
Highly Active Copolymerization of Ethylene and N-Acetyl-O-(ω-Alkenyl)-l-Tyrosine Ethyl Esters Catalyzed by Titanium Complex

 ,
,
Abstract
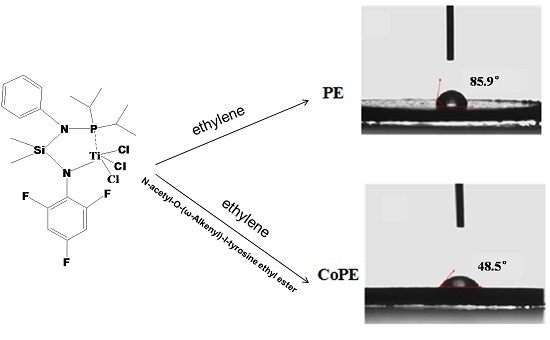
:
1. Introduction
2. Experimental
2.1. General Remarks
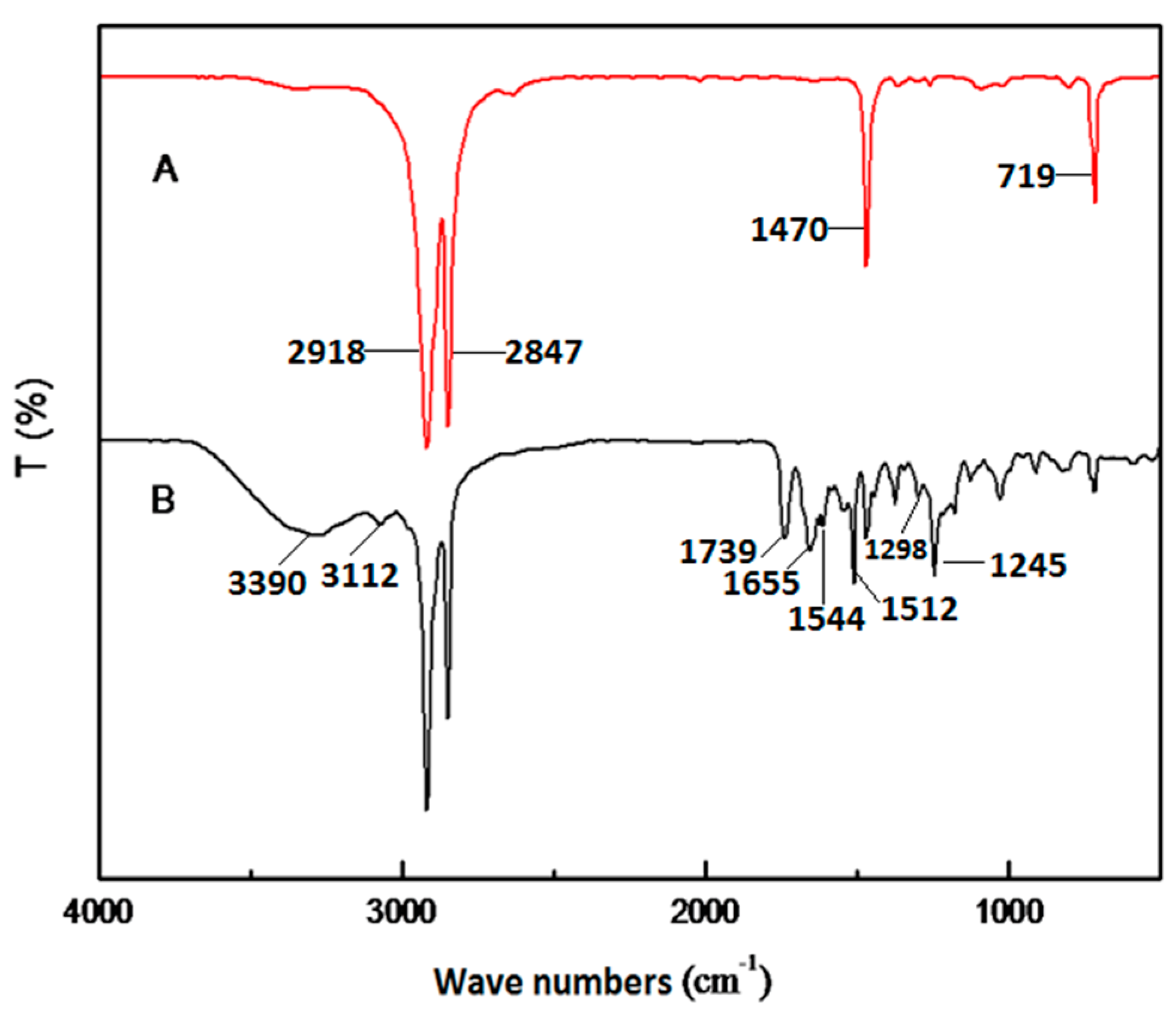
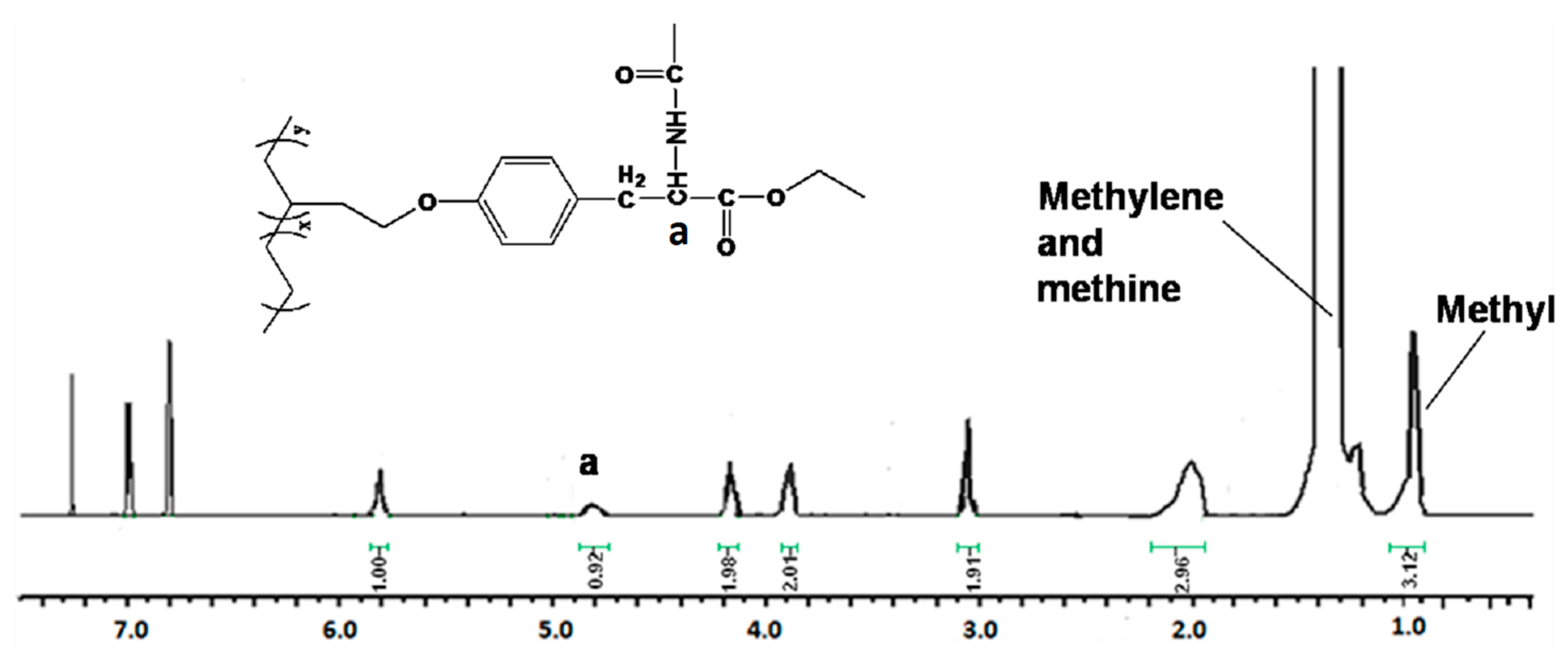
2.2. Characterization
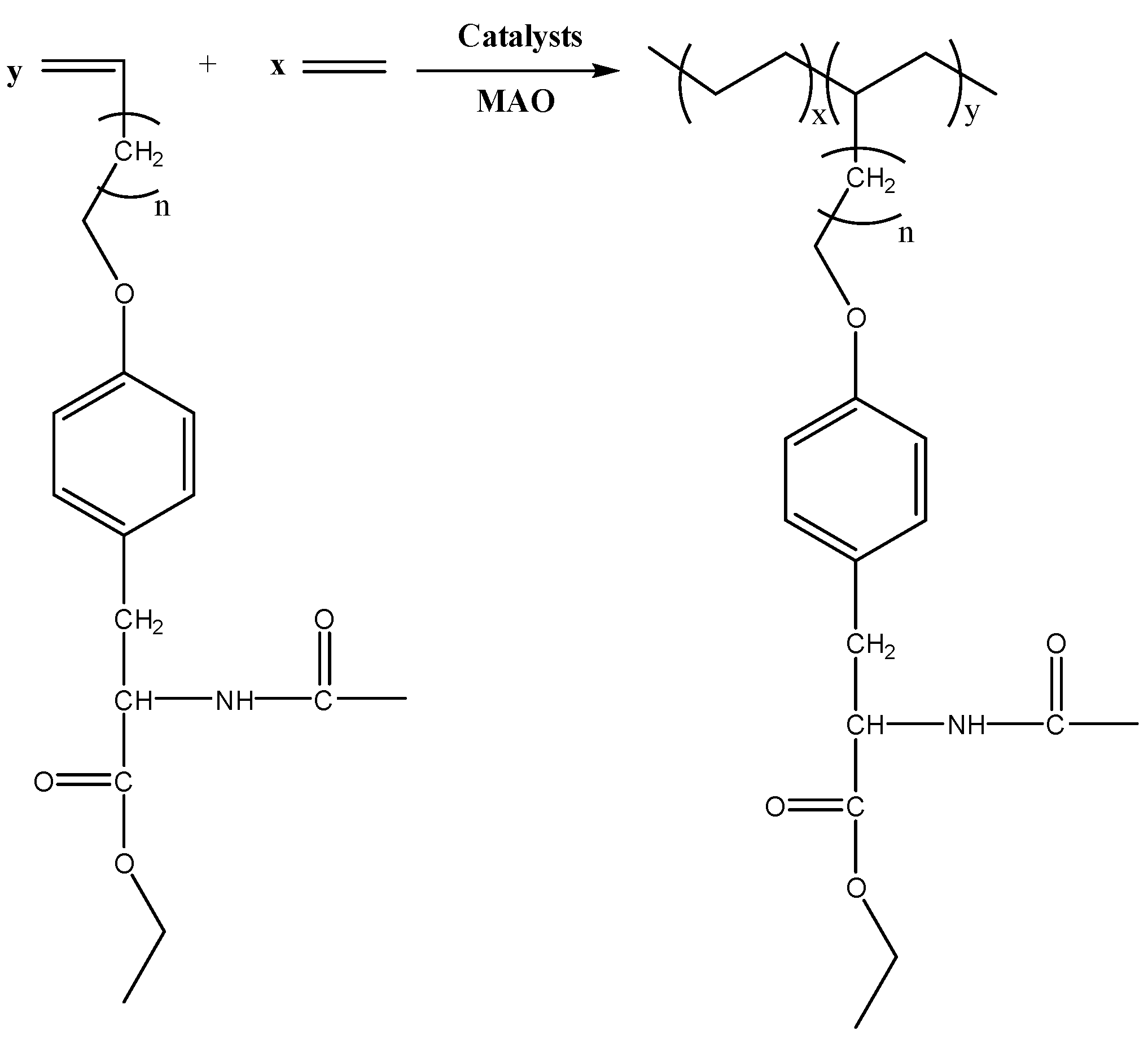
2.3. Synthesis of N-Acetyl-O-(ω-Alkenyl)-l-Tyrosine Ethyl Ester
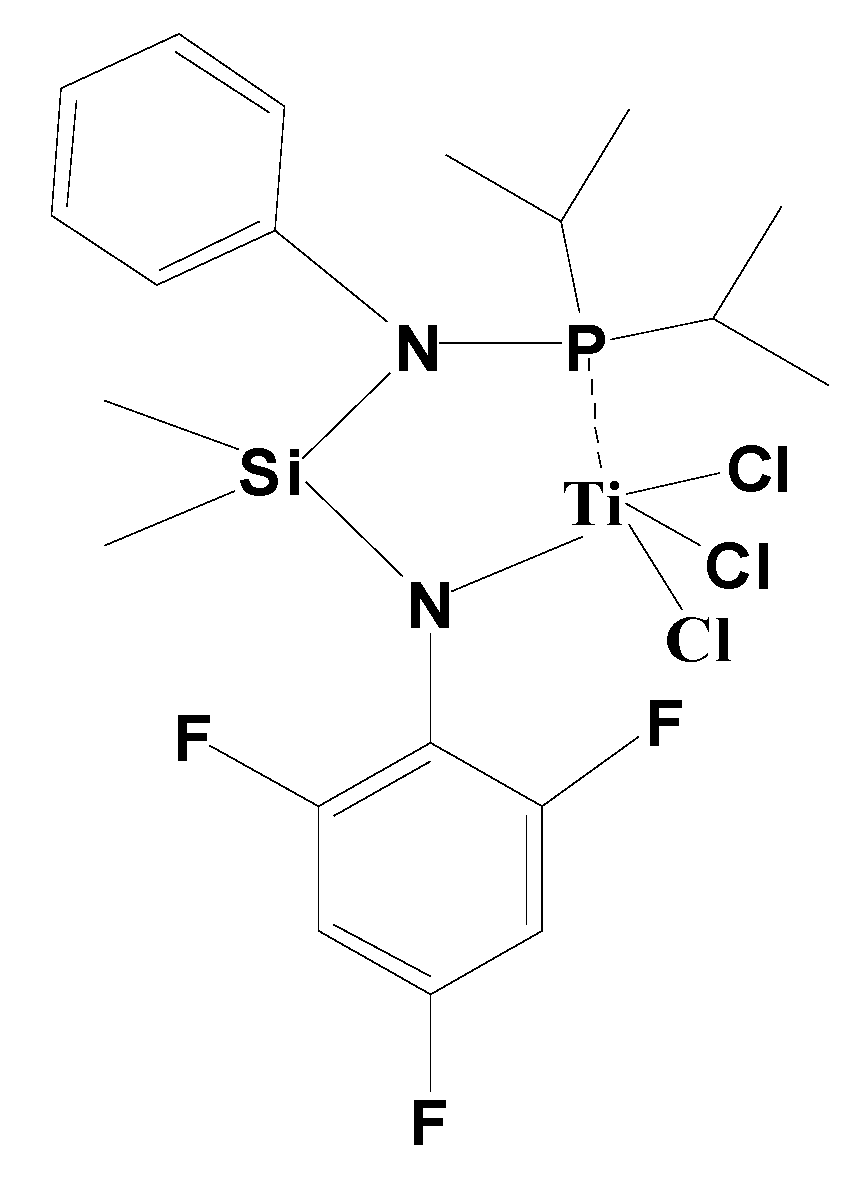
2.4. Synthesis of Catalyst Precursors
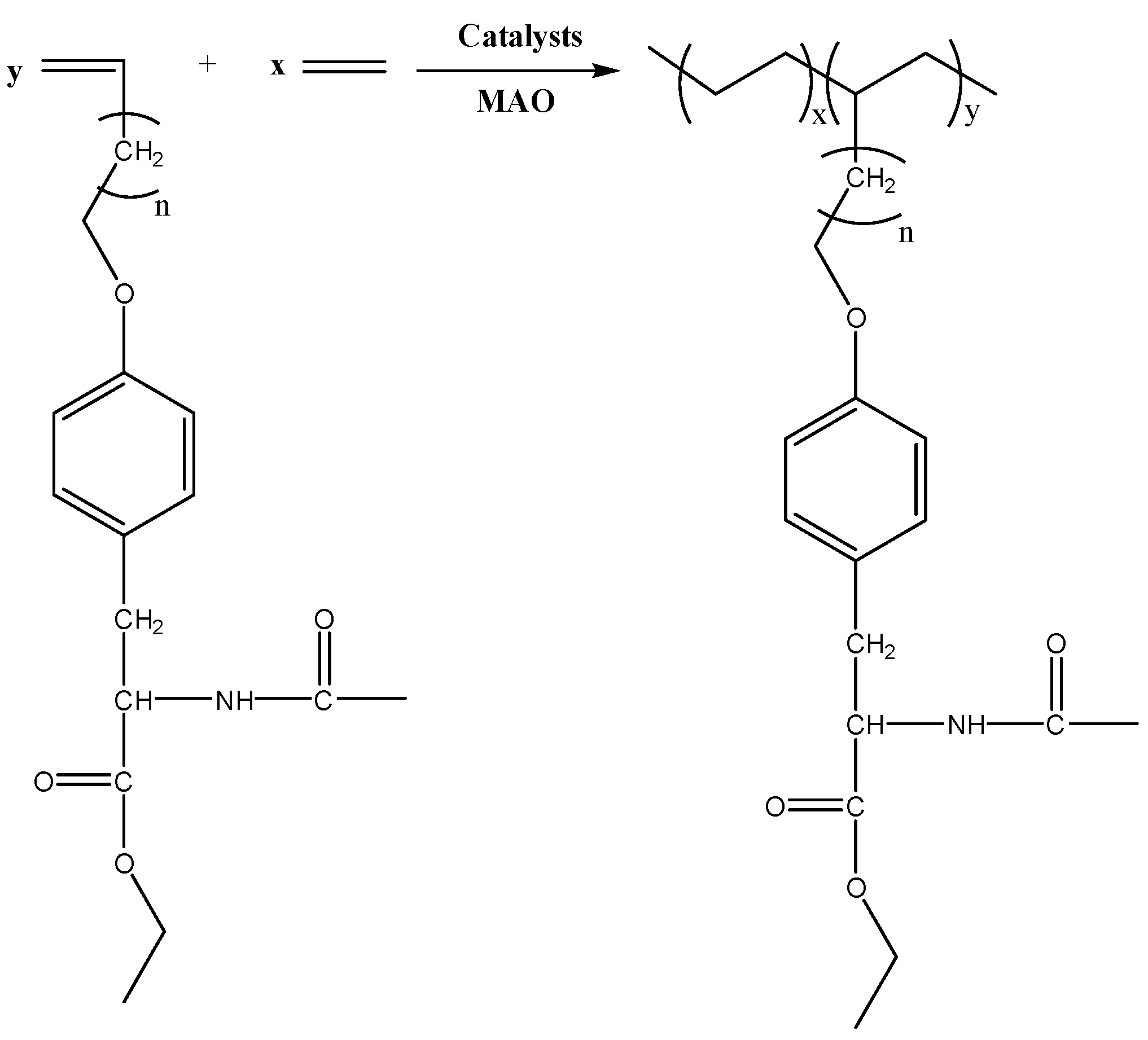
2.5. Polymerization Procedure
3. Results and Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Amin, S.B.; Marks, T.J. Alkenylsilane Effects on organotitanium-catalyzed ethylene polymerization. toward simultaneous polyolefin branch and functional group introduction. J. Am. Chem. Soc. 2006, 128, 4506–4507. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.W.; Zhang, M.; Sun, W.H. Imino-indolate half-titanocene chlorides: Synthesis and their ethylene (co-)polymerization. J. Polym. Sci. Part A 2009, 47, 357–372. [Google Scholar]
- Zhang, X.F.; Chen, S.T.; Li, H.Y.; Zhang, Z.C.; Lu, Y.Y.; Wu, C.H.; Hu, Y.L. Copolymerizations of ethylene and polar comonomers with bis(phenoxyketimine) group IV complexes: Effects of the central metal properties. J. Polym. Sci. Part A 2007, 45, 59–68. [Google Scholar]
