Enzyme-Catalyzed Modifications of Polysaccharides and Poly(ethylene glycol)


Abstract
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
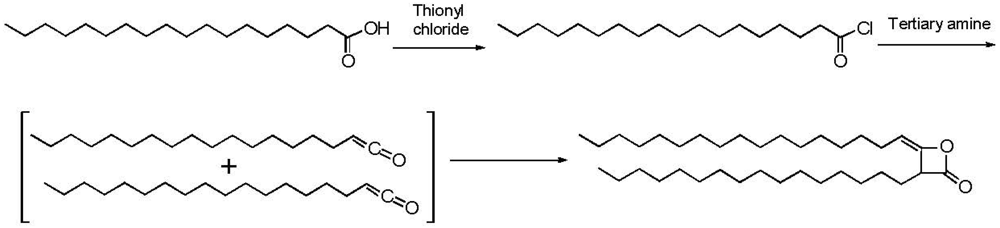{kind=link}
{kind=link}
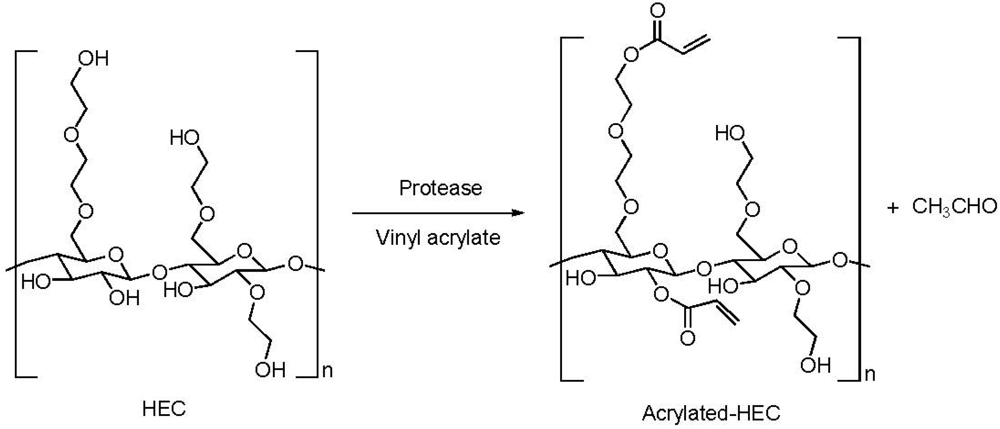{kind=link}
{kind=link}
{kind=link}
{kind=link}
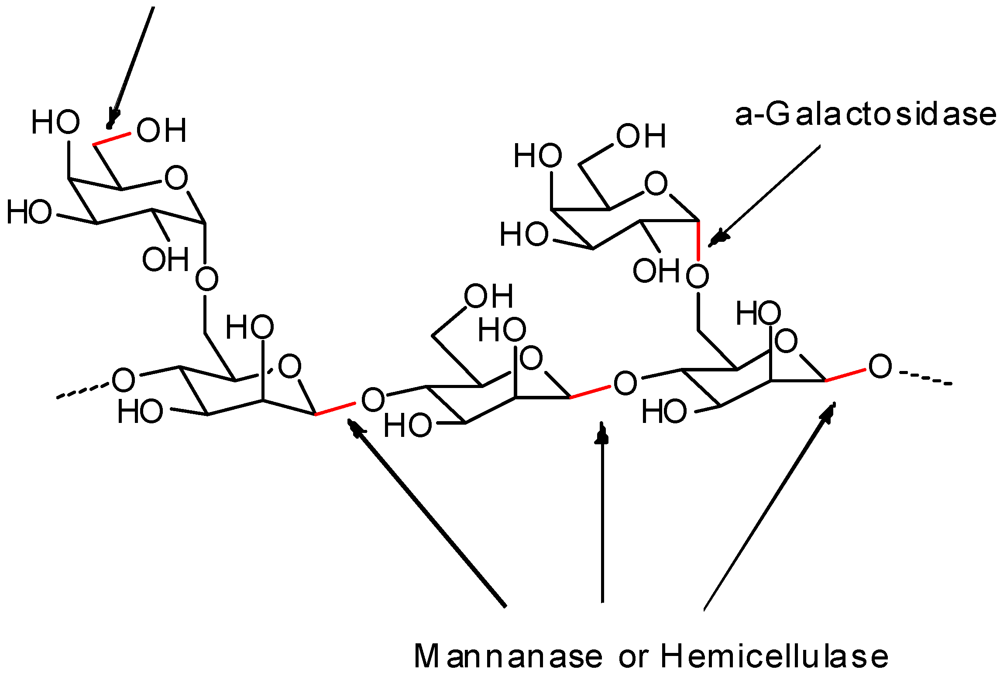{kind=link}
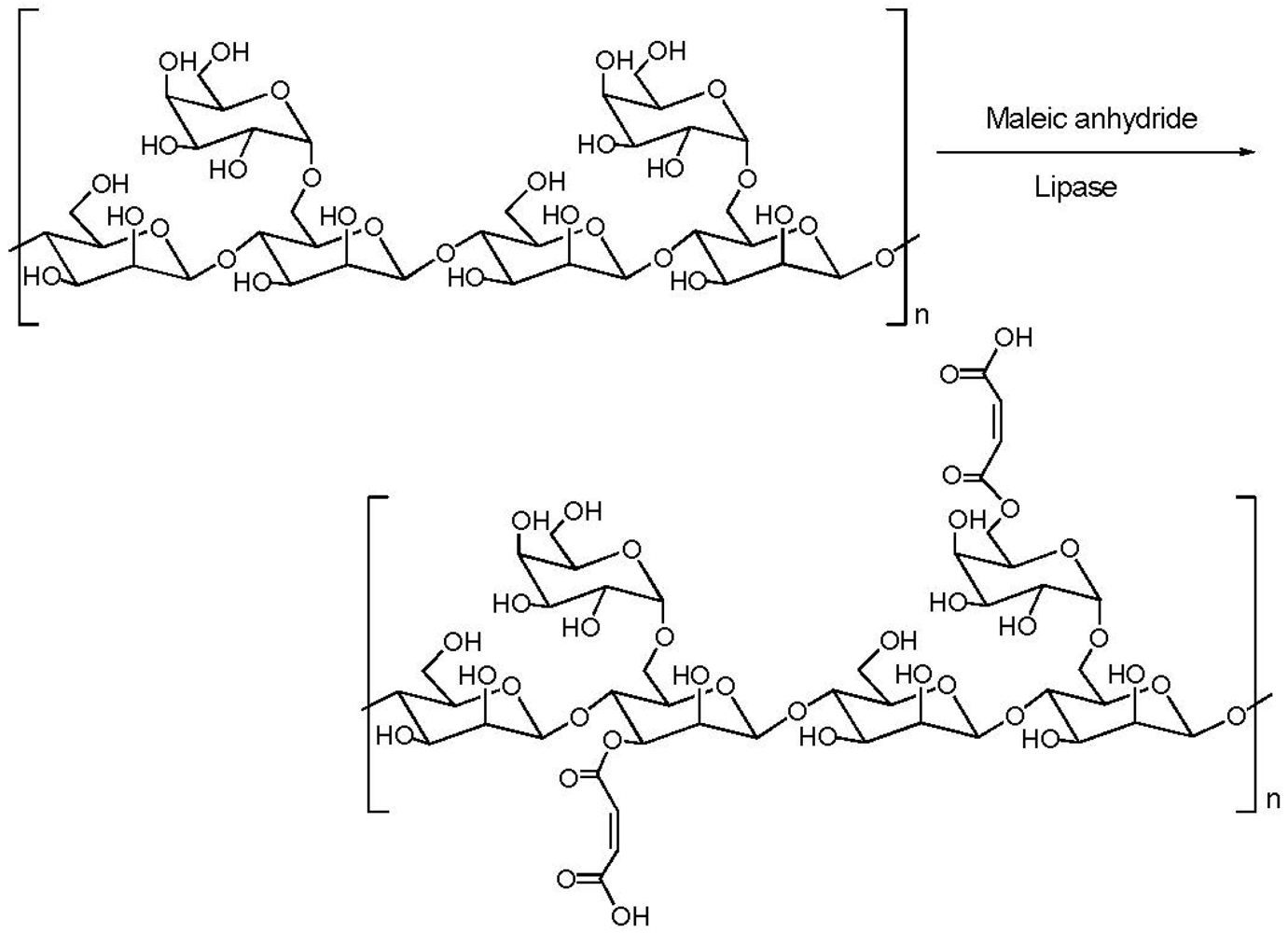{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
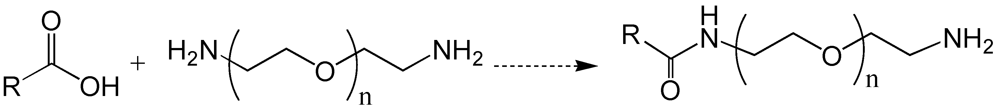{kind=link}
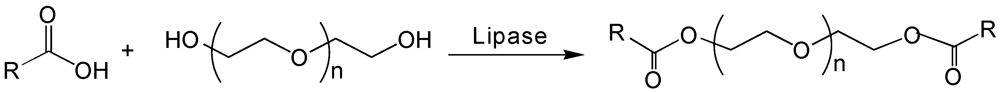{kind=link}
| Polymers | Structural Modification | Enzymatic Reaction | Property | Application |
|---|---|---|---|---|
| cellulosics | hydrolysis | oxidation | solubility | thickener |
| guar | - MW reduction | ester formation | rheology | gelling agent |
