Thermoresponsive Polymers for Biomedical Applications

Abstract
: Thermoresponsive polymers are a class of “smart” materials that have the ability to respond to a change in temperature; a property that makes them useful materials in a wide range of applications and consequently attracts much scientific interest. This review focuses mainly on the studies published over the last 10 years on the synthesis and use of thermoresponsive polymers for biomedical applications including drug delivery, tissue engineering and gene delivery. A summary of the main applications is given following the different studies on thermoresponsive polymers which are categorized based on their 3-dimensional structure; hydrogels, interpenetrating networks, micelles, crosslinked micelles, polymersomes, films and particles.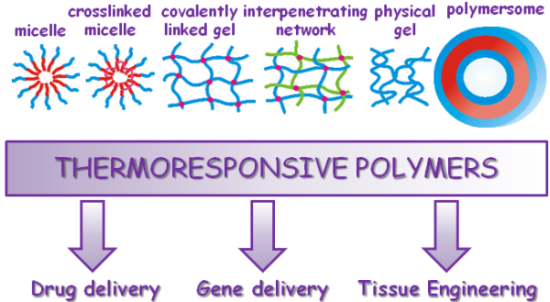
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
“Smart” materials, i.e., materials that have the ability to respond to external stimuli, represent one of the most exciting and immerging class of materials [1-9]. One of the main groups of “smart” materials is based on polymers because they are cheaper and more easily tailored than metals or ceramics. These polymeric “smart” materials can respond to stimuli such as pH, temperature, ionic strength, electric or magnetic field, light and/or chemical and biological stimuli and consequently have a wide range of applications that include sensors, drug delivery, gene delivery and tissue engineering [1-10].
This review focuses on polymers that respond to temperature, an easy external stimuli to apply. Specifically, it focuses on thermoresponsive polymers and their main biological related applications; drug delivery, gene delivery and tissue engineering. However, before the different applications and different thermoresponsive polymers are discussed, it is important to go through some basic terminologies.
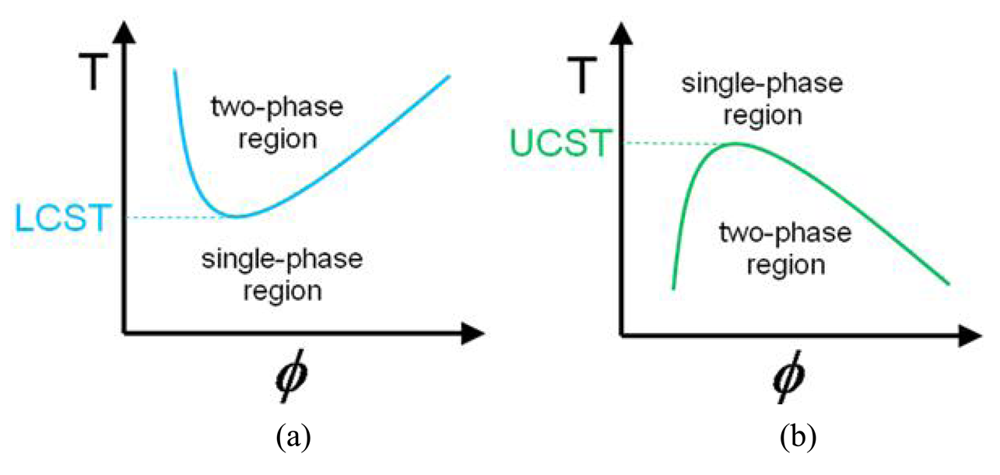
There are two main types of thermoresponsive polymers; the first present a lower critical solution temperature (LCST) while the second present an upper critical solution temperature (UCST). LCST and UCST are the respective critical temperature points below and above which the polymer and solvent are completely miscible as shown in Figure 1. Thus, for example, a polymer solution below the LCST is a clear, homogeneous solution while a polymer solution above the LCST appears cloudy (leading to LCST also being referred to as cloud point). This happens because it is energetically more favorable. In particular, considering the free energy of the system using the Gibbs equation ΔG = ΔH − TΔS (G: Gibbs free energy, H: enthalpy and S: entropy) the reason phase separation is more favorable when increasing the temperature is mostly due to the entropy of the system. Specifically, the main driving force is the entropy of the water, that when the polymer is not in solution the water is less ordered and has a higher entropy. This is also called the “hydrophobic effect” [11-13]. It is noteworthy that LCST is an entropically driven effect while UCST is an enthalpically driven effect [14].
Most of the studies discussed in this review use polymers that present an LCST. The most common of these is poly(N-isopropylacrylamide) (PNIPAAm) [1-4]. PNIPAAm has a lower critical solution temperature, LCST of around 32 °C, a very useful temperature for biomedical applications since it is close to the body temperature (37 °C). Adjustment of the LCST of PNIPAAm has been achieved by copolymerizing with hydrophilic or hydrophobic monomers rendering the overall hydrophilicity of the polymer higher or lower respectively [3,5,6,15]. Other polymers with thermoresponsive properties include poly(N,N-diethylacrylamide) (PDEAAm) with an LCST over the range of 25 to 32 °C, poly(N-vinlycaprolactam) (PVCL) [7,8,16] with an LCST between 25 and 35 °C, poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) [17-22] with an LCST of around 50 °C [21] and poly(ethylene glycol) (PEG), also called poly(ethylene oxide) (PEO) whose LCST is around 85 °C [23-26]. It should be noted the LCST is affected by the size of the side groups. Specifically, PEG methacrylate polymers (PEGMA), having a side-PEG chain of 2–10 ethylene oxide units (EO) < 10 present a lower critical solution temperature (LCST) [27] that varies depending on the length of the EO side chain [12]. However, it is important to note that the LCST of a polymer is dependent on molecular weight and architecture [19,28-31].
2. Applications
2.1. Delivery of Therapeutic Molecules
2.1.1. Drug Delivery
Drug delivery, as the name suggests, is the method or process of administering a pharmaceutical compound (drug) to achieve a therapeutic effect in humans or animals. Key factors are to deliver the drug to the right area, at the right time and at the right concentration. However there are many obstacles to achieving successful drug delivery. To name a few, these obstacles include the drugs': low solubility, environmental or enzymatic degradation, fast clearance rates from the body, non-specific toxicity and inability to cross the biological barriers [32,33]. In order to overcome these obstacles, drug delivery carriers are being used, most of which are based on polymers [32-34].
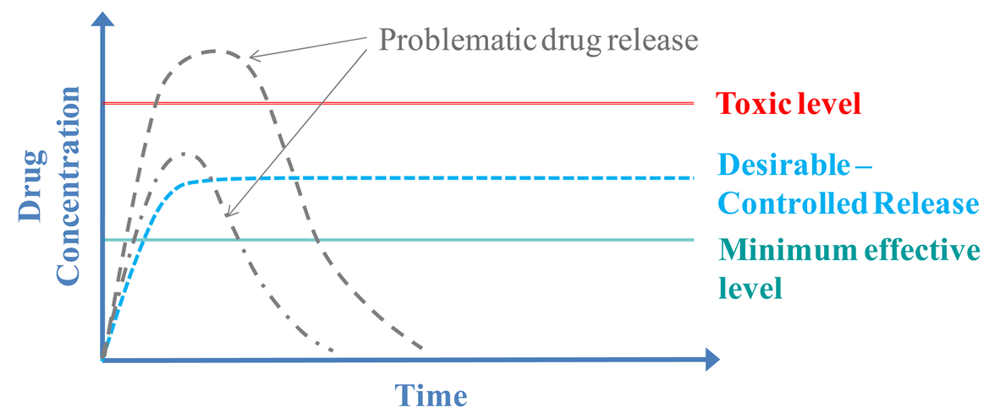
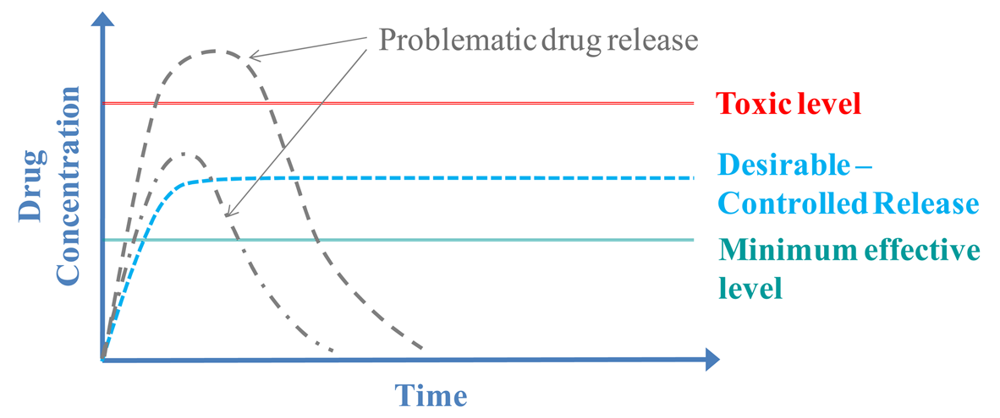
Nevertheless, even when using a carrier to deliver the drug it is difficult to have the desirable controlled release rate. Specifically, the delivery of a drug is often problematic because the concentration of the released drug at the target area is either too low or too high and is not delivered for the desirable amount of time, as shown with the gray dotted lines in the graph (Figure 2). Thus, the desirable-controlled drug release (shown with the blue line in the graph), that is often characteristic of a zero-order kinetic rate, is not easily achievable. Consequently “smart” polymeric carriers are used to deliver drugs. These carriers allow delivery of the drug at the right time and concentration by only releasing the drug in response to an external stimulus. For example the polymer chains of a carrier may expand as a result of the temperature increasing, thus enabling the drug to diffuse out and be released from the carrier [35]. Several studies aimed at controlled drug delivery using thermoresponsive polymer based carriers are discussed later in this review, categorized by their architecture/structural characteristics.