- Rodriguez, B.A.; Delferro, M.; Marks, T.J. Bimetallic effects for enhanced polar comonomer enchainment selectivity in catalytic ethylene polymerization. J. Am. Chem. Soc. 2009, 131, 5902–5919. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.L.; Dai, S.Y.; Chen, C.L. Ethylene polymerization and copolymerization with polar monomers by cationic phosphine phosphonic amide palladium complexes. ACS Catal. 2015, 5, 5932–5937. [Google Scholar] [CrossRef]
- Popeney, C.S.; Camacho, D.H.; Guan, Z.B. Efficient incorporation of polar comonomers in copolymerizations with ethylene using a cyclophane-based Pd(II) α-diimine catalyst. J. Am. Chem. Soc. 2007, 129, 10062–10063. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Zhang, K.J.; Ye, Z.B. One-pot synthesis of hyperbranched polyethylenes tethered with polymerizable methacryloyl groups via selective ethylene copolymerization with heterobifunctional comonomers by chain walking Pd–diimine catalysis. Macromolecules 2008, 41, 2290–2293. [Google Scholar] [CrossRef]
- Li, X.F.; Li, Y.G.; Li, Y.S. Copolymerization of ethylene with methyl methacrylate with neutral nickel(II) complexes bearing β-ketoiminato chelate ligands. Organometallics 2005, 24, 2502–2510. [Google Scholar] [CrossRef]
- Drent, E.; Dijk, R.V.; Ginkel, R.V.; Pugh, R.I. Palladium catalysed copolymerisation of ethene with alkylacrylates: Polar comonomer built into the linear polymer chain. Chem. Commun. 2002, 7, 744–745. [Google Scholar]