| starch | - side group cleavage | amidation | interfacial effects | emulsifier |
| pectin | addition | glycosylation | gel formation | flocculant |
| polyethers | polar substituent | hydrolysis | water retention | encapsulant |
| - charged functionality | tensile | stabilizer | ||
| - hydrophobe | texture | binder | ||
| - customized functionality | etc. | etc. |
2. Cellulosic Derivatives
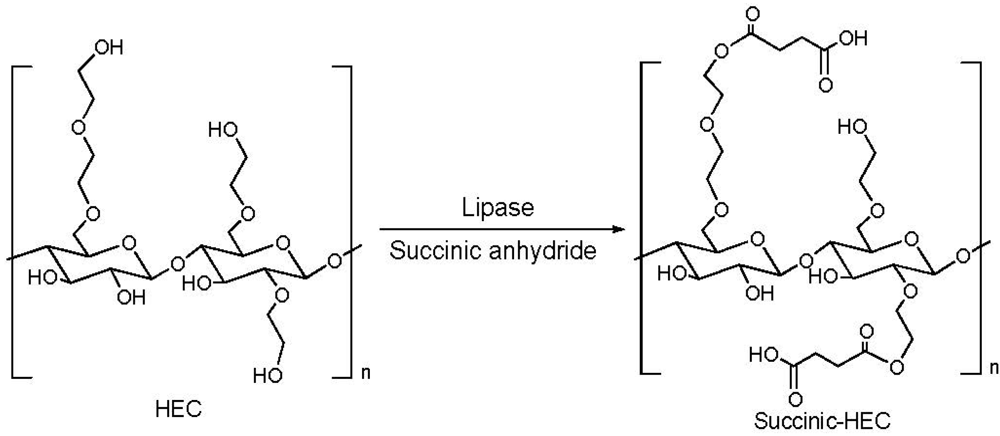
2.1. Addition of Polar Substituents
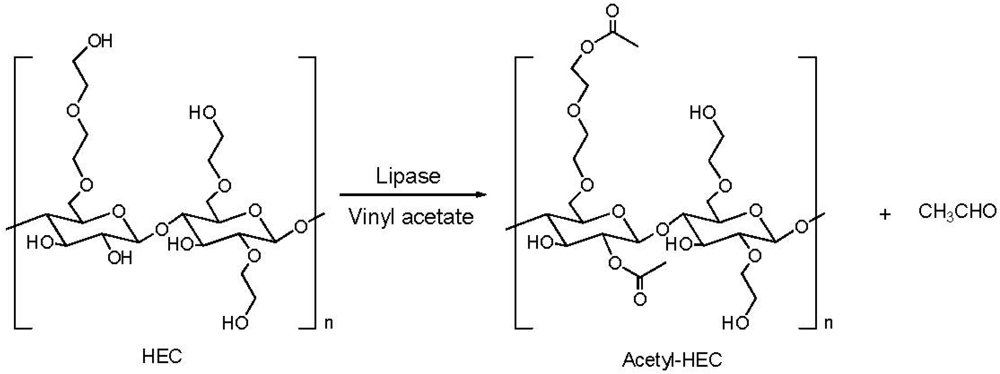
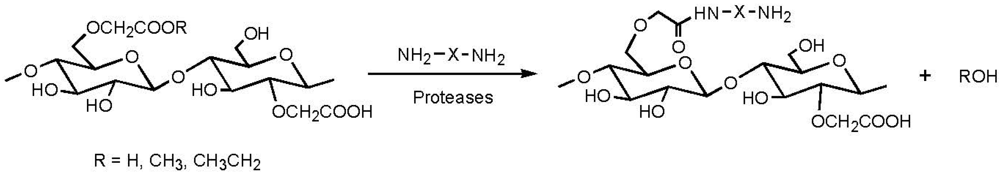
2.2. Addition of Charged Substituents
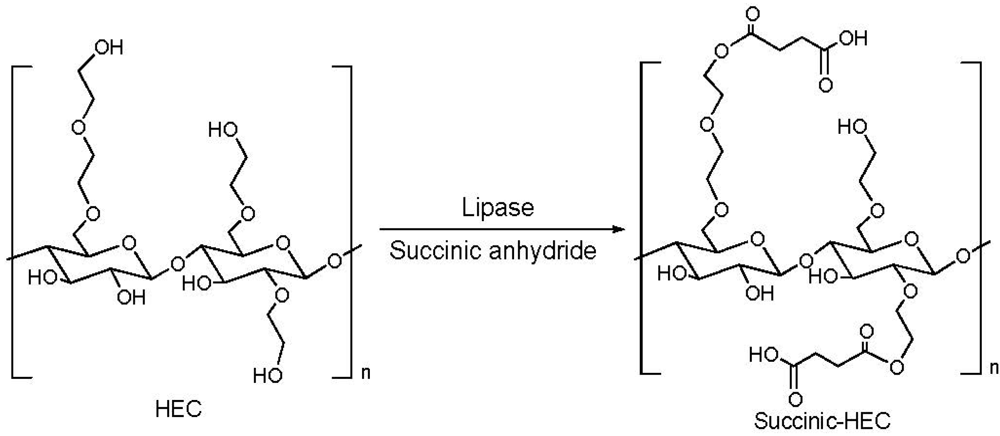
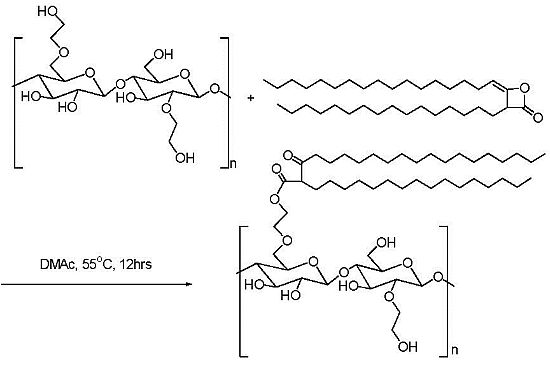
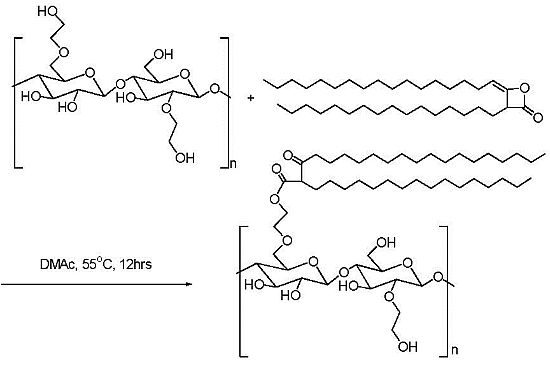
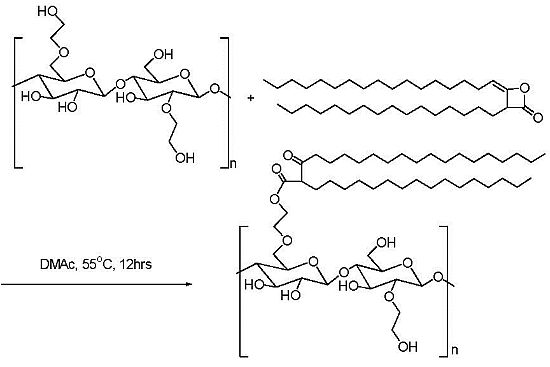
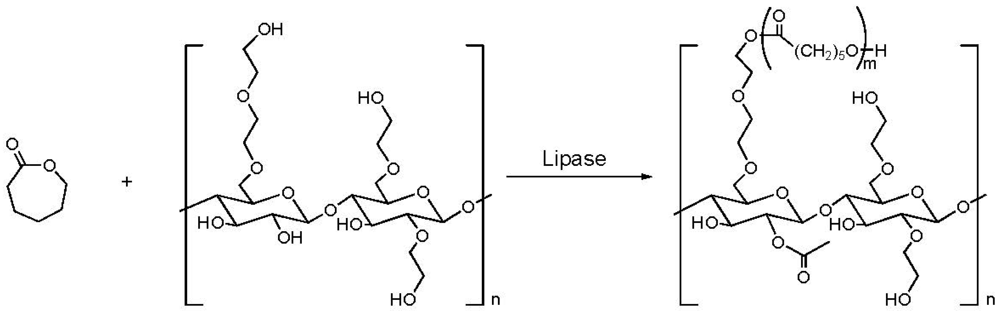
