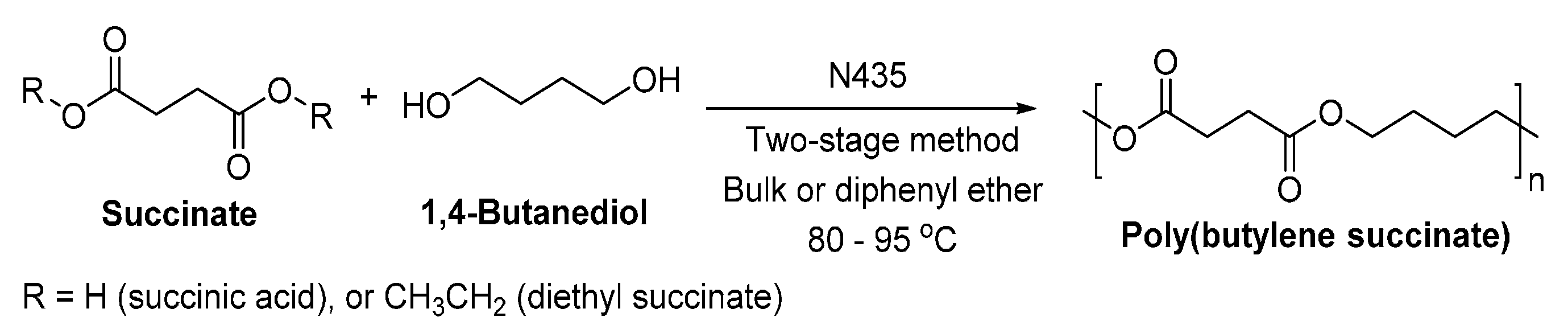
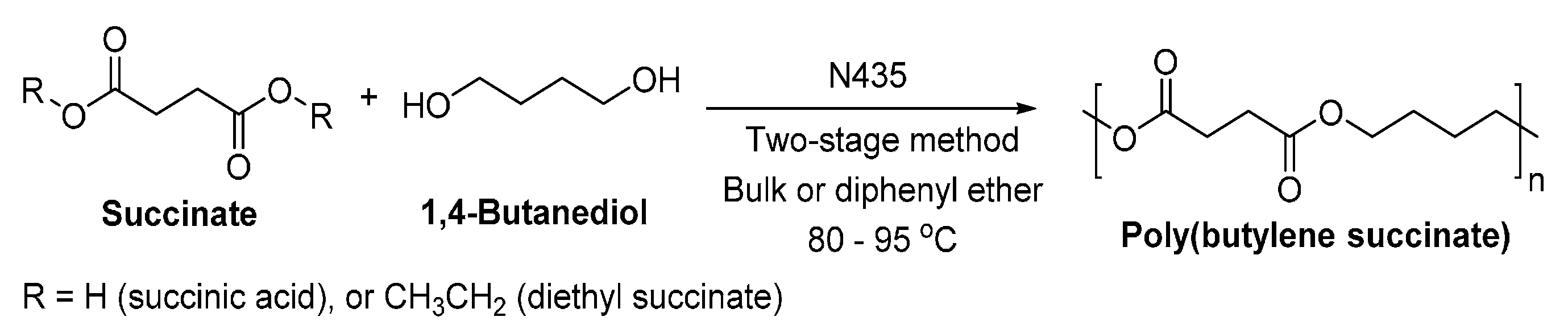
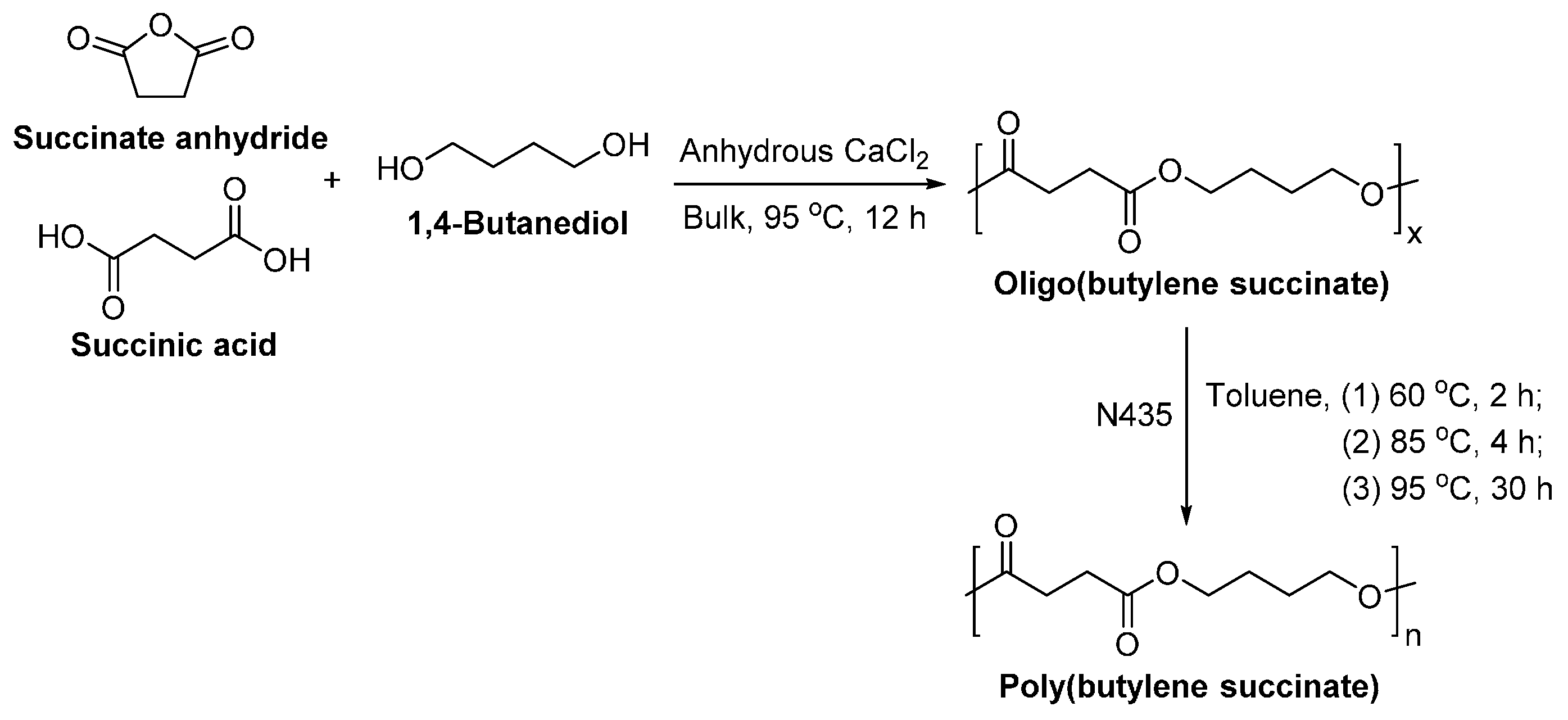
8.1.2. Poly(butylene succinate)
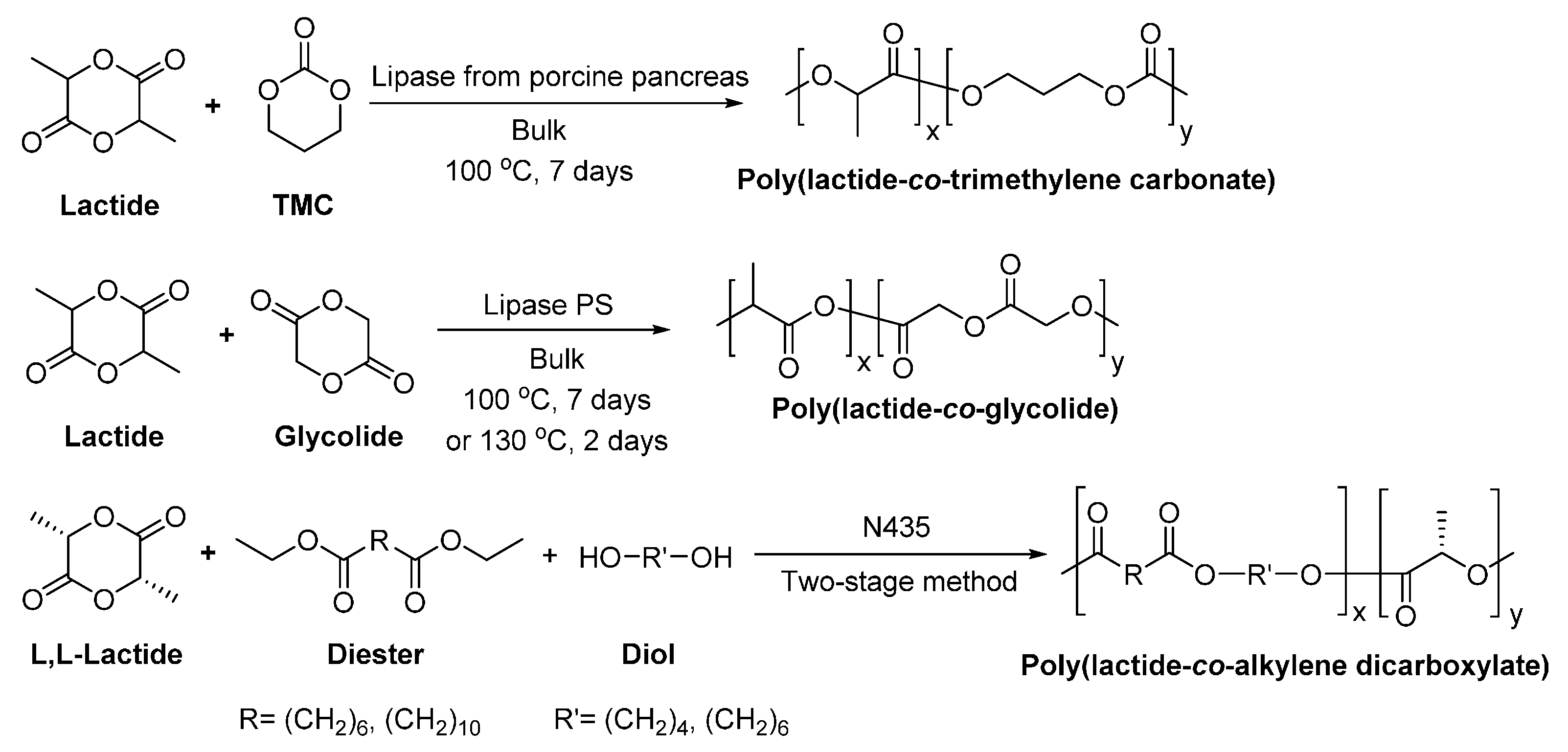
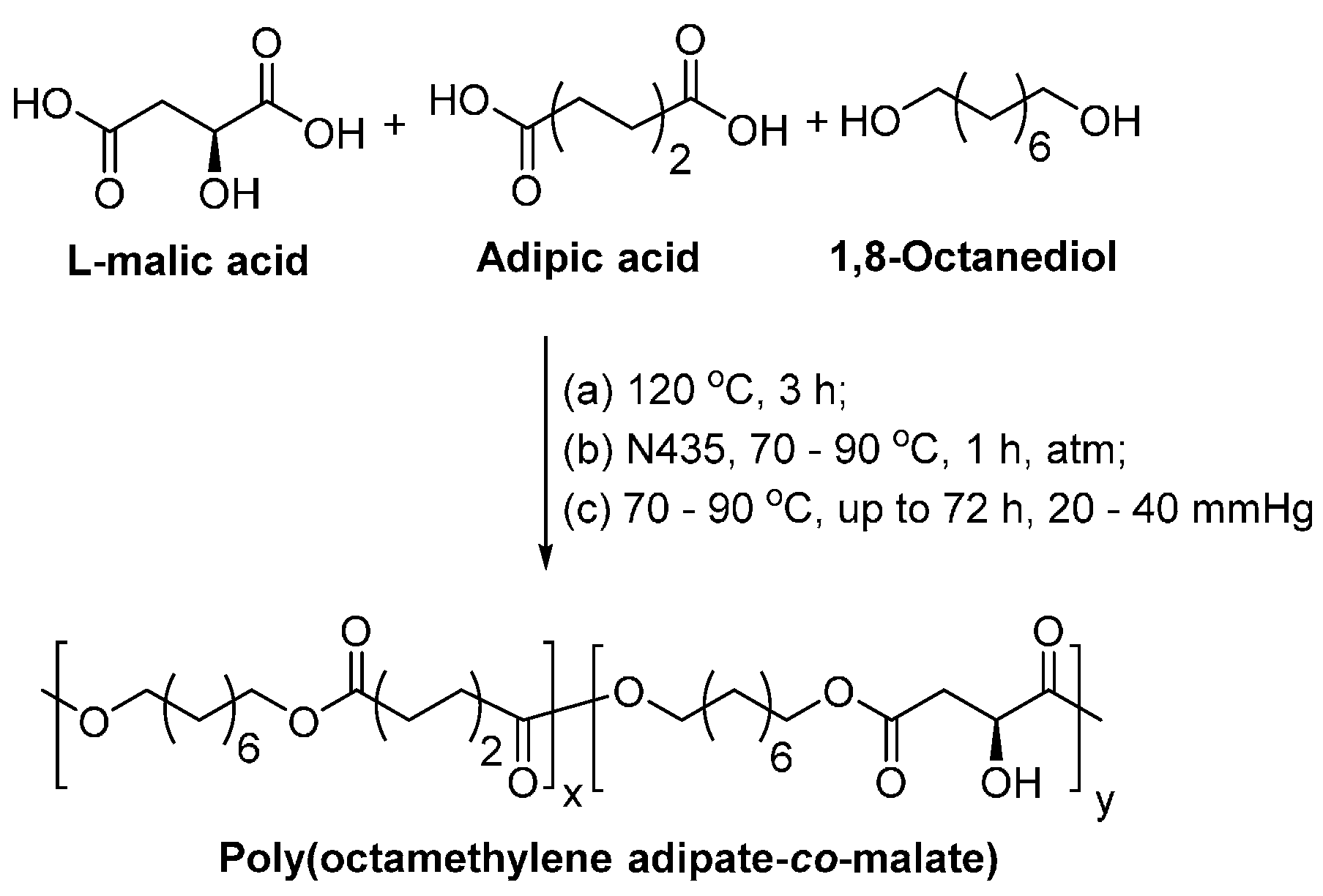
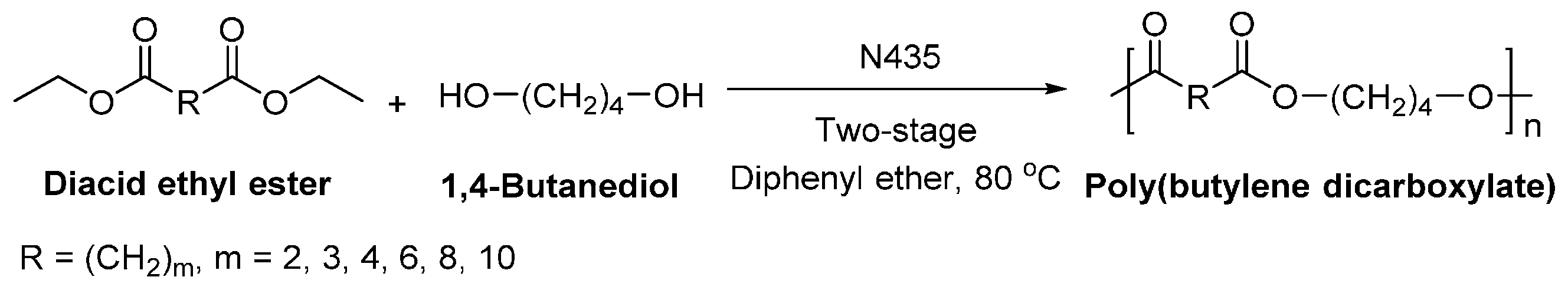
8.1.3. Other Biobased Aliphatic Polyesters
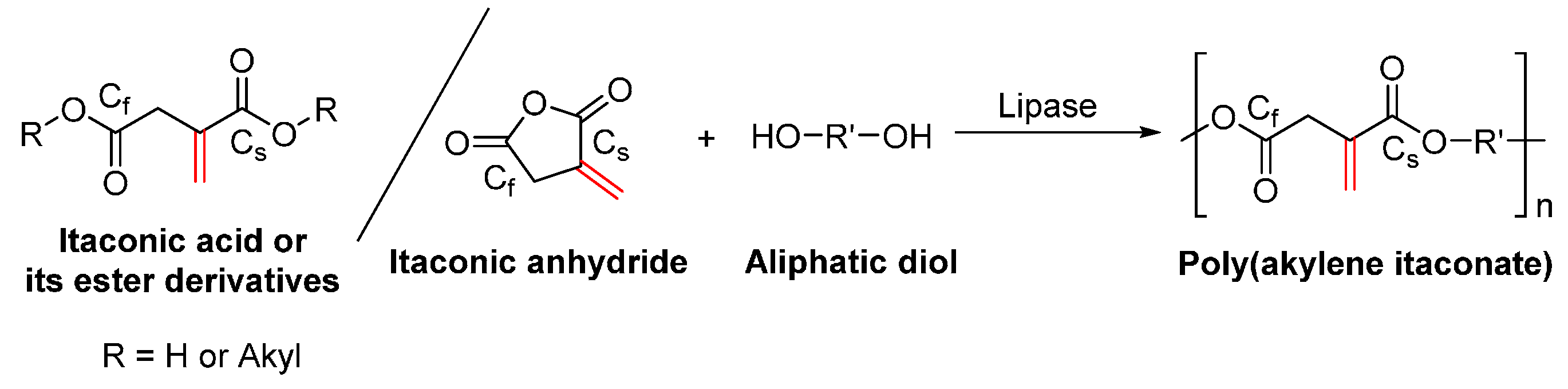
8.2. Biobased Unsaturated Aliphatic Polyesters
8.3. Polyesters Derived from Long Chain Fatty Acids and their Derivatives
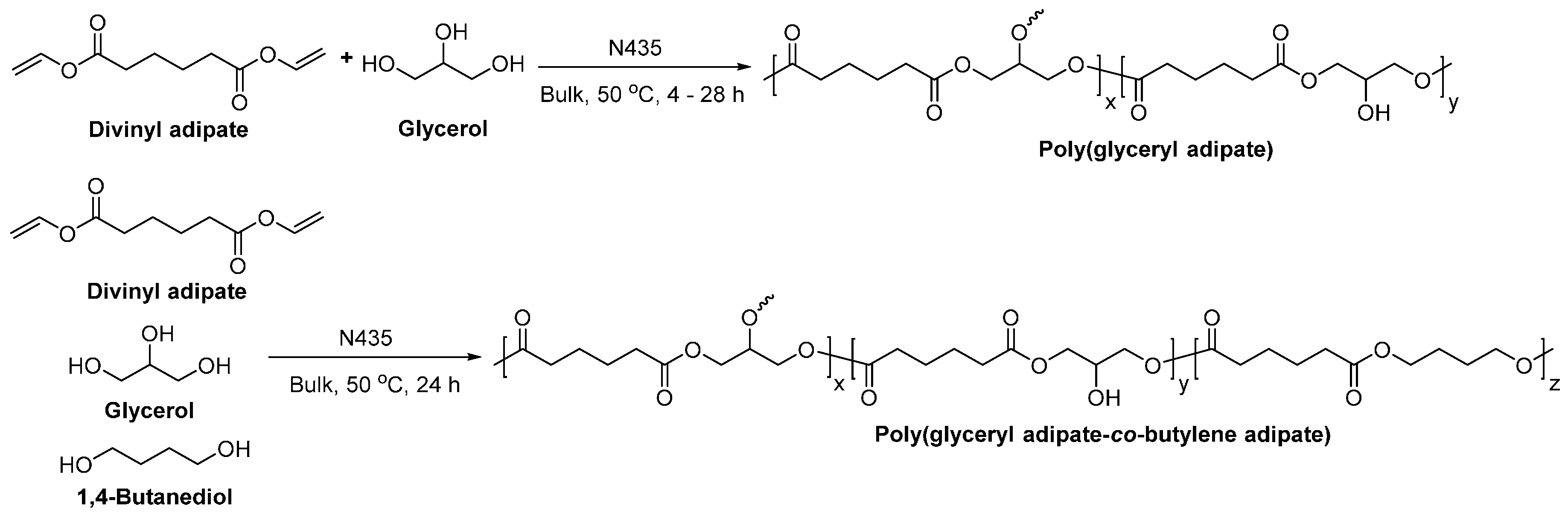
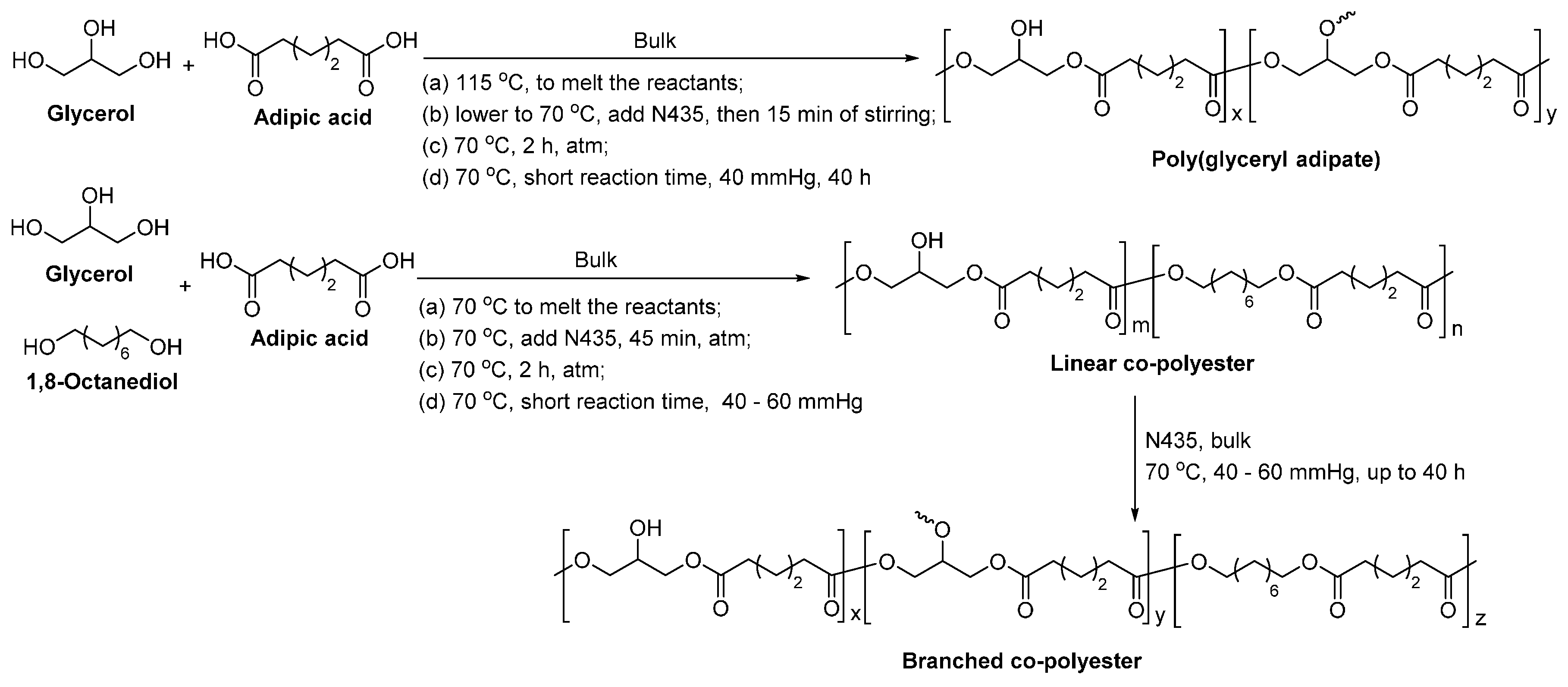
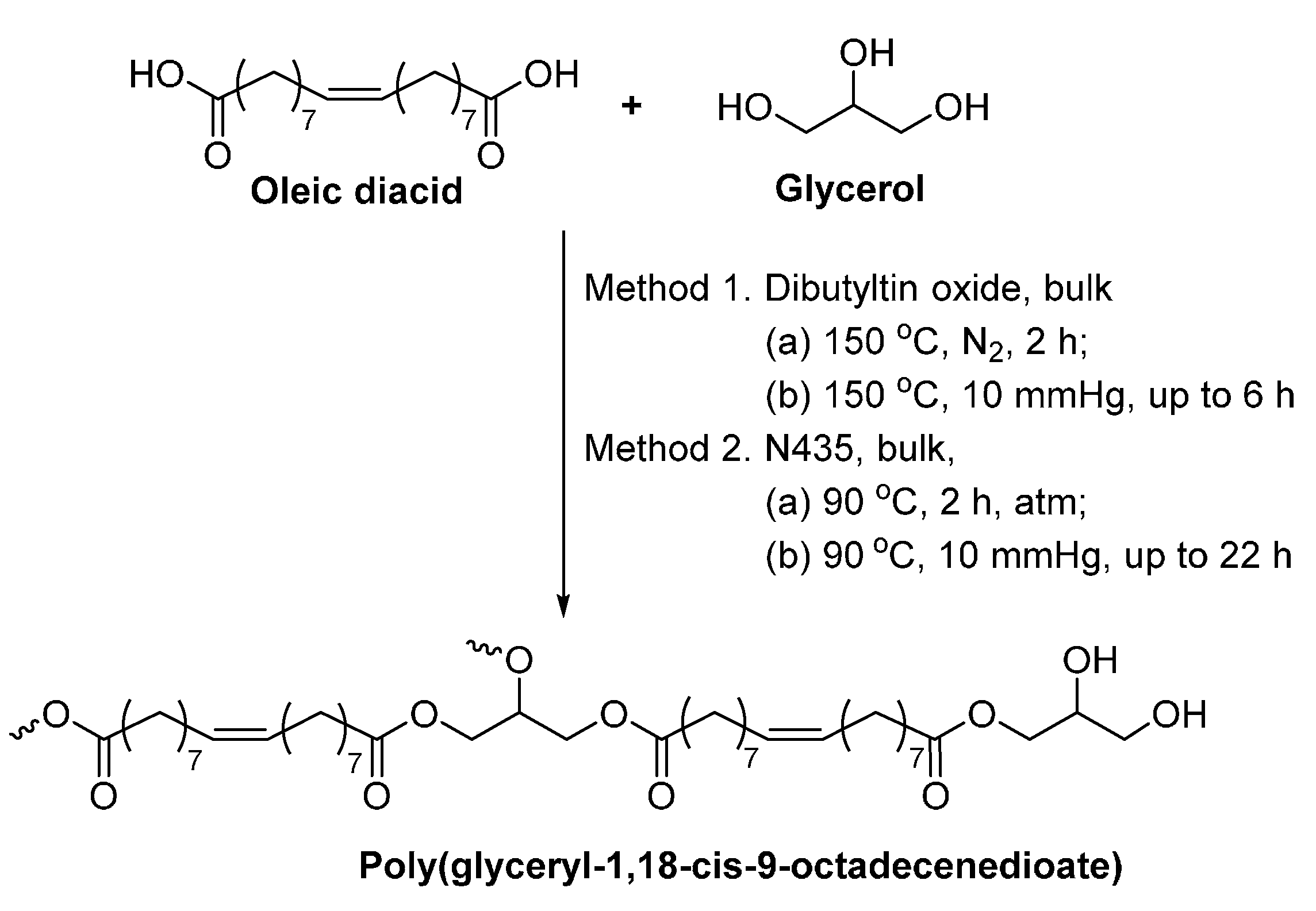
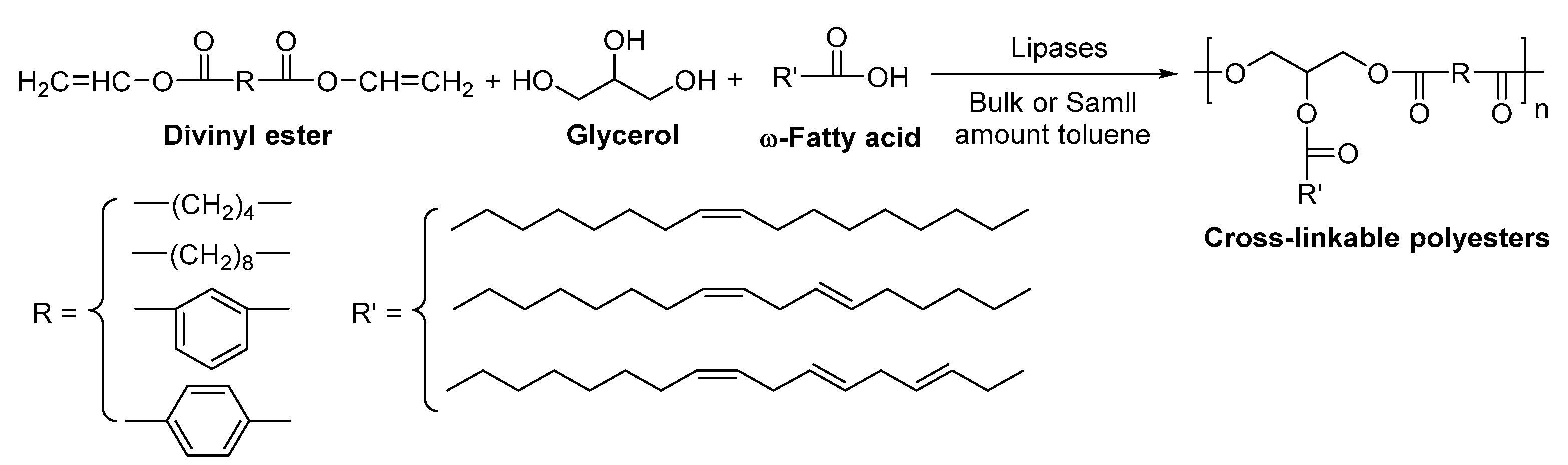
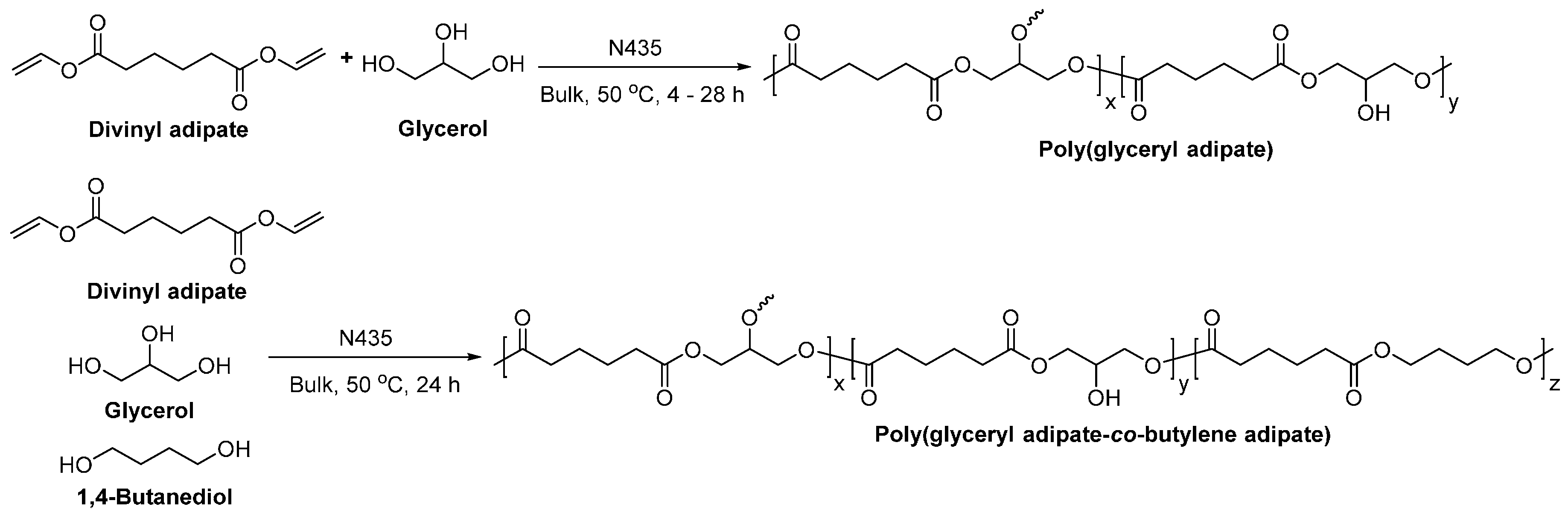
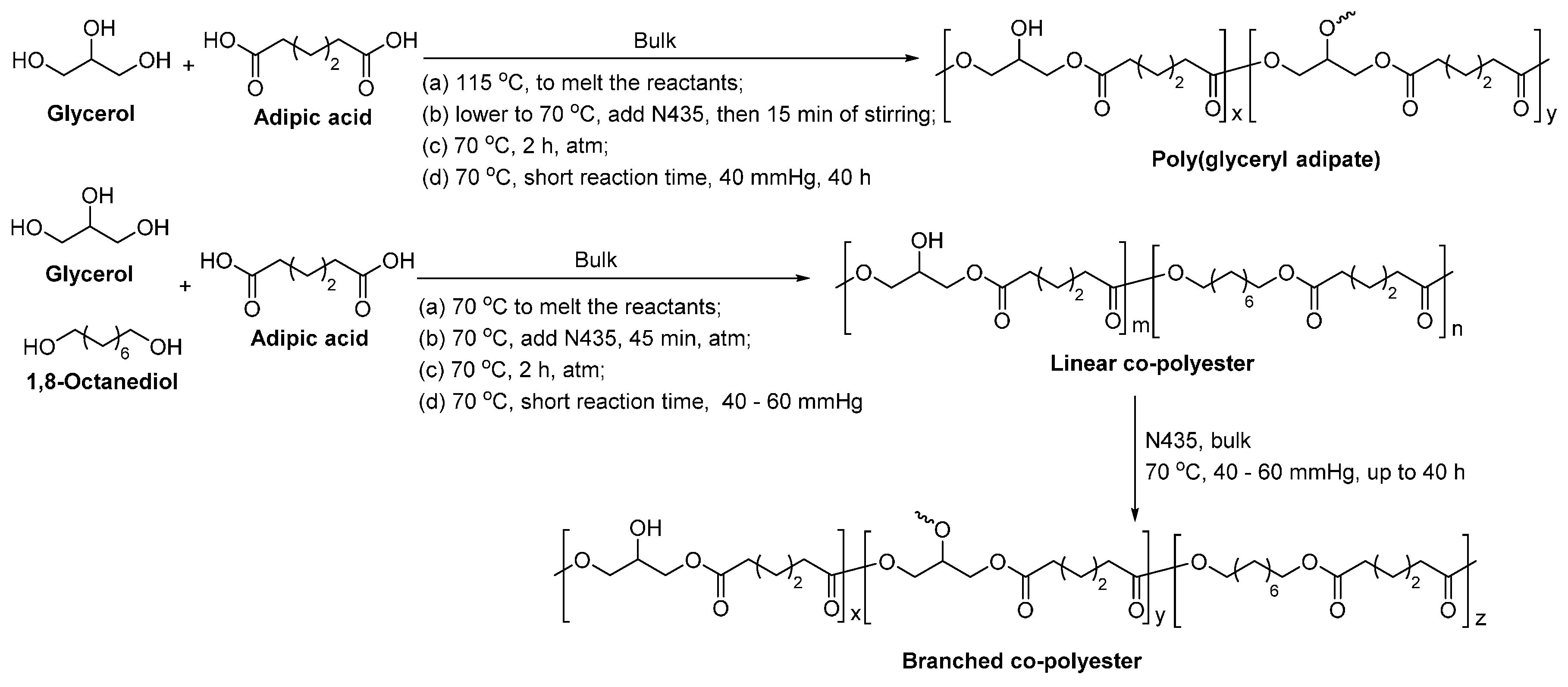
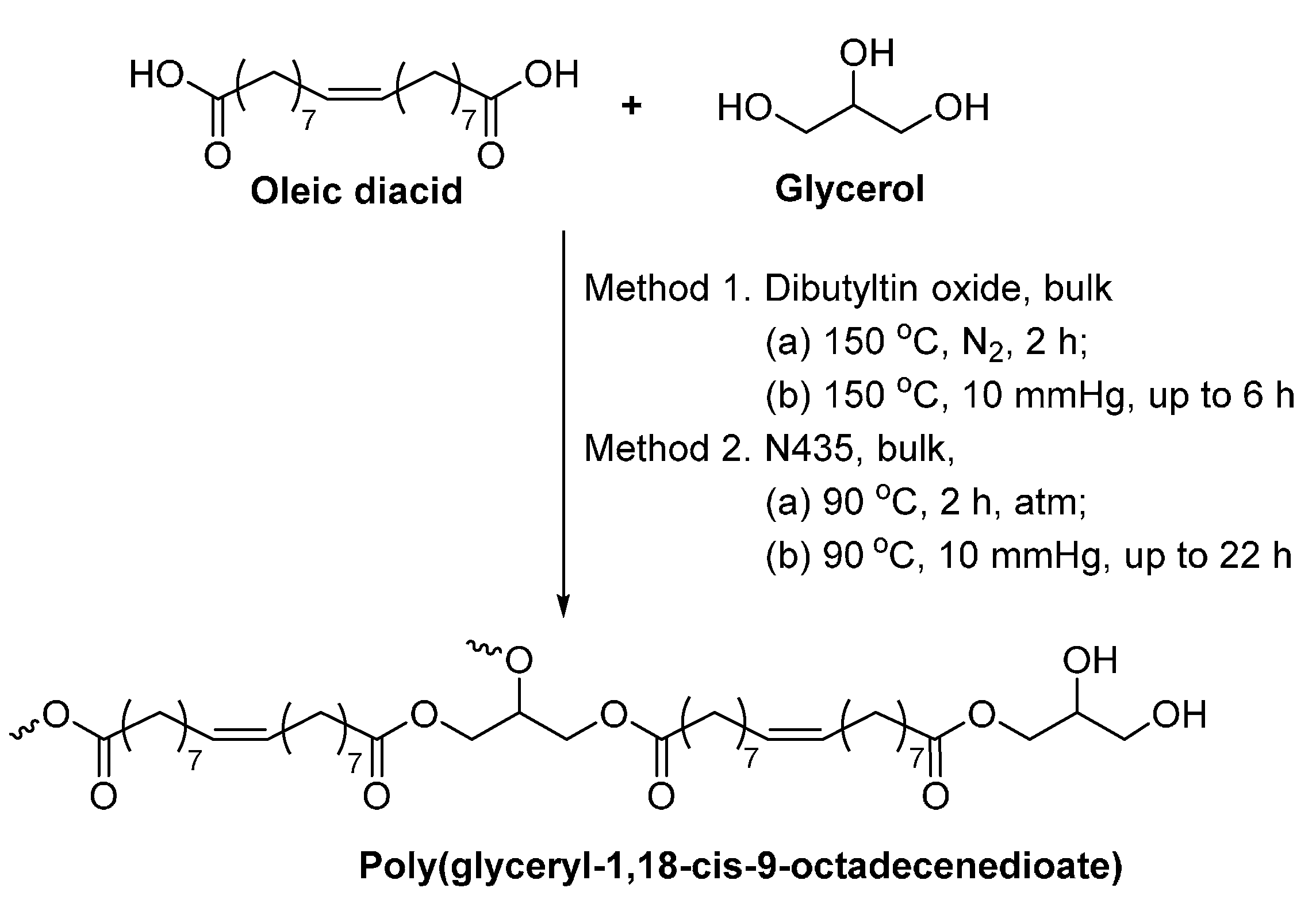
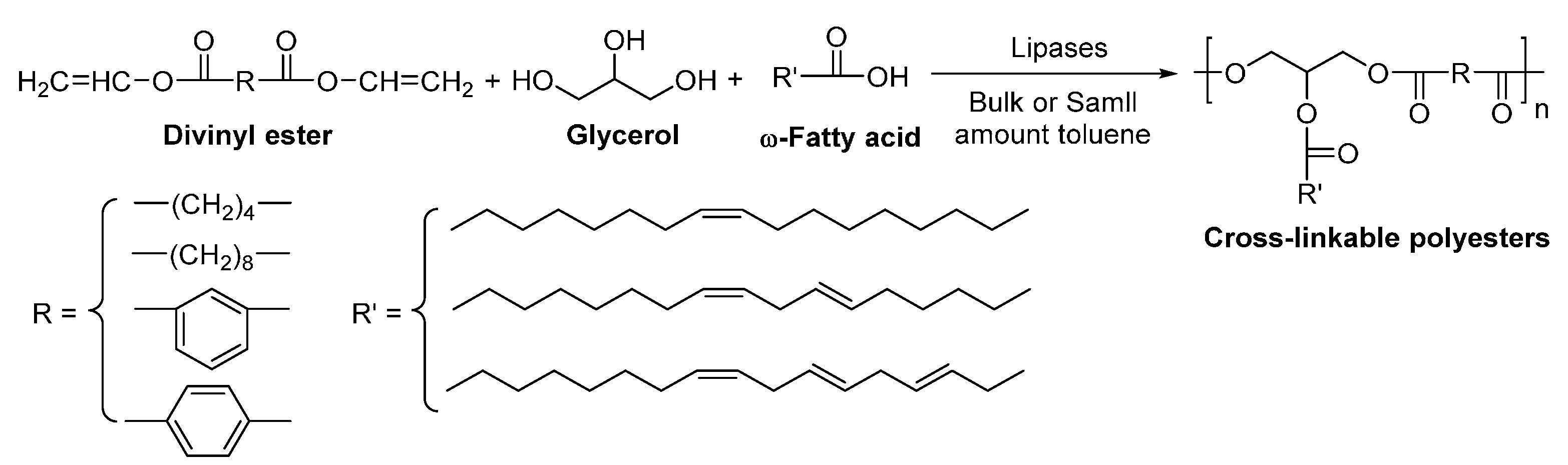
8.4. Glycerol-Based Polyesters
8.5. Sweet Polyesters Derived from Carbohydrates
8.5.1. Sugar and Sugar Alcohol-Based Polyesters
8.5.2. Polyesters Based on Rigid Sugar Derivatives
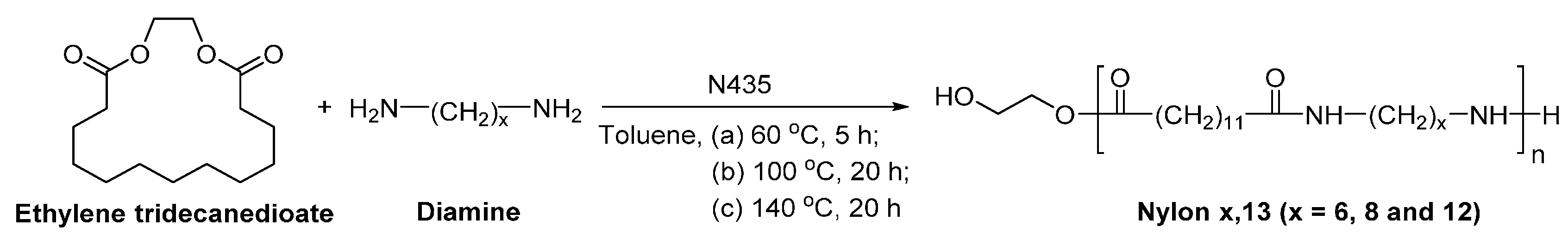
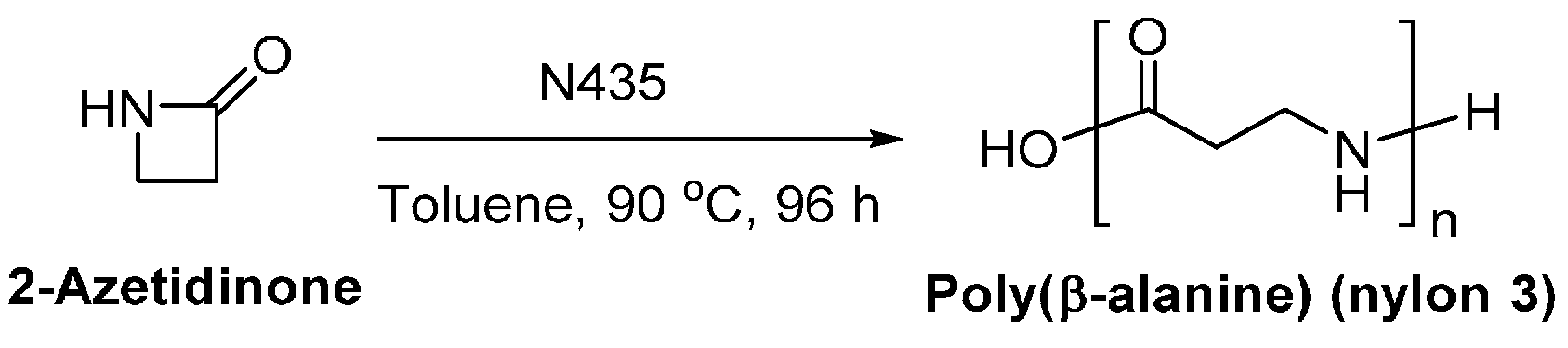
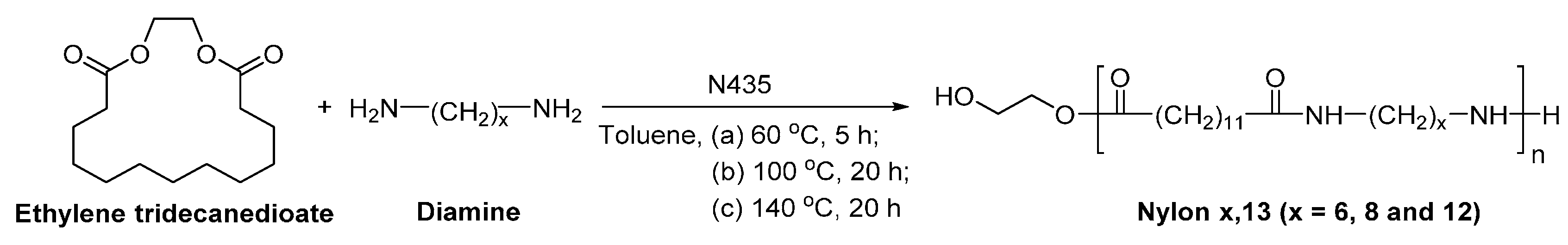
8.6. Biobased Polyamides
8.7. Biobased Furan Polyesters and Furan Polyamides
9. Conclusions and Outlook
- (1)
- the atom efficiency is low when ester derivatives rather than acids are used;
- (2)
- non-ecofriendly solvents including diphenyl ether and toluene are commonly used;
- (3)
- long polymerization times are required for achieving high molecular weights;
- (4)
- high reaction temperatures at around 100–140 °C were applied for enzymatic synthesis of polymers having a high Tm and low solubility; and the catalytic reactivity of enzymes decreases significantly at such elevated temperatures;
- (5)
- the price of enzyme catalysts is still quite high;
- (6)
- enzymatic polymerizations involving monomers with short chain length like 1,3-propanediol, monomers with secondary hydroxyl groups such as isosorbide and 2,3-butanediol, and polyols, generally result in low molecular weight products;
- (7)
- the purify and price of biobased monomers remain a concern;
- (8)
- last but certainly not least, only limited variety of biobased monomers are currently commercially available.
Acknowledgments
Conflicts of Interest
References
- Bower, D.I. An Introduction to Polymer Physics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- PlasticsEurope. Plastics-the Facts 2015; PlasticsEurope: Brussels, Belgium, 2015. [Google Scholar]
- Aeschelmann, F.; Carus, M. Bio-Based Building Blocks and Polymers in the World-Capacities, Production and Applications: Status Quo and Trends Towards 2020; nova-Institut GmbH: Hürth, Germany, 2015; pp. 1–500. [Google Scholar]
- Satyanarayana, K.G.; Arizaga, G.G.C.; Wypych, F. Biodegradable composites based on lignocellulosic fibers—An overview. Prog. Polym. Sci. 2009, 34, 982–1021. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Remzi Becer, C. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A. Monomers and macromonomers from renewable resources. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 1–33. [Google Scholar]
- Gandini, A.; Lacerda, T.M. From monomers to polymers from renewable resources: Recent advances. Prog. Polym. Sci. 2015, 48, 1–39. [Google Scholar] [CrossRef]
- Gandini, A.; Lacerda, T.M.; Carvalho, A.J.; Trovatti, E. Progress of polymers from renewable resources: Furans, vegetable oils, and polysaccharides. Chem. Rev. 2016, 116, 1637–1669. [Google Scholar] [CrossRef] [PubMed]
- Dove, A. Polymer science tries to make it easy to be green. Science 2012, 335, 1382–1384. [Google Scholar] [CrossRef]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Mathers, R.T. How well can renewable resources mimic commodity monomers and polymers? J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1–15. [Google Scholar] [CrossRef]
- Mülhaupt, R. Green polymer chemistry and bio-based plastics: Dreams and reality. Macromol. Chem. Phys. 2013, 214, 159–174. [Google Scholar] [CrossRef]
- Vilela, C.; Sousa, A.F.; Fonseca, A.C.; Serra, A.C.; Coelho, J.F.J.; Freire, C.S.R.; Silvestre, A.J.D. The quest for sustainable polyesters-insights into the future. Polym. Chem. 2014, 5, 3119–3141. [Google Scholar] [CrossRef]
- Galbis, J.A.; Garcia-Martin Mde, G.; de Paz, M.V.; Galbis, E. Synthetic polymers from sugar-based monomers. Chem. Rev. 2016, 116, 1600–1636. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.F.; Vilela, C.; Fonseca, A.C.; Matos, M.; Freire, C.S.R.; Gruter, G.-J.M.; Coelho, J.F.J.; Silvestre, A.J.D. Biobased polyesters and other polymers from 2,5-furandicarboxylic acid: A tribute to furan excellency. Polym. Chem. 2015, 6, 5961–5983. [Google Scholar] [CrossRef]
- Bell, S.L. Ihs Chemical Process Economics Program: Report 265a, Bio-Based Polymers; IHS Chemical: New York, NY, USA, 2013. [Google Scholar]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass: Volume I-Results of Screening for Potential Candidates from Sugars and Synthesis Gas; DOE/GO-102004-1992; Pacific Northwest National Laboratory and National Renewable Energy Laboratory: Oak Ridge, TN, USA, 2004; pp. 1–76. [Google Scholar]
- Corma, A.; Iborra, S.; Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 2007, 107, 2411–2502. [Google Scholar] [CrossRef] [PubMed]
- Holladay, J.E.; White, J.F.; Bozell, J.J.; Johnson, D. Top Value-Added Chemicals from Biomass-Volume II-Results of Screening for Potential Candidates from Biorefinery Lignin; PNNL-16983, Pacific Northwest National Laboratory, University of Tennessee, National Renewable Energy Laboratory: Oak Ridge, TN, USA, 2007; pp. 1–79. [Google Scholar]
- Gandini, A. Polymers from renewable resources: A challenge for the future of macromolecular materials. Macromolecules 2008, 41, 9491–9504. [Google Scholar] [CrossRef]
- Bozell, J.J.; Petersen, G.R. Technology development for the production of biobased products from biorefinery carbohydrates-the us department of energy’s “top 10” revisited. Green Chem. 2010, 12, 539–554. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- van Putten, R.-J.; van der Waal, J.C.; de Jong, E.; Rasrendra, C.B.; Heeres, H.J.; de Vries, J.G. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef] [PubMed]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Green and sustainable manufacture of chemicals from biomass: State of the art. Green Chem. 2014, 16, 950–963. [Google Scholar] [CrossRef]
- Delidovich, I.; Hausoul, P.J.; Deng, L.; Pfutzenreuter, R.; Rose, M.; Palkovits, R. Alternative monomers based on lignocellulose and their use for polymer production. Chem. Rev. 2016, 116, 1540–1599. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Wittmann, C. Advanced biotechnology: Metabolically engineered cells for the bio-based production of chemicals and fuels, materials, and health-care products. Angew. Chem. Int. Ed. 2015, 54, 3328–3350. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Song, C.W.; Shin, J.H.; Lee, S.Y. Biorefineries for the production of top building block chemicals and their derivatives. Metab. Eng. 2015, 28, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Dusselier, M.; Mascal, M.; Sels, B.F. Top chemical opportunities from carbohydrate biomass: A chemist’s view of the biorefinery. In Selective Catalysis for Renewable Feedstocks and Chemicals; Nicholas, K.M., Ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2014; Volume 353, pp. 1–40. [Google Scholar]
- Harmsen, P.F.H.; Hackmann, M.M.; Bos, H.L. Green building blocks for bio-based plastics. Biofuels Bioprod. Biorefin. 2014, 8, 306–324. [Google Scholar] [CrossRef]
- Taylor, R.; Nattrass, L.; Alberts, G.; Robson, P.; Chudziak, C.; Bauen, A.; Libelli, I.M.; Lotti, G.; Prussi, M.; Nistri, R.; et al. From the Sugar Platform to Biofuels and Biochemicals; contract No. ENER/C2/423-2012/SI2.673791; E4tech, RE-CORD and WUR: London, UK, 2015; pp. 1–183. [Google Scholar]
- De Jong, E.; Higson, A.; Walsh, P.; Wellisch, M. Bio-Based Chemicals: Value Added Products from Biorefineries; Avantium Chemicals (Netherlands), NNFCC (UK), Energy Research Group (Ireland), and Agriculture and Agri-Food Canada (Canada): Wageningen, The Netherlands, 2012. [Google Scholar]
- Golden, J.; Handfield, R. Why Biobased? Opportunities in the Emerging Bioeconomy; US Department of Agriculture, Office of Procurement and Property Management: Washington, DC, USA, 2014. [Google Scholar]
- Gross, R.A.; Kumar, A.; Kalra, B. Polymer synthesis by in vitro enzyme catalysis. Chem. Rev. 2001, 101, 2097–2124. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Uyama, H.; Kimura, S. Enzymatic polymerization. Chem. Rev. 2001, 101, 3793–3818. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S. Enzymatic synthesis of polyesters via ring-opening polymerization. In Enzyme-Catalyzed Synthesis of Polymers; Kobayashi, S., Ritter, H., Kaplan, D., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; Volume 194, pp. 95–132. [Google Scholar]
- Singh, A.; Kaplan, D.L. In vitro enzyme-induced vinyl polymerization. In Enzyme-Catalyzed Synthesis of Polymers; Kobayashi, S., Ritter, H., Kaplan, D., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; Volume 194, pp. 211–224. [Google Scholar]