2.1.2. Gene Delivery
Gene therapy aims at the treatment of many genetic diseases as it is a technique for correcting defective genes that are responsible for these genetic diseases. Specifically, the delivery of the appropriate, therapeutic gene (DNA) into the cells that will replace, repair or regulate the defective gene that causes the disease is a vital step for gene therapy. DNA, however, is a negatively charged, hydrophilic molecule; thus its delivery into the nucleus of the cell which requires it to pass through the also negatively charged and hydrophobic cell membrane is not feasible. Consequently, gene delivery carriers (also called vectors or vehicles) have been developed [36,37]. Nature's way to carry genes is viruses and these were the first carriers used for gene delivery [36,37]. However viruses have many disadvantages, the most severe of which is the immune response that they can cause and this is why non-viral carriers have been developed [36-41]. Many of these are polymer-based because polymers are cheaper and safer than viruses and also easier to tailor compared to other gene delivery carriers like liposomes [36-38,42].
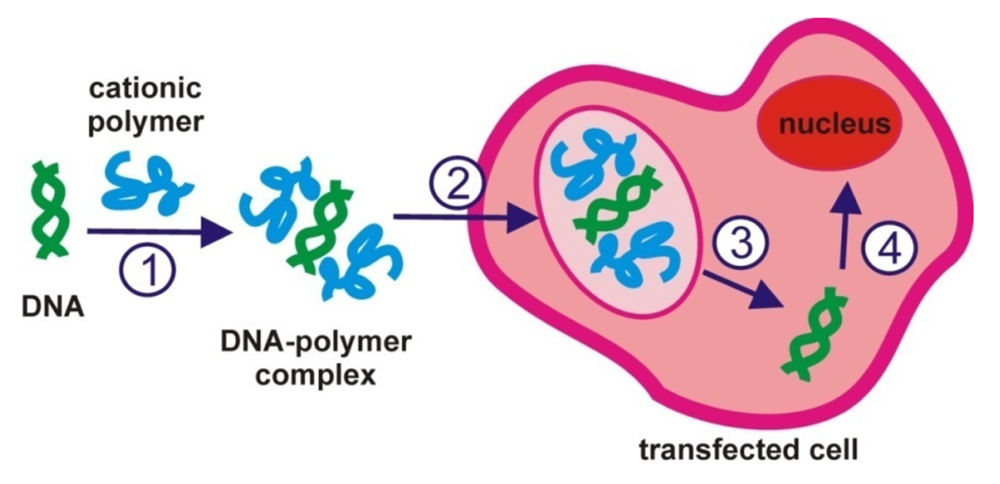
When using a polymeric carrier, the main steps of gene delivery (Figure 3), also called transfection, involve the: (1) DNA and polymer complexation; (2) addition of DNA/polymer complex (sometimes also called polyplex) onto cells for a period of time commonly called the transfection time; (3) removal of complex from the cells; (4) incubation time that is when the cells are left to incubate for a time period until results are observed. Complexation is usually performed at room temperature whilst the incubation and transfection periods are at 37 °C (body temperature that the cells need to be in order to survive). Interestingly, thermoresponsive polymers have been used to enhance the transfection efficiency by changing the temperature either during the complexation and/or during the incubation or transfection period. These studies are discussed below.
In gene delivery studies where thermoresponsive polymers were used the temperature at which one or two of the aforementioned steps were performed at was changed. In particular, in studies where polyethyleneimmine (PEI) with grafted PNIPAAm [43], chitosan grafted with PNIPAAm [44], linear and branched NIPAAm, DMAEMA and PEI polymers [2] and PEG polymers with grafted PEI chains [45] were used, the complexation and transfection temperature were changed to enhance the transfection efficiency. In other studies only the incubation or complexation temperature were varied using random terpolymers of P(NIPAAm-co-DMAEMA-co-BuMA) [18,46] or PNIPAAm copolymers [47], while both complexation and incubation temperature were varied using a polyarginine polymer conjugated with PNIPAAm [48].
In an interesting study by Zhou et al. using [poly(N,N-dimethylamino propyl acrylamide)-b-PNIPAAm]-star polymers, they used the polymers thermoresponsive ability in a different manner from the above studies [49]. The complexation was undertaken at room temperature which was below the polymer LCST and then the complex was deposited on a surface above the LCST [49]. On that surface, the cells were allowed to incubate at 37 °C [49]. The result of this was increased transfection to cells cultured on the surface compared to cells grown on a surface where DNA with no polymer was deposited [49].
2.2. Tissue Engineering
Tissue engineering as an interdisciplinary field that applies the principles of engineering and the life sciences towards the development of biological substitutes that restore or improve tissue function [50]. It aims to regenerate or replace biological damaged or diseased tissue or generate replacement organs for a wide range of medical conditions such as heart diseases, diabetes, cirrohosis, osteoarthritis, spinal cord injury and disfiguration [11,50-60].
The tissue engineering paradigm involves the use of a scaffold/material within which cells will be seeded and consequently tissue will mature. This requires the use of a biocompatible material/scaffold, usually natural materials like proteins or synthetic polymers, with the appropriate 3D structure that will provide sufficient mechanical support and has the ability to convey both nutrients and growth factors to encapsulated cells [54,56]. The use of synthetic polymers as this matrix forming material has received a lot of interest due to their advantages compared to natural products: they are not enzymatically degradable and it is easier to tailor their mechanical and chemical properties [54,56]. Synthetic polymers used in tissue engineering should be non-toxic, readily available and relatively inexpensive to produce.
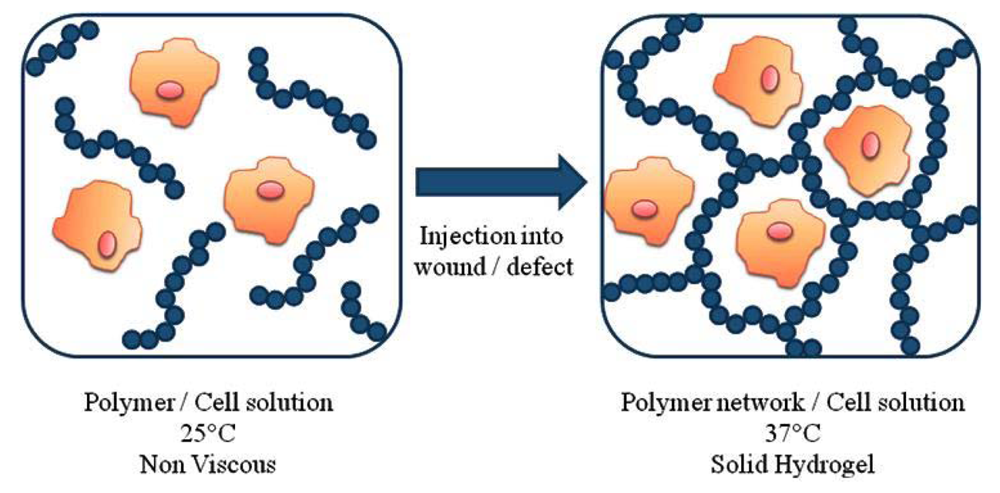
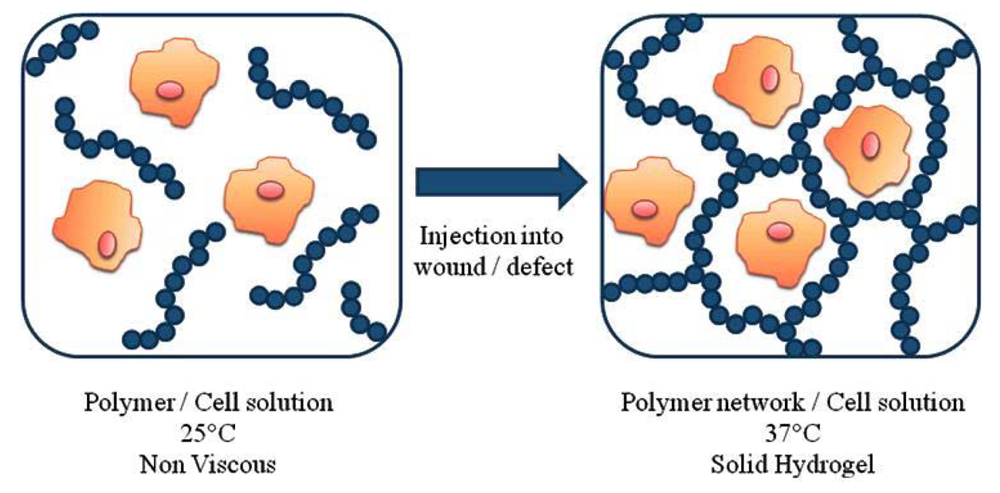
Thermoresponsive polymers in tissue engineering are commonly used in two situations: as substrates that enable the cell growth and proliferation and as injectable gels, for in situ of the scaffold. In the first application, the thermoresponsive ability of the polymers is used to regulate the cells' attachment and detachment from a surface [61-64]. In fact, in one study, the polymer surface was even reusable for repeated cell culture [65]. The second application involves the encapsulation of cells in a 3D structures in the body [19]. The in situ formation of cell/scaffold contrast compared to the in vitro formation of the construct allows the delivery of encapsulated cells, nutrients and growth factors to defects of any shape using minimally invasive techniques. The basic idea of the in situ formation is show in Figure 4. Specifically, the thermoresponsive polymer is mixed at room temperature with the cells and then injected into the body. Upon injection due to the temperature increase (to 37 °C) that is above the polymer's LCST, the polymer forms a physical gel. The cells are encapsulated within the 3D structure of the gel.
The studies that included thermoresponsive polymers for tissue engineering are discussed in the following sections, categorized by the polymers 3D structure.
3. Categories of Polymers Based on Their Architecture/Structure
3.1. Hydrogels
Gels are 3-dimensional polymeric networks. There are two main types of gels: physical gels and covalently linked gels. The latter, as the name suggests, are based on polymer chains that are linked together through covalent bonds at points that are called crosslinks. That is why this type of gels may also be referred to as crosslinked gels or covalently linked networks [66]. Physical gels, on the other hand, are formed by the physical entanglement of polymer chains and/or micelle ordering in solution and not from covalently linked polymer chains [67]. Both of these gels, crosslinked or physical, have the ability to swell in a solvent depending on their compatibility with the solvent. However their difference is that when a physical gel is in the appropriate solvent and it is given enough time and space it will dissolve in the solvent, whereas crosslinked gels will not.
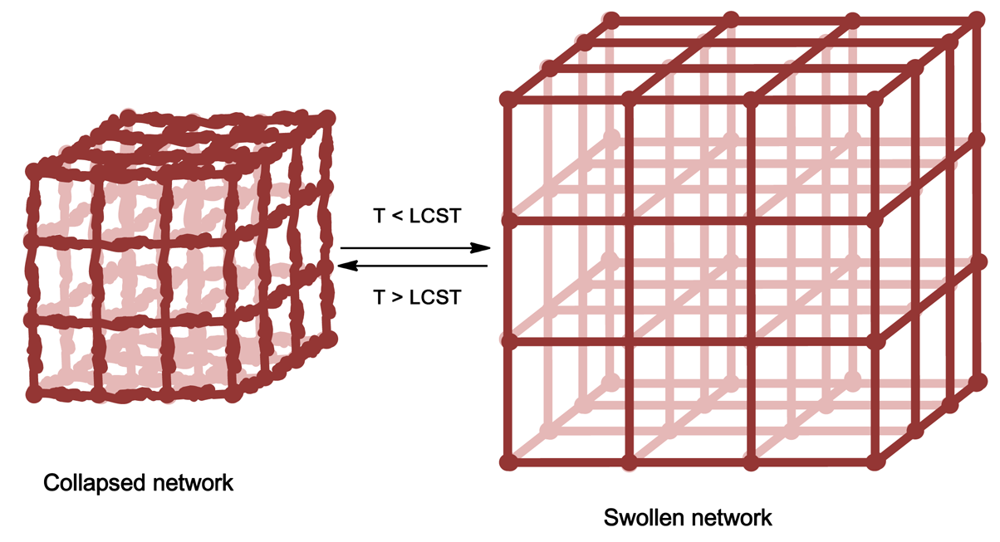
Hydrogels are polymer networks dispersed in water which form semi solid states containing upwards of 99% water w/w to polymer. These gels can be either covalently linked polymer networks or physical gels mentioned above. With reference to thermoresponsive polymers, covalently linked networks exhibit a change in their degree of swelling in response to temperature, Figure 5, whereas physical gels show a sol-gel transition [68].
With covalently linked networks, two approaches can be taken for drug delivery: (1) the networks can be formed in situ in response to chemical or photo induced crosslinking; or (2) the networks are formed before implantation to the body. Clearly, the first approach is more favorable due to its less invasive nature. In both of these systems, drugs are loaded into the hydrogel at room temperature. Upon heating to body temperature, the gels shrink and entrap the drug molecules limiting their release. This is achieved through a number of methods including the outer shell shrinking the fastest and forming a barrier, the pore size of the gel being reduced, and diffusion slowed due to reduced movement of water molecules. Incorporation of hydrophobic polymers into the network allows interaction between hydrophobic drugs also and greatly increases the overall strength of the gel.
PNIPAAm is one of the most intensely studied polymers in reference to biomedical applications due to its LCST being very close to body temperature and its fast on off switching. When crosslinked into hydrogels, the coil to globule transition causes a rapid decrease in the volume of the gel resulting in a fast release of entrapped drug and solvent followed by a more linear, diffusion controlled release [69-71]. Okuyama et al. wrote an introductory paper on the swelling kinetics of conetworks of NIPAAm with butyl methacrylate (BuMA), P(NIPAAm-co-BuMA), commenting on the need for zero order drug release profiles and found that after a burst release of drug from the outer part of the hydrogel a sustained release can be obtained [69].
Coughlan et al. showed the importance of understanding the nature of a loaded drug in a polymer network with the crosslinker concentration and drug interaction with the polymer having a large effect on the rate of release of drugs from PNIPAAm gels [72,73]. Another important consideration when studying these gels is the effect of salt on the LCST of the polymers. Jhon et al. studied this effect with PNIPAAm brushes and found salt to lower the LCST due to the Hofmeister effect of salt on the structure of water molecules [74].
Several PNIPAAm conetworks were synthesized by Jones et al. for the delivery of antimicrobial agents in conjunction with medical devices [70]. Conetworks of PNIPAAm and hydroxyethyl methacrylate (HEMA) showed the most promising results, presenting a LCST of 34 °C and suitable drug release and mechanical strength for in vivo application [70]. A similar study by Jones et al. previously noted the incorporation of zinc tetraphenylporphyrin (Zn-TPP) into PNIPAAm-co-PHEMA. They showed that above the LCST the Zn-TPP release rate was dramatically reduced. Once released, the Zn-TPP was free for photoinduced antimicrobiral action. Jones showed that, with careful tailoring, an on/off drug release can be achieved for various applications by controlling polymer composition and consequently the LCST [71].
Numerous other thermoresponsive monomers have been utilized for the preparation of hydrogels including poly(dimethyl acrylamide) (PDMAAm) [75], PEG [25,76,77]. Martellini et al. synthesized a PDMAAm-co-Poly(methoxyethyl acrylate) and showed that at body temperature this hydrogel releases drug following a Fickian diffusion process with a linear relationship in respect to the square root of time [75].
Another interesting approach utilizing biological structures for drug delivery has been taken by several groups using amino acid based hydrogels [78,79]. Yoshida et al. produced PNIPAAm hydrogels with polyamino acid crosslinked chains to produce thermoresponsive degradable hydrogels [78]. Hatefi and Amsden wrote a detailed review on biodegradable injectable drug delivery systems commenting on the numerous approaches and methods for in situ formation and drug release [35].
Rincon et al. and Bessa et al. synthesized elastin like polymers with polypeptide repeat units [79,80]. Elastin is a thermoresponsive polypeptide which has unique thermoresponsive ability. Below its transition temperature it is solvated and extended in solution, however, above its LCST of 30 °C, the chains fold to form a nanoparticle, in particular it forms nanoparticles which entrap molecules inside their structure, allowing them to be used as a drug delivery vector [79,80]. Bessa showed that bone morphogenetic proteins could be encapsulated this way and delivered in a sustained way for 14 days to promote bone formation [79]. Another interesting point is that these elastin particles are not recognized by the immune system giving a non toxic vector without further modification [79].
A biodegradable hydrogel was prepared by Xiao et al. comprising thermoresponsive PNIPAAm with cleavable lactic acid and dextran groups [81]. Similar work was done by Zhuo et al. grafting PNIPAAm and PVCL-HEMA onto a dextran chain to produce injectable and biodegradable hydrogels. This gel was capable of delivering drugs over several days with negligible cytotoxicity [82].
Merten et al. produced hydrogels from modified xyloglucan polymers and showed that the LCST can be altered by the removal of galactose rendering the polymer more hydrophobic [83].