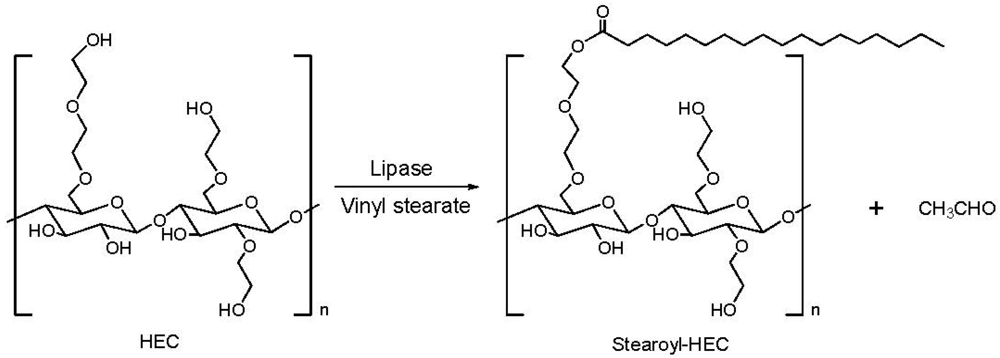
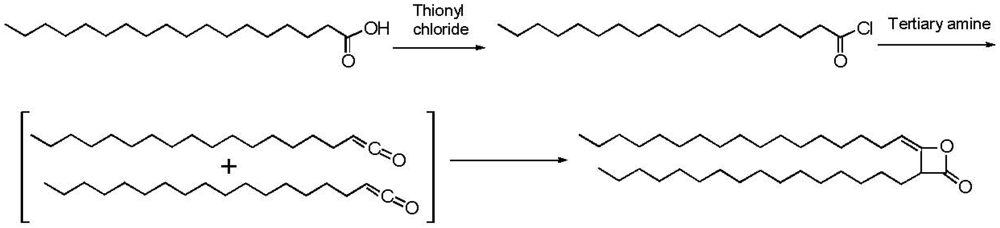
2.3. Addition of Hydrophobic Substituents
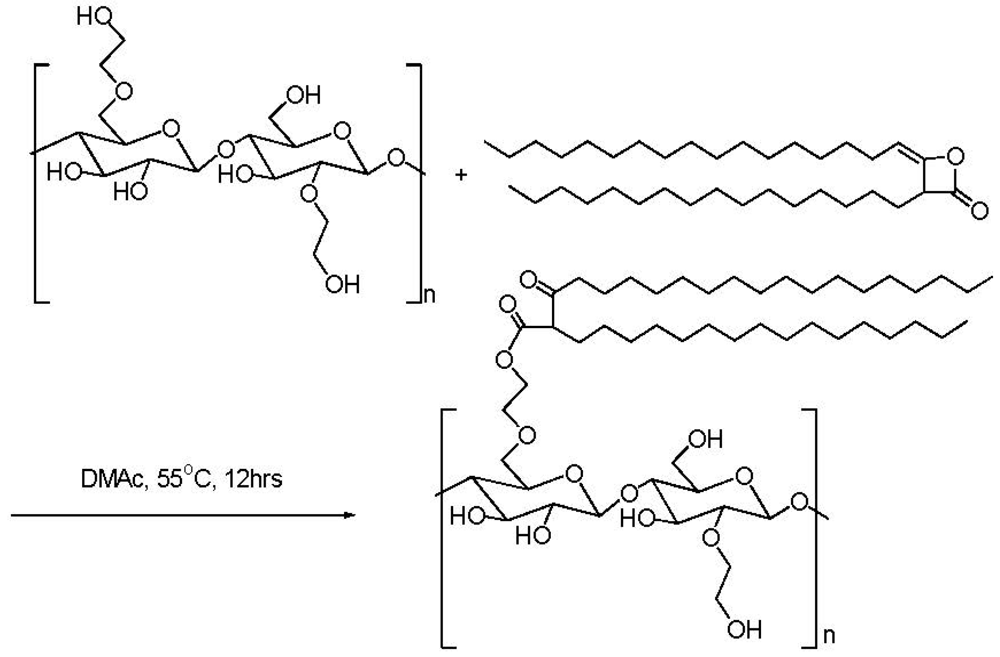
| HEC sample | Molecular weight | HEC BV (cps, % in water) | AKD/HEC Ester BV (cps, % in water) |
|---|---|---|---|
| 1 | 90,000 | <5 (2%) | 30 (2%) |
| 2 | 300,000 | 20 (1%) | 5,600 (1%) |
| 3 | 720,000 | 340 (1%) | 14,500 (1%) |
| 4 | 1,100,000 | 1,050 (1%) | 17,500 (1%) |
2.4. Addition of a Customized Functionality
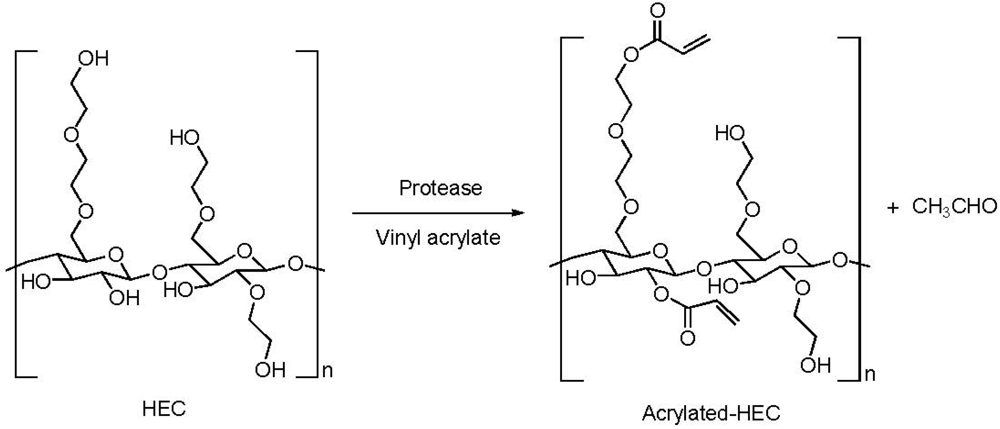
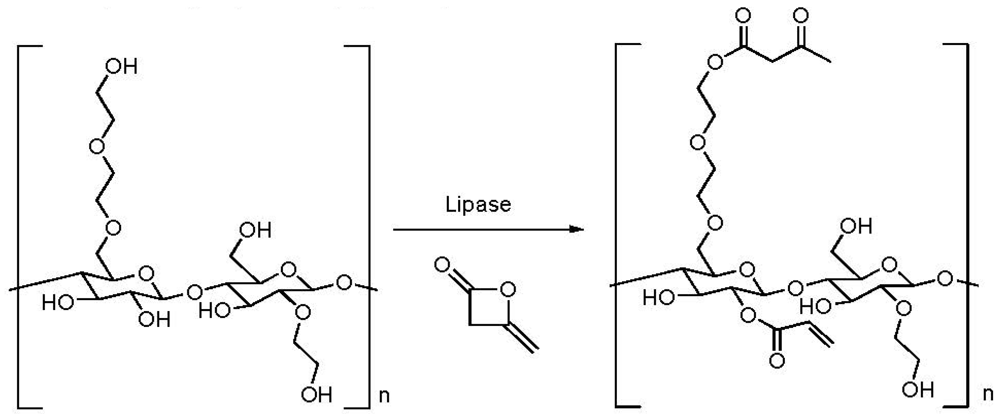
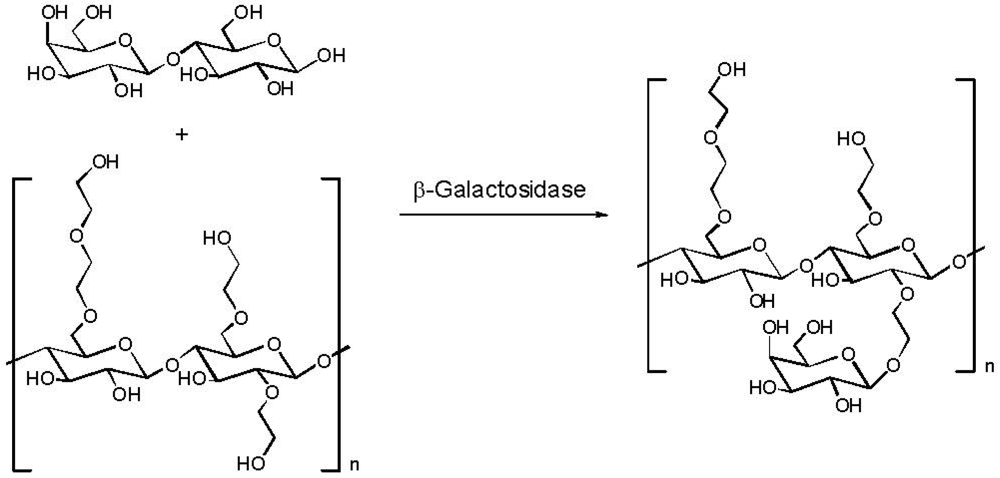
2.5. Reduction of Molecular Weight
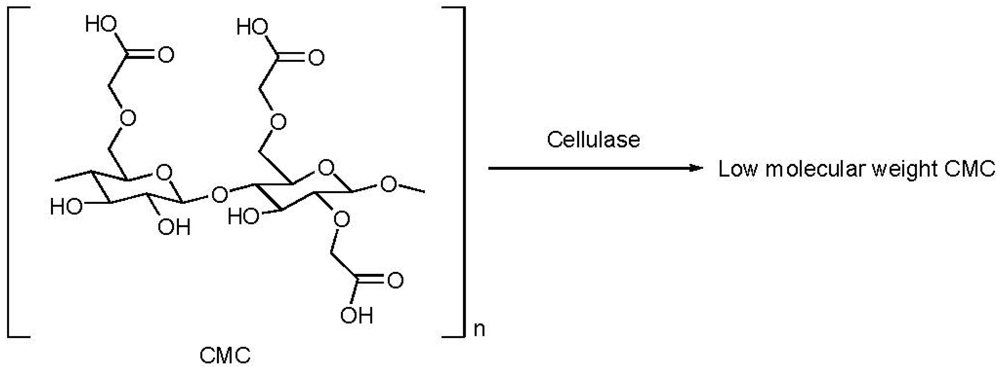
3. Guar
3.1. Hydrolysis Reactions
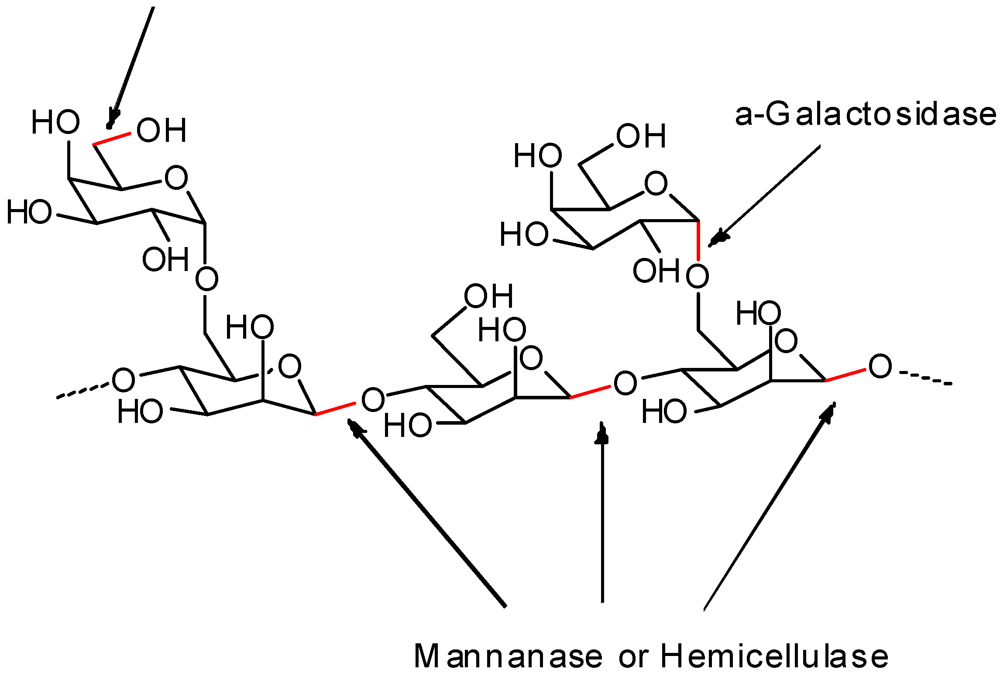
3.2. Addition of Charged Functionality
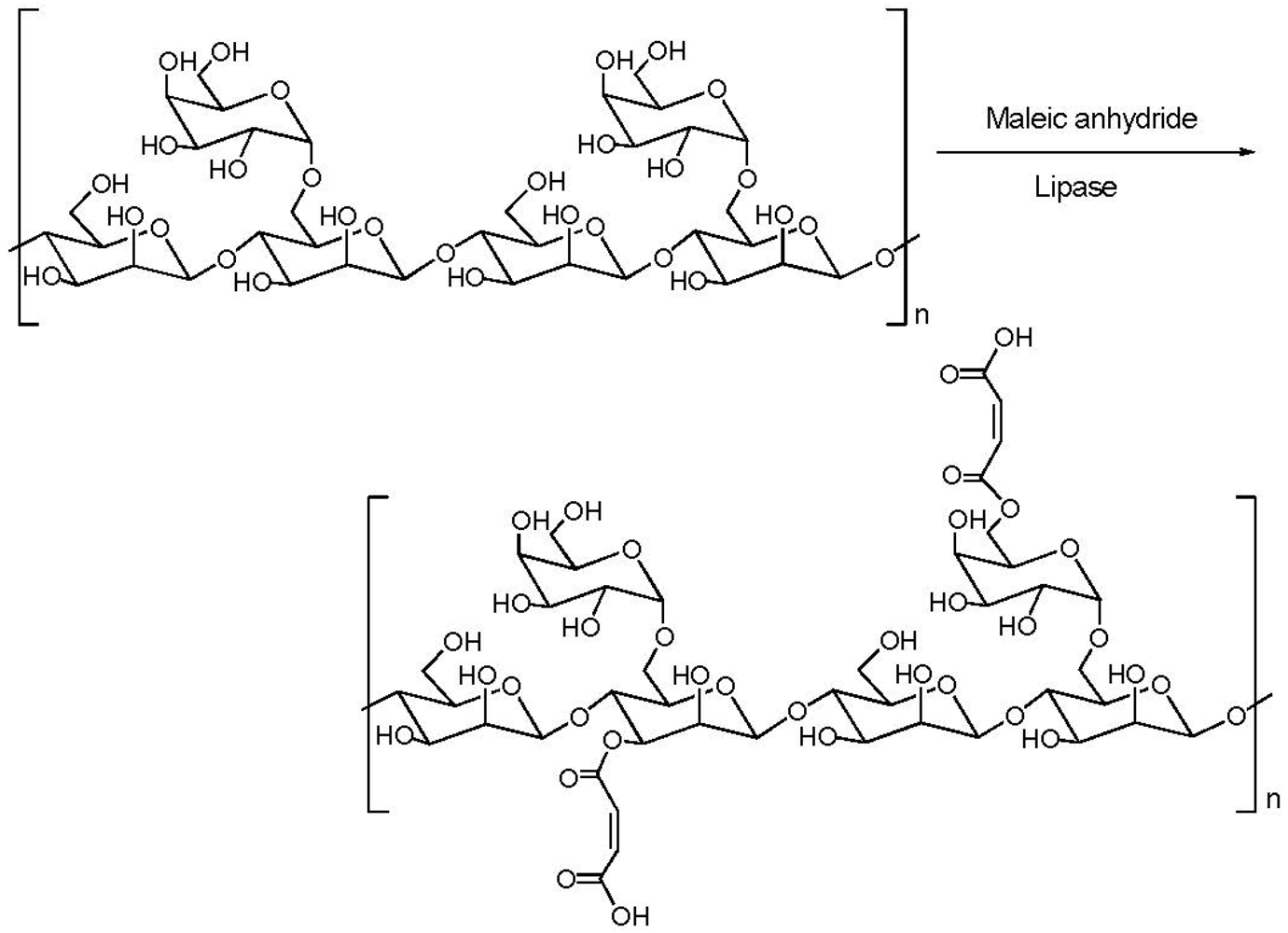
3.3. Addition of Hydrophobic Substitution
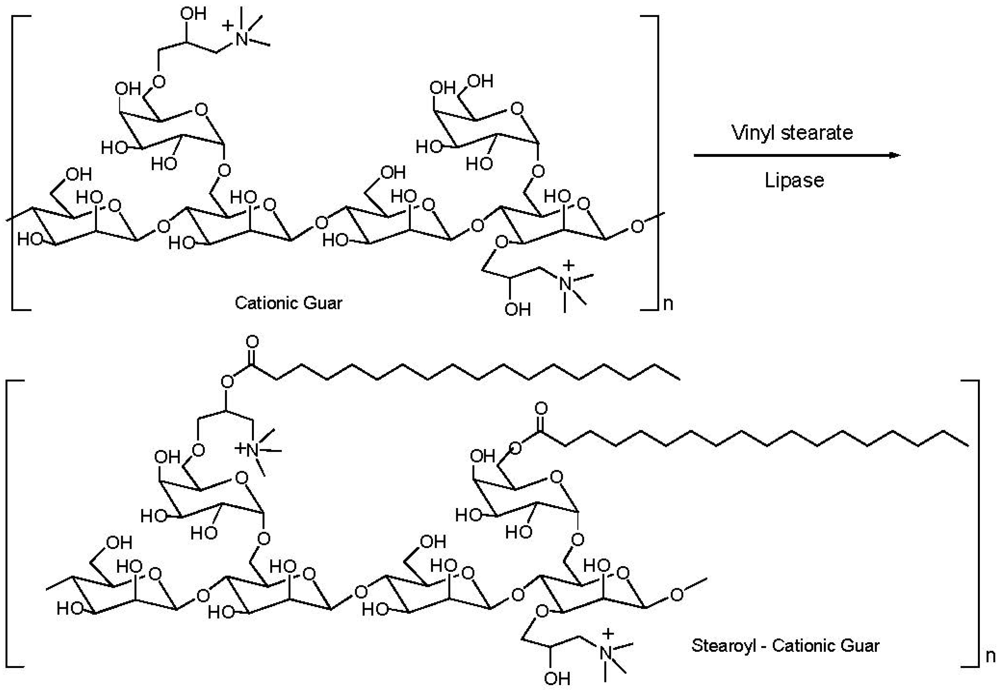
3.4. Addition of Customized Functionality
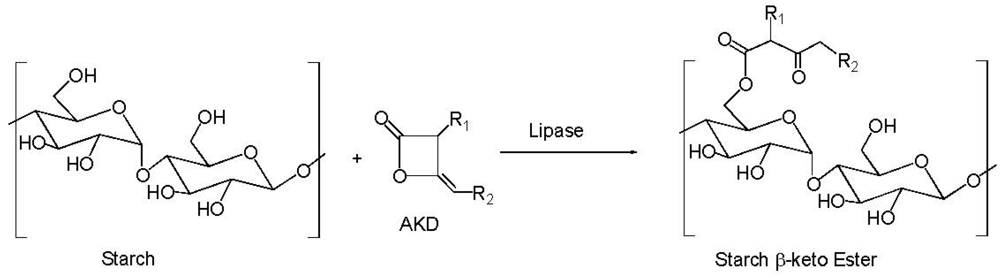
4. Starch
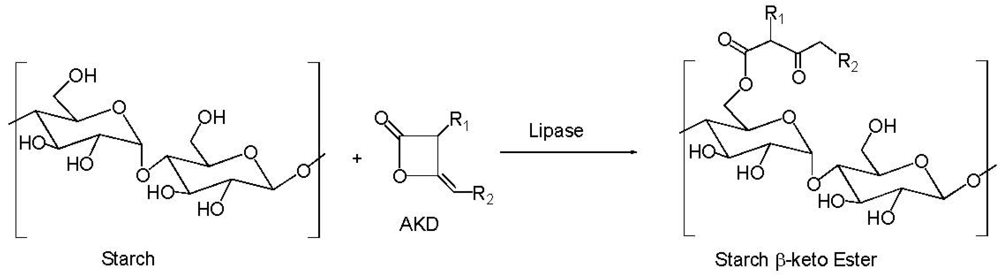
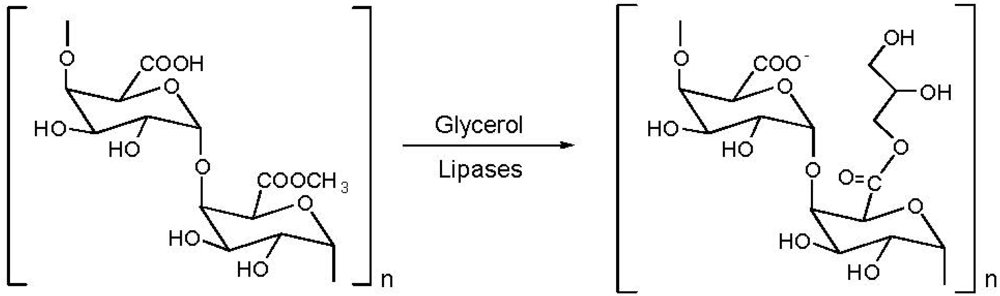
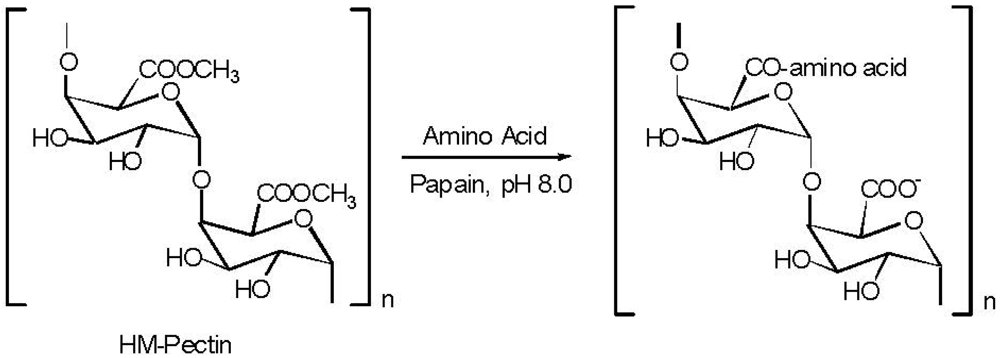
5. Pectin
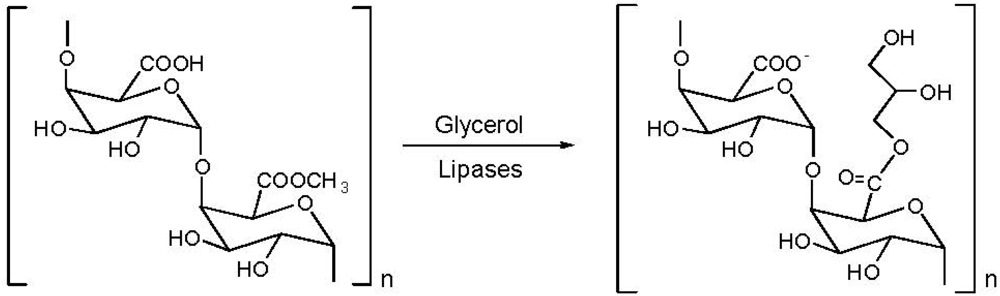
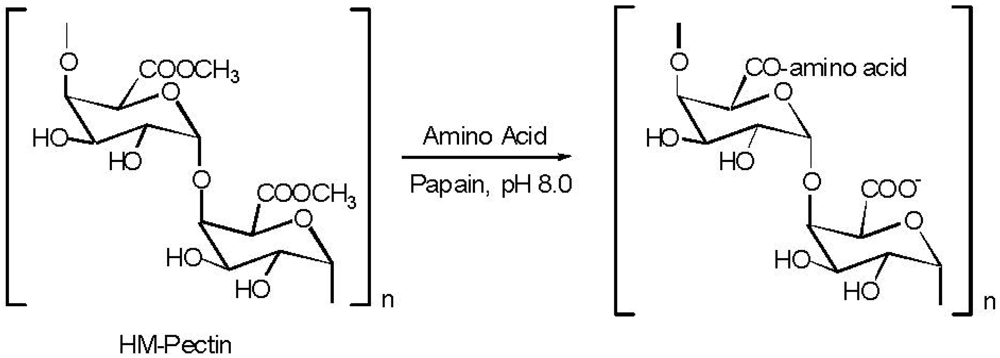
| Additive | Observation |
|---|---|
| Arginine | strong gel |
| Lysine | soft gel |
| Phenylalanine | cloudy liquid |
| Tyrosine | cloudy liquid |
| Tryptophan | cloudy liquid |
| Threonine | cloudy liquid |
| Serine | Liquid |
| Cysteine | Liquid |
| Alanine | Very soft gel after 3 h at rt. |
| 1-(3-Aminopropyl)imidazole | soft gel |
| Diethylene triamine | soft gel |
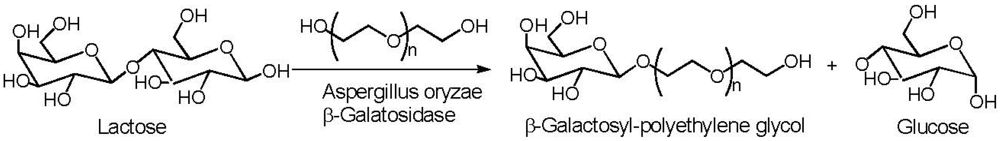
6. Poly(ethylene glycol)
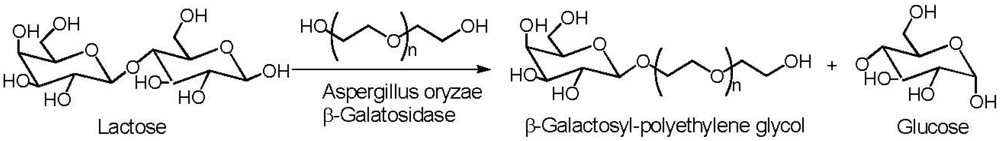
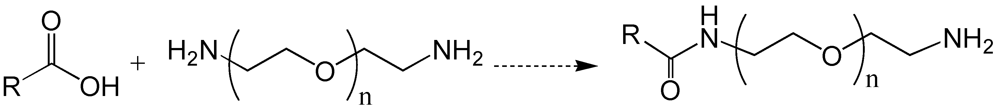
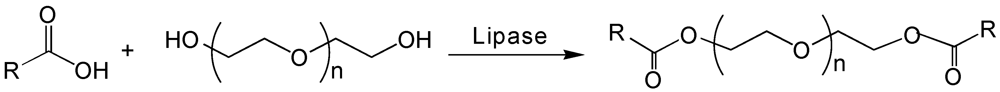
7. Conclusions
- (a) Most of the polysaccharide modifications need not be carried out to very high DS to give the desired end-use properties; thus, enzymatic reactions do not necessarily have to entail high conversion yields to be useful. For example, hydrophobic modification of HEC and guar only needs to be carried out to DS of 0.1 (or less) to produce a significant effect on physical properties, such as high viscosity and shear thinning behavior. Similarly, the DS for the amino acid or diamine substitution can be 0.2 (or less) to induce the formation of pectin gels.
- (b) If a desired polysaccharide modification is needed, a scouting study is usually first carried out to check for the feasibility with specific substrates and enzymes. In order to achieve best results, the modification reaction needs to be optimized with particular care paid to the choice of the enzyme and the solvent medium.
- (c) Most solvent media used are polar aprotic solvents, water, or mixed solvents. Preferably both the starting material and the final product are either soluble or swellable in the solvent media. Examples of often used solvents are DMF, DMAc, N-methyl pyrrolidone, butyl methyl ether, or aqueous buffer. If an aqueous buffer is used, the pH needs to be optimized for the reaction. (Note that each enzyme usually has its own optimal operating pH range.) Sometimes it is possible to carry out the enzymatic reaction without a solvent (e.g., HEC-caprolactone and pectin-glycerol); in such cases, the enzymes used for the reaction should be mostly dry but contain a trace amount of water for optimal results. A good way is to use a freeze dried enzyme for the modification reaction.
- (d) If a new enzymatic reaction is being attempted, it would be useful initially to use an artificially high level of an enzyme (as much as 10%–20% of the weight of the polysaccharide) together with different stoichiometries of the reactants. If the reaction is shown to work, then the enzyme level can be scaled back later in an optimization process. The stoichiometry of the reactants can also be optimized.
- (e) The reaction temperature can be varied in the optimization process. Note that most enzymes undergo increasing degrees of denaturation at higher temperatures (approximately 70 °C or higher). This upper temperature limit can be raised for special enzymes from extremophiles or from directed evolution, up to 90 °C in some cases.
- (f) Some enzymatic reactions involve the co-generation of a small molecule as a byproduct (e.g., acetaldehyde, methanol, and water). For these reactions, often it is possible to increase the reaction rate or yield by pulling a vacuum or decreasing the pressure in the reaction vessel in order to remove the small molecule. In the case of water, an alternative is to use a dehydrating agent (e.g., molecular sieves) in the reaction mixture.
- (g) Some enzymes can be expensive. In order to decrease the cost, the enzyme can be immobilized on a solid support and reused. Some commercial enzymes are already available on a solid support (e.g., Novozym® 435 lipase).
Acknowledgments
References
- Whistler, R.L.; BeMiller, J.N. Industrial Gums, 3rd ed; Academic Press: San Diego, CA, USA, 1993. [Google Scholar]
- Stephen, A.M.; Phillips, G.O.; Williams, P.A. Food Polysaccharides and Their Applications, 2nd ed; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Cheng, H.N.; Cote, G.L.; Baianu, I.C. Application of Polymers in Foods (Macromolecular Symposia 140); Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Heinze, T.J.; Glasser, W.G. Cellulose Derivatives. Modification, Characterization, and Nanostructures (ACS Symp. Ser. 688); American Chemical Society: Washington, DC, USA, 1998. [Google Scholar]
- Heinze, T.; Libert, T.; Koschella, A. Esterification of Polysaccharides; Springer-Verlag: Berlin, Germany, 2006. [Google Scholar]
- Ashland. Available online: http://www.ashland.com (accessed on 9 March 2012).
- Dow Wolff Cellulosics. Available online: http://www.dow.com/dowwolff/en/ (accessed on 9 March 2012).
- Loos, K. Biocatalysis in Polymer Chemistry; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Cheng, H.N.; Gross, R.A. Green Polymer Chemistry: Biocatalysis and Biomaterials (ACS Symp. Ser. 1043); American Chemical Society: Washington, DC, USA, 2010. [Google Scholar]
- Cheng, H.N.; Gross, R.A. Polymer Biocatalysis and Biomaterials II (ACS Symp. Ser. 999); American Chemical Society: Washington, DC, USA, 2008. [Google Scholar]
- Roberts, S.M. Biocatalysis for Fine Chemicals Synthesis; Wiley: Chichester, UK, 1999. [Google Scholar]
- Faber, K. Biotransformations in Organic Chemistry, 3rd ed; Springer: Berlin, Germany, 1997. [Google Scholar]
- Drauz, K.; Waldemann, H. Enzyme Catalysis in Organic Synthesis; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Gu, Q.-M.; Cheng, H.N. Enzyme-catalyzed condensation reactions for polymer modification. ACS Symp. Ser. 2005, 900, 427–436. [Google Scholar]
- Gu, Q.-M.; Cheng, H.N. Enzyme-Catalyzed reactions of polysaccharides. ACS Symp. Ser. 2003, 840, 203–216. [Google Scholar]
- Gubitz, G.M.; Paulo, A.C. New substrates for reliable enzymes: Enzymatic modification of polymers. Curr. Opin. Biotechnol. 2003, 14, 577–582. [Google Scholar] [CrossRef]
- McCleary, B.V. Enzymatic modification of plant polysaccharides. Int. J. Biol. Macromol. 1986, 8, 349–354. [Google Scholar] [CrossRef]
- Li, J.; Xie, W.; Cheng, H.N.; Nickol, R.G.; Wang, P.G. Polycaprolactone-modified hydroxyethylcellulose films prepared by lipase-catalyzed ring-opening polymerization. Macromolecules 1999, 32, 2789–2792. [Google Scholar]
- Hu, S.; Gao, W.; Kumar, R.; Gross, R.A.; Gu, Q.-M.; Cheng, H.N. Lipase-mediated selective TEMPO oxidation of hydroxyethylcellulose. ACS Symp. Ser. 2003, 840, 253–264. [Google Scholar]
- Cheng, H.N.; Gu, Q.-M. Enzymatic modifications of water-soluble polymers. ACS Polym. Prepr. 2000, 41, 1873–1874. [Google Scholar]
- Glass, J.E. Associative Polymers in Aqueous Media (ACS Symp. Ser. 765); American Chemical Society: Washington, DC, USA, 2000. [Google Scholar]
- Landoll, L.M. Nonionic polymer surfactants. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 443–455. [Google Scholar] [CrossRef]
- Emmons, W.D.; Stevens, T.E. Polyurethane thickeners in latex compositions. U.S. Patent 4,079,028, 14 March 1978. [Google Scholar]
- Lundberg, D.J.; Brown, R.G.; Glass, J.E.; Eley, R.R. Synthesis, characterization, and solution rheology of model hydrophobically-modified, water-soluble ethoxylated urethanes. Langmuir 1994, 10, 3027–3034. [Google Scholar] [CrossRef]
- Shay, G.D.; Eldridge, E.; Kail, E. Alkali soluble latex thickeners. U.S. Patent 4,514,552, 30 April 1985. [Google Scholar]
- Shay, G.D.; Kravitz, F.K.; Brizgys, P.V.; Kersten, M.A. Production of alkali-soluble, carboxyl-functional aqueous emulsion thickeners. U.S. Patent 4,801,671, 31 January 1989. [Google Scholar]
- Shay, G.D.; Kravitz, F.K.; Brizgys, P.V. Effects of process variables on the emulsion and solution properties of hydrophobically modified alkali-swellable emulsion thickeners. ACS Symp. Ser. 1991, 462, 121–141. [Google Scholar]
- Gu, Q.-M. Lipase-catalyzed grafting reactions on polysaccharides. ACS Symp. Ser. 2003, 840, 243–247. [Google Scholar]
- Reynolds, W.F. The Sizing of Paper, 2nd ed; TAPPI: Atlanta, GA, USA, 1989. [Google Scholar]
- Lindstrom, T.; Larsson, P.T. Alkyl ketene dimer (AKD) sizing—a review. Nordic Pulp Paper Res. J. 2008, 23, 202–209. [Google Scholar]
- Cheng, H.N.; Gu, Q.-M. Esterified polysaccharide products with ketene dimer by beta-lactone ring opening. U.S. Patent 6,528,643, 4 March 2003. [Google Scholar]
- Gu, Q.-M.; Cheng, H.N. Enzyme-catalyzed esterification of cellulosics, guar, and polyethers. ACS Polym. Prepr. 2005, 46, 30–31. [Google Scholar]
- Cheng, H.N.; Gu, Q.-M.; Qiao, L. Reactions of enzymes with non-substrate polymers. ACS Symp. Ser. 2005, 900, 267–278. [Google Scholar]
- Wang, P.G.; Xie, W.; Qiao, L.; Nickol, R.G.; Cheng, H.N. Actoacetylated saccharides and process of making the same. U.S. Patent 6,528,644, 4 March 2003. [Google Scholar]
- Gu, Q.-M. Enzyme-mediated reactions of oligosaccharides and polysaccharides. ACS Symp. Ser. 2001, 786, 184–194. [Google Scholar]
- Li, J.; Cheng, H.N.; Nickol, R.G.; Wang, P.G. Enzymatic modification of hydroxyethylcellulose by transgalactosylation with beta-galactosidase. Carbohydr. Res. 1999, 316, 133–137. [Google Scholar] [CrossRef]
- Cheng, H.N.; Li, J.; Nickol, R.G.; Wang, P.G. Galactosylated hydroxyalkyl polysaccharides and their derivatives and enzymatic process for manufacture. U.S. Patent 6,433,161, 13 August 2002. [Google Scholar]
- Vink, H. Degradation of some polymers in aqueous solutions. Makromol. Chem. 1963, 67, 105–123. [Google Scholar] [CrossRef]
- Klemm, D.; Philipp, B.; Heinze, T.; Heinze, U.; Wagenknecht, W. Comprehensive Cellulose Chemistry; Wiley-VCH: Weinheim, Germany, 1998. [Google Scholar]
- Ott, E.; Spurlin, H.M.; Grafflin, M.W. Cellulose and Cellulose Derivatives; Interscience: New York, NY, USA, 1954. [Google Scholar]
- Saake, B.; Horner, S.; Puls, J. Progress in the enzymatic hydrolysis of cellulose derivatives. ACS Symp. Ser. 1998, 688, 201–216. [Google Scholar]
- Sau, A.C. Biostable water-borne paints and processes for their preparation. U.S. Patent 5,879,440, 8 March 1999. [Google Scholar]
- Tayal, A.; Kelly, R.M.; Khan, S.A. Rheology and molecular weight changes during enzymatic degradation of a water-soluble polymer. Macromolecules 1999, 32, 294–300. [Google Scholar] [CrossRef]
- Cheng, Y.; Prud’homme, R.K. Reaction-diffusion of enzyme molecules in biopolymer matrices. ACS Symp. Ser. 2003, 840, 265–284. [Google Scholar]
- Mahammad, S.; Prudhomme, R.K.; Roberts, G.W.; Khan, S.A. Kinetics of enzymatic depolymerizaton of guar galactomannan. Biomacromolecules 2006, 7, 2583–2590. [Google Scholar] [CrossRef]
- Cheng, H.N.; Gu, Q.-M. Biotransformation of Polysaccharides. In Glycochemistry: Principles, Synthesis, and Applications; Wang, P.G., Bertozzi, C.R., Eds.; Marcel Dekker: New York, NY, USA, 2001; pp. 567–579. [Google Scholar]
- MacCleary, B.V.; Critchley, P.; Bulpin, P.V. Process of polysaccharides. U.S. Patent 5,234,825, 10 August 1993. [Google Scholar]
- Bulpin, P.V.; Gidley, M.J.; Jeffcoat, R.; Underwood, D.R. Development of a biotechnological process for the modification of galactomannan polymers with plant α-galactosidase. Carbohydr. Polym. 1990, 12, 155–168. [Google Scholar] [CrossRef]
- Shobha, M.S.; Tharanathan, R.N. Rheological behavior of pullulanase-treated guar galactomannan on co-gelation with xanthan. Food Hydr. 2009, 23, 749–754. [Google Scholar] [CrossRef]
- Shobha, M.S.; Tharanathan, R.N. Nonspecific activity of Bacillus acidopullulyticus pullulanase on debranching of guar galactomannan. J. Agric. Food. Chem. 2008, 56, 10858–10864. [Google Scholar]
- Shobha, M.S.; Kumar, A.B.V.; Tharanathan, R.N.; Koka, R.; Gaonkar, A.K. Modification of guar galactomannan with the aid of Aspergillus niger pectinase. Carbohydr. Polym. 2005, 62, 267–273. [Google Scholar] [CrossRef]
- Mahammad, S.; Comfort, D.A.; Kelly, R.M.; Khan, S.A. Rheological properties of guar galactomannan solutions during hydrolysis with galactomannanase and alpha-galactosidase enzyme mixtures. Biomacromolecules 2007, 8, 949–956. [Google Scholar] [CrossRef]
- Crescenzi, V.; Dentini, M.; Risica, D.; Spadoni, S.; Skjak-Braek, G.; Capitani, D.; Mannina, L. Viel, S. C(6)-oxidation followed by C(5)-epimerization of guar gum studied by high field NMR. Biomacromolecules 2005, 5, 537–546. [Google Scholar]
- Yalpani, M.; Hall, L.D. Some chemical and analytical aspects of polysaccharide modifications. II. A high-yielding, specific method for the chemical derivatization of galactose-containing polysaccharides: Oxidation with galactose oxidase followed by reductive amination. J. Polym. Sci. Polym. Chem. Ed. 1982, 20, 3399–3420. [Google Scholar] [CrossRef]
- Frollini, E.; Reed, W.F.; Milas, M.; Rinaudo, M. Polyelectrolytes from polysaccharides: Selective oxidation of guar gum - a revisited reaction. Carbohydr. Polym. 1995, 27, 129–135. [Google Scholar] [CrossRef]
- Chiu, C.-W.; Jeffcoat, R.; Henley, M.; Peek, L. Aldehyde cationic derivatives of galactose containing polysaccharides used as paper strength additives. U.S. Patent 5,554,745, 10 September 1996. [Google Scholar]
- Brady, R.L.; Leibfried, R.L. Use of oxidation promoting chemicals in the oxidation oxidizable galactose type of alcohol configuration containing polymer. U.S. Patent 6,022,717, 8 February 2000. [Google Scholar]
- Brady, R.L.; Leibfried, R.L.; Nguyen, T.T. Oxidation in solid state of oxidizable galactose type of alcohol configuration containing polymer. U.S. Patent 6,124,124, 26 September 2000. [Google Scholar]
- Brady, R.L.; Leibfried, R.L.; Nguyen, T.T. Paper having improved strength characteristics and process for making same. U.S. Patent 6,179,962, 30 January 2001. [Google Scholar]
- Lavazza, M.; Formantici, C.; Langella, V.; Monti, D.; Pfeiffer, U.; Galante, Y.M. Oxidation of galactomannan by laccase plus TEMPO yields an elastic gel. J. Biotechnol. 2011, 156, 108–116. [Google Scholar]
- Parikka, K.; Lappanen, A.S.; Pitkanen, L.; Ruenanen, M.; Willfor, S.; Tenkanen, M. Oxidation of polysaccharides by galactose oxidase. J. Agric. Food Chem. 2010, 58, 262–271. [Google Scholar]
- Godfrey, T.; West, S. Industrial Enzymology, 2nd ed; Stockton Press: New York, NY, USA, 1996; pp. 227–264. [Google Scholar]
- White, J.S.; White, D.C. Source Book of Enzymes; CRC Press: New York, NY, USA, 1997. [Google Scholar]
- Abbas, K.A.; Kalil, S.K.; Hussin, A.S.M. Modified starches and their usages in selected food products: A review study. J. Agric. Sci. 2010, 2, 90–100. [Google Scholar]
- Zhang, H.X.; Jin, Z.Y. Preparation of products rich in resistant starch from maize starch by an enzymatic method. Carbohydr. Polym. 2011, 86, 1610–1614. [Google Scholar] [CrossRef]
- Lee, K.Y.; Lee, S.; Lee, H.G. Effect of the degree of enzymatic hydrolysis on the physicochemical properties and in vitro digestibility of rice starch. Food Sci. Biotechnol. 2010, 19, 1333–1340. [Google Scholar] [CrossRef]
- Khatoon, S.; Sreerama, Y.N.; Raghavendra, D.; Hhattacharya, S.; Bhat, K.K. Properties of enzyme modified corn, rice and tapioca starches. Food Res. Int. 2009, 42, 1426–1433. [Google Scholar] [CrossRef]
- Karim, A.A.; Sufha, E.H.; Zaidul, I.S.M. Dual modification of starch via partial enzymatic hydrolysis in the granular state and subsequent hydroxypropylation. J. Agric. Food Chem. 2008, 56, 10901–10907. [Google Scholar] [CrossRef]
- Qiao, L.; Gu, Q.-M.; Cheng, H.N. Enzyme-catalyzed synthesis of hydrophobically modified starch. Carbohydr. Polym. 2006, 66, 135–140. [Google Scholar] [CrossRef]
- Alissandratos, A.; Baudendistel, N.; Flitsch, S.L.; Hauer, B.; Halling, P.J. Lipase-catalysed acylation of starch and determination of the degree of substitution by methanolysis and GC. BMC Biotechnol. 2010, 10, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Rajan, A.; Sudha, J.D.; Abraham, T.E. Enzymatic modification of cassava starch by fungal lipase. Ind. Crops Prod. 2008, 27, 50–59. [Google Scholar] [CrossRef]
- Rajan, A.; Prasad, V.S.; Abraham, T.E. Enzymatic esterification of starch using recovered coconut oil. Int. J. Biol. Macromol. 2006, 39, 265–272. [Google Scholar] [CrossRef]
- Chakraborty, S.; Sahoo, B.; Teraoka, I.; Miller, L.M.; Gross, R.A. Enzyme-catalyzed regioselective modification of starch nanoparticles. Macromolecules 2005, 38, 61–68. [Google Scholar]
- Alkorta, I.; Garbisu, C.; Llama, M.J.; Serra, J.L. Industrial applications of pectic enzymes: A review. Process Biochem. 1998, 33, 21–28. [Google Scholar] [CrossRef]
- Kashyap, D.R.; Vohra, P.K.; Chopra, S.; Tewari, R. Applications of pectinases in the commercial sector: A review. Bioresour. Technol. 2001, 77, 215–227. [Google Scholar] [CrossRef]
- Pedrolli, D.B.; Monteiro, A.C.; Gomes, E.; Carmona, E.C. Pectin and pectinases: Production, characterization and industrial application of microbial pectinolytic enzymes. Open Biotechnol. J. 2009, 3, 9–18. [Google Scholar] [CrossRef]
- Wang, Q.; Fan, X.R.; Hua, Z.Z.; Gao, W.D.; Chen, J. Degradation kinetics of pectins by an alkaline pectinase in bioscouring of cotton fabrics. Carbohydr. Polym. 2007, 67, 572–575. [Google Scholar] [CrossRef]
- Hoondal, G.; Tiwari, R.; Tewari, R.; Dahiya, N.; Beg, Q. Microbial alkaline pectinases and their industrial applications: A review. Appl. Microbiol. Biotechnol. 2002, 59, 409–418. [Google Scholar] [CrossRef]
- Videcoq, P.; Garnier, C.; Robert, P.; Bonnin, E. Influence of calcium on pectin methylesterase behavior in the presence of medium methylated pectins. Carbohydr. Polym. 2011, 86, 1657–1664. [Google Scholar] [CrossRef]
- Cameron, R.G.; Luzio, G.A.; Vasu, P.; Savary, B.J.; Williams, M.A.K. Enzymatic modification of a model homogalacturonan with the thermally tolerant pectin methylesterase from citrus: 1. Nanostructural characterization, enzyme mode of action, and effect of pH. J. Agric. Food. Chem. 2011, 59, 2717–2724. [Google Scholar] [CrossRef]
- Jolie, R.P.; Duveter, T.; van Loey, A.M.; Hendrickx, M.E. Pectin methyesterase and its proteinaceous inhibitor: A review. Carbohydr. Res. 2010, 34, 2583–2595. [Google Scholar]
- Pelloux, J.; Rustérucci, C.; Mellerowicz, E.J. New insights into pectin methylesterase structure and function. Trends Plant Sci. 2007, 12, 267–277. [Google Scholar] [CrossRef]
- Andrade, M.V.V.; Delatorre, A.B.; Silvania Alves Ladeira, S.A.; Martins, M.L.L. Production and partial characterization of alkaline polygalacturonase secreted by thermophilic Bacillus sp. SMIA-2 under submerged culture using pectin and corn steep liquor. Ciênc. Tecnol. Aliment. Camp. 2011, 31, 204–208. [Google Scholar] [CrossRef]
- Kiss, K.; Nemesthothy, N.; Gubicza, L.; Belafi-Bako, K. Vacuum assisted membrane bioreactor for enzymatic hydrolysis of pectin from various agro-wastes. Desalination 2009, 241, 29–33. [Google Scholar] [CrossRef]
- van Alebeek, G.; Christensen, T.; Schols, H.A.; Mikkelsen, J.D.; Voragen, A.G.J. Mode of action of pectin lyase A of Aspergillus niger on differently C(6)-substituted oligogalacturonides. J. Biol. Chem. 2002, 277, 25929–25936. [Google Scholar]
- Yadav, S.; Yadav, P.K.; Yadav, D.; Yadav, K.D.S. Pectin lyase: A review. Proc. Biochem. 2009, 44, 1–10. [Google Scholar] [CrossRef]
- Funami, T.; Nakauma, M.; Ishihara, S.; Tanaka, R.; Inoue, T.; Phillips, G.O. Structural modificationsof sugar beet pectin and the relationship of structure to functionality. Food Hydr. 2011, 25, 221–229. [Google Scholar] [CrossRef]
- Nuñez, A.; Fishman, M.L.; Fortis, L.L.; Cooke, P.H.; Hotchkiss, A.T. Identification of extensin protein associated with sugar beet pectin. J. Agric. Food Chem. 2009, 57, 10951–10958. [Google Scholar]
- Zeeb, B.; Fischer, L.; Weiss, J. Crosslinking of interfacial layers affects the salt and temperature stability of multilayered emulsions consisting of fish gelatin and sugar beet pectin. J. Agric. Food. Chem. 2011, 59, 10546–10555. [Google Scholar]
- Norsker, M.; Jensen, M.; Adler-Nissen, J. Enzymatic gelation of sugar beet pectin in food products. Food Hydr. 2000, 14, 237–243. [Google Scholar] [CrossRef]
- Cheng, H.N.; Gu, Q.-M.; Nickol, R.G. Amine-modified polymers, especially polysaccharides and pectins, and preparation thereof for improved gelation. U.S. Patent 6,159,721, 12 December 2000. [Google Scholar]
- Gu, Q.-M.; Nickol, R.G.; Cheng, H.N. Enzyme-catalyzed modification of pectin. ACS Polym. Prepr. 2003, 44, 608–609. [Google Scholar]
- Kuznetsova, E.; Proudfoot, M.; Sanders, S.A.; Reinking, J.; Savchenko, A.; Arrowsmith, C.H.; Edwards, A.M.; Yakunin, A.F. Enzyme genomics: Application of general enzymatic screens to discover new enzymes. FEMS Microbiol. Rev. 2005, 29, 263–279. [Google Scholar]
- Faber, K.; Kroutil, W. New enzymes for biotransformations. Curr. Opin. Chem. Biol. 2005, 9, 181–187. [Google Scholar] [CrossRef]
- Haki, G.D.; Rakshit, S.K. Developments in industrially important thermostable enzymes: A review. Bioresour. Technol. 2003, 89, 17–34. [Google Scholar] [CrossRef]
- Cherry, J.R.; Fidantsef, A.L. Directed evolution of industrial enzymes: An update. Curr. Opin. Biotechnol. 2003, 14, 438–443. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Pohl, M. Improved biocatalysts by directed evolution and rational protein design. Curr. Opin. Chem. Biol. 2001, 5, 137–143. [Google Scholar] [CrossRef]
- Qi, D.; Tann, C.-M.; Haring, D.; Distefano, M.D. Generation of new enzymes via covalent modification of existing proteins. Chem. Rev. 2001, 101, 3081–3112. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cheng, H.N.; Gu, Q.-M. Enzyme-Catalyzed Modifications of Polysaccharides and Poly(ethylene glycol). Polymers 2012, 4, 1311-1330. https://doi.org/10.3390/polym4021311
Cheng HN, Gu Q-M. Enzyme-Catalyzed Modifications of Polysaccharides and Poly(ethylene glycol). Polymers. 2012; 4(2):1311-1330. https://doi.org/10.3390/polym4021311
Chicago/Turabian StyleCheng, H. N., and Qu-Ming Gu. 2012. "Enzyme-Catalyzed Modifications of Polysaccharides and Poly(ethylene glycol)" Polymers 4, no. 2: 1311-1330. https://doi.org/10.3390/polym4021311
APA StyleCheng, H. N., & Gu, Q.-M. (2012). Enzyme-Catalyzed Modifications of Polysaccharides and Poly(ethylene glycol). Polymers, 4(2), 1311-1330. https://doi.org/10.3390/polym4021311