- Uyama, H.; Kobayashi, S. Enzymatic synthesis of polyesters via polycondensation. In Enzyme-Catalyzed Synthesis of Polymers; Kobayashi, S., Ritter, H., Kaplan, D., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2006; Volume 194, pp. 133–158. [Google Scholar]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.A.; Ganesh, M.; Lu, W. Enzyme-catalysis breathes new life into polyester condensation polymerizations. Trends Biotechnol. 2010, 28, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Miletić, N.; Loos, K.; Gross, R.A. Enzymatic polymerization of polyester. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 83–129. [Google Scholar]
- Kobayashi, S. Green polymer chemistry: Recent developments. In Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II; Percec, V., Ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2013; Volume 262, pp. 141–166. [Google Scholar]
- Kobayashi, S. Enzymatic ring-opening polymerization and polycondensation for the green synthesis of polyesters. Polym. Adv. Technol. 2015, 26, 677–686. [Google Scholar] [CrossRef]
- Díaz, A.; Katsarava, R.; Puiggalí, J. Synthesis, properties and applications of biodegradable polymers derived from diols and dicarboxylic acids: From polyesters to poly(ester amide)s. Int. J. Mol. Sci. 2014, 15, 7064–7123. [Google Scholar] [CrossRef] [PubMed]
- Hillmyer, M.A.; Tolman, W.B. Aliphatic polyester block polymers: Renewable, degradable, and sustainable. Acc. Chem. Res. 2014, 47, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Vert, M. Aliphatic polyesters: Great degradable polymers that cannot do everything. Biomacromolecules 2004, 6, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Grand View Research. Lactic Acid and Poly Lactic Acid (Pla) Market Analysis by Application (Packaging, Agriculture, Transport, Electronics, Textiles) and Segment Forecasts to 2020; Grand View Research, Inc.: San Francisco, CA, USA, 2014. [Google Scholar]
- Lunt, J. Marketplace Opportunities for Integration of Biobased and Conventional Plastics; Agricultural Utilization Research Institute, Minnesota Corn Research & Promotion Council, and Minnesota Soybean Research & Promotion Council: Mankato, MN, USA, 2014; pp. 1–115. [Google Scholar]
- Albertsson, A.-C.; Varma, I. Aliphatic polyesters: Synthesis, properties and applications. In Degradable Aliphatic Polyesters; Springer-Verlag: Berlin/Heidelberg, Germany, 2002; Volume 157, pp. 1–40. [Google Scholar]
- Seyednejad, H.; Ghassemi, A.H.; van Nostrum, C.F.; Vermonden, T.; Hennink, W.E. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J. Control. Release 2011, 152, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Vert, M.; Li, S.M.; Spenlehauer, G.; Guerin, P. Bioresorbability and biocompatibility of aliphatic polyesters. J. Mater. Sci. Mater. Med. 1992, 3, 432–446. [Google Scholar] [CrossRef]
- Biron, M. The plastics industry: Economic overview. In Thermoplastics and Thermoplastic Composites; Biron, M., Ed.; Elsevier: Oxford, UK, 2007; pp. 33–153. [Google Scholar]
- Merchant Research & Consulting. Polyethylene Terephthalate (PET): 2014 World Market Outlook and Forecast up to 2018; Merchant Research & Consulting, Ltd.: Birmingham, UK, 2014. [Google Scholar]
- Samui, A.B.; Rao, V.S. Liquid crystal polyesters. In Handbook of Engineering and Speciality Thermoplastics; Scrivener Publishing LLC and John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 271–347. [Google Scholar]
- Honkhambe, P.N.; Biyani, M.V.; Bhairamadgi, N.S.; Wadgaonkar, P.P.; Salunkhe, M.M. Synthesis and characterization of new aromatic polyesters containing pendent naphthyl units. J. Appl. Polym. Sci. 2010, 117, 2545–2552. [Google Scholar] [CrossRef]
- Santhana Gopala Krishnan, P.; Kulkarni, S.T. Polyester resins. In Polyesters and Polyamides; Deopura, B.L., Alagirusamy, R., Joshi, M., Gupta, B., Eds.; Woodhead Publishing: Cambridge, UK, 2008; pp. 3–40. [Google Scholar]
- Madec, P.-J.; Maréchal, E. Kinetics and mechanisms of polyesterifications II. Reactions of diacids with diepoxides. In Analysis/Reactions/Morphology; Springer-Verlag: Berlin/Heidelberg, Germany, 1985; Volume 71, pp. 153–228. [Google Scholar]
- Yao, K.; Tang, C. Controlled polymerization of next-generation renewable monomers and beyond. Macromolecules 2013, 46, 1689–1712. [Google Scholar] [CrossRef]
- Niaounakis, M. Introduction. In Biopolymers: Processing and Products; Niaounakis, M., Ed.; William Andrew Publishing: Oxford, UK, 2015; pp. 1–77. [Google Scholar]
- Babu, R.; O’Connor, K.; Seeram, R. Current progress on bio-based polymers and their future trends. Prog. Biomater. 2013, 2, 8. [Google Scholar] [CrossRef]
- OECD. Biobased Chemicals and Bioplastics: Finding the Right Policy Balance. In Technology and Industry Policy Papers No. 17; OECD Science: Paris, France, 2014; pp. 1–96. [Google Scholar]
- Dammer, L.; Carus, M.; Raschka, A.; Scholz, L. Market Developments of and Opportunities for Biobased Products and Chemicals; Nova-Institute for Ecology and Innovation: Hürth, Germany, 2013; pp. 1–69. [Google Scholar]
- Siegenthaler, K.O.; Künkel, A.; Skupin, G.; Yamamoto, M. Ecoflex® and ecovio®: Biodegradable, performance-enabling plastics. In Synthetic Biodegradable Polymers; Rieger, B., Künkel, A., Coates, G.W., Reichardt, R., Dinjus, E., Zevaco, T.A., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2012; Volume 245, pp. 91–136. [Google Scholar]
- Jacquel, N.; Saint-Loup, R.; Pascault, J.-P.; Rousseau, A.; Fenouillot, F. Bio-based alternatives in the synthesis of aliphatic–aromatic polyesters dedicated to biodegradable film applications. Polymer 2015, 59, 234–242. [Google Scholar] [CrossRef]
- Fink, J.K. An overview of methods and standards. In The Chemistry of Bio-Based Polymers; Scrivener Publishing LLC and John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 1–41. [Google Scholar]
- Gohil, R.M.; Hasty, N.M.; Hayes, R.A.; Kurian, J.V.; Liang, Y.; Stancik, E.J.; Strukelj, M.; Tseng, S.Y.T. Aliphatic-Aromatic Polyesters, and Articles Made Therefrom. U.S. Patent US788 US7888405 B2, 15 Feburary 2011. [Google Scholar]
- Zini, E.; Scandola, M. Green composites: An overview. Polym. Compos. 2011, 32, 1905–1915. [Google Scholar] [CrossRef]
- Deopura, B.L. Polyamide fibers. In Polyesters and Polyamides; Deopura, B.L., Alagirusamy, R., Joshi, M., Gupta, B., Eds.; Woodhead Publishing: Cambridge, UK, 2008; pp. 41–61. [Google Scholar]
- Schlack, P. Preparation of Polyamides. U.S. Patent US2241321 A, 3 June 1941. [Google Scholar]
- Schlack, P. Verfahren zur Herstellung Verformbarer Hochmolekularer Polyamide. German Patent DE748253 (C), 30 October 1944. [Google Scholar]
- Brehmer, B. Polyamides from biomass derived monomers. In Bio-Based Plastics; Kabasci, S., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2013; pp. 275–293. [Google Scholar]
- Marchildon, K. Polyamides-still strong after seventy years. Macromol. React. Eng. 2011, 5, 22–54. [Google Scholar] [CrossRef]
- Zhang, G.; Zhou, Y.X.; Li, Y.; Wang, X.J.; Long, S.R.; Yang, J. Investigation of the synthesis and properties of isophorone and ether units based semi-aromatic polyamides. RSC Adv. 2015, 5, 49958–49967. [Google Scholar] [CrossRef]
- García, J.M.; García, F.C.; Serna, F.; de la Peña, J.L. Aromatic polyamides (aramids). In Handbook of Engineering and Specialty Thermoplastics; Thomas, S., Visakh, P.M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Volume 4, pp. 141–181. [Google Scholar]
- García, J.M.; García, F.C.; Serna, F.; de la Peña, J.L. High-performance aromatic polyamides. Prog. Polym. Sci. 2010, 35, 623–686. [Google Scholar] [CrossRef]
- Hong, M.; Chen, E.Y.X. Coordination ring-opening copolymerization of naturally renewable α-methylene-γ-butyrolactone into unsaturated polyesters. Macromolecules 2014, 47, 3614–3624. [Google Scholar] [CrossRef]
- Martin, C.H.; Dhamankar, H.; Tseng, H.-C.; Sheppard, M.J.; Reisch, C.R.; Prather, K.L.J. A platform pathway for production of 3-hydroxyacids provides a biosynthetic route to 3-hydroxy-γ-butyrolactone. Nat. Commun. 2013, 4, 1414. [Google Scholar] [CrossRef] [PubMed]
- Rouhi, A.M. Custom chemicals. Chem. Eng. News Arch. 2003, 81, 55–73. [Google Scholar] [CrossRef]
- KWAK, B.-S. Development of chiral pharmaceutical fine chemicals through technology fusion. Chim. Oggi 2003, 21, 23–26. [Google Scholar]
- Dhamankar, H.; Tarasova, Y.; Martin, C.H.; Prather, K.L.J. Engineering E. Coli for the biosynthesis of 3-hydroxy-γ-butyrolactone (3HBL) and 3,4-dihydroxybutyric acid (3,4-DHBA) as value-added chemicals from glucose as a sole carbon source. Metab. Eng. 2014, 25, 72–81. [Google Scholar] [PubMed]
- Chen, T.N.; Qin, Z.F.; Qi, Y.Q.; Deng, T.S.; Ge, X.J.; Wang, J.G.; Hou, X.L. Degradable polymers from ring-opening polymerization of α-angelica lactone, a five-membered unsaturated lactone. Polym. Chem. 2011, 2, 1190–1194. [Google Scholar] [CrossRef]
- Buntara, T.; Noel, S.; Phua, P.H.; Melián-Cabrera, I.; delVries, J.G.; Heeres, H.J. Caprolactam from renewable resources: Catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew. Chem. Int. Ed. 2011, 50, 7083–7087. [Google Scholar] [CrossRef] [PubMed]
- Raoufmoghaddam, S.; Rood, M.T.M.; Buijze, F.K.W.; Drent, E.; Bouwman, E. Catalytic conversion of γ-valerolactone to ε-caprolactam: Towards nylon from renewable feedstock. ChemSusChem 2014, 7, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.W. Synthesis of Caprolactam from Lysine. International Patent WO2005123669A1, 29 December 2005. [Google Scholar]