Zero order drug release was achieved by Kan et al. by incorporating a thermoresponsive polymer, PEG-b-PLGA-b-PEG, into an oil in water emulsion [84]. The water phase formed a hydrogel over a 20–30 °C range, above the polymer LCST, entrapping the oil droplets. Drug release form these droplets showed a controlled rate with no burst release [84].
Hydrogels of PEGMA containing iron oxide particles were synthesized by Meenach et al. for possible drug delivery applications [85]. These hydrogels showed thermoresponsive ability with a deswelling of the gels upon increasing the temperature. Moreover the gels showed a magnetic ability by internally heating upon application of an alternating magnetic field leading to rapid heating and deswelling [85].
Papaphilippou et al. prepared PEGMA hydrogels with superparamagnetic properties by incorpartion of magnetite nanoparticles during the polymerization [86]. These hydrogels showed a deswelling on increasing temperature and also a tunable superparamagnetic character dependant on the amount of magnetite incorporated [86].
Wang et al. produced a series of degradable PEG-alt-Thiol hydrogels with varying LCSTs for tissue engineering [87]. The thiol groups provided suitable sites for degradation of the hydrogels in vivo allowing for a series of biocompatible and degradable cell matrices [87]. Conjugation of Pluronics® to peptide moieties enhances cell interaction increasing adhesion and growth within the tissue scaffold [88]. PoligoGel® is similar to Pluronics® utilizing PEG and PPG blocks and has been shown to be non toxic to cells with an LCST near body temperature [89].
Polymers of different architectures have also been used as injectable gels for tissue engineering. Kwon et al. produced physical gels of PNIPAAm-b-PEG with linear and star architectures [90] while Kirklan et al. used ABA triblock copolymers of PNIPAAm (block A) and poly(N,N-dimethylacrylamide) (PDMAAm, block B) [91].
Stile and Healy produced peptide modified crosslinked PNIPAAm hydrogels pliable enough below their LCST able to be injected though a small aperture [4]. They found that modification with peptides gives much greater cell proliferation throughout the gels [4].
Conetworks of PNIPAAm, PHEMA and a lactic acid monomer were synthesized by Ma et al. and found to exhibit LCSTs of 10–20 °C with PNIPAAm contents of 80% or more [92]. The gels had high tensile strength and degraded over several months with no cytotoxic byproducts when used in tissue engineering [92].
A thermoresponsive methylcellulose based hydrogel was prepared by Stabenfeldt et al. [93]. Methylcellulose was conjugated to Laminin protein to produce a physically gelling hydrogel capable of producing a suitable environment for neural tissue growth when injected into a defect in the central nervous system [93]. They demonstrated that the addition of the protein conjugate greatly improved the cell viability and adhesion [93].
Research by Ratner et al. used templating methods to produce crosslinked PNIPAAm hydrogels with a defined pore size [94]. Switching of the pore size with temperature allowed for encapsulation of cells within the gel and subsequent uses for tissue engineering [94].
3.1.1. Interpenetrating Networks
Another group of crosslinked hydrogels are the interpenetrating networks (IPN). Interpenetrating networks, according to IUPAC, consist of two covalently linked polymer networks which are bound together by physical entanglement as opposed to covalent bonds. Specifically, this requires the polymerization of both networks simultaneously and results in two intermixed networks that can only be separated by breaking bonds. These materials are of interest due to their ability to introduce new properties when the networks interact [95,96] or two different properties when acting independently.
In contrast to the studies discussed above, where the drug release was slowed by the temperature increase, Shin et al. demonstrated a hydrogel with a positive thermoresponse, i.e., drug release increases with increasing temperature [95]. Specifically, an interpenetrating network of polyacrylic acid (PAA) and polyacrylamide (PAAm) forms hydrogels that swell above their upper critical solution temperature, UCST, due to hydrogen bonding between the two different networks being disrupted at higher temperatures allowing the networks to swell [95]. This preliminary work showed the possibility of increasing drug release with increased temperature [95]. Recent work on the same IPN with grafted β-cyclodextrin showed a faster thermoresponse and lower UCST (35 °C) and a lowered effect of salt on the swelling [96]. Incorporation of a model drug, ibuprofen, showed a positive drug release with a controlled rate above and below the UCST [96].
Semi interpenetrating networks (SIPN) differ from IPNs in the sense that they are not entwined networks. SIPNs consist of a crosslinked network with linear or branched polymer chains penetrating them. An example of this is the SIPN synthesized by Mundargi et al. formed from crosslinked gellan gum microspheres with interpenetrating PNIPAAm chains [95]. Drug loading into the microspheres allows for pulsatile drug release. Specifically release below the LCST is greater due to swelling of the spheres and greater water transport through the spheres [97].
Kim et al. produced photodegradable injectable seminterpenetrating networks of PNIPAAm-co-PAAc with linear PAAc-co-peptide chains and showed the degradation to be dependent on peptide crosslinks and PAAc-co-peptide concentrations [98]. They also showed the degradation products to be relatively non cytotoxic demonstrating a promising tissue engineering scaffold [98].
3.1.2. Micelles
Combining hydrophilic and hydrophobic monomers into block copolymers allows the formation of ordered structures in solution, the most common of these being the micelle. Micelles are useful for encapsulating hydrophobic drugs and delivering them into an aqueous environment.
In 1998 Chung et al. showed the effect of attaching hydrophilic or hydrophobic moieties to the terminal end of a PNIPAAm [99]. The LCST and the rate of the phase transition dramatically change depending on the nature of the end moieties [99]. More specifically, hydrophilic monomers stabilized the chain and raised the LCST and the inverse was seen when hydrophobising the chain [99]. This effect however was less pronounced when the hydrophobic modification was undertaken using a long hydrophobic chain [99]. This was found to be due to the internalization of the hydrophobic ends into micelles in solution allowing the structures to exhibit the phase transition of PNIPAAm alone [99].
Several groups have focused on using PNIPAAm as the thermoresponsive block in the formation of thermoresponsive micelles [100-104]. Akimoto et al. produced micelles of P(NIPAAm-co-DMAAm)-b-PLA, where PLA was poly(lactic acid), and showed that these micelles were able to internalize into cells above their LCST, specifically due to the increased interaction between the hydrated NIPAAm outer sphere and the cells [105]. Interestingly, they also showed that fictionalization of the PNIPAAm chain ends with thiol groups can increase this interaction and internalization, the results show promising methods for intracellular delivery of therapeutic molecules [100].
Diblock copolymers of PNIPAAm and poly(methyl methacrylate) (PMMA) or 10-undecenoic acid were synthesized by Wei et al. [103,104]. The copolymers were found to form micelles in water capable of releasing the encapsulated drug in relation to temperature [103,104]. At temperatures below the LCST, drug release stops after the initial burst release [103,104]. This is due to the hydrated PNIPAAm chains limiting drug release [103,104]. Heating above the LCST causes a big structural change in the micelle with the PNIPAAm collapsing and becoming hydrophobic, allowing release of the contained drug with an on/off drug switching [103,104].
Degradable copolymers of poly(NIPAAm-co-HPMAm-lactate)-b-PEG [106] and PEG-b-(HEMAm-lactate) [107] were shown by Hennink et al. to form micelles above a critical micelle temperature dependant on the polymer LCST [106,107]. Cleavage of the lactate moiety in vitro or in vivo causes an increase in the polymer LCST and so a destabilization of the micelles, releasing any encapsulated drug [106,107].
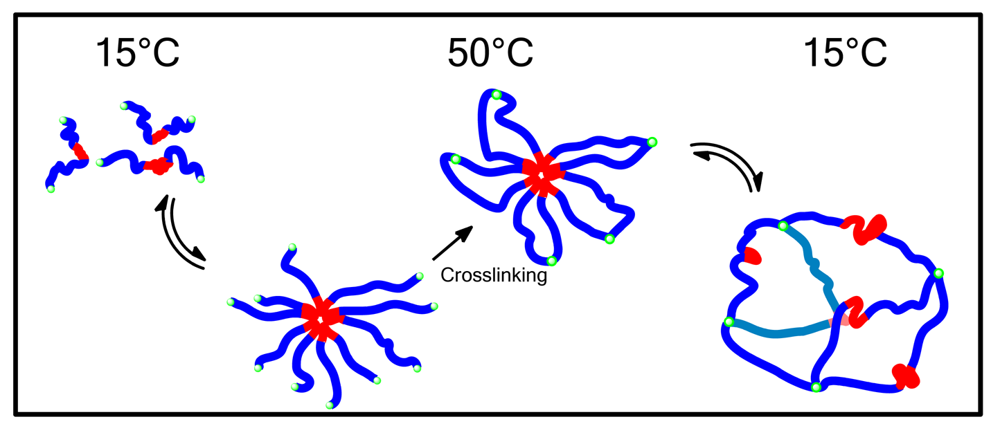
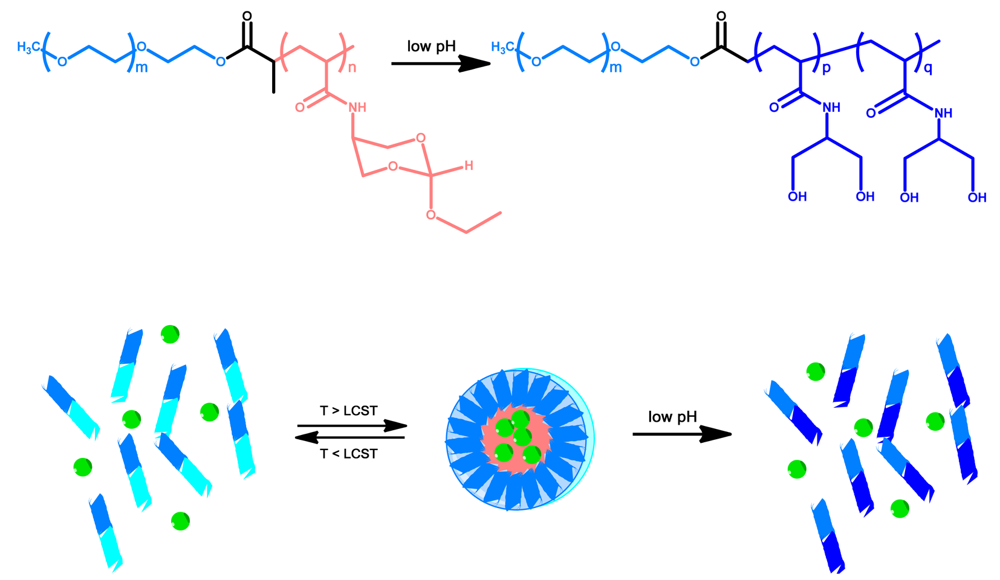
Preparation of thermoresponsive micelles is not limited to PNIPAAm. Figure 6 above shows a block copolymer synthesized by Huang et al., utilizing an orthoester containing acrylamide block which becomes hydrophobic above its LCST, and a hydrophilic PEG block [108]. This copolymer formed micelles above its LCST but also showed pH responsive degradation giving another option for drug release [108]. Polymers with similar properties were produced by Hruby et al. based on poly oxazolines [109] and Wang et al. based on PEO [110].
The thermoresponsive properties of terpolymers of PEGMA-b-(DMAEMA-co-MMA) were shown to have an interesting pH and temperature sensitivity [111]. Polymers transitioned from molecularly dissolved at low temperature and low pH, to micelles with either a PEGMA core upon increasing temperature or a DMAEMA core upon increasing pH [111].
San Miguel et al. synthesized ABA triblock copolymers of PDMAEMA (block A) and PVCL (block B) and demonstrated the formation of thermoresponsive micelles with sustained drug release in vitro [17].
Wei et al. synthesized a thermoresponsive star block copolymer based on L-Lactide and NIPAAm [112]. These star polymers were found to self assemble into large micelle structures in water which showed a fast on/off drug switching with temperature [112].
3.1.3. Cross Linked Micelles
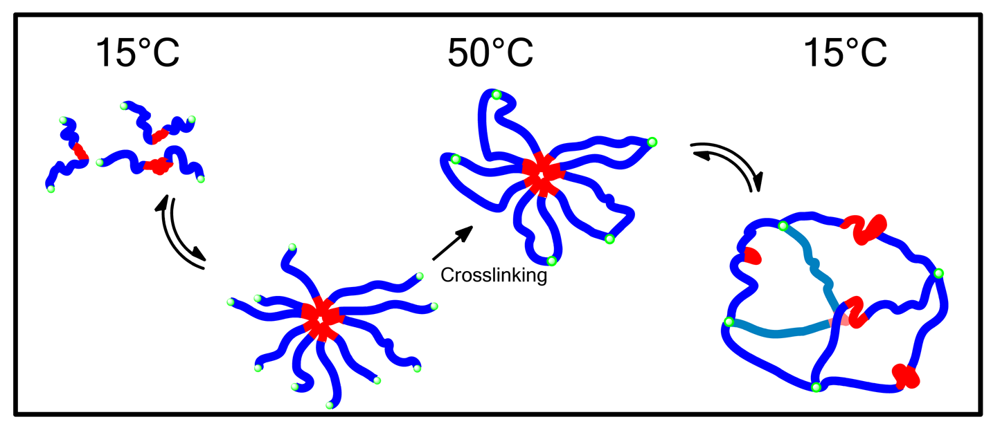
The formation of micelles in solution is a reversible and dynamic process; this can be overcome by crosslinking polymer chains once micellization has occurred. Selective crosslinking of specific blocks within the micelle can lead to interesting structures with varied properties. A review of non thermoresponsive shell crosslinked micelles was published by Armes et al., it focused on the crosslinking of the middle block of ABC triblock copolymers [113]. This modification allowed for synthesis without the need for low concentrations as no intermicellar crosslinking took place [113].
Wei et al. produced a number of shell crosslinked hybrid micelles using inorganic based crosslinks and PNIPAAm thermoresponsive groups [114,115]. They found drug release to be sustained for around 100 hours followed by a slow leveling off of the release rate [114,115]. The shell crosslinked layer slowed the release of drug by entrapment but allowed an increase in the release with increased temperature [114,115].
Cohn et al. utilized the formation of micelles above the LCST of PEG-b-PPG-b-PEG triblock copolymers, where PPG was the hydrophobic poly(propylene glycol) block, also called poly(propylene oxide) (PPO), to form functional nanoshells, Figure 7 [116], by incorporating a methacrylate group onto each end of the block copolymers and crosslinking them while in a micellar configuration a collapsible and expandable shell structure was formed with an extremely large void spaces capable of encapsulating numerous moieties [116].
3.1.4. Polymersomes
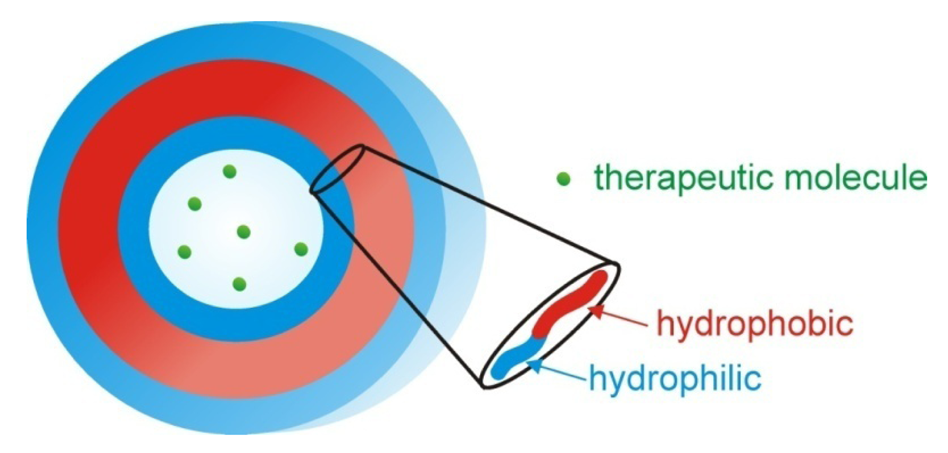
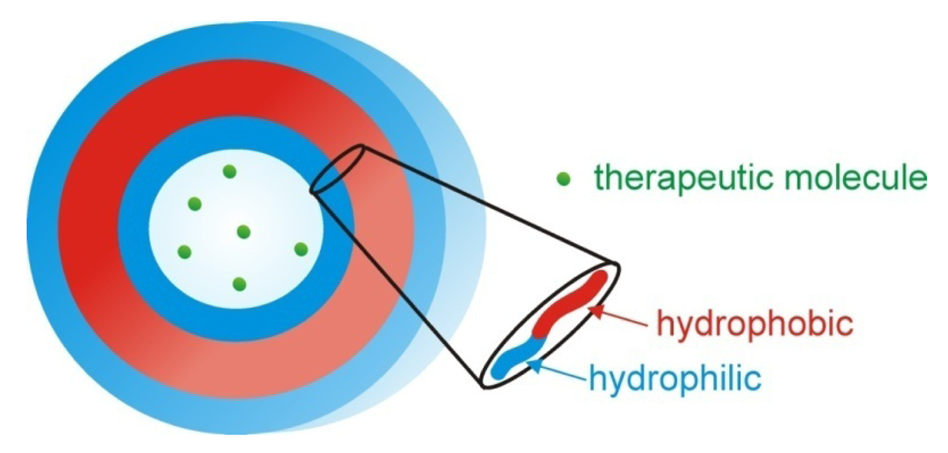
Polymersomes, another class of self assembled structures, have also been used for drug and DNA delivery. They are based on self assembled amphiphilic block copolymers with a hydrophilic interior and exterior (Figure 8). They provide an advantage of the ability to encapsulate hydrophilic molecules in their interior protected from the outside environment; compared to micelles which preferentially encapsulate hydrophobic molecules. Reviews on this subject that include thermoresponsive polymersomes have been published previously by Meng et al. [117], Du and O'Reilly [118] and Kim et al. [119].