- Kochi, T.; Noda, S.; Nozaki, K. Formation of linear copolymers of ethylene and acrylonitrile catalyzed by phosphine sulfonate palladium complexes. J. Am. Chem. Soc. 2007, 129, 8948–8949. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kanazawa, M.; Munakata, K.; Kuroda, J.; Okumura, Y.; Nozaki, K. Coordination—insertion copolymerization of allyl monomers with ethylene. J. Am. Chem. Soc. 2011, 133, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Shen, Z.L.; Jordan, R.F. Copolymerization of ethylene and vinyl fluoride by (phosphine-sulfonate)Pd(Me)(py) catalysts. J. Am. Chem. Soc. 2007, 129, 15450–15451. [Google Scholar] [CrossRef] [PubMed]
- Skupov, K.M.; Piche, L.; Claverie, J.P. Linear polyethylene with tunable surface properties by catalytic copolymerization of ethylene with N-vinyl-2-pyrrolidinone and N-isopropylacrylamide. Macromolecules 2008, 41, 2309–2310. [Google Scholar] [CrossRef]
- Ma, L.F.; Sheng, Y.P.; Huang, Q.G.; Zhao, Y.F.; Deng, K.X.; Li, J.L.; Yang, W.T. A kind of novel nonmetallocene catalysts for ethylene polymerization. J. Polym. Sci. Part A 2008, 46, 33–37. [Google Scholar]
- Ma, L.F.; Wang, H.L.; Yi, J.J.; Huang, Q.G.; Gao, K.J.; Yang, W.T. Copolymerization of ethylene with 1-hexene promoted by novel multi-chelated non-metallocene complexes with imine bridged imidazole ligand. J. Polym. Sci. Part A 2010, 48, 417–424. [Google Scholar]
- Zhang, X.L.; Liu, Z.; Yi, J.J.; Huang, H.B.; Dou, X.L.; Zhen, H.P.; Huang, Q.G.; Gao, K.J.; Zhang, M.G.; Yang, W.T. The investigation of novel non-metallocene catalysts with phenoxy-imine ligands for ethylene (co-)polymerization. Polym. Int. 2013, 62, 419–426. [Google Scholar] [CrossRef]
- Zhang, X.L.; Liu, Z.; Yi, J.J.; Li, F.J.; Huang, H.B.; Liu, W.; Zhen, H.P.; Huang, Q.G.; Gao, K.J.; Yang, W.T. Copolymerization of ethylene with acrylonitrile promoted by novel nonmetallocene catalysts with phenoxy-imine ligands. J. Polym. Sci. Part A 2012, 50, 2068–2074. [Google Scholar] [CrossRef]
- Grodzinski, J.J. Biomedical application of functional polymers. React. Funct. Polym. 1999, 39, 99–138. [Google Scholar] [CrossRef]
- Maynard, H.D.; Okada, S.Y.; Grubbs, R.H. Synthesis of norbornenyl polymers with bioactive oligopeptides by ring-opening metathesis polymerization. Macromolecules 2000, 33, 6239–6248. [Google Scholar] [CrossRef]
- Maynard, H.D.; Okada, S.Y.; Grubbs, R.H. Inhibition of cell adhesion to fibronectin by oligopeptide-substituted polynorbornenes. J. Am. Chem. Soc. 2001, 123, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Gestwicki, J.E.; Cairo, C.W.; Strong, L.E.; Oetjen, K.A.; Kiessling, L.L. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J. Am.Chem. Soc. 2002, 124, 14922–14933. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.E.; Wagener, K.B. ADMET synthesis of polyolefins targeted for biological applications. Macromolecules 2004, 37, 1180–1189. [Google Scholar] [CrossRef]
- Ayres, L.; Vos, M.R.J.; Adams, P.J.H.; Shklyarevskiy, O.I.; van Hest, J.C.M. Elastin-based side-chain polymers synthesized by ATRP. Macromolecules 2003, 36, 5967–5973. [Google Scholar] [CrossRef]
- Malinova, V.; Rieger, B. Synthesis of functional poly (1,4-ketone) s bearing bioactive moieties by Pd-catalyzed insertion polymerization. Biomacromolecules 2006, 7, 2931–2936. [Google Scholar] [CrossRef] [PubMed]
- Moatsou, D.; Li, J.; Ranji, A.; Pitto-Barry, A.; Ntai, I.; Jewett, M.C.; O’Reilly, R.K. Self-assembly of temperature-responsive protein–polymer bioconjugates. Bioconj. Chem. 2015, 26, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shi, X.H.; Chen, Y.; Li, H.M.; Zhang, R.C.; Yi, J.J.; Wang, J.; Huang, Q.G.; Yang, W.T. Copolymerization of ethylene and vinyl amino acidic ester catalyzed by early transition metal complexes. Catalysts 2015, 5, 1831–1845. [Google Scholar] [CrossRef]
- Wang, J.; Nan, F.; Guo, J.P.; Wang, J.; Shi, X.H.; Huang, H.B.; Huang, Q.G.; Yang, W.T. Copolymers of ethylene and vinyl amino acidic ester with high molecular weight prepared by non-metallocene catalysts. Catal. Lett. 2016. [Google Scholar] [CrossRef]
- Fan, X.Y. Application of fourier transform infrared spectroscopy in life science. Life Sci. Res. 2003, 7, 83–87. [Google Scholar]
- Senak, L.; Ju, Z.M.; NOY, N.; Callender, R.; Manor, D. The Interactions between cellular retinol-binding protein (CRBP-I) and retinal: A vibrational spectroscopic study. Biospectroscopy 1997, 3, 131–142. [Google Scholar] [CrossRef]
- Randall, J.C. A review of high resolution liquid 13carbon nuclear magnetic resonance characterizations of ethylen-based polymers. J. Macromol. Sci. Part C 1989, 29, 201–317. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
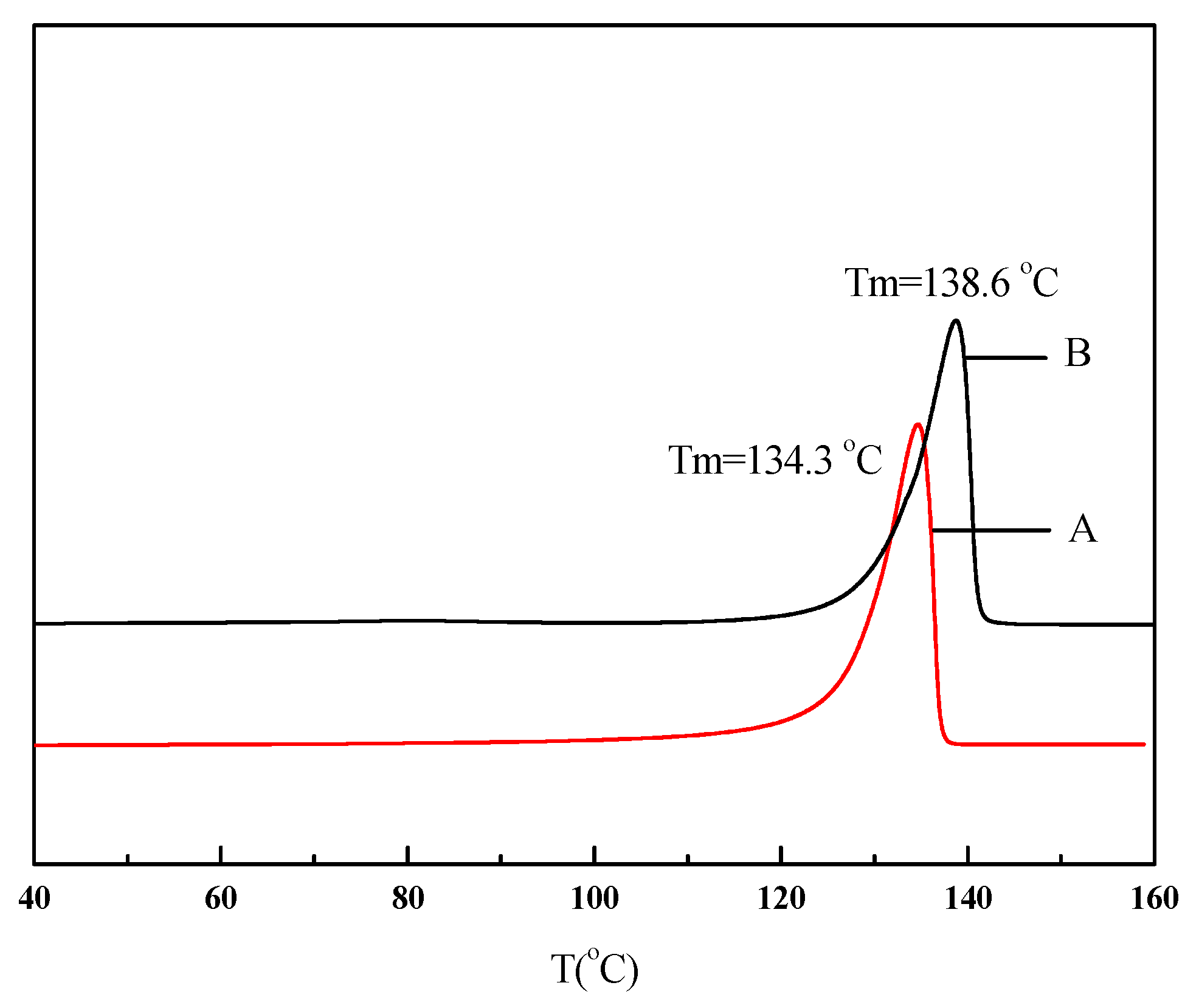
| Run | Comonomer | Comon a (g/L) | A b (×104) | Tm c (°C) | N-Cont d (mol%) | Mw e (×105) | Mw/Mn e |
|---|---|---|---|---|---|---|---|
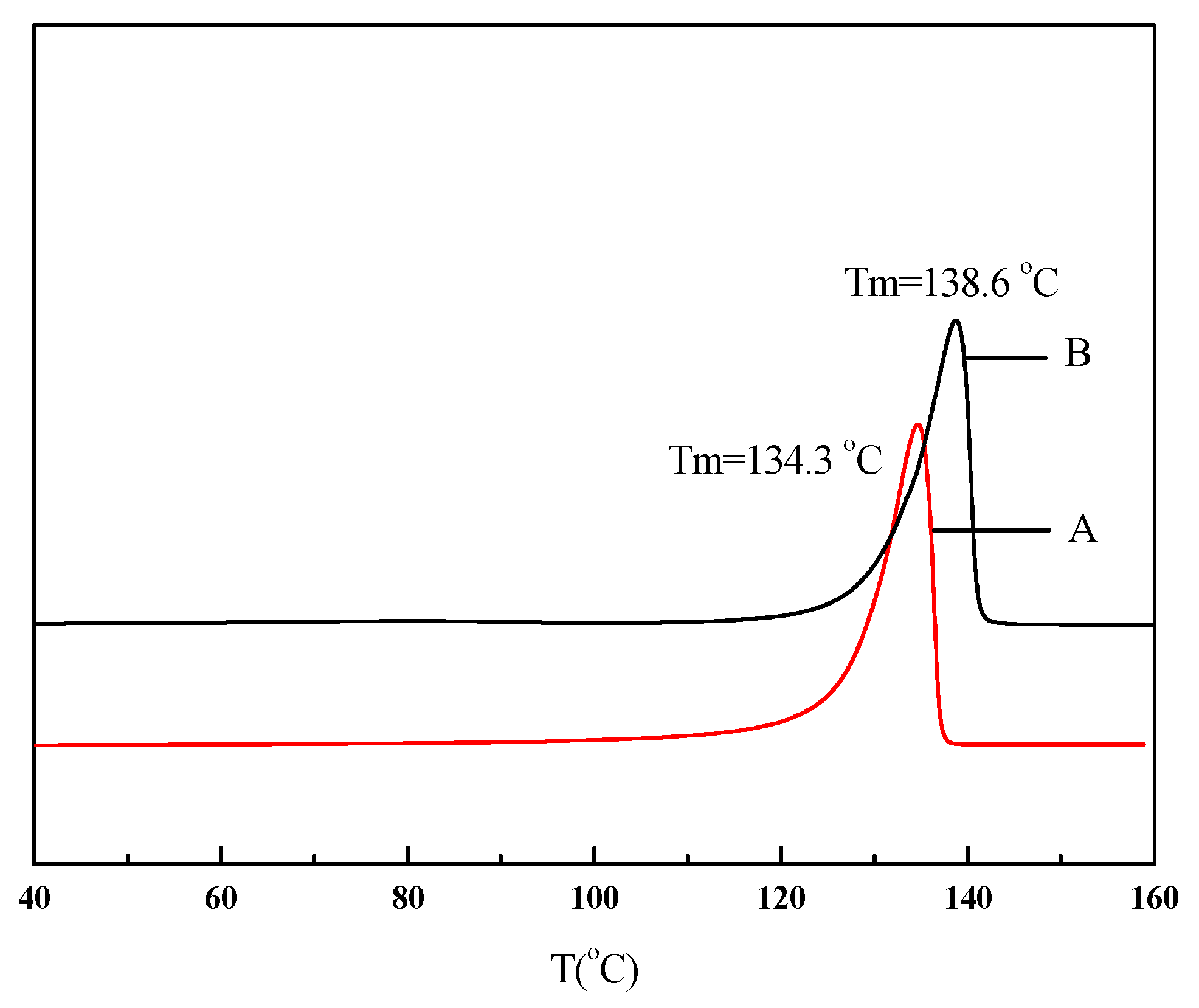
| 1 | None | 0 | 170 | 134.3 | 0 | 8.27 | 1.77 |
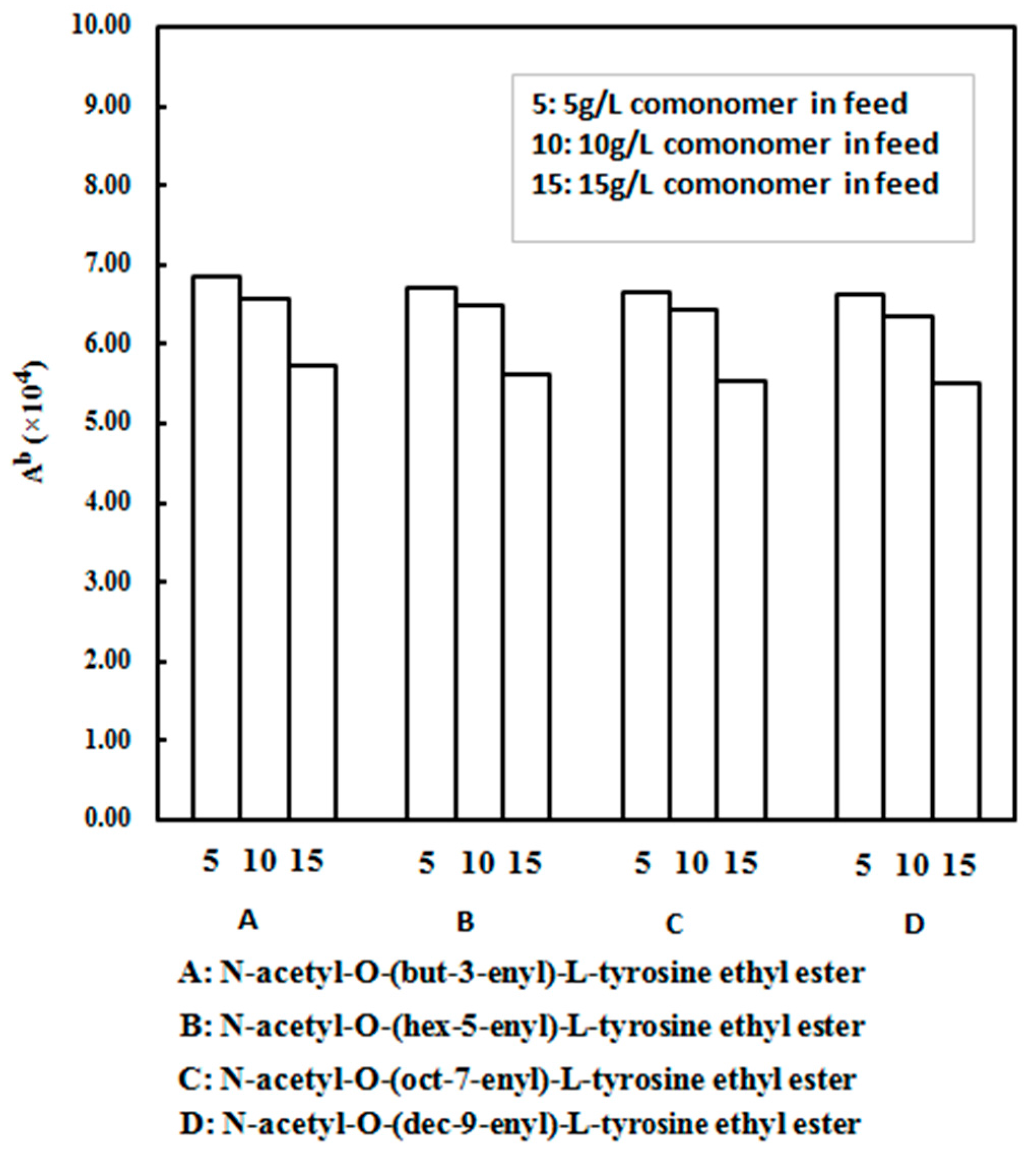
| 2 | N-acetyl-O-(but-3-enyl)-l-tyrosine ethyl ester | 5 | 6.86 | 137.9 | 1.16 | 2.75 | 2.88 |
| 3 | N-acetyl-O-(but-3-enyl)-l-tyrosine ethyl ester | 10 | 6.57 | 138.4 | 2.24 | 2.76 | 2.87 |
| 4 | N-acetyl-O-(but-3-enyl)-l-tyrosine ethyl ester | 15 | 5.73 | 139.0 | 2.65 | 2.85 | 2.91 |
| 5 | N-acetyl-O-(hex-5-enyl)-l-tyrosine ethyl ester | 5 | 6.71 | 138.0 | 1.13 | 2.72 | 2.85 |
| 6 | N-acetyl-O-(hex-5-enyl)-l-tyrosine ethyl ester | 10 | 6.49 | 138.6 | 2.19 | 2.74 | 2.86 |
| 7 | N-acetyl-O-(hex-5-enyl)-l-tyrosine ethyl ester | 15 | 5.61 | 139.3 | 2.60 | 2.87 | 2.89 |
| 8 | N-acetyl-O-(oct-7-enyl)-l-tyrosine ethyl ester | 5 | 6.67 | 137.8 | 1.12 | 2.76 | 2.87 |
| 9 | N-acetyl-O-(oct-7-enyl)-l-tyrosine ethyl ester | 10 | 6.43 | 138.5 | 2.21 | 2.79 | 2.85 |
| 10 | N-acetyl-O-(oct-7-enyl)-l-tyrosine ethyl ester | 15 | 5.53 | 139.2 | 2.59 | 2.89 | 2.90 |
| 11 | N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester | 5 | 6.63 | 137.7 | 1.11 | 2.73 | 2.86 |
| 12 | N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester | 10 | 6.34 | 138.4 | 2.28 | 2.70 | 2.79 |
| 13 | N-acetyl-O-(dec-9-enyl)-l-tyrosine ethyl ester | 15 | 5.50 | 139.1 | 2.57 | 2.82 | 2.88 |
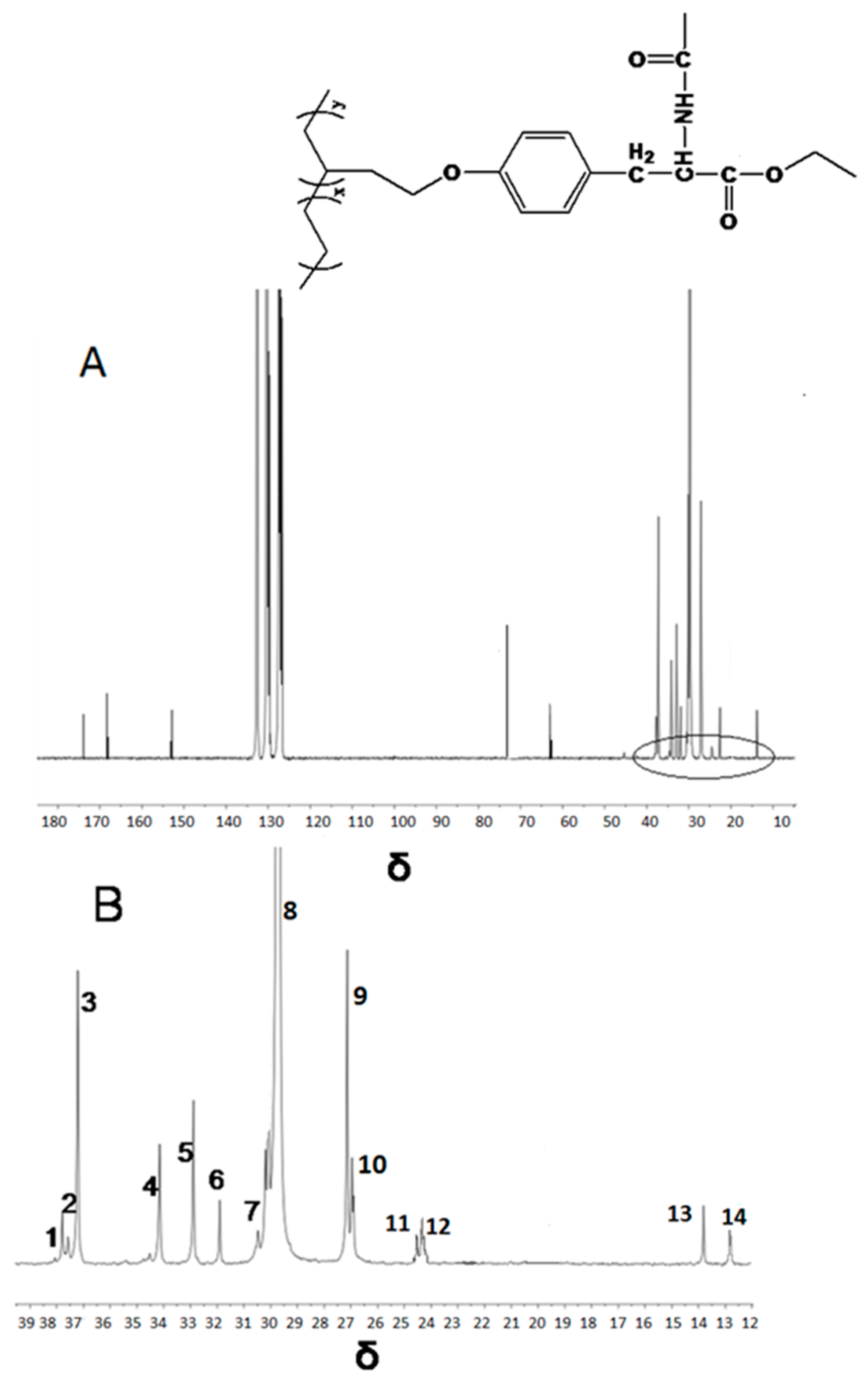
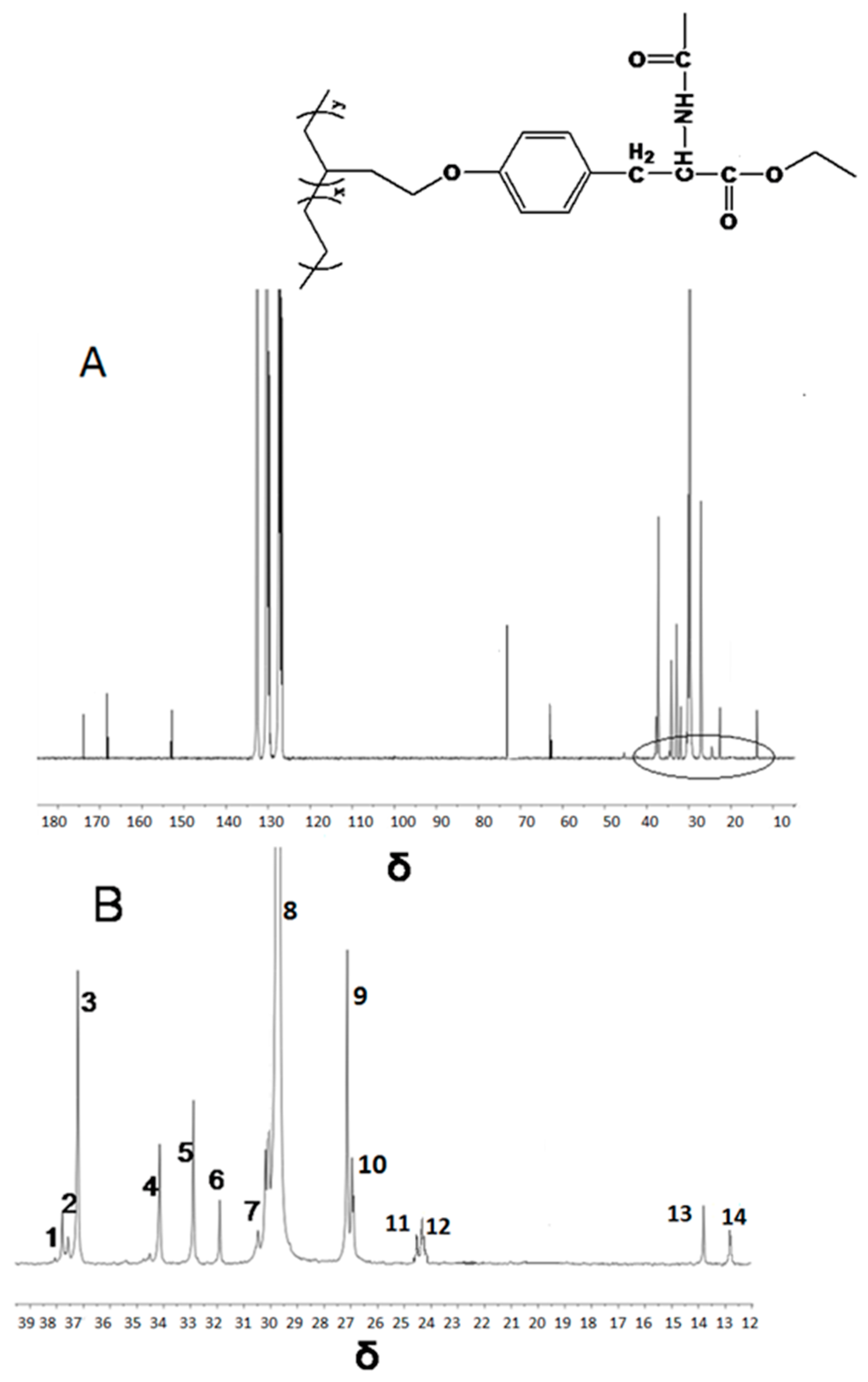
| Peak NO. | Carbon Assignment | Sequence Assignment | Chemical Shift | |
|---|---|---|---|---|
| Calculated | Found | |||
| 1 | αα | EBBE | 38.03 | 37.91 |
| 2 | Methine | EBBE | 37.69 | 37.66 |
| 3 | Methine | EBB+BBE | 37.01 | 37.15 |
| 4 | αδ+ | BBEE+EEBB | 34.01 | 34.12 |
| 5 | αδ+ | EBEE+EEBE | 32.91 | 32.87 |
| 6 | γγ | BEEB | 30.92 | 31.87 |
| 7 | γδ+ | BEEE+EEEB | 30.47 | 30.50 |
| 8 | δ+δ+ | (EEE)n | 29.98 | 29.78 |
| 9 | βδ+ | ETEE+EETE | 27.27 | 26.90 |
| 10 | 2B2+ | EBB+BBE | 26.80 | 26.86 |
| 11 | ββ | EBEBE | 24.54 | 24.50 |
| 12 | ββ | BBEBE+BBEBB | 24.39 | 24.32 |
| 13 | Methyl | EBE | 13. 80 | 13.86 |
| 14 | Methyl | BBE+EBB | 12.41 | 12.60 |
| Content of Sequences | Mole Fractions | Reactivity Ratio | |||||||
|---|---|---|---|---|---|---|---|---|---|
| [EBE] | [EBB] | [BBB] | [BEB] | [EEB] | [EEE] | [E] | [B] | rE | rB |
| 2.163 | 0.008 | 0 | 0.072 | 4.326 | 93.431 | 0.9778 | 0.0222 | 149.31 | 0.083 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Li, H.; Zhang, R.; Shi, X.; Yi, J.; Wang, J.; Huang, Q.; Yang, W. Highly Active Copolymerization of Ethylene and N-Acetyl-O-(ω-Alkenyl)-l-Tyrosine Ethyl Esters Catalyzed by Titanium Complex. Polymers 2016, 8, 64. https://doi.org/10.3390/polym8030064
Wang J, Li H, Zhang R, Shi X, Yi J, Wang J, Huang Q, Yang W. Highly Active Copolymerization of Ethylene and N-Acetyl-O-(ω-Alkenyl)-l-Tyrosine Ethyl Esters Catalyzed by Titanium Complex. Polymers. 2016; 8(3):64. https://doi.org/10.3390/polym8030064
Chicago/Turabian StyleWang, Jing, Hongming Li, Runcong Zhang, Xianghui Shi, Jianjun Yi, Jian Wang, Qigu Huang, and Wantai Yang. 2016. "Highly Active Copolymerization of Ethylene and N-Acetyl-O-(ω-Alkenyl)-l-Tyrosine Ethyl Esters Catalyzed by Titanium Complex" Polymers 8, no. 3: 64. https://doi.org/10.3390/polym8030064
APA StyleWang, J., Li, H., Zhang, R., Shi, X., Yi, J., Wang, J., Huang, Q., & Yang, W. (2016). Highly Active Copolymerization of Ethylene and N-Acetyl-O-(ω-Alkenyl)-l-Tyrosine Ethyl Esters Catalyzed by Titanium Complex. Polymers, 8(3), 64. https://doi.org/10.3390/polym8030064