- van Haveren, J.; Scott, E.L.; Sanders, J. Bulk chemicals from biomass. Biofuels Bioprod. Biorefin. 2008, 2, 41–57. [Google Scholar] [CrossRef]
- Jansen, M.L.A.; van Gulik, W.M. Towards large scale fermentative production of succinic acid. Curr. Opin. Biotechnol. 2014, 30, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Nattrass, L.; Aylott, M.; Higson, A. Nnfcc Renewable Chemicals Fact Sheet: Succinic Acid; NNFCC: York, UK, 2013. [Google Scholar]
- MuralidharaRao, D.; Hussain, S.M.D.J.; Rangadu, V.P.; Subramanyam, K.; Krishna, G.S.; Swamy, A.V.N. Fermentatative production of itaconic acid by aspergillus terreus using jatropha seed cake. Afr. J. Biotechnol. 2007, 6, 2140–2142. [Google Scholar]
- Steiger, M.G.; Blumhoff, M.L.; Mattanovich, D.; Sauer, M. Biochemistry of microbial itaconic acid production. Front. Microbiol. 2013, 4, 23. [Google Scholar] [CrossRef] [PubMed]
- Okabe, M.; Lies, D.; Kanamasa, S.; Park, E.Y. Biotechnological production of itaconic acid and its biosynthesis in aspergillus terreus. Appl. Microbiol. Biotechnol. 2009, 84, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Beerthuis, R.; Rothenberg, G.; Shiju, N.R. Catalytic routes towards acrylic acid, adipic acid and ε-caprolactam starting from biorenewables. Green Chem. 2015, 17, 1341–1361. [Google Scholar] [CrossRef]
- Bart, J.C.J.; Cavallaro, S. Transiting from adipic acid to bioadipic acid. Part ii. Biosynthetic pathways. Ind. Eng. Chem. Res. 2015, 54, 567–576. [Google Scholar] [CrossRef]
- Bart, J.C.J.; Cavallaro, S. Transiting from adipic acid to bioadipic acid. 1, petroleum-based processes. Ind. Eng. Chem. Res. 2015, 54, 1–46. [Google Scholar]
- Deng, Y.; Ma, L.; Mao, Y. Biological production of adipic acid from renewable substrates: Current and future methods. Biochem. Eng. J. 2016, 105, 16–26. [Google Scholar] [CrossRef]
- Ayorinde, F.; Osman, G.; Shepard, R.; Powers, F. Synthesis of azelaic acid and suberic acid fromvernonia galamensis oil. J. Am. Oil Chemists’ Soc. 1988, 65, 1774–1777. [Google Scholar] [CrossRef]
- Metzger, J.O. Fats and oils as renewable feedstock for chemistry. Eur. J. Lipid Sci. Technol. 2009, 111, 865–876. [Google Scholar] [CrossRef]
- Frost, J.W.; Millis, J.; Tang, Z. Methods for Producing Dodecanedioic Acid and Derivatives Thereof. International Patent WO2010085712 A2, 29 July 2010. [Google Scholar]
- Sun, X.; Shen, X.; Jain, R.; Lin, Y.; Wang, J.; Sun, J.; Wang, J.; Yan, Y.; Yuan, Q. Synthesis of chemicals by metabolic engineering of microbes. Chem. Soc. Rev. 2015, 44, 3760–3785. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, P. The Environmental Performance of Biobased 1, 3-propanediol Production from Glycerol Compared to Conventional Production Pathways-A Life Cycle Assessment. Master’s Thesis, University of Utrecht, Utrecht, The Netherlands, 2014. [Google Scholar]
- Przystalowska, H.; Lipinski, D.; Slomski, R. Biotechnological conversion of glycerol from biofuels to 1,3-propanediol using escherichia coil. Acta Biochim. Pol. 2015, 62, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, G.P.; Contiero, J.; Avila Neto, P.M.; Bolner de Lima, C.J. 1,3-propanediol: Production, applications and biotechnological potential. Quim. Nova 2014, 37, 527–534. [Google Scholar] [CrossRef]
- Lee, C.S.; Aroua, M.K.; Daud, W.M.A.W.; Cognet, P.; Peres-Lucchese, Y.; Fabre, P.L.; Reynes, O.; Latapie, L. A review: Conversion of bioglycerol into 1,3-propanediol via biological and chemical method. Renew. Sustain. Energy Rev. 2015, 42, 963–972. [Google Scholar] [CrossRef]
- Yim, H.; Haselbeck, R.; Niu, W.; Pujol-Baxley, C.; Burgard, A.; Boldt, J.; Khandurina, J.; Trawick, J.D.; Osterhout, R.E.; Stephen, R.; et al. Metabolic engineering of escherichia coli for direct production of 1,4-butanediol. Nat. Chem. Biol. 2011, 7, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Bibolet, E.R.; Fernando, G.E.; Shah, S.M. Renewable 1, 4-Butanediol; University of Pennsylvania: Philadelphia, PA, USA, 2011. [Google Scholar]
- DSM Engineering Plastics. Achieving Higher Bio-Based Content in Dsm Arnitel® Copolyesters; DSM: Heerlen, The Netherlands, 2013. [Google Scholar]
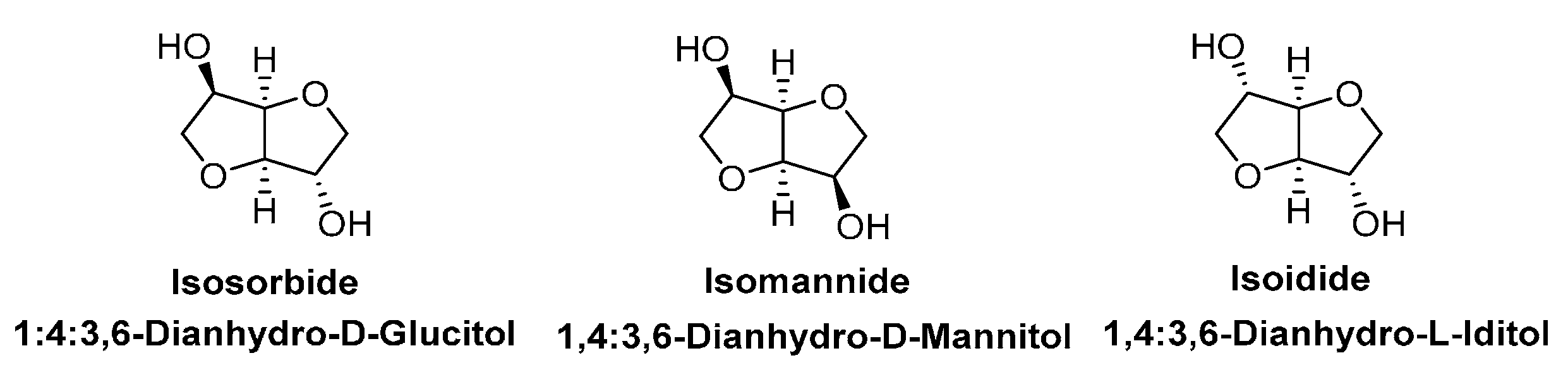
- Rose, M.; Palkovits, R. Isosorbide as a renewable platform chemical for versatile applicationsuquo vadis? Chemsuschem 2012, 5, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35, 578–622. [Google Scholar] [CrossRef]
- Celinska, E.; Grajek, W. Biotechnological production of 2,3-butanediol-current state and prospects. Biotechnol. Adv. 2009, 27, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Dias, E.L.; Shoemaker, J.A.W.; Boussie, T.R.; Murphy, V.J. Process for Production of Hexamethylenediamine from Carbohydrate-Containing Materials and Intermediates Therefor. U.S. Patent US8853458 B2, 7 October 2014. [Google Scholar]
- Hoekman, S.K.; Broch, A.; Robbins, C.; Ceniceros, E.; Natarajan, M. Review of biodiesel composition, properties, and specifications. Renew. Sustain. Energy Rev. 2012, 16, 143–169. [Google Scholar] [CrossRef]
- Galbis, J.A.; Garcia-Martin, M.G. Synthetic polymers from readily available monosaccharides. In Carbohydrates in Sustainable Development II: A Mine for Functional Molecules and Materials; Rauter, A.P., Vogel, P., Queneau, Y., Eds.; Springer-Verlag Berlin: Berlin, Germany, 2010; Volume 295, pp. 147–176. [Google Scholar]
- Qian, Z.-G.; Xia, X.-X.; Lee, S.Y. Metabolic engineering of Escherichia coli for the production of putrescine: A four carbon diamine. Biotechnol. Bioeng. 2009, 104, 651–662. [Google Scholar] [PubMed]
- Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. Amines, aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Elvers, B., Hawkins, S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 1996; pp. 1–54. [Google Scholar]
- Becker, J.; Wittmann, C. Bio-based production of chemicals, materials and fuels–corynebacterium glutamicum as versatile cell factory. Curr. Opin. Biotechnol. 2012, 23, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, Y.H.; Shin, J.-H.; Bhatia, S.K.; Sathiyanarayanan, G.; Seo, H.-M.; Choi, K.Y.; Yang, Y.-H.; Park, K. Optimization of direct lysine decarboxylase biotransformation for cadaverine production with whole cell biocatalysts at high substrate concentration. J. Microbiol. Biotechnol. 2015, 25, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Boussie, T.R.; Dias, E.L.; Fresco, Z.M.; Murphy, V.J. Production of Adipic Acid and Derivatives from Carbohydrate-Containing Materials. U.S. Patent US8501989 B2, 6 August 2013. [Google Scholar]
- Tilley, T.G. Ueber die einwirkung der salpetersäure auf das ricinusöl. Justus Liebigs Ann. Chem. 1841, 39, 160–168. [Google Scholar] [CrossRef]
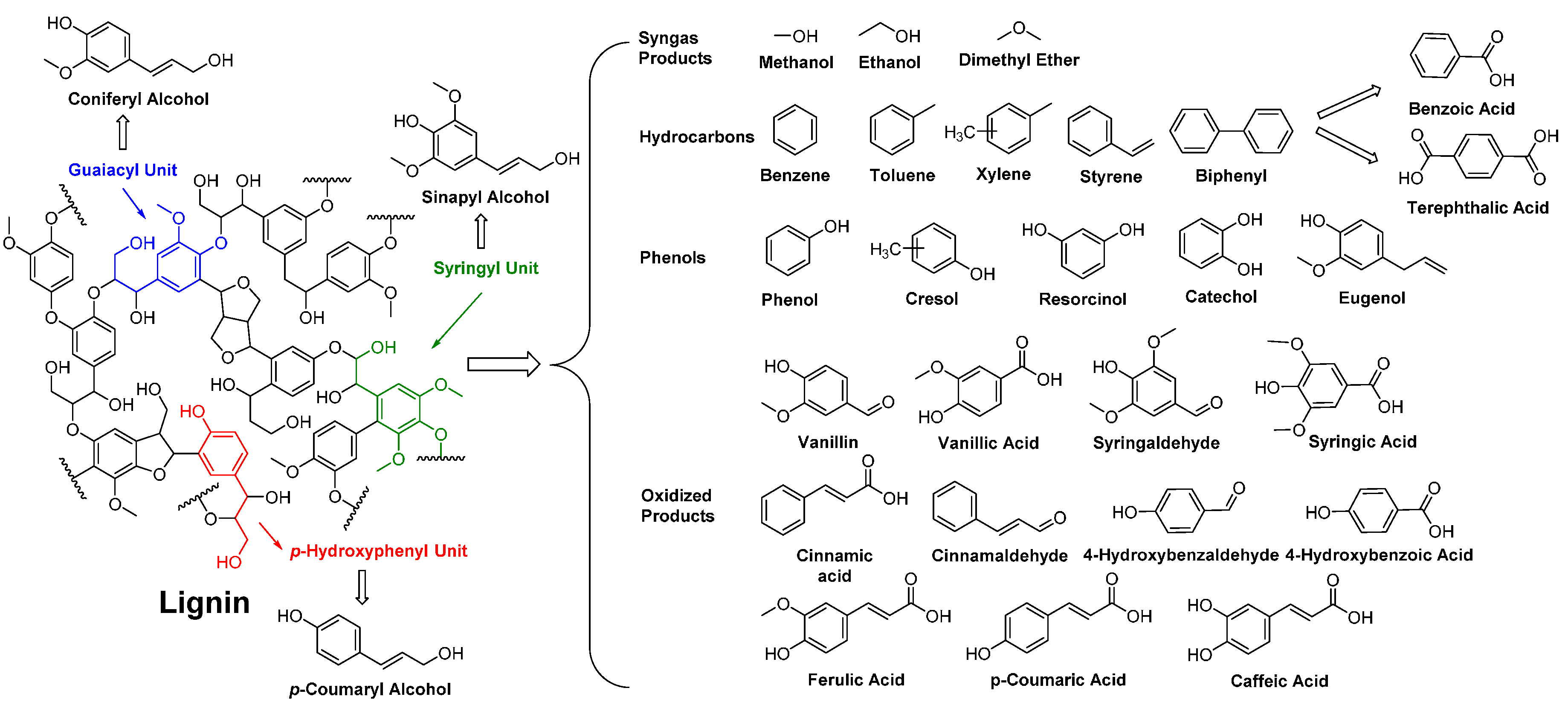
- Hatakeyama, H.; Hatakeyama, T. Lignin structure, properties, and applications. In Biopolymers; Abe, A., Dusek, K., Kobayashi, S., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2010; Volume 232, pp. 1–63. [Google Scholar]
- Pandey, M.P.; Kim, C.S. Lignin depolymerization and conversion: A review of thermochemical methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Pearl, I.A. Vanillin from lignin materials. J. Am. Chem. Soc. 1942, 64, 1429–1431. [Google Scholar] [CrossRef]
- Tuck, C.O.; Pérez, E.; Horváth, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Voitl, T.; von Rohr, P.R. Demonstration of a process for the conversion of kraft lignin into vanillin and methyl vanillate by acidic oxidation in aqueous methanol. Ind. Eng. Chem. Res. 2010, 49, 520–525. [Google Scholar] [CrossRef]
- Priefert, H.; Rabenhorst, J.; Steinbüchel, A. Biotechnological production of vanillin. Appl. Microbiol. Biotechnol. 2001, 56, 296–314. [Google Scholar] [CrossRef] [PubMed]
- Pinto, P.C.R.; Costa, C.E.; Rodrigues, A.E. Oxidation of lignin from eucalyptus globulus pulping liquors to produce syringaldehyde and vanillin. Ind. Eng. Chem. Res. 2013, 52, 4421–4428. [Google Scholar] [CrossRef]
- Havkin-Frenkel, D.; Belanger, F.C. Biotechnological production of vanillin. In Biotechnology in Flavor Production; Havkin-Frenkel, D., Belanger, A.C., Eds.; Blackwell Publishing Ltd.: Oxford, UK, 2009; pp. 83–103. [Google Scholar]
- Llevot, A.; Grau, E.; Carlotti, S.; Grelier, S.; Cramail, H. Renewable (semi)aromatic polyesters from symmetrical vanillin-based dimers. Polym. Chem. 2015, 6, 6058–6066. [Google Scholar] [CrossRef]
- Fache, M.; Darroman, E.; Besse, V.; Auvergne, R.; Caillol, S.; Boutevin, B. Vanillin, a promising biobased building-block for monomer synthesis. Green Chem. 2014, 16, 1987–1998. [Google Scholar] [CrossRef]
- Fache, M.; Boutevin, B.; Caillol, S. Vanillin, a key-intermediate of biobased polymers. Eur. Polym. J. 2015, 68, 488–502. [Google Scholar] [CrossRef]
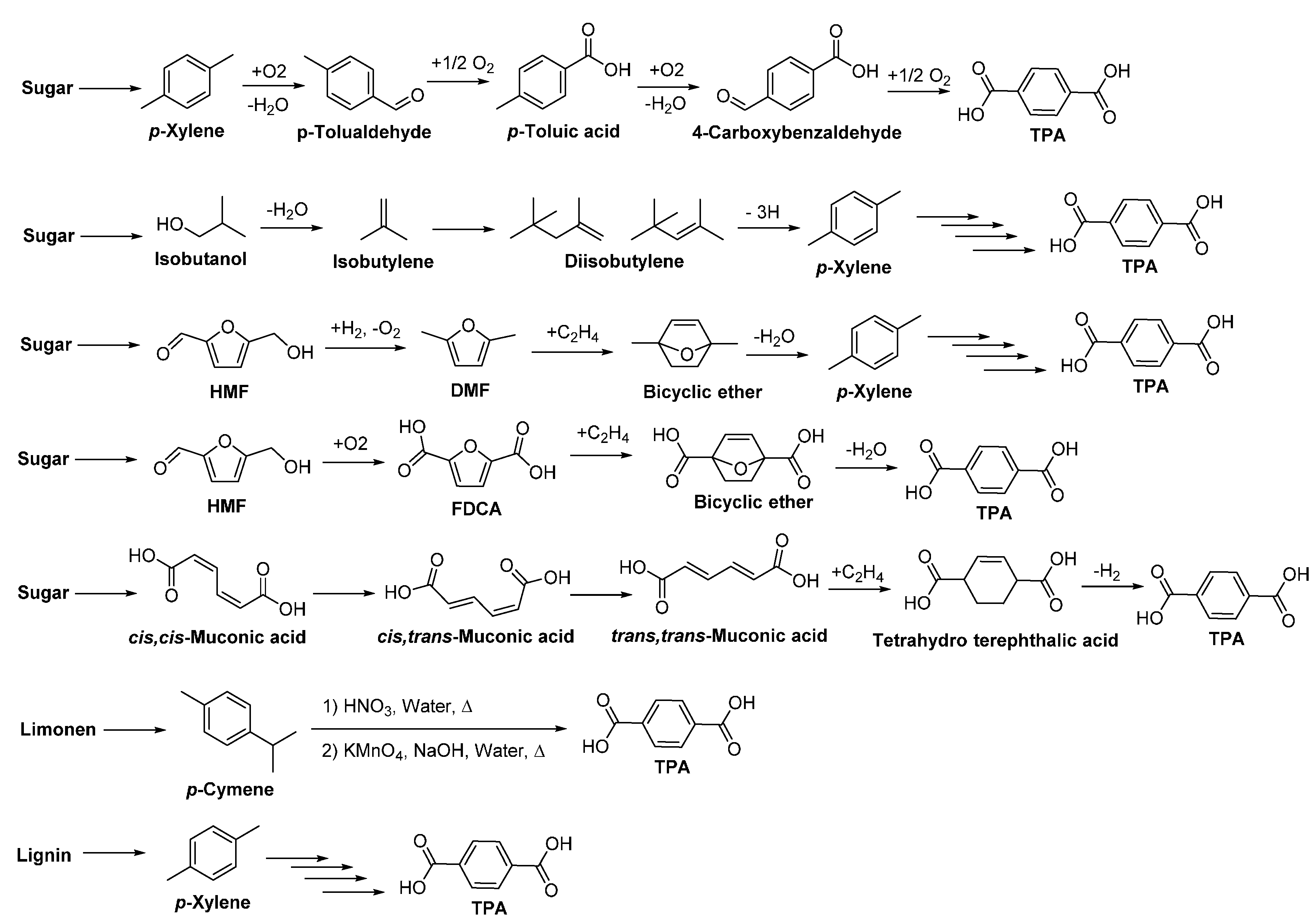
- Mialon, L.; Pemba, A.G.; Miller, S.A. Biorenewable polyethylene terephthalate mimics derived from lignin and acetic acid. Green Chem. 2010, 12, 1704–1706. [Google Scholar] [CrossRef]
- Mialon, L.; Vanderhenst, R.; Pemba, A.G.; Miller, S.A. Polyalkylenehydroxybenzoates (PAHBs): Biorenewable aromatic/aliphatic polyesters from lignin. Macromol. Rapid Commun. 2011, 32, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Berti, C.; Fiorini, M.; Binassi, E.; Mazzacurati, M.; Vannini, M.; Karanam, S. Synthesis and radiocarbon evidence of terephthalate polyesters completely prepared from renewable resources. Green Chem. 2011, 13, 2543–2548. [Google Scholar] [CrossRef]
- Shiramizu, M.; Toste, F.D. On the diels-alder approach to solely biomass-derived polyethylene terephthalate (pet): Conversion of 2,5-dimethylfuran and acrolein into p-xylene. Chem. Eur. J. 2011, 17, 12452–12457. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-T.; Wang, Z.; Gilbert, C.J.; Fan, W.; Huber, G.W. Production of p-xylene from biomass by catalytic fast pyrolysis using ZSM-5 catalysts with reduced pore openings. Angew. Chem. Int. Ed. 2012, 51, 11097–11100. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.W.; Guironnet, D.; Findlater, M.; Brookhart, M. Synthesis of p-xylene from ethylene. J. Am. Chem. Soc. 2012, 134, 15708–15711. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Chang, C.-C.; Do, P.; Nikbin, N.; Caratzoulas, S.; Vlachos, D.G.; Lobo, R.F.; Fan, W.; Dauenhauer, P.J. Cycloaddition of biomass-derived furans for catalytic production of renewable p-xylene. ACS Catal. 2012, 2, 935–939. [Google Scholar] [CrossRef]
- Agirrezabal-Telleria, I.; Gandarias, I.; Arias, P.L. Heterogeneous acid-catalysts for the production of furan-derived compounds (furfural and hydroxymethylfurfural) from renewable carbohydrates: A review. Catal. Today 2014, 234, 42–58. [Google Scholar] [CrossRef]
- Collias, D.I.; Harris, A.M.; Nagpal, V.; Cottrell, I.W.; Schultheis, M.W. Biobased terephthalic acid technologies: A literature review. Ind. Biotechnol. 2014, 10, 91–105. [Google Scholar] [CrossRef]
- Tachibana, Y.; Kimura, S.; Kasuya, K.-I. Synthesis and verification of biobased terephthalic acid from furfural. Sci. Rep. 2015, 5, 8249. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Lange, A.; Fabarius, J.; Wittmann, C. Top value platform chemicals: Bio-based production of organic acids. Curr. Opin. Biotechnol. 2015, 36, 168–175. [Google Scholar] [CrossRef] [PubMed]
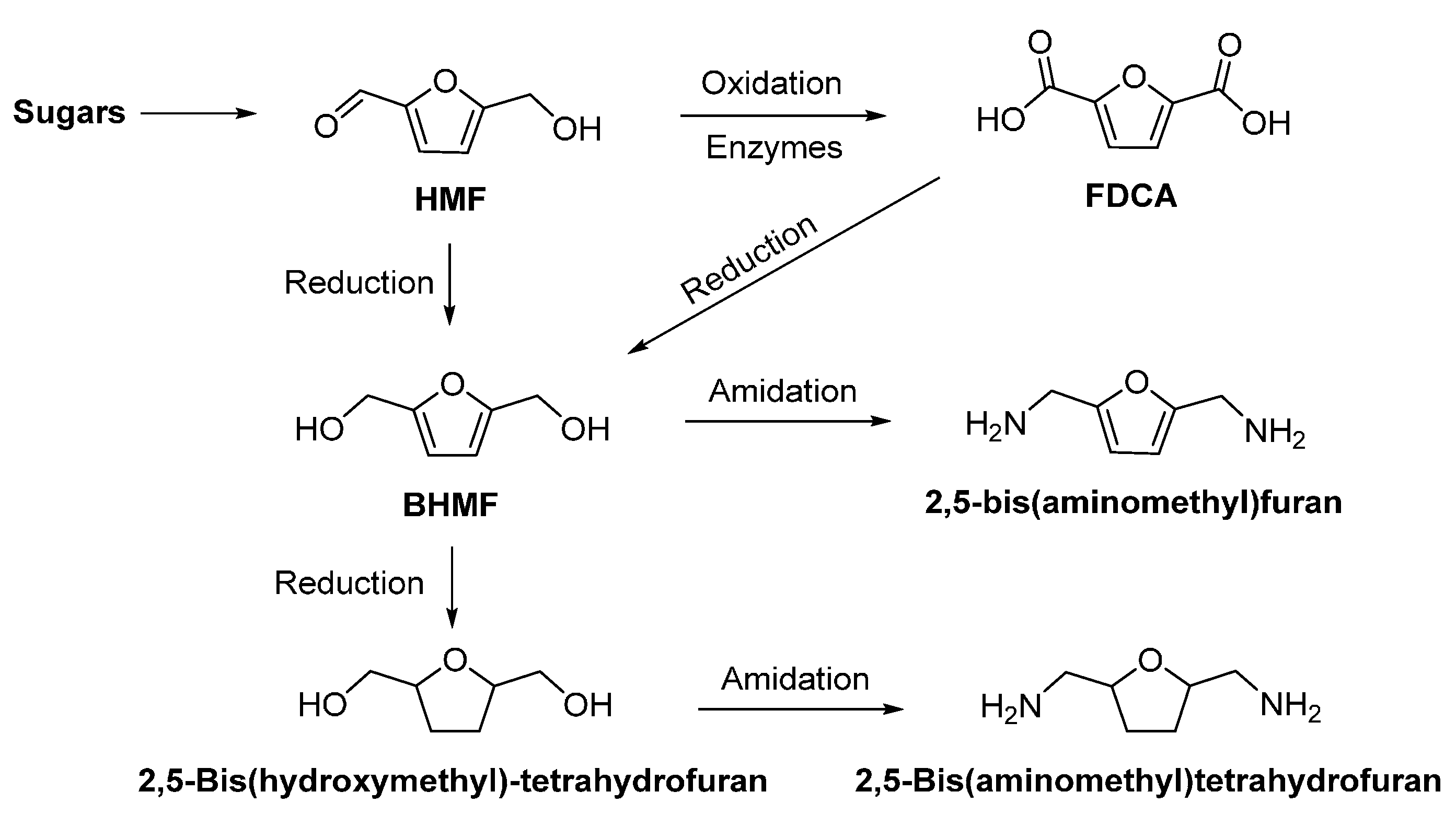
- Dijkman, W.P.; Groothuis, D.E.; Fraaije, M.W. Enzyme-catalyzed oxidation of 5-hydroxymethylfurfural to furan-2,5-dicarboxylic acid. Angew. Chem. Int. Ed. 2014, 53, 6515–6518. [Google Scholar] [CrossRef] [PubMed]
- Jong, E.d.; Dam, M.A.; Sipos, L.; Gruter, G.J.M. Furandicarboxylic acid (FDCA), a versatile building block for a very interesting class of polyesters. In Biobased Monomers, Polymers, and Materials; American Chemical Society: Washington, DC, USA, 2012; Volume 1105, pp. 1–13. [Google Scholar]
- Cherubini, F.; Strømman, A.H. Chemicals from lignocellulosic biomass: Opportunities, perspectives, and potential of biorefinery systems. Biofuels Bioprod. Biorefin. 2011, 5, 548–561. [Google Scholar] [CrossRef]
- Datta, R.; Tsai, S.-P.; Bonsignore, P.; Moon, S.-H.; Frank, J.R. Technological and economic potential of poly(lactic acid) and lactic acid derivatives. FEMS Microbiol. Rev. 1995, 16, 221–231. [Google Scholar] [CrossRef]
- Onda, A. Production of lactic acid from sugars by homogeneous and heterogeneous catalysts. In Application of Hydrothermal Reactions to Biomass Conversion; Jin, F., Ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2014; pp. 83–107. [Google Scholar]
- John, R.; Nampoothiri, K.M.; Pandey, A. Fermentative production of lactic acid from biomass: An overview on process developments and future perspectives. Appl. Microbiol. Biotechnol. 2007, 74, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Tanaka, T.; Ogino, C.; Fukuda, H.; Kondo, A. Biotechnological production of enantiomeric pure lactic acid from renewable resources: Recent achievements, perspectives, and limits. Appl. Microbiol. Biotechnol. 2010, 85, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Della Pina, C.; Falletta, E.; Rossi, M. A green approach to chemical building blocks. The case of 3-hydroxypropanoic acid. Green Chem. 2011, 13, 1624–1632. [Google Scholar]
- Kumar, V.; Ashok, S.; Park, S. Recent advances in biological production of 3-hydroxypropionic acid. Biotechnol. Adv. 2013, 31, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Valdehuesa, K.N.G.; Liu, H.W.; Nisola, G.M.; Chung, W.J.; Lee, S.H.; Park, S.J. Recent advances in the metabolic engineering of microorganisms for the production of 3-hydroxypropionic acid as C3 platform chemical. Appl. Microbiol. Biotechnol. 2013, 97, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.F.; Gandini, A.; Silvestre, A.J.D.; Neto, C.P.; Pinto, J.; Eckerman, C.; Holmbom, B. Novel suberin-based biopolyesters: From synthesis to properties. J. Polym. Sci. Part A Polym. Chem. 2011, 49, 2281–2291. [Google Scholar] [CrossRef]
- Graça, J. Suberin: The biopolyester at the frontier of plants. Front. Chem. 2015, 3, 62. [Google Scholar] [CrossRef] [PubMed]
- Biermann, U.; Bornscheuer, U.; Meier, M.A.; Metzger, J.O.; Schafer, H.J. Oils and fats as renewable raw materials in chemistry. Angew. Chem. Int. Ed. 2011, 50, 3854–3871. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Larock, R.C. Novel polymeric materials from vegetable oils and vinyl monomers: Preparation, properties, and applications. ChemSusChem 2009, 2, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, M.; Mozhaev, V.; Rich, J.; Khmelnitsky, Y. Lipase-catalyzed transamidation of non-activated amides in organic solvent. Biotechnol. Lett. 2000, 22, 1419–1422. [Google Scholar] [CrossRef]
- Gotor, V. Non-conventional hydrolase chemistry: Amide and carbamate bond formation catalyzed by lipases. Biorg. Med. Chem. 1999, 7, 2189–2197. [Google Scholar] [CrossRef]
- Hari Krishna, S.; Karanth, N.G. Lipases and lipase-catalyzed esterification reactions in nonaqueous media. Catal. Rev. 2002, 44, 499–591. [Google Scholar] [CrossRef]
- Lundberg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Catalytic amide formation from non-activated carboxylic acids and amines. Chem. Soc. Rev. 2014, 43, 2714–2742. [Google Scholar] [CrossRef] [PubMed]
- Gotor-Fernández, V.; Vicente, G. Use of lipases in organic synthesis. In Industrial Enzymes: Structure, Function and Applications; Polaina, J., MacCabe, A., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2007; pp. 301–315. [Google Scholar]
- Laane, C.; Boeren, S.; Vos, K.; Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 1987, 30, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gross, R.A. Candida antartica lipase B catalyzed polycaprolactone synthesis: Effects of organic media and temperature. Biomacromolecules 2000, 1, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Mahapatro, A.; Kalra, B.; Kumar, A.; Gross, R.A. Lipase-catalyzed polycondensations: Effect of substrates and solvent on chain formation, dispersity, and end-group structure. Biomacromolecules 2003, 4, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.; Liu, T.; Yan, J.; Kobayashi, S. Enzymatic polymerizations. In Encyclopedia of Polymeric Nanomaterials; Kobayashi, S., Müllen, K., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2015; pp. 1–10. [Google Scholar]
- Mallakpour, S.; Rafiee, Z. Ionic liquids as environmentally friendly solvents in macromolecules chemistry and technology, part I. J. Polym. Environ. 2011, 19, 447–484. [Google Scholar] [CrossRef]
- Lozano, P. Enzymes in neoteric solvents: From one-phase to multiphase systems. Green Chem. 2010, 12, 555–569. [Google Scholar] [CrossRef]
- Lozano, P.; Garcia-Verdugo, E.; V. Luis, S.; Pucheault, M.; Vaultier, M. (Bio)catalytic continuous flow processes in scco2 and/or ils: Towards sustainable (bio)catalytic synthetic platforms. Curr. Org. Synth. 2011, 8, 810–823. [Google Scholar] [CrossRef]
- Fan, Y.X.; Qian, J.Q. Lipase catalysis in ionic liquids/supercritical carbon dioxide and its applications. J. Mol. Catal. B-Enzym. 2010, 66, 1–7. [Google Scholar] [CrossRef]
- Matsuda, T. Recent progress in biocatalysis using supercritical carbon dioxide. J. Biosci. Bioeng. 2013, 115, 233–241. [Google Scholar] [CrossRef] [PubMed]
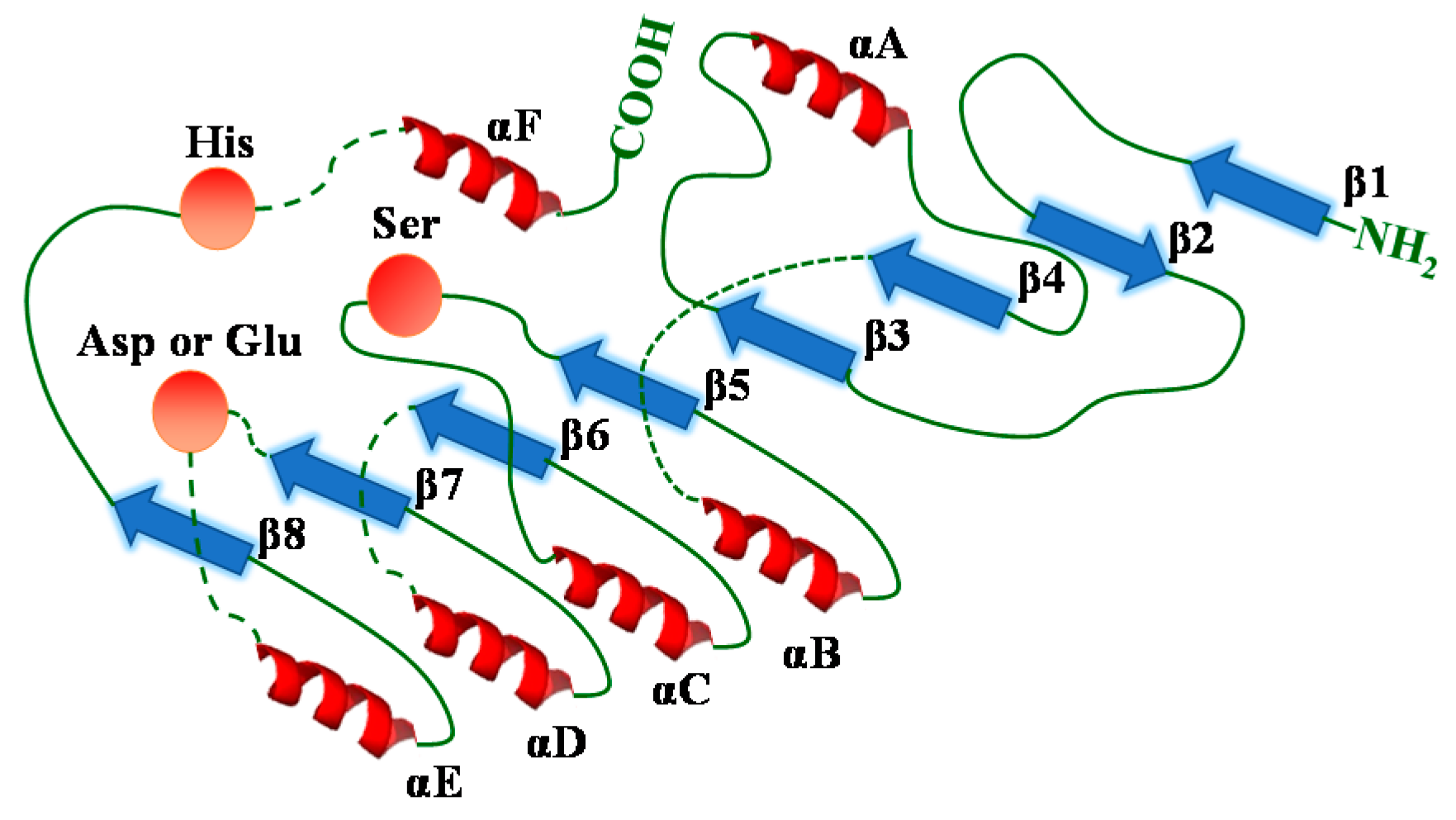
- Nardini, M.; Dijkstra, B.W. A/β hydrolase fold enzymes: The family keeps growing. Curr. Opin. Struct. Biol. 1999, 9, 732–737. [Google Scholar] [CrossRef]
- Hotelier, T.; Renault, L.; Cousin, X.; Negre, V.; Marchot, P.; Chatonnet, A. Esther, the database of the α/β-hydrolase fold superfamily of proteins. Nucleic Acids Res. 2004, 32, D145–D147. [Google Scholar] [CrossRef] [PubMed]
- Bezborodov, A.M.; Zagustina, N.A. Lipases in catalytic reactions of organic chemistry. Appl. Biochem. Microbiol. 2014, 50, 313–337. [Google Scholar] [CrossRef]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J.; et al. The α/β hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Anobom, C.D.; Pinheiro, A.S.; De-Andrade, R.A.; Aguieiras, E.C.G.; Andrade, G.C.; Moura, M.V.; Almeida, R.V.; Freire, D.M. From structure to catalysis: Recent developments in the biotechnological applications of lipases. BioMed Res. Int. 2014, 2014, 684506–684506. [Google Scholar] [CrossRef] [PubMed]
- Casas-Godoy, L.; Duquesne, S.; Bordes, F.; Sandoval, G.; Marty, A. Lipases: An overview. In Lipases and Phospholipases; Sandoval, G., Ed.; Humana Press: New York, NY, USA, 2012; Volume 861, pp. 3–30. [Google Scholar]
- Pleiss, J.; Fischer, M.; Schmid, R.D. Anatomy of lipase binding sites: The scissile fatty acid binding site. Chem. Phys. Lipids 1998, 93, 67–80. [Google Scholar] [CrossRef]
- Stergiou, P.Y.; Foukis, A.; Filippou, M.; Koukouritaki, M.; Parapouli, M.; Theodorou, L.G.; Hatziloukas, E.; Afendra, A.; Pandey, A.; Papamichael, E.M. Advances in lipase-catalyzed esterification reactions. Biotechnol. Adv. 2013, 31, 1846–1859. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, K.E.; Dijkstra, B.W.; Reetz, M.T. Bacterial biocatalysts: Molecular biology, three-dimensional structures, and biotechnological applications of lipases. Annu. Rev. Microbiol. 1999, 53, 315–351. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, V.; Bidmanova, S.; Koudelakova, T.; Prokop, Z.; Chaloupkova, R.; Damborsky, J. Strategies for stabilization of enzymes in organic solvents. ACS Catal. 2013, 3, 2823–2836. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Fernandez-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Adlercreutz, P. Immobilisation and application of lipases in organic media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef] [PubMed]
- Miletic, N.; Rohandi, R.; Vukovic, Z.; Nastasovic, A.; Loos, K. Surface modification of macroporous poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) resins for improved candida antarctica lipase b immobilization. React. Funct. Polym. 2009, 69, 68–75. [Google Scholar] [CrossRef]
- Miletić, N.; Vuković, Z.; Nastasović, A.; Loos, K. Macroporous poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) resins—Versatile immobilization supports for biocatalysts. J. Mol. Catal. B-Enzym. 2009, 56, 196–201. [Google Scholar] [CrossRef]
- Miletic, N.; Abetz, V.; Ebert, K.; Loos, K. Immobilization of candida antarctica lipase B on polystyrene nanoparticles. Macromol. Rapid Commun. 2010, 31, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Miletic, N.; Fahriansyah; Nguyen, L.T.T.; Loos, K. Formation, topography and reactivity of candida antarctica lipase B immobilized on silicon surface. Biocatal. Biotransform. 2010, 28, 357–369. [Google Scholar] [CrossRef]
- Miletic, N.; Nastasovic, A.; Loos, K. Immobilization of biocatalysts for enzymatic polymerizations: Possibilities, advantages, applications. Bioresour. Technol. 2012, 115, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Koike, H.; Koyama, Y.; Hagiwara, H.; Ito, E.; Fukuoka, T.; Imura, T.; Machida, M.; Kitamoto, D. Genome sequence of the basidiomycetous yeast pseudozyma antarctica T-34, a producer of the glycolipid biosurfactants mannosylerythritol lipids. Genome Announc. 2013, 1, e0006413. [Google Scholar] [CrossRef] [PubMed]
- Uppenberg, J.; Hansen, M.T.; Patkar, S.; Jones, T.A. The sequence, crystal structure determination and refinement of two crystal forms of lipase b from candida antarctica. Structure 1994, 2, 293–308. [Google Scholar] [CrossRef]
- Cygler, M.; Schrag, J.D. Structure as basis for understanding interfacial properties of lipases. In Lipases, Part A: Biotechnology; Rubin, B., Dennis, E.A., Eds.; Elsevier Academic Press Inc.: San Diego, CA, USA, 1997; Volume 284, pp. 3–27. [Google Scholar]
- Stauch, B.; Fisher, S.J.; Cianci, M. Open and closed states of candida antarctica lipase b: Protonation and the mechanism of interfacial activation. J. Lipid Res. 2015, 56, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Zisis, T.; Freddolino, P.L.; Turunen, P.; van Teeseling, M.C.F.; Rowan, A.E.; Blank, K.G. Interfacial activation of candida antarctica lipase b: Combined evidence from experiment and simulation. Biochemistry 2015, 54, 5969–5979. [Google Scholar] [CrossRef] [PubMed]
- Skjøt, M.; De Maria, L.; Chatterjee, R.; Svendsen, A.; Patkar, S.A.; Østergaard, P.R.; Brask, J. Understanding the plasticity of the α/β hydrolase fold: Lid swapping on the candida antarctica lipase b results in chimeras with interesting biocatalytic properties. ChemBioChem 2009, 10, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Martinelle, M.; Holmquist, M.; Hult, K. On the interfacial activation of Candida antarctica lipase a and b as compared with Humicola lanuginosa lipase. Biochim. Biophys. Acta 1995, 1258, 272–276. [Google Scholar] [CrossRef]
- Takwa, M. Lipase Specificity and Selectivity: Engineering, Kinetics and Applied Catalysis. Ph.D. Thesis, KTH, Royal Institute of Technology, Stockholm, Sweden, 2010. [Google Scholar]
- Tufvesson, P.; Törnvall, U.; Carvalho, J.; Karlsson, A.J.; Hatti-Kaul, R. Towards a cost-effective immobilized lipase for the synthesis of specialty chemicals. J. Mol. Catal. B-Enzym. 2011, 68, 200–205. [Google Scholar] [CrossRef]
- Mei, Y.; Miller, L.; Gao, W.; Gross, R.A. Imaging the distribution and secondary structure of immobilized enzymes using infrared microspectroscopy. Biomacromolecules 2003, 4, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Lozano, P.; De Diego, T.; Carrie, D.; Vaultier, M.; Iborra, J.L. Lipase catalysis in ionic liquids and supercritical carbon dioxide at 150 °C. Biotechnol. Prog. 2003, 19, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Ragupathy, L.; Ziener, U.; Dyllick-Brenzinger, R.; von Vacano, B.; Landfester, K. Enzyme-catalyzed polymerizations at higher temperatures: Synthetic methods to produce polyamides and new poly(amide-co-ester)s. J. Mol. Catal. B-Enzym. 2012, 76, 94–105. [Google Scholar] [CrossRef]
- Frampton, M.B.; Zelisko, P.M. Synthesis of lipase-catalysed silicone-polyesters and silicone-polyamides at elevated temperatures. Chem. Commun. 2013, 49, 9269–9271. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Shoda, S.; Uyama, H. Enzymatic polymerization and oligomerization. In Polymer Synthesis/Polymer Engineering; Springer-Verlag: Berlin/Heidelberg, Germany, 1995; Volume 121, pp. 1–30. [Google Scholar]
- Loos, K. Preface. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. i–xxix. [Google Scholar]
- Hollmann, F. Enzymatic polymerization of vinyl polymers. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 143–163. [Google Scholar]
- van der Vlist, J.; Loos, K. Enzymatic polymerizations of polysaccharides. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 211–246. [Google Scholar]
- van der Vlist, J.; Palomo Reixach, M.; van der Maarel, M.; Dijkhuizen, L.; Schouten, A.J.; Loos, K. Synthesis of branched polyglucans by the tandem action of potato phosphorylase and deinococcus geothermalis glycogen branching enzyme. Macromol. Rapid Commun. 2008, 29, 1293–1297. [Google Scholar] [CrossRef]
- Ciric, J.; Loos, K. Synthesis of branched polysaccharides with tunable degree of branching. Carbohydr. Polym. 2013, 93, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Ciric, J.; Petrovic, D.M.; Loos, K. Polysaccharide biocatalysis: From synthesizing carbohydrate standards to establishing characterization methods. Macromol. Chem. Phys. 2014, 215, 931–944. [Google Scholar] [CrossRef]
- Cheng, H.N. Enzyme-catalyzed synthesis of polyamides and polypeptides. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 131–141. [Google Scholar]
- Stavila, E.; Loos, K. Synthesis of polyamides and their copolymers via enzymatic polymerization. J. Renew. Mater. 2015, 3, 268–280. [Google Scholar] [CrossRef]
- Linares, G.G.; Baldessari, A. Lipases as efficient catalysts in the synthesis of monomers and polymers with biomedical applications. Curr. Org. Chem. 2013, 17, 719–743. [Google Scholar] [CrossRef]
- Okumura, S.; Iwai, M.; Tominaga, Y. Synthesis of ester oligomer by aspergillus-niger lipase. Agric. Biol. Chem. 1984, 48, 2805–2808. [Google Scholar] [CrossRef]
- Knani, D.; Gutman, A.L.; Kohn, D.H. Enzymatic polyesterification in organic media. Enzyme-catalyzed synthesis of linear polyesters. I. Condensation polymerization of linear hydroxyesters. II. Ring-opening polymerization of ϵ-caprolactone. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 1221–1232. [Google Scholar]
- Uyama, H.; Kobayashi, S. Enzymatic ring-opening polymerization of lactones catalyzed by lipase. Chem. Lett. 1993, 22, 1149–1150. [Google Scholar] [CrossRef]
- Bisht, K.S.; Henderson, L.A.; Gross, R.A.; Kaplan, D.L.; Swift, G. Enzyme-catalyzed ring-opening polymerization of ω-pentadecalactone. Macromolecules 1997, 30, 2705–2711. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, D.; Liu, C.B.; Zhao, Z.H.; Yang, Y.; Li, Q.S. Lipase/esterase-catalyzed synthesis of aliphatic polyesters via polycondensation: A review. Process Biochem. 2012, 47, 1027–1036. [Google Scholar] [CrossRef]
- Shoda, S.; Uyama, H.; Kadokawa, J.; Kimura, S.; Kobayashi, S. Enzymes as green catalysts for precision macromolecular synthesis. Chem. Rev. 2016, 116, 2307–2413. [Google Scholar] [CrossRef] [PubMed]
- Binns, F.; Harffey, P.; Roberts, S.M.; Taylor, A. Studies leading to the large scale synthesis of polyesters using enzymes. J. Chem. Soc. Perkin Trans. 1999, 1, 2671–2676. [Google Scholar] [CrossRef]
- Duda, A.; Kowalski, A.; Penczek, S.; Uyama, H.; Kobayashi, S. Kinetics of the ring-opening polymerization of 6-, 7-, 9-, 12-, 13-, 16-, and 17-membered lactones. Comparison of chemical and enzymatic polymerizations. Macromolecules 2002, 35, 4266–4270. [Google Scholar]
- Park, H.G.; Chang, H.N.; Dordick, J.S. Enzymatic synthesis of various aromatic polyesters in anhydrous organic solvents. Biocatalysis 1994, 11, 263–271. [Google Scholar] [CrossRef]
- Mezoul, G.; Lalot, T.; Brigodiot, M.; Maréchal, E. Enzyme-catalyzed syntheses of poly(1,6-hexanediyl isophthalate) and poly(1,6-hexanediyl terephthalate) in organic medium. Polym. Bull. 1996, 36, 541–548. [Google Scholar] [CrossRef]
- Linko, Y.Y.; Lamsa, M.; Wu, X.Y.; Uosukainen, E.; Seppala, J.; Linko, P. Biodegradable products by lipase biocatalysis. J. Biotechnol. 1998, 66, 41–50. [Google Scholar] [CrossRef]
- Wu, X.Y.; Linko, Y.Y.; Seppälä, J.; Leisola, M.; Linko, P. Lipase-catalyzed synthesis of aromatic polyesters. J. Ind. Microbiol. Biotechnol. 1998, 20, 328–332. [Google Scholar] [CrossRef]
- Rodney, R.L.; Allinson, B.T.; Beckman, E.J.; Russell, A.J. Enzyme-catalyzed polycondensation reactions for the synthesis of aromatic polycarbonates and polyesters. Biotechnol. Bioeng. 1999, 65, 485–489. [Google Scholar] [CrossRef]
- Uyama, H.; Yaguchi, S.; Kobayashi, S. Enzymatic synthesis of aromatic polyesters by lipase-catalyzed polymerization of dicarboxylic acid divinyl esters and glycols. Polym. J. 1999, 31, 380–383. [Google Scholar] [CrossRef]
- Park, H.G.; Chang, H.N.; Dordick, J.S. Chemoenzymatic synthesis of sucrose-containing aromatic polymers. Biotechnol. Bioeng. 2001, 72, 541–547. [Google Scholar] [CrossRef]
- Lavalette, A.; Lalot, T.; Brigodiot, M.; Maréchal, E. Lipase-catalyzed synthesis of a pure macrocyclic polyester from dimethyl terephthalate and diethylene glycol. Biomacromolecules 2002, 3, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Tyagi, R.; Parmar, V.S.; Samuelson, L.A.; Kumar, J.; Watterson, A.C. Biocatalytic “Green” Synthesis of peg-based aromatic polyesters: Optimization of the substrate and reaction conditions. Green Chem. 2004, 6, 516–520. [Google Scholar] [CrossRef]
- Poojari, Y.; Clarson, S.J. Lipase-catalyzed synthesis and properties of silicone aromatic polyesters and silicone aromatic polyamides. Macromolecules 2010, 43, 4616–4622. [Google Scholar] [CrossRef]
- Qian, X.Q.; Wu, Q.; Xu, F.L.; Lin, X.F. Lipase-catalyzed synthesis of polymeric prodrugs of nonsteroidal anti-inflammatory drugs. J. Appl. Polym. Sci. 2013, 128, 3271–3279. [Google Scholar] [CrossRef]
- Wallace, J.S.; Morrow, C.J. Biocatalytic synthesis of polymers. II. Preparation of [aa–bb]x polyesters by porcine pancreatic lipase catalyzed transesterification in anhydrous, low polarity organic solvents. J. Polym. Sci. Part A Polym. Chem. 1989, 27, 3271–3284. [Google Scholar]
- Fukuda, S.; Matsumura, S. Enzymatic synthesis and chemical recycling of aromatic polyesters via cyclic oligomers. Kobunshi Ronbunshu 2011, 68, 332–340. [Google Scholar] [CrossRef]
- Cheng, H.N.; Gu, Q.M.; Maslanka, W.W. Enzyme-Catalyzed Polyamides and Compositions and Processes of Preparing and Using the Same. U.S. Patent US6677427 B1, 13 January 2004. [Google Scholar]
- Qu-Ming, G.; Maslanka, W.W.; Cheng, H.N. Enzyme-catalyzed polyamides and their derivatives. In Polymer Biocatalysis and Biomaterials II; American Chemical Society: Washington, DC, USA, 2008; Volume 999, pp. 309–319. [Google Scholar]
- Kong, X.M.; Yamamoto, M.; Haring, D. Method for Producing an Aqueous Polyamide Dispersion. U.S. Patent US20080167418 A1, 10 June 2008. [Google Scholar]
- Schwab, L.W.; Kroon, R.; Schouten, A.J.; Loos, K. Enzyme-catalyzed ring-opening polymerization of unsubstituted β-lactam. Macromol. Rapid Commun. 2008, 29, 794–797. [Google Scholar] [CrossRef]
- Baum, I.; Elsässer, B.; Schwab, L.W.; Loos, K.; Fels, G. Atomistic model for the polyamide formation from β-lactam catalyzed by candida antarctica lipase b. ACS Catal. 2011, 1, 323–336. [Google Scholar] [CrossRef]
- Stavila, E.; Arsyi, R.Z.; Petrovic, D.M.; Loos, K. Fusarium solani pisi cutinase-catalyzed synthesis of polyamides. Eur. Polym. J. 2013, 49, 834–842. [Google Scholar] [CrossRef]
- Stavila, E.; Loos, K. Synthesis of lactams using enzyme-catalyzed aminolysis. Tetrahedron Lett. 2013, 54, 370–372. [Google Scholar] [CrossRef]
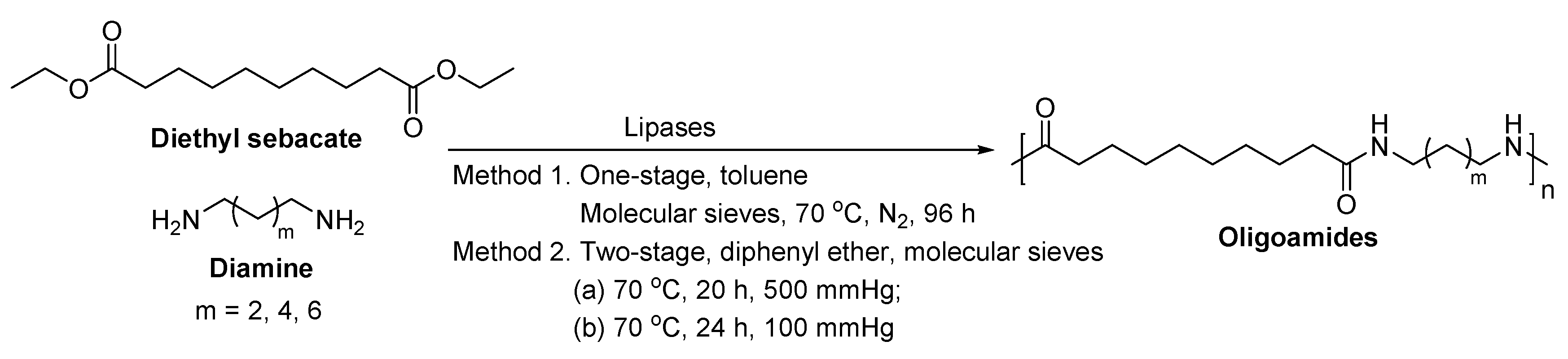
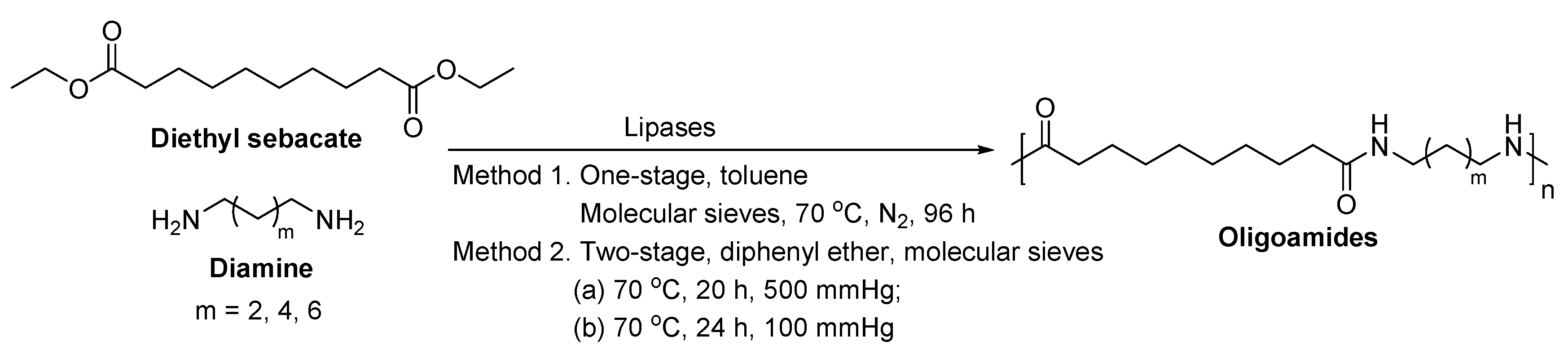
- Stavila, E.; Alberda van Ekenstein, G.O.R.; Loos, K. Enzyme-catalyzed synthesis of aliphatic-aromatic oligoamides. Biomacromolecules 2013, 14, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Stavila, E.; Alberda van Ekenstein, G.O.R.; Woortman, A.J.J.; Loos, K. Lipase-catalyzed ring-opening copolymerization of epsilon-caprolactone and beta-lactam. Biomacromolecules 2014, 15, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Mabuchi, K.; Toshima, K. Lipase-catalyzed ring-opening polymerization of lactide. Macromol. Rapid Commun. 1997, 18, 477–482. [Google Scholar] [CrossRef]
- Matsumura, S.; Mabuchi, K.; Toshima, K. Novel ring-opening polymerization of lactide by lipase. Macromol. Symp. 1998, 130, 285–304. [Google Scholar] [CrossRef]
- Fujioka, M.; Hosoda, N.; Nishiyama, S.; Noguchi, H.; Shoji, A.; Kumar, D.S.; Katsuraya, K.; Ishii, S.; Yoshida, Y. One-pot enzymatic synthesis of poly(l,l-lactide) by immobilized lipase catalyst. Sen’i Gakkaishi 2006, 62, 63–65. [Google Scholar] [CrossRef]
- Hans, M.; Keul, H.; Moeller, M. Ring-opening polymerization of dd-lactide catalyzed by novozyme 435. Macromol. Biosci. 2009, 9, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arrazola, R.; Lopez-Guerrero, D.A.; Gimeno, M.; Barzana, E. Lipase-catalyzed synthesis of poly-l-lactide using supercritical carbon dioxide. J. Supercrit. Fluids 2009, 51, 197–201. [Google Scholar] [CrossRef]
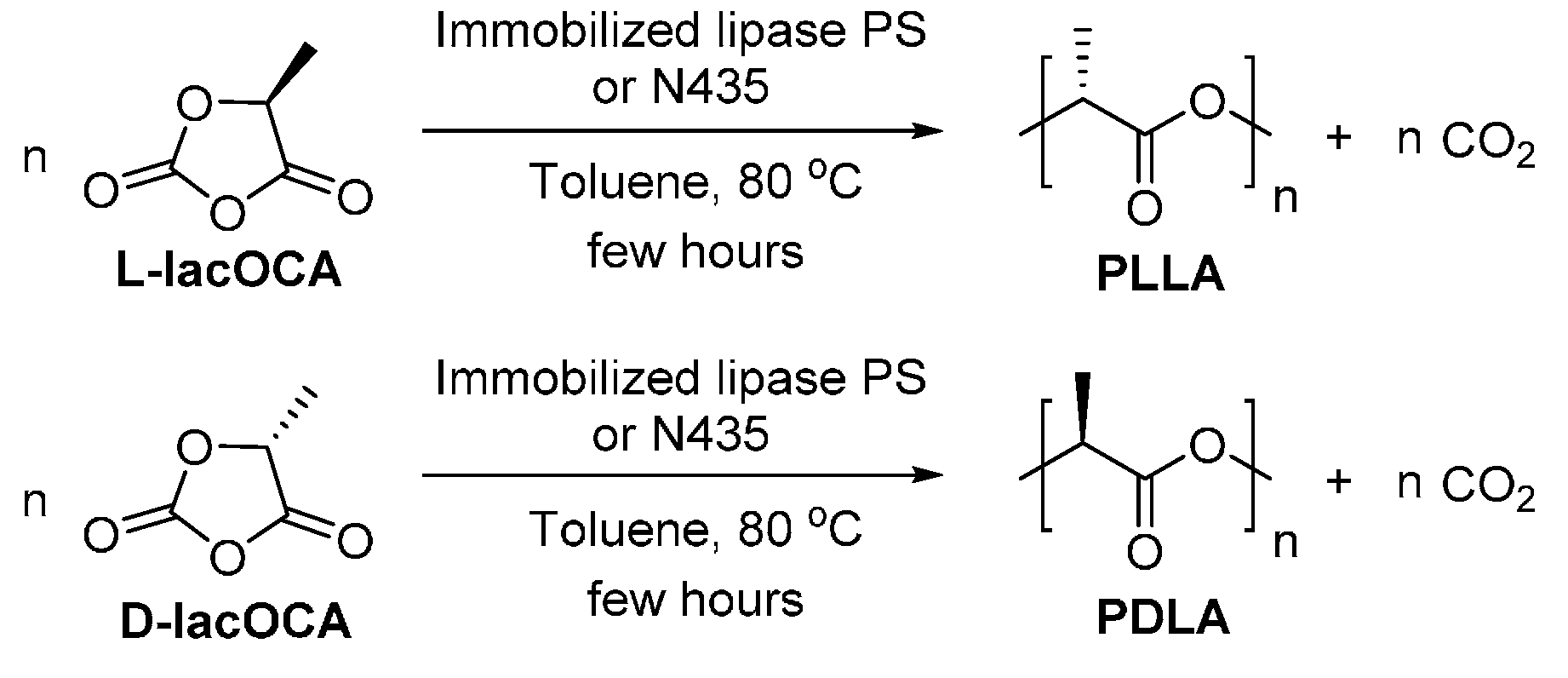
- Bonduelle, C.; Martin-Vaca, B.; Bourissou, D. Lipase-catalyzed ring-opening polymerization of the o-carboxylic anhydride derived from lactic acid. Biomacromolecules 2009, 10, 3069–3073. [Google Scholar] [CrossRef] [PubMed]
- Numata, K.; Srivastava, R.K.; Finne-Wistrand, A.; Albertsson, A.-C.; Doi, Y.; Abe, H. Branched poly(lactide) synthesized by enzymatic polymerization: Effects of molecular branches and stereochemistry on enzymatic degradation and alkaline hydrolysis. Biomacromolecules 2007, 8, 3115–3125. [Google Scholar] [CrossRef] [PubMed]
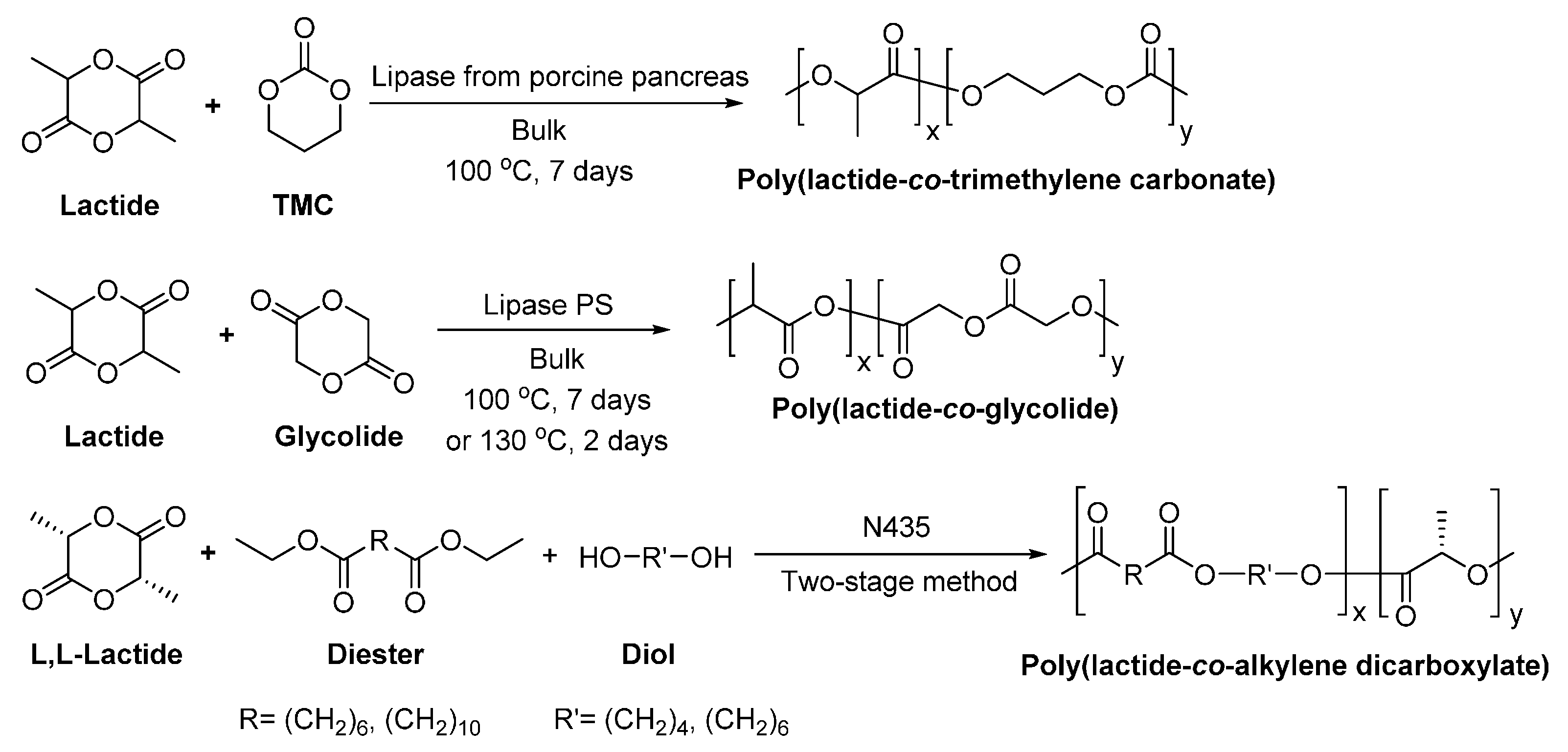
- Matsumura, S.; Tsukada, K.; Toshima, K. Novel lipase-catalyzed ring-opening copolymerization of lactide and trimethylene carbonate forming poly(ester carbonate)s. Int. J. Biol. Macromol. 1999, 25, 161–167. [Google Scholar] [CrossRef]
- Huijser, S.; Staal, B.B.P.; Huang, J.; Duchateau, R.; Koning, C.E. Topology characterization by MALDI-ToF-MS of enzymatically synthesized poly(lactide-co-glycolide). Biomacromolecules 2006, 7, 2465–2469. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Zhang, J. Lipase-catalyzed synthesis of aliphatic polyesters via copolymerization of lactide with diesters and diols. Polymer 2013, 54, 6105–6113. [Google Scholar] [CrossRef]
- Takiyama, E.; Fujimaki, T. synthesis. In Studies in Polymer Science; Yoshiharu, D., Kazuhiko, F., Eds.; Elsevier: Amsterdam, The Netherlands, 1994; Volume 12, pp. 150–174. [Google Scholar]
- Azim, H.; Dekhterman, A.; Jiang, Z.; Gross, R.A. Candida antarctica lipase B-catalyzed synthesis of poly(butylene succinate): Shorter chain building blocks also work. Biomacromolecules 2006, 7, 3093–3097. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, S.; Toshima, K.; Matsumura, S. New strategy for enzymatic synthesis of high-molecular-weight poly(butylene succinate) via cyclic oligomers. Macromol. Rapid Commun. 2006, 27, 203–207. [Google Scholar] [CrossRef]
- Ren, L.W.; Wang, Y.S.; Ge, J.; Lu, D.N.; Liu, Z. Enzymatic synthesis of high-molecular-weight poly(butylene succinate) and its copolymers. Macromol. Chem. Phys. 2015, 216, 636–640. [Google Scholar] [CrossRef]
- Uyama, H.; Inada, K.; Kobayashi, S. Lipase-catalyzed synthesis of aliphatic polyesters by polycondensation of dicarboxylic acids and glycols in solvent-free system. Polym. J. 2000, 32, 440–443. [Google Scholar] [CrossRef]
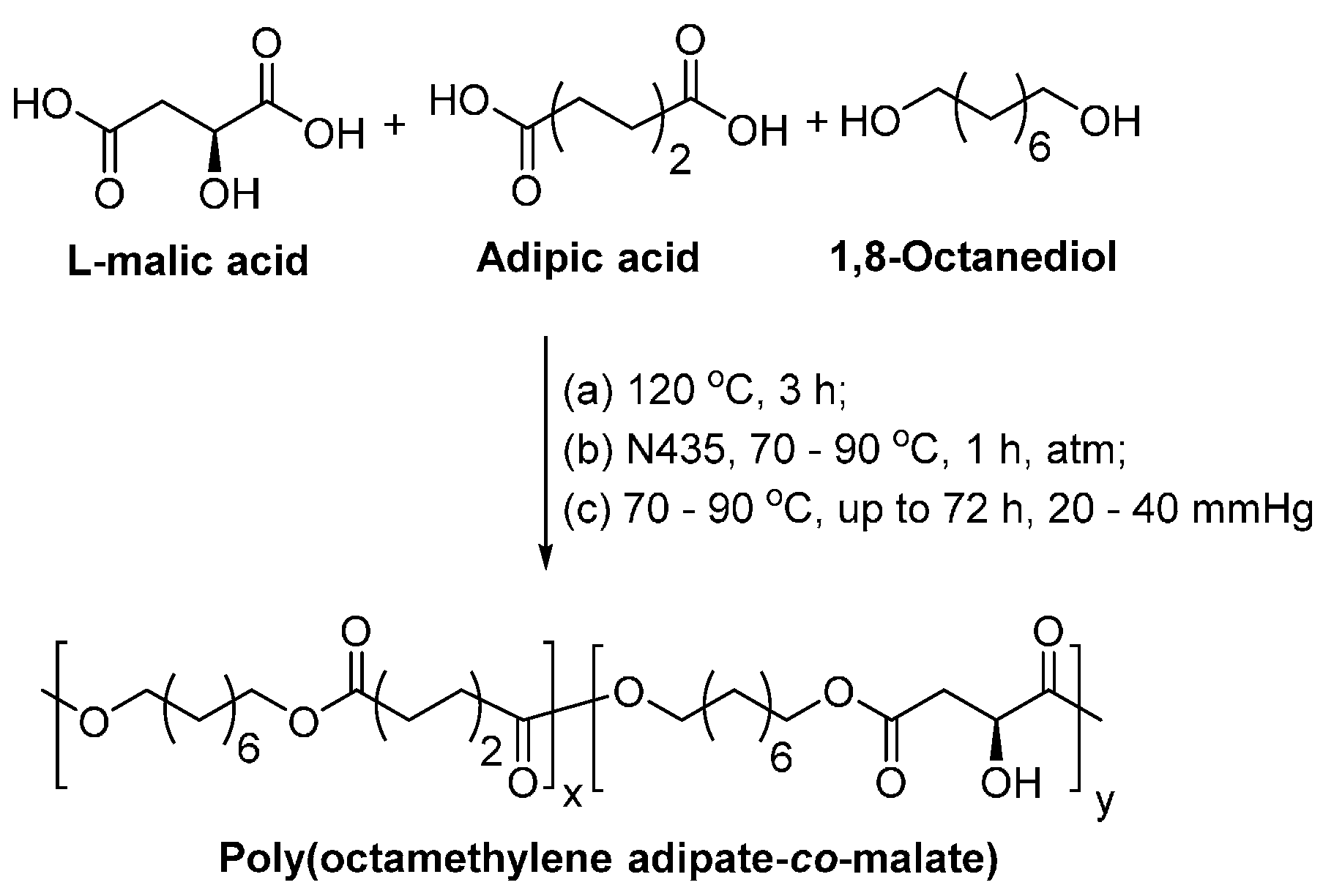
- Li, G.J.; Yao, D.H.; Zong, M.H. Lipase-catalyzed synthesis of biodegradable copolymer containing malic acid units in solvent-free system. Eur. Polym. J. 2008, 44, 1123–1129. [Google Scholar] [CrossRef]
- Liu, W.H.; Chen, B.Q.; Wang, F.; Tan, T.W.; Deng, L. Lipase-catalyzed synthesis of aliphatic polyesters and properties characterization. Process Biochem. 2011, 46, 1993–2000. [Google Scholar] [CrossRef]
- Liu, W.; Wang, F.; Tan, T.; Chen, B. Lipase-catalyzed synthesis and characterization of polymers by cyclodextrin as support architecture. Carbohydr. Polym. 2013, 92, 633–640. [Google Scholar] [CrossRef] [PubMed]
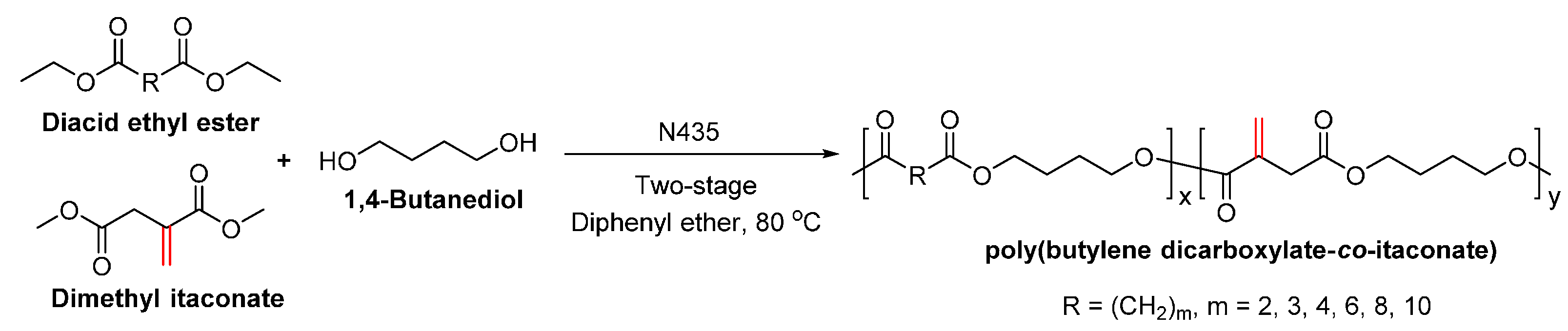
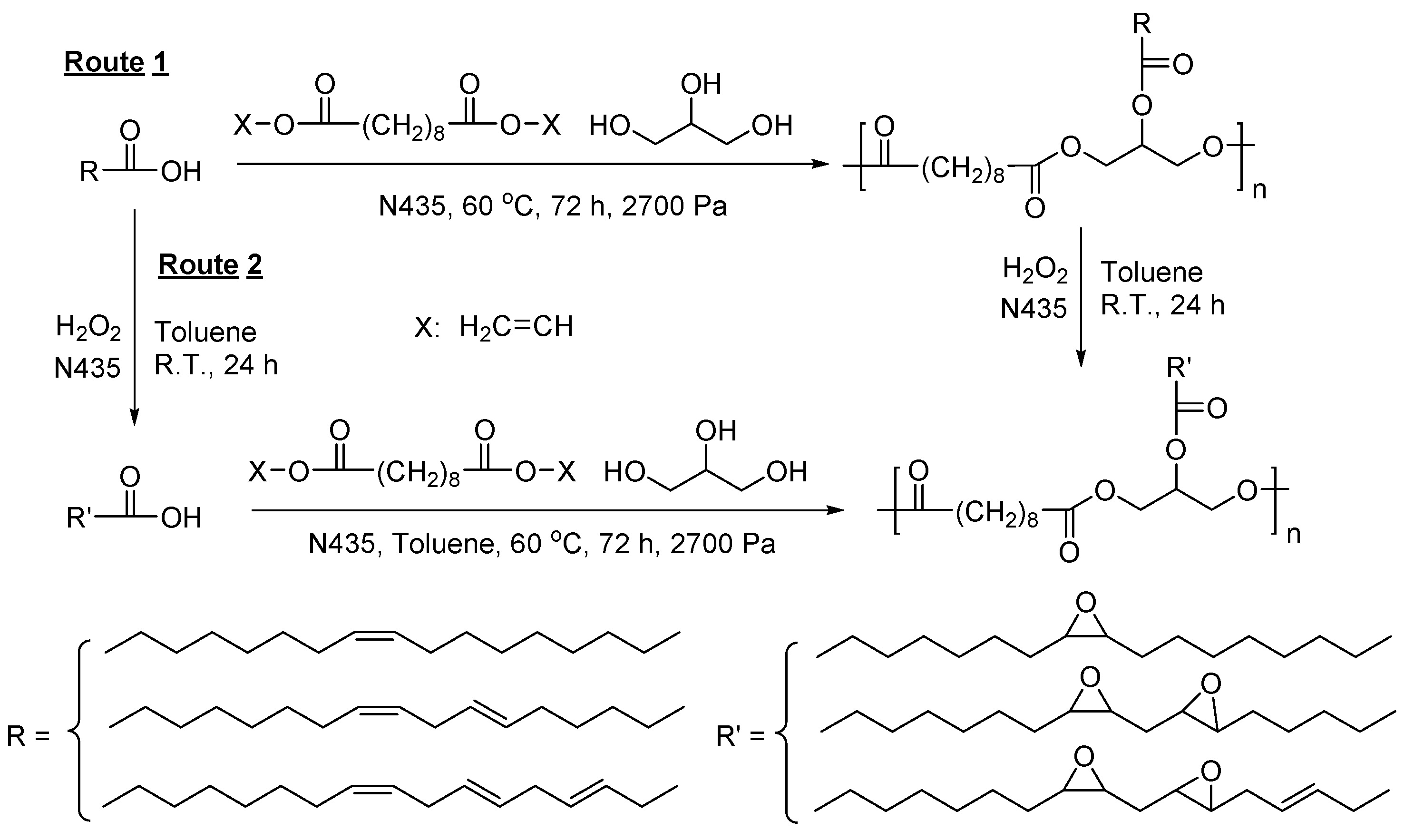
- Jiang, Y.; Woortman, A.J.J.; Alberda van Ekenstein, G.O.R.; Loos, K. Environmentally benign synthesis of saturated and unsaturated aliphatic polyesters via enzymatic polymerization of biobased monomers derived from renewable resources. Polym. Chem. 2015, 6, 5451–5463. [Google Scholar] [CrossRef]
- Curia, S.; Barclay, A.F.; Torron, S.; Johansson, M.; Howdle, S.M. Green process for green materials: Viable low-temperature lipase-catalysed synthesis of renewable telechelics in supercritical CO2. Philos. Trans. R. Soc. A 2015, 373. [Google Scholar] [CrossRef] [PubMed]
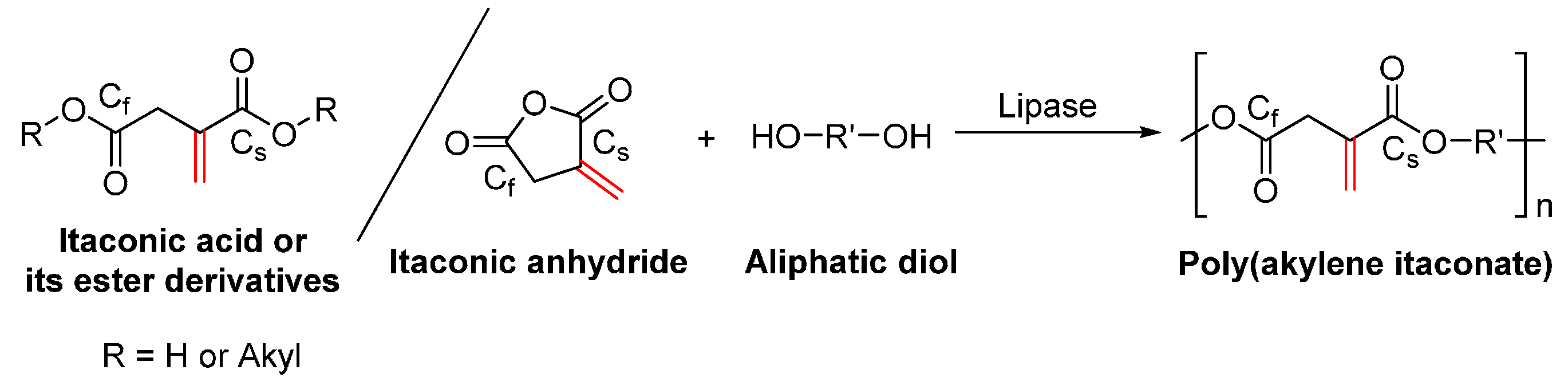
- Corici, L.; Pellis, A.; Ferrario, V.; Ebert, C.; Cantone, S.; Gardossi, L. Understanding potentials and restrictions of solvent-free enzymatic polycondensation of itaconic acid: An experimental and computational analysis. Adv. Synth. Catal. 2015, 357, 1763–1774. [Google Scholar] [CrossRef]
- Pellis, A.; Corici, L.; Sinigoi, L.; D’Amelio, N.; Fattor, D.; Ferrario, V.; Ebert, C.; Gardossi, L. Towards feasible and scalable solvent-free enzymatic polycondensations: Integrating robust biocatalysts with thin film reactions. Green Chem. 2015, 17, 1756–1766. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Tanha, M.; Hult, A.; Okuda, T.; Ohara, H.; Kobayashi, S. Green polymer chemistry: Lipase-catalyzed synthesis of bio-based reactive polyesters employing itaconic anhydride as a renewable monomer. Polym. J. 2014, 46, 2–13. [Google Scholar] [CrossRef]
- Barrett, D.G.; Merkel, T.J.; Luft, J.C.; Yousaf, M.N. One-step syntheses of photocurable polyesters based on a renewable resource. Macromolecules 2010, 43, 9660–9667. [Google Scholar] [CrossRef]
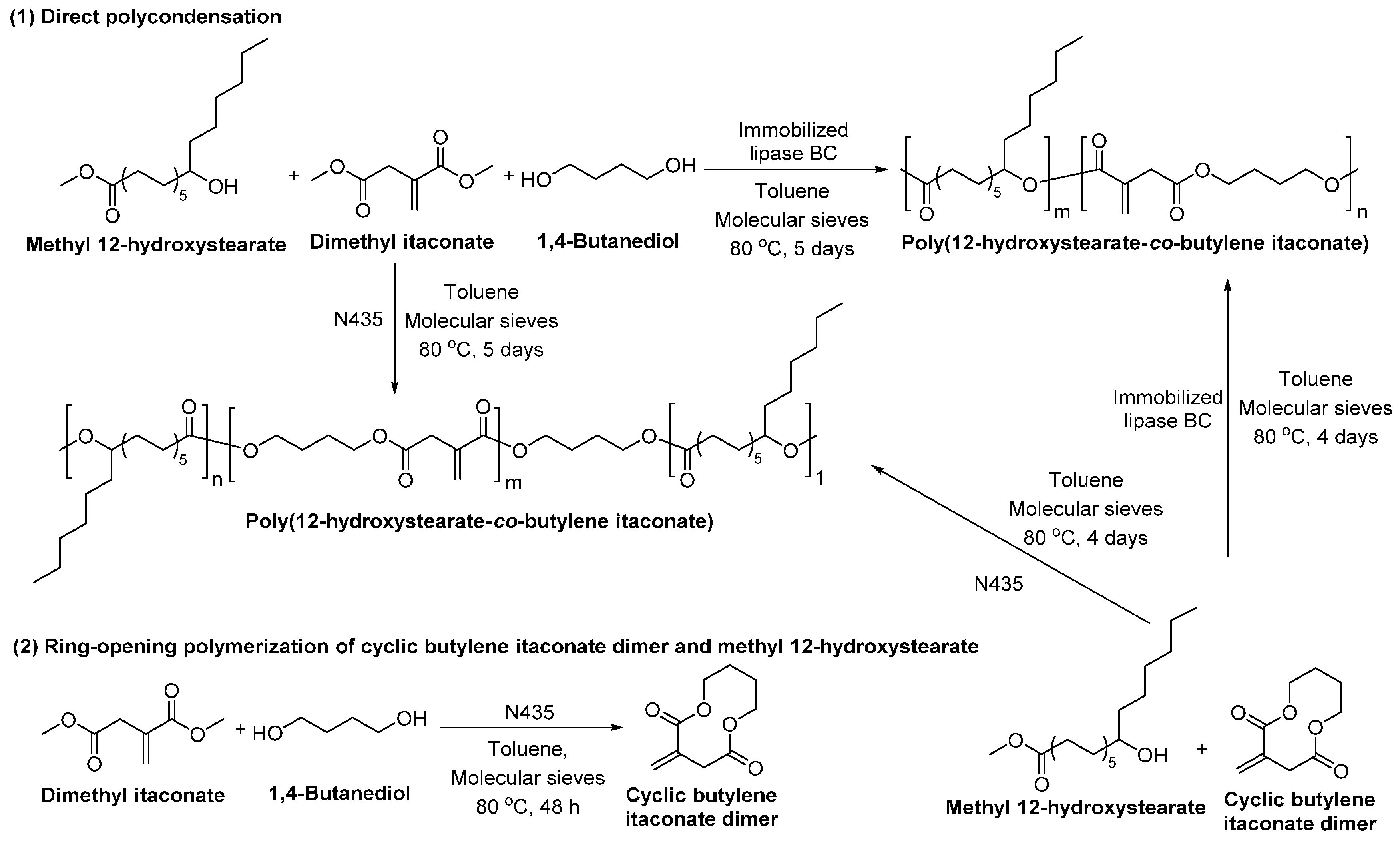
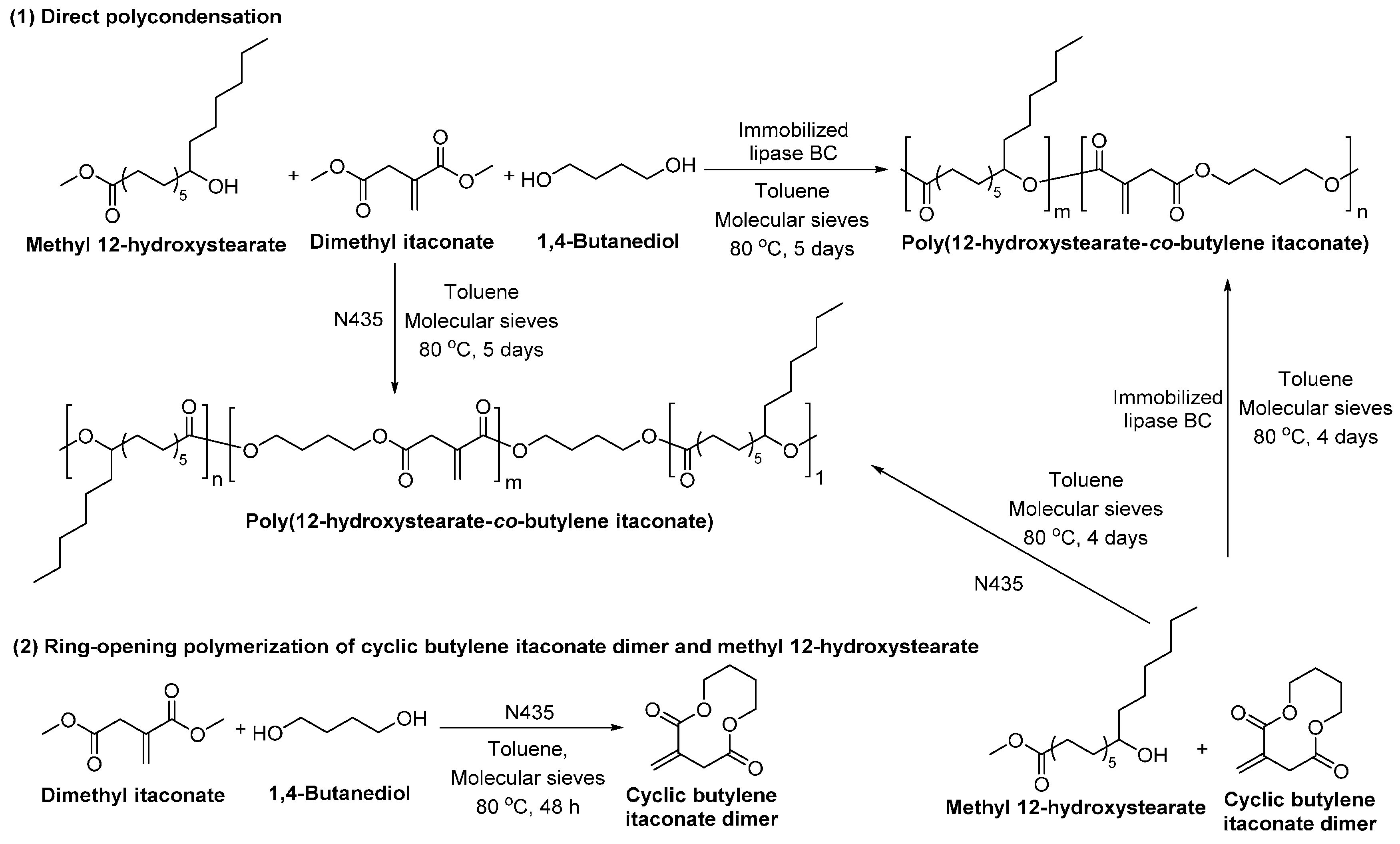
- Mayumi, Y.; Hiroki, E.; Shuichi, M. Enzymatic synthesis and properties of novel biobased elastomers consisting of 12-hydroxystearate, itaconate and butane-1,4-diol. In Green Polymer Chemistry: Biocatalysis and Biomaterials; American Chemical Society: Washington, DC, USA, 2010; Volume 1043, pp. 237–251. [Google Scholar]
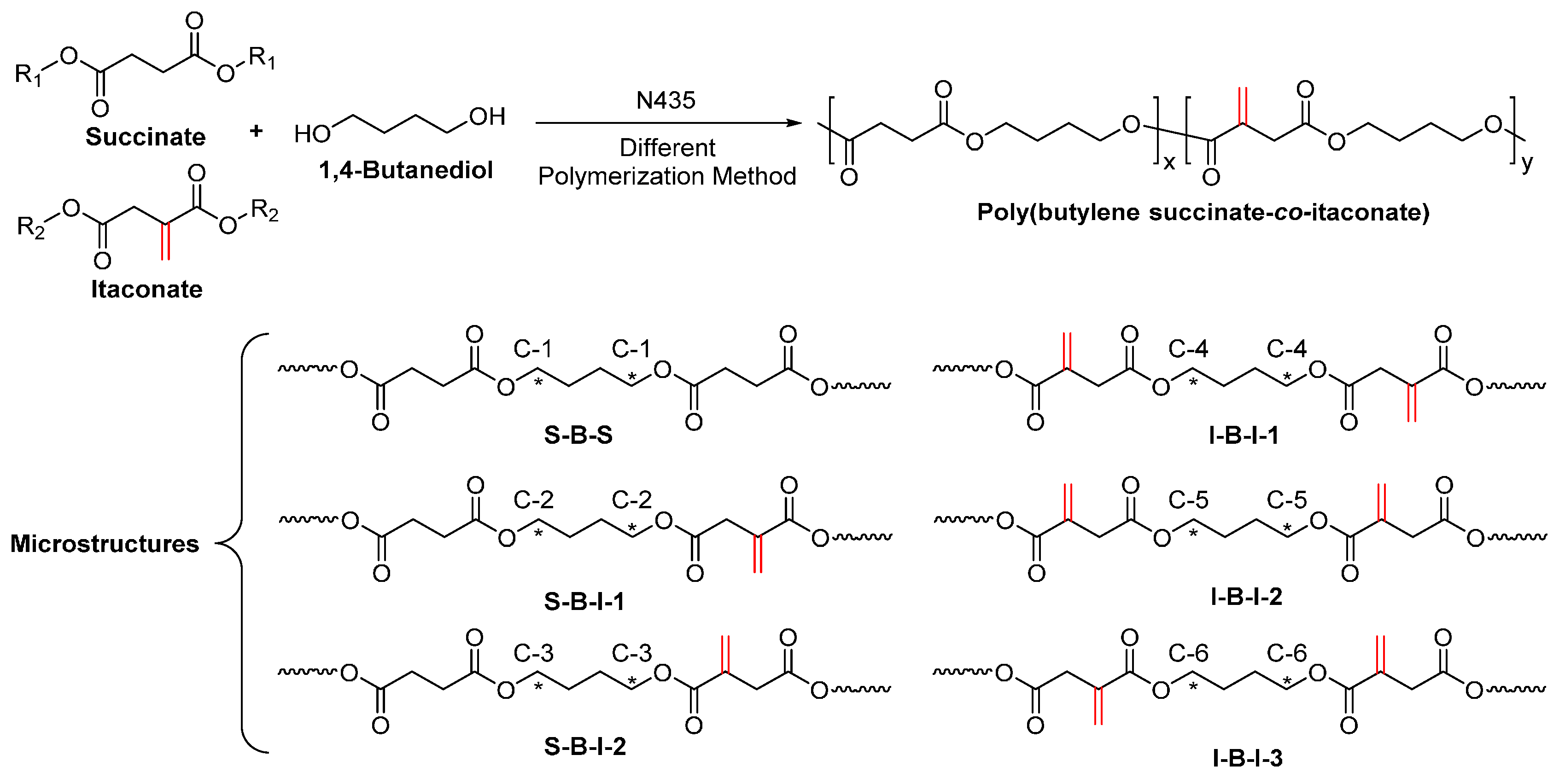
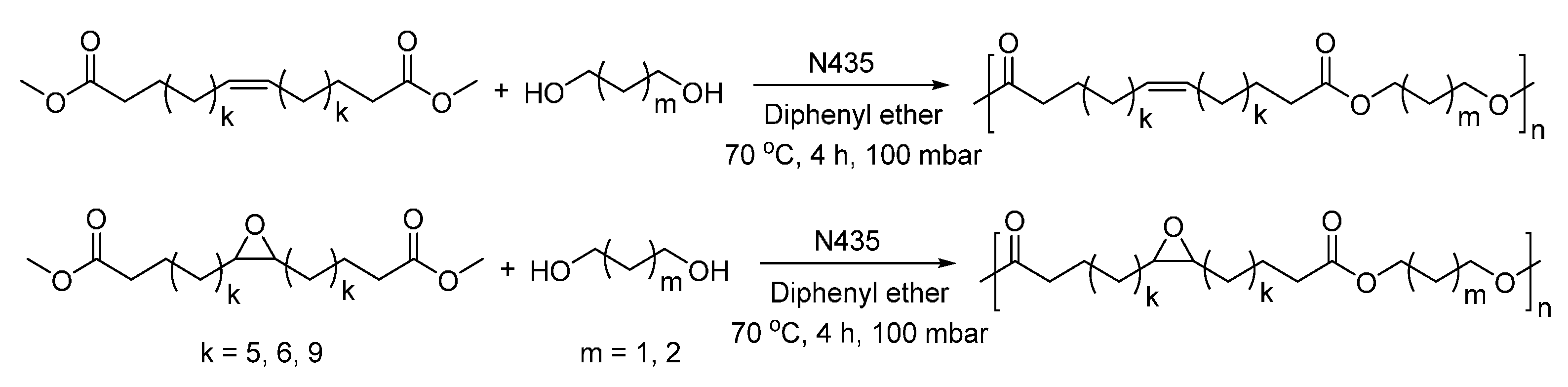
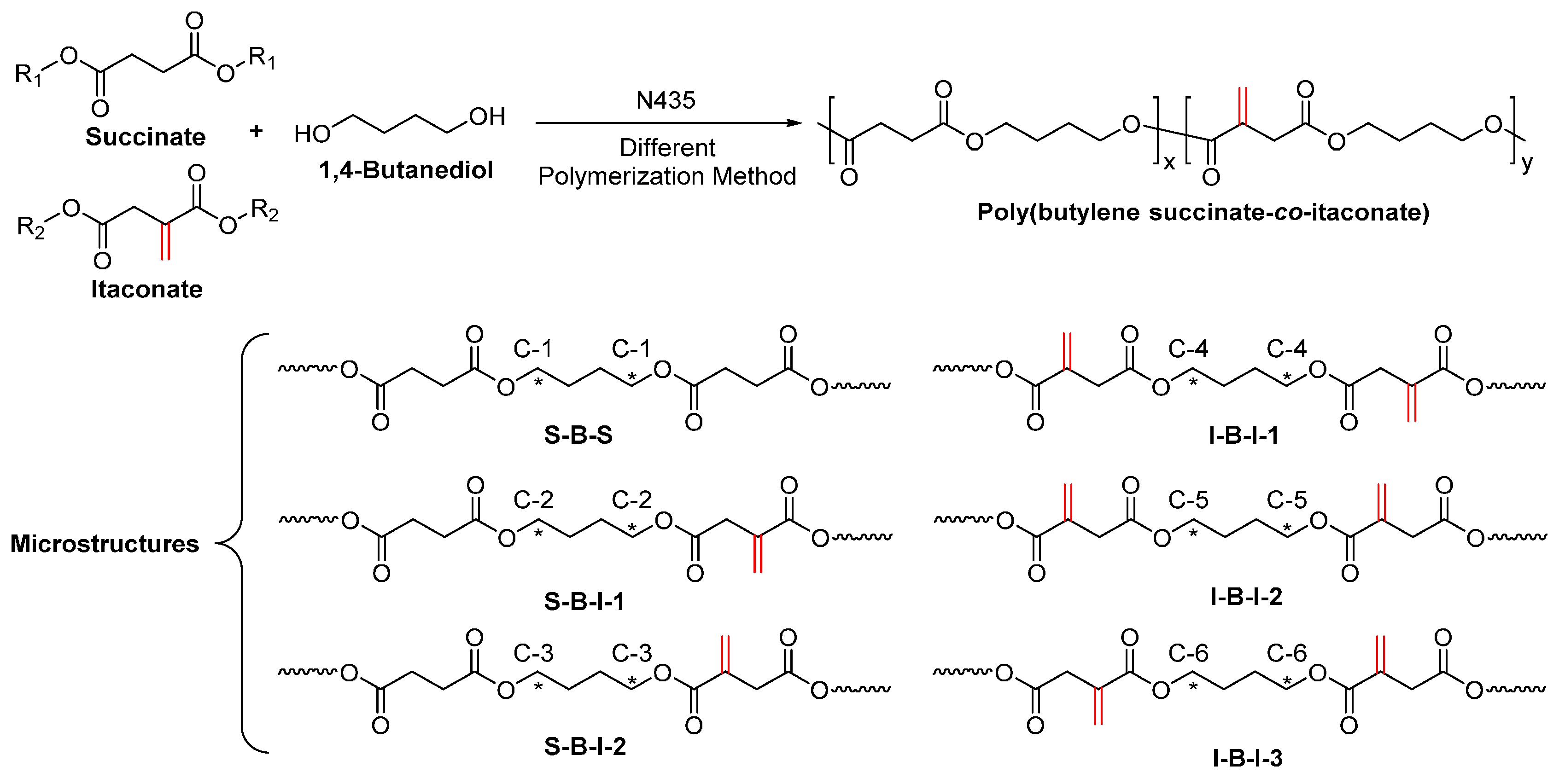
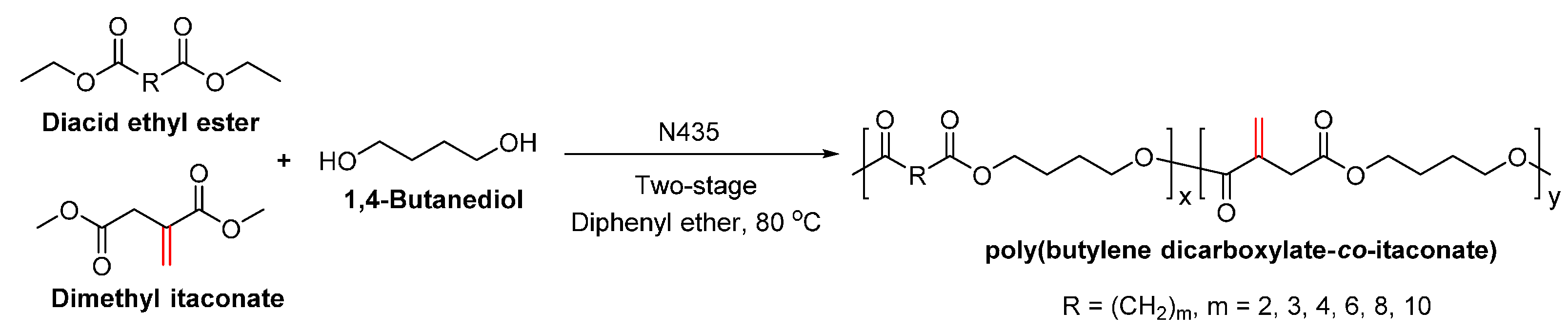
- Jiang, Y.; Woortman, A.J.J.; Alberda van Ekenstein, G.O.R.; Loos, K. Enzyme-catalyzed synthesis of unsaturated aliphatic polyesters based on green monomers from renewable resources. Biomolecules 2013, 3, 461–480. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Alberda van Ekenstein, G.O.R.; Woortman, A.J.J.; Loos, K. Fully biobased unsaturated aliphatic polyesters from renewable resources: Enzymatic synthesis, characterization, and properties. Macromol. Chem. Phys. 2014, 215, 2185–2197. [Google Scholar] [CrossRef]
- Biermann, U.; Friedt, W.; Lang, S.; Lühs, W.; Machmüller, G.; Metzger, J.O.; Rüsch gen. Klaas, M.; Schäfer, H.J.; Schneider, M.P. New syntheses with oils and fats as renewable raw materials for the chemical industry. Angew. Chem. Int. Ed. 2000, 39, 2206–2224. [Google Scholar] [CrossRef]
- Lligadas, G.; Ronda, J.C.; Galia, M.; Cadiz, V. Renewable polymeric materials from vegetable oils: A perspective. Mater. Today 2013, 16, 337–343. [Google Scholar] [CrossRef]
- Alam, M.; Akra, D.; Sharmin, E.; Zafar, F.; Ahmad, S. Vegetable oil based eco-friendly coating materials: A review article. Arabian J. Chem. 2014, 7, 469–479. [Google Scholar] [CrossRef]
- Miao, S.; Wang, P.; Su, Z.; Zhang, S. Vegetable-oil-based polymers as future polymeric biomaterials. Acta Biomater. 2014, 10, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Ebata, H.; Toshima, K.; Matsumura, S. Lipase-catalyzed synthesis and curing of high-molecular-weight polyricinoleate. Macromol. Biosci. 2007, 7, 798–803. [Google Scholar] [CrossRef] [PubMed]
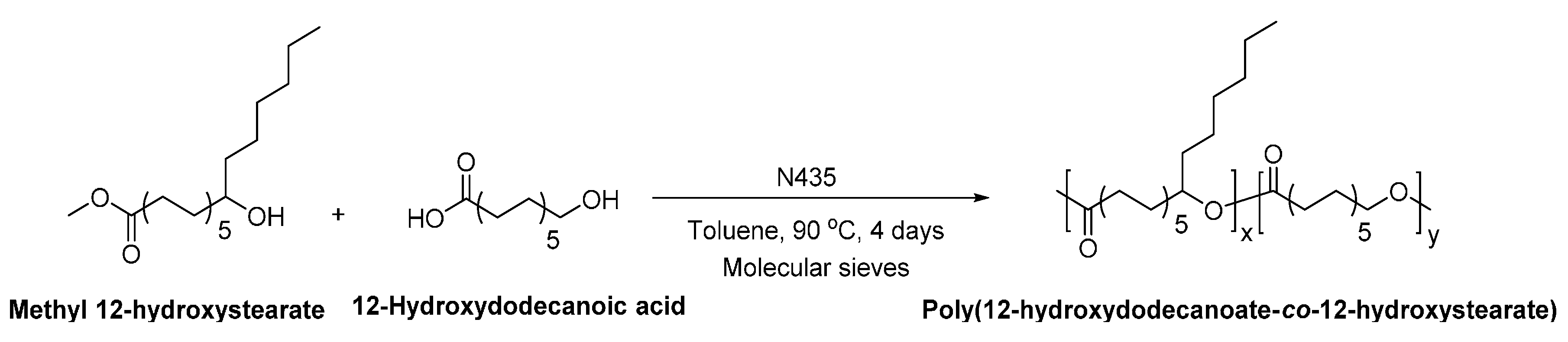
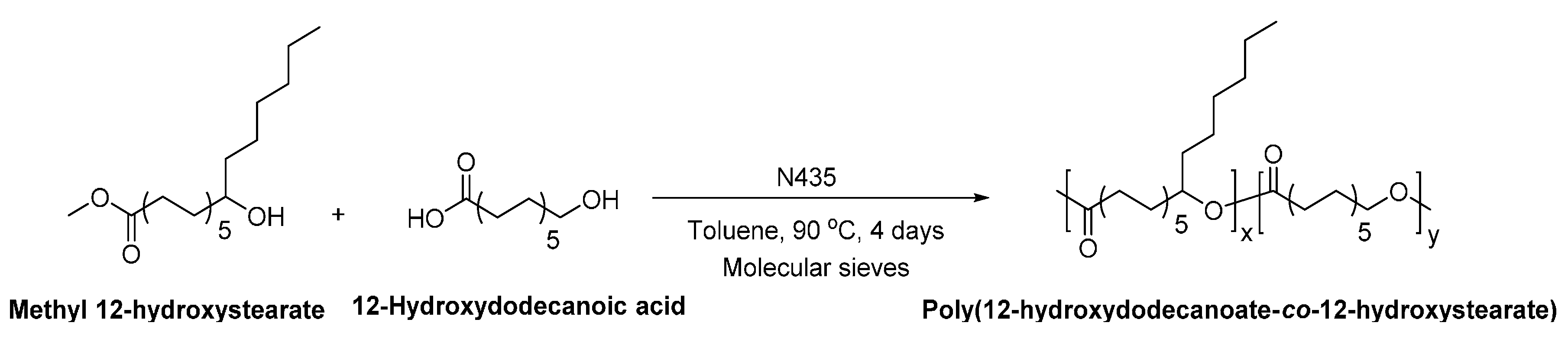
- Ebata, H.; Toshima, K.; Matsumura, S. Lipase-catalyzed synthesis and properties of poly[(12-hydroxydodecanoate)-co-(12-hydroxystearate)] directed towards novel green and sustainable elastomers. Macromol. Biosci. 2008, 8, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Warwel, S.; Demes, C.; Steinke, G. Polyesters by lipase-catalyzed polycondensation of unsaturated and epoxidized long-chain alpha,omega-dicarboxylic acid methyl esters with diols. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 1601–1609. [Google Scholar] [CrossRef]
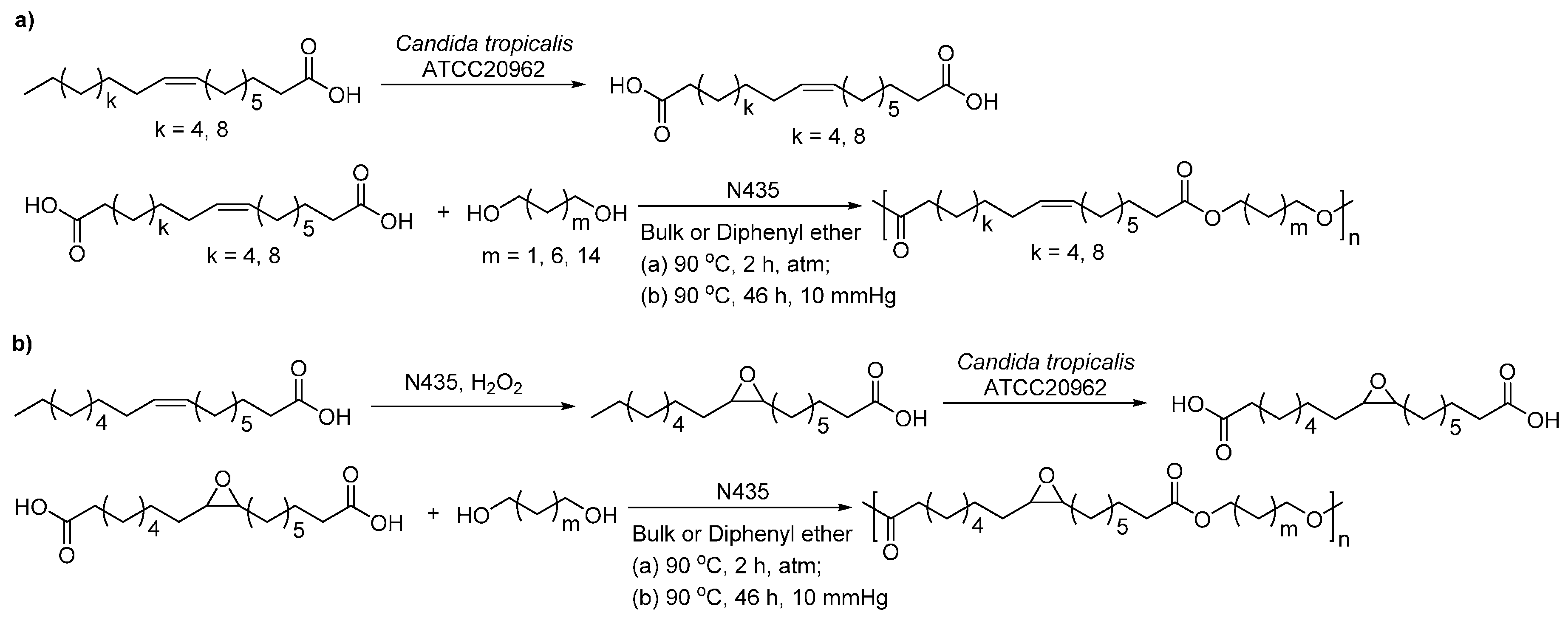
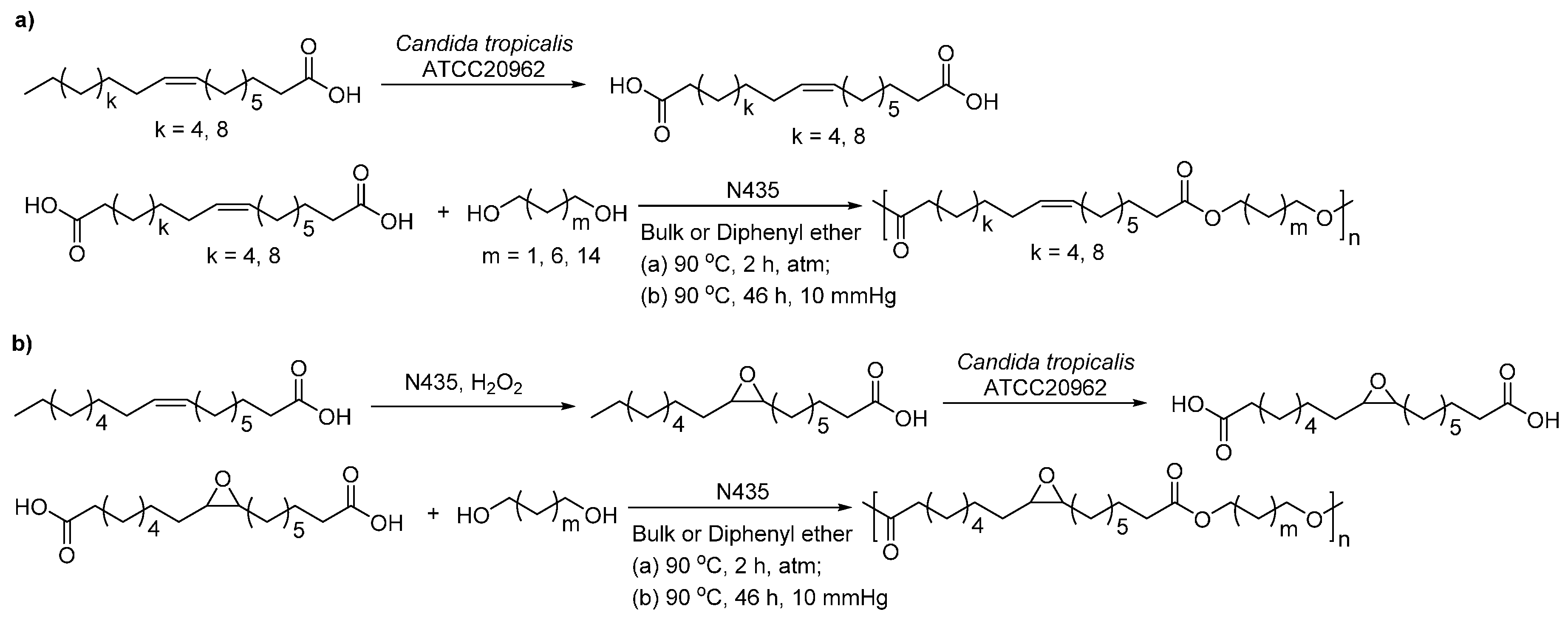
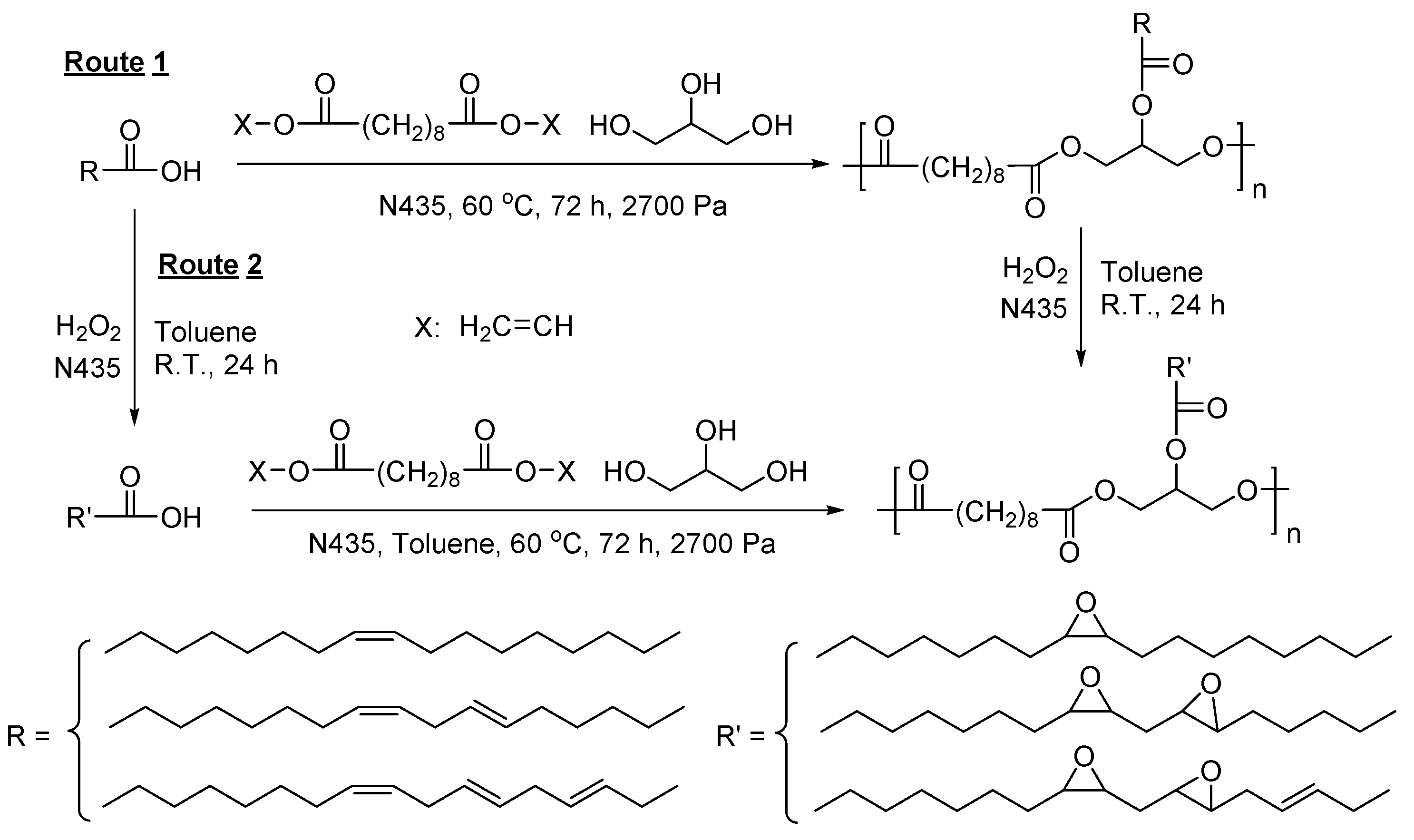
- Yang, Y.X.; Lu, W.H.; Zhang, X.Y.; Xie, W.C.; Cai, M.M.; Gross, R.A. Two-step biocatalytic route to biobased functional polyesters from omega-carboxy fatty acids and diols. Biomacromolecules 2010, 11, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Beisson, F.; Li-Beisson, Y.; Pollard, M. Solving the puzzles of cutin and suberin polymer biosynthesis. Curr. Opin. Plant Biol. 2012, 15, 329–337. [Google Scholar] [CrossRef] [PubMed]
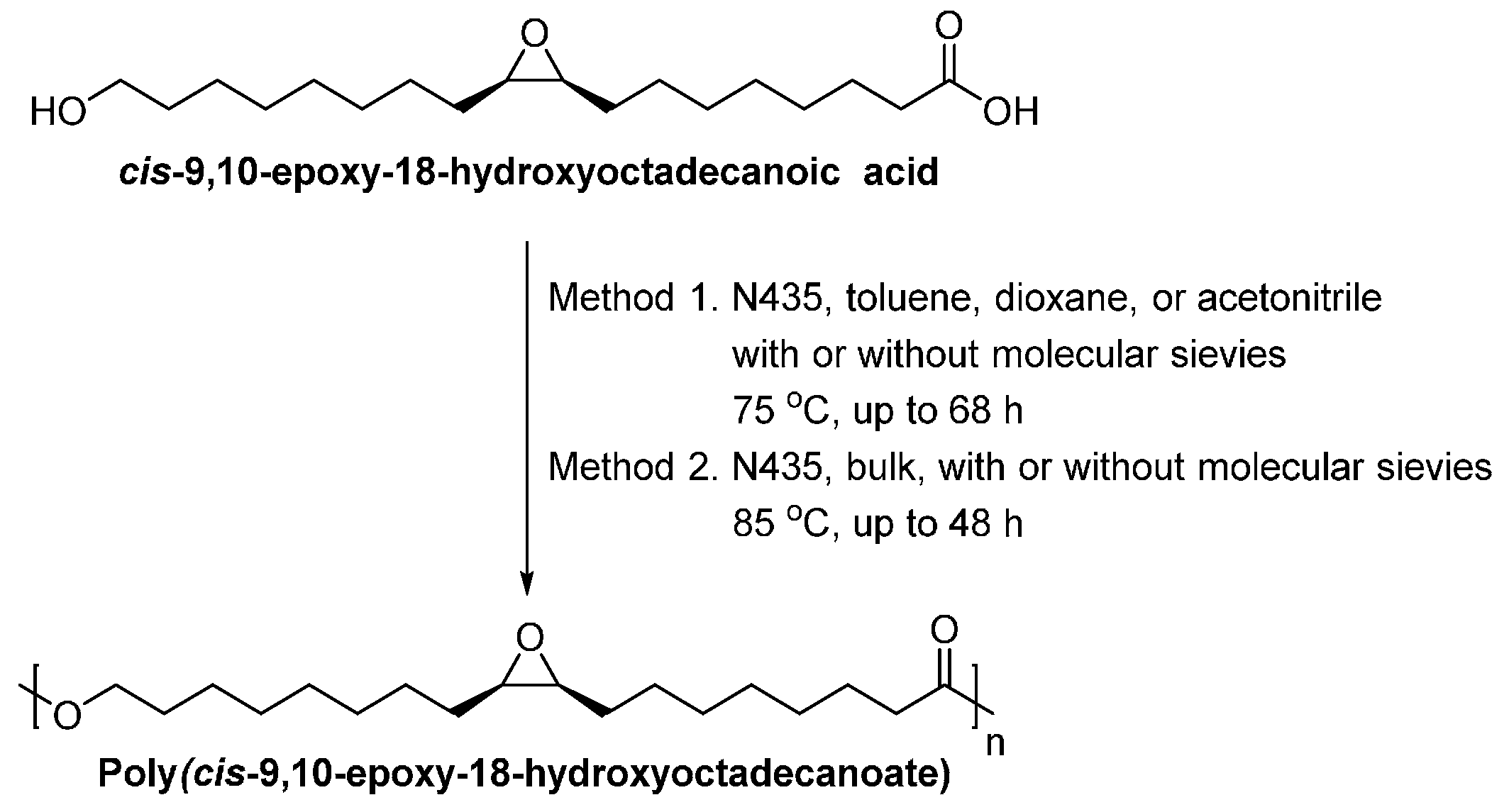
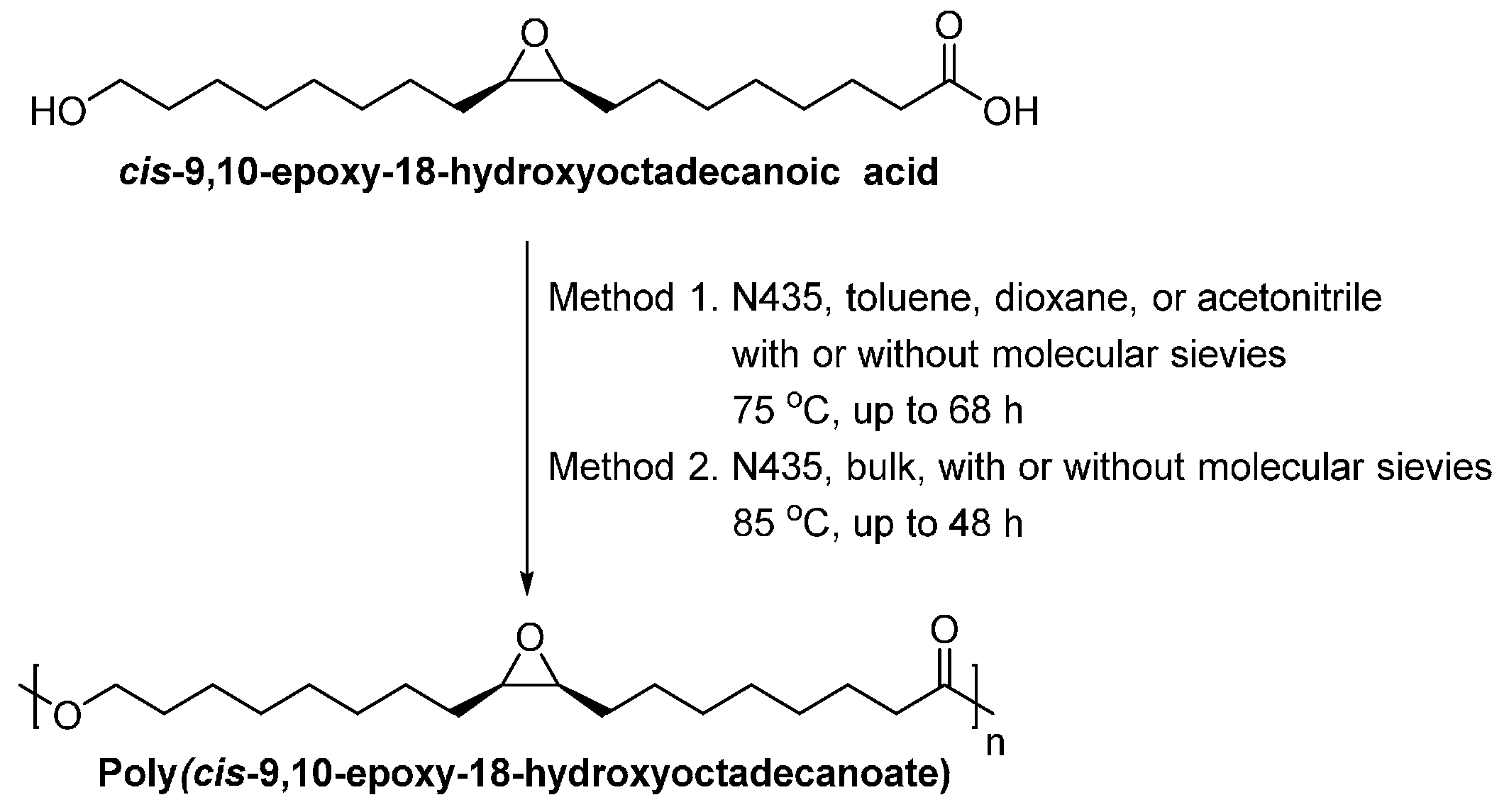
- Olsson, A.; Lindstrom, M.; Iversen, T. Lipase-catalyzed synthesis of an epoxy-functionalized polyester from the suberin monomer cis-9,10-epoxy-18-hydroxyoctadecanoic acid. Biomacromolecules 2007, 8, 757–760. [Google Scholar] [CrossRef] [PubMed]
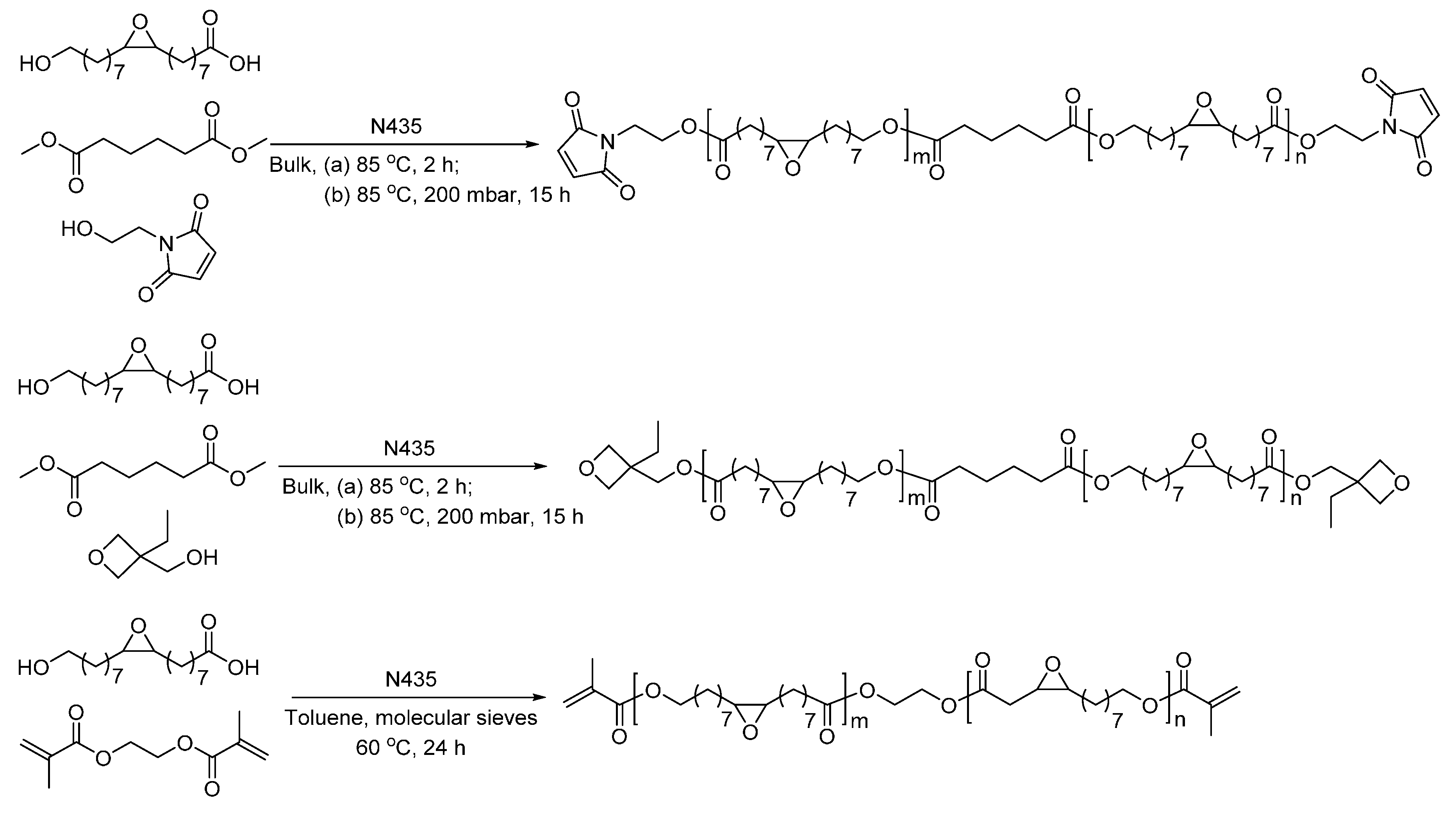
- Torron, S.; Johansson, M. Oxetane-terminated telechelic epoxy-functional polyesters as cationically polymerizable thermoset resins: Tuning the reactivity with structural design. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 2258–2266. [Google Scholar] [CrossRef]
- Semlitsch, S.; Torron, S.; Johansson, M.; Martinelle, M. Enzymatic catalysis as a versatile tool for the synthesis of multifunctional, bio-based oligoester resins. Green Chem. 2016, 18, 1923–1929. [Google Scholar] [CrossRef]
- Zhang, H.; Grinstaff, M.W. Recent advances in glycerol polymers: Chemistry and biomedical applications. Macromol. Rapid Commun. 2014, 35, 1906–1924. [Google Scholar] [CrossRef] [PubMed]
- Kline, B.J.; Beckman, E.J.; Russell, A.J. One-step biocatalytic synthesis of linear polyesters with pendant hydroxyl groups. J. Am. Chem. Soc. 1998, 120, 9475–9480. [Google Scholar] [CrossRef]
- Kumar, A.; Kulshrestha, A.S.; Gao, W.; Gross, R.A. Versatile route to polyol polyesters by lipase catalysis. Macromolecules 2003, 36, 8219–8221. [Google Scholar] [CrossRef]
- Fu, H.; Kulshrestha, A.S.; Gao, W.; Gross, R.A.; Baiardo, M.; Scandola, M. Physical characterization of sorbitol or glycerol containing aliphatic copolyesters synthesized by lipase-catalyzed polymerization. Macromolecules 2003, 36, 9804–9808. [Google Scholar] [CrossRef]
- Kulshrestha, A.S.; Gao, W.; Gross, R.A. Glycerol copolyesters: Control of branching and molecular weight using a lipase catalyst. Macromolecules 2005, 38, 3193–3204. [Google Scholar] [CrossRef]
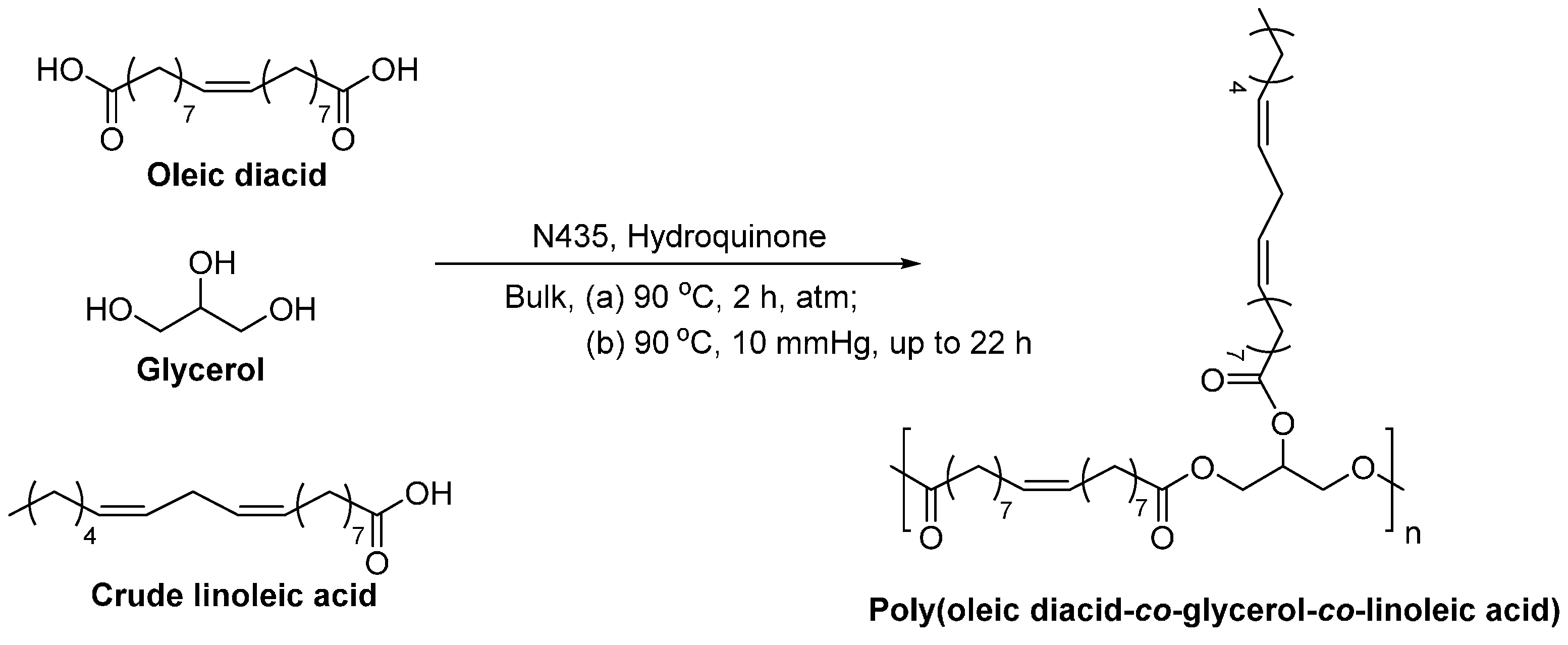
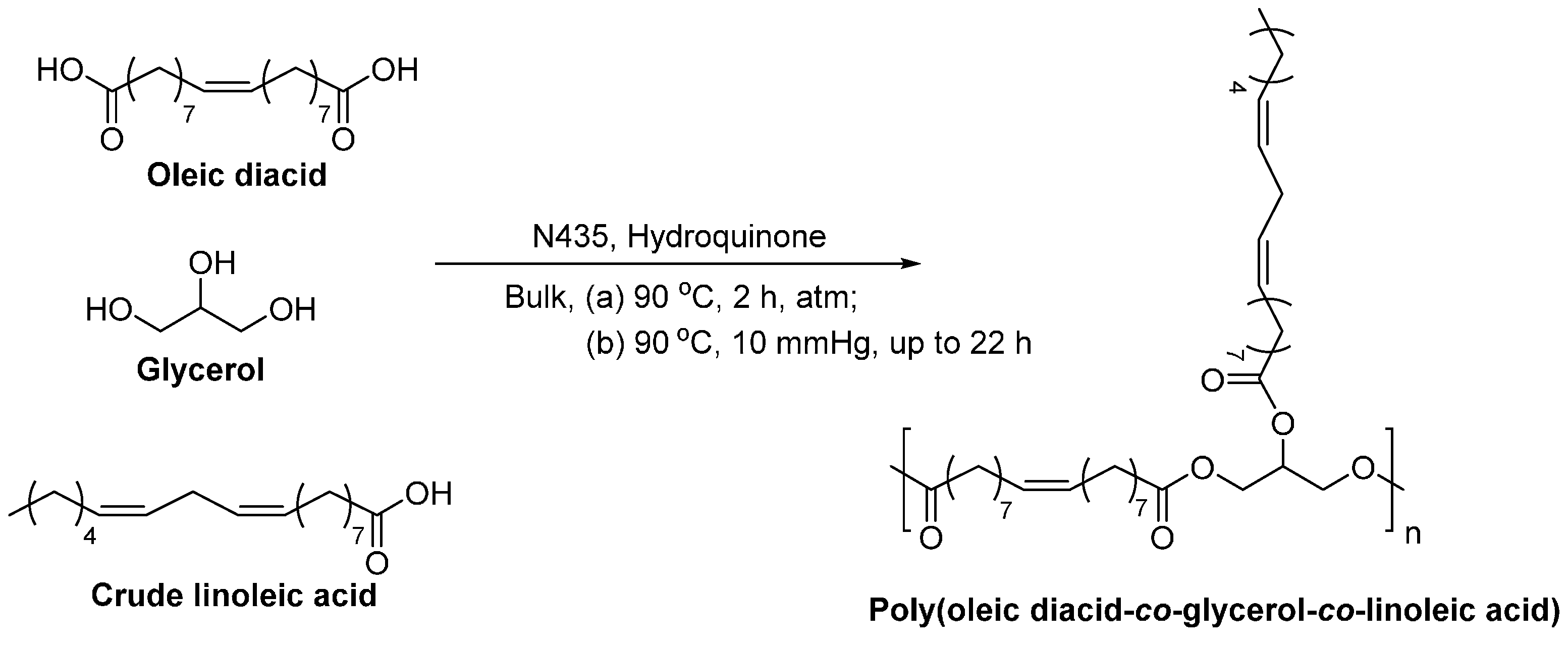
- Yang, Y.X.; Lu, W.H.; Cai, J.L.; Hou, Y.; Ouyang, S.Y.; Xie, W.C.; Gross, R.A. Poly(oleic diacid-co-glycerol): Comparison of polymer structure resulting from chemical and lipase catalysis. Macromolecules 2011, 44, 1977–1985. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Uyama, H.; Kobayashi, S. Enzymatic synthesis of cross-linkable polyesters from renewable resources. Biomacromolecules 2001, 2, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Uyama, H.; Kobayashi, S. Enzymatic synthesis and curing of biodegradable crosslinkable polyesters. Macromol. Biosci. 2002, 2, 329–335. [Google Scholar] [CrossRef]
- Uyama, H.; Kuwabara, M.; Tsujimoto, T.; Kobayashi, S. Enzymatic synthesis and curing of biodegradable epoxide-containing polyesters from renewable resources. Biomacromolecules 2003, 4, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-R.; Spinella, S.; Xie, W.; Cai, J.; Yang, Y.; Wang, Y.-Z.; Gross, R.A. Polymeric triglyceride analogs prepared by enzyme-catalyzed condensation polymerization. Eur. Polym. J. 2013, 49, 793–803. [Google Scholar] [CrossRef]
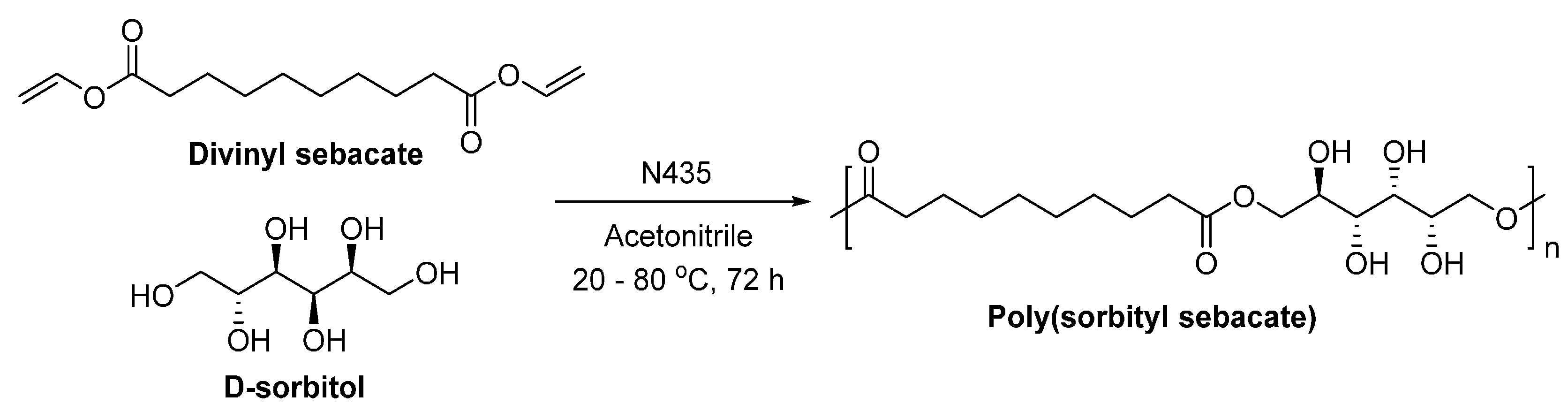
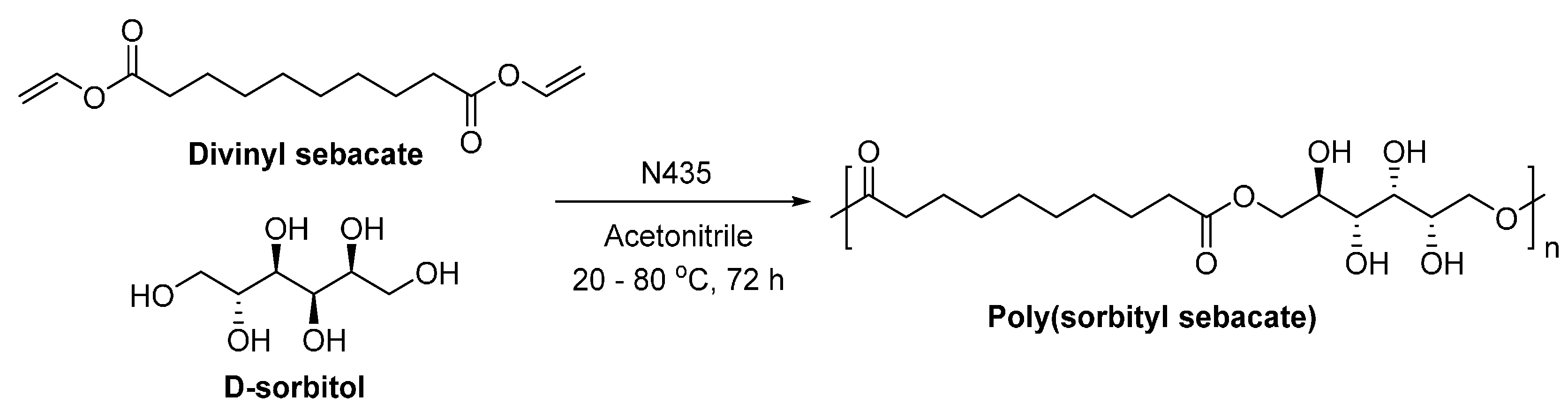
- Uyama, H.; Klegraf, E.; Wada, S.; Kobayashi, S. Regioselective polymerization of sorbitol and divinyl sebacate using lipase catalyst. Chem. Lett. 2000, 29, 800–801. [Google Scholar] [CrossRef]
- Kim, D.Y.; Dordick, J.S. Combinatorial array-based enzymatic polyester synthesis. Biotechnol. Bioeng. 2001, 76, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.; Wei, G.; Ankur, K.; Richard, A.G. “Sweet polyesters”: Lipase-catalyzed condensation-polymerizations of alditols. In Polymer Biocatalysis and Biomaterials II; American Chemical Society: Washington, DC, USA, 2008; Volume 999, pp. 275–284. [Google Scholar]
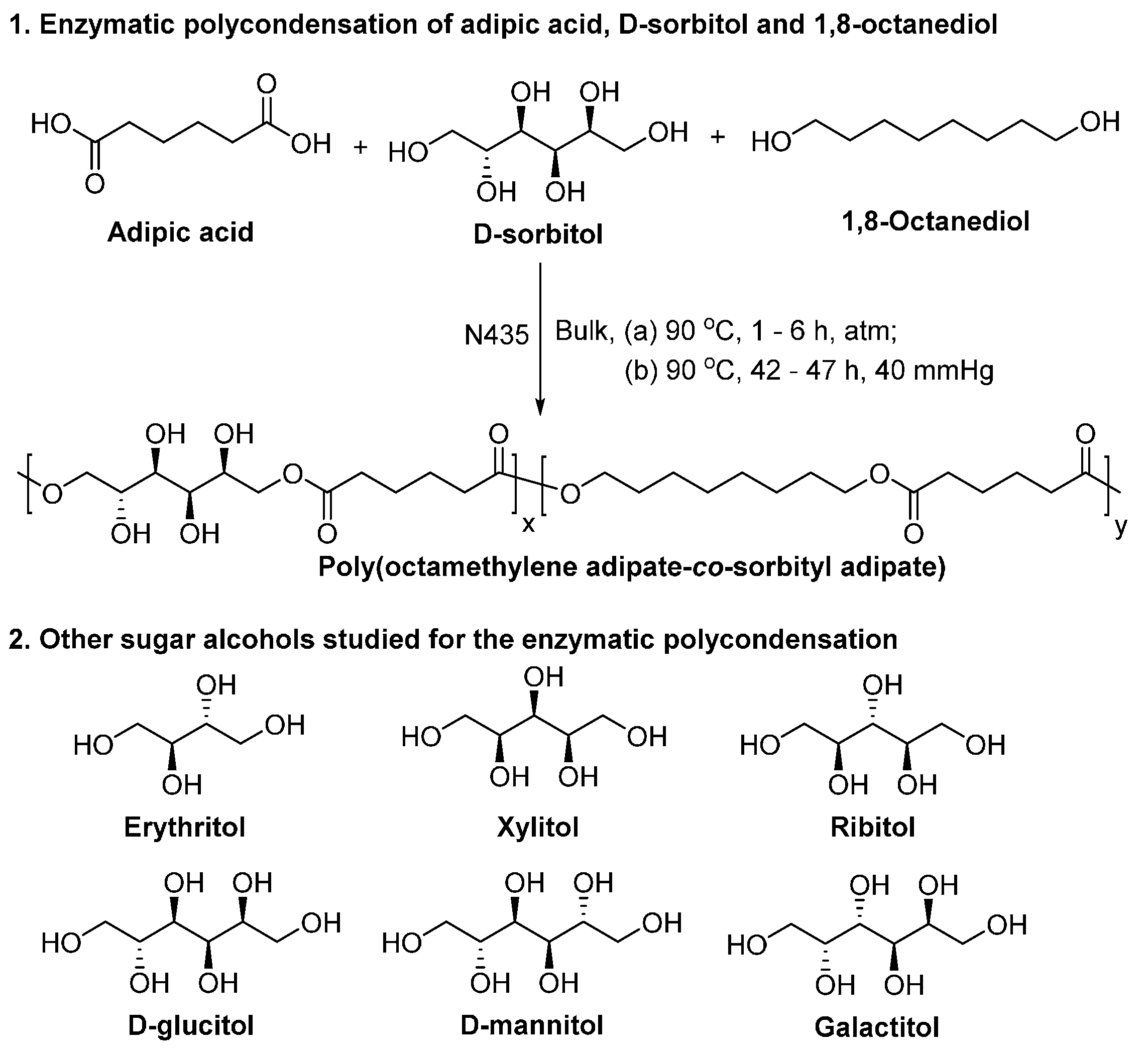
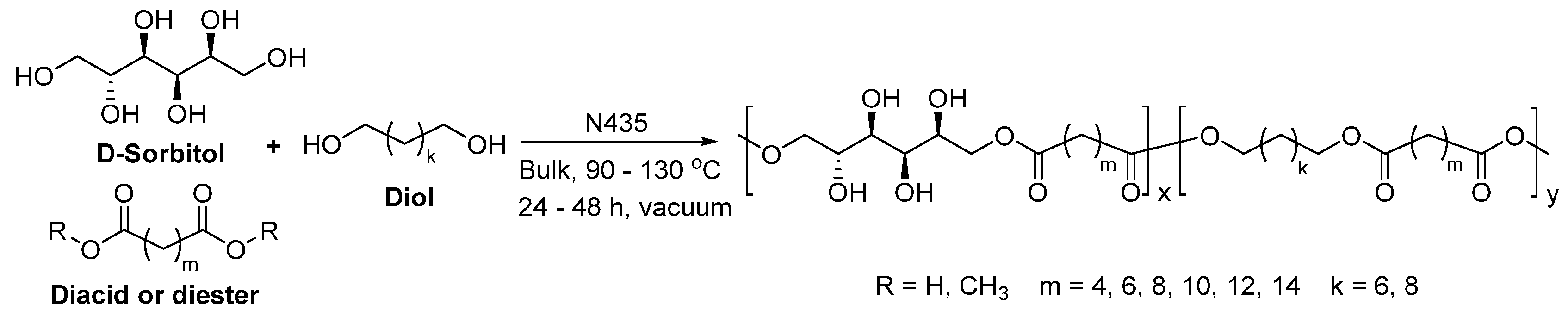
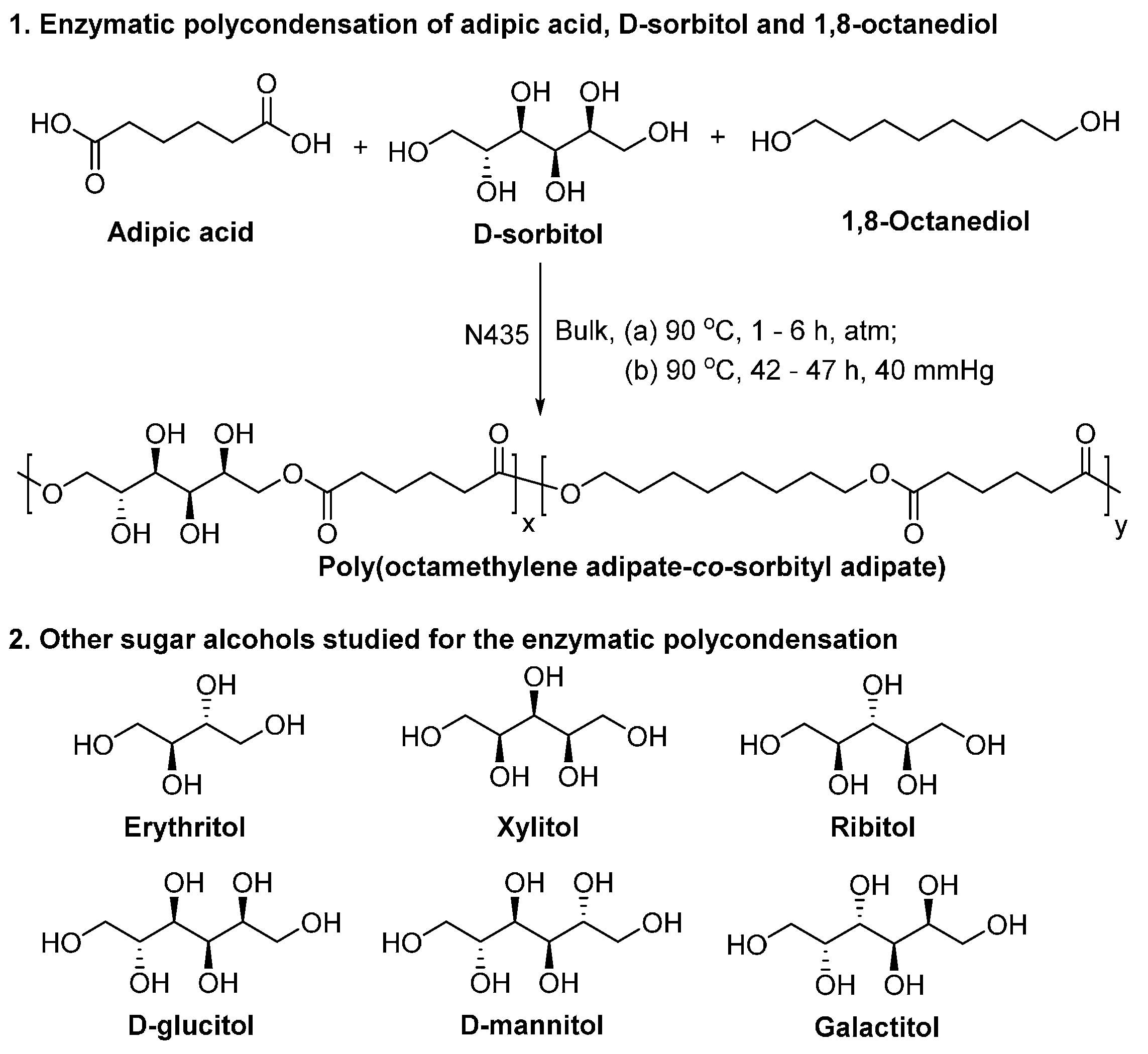
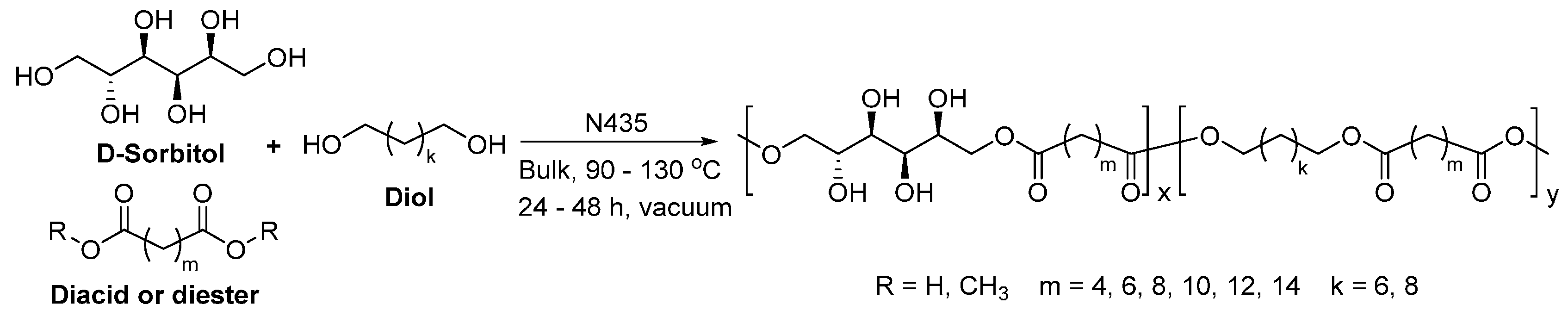
- Gustini, L.; Noordover, B.A.J.; Gehrels, C.; Dietz, C.; Koning, C.E. Enzymatic synthesis and preliminary evaluation as coating of sorbitol-based, hydroxy-functional polyesters with controlled molecular weights. Eur. Polym. J. 2015, 67, 459–475. [Google Scholar] [CrossRef]
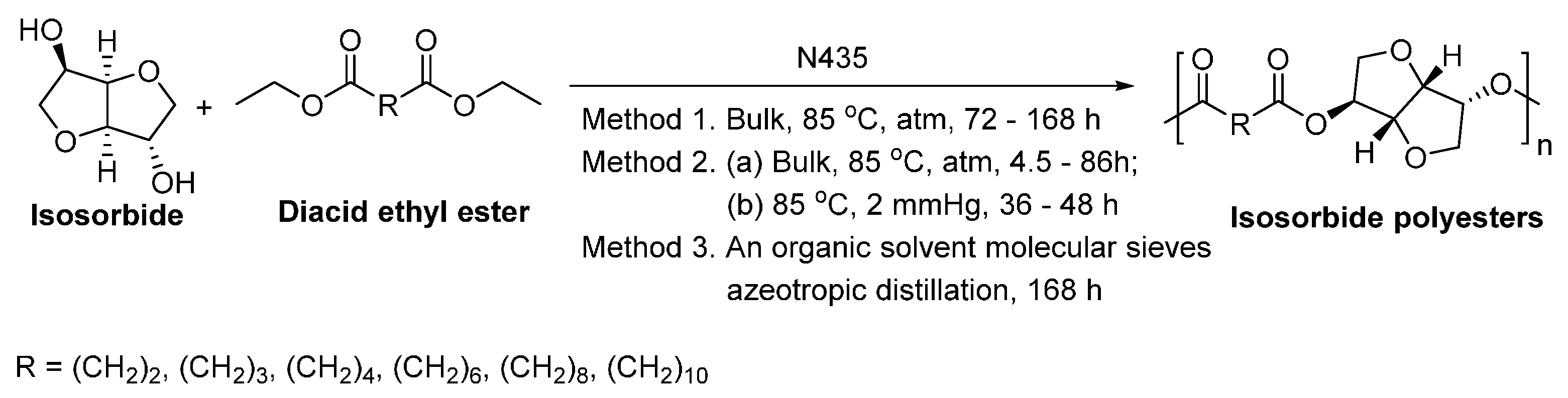
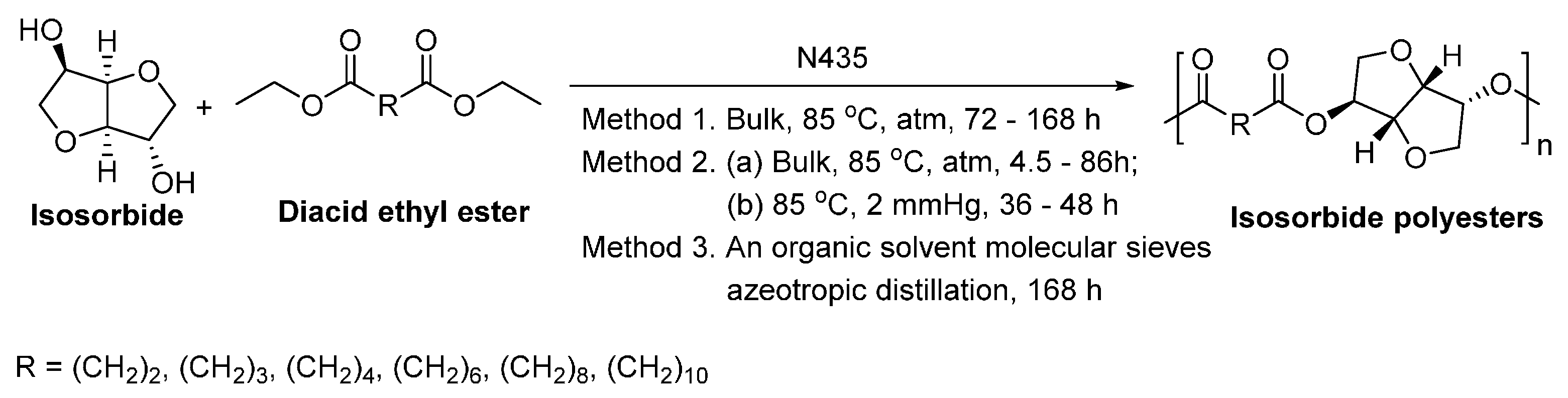
- Juais, D.; Naves, A.F.; Li, C.; Gross, R.A.; Catalani, L.H. Isosorbide polyesters from enzymatic catalysis. Macromolecules 2010, 43, 10315–10319. [Google Scholar] [CrossRef]
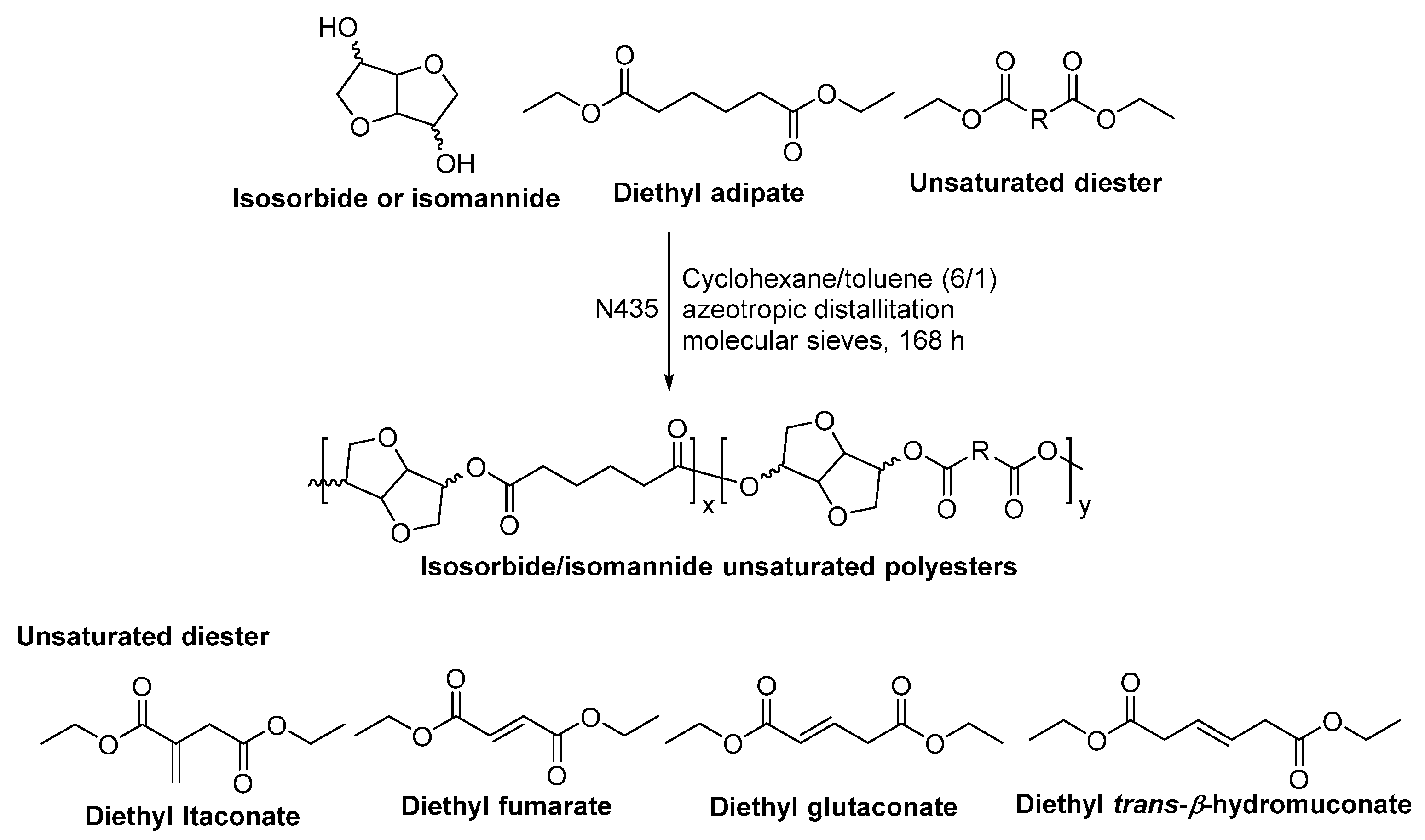
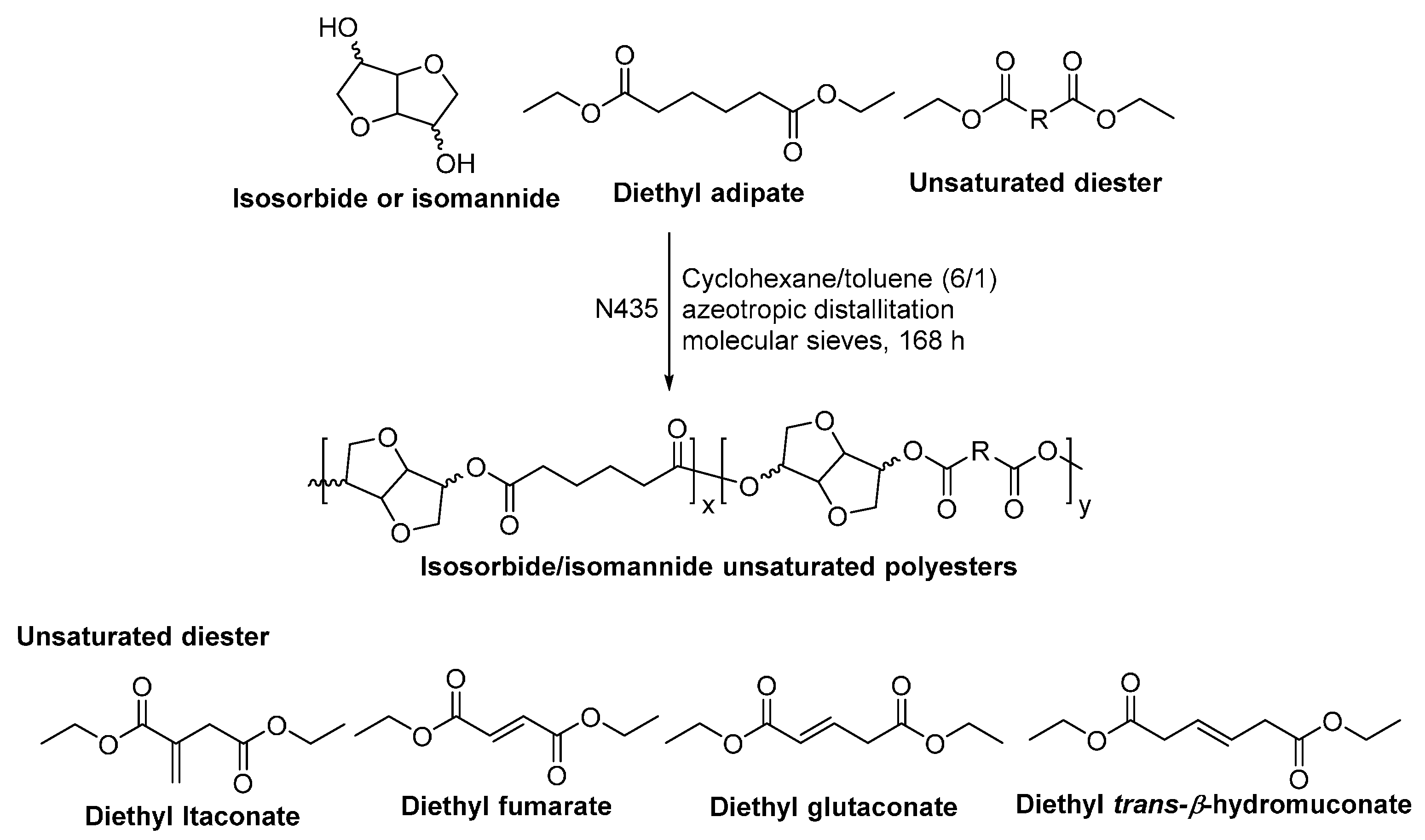
- Naves, A.F.; Fernandes, H.T.C.; Immich, A.P.S.; Catalani, L.H. Enzymatic syntheses of unsaturated polyesters based on isosorbide and isomannide. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 3881–3891. [Google Scholar] [CrossRef]
- Habeych, D.I.; Juhl, P.B.; Pleiss, J.; Vanegas, D.; Eggink, G.; Boeriu, C.G. Biocatalytic synthesis of polyesters from sugar-based building blocks using immobilized candida antarctica lipase B. J. Mol. Catal. B-Enzym. 2011, 71, 1–9. [Google Scholar] [CrossRef]
- Japu, C.; Martinez de Ilarduya, A.; Alla, A.; Jiang, Y.; Loos, K.; Munoz-Guerra, S. Copolyesters made from 1,4-butanediol, sebacic acid, and d-glucose by melt and enzymatic polycondensation. Biomacromolecules 2015, 16, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.A.; Kelly, J.E. Polyesters of 2,5-disubstituted furan and tetrahydrofuran. In Presented at the 168th ACS National Meeting, Atlantic, NJ, USA, 8–13 September 1974.
- Moore, J.A.; Kelly, J.E. Polyesters derived from furan and tetrahydrofuran nuclei. Macromolecules 1978, 11, 568–573. [Google Scholar] [CrossRef]
- Storbeck, R.; Ballauff, M. Synthesis and properties of polyesters based on 2,5-furandicarboxylic acid and 1,4:3,6-dianhydrohexitols. Polymer 1993, 34, 5003–5006. [Google Scholar] [CrossRef]
- Khrouf, A.; Boufi, S.; El Gharbi, R.; Belgacem, N.M.; Gandini, A. Polyesters bearing furan moieties: 1. Polytransesterification involving difuranic diesters and aliphatic diols. Polym. Bull. 1996, 37, 589–596. [Google Scholar] [CrossRef]
- Okada, M.; Tachikawa, K.; Aoi, K. Biodegradable polymers based on renewable resources. II. Synthesis and biodegradability of polyesters containing furan rings. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 2729–2737. [Google Scholar]
- Burgess, S.K.; Karvan, O.; Johnson, J.R.; Kriegel, R.M.; Koros, W.J. Oxygen sorption and transport in amorphous poly(ethylene furanoate). Polymer 2014, 55, 4748–4756. [Google Scholar] [CrossRef]
- Burgess, S.K.; Leisen, J.E.; Kraftschik, B.E.; Mubarak, C.R.; Kriegel, R.M.; Koros, W.J. Chain mobility, thermal, and mechanical properties of poly(ethylene furanoate) compared to poly(ethylene terephthalate). Macromolecules 2014, 47, 1383–1391. [Google Scholar] [CrossRef]
- Hopff, H.; Krieger, A. Uber decarboxylierung und dissoziation heterocyclischer dicarbonsauren. Helv. Chim. Acta 1961, 44, 1058–1063. [Google Scholar] [CrossRef]
- Hopff, H.; Krieger, A. Über polyamide aus heterocyclischen dicarbonsäuren. Die Makromol. Chem. 1961, 47, 93–113. [Google Scholar] [CrossRef]
- Heertjes, P.; Kok, G. Polycondensation products of 2, 5-furandicarboxylic acid. Delft Prog. Rep. Ser. A 1974, 1, 59–63. [Google Scholar]
- Habeych N., D.I. Biocatalytic Synthesis of Cyclic Ester Oligomers from Biobased Building Blocks. Ph.D. Thesis, Wageningen University, Wageningen, The Netherland, 2011. [Google Scholar]
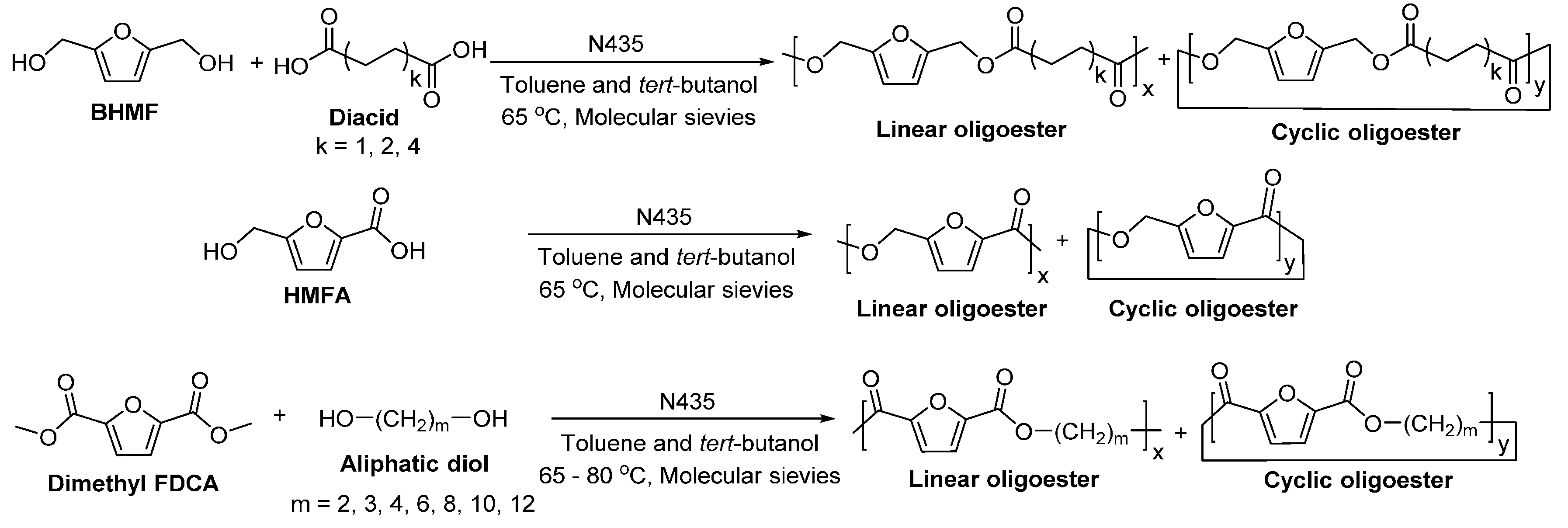
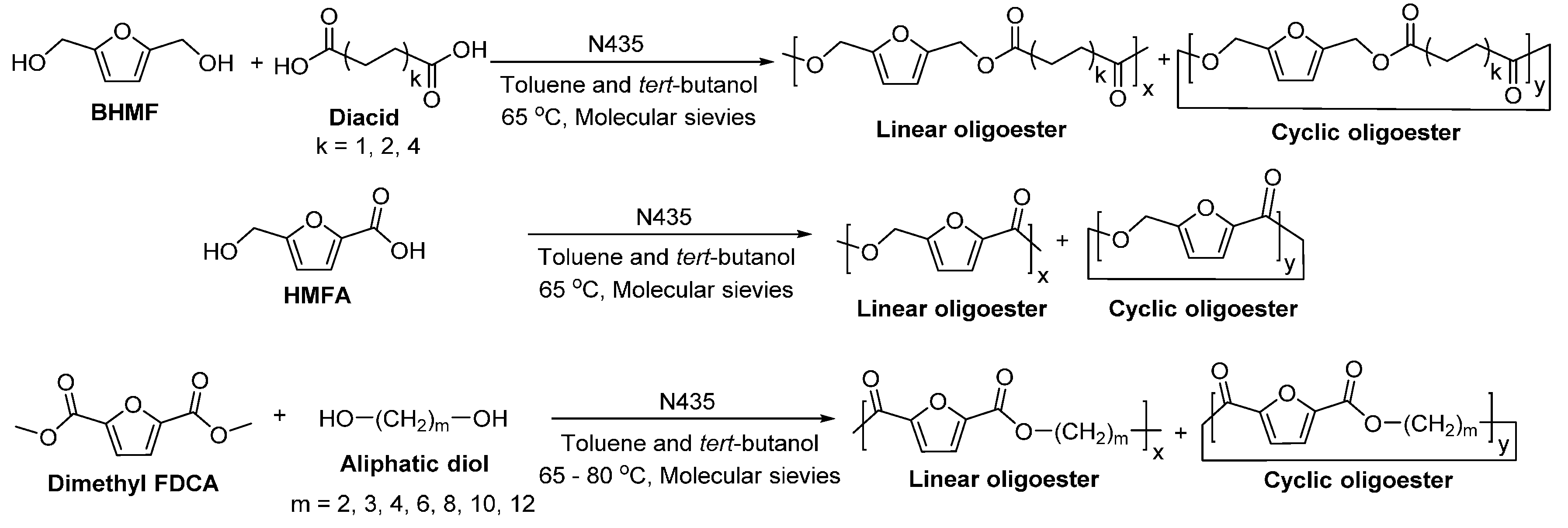
- Cruz-Izquierdo, Á.; van den Broek, L.A.M.; Serra, J.L.; Llama, M.J.; Boeriu, C.G. Lipase-catalyzed synthesis of oligoesters of 2,5-furandicarboxylic acid with aliphatic diols. Pure Appl. Chem. 2015, 87, 59–69. [Google Scholar] [CrossRef]
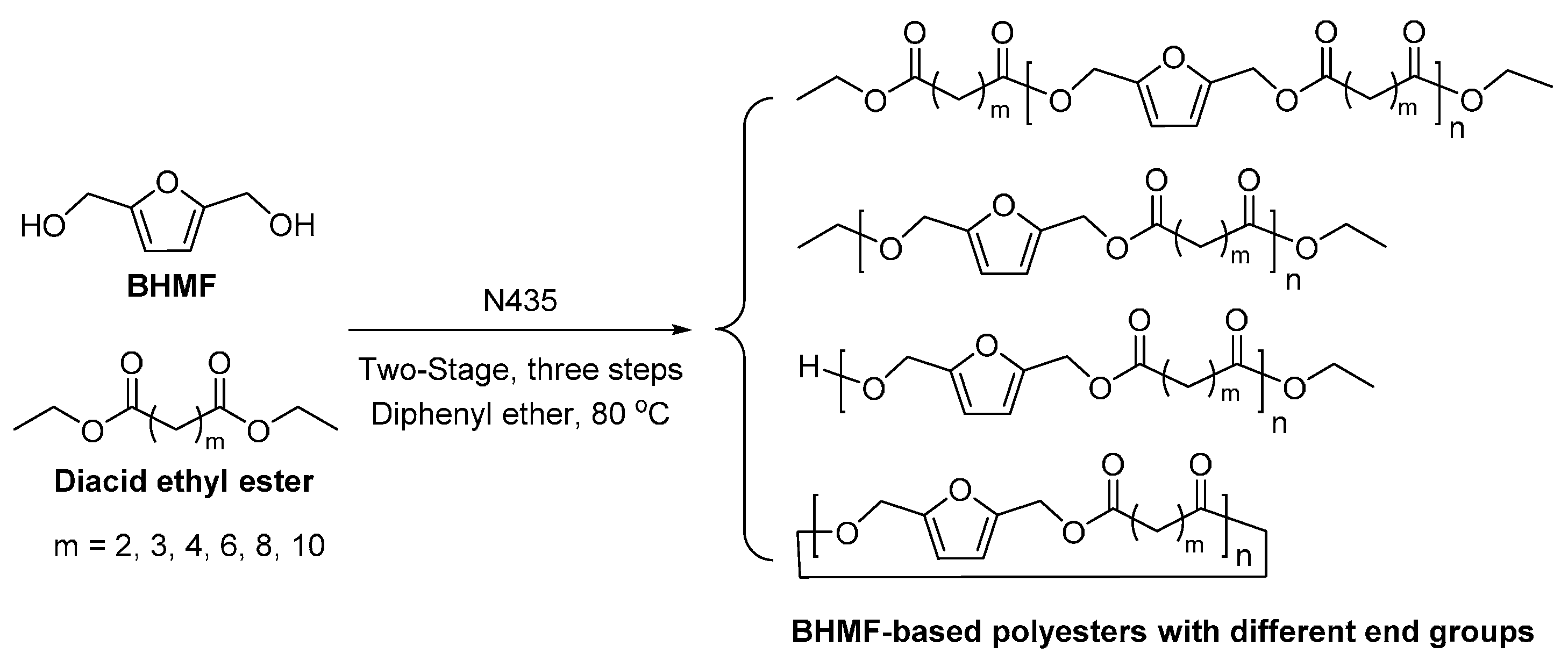
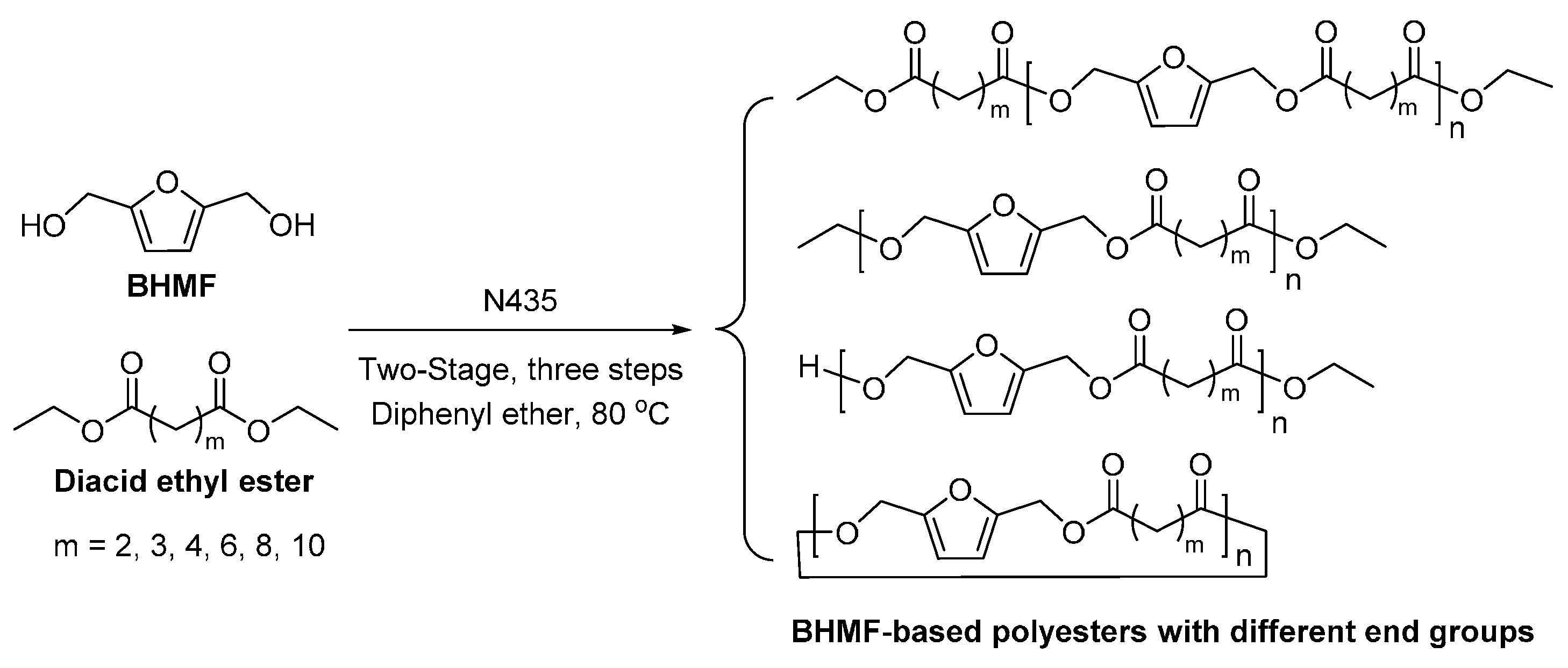
- Jiang, Y.; Woortman, A.J.J.; Alberda van Ekenstein, G.O.R.; Petrovic, D.M.; Loos, K. Enzymatic synthesis of biobased polyesters using 2,5-bis(hydroxymethyl)furan as the building block. Biomacromolecules 2014, 15, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
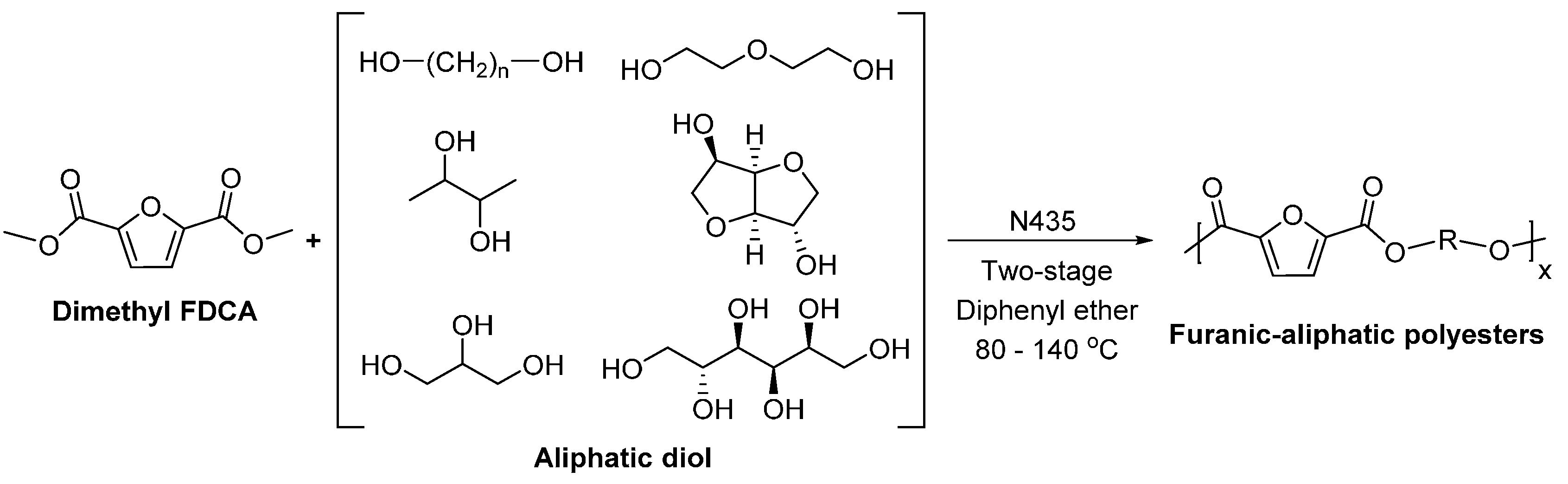
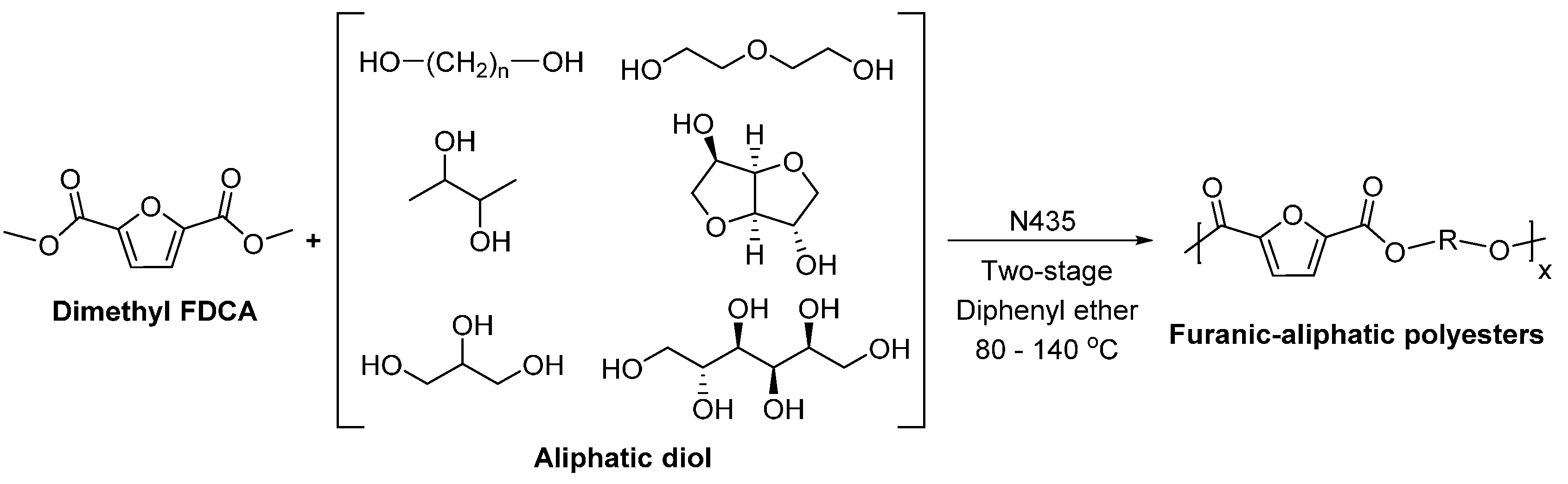
- Jiang, Y.; Woortman, A.J.J.; Alberda van Ekenstein, G.O.R.; Loos, K. A biocatalytic approach towards sustainable furanic–aliphatic polyesters. Polym. Chem. 2015, 6, 5198–5211. [Google Scholar] [CrossRef]
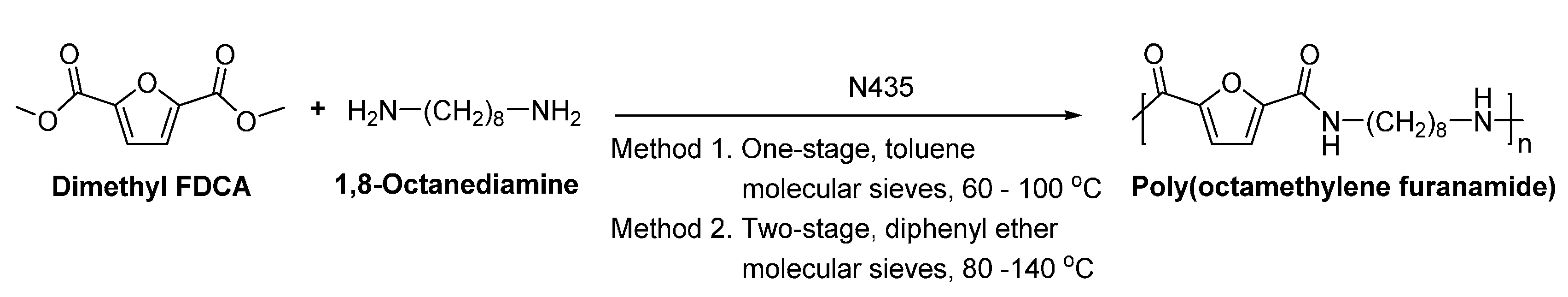
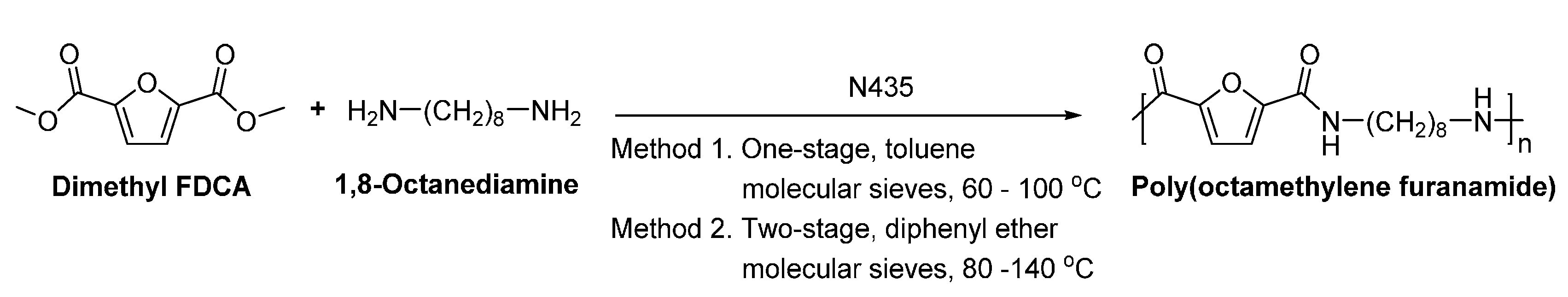
- Jiang, Y.; Maniar, D.; Woortman, A.J.J.; Alberda van Ekenstein, G.O.R.; Loos, K. Enzymatic polymerization of furan-2,5-dicarboxylic acid-based furanic-aliphatic polyamides as sustainable alternatives to polyphthalamides. Biomacromolecules 2015, 16, 3674–3685. [Google Scholar] [CrossRef] [PubMed]

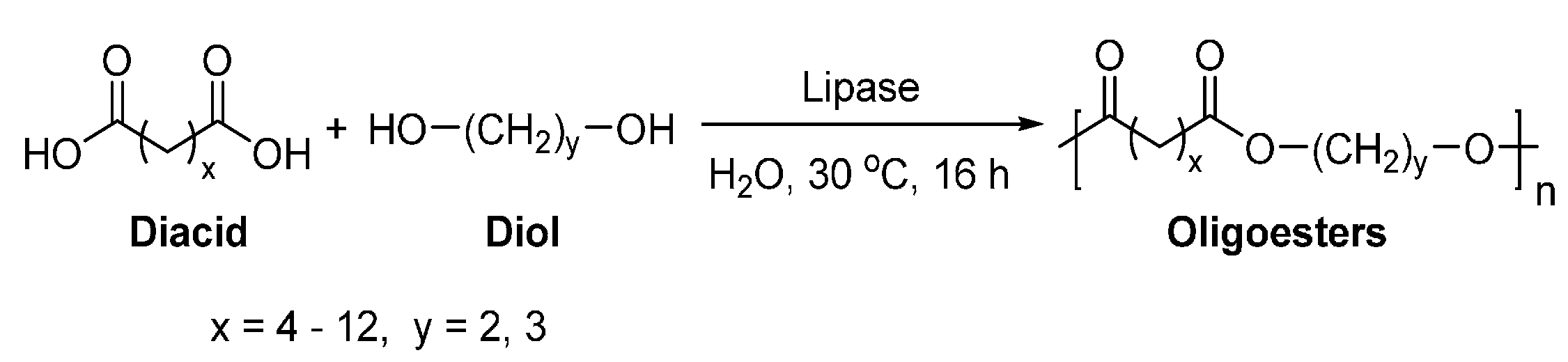
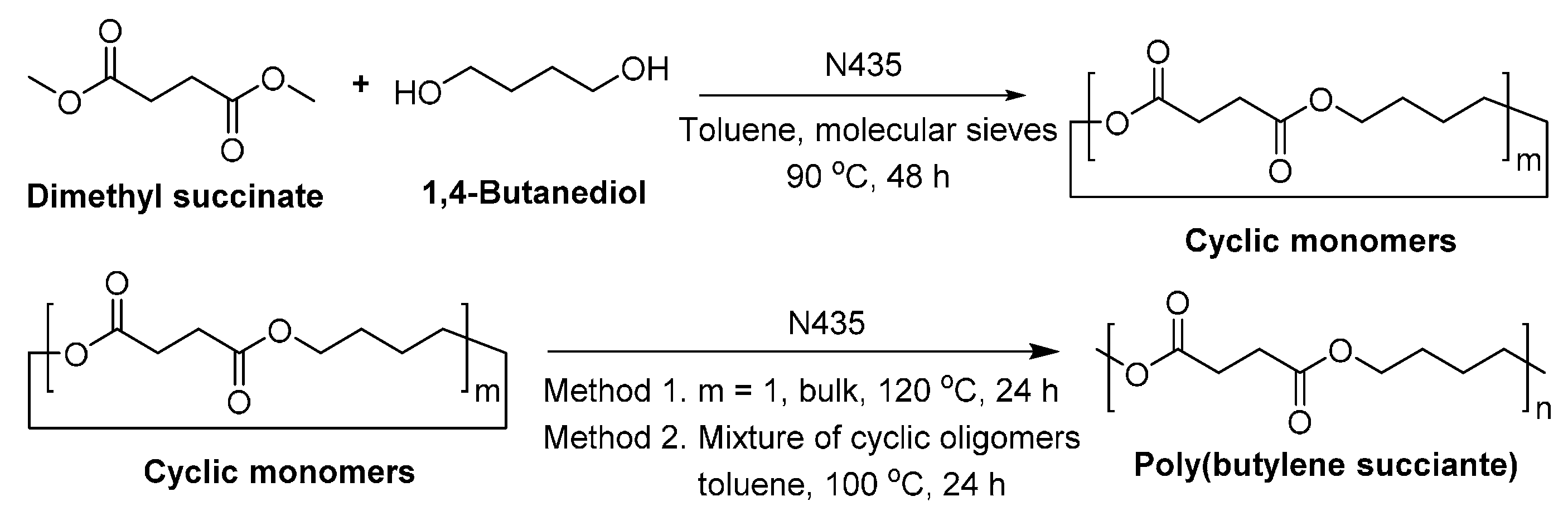


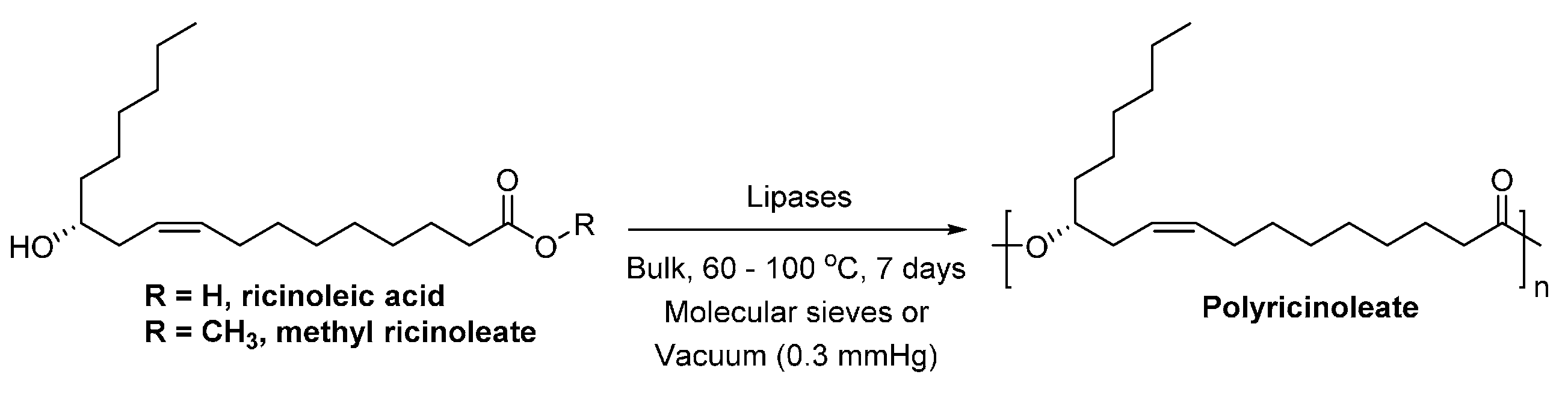
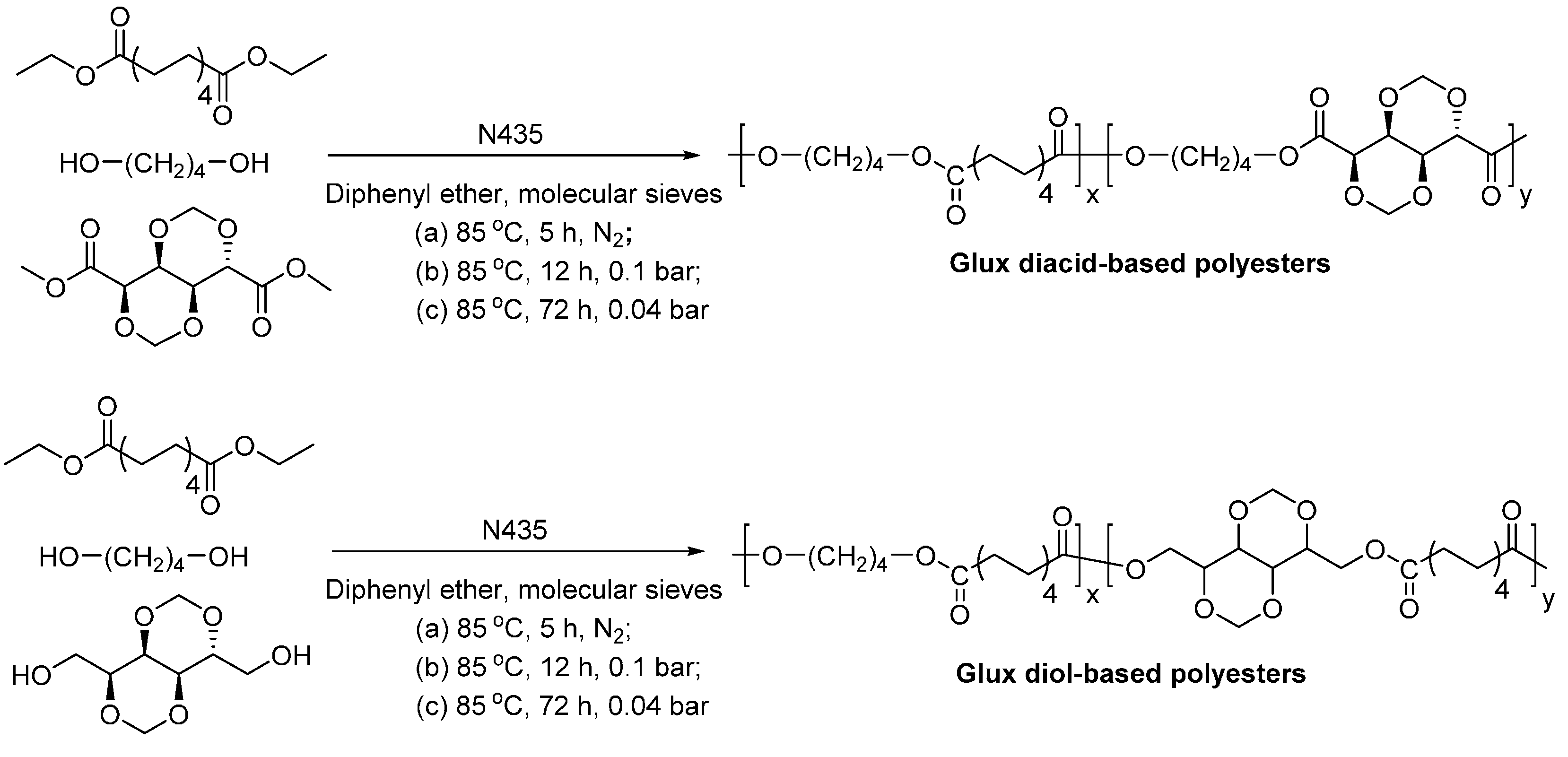
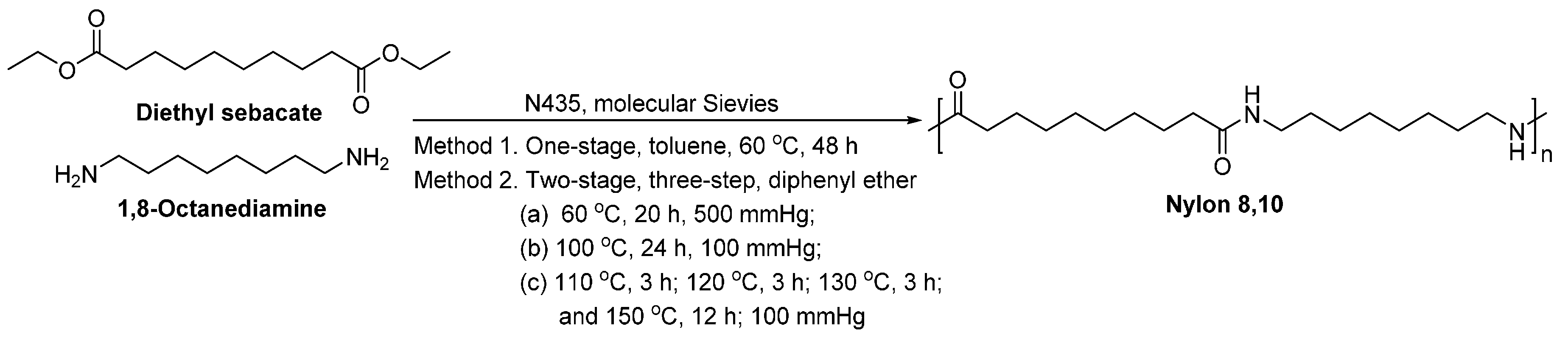

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
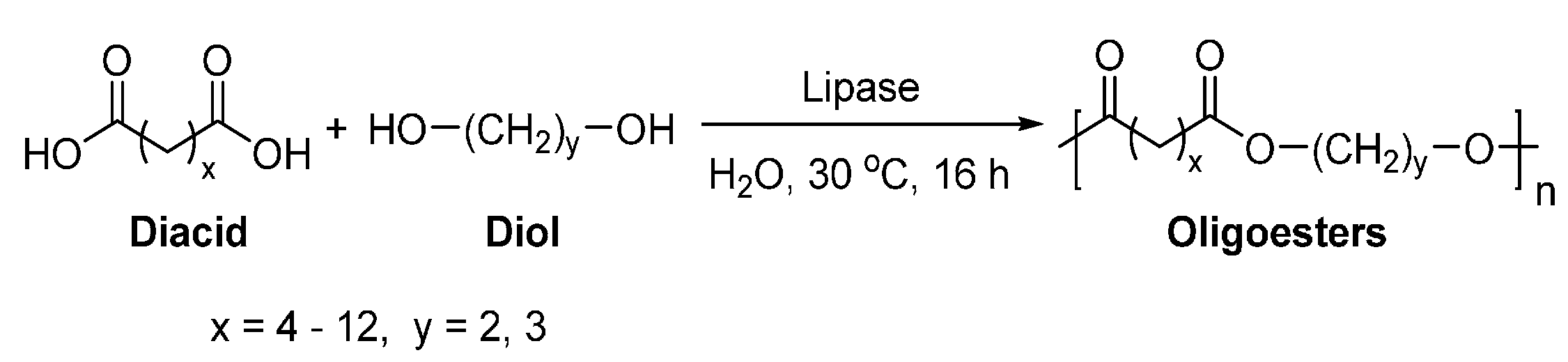{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
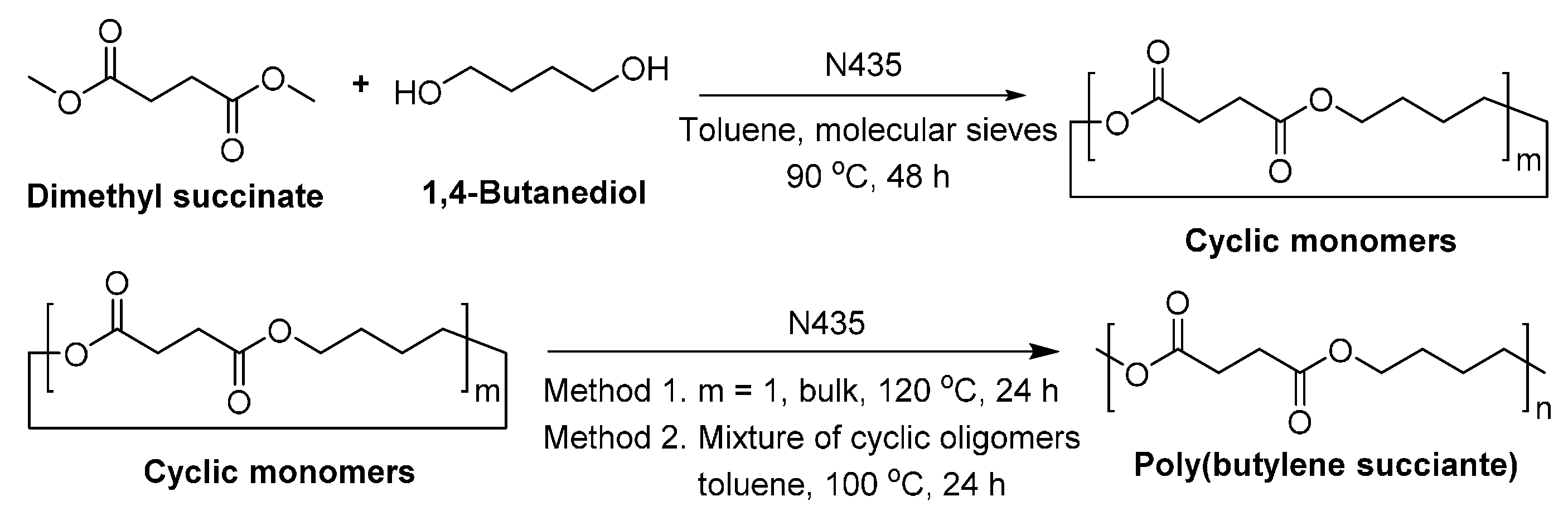{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
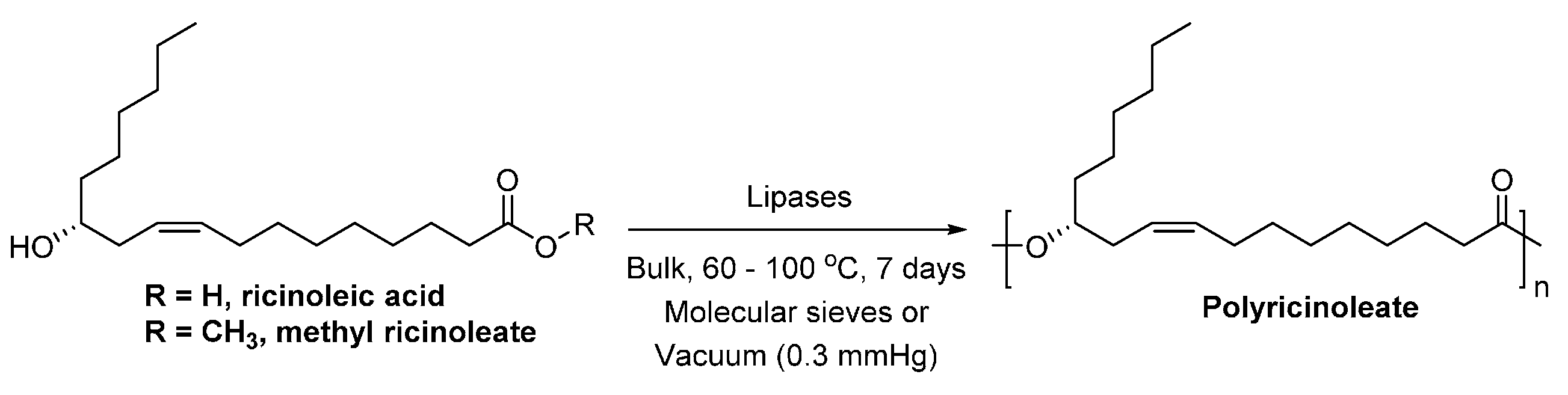{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
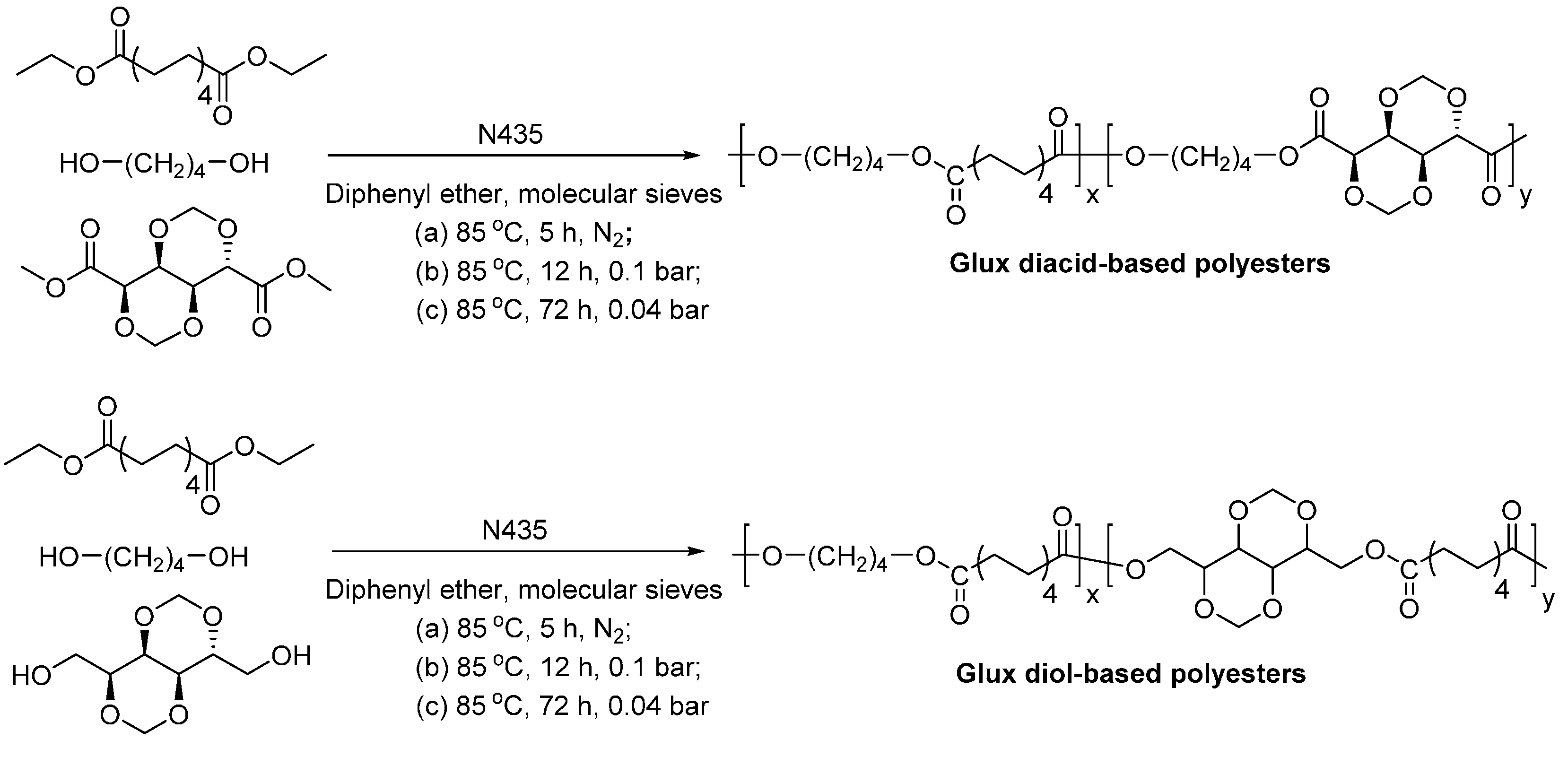{kind=link}
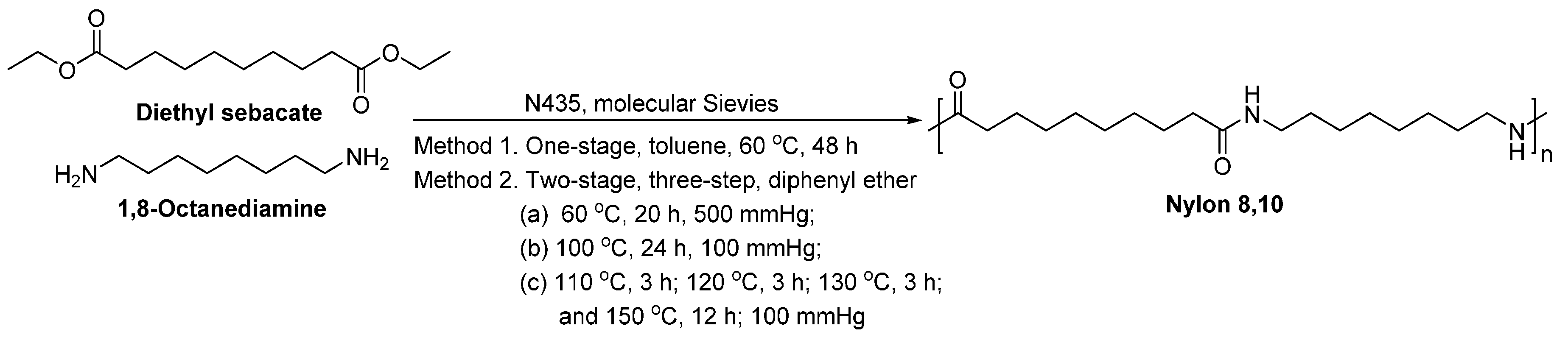{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biobased Polyester | Biosourcing (%) a | Manufacturer | Trademark |
|---|---|---|---|
| PLA | up to 100 | NatureWorks (Minnetonka, MN, USA) | Ingeo™, NatureWorks® |
| Synbra (Etten-Leur, The Netherlands) | BioFoam® | ||
| Zhejiang Hisun Biomaterials Biological Engineering (Taizhou, Zhejiang, China) | REVODE 100 and 200 series | ||
| Nantong Jiuding Biological Engineering (Rugao, Jiangsu, China) | - | ||
| Teijin (Chiyoda, Tokyo, Japan, Japan) | BIOFRONT™ | ||
| Mitsui Chemicals (Minato, Tokyo, Japan) | LACEA® | ||
| Futerro (Celles, Belgium) | Futerro® | ||
| Corbion Purac (Amsterdam, The Netherlands) | LX175, L175, L130, L105, D070 | ||
| PHAs | 100 | Metabolix (Cambridge, MA, USA) and ADM (Decatur, IL, USA) | Mirel™ |
| MHG (Bainbridge, GA, USA) | Nodax™ | ||
| Bio-on (San Giorgio di Piano, Bologna, Italy) | MINERV-PHA™ | ||
| Tianjin Green Biosciences (Tianjin, China) | GreenBio | ||
| Kaneka (Tokyo, Japan) | Kaneka PHBH | ||
| Tianan Biological Materials (Ningbo, Zhejiang, China) | ENMAT™ | ||
| PHB Industrial S/A (Serrana, Brazil) | BIOCYCLE® | ||
| PBS | 50 | PTT MCC Biochem ( Chatuchak, Bangkok, Thailand) | BioPBS™ |
| Showa Denko K.K. (Tokyo, Japan) | Bionolle™ | ||
| Mitsubishi Chemical (Chiyoda-ku, Tokyo, Japan) | GS Pla® | ||
| PEF | 100 | Avantium (Geleen, The Netherlands) | - |
| PET | up to 30 | Coca Cola (Atlanta, GA, USA) | PlantBottle™ |
| Toyota Tsusho Corporation (Nagoya, Aichi Prefecture, Japan) | GLOBIO® | ||
| PTT | 37 | DuPont (Wilmington, DE, USA) | Sorona® |
| up to 35 | Biomax® | ||
| PBAT | 30–70 | Novamont (Novara, Italy) | Origo-Bi™ |
| BASF (Ludwigshafen, Germany) | Ecoflex® FS | ||
| Co-polyester | 9–30 | SK Chemicals (Seongnam-si, Gyeonggi-do, Korea) | ECOZEN® |
| Co-polyester | - | DuPont (Wilmington, DE, USA) | Biomax® |
| Biobased Polyamide | Biosourcing (%) a | Manufacturer | Trademark |
|---|---|---|---|
| Nylon 4,10 (PA 4,10) | 100 | DSM (Heerlen, The Netherlands) | EcoPaXX® |
| Nylon 6,10 (PA 6,10) | 63 | BASF (Ludwigshafen, Germany) | Ultramid® S Balance |
| EMS-GRIVORY (Domat/Ems, Switzerland) | Grilamid® 2S | ||
| Evonik (Essen, Germany) | VESTAMID® Terra HS | ||
| Solvay (Rhodia) (Brussels, Belgium) | Technyl® eXten | ||
| DuPont (Wilmington, DE, USA) | Zytel® RS LC3030 | ||
| Arkema (Colombes, France) | Rilsan® S | ||
| Suzhou Hipro Polymers (Suzhou, Jiangsu, China) | Hiprolon® 70 | ||
| Nylon 10,10 (PA 10,10) | 100 | EMS-GRIVORY (Domat/Ems, Switzerland) | Grilamid® 1S |
| Evonik (Essen, Germany) | VESTAMID® Terra DS | ||
| DuPont (Wilmington, DE, USA) | Zytel® RS LC1000 | ||
| Arkema (Colombes, France) | Rilsan® T | ||
| Suzhou Hipro Polymers (Suzhou, Jiangsu, China) | Hiprolon® 200, Hiprolon® 211 | ||
| Nylon 10,12 (PA 10,12) | 45 | Evonik (Essen, Germany) | VESTAMID® Terra DD |
| Suzhou Hipro Polymers (Suzhou, Jiangsu, China) | Hiprolon® 400 | ||
| Nylon 11 (PA 11) | 100 | Arkema (Colombes, France) | Rilsan® PA11 |
| Suzhou Hipro Polymers (Suzhou, Jiangsu, China) | Hiprolon® 11 | ||
| PA 10,T | 50 | EMS-GRIVORY (Domat/Ems, Switzerland) | Grilamid® HT3 |
| Evonik (Essen, Germany) | VESTAMID® HTplus M3000 | ||
| Polyphthalamide (PPA) | >70 | Arkema (Colombes, France) | Rilsan® HT |
| Transparent polyamide | 54 | Arkema (Colombes, France) | Rilsan® Clear G830 Rnew |
| Co-polyamide | Tailored, up to 100 | Arkema (Colombes, France) | Platamid® Rnew |
| Polyamide | High Bio-Content | EMS-GRIVORY (Domat/Ems, Switzerland) | Grilamid® TR |
| Enzyme class | Reaction catalyzed | Typical enzymes | Typical polymers |
|---|---|---|---|
| EC 1. Oxidoreductases | Oxidation/Reduction AH2 + B A + BH2 | Peroxidase Laccase | Polyanilines Polyphenol Polystyrenes Poly(methyl methacrylate) |
| EC 2. Transferases | Group transfer A-X + B A + B-X | PHA synthase Hyaluronan synthase Phosphorylase | Polyesters Hyaluronan Amylose |
| EC 3. Hydrolases | Hydrolysis by H2O A-B + H2O AH + BOH | Lipase Cellulase Hyaluronidase Papain | Polyesters Polyamides Cellulose (Oligo)peptides Glycosaminoglycan |
| EC 6. Ligases | Bond formation requiring triphosphate 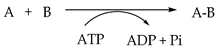 | Cyanophycin synthetase | Cyanophycin |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; Loos, K. Enzymatic Synthesis of Biobased Polyesters and Polyamides. Polymers 2016, 8, 243. https://doi.org/10.3390/polym8070243
Jiang Y, Loos K. Enzymatic Synthesis of Biobased Polyesters and Polyamides. Polymers. 2016; 8(7):243. https://doi.org/10.3390/polym8070243
Chicago/Turabian StyleJiang, Yi, and Katja Loos. 2016. "Enzymatic Synthesis of Biobased Polyesters and Polyamides" Polymers 8, no. 7: 243. https://doi.org/10.3390/polym8070243
APA StyleJiang, Y., & Loos, K. (2016). Enzymatic Synthesis of Biobased Polyesters and Polyamides. Polymers, 8(7), 243. https://doi.org/10.3390/polym8070243