A diblock copolymer, synthesized by Qin et al., comprising of PEG-b-PNIPAAm with 7–20% PEG content spontaneously formed polymersomes in water at temperatures of 37 °C and above. These polymersomes would allow for encapsulation of both hydrophilic molecules within the interior, and hydrophobic molecules in the membrane promoting several possible therapeutic applications [120].
Polymersomes based on copolymers of PNIPAAm and poly(N-(3-aminopropyl)methacrylamide hydrochloride) (PAMPA) [121], PNIPAAm and PEG [122], P(LA-star-b-NIPAAm) [123] and hyperbranched poly[3-ethyl-3-(hydroxymethyl)oxetane]-g-PEG [124] where also studied, where NIPAAm or PEG where the thermoresponsive blocks.
McCormick et al. and Qin et al. utilized the LCST of a thermoresponsive block to direct the formation of polymersomes [121,122]. McCormick used the LCST of PNIPAAm to induce formation of polymersomes and crosslinked the shell by ionic crosslinking using the polyelectrolyte PAMPA [121]. Non-crosslinked polymersomes by Qin et al. were synthesized using PEG and PNIPAAm [122]. Above the LCST of PNIPAAM the polymersomes formed spontaneously, encapsulating drugs loaded into the solution beforehand [122]. These polymersomes showed no drug release until further cooling to destabilize them [122].
Expanding on the research by Qin et al., several other studies have utilized the thermoresponsive ability of polymers to aid the release from polymersomes. Star polymers synthesized by Wei et al. of a hydrophobic PLA block and thermoresponsive PNIPAAm blocks spontaneously formed polymersomes in solution [123]. Heating above the LCST hydrophobized the PNIPAAm blocks releasing any encapsulated drugs [123]. Star copolymers having a hydrophobic hyperbranched poly[3-ethyl-3-(hydroxymethyl)oxetane] (HBPO) core and many hydrophilic PEG arms were synthesized by Zhou et al. [124]. These polymers form polymersomes below their LCST, however, above the LCST the polymersomes destabilize undergoing a “vesicle-to-membrane” transformation and releasing any encapsulate [124].
In a recent interesting study, Agut et al. investigated both the pH and temperature affect on PDMAEMA containing polymersomes [125]. At low pH, the DMAEMA units were protonated and polymersomes formed due to electrostatic interactions [125]. Upon increasing the pH the mechanism changed with polymersomes only forming with increasing temperatures above the LCST of the DMAEMA [125].
3.1.5. Films
Copolymer films of PNIPAAm and poly(N-butylacrylamide) were shown by Wilson et al. and Doorty et al. to give a sustained release of drugs from the film over a considerable time period [3,126]. They showed the released amounts of drug loaded at room temperature to be inversely proportional to the hydrophobic monomer content once heated to 37 °C [3,126].
Dinarvand et al. investigated the possibility of using a copolymer of PNIPAAm with PAAm as a stimuli responsive membrane for the control of permeation of molecules for numerous applications like drug delivery [127]. They found that an increasing temperature above the LCST reduces the transport through the membrane by collapse of the PNIPAAm structure [127].
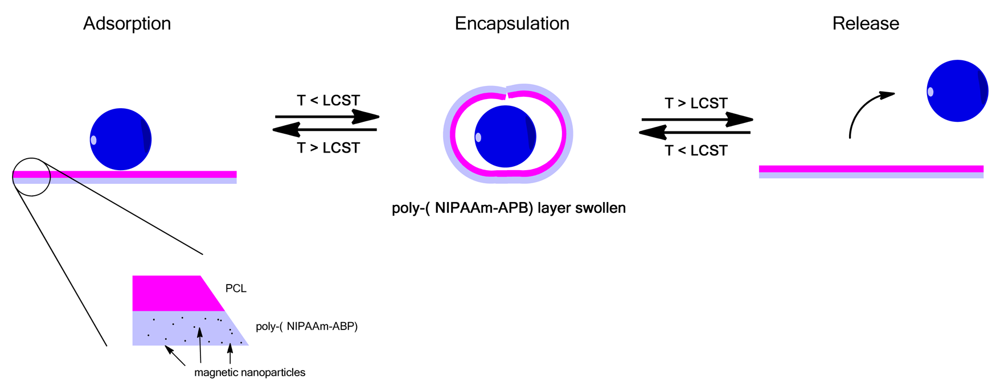
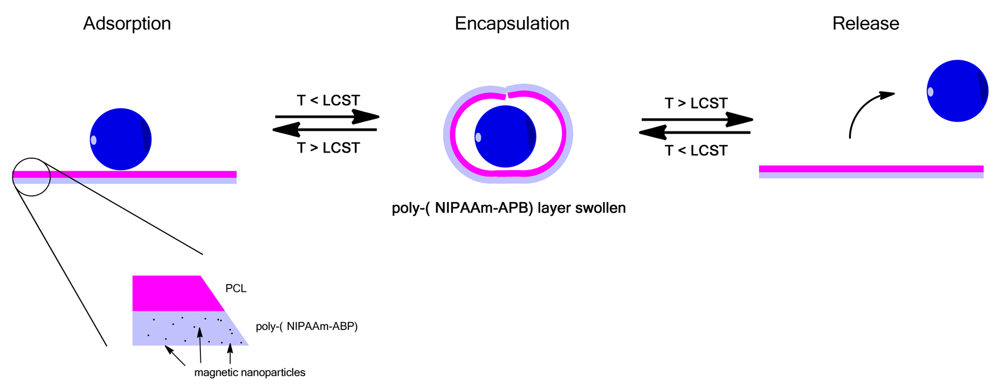
An interesting use of thermoresponsive polymer films was shown by Zakharchenko et al. where a bilayer of PVCL on top of PNIPAAm with encapsulated magnetic nanoparticles was prepared [128]. At temperatures greater than the LCST the films were flat and allowed for adsorption of nanoparticles, cells or drugs onto the surface, Figure 9, upon cooling the films rolled up entrapping the absorbed particles which could then be release by heating again [128]. This is a novel approach for the encapsulation and release of nanoparticles and cells with the addition of the magnetic particles allowing manipulation of the films by an external field [128].
Sukhishvil and Zhu showed a very interesting approach to film preparation using the assembly of diblock copolymer micelles [129]. A block copolymer consisting of PNIPAAm and poly(N-vinylpyrrolidone) (PVPON) was prepared [129]. These polymers formed micelles above the LCST of PNIPAAm with PNIPAAm cores [129]. Assembly of these micelles onto a substrate was achieved layer by layer with incorporation of poly(methacrylic acid) (PMAA) between the micelles [129]. Upon cooling, hydrogen bonding between the PMAA and PVPON in the micelle shells stopped the disassembly of the micelles [129]. These films showed good incorporation of hydrophobic drugs with a slower release above the LCST due to collapse of drug containing PNIPAAm cores [129].
Schmaljohann produced a series of graft copolymers of PNIPAAm, PDMAAm and PEG which showed suitable swelling/deswelling and surface changes for cell attachment and detachment in response to temperature [130]. Membranes of triblock copolymers of PNIPAAm-b-PVCL-b-PNIPAAm produced by Li et al. also displayed cell attachment and detachment in response to temperatures above and below the LCST, respectively [131].
Abraham et al. produced a P(NIPAAm-co-MMA) polymers and grafted this with phosphorylate HEMA [132]. Cells were found to adhere well to the graft copolymer and spread across the surface due to the phosphate moieties [132]. Upon cooling below the LCST, whole cell sheets were released from the surface [132].
Previously, the conjugation of insulin to PNIPAAm was studied by Chen et al. and, more recently, Hatakeyama demonstrated that grafting of the polymer to a surface allows cell growth at insulin levels 10 times lower than free insulin and allows for easy detachment when polymer is cooled below its LCST [133,134].
Ito et al. prepare plates of polystyrene grafted with PNIPAAm copolymers using a mask to create a micropatterned surface [135,136]. Cell growth and selected cell detachment was shown to be achievable with this approach [135,136]. Tsuda et al. also utilized patterning for cell culture by combing a patterned surface of PNIPAAm and PNIPAAm-graft-PBuMA with two different LCSTs. This substrate was able to selectively adhere cells at different temperatures allowing formation of cell sheet monolayers comprising two cell types in an ordered pattern [137]. A review on micro-patterning using stimuli responsive polymers by Ito gives a much more extensive overview of the subject [138].
The encapsulation of biological substrates into a PNIPAAm layer for controlled release during cell growth and also showed the possible release of whole cell sheets from the PNIPAAm grafted plates [139]. A review on cell sheet engineering by Kikuchi and Okano also showed the release of cell sheets from PNIPAAm films and the dependence on film thickness [140-142]. Films 20–30 nm show the best cell adhesion and detachment [140-142]. However films with thicknesses greater than 30 nm have minimal cell adhesion [140-142]. This is due to the hydration of the outer PNIPAAm layer being too hydrophilic and hence support less protein and cell adhesion [140-142]. This problem was addressed by Moran et al. who produced films of thicknesses greater than 30nm and coated them with cell adhesion promoters such as collagen of laminin [143]. Cells successfully adhered to these films and the thermal detachment of cells was not affected [143]. A further solution to this was demonstrated by Shimizu et al. who treated the PNIPAAm surface with an oxygen plasma which roughened the surface and removed its thermoresponsive ability making it hydrophobic at all temperature. Release of the cell layer at low temperatures takes the hydrophobic surface layer of PNIPAAm with it resulting in cell sheets with an increased mechanical strength [1].
Cheng et al. showed the production of plasma polymerized PNIPAAm films onto microheater arrays produced using photolithography [144]. This method allows for localized heating and specific area detachment of cells with many possible applications [144].
An interesting 3D cell culture method was envisaged by Poon et al. They synthesized chitosan-graft-PEG-graft-methacrylate copolymers which gelled at 37 °C but were also UV curable [145]. Cells were encapsulated layer by layer by heating each layer above the LCST and the final construct was cured with UV to enable cooling below the LCST without hydrogel breakdown [145].
3.1.6. Particles
Li et al. synthesized nanoparticles of thermoresponsive polymers by fast heating of a solution of the thermoresponsive polymer P(PEGMA-co-Boc-Cyst-MMAm) [146]. The particles produced were dependant on the heating rate during formation [147]. These particles possessed sensitivity to a reducing environment, such as the intracellular cytoplasm, by reduction of the disulfide bonds in the polymer chain resulting in breakdown of the nanoparticles [147]. This would allow for intracellular delivery of numerous therapeutic molecules [146].
Much work has been done on the modification of inorganic particles with thermoresponsive polymers [148-154]. Zhang et al. coated insoluble nanoparticles with PNIPAAm rendering them stable in aqueous solutions with temperature dependant solution properties and suggested uses in drug delivery and biological sensing [148]. Several groups have shown the formation of a novel drug delivery device with magnetic targeting capability by coating a magnetic nanoparticle, conjugated to active drug, with a thermoresponsive polymer. Upon heating above the LCST, the coating shrinks and releases the active drug [149,151,152,155,156]. Purushotham also showed the possibility of using a magnetic field to heat the composite and hence release entrapped drugs form the thermoresponsive coating [150]. Work by Papaphilippou et al. has involved the preparation of hybrid micelles utilizing a thermoresponsive PEGMA containing polymer and a magnetite nanoparticle core [154]. These micelles showed a thermoresponsive ability with LCST of 60 °C and a magnetic ability with future prospects aiming controlled drug delivery [154].
Pitch et al. demonstrated the stabilisation of magnetite nanoparticles by a thermoresponsive polymer [157]. In particular, microgels containing thermoresponsive PVCL and a hydrophobic monomer, acetoxy ethylmethacrylate, were prepared and loaded with the nanoparticles. These microgels showed thermoresponsive swelling/deswelling whilst stabilizing the suspension of nanoparticles [157].
A comprehensive review of combining magnetic particles and thermoresponsive polymers was written recently by Yuan et al. [158].
Das et al. showed another approach to polymer nanoparticle composites [159]. By incorporating gold nanorods into NIPAAm microgels, photo-induced heating of the gold nanorods provided enough heat to induce the phase transition of the microgel providing a possible photoinduced drug release [159]. With similar work on grafting NIPAAm onto gold nanoparticles being done by Pérez-Luna et al. [160].
Nanoparticles of crosslinked P(NIPAAm-co-AAm) were prepared by Fundueanue et al. by dispersion of the polymer in mineral oil and crosslinking with gluteraldehyde [161,162]. When prepared below the LCST, these particles were found to have a uniform pore size and sharp thermoresponse with a large increase in pore size when brought to body temperature showing a promising possibility for drug encapsulation and delivery [161,162].
Jun Wang et al. have produced a series of thermoresponsive micellar nanoparticles from PVCL and polyphosphoester and examined their cell change in size with temperature [163,164]. They found that as the temperature was increased above the LCST the micelles became more hydrophobic and formed aggregates in a reversible way [163,164]. The micelles were found to be both biodegradable and non cytotoxic showing a promising drug delivery vector [163,164].
Another class of nanoparticles are microgels. Microgels are microscopic sized crosslinked hydrogel particles with similar properties to crosslinked hydrogels. The advantage of a microgel is that, although it is crosslinked outside of the body, it is still easy to insert it into the body via injection. Vihola et al. synthesized PVCL and PVCL-graft-PEG microgels were formed by heating the polymer above its LCST and using salicylic acid as a crosslinker [7]. The salicylic acid formed hydrogen bonds between the polymer chains forming a physical hydrogel [7]. By adding a solution of polymer and drug to a solution containing the crosslinker at temperatures greater the LCST, hydrogel particles were formed which showed sustained release [7]. Interestingly, the PEG graft copolymers showed a slower drug release due to an increase in hydrogen bonding and hence increase packing from the PEG chains [7].
4. Conclusions
To summarize, thermoresponsive polymers have found biomedical uses in many forms; from tissue engineering to the delivery of therapeautic molecules such as drugs or genes. The use of preloaded polymer networks aiming at the release of drugs at body temperature has almost become a commonly used study with more recent work focusing on polymer networks as a 3D structure for tissue engineering and cell culture. The use of micelles for drug delivery has been studied extensively with the architecture of the micelles used becoming more complex and more responsive utilizing, among others, cross linked micelles, polymersomes, nanocages and nanoparticles. All of the recent advances in thermoresponsive polymers point to an area of chemistry that both is growing in popularity, but also vast in the scope to be covered and will be an area to watch in the future. Thermoresponsive polymers and their use in biomedical applications will have a definite impact in the medical field.


Referneces
- Shimizu, K.; Fujita, H.; Nagamori, E. Oxygen plasma-treated thermoresponsive polymer surfaces for cell sheet engineering. Biotechnol. Bioeng. 2010, 106, 303–310. [Google Scholar]
- Twaites, B.R.; Alarcon, C.D.H.; Lavigne, M.; Saulnier, A.; Pennadam, S.S.; Cunliffe, D.; Gorecki, D.C.; Alexander, C. Thermoresponsive polymers as gene delivery vectors: Cell viability, DNA transport and transfection studies. J. Control. Release 2005, 108, 472–483. [Google Scholar]
- Doorty, K.B.; Golubeva, T.A.; Gorelov, A.V.; Rochev, Y.A.; Allen, L.T.; Dawson, K.A.; Gallagher, W.M.; Keenan, A.K. Poly(N-isopropylacrylamide) co-polymer films as potential vehicles for delivery of an antimitotic agent to vascular smooth muscle cells. Cardiovasc. Pathol. 2003, 12, 105–110. [Google Scholar]
- Stile, R.A.; Healy, K.E. Thermo-responsive peptide-modified hydrogels for tissue regeneration. Biomacromolecules 2001, 2, 185–194. [Google Scholar]
- Hacker, M.C.; Klouda, L.; Ma, B.B.; Kretlow, J.D.; Mikos, A.G. Synthesis and characterization of injectable, thermally and chemically gelable, amphiphilic poly(N-isopropylacrylamide)-based macromers. Biomacromolecules 2008, 9, 1558–1570. [Google Scholar]
- Feil, H.; Bae, Y.H.; Feijen, J.; Kim, S.W. Effect of comonomer hydrophilicity and ionization on the lower critical solution temperature of N-isopropylacrylamide copolymers. Macromolecules 1993, 26, 2496–2500. [Google Scholar]
- Vihola, H.; Laukkanen, A.; Tenhu, H.; Hirvonen, J. Drug release characteristics of physically cross-linked thermosensitive poly(N-vinylcaprolactam) hydrogel particles. J. Pharm. Sci. 2008, 97, 4783–4793. [Google Scholar]
- Vihola, H.; Marttila, A.K.; Pakkanen, J.S.; Andersson, M.; Laukkanen, A.; Kaukonen, A.M.; Tenhu, H.; Hirvonen, J. Cell-polymer interactions of fluorescent polystyrene latex particles coated with thermosensitive poly(N-isopropylacrylamide) and poly(N-vinylcaprolactam) or grafted with poly(ethylene oxide)-macromonomer. Int. J. Pharm. 2007, 343, 238–246. [Google Scholar]
- Pasparakis, G.; Vamvakaki, M. Multiresponsive polymers: Nano-sized assemblies, stimuli-sensitive gels and smart surfaces. Polym. Chem. 2011, 2, 1234–1248. [Google Scholar]
- Liu, F.; Urban, M.W. Recent advances and challenges in designing stimuli-responsive polymers. Prog. Polym. Sci. 2010, 35, 3–23. [Google Scholar]
- Klouda, L.; Mikos, A.G. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. [Google Scholar]
- Lutz, J.-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci. Part A 2008, 46, 3459–3470. [Google Scholar]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A view of the hydrophobic effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar]
- Vasile, C.; Kulshreshtha, A.K. Handbook of Polymer Blends and Composites; Rapra Technology Ltd.: Shawbury, UK, 2003. [Google Scholar]
- Yin, X.; Hoffman, A.S.; Stayton, P.S. Poly(N-isopropylacrylamide-co-propylacrylic acid) copolymers that respond sharply to temperature and pH. Biomacromolecules 2006, 7, 1381–1385. [Google Scholar]
- Vihola, H.; Laukkanen, A.; Valtola, L.; Tenhu, H.; Hirvonen, J. Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam). Biomaterials 2005, 26, 3055–3064. [Google Scholar]
- San Miguel, V.; Limer, A.J.; Haddleton, D.M.; Catalina, F.; Peinado, C. Biodegradable and thermoresponsive micelles of triblock copolymers based on 2-(N,N-dimethylamino)ethyl methacrylate and epsilon-caprolactone for controlled drug delivery. Eur. Polym. J. 2008, 44, 3853–3863. [Google Scholar]
- Takeda, N.; Nakamura, E.; Yokoyama, M.; Okano, T. Temperature-responsive polymeric carriers incorporating hydrophobic monomers for effective transfection in small doses. J. Control. Release 2004, 95, 343–355. [Google Scholar]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive terpolymers based on methacrylate monomers: Effect of architecture and composition. J. Polym. Sci. Part A 2010, 48, 775–783. [Google Scholar]
- Fraylich, M.R.; Liu, R.; Richardson, S.M.; Baird, P.; Hoyland, J.; Freemont, A.J.; Alexander, C.; Shakesheff, K.; Cellesi, F.; Saunders, B.R. Thermally-triggered gelation of plga dispersions: Towards an injectable colloidal cell delivery system. J. Colloid Interface Sci. 2010, 344, 61–69. [Google Scholar]
- Butun, V.; Armes, S.P.; Billingham, N.C. Synthesis and aqueous solution properties of near-monodisperse tertiary amine methacrylate homopolymers and diblock copolymers. Polymer 2001, 42, 5993–6008. [Google Scholar]
- Raduan, N.H.; Horozov, T.S.; Georgiou, T.K. “Comb-like” non-ionic polymeric macrosurfactants. Soft Matter 2010, 6, 2321–2329. [Google Scholar]
- Kim, M.S.; Hyun, H.; Seo, K.S.; Cho, Y.H.; Lee, J.W.; Lee, C.R.; Khang, G.; Lee, H.B. Preparation and characterization of mpeg-pcl diblock copolymers with thermo-responsive sol-gel-sol phase transition. J. Polym. Sci. Part A 2006, 44, 5413–5423. [Google Scholar]
- Hu, Z.B.; Cai, T.; Chi, C.L. Thermoresponsive oligo(ethylene glycol)-methacrylate-based polymers and microgels. Soft Matter 2010, 6, 2115–2123. [Google Scholar]
- Yoon, J.A.; Gayathri, C.; Gil, R.R.; Kowalewski, T.; Matyjaszewski, K. Comparison of the thermoresponsive deswelling kinetics of poly(2-(2-methoxyethoxy)ethyl methacrylate) hydrogels prepared by atrp and frp. Macromolecules 2010, 43, 4791–4797. [Google Scholar]
- Becer, C.R.; Hahn, S.; Fijten, M.W.M.; Thijs, H.M.L.; Hoogenboom, R.; Schubert, U.S. Libraries of methacrylic acid and oligo(ethylene glycol) methacrylate copolymers with lcst behavior. J. Polym. Sci. Part A 2008, 46, 7138–7147. [Google Scholar]
- Lutz, J.-F.; Hoth, A. Preparation of ideal peg analogues with a tunable thermosensitivity by controlled radical copolymerization of 2-(2-methoxyethoxy)ethyl methacrylate and oligo(ethylene glycol) methacrylate. Macromolecules 2005, 39, 893–896. [Google Scholar]
- Li, W.; Wu, D.L.; Schluter, A.D.; Zhang, A.F. Synthesis of an oligo(ethylene glycol)-based third-generation thermoresponsive dendronized polymer. J. Polym. Sci. Part A 2009, 47, 6630–6640. [Google Scholar]
- Kojima, C.; Yoshimura, K.; Harada, A.; Sakanishi, Y.; Kono, K. Temperature-sensitive hyperbranched poly(glycidol)s with oligo(ethylene glycol) monoethers. J. Polym. Sci. Part A 2010, 48, 4047–4054. [Google Scholar]
- Li, W.; Zhang, A.; Schluter, A.D. Thermoresponsive dendronized polymers with tunable lower critical solution temperatures. Chem. Commun. 2008, 5523–5525. [Google Scholar]
- Li, W.; Zhang, A.; Feldman, K.; Walde, P.; Schluter, A.D. Thermoresponsive dendronized polymers. Macromolecules 2008, 41, 3659–3667. [Google Scholar]
- Juillerat-Jeanneret, L. The targeted delivery of cancer drugs across the blood-brain barrier: Chemical modifications of drugs or drug-nanoparticles? Drug Discov. Today 2008, 13, 1099–1106. [Google Scholar]
- Goodman, T.T.; Ng, C.P.; Pun, S.H. 3-D tissue culture systems for the evaluation and optimization of nanoparticle-based drug carriers. Bioconjugate Chem. 2008, 19, 1951–1959. [Google Scholar]
- Ganta, S.; Devalapally, H.; Shahiwala, A.; Amiji, M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J. Control. Release 2008, 126, 187–204. [Google Scholar]
- Hatefi, A.; Amsden, B. Biodegradable injectable in situ forming drug delivery systems. J. Control. Release 2002, 80, 9–28. [Google Scholar]
- Merdan, T.; Kopecek, J.; Kissel, T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer. Adv. Drug Deliv. Rev. 2002, 54, 715–758. [Google Scholar]
- Felgner, P.L. Nonviral strategies for gene therapy. Sci. Am. 1997, 276, 102–106. [Google Scholar]
- Han, S.; Mahato, R.I.; Sung, Y.K.; Kim, S.W. Development of biomaterials for gene therapy. Mol. Ther. 2000, 2, 302–317. [Google Scholar]
- Godbey, W.T.; Mikos, A.G. Recent progress in gene delivery using non-viral transfer complexes. J. Control. Release 2001, 72, 115–125. [Google Scholar]
- Crommelin, D.J.A.; Storm, G.; Jiskoot, W.; Stenekes, R.; Mastrobattista, E.; Hennink, W.E. Nanotechnological approaches for the delivery of macromolecules. J. Control. Release 2003, 87, 81–88. [Google Scholar]
- Jagur-Grodzinski, J. Biomedical applications of polymers 2001–2002. E-Polymers 2003, 12, 1–38. [Google Scholar]
- Kabanov, A.V. Taking polycation gene delivery systems from in vitro to in vivo. Pharm. Sci. Technol. Today 1999, 2, 365–372. [Google Scholar]
- Lavigne, M.D.; Pennadam, S.S.; Ellis, J.; Alexander, C.; Gorecki, D.C. Enhanced gene expression through temperature profile-induced variations in molecular architecture of thermoresponsive polymer vectors. J. Gene. Med. 2007, 9, 44–54. [Google Scholar]
- Mao, Z.W.; Ma, L.; Yan, J.; Yan, M.; Gao, C.Y.; Shen, J.C. The gene transfection efficiency of thermoresponsive N,N,N-trimethyl chitosan chloride-g-poly(N-isopropylacrylamide) copolymer. Biomaterials 2007, 28, 4488–4500. [Google Scholar]
- Yang, J.H.; Zhang, P.; Tang, L.; Sun, P.; Liu, W.G.; Zuo, A.J.; Liang, D.C. Temperature-tuned DNA condensation and gene transfection by pei-g-(pmeo(2)ma-b-phema) copolymer-based nonviral vectors. Biomaterials 2010, 31, 144–155. [Google Scholar]
- Kurisawa, M.; Yokoyama, M.; Okano, T. Gene expression control by temperature with thermo-responsive polymeric gene carriers. J. Control. Release 2000, 69, 127–137. [Google Scholar]
- Twaites, B.R.; Alarcon, C.D.; Cunliffe, D.; Lavigne, M.; Pennadam, S.; Smith, J.R.; Gorecki, D.C.; Alexander, C. Thermo and pH responsive polymers as gene delivery vectors: Effect of polymer architecture on DNA complexation in vitro. J. Control. Release 2004, 97, 551–566. [Google Scholar]
- Cheng, N.; Liu, W.G.; Cao, Z.Q.; Ji, W.H.; Liang, D.C.; Guo, G.; Zhang, J.Y. A study of thermoresponsive poly(N-isopropylacrylamide)/polyarginine bioconjugate non-viral transgene vectors. Biomaterials 2006, 27, 4984–4992. [Google Scholar]
- Zhou, Y.M.; Ishikawa, A.; Okahashi, R.; Uchida, K.; