
Applications of Solid-State NMR Spectroscopy for the Study of Lipid Membranes with Polyphilic Guest (Macro)Molecules


Abstract
:
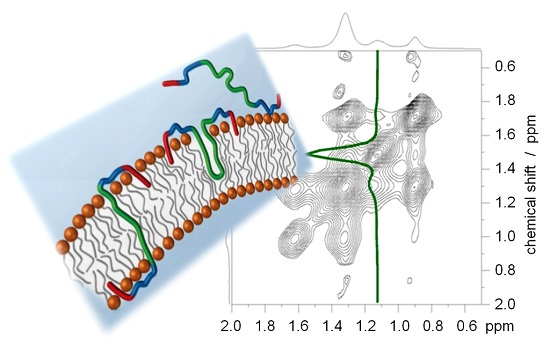{kind=link}
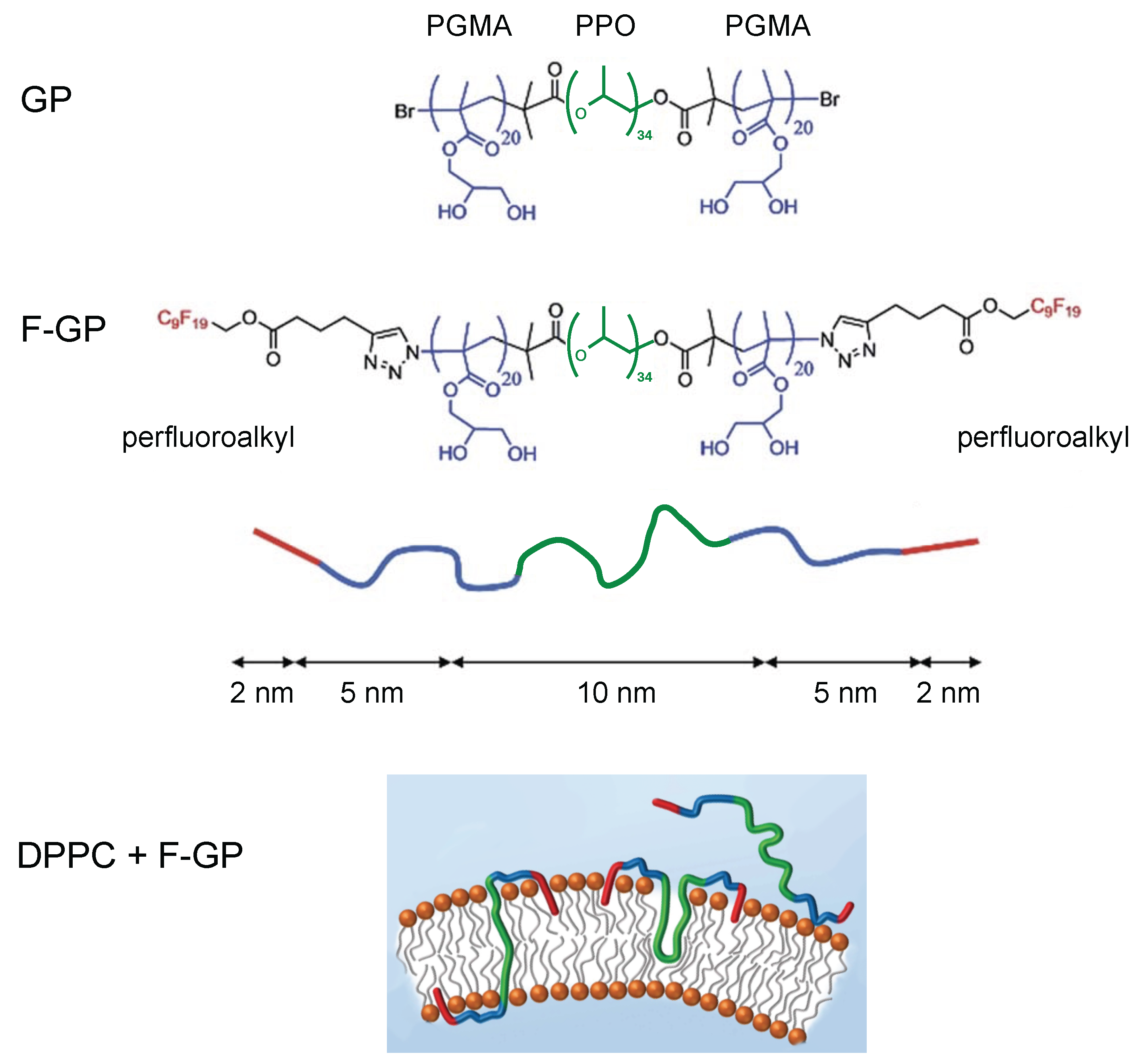{kind=link}
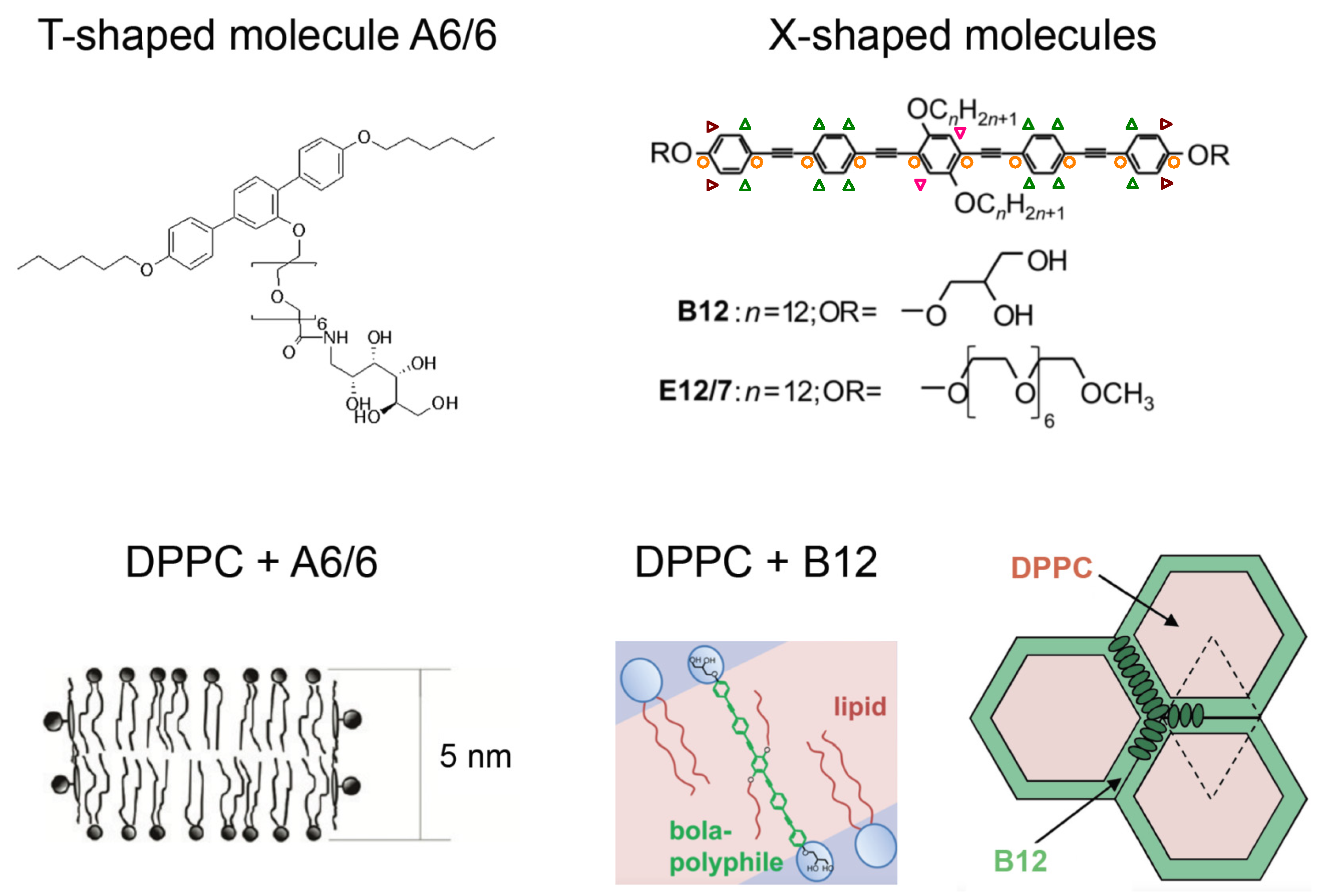{kind=link}
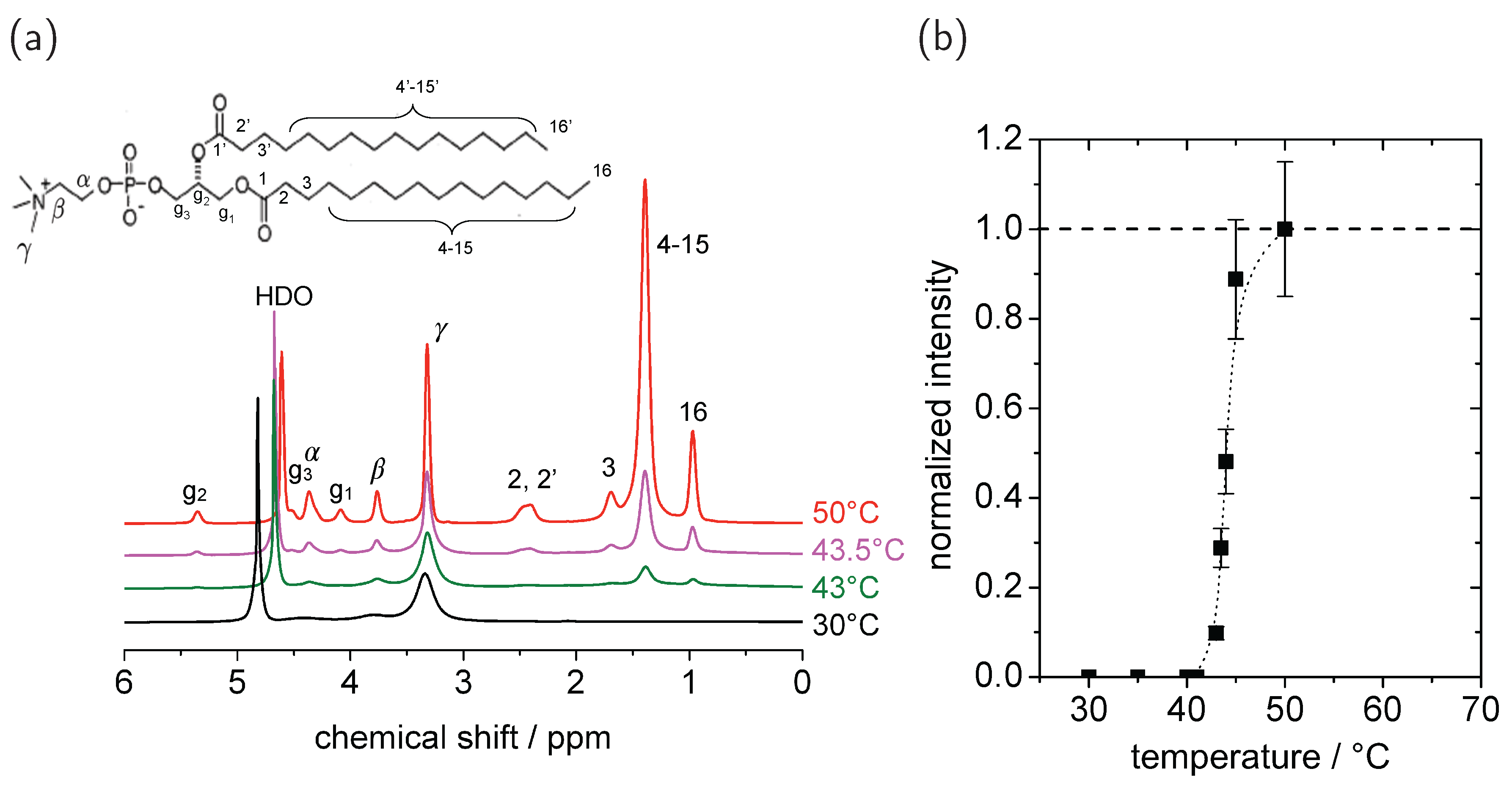{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. NMR Basics and Applications to Lipid Systems
2.1. Interactions in SSNMR
2.2. High-Resolution SSNMR
2.3. Overview of SSNMR Applications to Lipid Membranes
2.4. Experimental Aspects
3. High-Resolution SSNMR Applications to Lipid Membranes
3.1. Quantitative 1H MAS Spectra
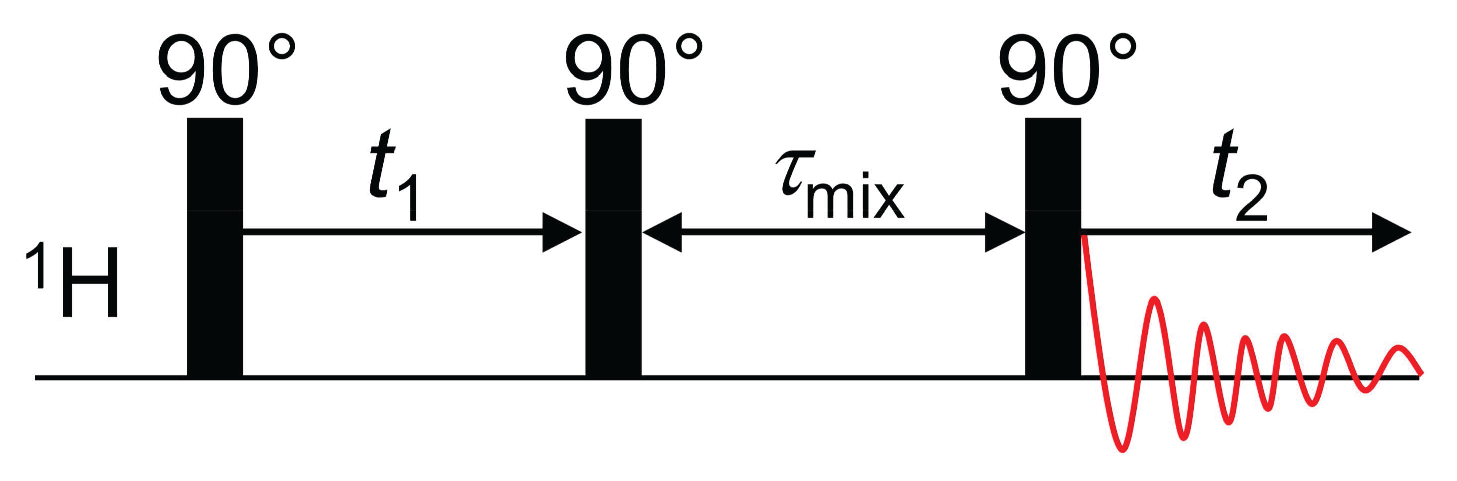
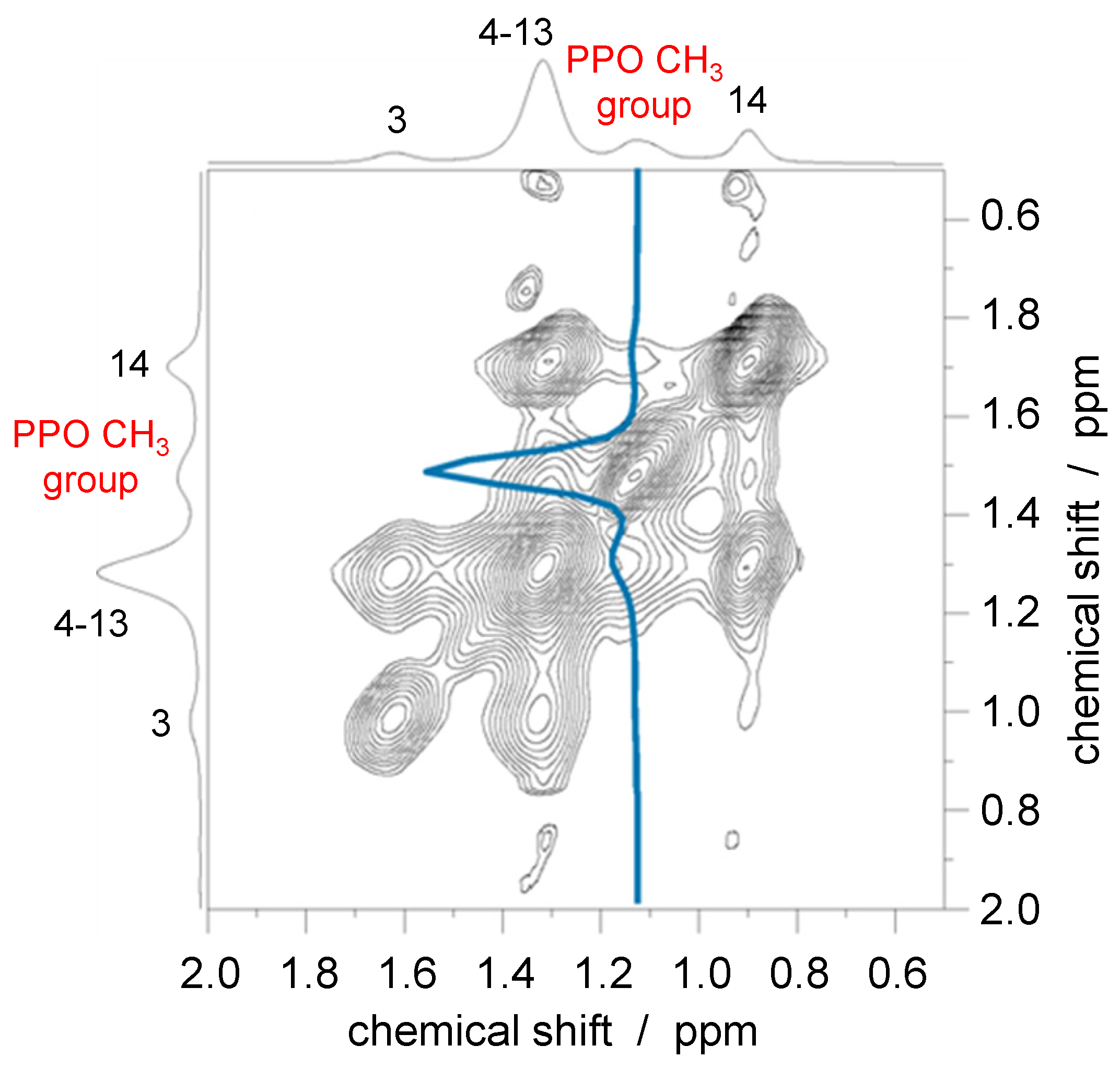
3.2. Nuclear Overhauser Enhancement Spectroscopy
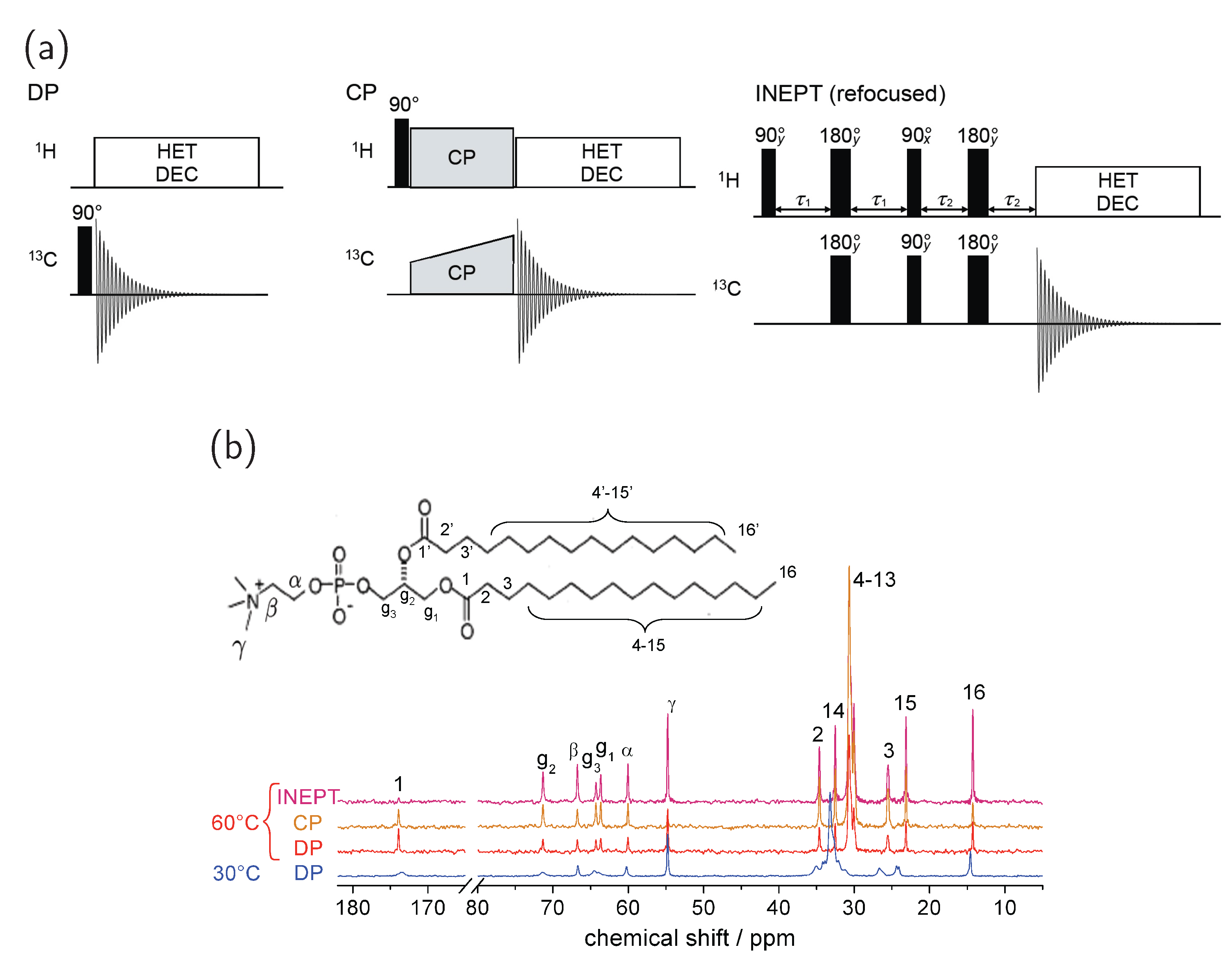
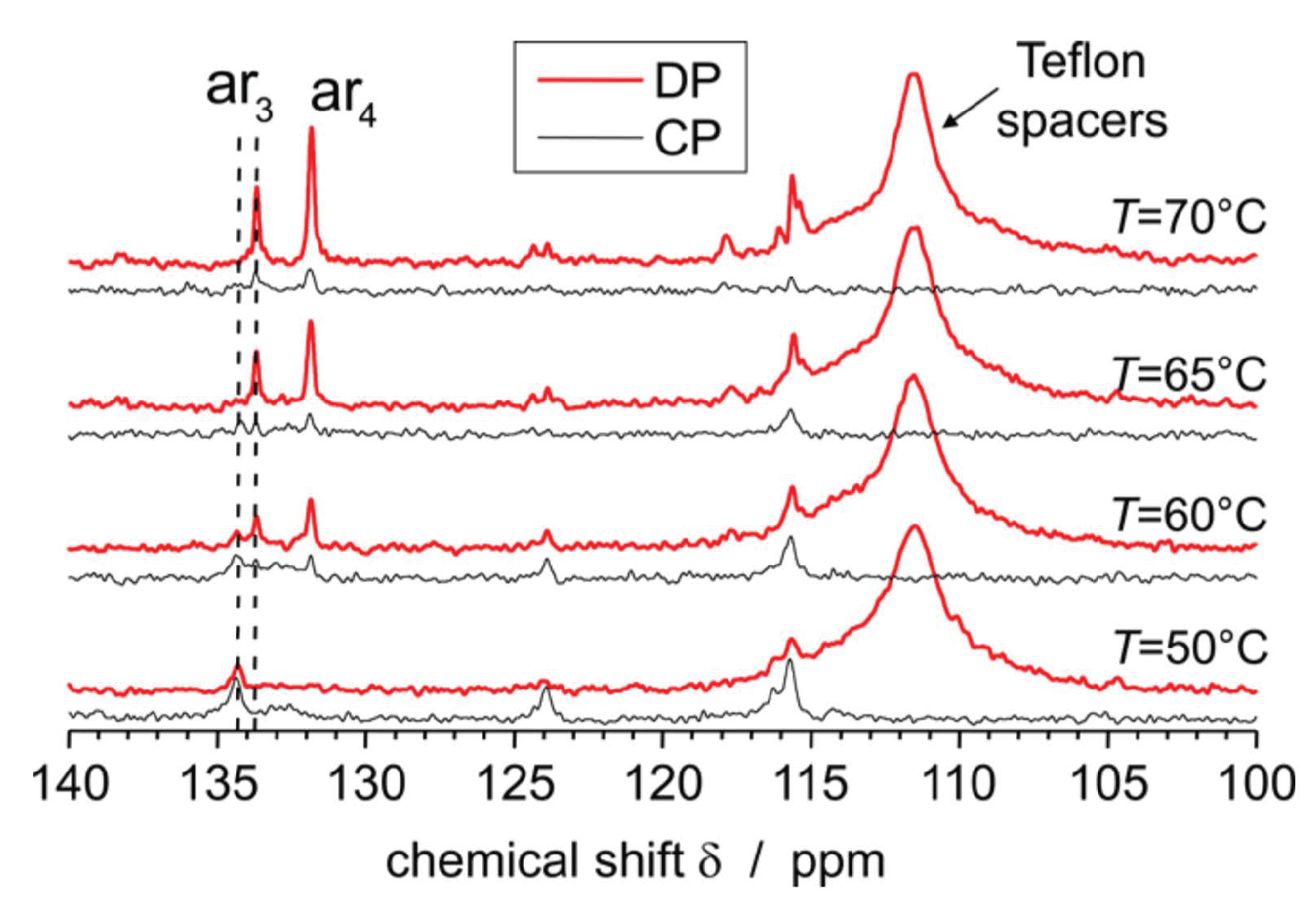
3.3. 13C MAS Spectra
4. Dynamic Order Parameters in Lipid Membranes
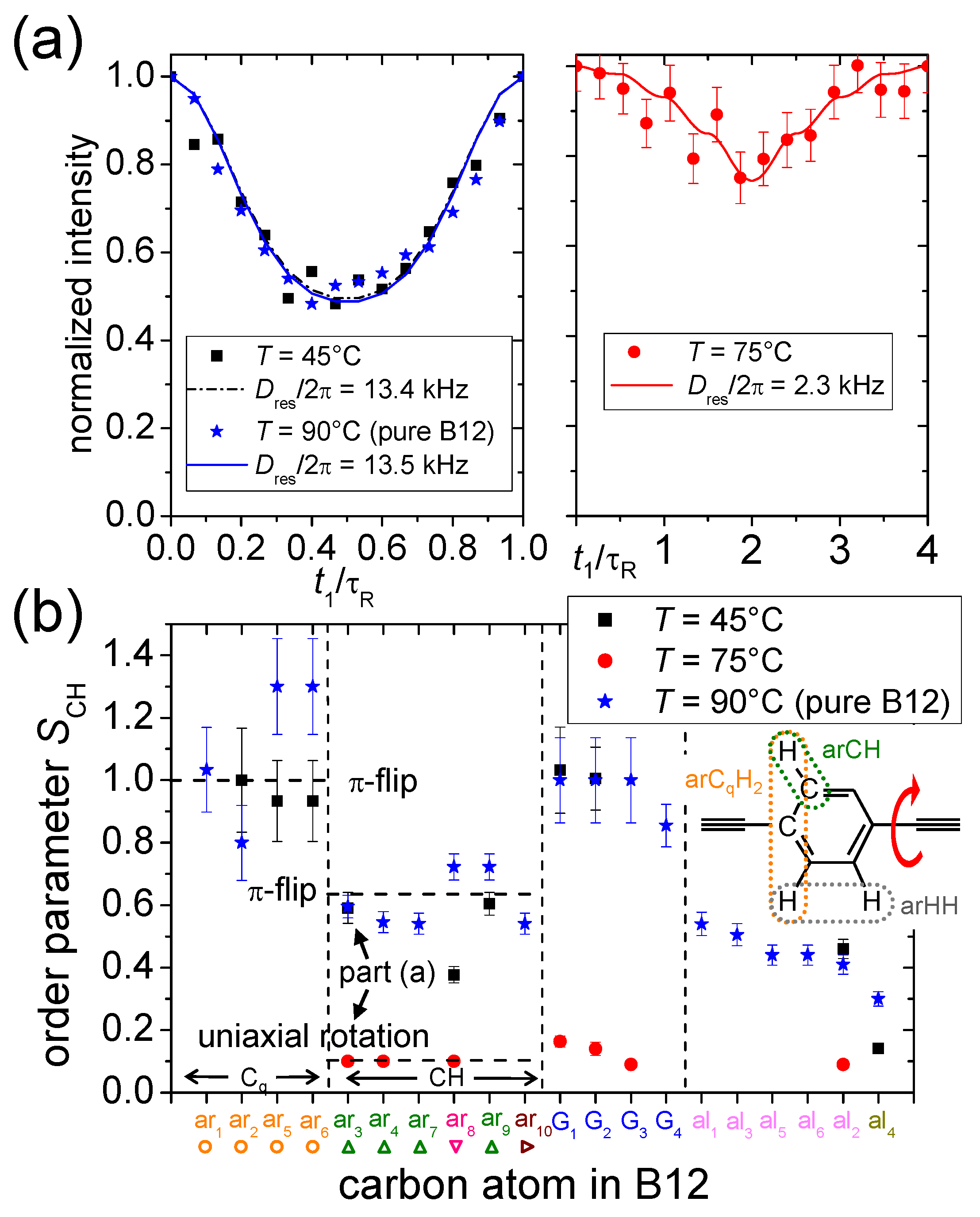
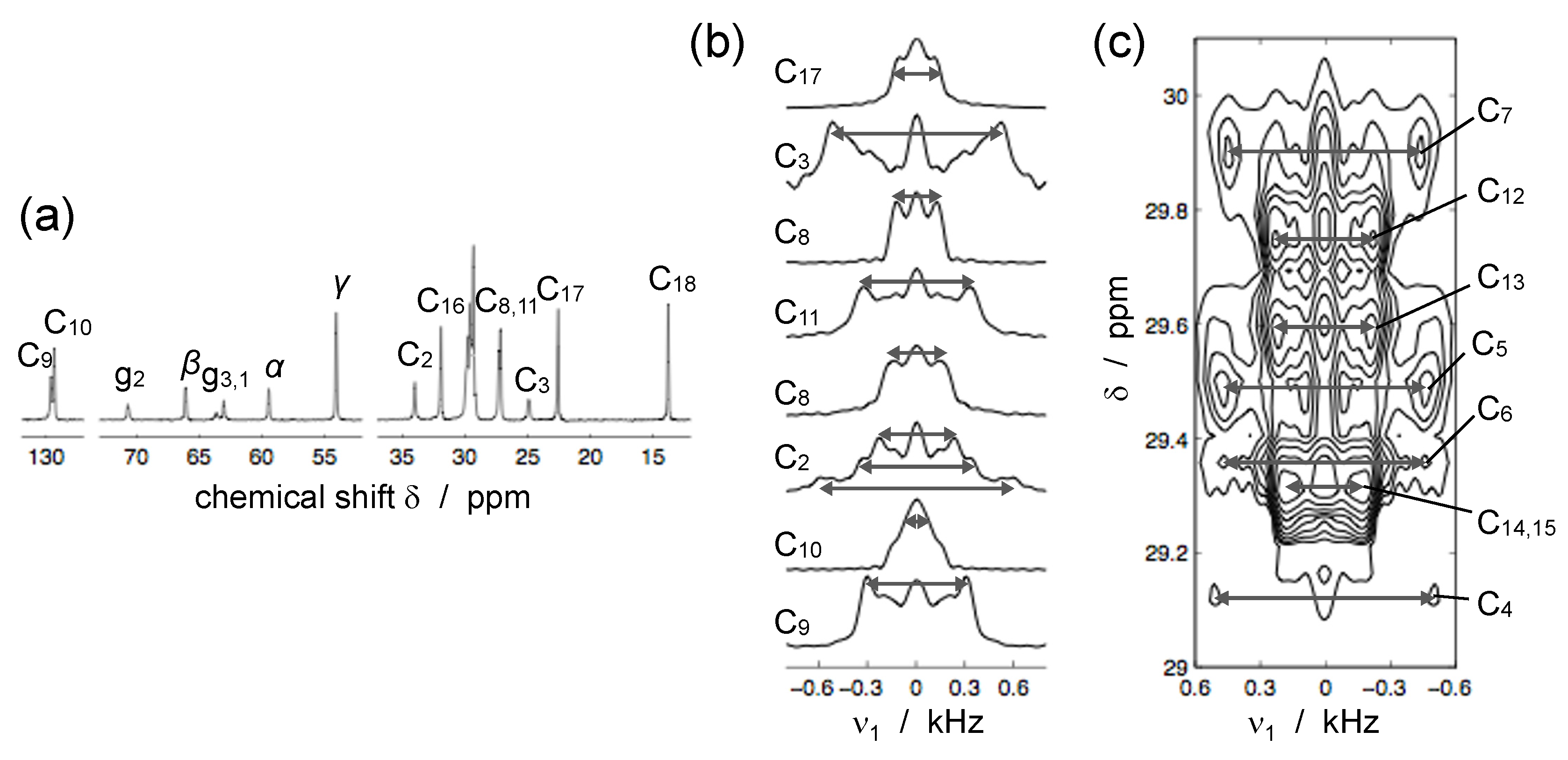
4.1. C–H Order Parameters
4.2. SLF NMR Spectroscopy
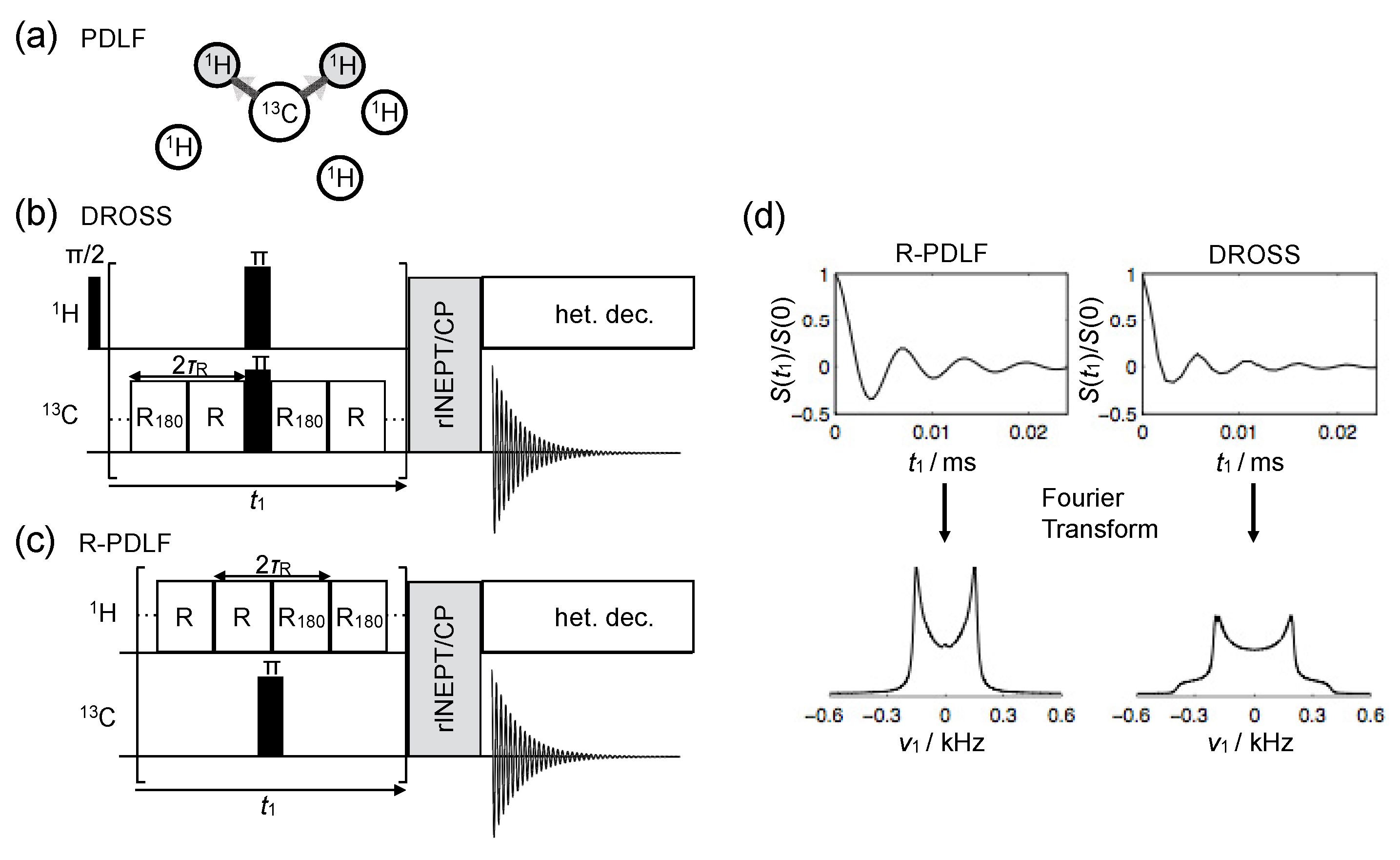
4.2.1. Carbon-Detected Local Field NMR
4.2.2. Proton-Detected Local Field NMR
4.3. Intermediate Motional Regime
4.4. H–H Order Parameters
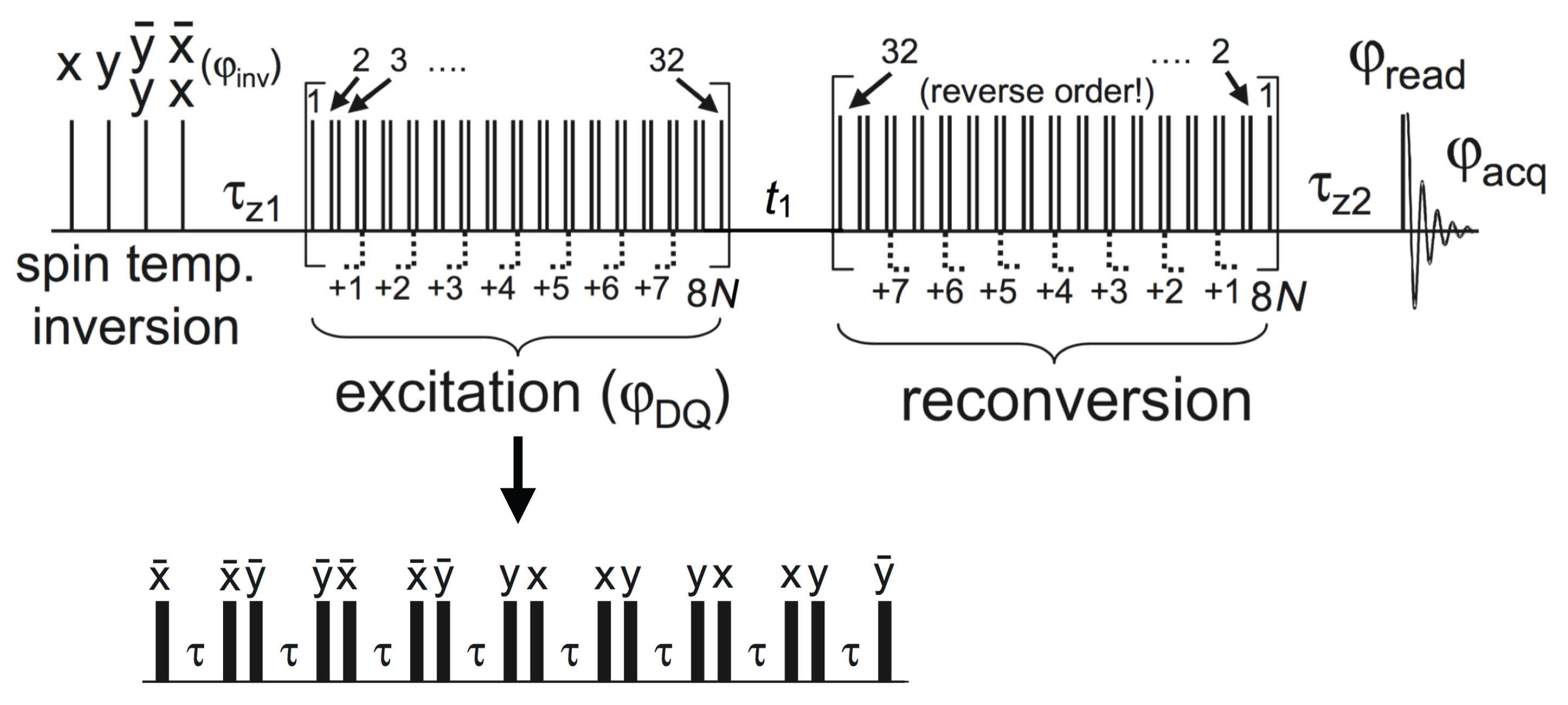
4.4.1. Experimental Considerations
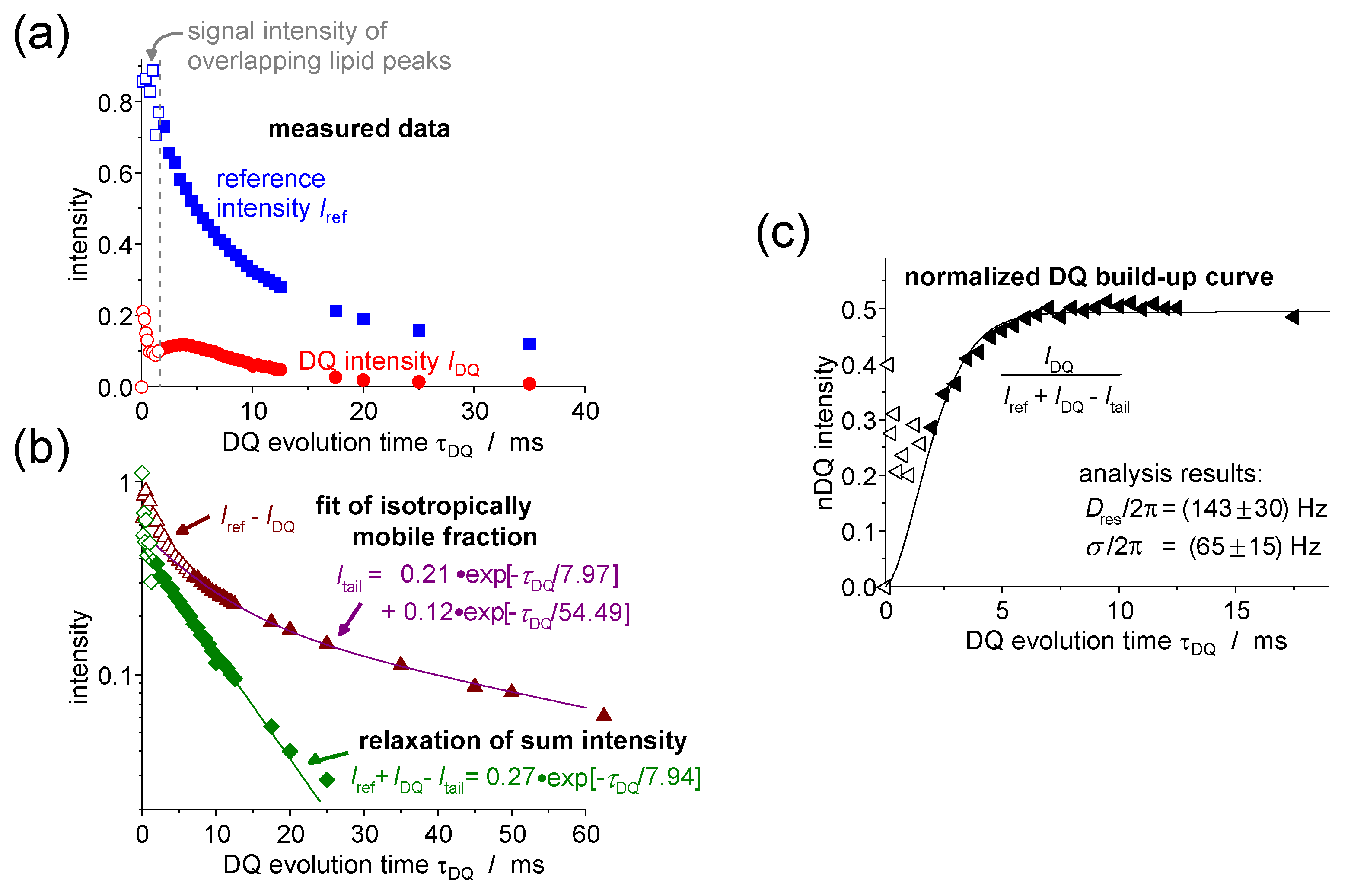
4.4.2. DQ Build-Up Curve Analysis
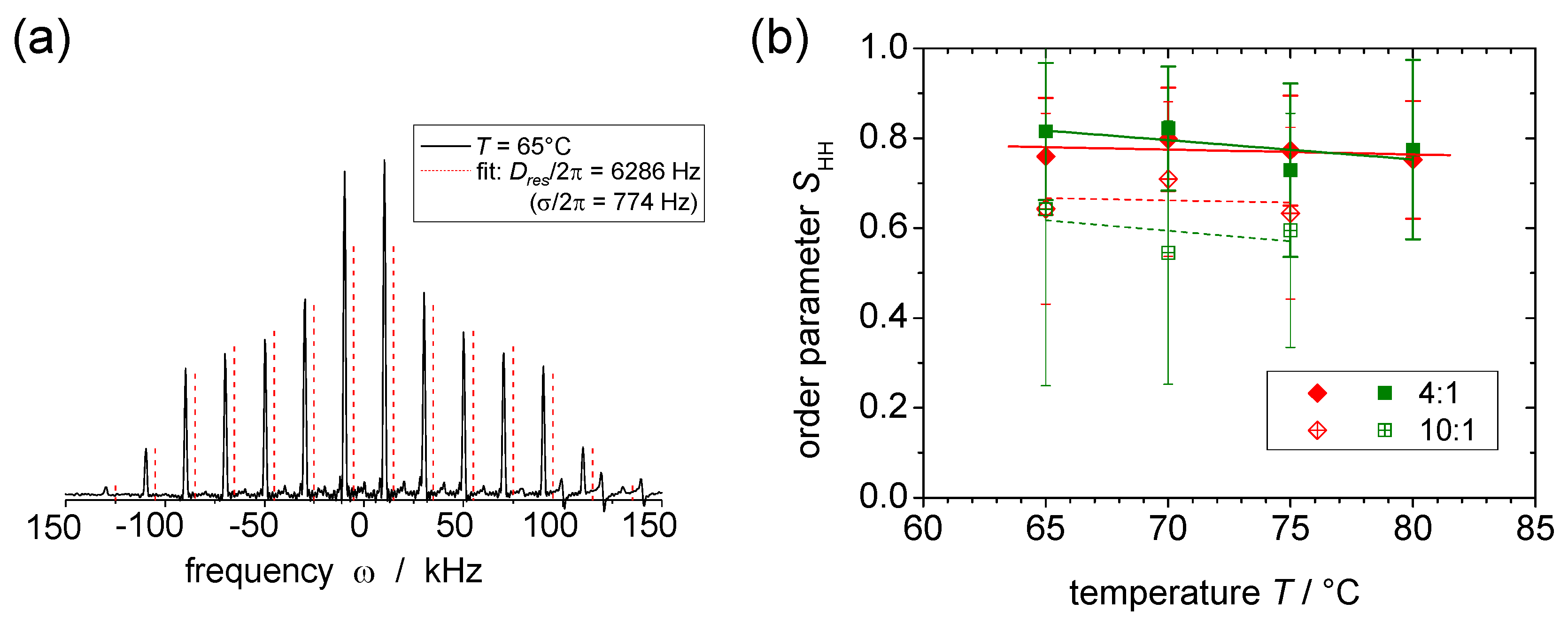
4.4.3. DQ Sideband-Pattern Analysis
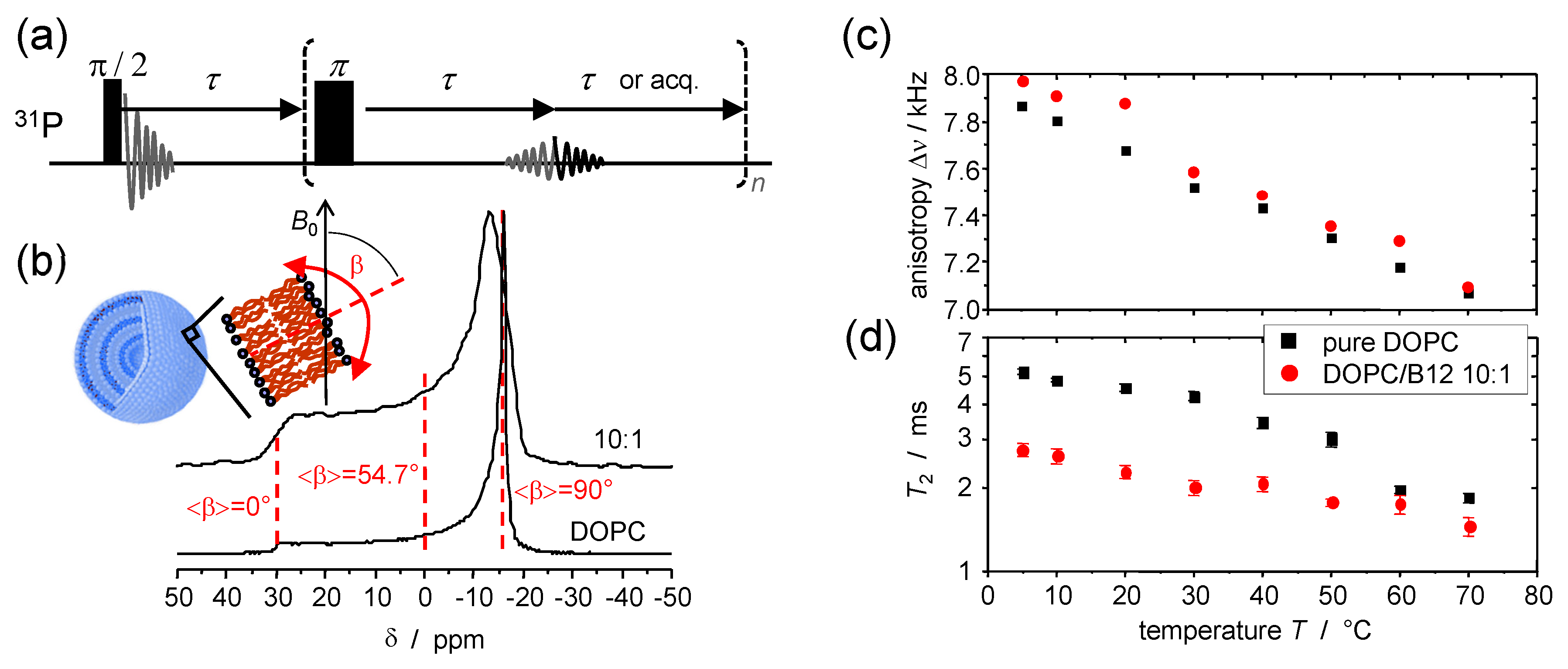
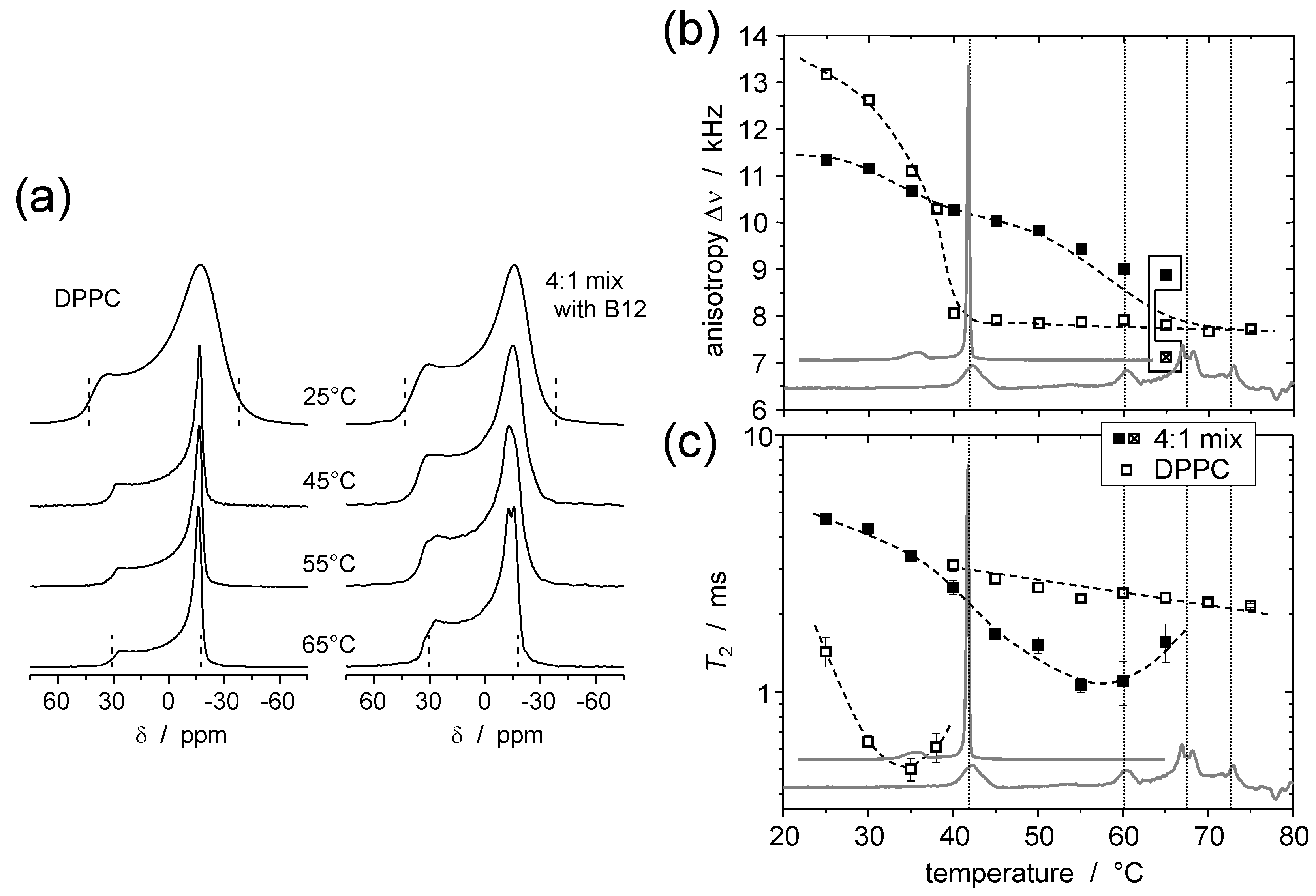
5. Static 31P NMR as a Probe of Headgroup Mobility
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nicolson, G.L. The Fluid-Mosaic Model of Membrane Structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta 2014, 1838, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Gon˜i, F.M. The basic structure and dynamics of cell membranes: An update of the Singer–Nicolson model. Biochim. Biophys. Acta 2014, 1838, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Toppozini, L.; Meinhardt, S.; Armstrong, C.L.; Yamani, Z.; Kučerka, N.; Schmid, F.; Rheinstädter, M.C. Structure of Cholesterol in Lipid Rafts. Phys. Rev. Lett. 2014, 113, 228101. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Matile, S. Synthetic Ion Channels. Langmuir 2013, 29, 9031–9040. [Google Scholar] [CrossRef] [PubMed]
- Busschaert, N.; Gale, P.A. Small-Molecule Lipid-Bilayer Anion Transporters for Biological Applications. Angew. Chem. Int. Ed. 2013, 52, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Widanapathirana, L.; Zhao, Y.; Hong, M. Aggregation and Dynamics of Oligocholate Transporters in Phospholipid Bilayers Revealed by Solid-State NMR Spectroscopy. Langmuir 2012, 28, 17071–17078. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, D.A.; Sosnik, A. Poly(ethylene oxide)–poly(propylene oxide) block copolymer micelles as drug delivery agents: Improved hydrosolubility, stability and bioavailability of drugs. Eur. J. Pharm. Biopharm. 2007, 66, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V. Pluronic® block copolymers for overcoming drug resistance in cancer. Adv. Drug Deliv. Rev. 2002, 54, 759–779. [Google Scholar] [CrossRef]
- Firestone, M.A.; Wolf, A.C.; Seifert, S. Small-Angle X-ray Scattering Study of the Interaction of Poly(ethylene oxide)-b-Poly(propylene oxide)-b-Poly(ethylene oxide) Triblock Copolymers with Lipid Bilayers. Biomacromolecules 2003, 4, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Maskarinec, S.A.; Wu, G.; Lee, K.Y.S. Membrane Sealing by Polymers. Ann. N. Y. Acad. Sci. 2005, 1066, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Williamson, P.T.F. Solid-State NMR for the Analysis of High-Affinity Ligand/Receptor Interactions. Concepts Magn. Reson. A 2009, 34A, 144–172. [Google Scholar] [CrossRef]
- Amado, E.; Augsten, C.; Mäder, K.; Blume, A.; Kressler, J. Amphiphilic Water Soluble Triblock Copolymers Based on Poly(2,3-dihydroxypropyl methacrylate) and Poly(propylene oxide): Synthesis by Atom Transfer Radical Polymerization and Micellization in Aqueous Solutions. Macromolecules 2006, 39, 9486–9496. [Google Scholar] [CrossRef]
- Schwieger, C.; Achilles, A.; Scholz, S.; Rüger, J.; Bacia, K.; Saalwächter, K.; Kressler, J.; Blume, A. Binding of Amphiphilic and Triphilic Block Copolymers to Lipid Model Membranes: The Role of Perfluorinated Moieties. Soft Matter 2014, 10, 6147–6160. [Google Scholar] [CrossRef] [PubMed]
- Kyeremateng, S.O.; Amado, E.; Blume, A.; Kressler, J. Synthesis of ABC and CABAC Triphilic Block Copolymers by ATRP Combined with ’Click’ Chemistry. Macromol. Rapid Commun. 2008, 29, 1140–1146. [Google Scholar] [CrossRef]
- Amado, E.; Kerth, A.; Blume, A.; Kressler, J. Infrared Reflection Absorption Spectroscopy Coupled with Brewster Angle Microscopy for Studying Interactions of Amphiphilic Triblock Copolymers with Phospholipid Monolayers. Langmuir 2008, 24, 10041–10053. [Google Scholar] [CrossRef] [PubMed]
- Amado, E.; Kerth, A.; Blume, A.; Kressler, J. Phospholipid crystalline clusters induced by adsorption of novel amphiphilic triblock copolymers to monolayers. Soft Matter 2009, 5, 669–675. [Google Scholar] [CrossRef]
- Chen, B.; Baumeister, U.; Pelzl, G.; Das, M.K.; Zeng, X.B.; Ungar, G.; Tschierske, C. Carbohydrate rod conjugates: Ternary rod-coil molecules forming complex liquid crystal structures. J. Am. Chem. Soc. 2005, 127, 16578–16591. [Google Scholar] [CrossRef] [PubMed]
- Scholtysek, P.; Achilles, A.; Hoffmann, C.V.; Lechner, B.D.; Meister, A.; Tschierske, C.; Saalwächter, K.; Edwards, K.; Blume, A. A T-Shaped Amphiphilic Molecule Forms Closed Vesicles in Water and Bicelles in Mixtures with a Membrane Lipid. J. Phys. Chem. B 2012, 116, 4871–4878. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Ebert, H.; Lechner, B.D.; Lange, F.; Achilles, A.; Bärenwald, R.; Poppe, S.; Blume, A.; Saalwächter, K.; Tschierske, C.; et al. Dendritic domains with hexagonal symmetry formed by X-shaped bolapolyphiles in lipid membranes. Chem. Eur. J. 2015, 21, 8840–8850. [Google Scholar] [CrossRef] [PubMed]
- Achilles, A.; Bärenwald, R.; Lechner, B.D.; Werner, S.; Ebert, H.; Tschierske, C.; Blume, A.; Bacia, K.; Saalwächter, K. Self-Assembly of X-Shaped Bolapolyphiles in Lipid Membranes: Solid-State NMR Investigations. Langmuir 2016, 32, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H. The Description of Membrane Lipid Conformation, Order and Dynamics by 2H-NMR. Biochim. Biophys. Acta 1983, 737, 117–171. [Google Scholar] [CrossRef]
- Wennerström, H. Proton nuclear magnetic resonance lineshapes in lamellar liquid crystals. Chem. Phys. Lett. 1973, 18, 41–44. [Google Scholar] [CrossRef]
- De Angelis, A.A.; Opella, S.J. Bicelle samples for solid-state NMR of membrane proteins. Nat. Protoc. 2007, 2, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.E.; Rienstra, C.M.; Auger, M.; Lakshmi, K.V.; Griffin, R.G. Heteronuclear decoupling in rotating solids. J. Chem. Phys. 1995, 103, 6951–6958. [Google Scholar] [CrossRef]
- Fung, B.M.; Khitrin, A.K.; Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 2000, 142, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Yao, X.; Hong, M. Membrane Protein Topology Probed by 1H Spin Diffusion from Lipids Using Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2002, 124, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yao, H.; Hong, M. Determining the depth of insertion of dynamically invisible membrane peptides by gel-phase 1H spin diffusion heteronuclear correlation NMR. J. Biomol. NMR 2013, 56, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.F.; Ribeiro, A.A.; Williams, G.D. New view of lipid bilayer dynamics from 2H and 13C NMR relaxation time measurements. Proc. Natl. Acad. Sci. USA 1983, 80, 4325–4329. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.M.; Ollila, O.H.S.; Pigliapochi, R.; Dabkowska, A.P.; Topgaard, D. Model-free estimation of the effective correlation time for C–H bond reorientation in amphiphilic bilayers: 1H–13C solid-state NMR and MD simulations. J. Chem. Phys. 2015, 142, 044905. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Yarger, J.L.; Holland, G.P. 2H–13C HETCOR MAS NMR for indirect detection of 2H quadrupole patterns and spin–lattice relaxation rates. J. Magn. Reson. 2013, 226, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Rienstra, C.M. Site-Specific Internal Motions in GB1 Protein Microcrystals Revealed by 3D 2H–13C–13C Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2016, 138, 4105–4119. [Google Scholar] [CrossRef] [PubMed]
- Dolainsky, C.; Möps, A.; Bayerl, T.M. Transverse relaxation in supported and nonsupported phospholipid model membranes and the influence of ultraslow motions: A 31P-NMR study. J. Chem. Phys. 1993, 98, 1712–1720. [Google Scholar] [CrossRef]
- Kothe, G.; Heaton, N. Slow & Ultraslow Motions in Biology. In Encyclopedia of Nucleaner Magnetic Resonance; Grant, D.M., Harris, R.K., Eds.; John Wiley & Sons: Chichester, UK, 1996; Volume 7, pp. 4436–4444. [Google Scholar]
- Bloom, M.; Sternin, E. Transverse Nuclear Spin Relaxation in Phospholipid Bilayer Membranes. Biochemistry 1987, 26, 2101–2105. [Google Scholar] [CrossRef]
- Furó, I.; Dvinskikh, S.V. NMR methods applied to anisotropic diffusion. Magn. Reson. Chem. 2002, 40, S3–S14. [Google Scholar] [CrossRef]
- Lindblom, G.; Orädd, G. Lipid lateral diffusion and membrane heterogeneity. Biochim. Biophys. Acta 2009, 1788, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.M.; Saleem, Q.; Lai, A.; Morales, H.H. NMR methods for measuring lateral diffusion in membranes. Chem. Phys. Lipids 2013, 166, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Rohr, K.; Spiess, H.W. Multidimensional Solid-State NMR and Polymers; Academic Press: London, UK, 1994. [Google Scholar]
- Bielecki, A.; Burum, D.P. Temperature Dependence of 207Pb MAS Spectra of Solid Lead Nitrate. An Accurate, Sensitive Thermometer for Variable-Temperature MAS. J. Magn. Reson. A 1995, 116, 215–220. [Google Scholar] [CrossRef]
- Dvinskikh, S.V.; Castro, V.; Sandström, D. Heating caused by radiofrequency irradiation and sample rotation in 13C magic angle spinning NMR studies of lipid bilayers. Magn. Reson. Chem. 2004, 42, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.; Bowers, J.; Shan, X.; Moran, L.; Oldfield, E.; Moscarello, M.A. Some New Developments in Solid-state Nuclear Magnetic Resonance Spectroscopic Studies of Lipids and Biological Membranes, including the Effects of Cholesterol in Model and Natural Systems. J. Chem. Soc. Faraday Trans. 1 1988, 84, 3821–3849. [Google Scholar] [CrossRef]
- Forbes, J.; Husted, C.; Oldfield, E. High-Field, High-Resolution Proton “Magic-Angle” Sample-Spinning Nuclear Magnetic Resonance Spectroscopic Studies of Gel and Liquid Crystalline Lipid Bilayers and the Effects of Cholesterol. J. Am. Chem. Soc. 1988, 110, 1059–1065. [Google Scholar] [CrossRef]
- Davis, J.H.; Auger, M.; Hodges, R.S. High Resolution 1H Nuclear Magnetic Resonance of a Transmembrane Peptide. Biophys. J. 1995, 69, 1917–1932. [Google Scholar] [CrossRef]
- Birdsall, N.J.M.; Feeney, J.; Lee, A.G.; Levine, Y.K.; Metcalfe, J.C. Dipalmitoyl-lecithin: Assignment of the 1H and 13C Nuclear Magnetic Resonance Spectra, and Conformational Studies. J. Chem. Soc. Perkin Trans. 2 1972, 1441–1445. [Google Scholar] [CrossRef]
- Jeener, J.; Meier, B.H.; Bachmann, P.; Ernst, R.R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- Scheidt, H.; Huster, D. The interaction of small molecules with phospholipid membranes studied by 1H NOESY NMR under magic-angle spinning. Acta Pharmacol. Sin. 2008, 29, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Arnold, K.; Gawrisch, K. Investigation of Lipid Organization in Biological Membranes by Two-Dimensional Nuclear Overhauser Enhancement Spectroscopy. J. Phys. Chem. B 1999, 103, 243–251. [Google Scholar] [CrossRef]
- Huster, D.; Gawrisch, K. NOESY NMR Crosspeaks between Lipid Headgroups and Hydrocarbon Chains: Spin Diffusion or Molecular Disorder? J. Am. Chem. Soc. 1999, 121, 1992–1993. [Google Scholar] [CrossRef]
- Holte, L.L.; Gawrisch, K. Determining Ethanol Distribution in Phospholipid Multilayers with MAS-NOESY Spectra. Biochemistry 1997, 36, 4669–4674. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.E.; Brown, C.A.; Nizza, D.T.; Gawrisch, K. Nuclear Overhauser Enhancement Spectroscopy Cross-Relaxation Rates and Ethanol Distribution across Membranes. Biophys. J. 2002, 82, 1396–1404. [Google Scholar] [CrossRef]
- Scheidt, H.A.; Pampel, A.; Nissler, L.; Gebhardt, R.; Huster, D. Investigation of the membrane localization and distribution of flavonoids by high-resolution magic angle spinning NMR spectroscopy. Biochim. Biophys. Acta 2004, 1663, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Gaede, H.C.; Yau, W.M.; Gawrisch, K. Electrostatic Contributions to Indole-Lipid Interactions. J. Phys. Chem. B 2005, 109, 13014–13023. [Google Scholar] [CrossRef] [PubMed]
- Siarheyeva, A.; Lopez, J.J.; Glaubitz, C. Localization of Multidrug Transporter Substrates within Model Membranes. Biochemistry 2006, 45, 6203–6211. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Vogel, A.; Katzka, C.; Scheidt, H.A.; Binder, H.; Dante, S.; Gutberlet, T.; Zschörnig, O.; Waldmann, H.; Arnold, K. Membrane Insertion of a Lipidated Ras Peptide Studied by FTIR, Solid-State NMR, and Neutron Diffraction Spectroscopy. J. Am. Chem. Soc. 2003, 125, 4070–4079. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, A.E. NMR Spectroscopy and Polymer Microstructure: The Conformational Connection; VCH: Weinheim, Germany, 1989. [Google Scholar]
- Peersen, O.B.; Wu, X.; Kustanovich, I.; Smith, S.O. Variable-Amplitude Cross-Polarization MAS NMR. J. Magn. Reson. A 1993, 104, 334–339. [Google Scholar] [CrossRef]
- Metz, G.; Wu, X.; Smith, S.O. Ramped-Amplitude Cross Polarization in Magic-Angle-Spinning NMR. J. Magn. Reson. A 1994, 110, 219–227. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of Nuclear Magnetic Resonance Signals by Polarization Transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Gross, J.D.; Costa, P.R.; Dubacq, J.P.; Warschawski, D.E.; Lirsac, P.N.; Devaux, P.F.; Griffin, R.G. Multidimensional NMR in Lipid Systems. Coherence Transfer through J Couplings under MAS. J. Magn. Reson. B 1995, 106, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Nowacka, A.; Mohr, P.C.; Norrman, J.; Martin, R.W.; Topgaard, D. Polarization Transfer Solid-State NMR for Studying Surfactant Phase Behavior. Langmuir 2010, 26, 16848–16856. [Google Scholar] [CrossRef] [PubMed]
- Warschawski, D.E.; Devaux, P.F. Polarization Transfer in Lipid Membranes. J. Magn. Reson. 2000, 145, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Seelig, J. Deuterium Magnetic Resonance—Theory and Application to Lipid Membranes. Q. Rev. Biophys. 1977, 10, 353–418. [Google Scholar] [CrossRef] [PubMed]
- Leftin, A.; Brown, M.F. An NMR database for simulations of membrane dynamics. Biochim. Biophys. Acta 2011, 1808, 818–839. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.M.; Coreta-Gomes, F.; Ollila, O.H.S.; Moreno, M.J.; Vaz, W.L.C.; Topgaard, D. Cholesterol and POPC segmental order parameters in lipid membranes: solid state 1H–13C NMR and MD simulation studies. Phys. Chem. Chem. Phys. 2013, 15, 1976–1989. [Google Scholar] [CrossRef] [PubMed]
- Ollila, O.H.S.; Pabst, G. Atomistic resolution structure and dynamics of lipid bilayers in simulations and experiments. Biochim. Biophys. Acta 2016, 1858, 2512–2528. [Google Scholar] [CrossRef] [PubMed]
- Petrache, H.I.; Dodd, S.W.; Brown, M.F. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys. J. 2000, 79, 3172–3192. [Google Scholar] [CrossRef]
- Tulloch, A.P. Synthesis, analysis and application of specifically deuterated lipids. Prog. Lipid Res. 1983, 22, 235–256. [Google Scholar] [CrossRef]
- Vist, M.R.; Davis, J.H. Phase equilibria of cholesterol dipalmitoylphosphatidylcholine mixtures—2H nuclear magnetic resonance and differential scanning calorimetry. Biochemistry 1990, 29, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Clair, J.J.; Juhasz, J. Phase Equilibria in DOPC/DPPC-d62/Cholesterol Mixtures. Biophys. J. 2009, 96, 521–539. [Google Scholar] [CrossRef] [PubMed]
- Hester, R.K.; Ackerman, J.L.; Neff, B.L.; Waugh, J.S. Separated local field spectra in NMR—Determination of structure of solids. Phys. Rev. Lett. 1976, 36, 1081–1083. [Google Scholar] [CrossRef]
- Duer, M.J. Introduction to Solid-State NMR Spectroscopy; Blackwell: Oxford, UK, 2004. [Google Scholar]
- Reichert, D.; Saalwächter, K. Dipolar Coupling: Molecular-Level Mobility. In NMR Crystallography; Harris, R.K., Wasylishen, R.E., Duer, M.J., Eds.; John Wiley & Sons: Chichester, UK, 2009; pp. 177–193. [Google Scholar]
- Munowitz, M.; Griffin, R.G.; Bodenhausen, G.; Huang, T.H. Two-Dimensional Rotational Spin-Echo Nuclear Magnetic Resonance in Solids: Correlation of Chemical Shift and Dipolar Interaction. J. Am. Chem. Soc. 1981, 103, 2529–2533. [Google Scholar] [CrossRef]
- Hong, M.; Gross, J.D.; Griffin, R.G. Site-Resolved Determination of Peptide Torsion Angle Φ from the Relative Orientations of Backbone N–H and C–H Bonds by Solid-State NMR. J. Phys. Chem. B 1997, 101, 5869–5874. [Google Scholar] [CrossRef]
- Cobo, M.F.; Achilles, A.; Reichert, D.; deAzevedo, E.R.; Saalwächter, K. Recoupled separated-local-field experiments and applications to study intermediate-regime molecular motions. J. Magn. Reson. 2012, 221, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kurz, R.; Cobo, M.F.; de Azevedo, E.R.; Sommer, M.; Wicklein, A.; Thelakkat, M.; Hempel, G.; Saalwächter, K. Avoiding Bias Effects in NMR Experiments for Heteronuclear Dipole–Dipole Coupling Determinations: Principles and Application to Organic Semiconductor Materials. ChemPhysChem 2013, 14, 3146–3155. [Google Scholar] [CrossRef] [PubMed]
- Hackel, C.; Zinkevich, T.; Belton, P.; Achilles, A.; Reichert, D.; Krushelnitsky, A. The trehalose coating effect on the internal protein dynamics. Phys. Chem. Chem. Phys. 2012, 14, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Gullion, T.; Schaefer, J. Rotational-Echo Double-Resonance NMR. J. Magn. Reson. 1989, 81, 196–200. [Google Scholar] [CrossRef]
- Poger, D.; Caron, B.; Mark, A.E. Validating lipid force fields against experimental data: Progress, challenges and perspectives. Biochim. Biophys. Acta 2016, 1858, 1556–1565. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.K.; Zhang, Y.; Schmidt-Rohr, K.; Hong, M. pH-Dependent Conformation, Dynamics, and Aromatic Interaction of the Gating Tryptophan Residue of the Influenza M2 Proton Channel from Solid-State NMR. Biophys. J. 2013, 104, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Reuther, G.; Roark, M.B.; Tan, K.; Waldmann, H.; Feller, S.E.; Huster, D. Backbone conformational flexibility of the lipid modified membrane anchor of the human N-Ras protein investigated by solid-state NMR and molecular dynamics simulation. Biochim. Biophys. Acta 2010, 1798, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.; Su, Y.; Hong, M. High-Resolution Orientation and Depth of Insertion of the Voltage-Sensing S4 Helix of a Potassium Channel in Lipid Bilayers. J. Mol. Biol. 2010, 401, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Edén, M.; Levitt, M.H. Recoupling of heteronuclear dipolar interactions in solid-state NMR using symmetry-based pulse sequences. Chem. Phys. Lett. 2001, 342, 353–361. [Google Scholar] [CrossRef]
- Nakai, T.; Terao, T. Measurements of heteronuclear dipolar powder patterns due only to directly bonded couplings. Magn. Reson. Chem. 1992, 30, 42–44. [Google Scholar] [CrossRef]
- Elena, B.; Lesage, A.; Steuernagel, S.; Böckmann, A.; Emsley, L. Proton to Carbon-13 INEPT in Solid-State NMR Spectroscopy. J. Chem. Am. Soc. 2005, 127, 17296–17302. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.; Rasmussen, J.T.; Nielsen, N.C. SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson. 2000, 147, 296–330. [Google Scholar] [CrossRef] [PubMed]
- Caldarelli, S.; Hong, M.; Emsley, L.; Pines, A. Measurement of carbon-proton dipolar couplings in liquid crystals by local dipolar field NMR spectroscopy. J. Phys. Chem. 1996, 100, 18696–18701. [Google Scholar] [CrossRef]
- Hong, M.; Schmidt-Rohr, K.; Nanz, D. Study of phospholipid structure by 1H, 13C, 31P dipolar couplings from two-dimensional NMR. Biophys. J. 1995, 69, 1939–1950. [Google Scholar] [CrossRef]
- Gross, J.D.; Warschawski, D.E.; Griffin, R.G. Dipolar Recoupling in MAS NMR: A Probe for Segmental Order in Lipid Bilayers. J. Chem. Am. Soc. 1997, 119, 796. [Google Scholar] [CrossRef]
- Dvinskikh, S.V.; Castro, V.; Sandström, D. Efficient solid-state NMR methods for measuring heteronuclear dipolar couplings in unoriented lipid membrane systems. Phys. Chem. Chem. Phys. 2005, 7, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Dvinskikh, S.V.; Zimmermann, H.; Maliniak, A.; Sandström, D. Measurements of motionally averaged heteronuclear dipolar couplings in MAS NMR using R-type recoupling. J. Magn. Reson. 2004, 168, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Warschawski, D.E.; Devaux, P.F. Order parameters of unsaturated phospholipids in membranes and the effect of cholesterol: a 1H–13C solid-state NMR study at natural abundance. Eur. Biophys. J. 2005, 34, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.M.; Sood, R.; Bärenwald, R.; Carlström, G.; Topgaard, D.; Saalwächter, K.; Kinnunen, P.K.J.; Ollila, O.H.S. Acyl Chain Disorder and Azelaoyl Orientation in Lipid Membranes Containing Oxidized Lipids. Langmuir 2016, 32, 6524–6533. [Google Scholar] [CrossRef] [PubMed]
- Spiess, H.W. Molecular dynamics of solid polymers as revealed by deuteron NMR. Colloid Polym. Sci. 1983, 261, 193–209. [Google Scholar] [CrossRef]
- DeAzevedo, E.R.; Saalwächter, K.; Pascui, O.; de Souza, A.A.; Bonagamba, T.J.; Reichert, D. Intermediate motions as studied by solid-state separated local field NMR experiments. J. Chem. Phys. 2008, 128, 104505. [Google Scholar] [CrossRef] [PubMed]
- Cobo, M.F.; Reichert, D.; Saalwächter, K.; deAzevedo, E.R. A double-component Anderson–Weiss approach for describing NMR signals of mobile SIn units: Application to constant-time DIPSHIFT experiments. J. Magn. Reson. 2014, 248, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Feike, M.; Demco, D.E.; Graf, R.; Gottwald, J.; Hafner, S.; Spiess, H.W. Broadband Multiple-Quantum NMR Spectroscopy. J. Magn. Reson. 1996, 122, 214–221. [Google Scholar] [CrossRef]
- Gullion, T.; Baker, D.B.; Conradi, M.S. New, Compensated Carr-Purcell Sequences. J. Magn. Reson. 1990, 89, 479–484. [Google Scholar] [CrossRef]
- Saalwächter, K.; Lange, F.; Matyjaszewski, K.; Huang, C.F.; Graf, R. BaBa-xy16: Robust and broadband homonuclear DQ recoupling for applications in rigid and soft solids up to the highest MAS frequencies. J. Magn. Reson. 2011, 212, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Saalwächter, K. Proton Multiple-Quantum NMR for the Study of Chain Dynamics and Structural Constraints in Polymeric Soft Materials. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 1–35. [Google Scholar] [CrossRef]
- Saalwächter, K. Robust NMR Approaches for the Determination of Homonuclear Dipole–Dipole Coupling Constants in Studies of Solid Materials and Biomolecules. ChemPhysChem 2013, 14, 3000–3014. [Google Scholar] [CrossRef] [PubMed]
- Geen, H.; Titman, J.J.; Gottwald, J.; Spiess, H.W. Spinning Sidebands in the Fast-MAS Multiple-Quantum Spectra of Protons in Solids. J. Magn. Reson. A 1995, 114, 264–267. [Google Scholar] [CrossRef]
- Chassé, W.; Valentín, J.L.; Genesky, G.D.; Cohen, C.; Saalwächter, K. Precise dipolar coupling constant distribution analysis in proton multiple-quantum NMR of elastomers. J. Chem. Phys. 2011, 134, 044907. [Google Scholar] [CrossRef] [PubMed]
- Saalwächter, K.; Herrero, B.; López-Manchado, M.A. Chain order and crosslink density of elastomers as investigated by proton multiple-quantum NMR. Macromolecules 2005, 38, 9650–9660. [Google Scholar] [CrossRef]
- Kuhn, W.; Grün, F. Beziehungen zwischen elastischen Konstanten und Dehnungsdoppelbrechung hochelastischer Stoffe. Kolloid Z. 1942, 101, 248–271. [Google Scholar] [CrossRef]
- Sommer, J.U.; Chassé, W.; Valentín, J.L.; Saalwächter, K. Effect of excluded volume on segmental orientation correlations in polymer chains. Phys. Rev. E 2008, 78, 051803. [Google Scholar] [CrossRef] [PubMed]
- Kučerka, N.; Liu, Y.; Chu, N.; Petrache, H.I.; Tristram-Nagle, S.; Nagle, J.F. Structure of Fully Hydrated Fluid Phase DMPC and DLPC Lipid Bilayers Using X-Ray Scattering from Oriented Multilamellar Arrays and from Unilamellar Vesicles. Biophys. J. 2005, 88, 2626–2637. [Google Scholar] [CrossRef] [PubMed]
- Dufourc, E.J.; Mayer, C.; Stohrer, J.; Althoff, G.; Kothe, G. Dynamics of phosphate head groups in biomembranes. Comprehensive analysis using phosphorus-31 nuclear magnetic resonance lineshape and relaxation time measurements. Biophys. J. 1992, 61, 42–57. [Google Scholar] [CrossRef]
- Gustafsson, S.; Halle, B. Spin relaxation by collective director fluctuations and molecular diffusion in lamellar phases. Continuum theory of relaxation anisotropy and dispersion. J. Chem. Phys. 1997, 106, 9337–9352. [Google Scholar] [CrossRef]
- Frezzato, D.; Kothe, G.; Moro, G.J. Transverse Nuclear Spin Relaxation Due to Director Fluctuations in Liquid Crystals—A Slow-Motional Theory. J. Phys. Chem. B 2001, 105, 1281–1292. [Google Scholar] [CrossRef]
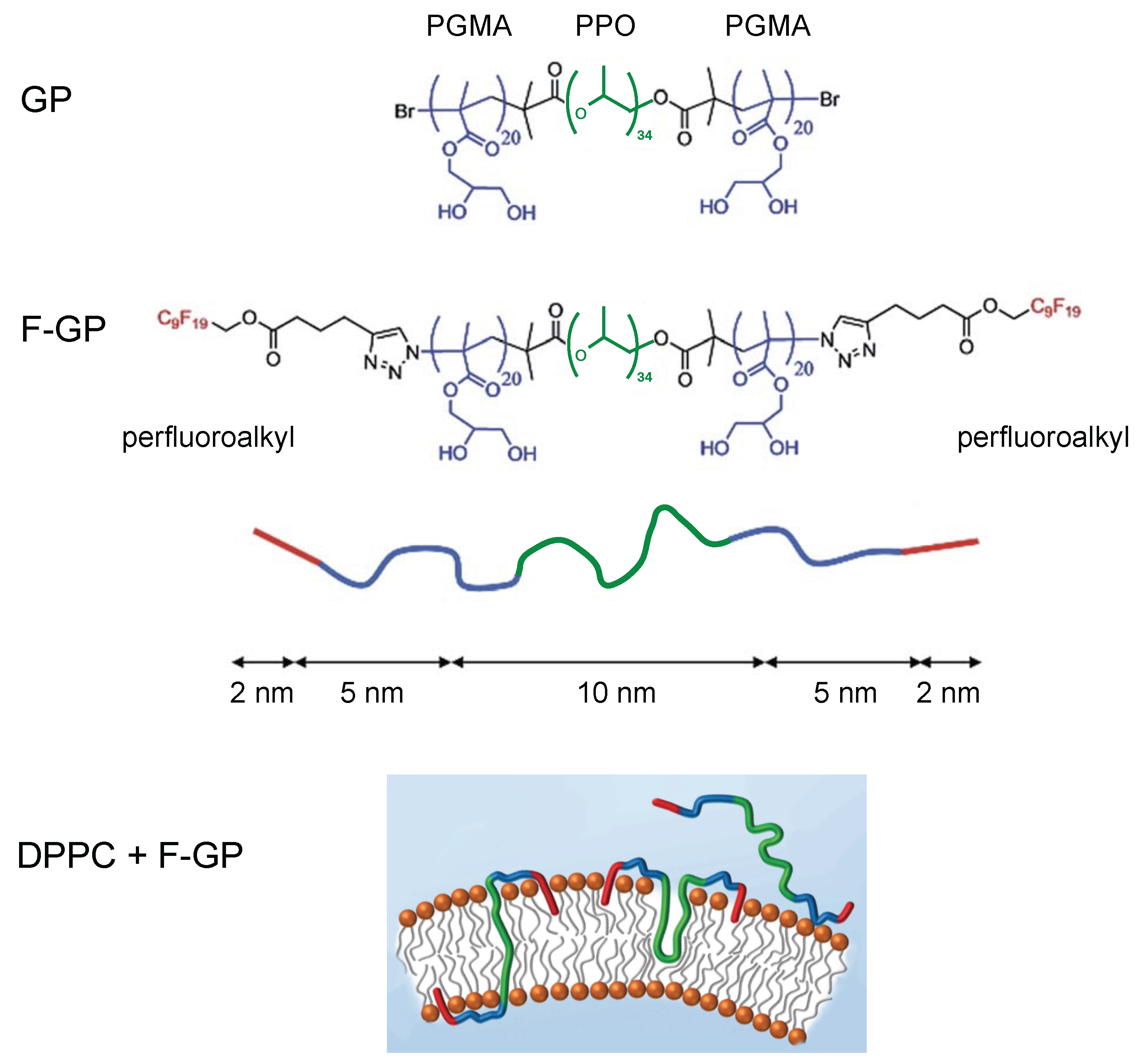
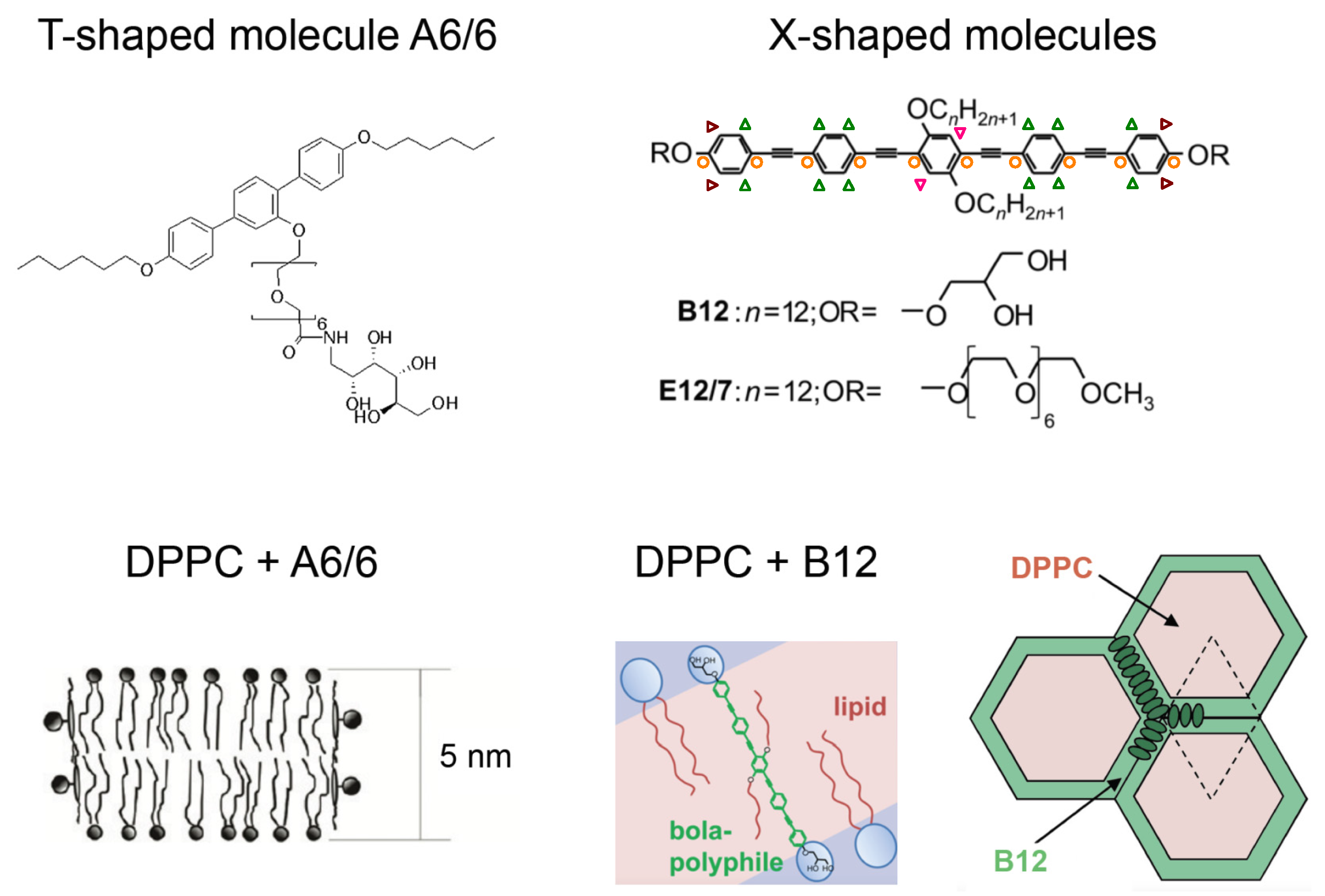
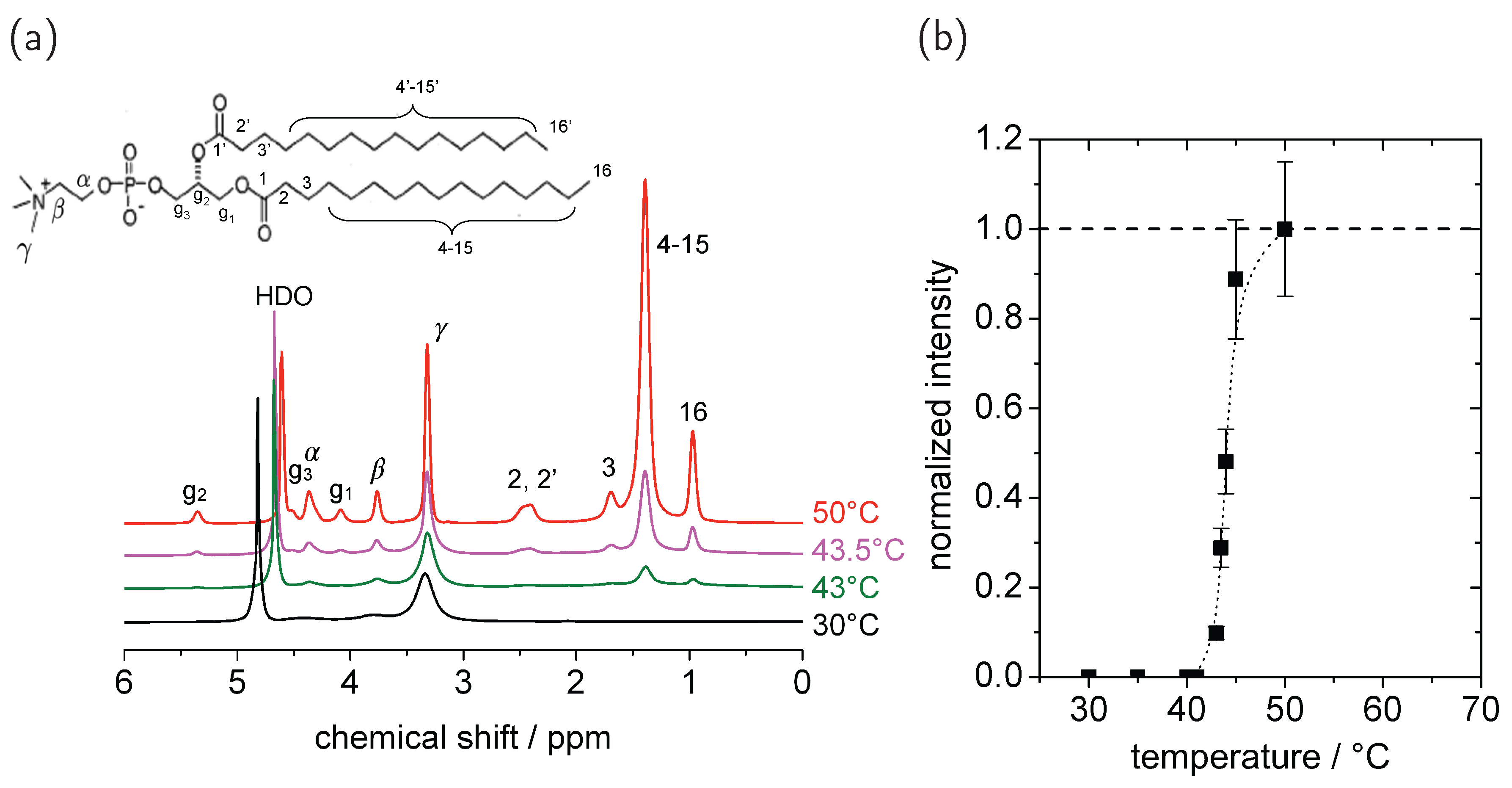
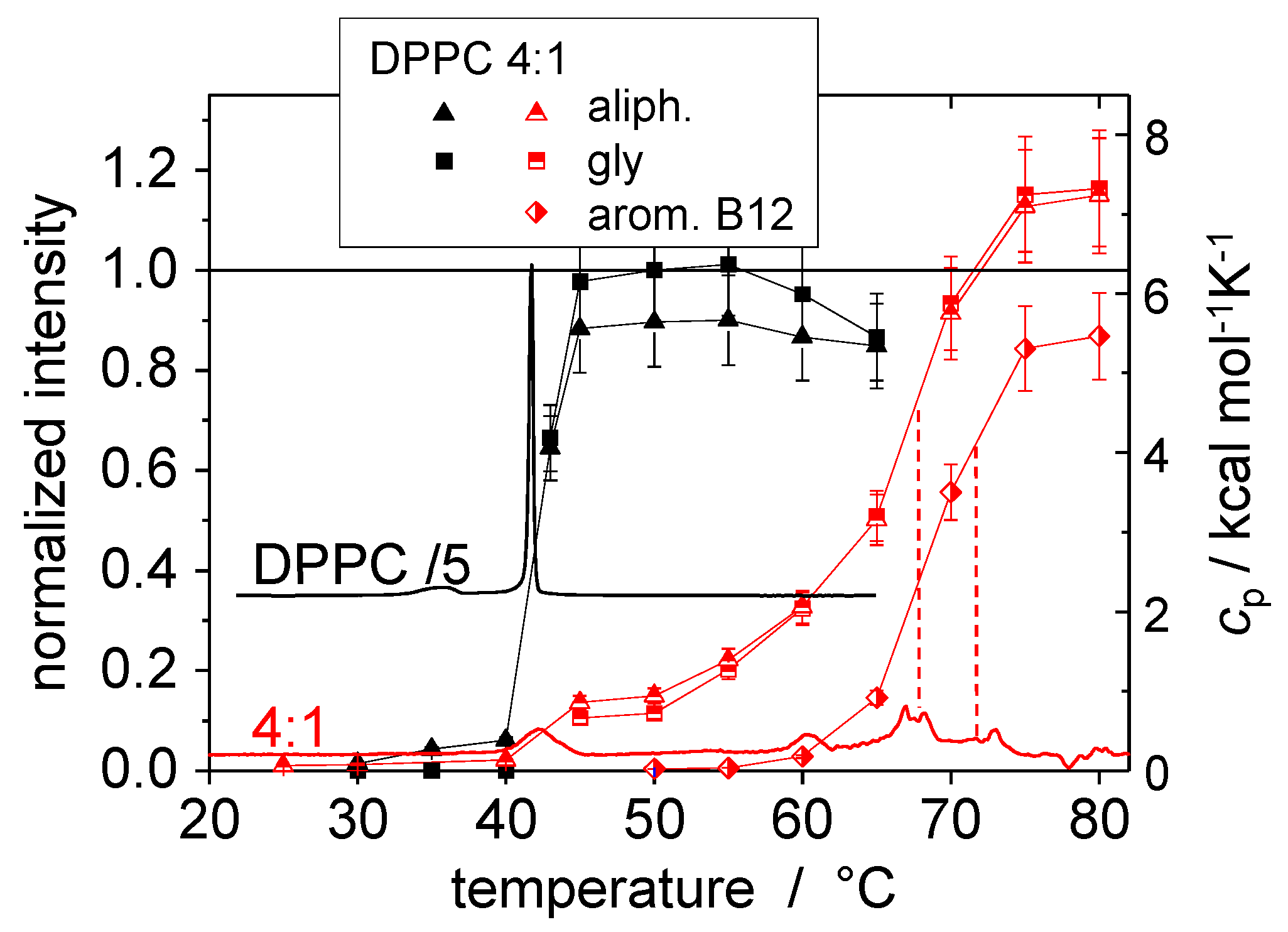
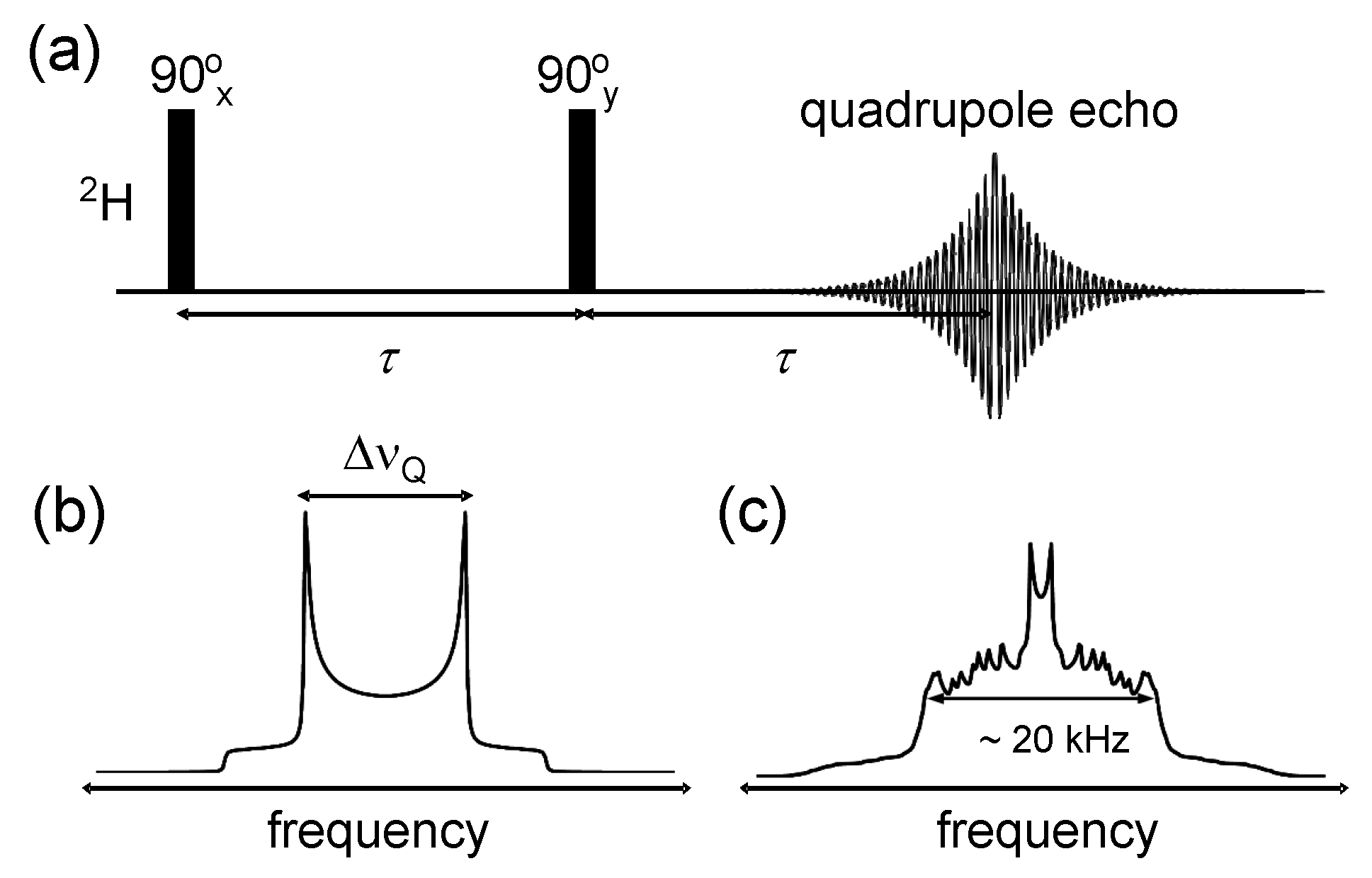
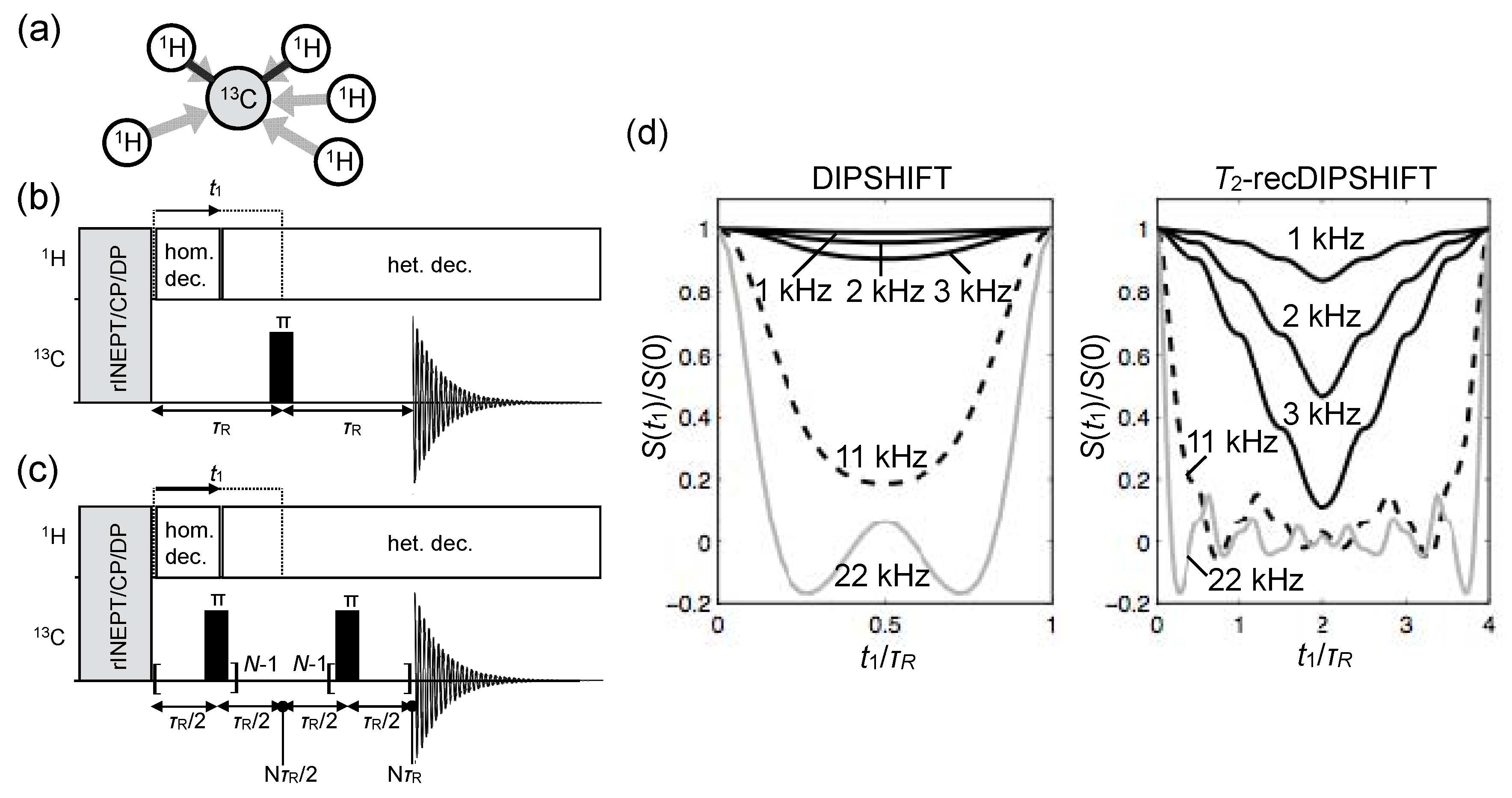
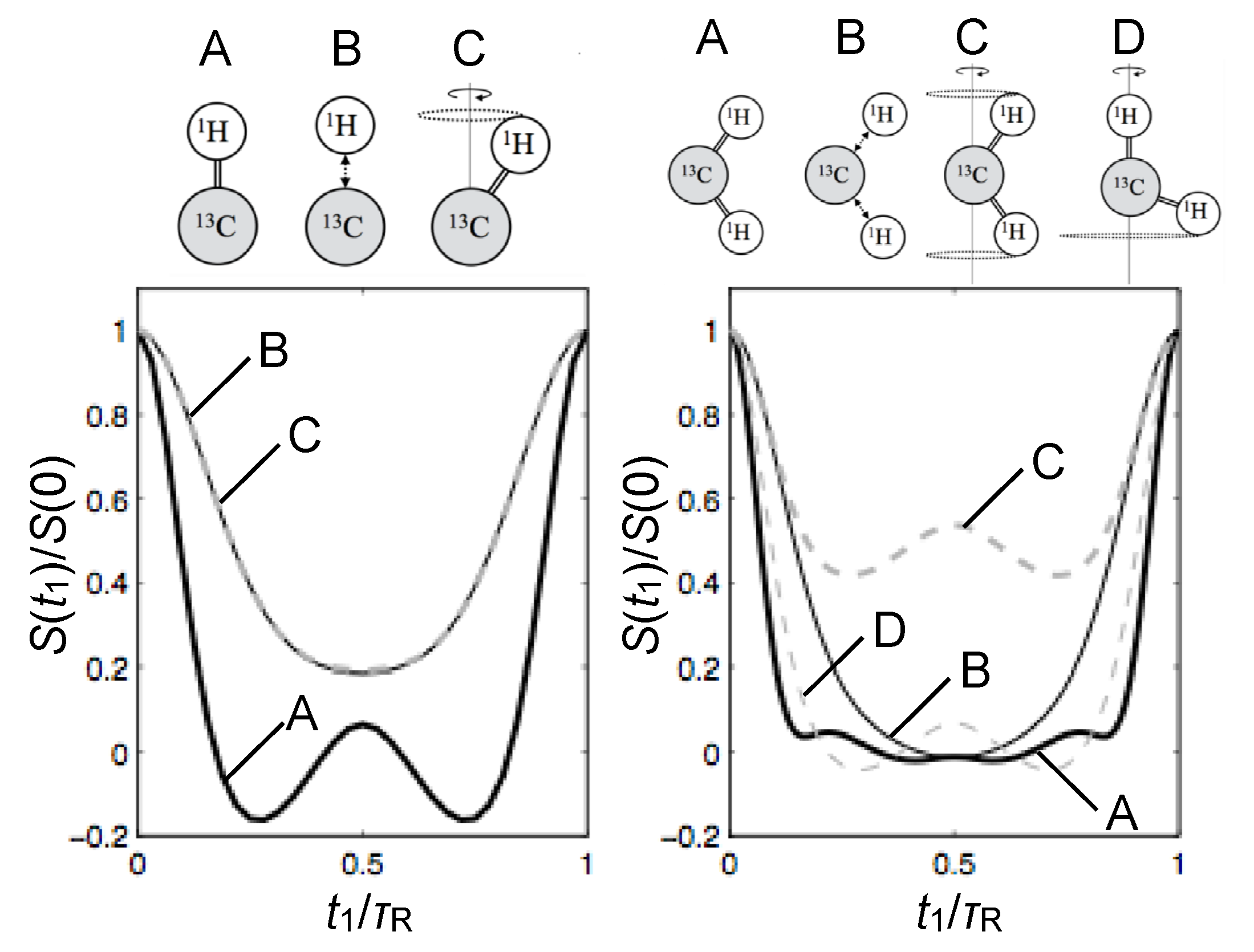

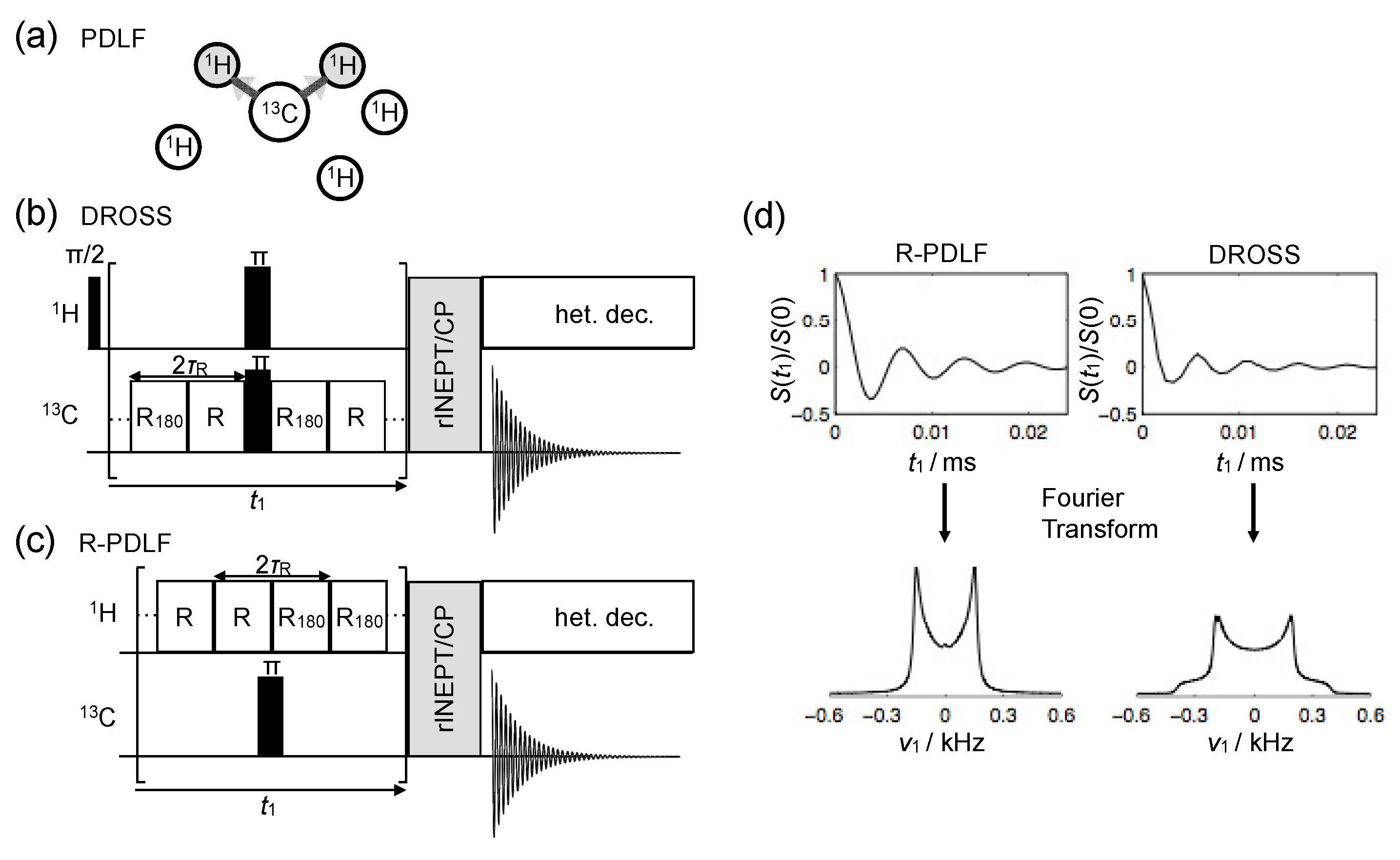
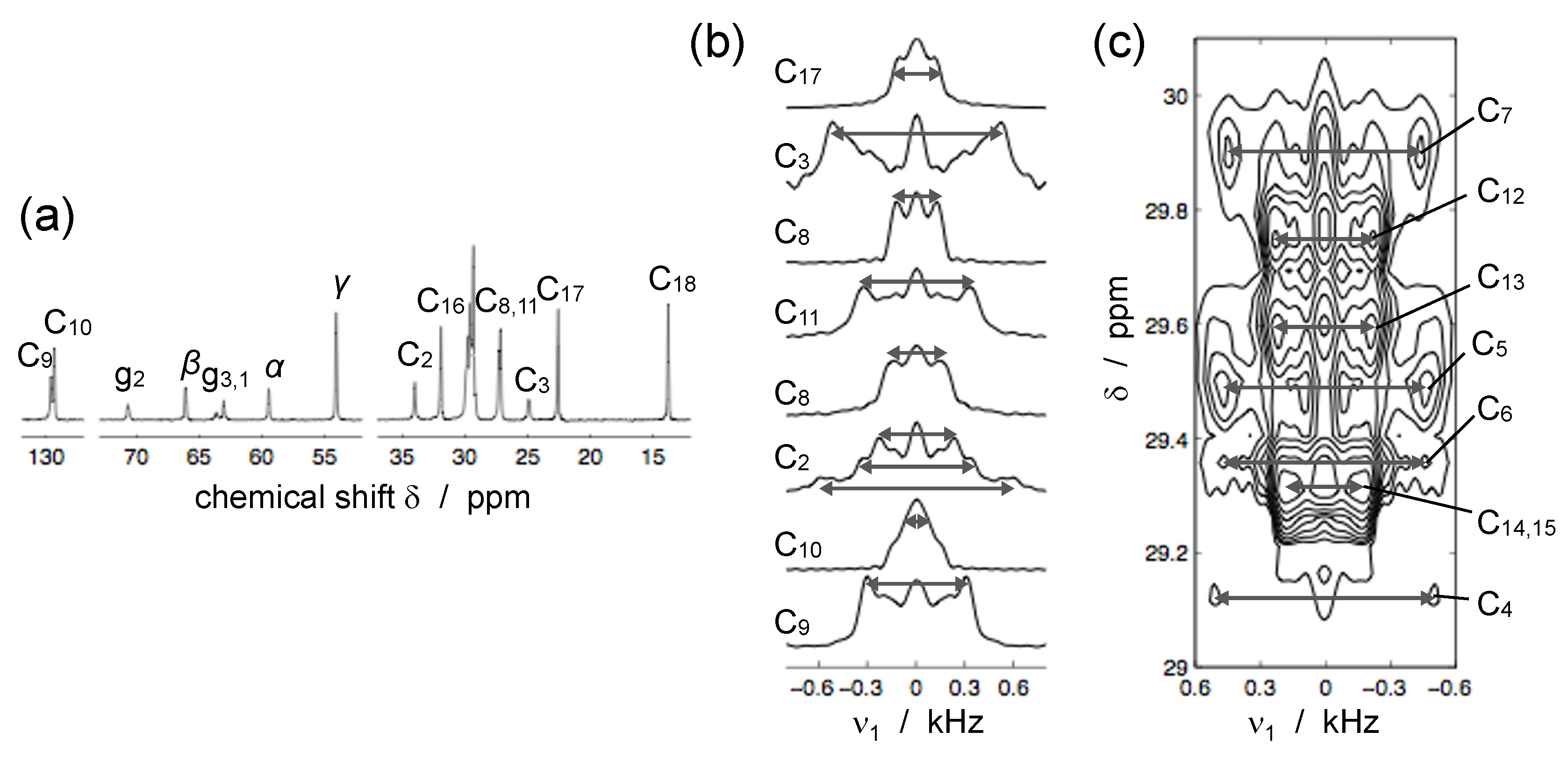
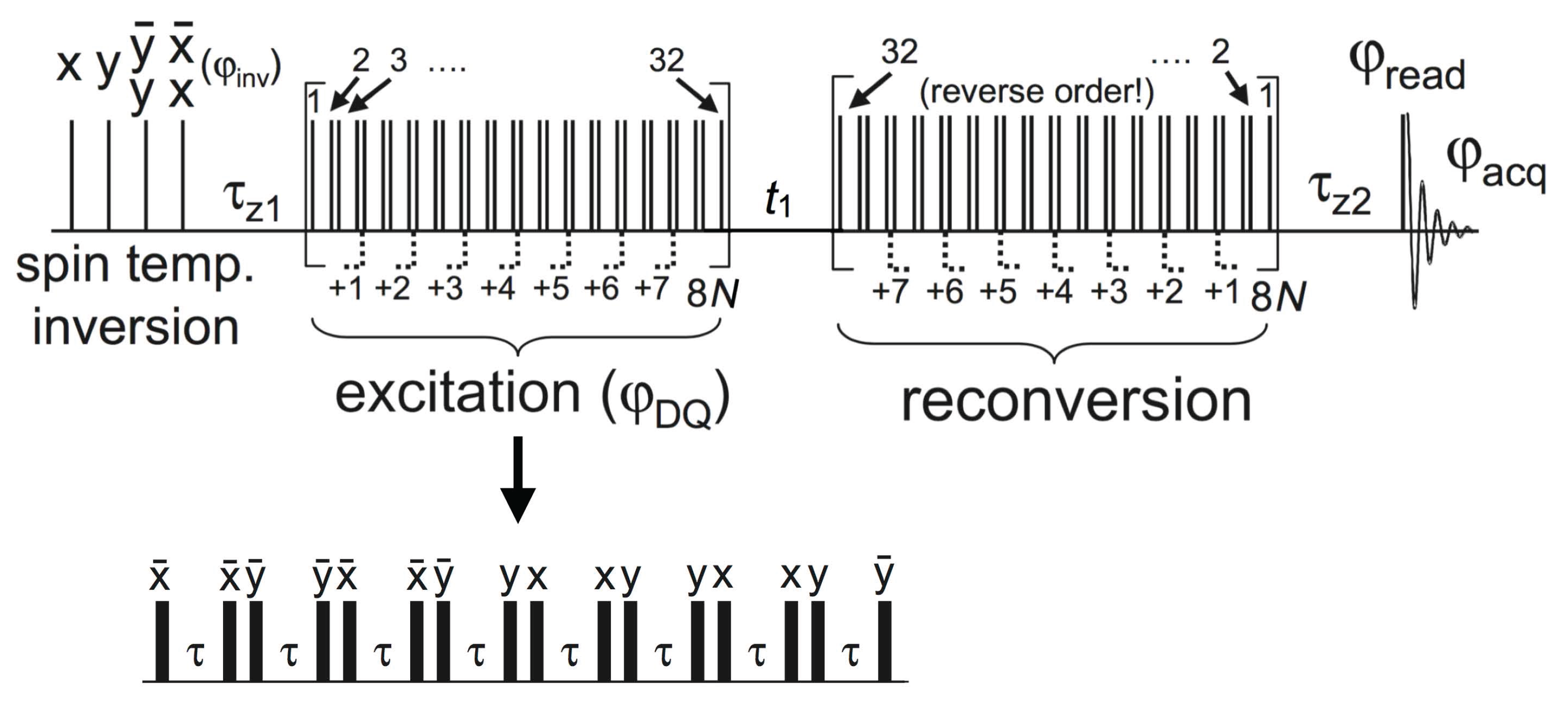
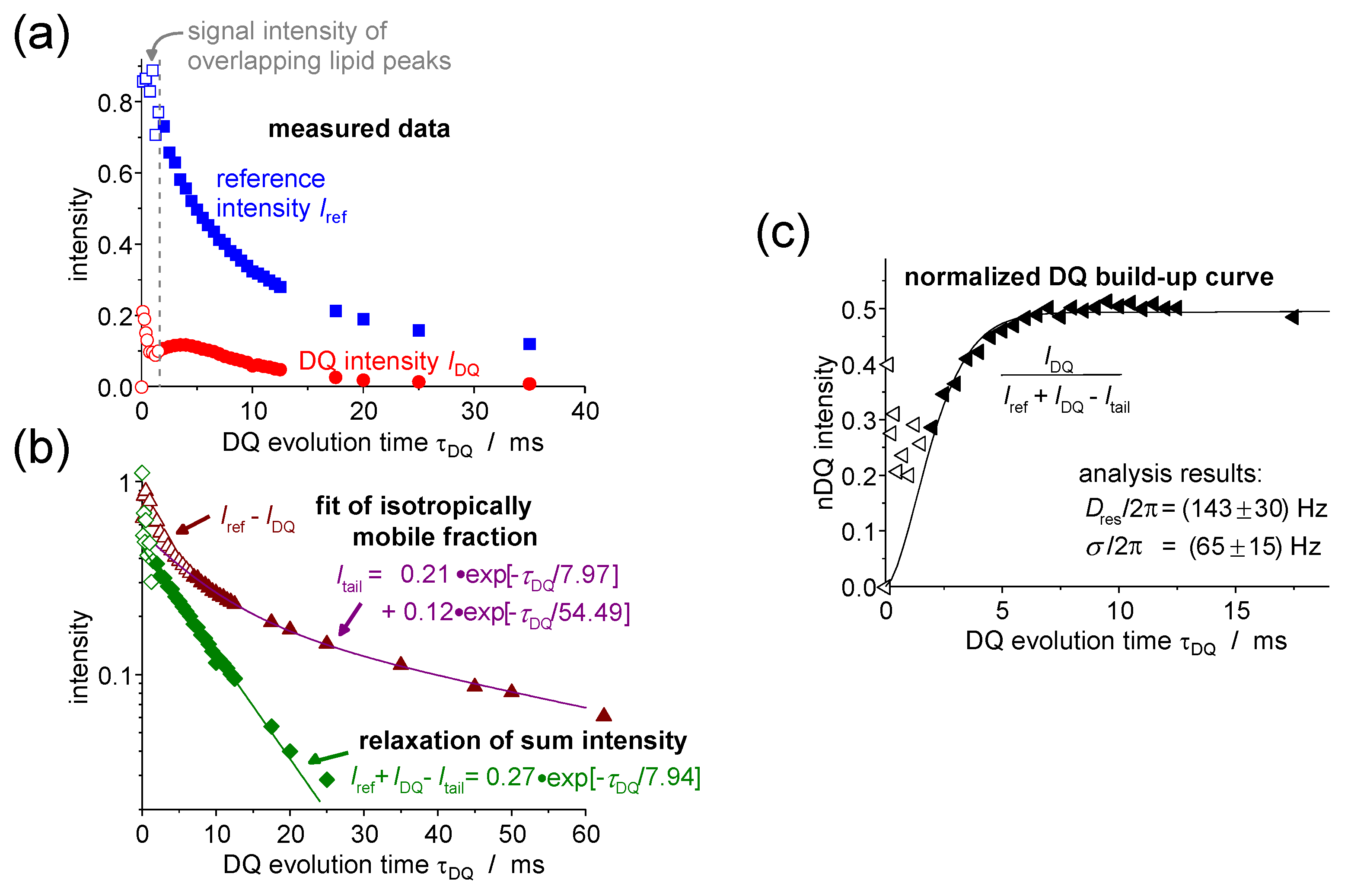
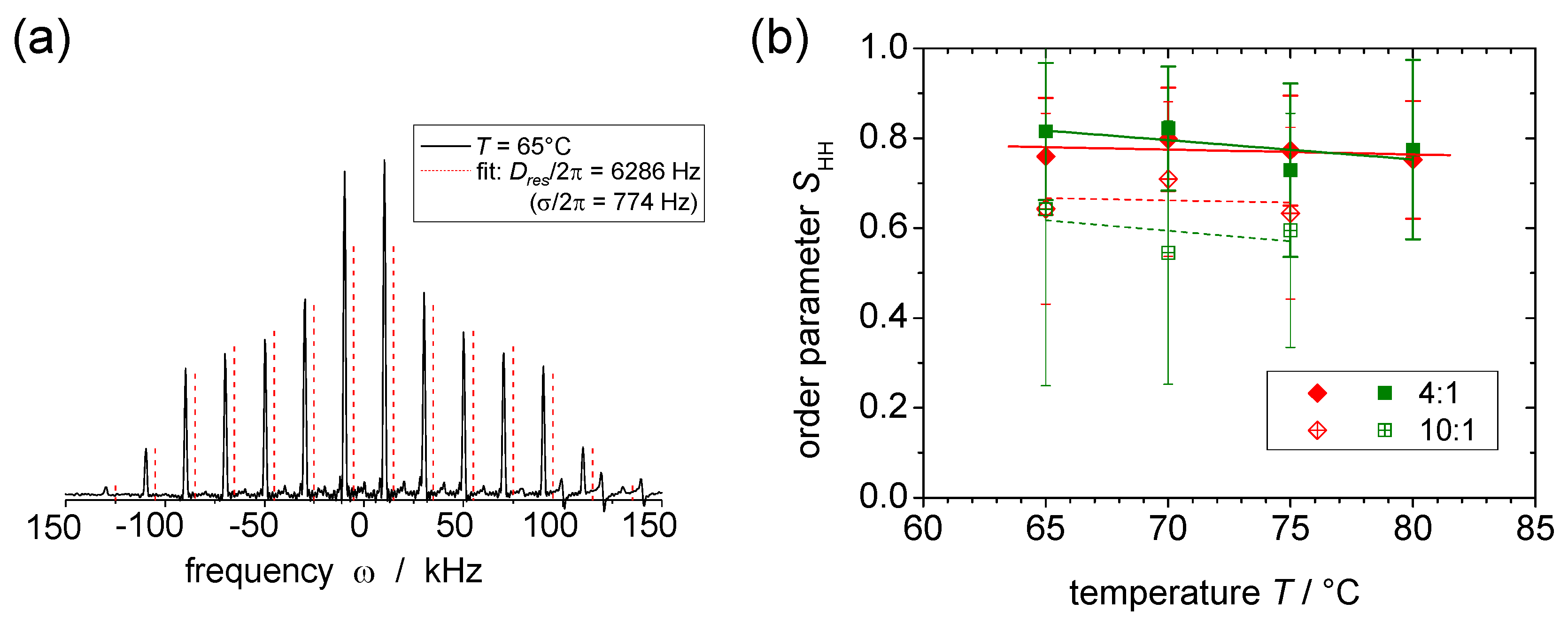
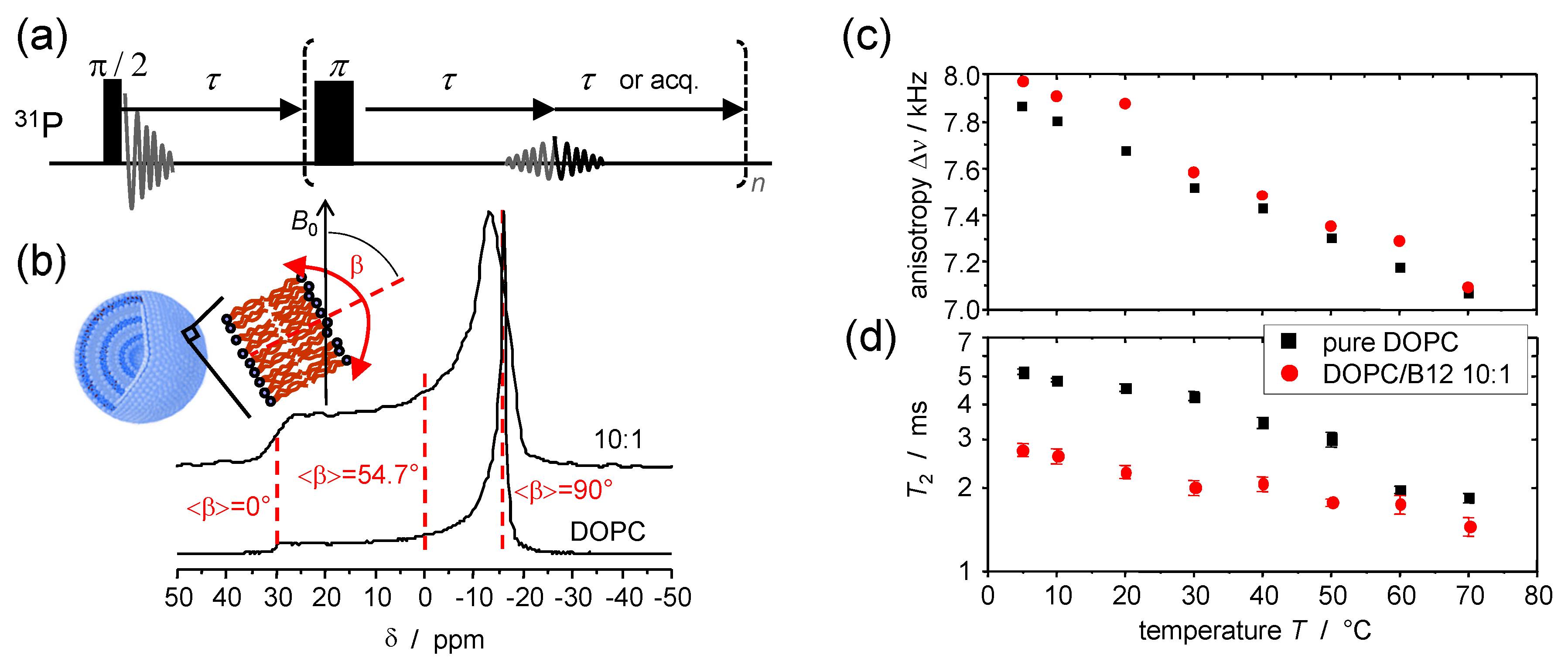
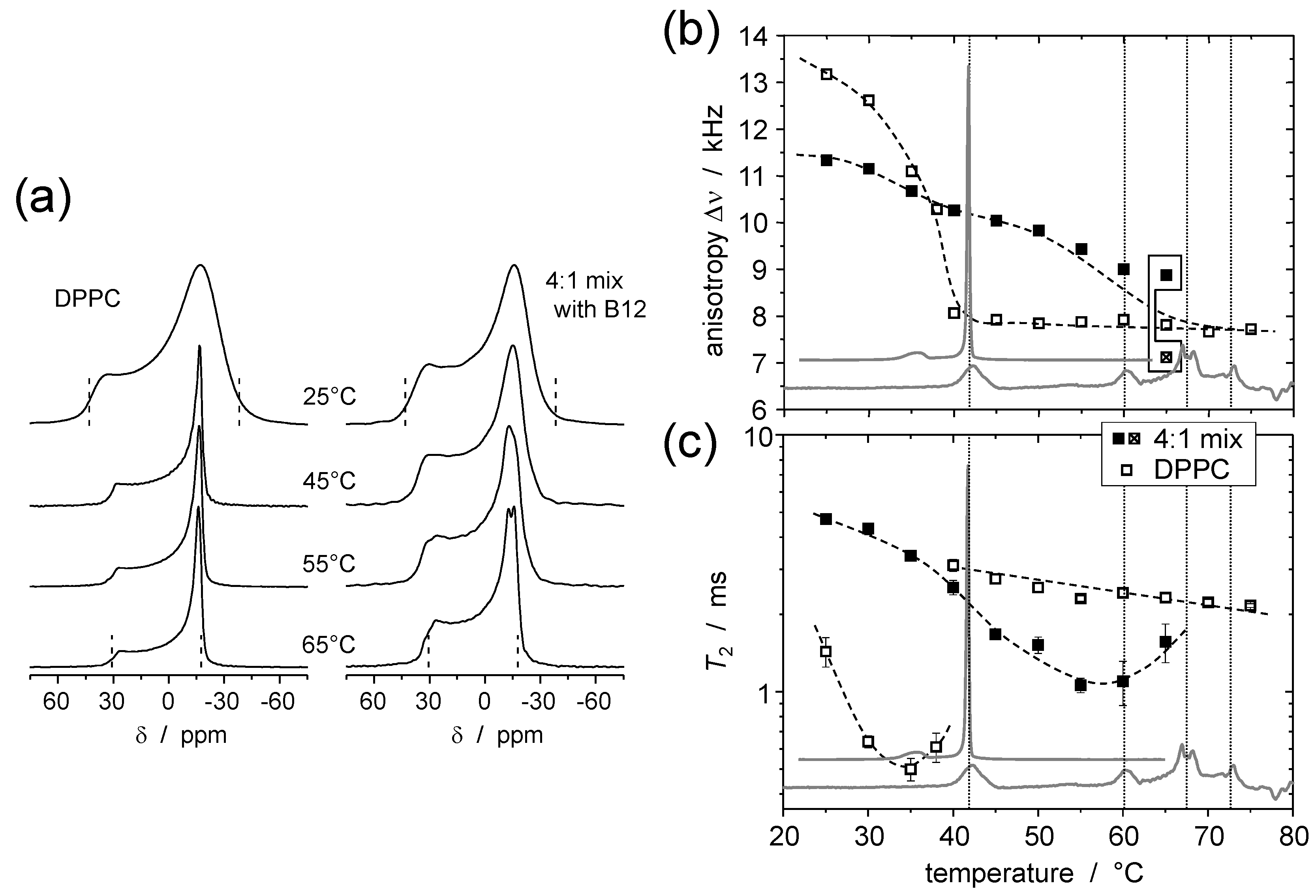
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bärenwald, R.; Achilles, A.; Lange, F.; Ferreira, T.M.; Saalwächter, K. Applications of Solid-State NMR Spectroscopy for the Study of Lipid Membranes with Polyphilic Guest (Macro)Molecules. Polymers 2016, 8, 439. https://doi.org/10.3390/polym8120439
Bärenwald R, Achilles A, Lange F, Ferreira TM, Saalwächter K. Applications of Solid-State NMR Spectroscopy for the Study of Lipid Membranes with Polyphilic Guest (Macro)Molecules. Polymers. 2016; 8(12):439. https://doi.org/10.3390/polym8120439
Chicago/Turabian StyleBärenwald, Ruth, Anja Achilles, Frank Lange, Tiago Mendes Ferreira, and Kay Saalwächter. 2016. "Applications of Solid-State NMR Spectroscopy for the Study of Lipid Membranes with Polyphilic Guest (Macro)Molecules" Polymers 8, no. 12: 439. https://doi.org/10.3390/polym8120439
APA StyleBärenwald, R., Achilles, A., Lange, F., Ferreira, T. M., & Saalwächter, K. (2016). Applications of Solid-State NMR Spectroscopy for the Study of Lipid Membranes with Polyphilic Guest (Macro)Molecules. Polymers, 8(12), 439. https://doi.org/10.3390/polym8120439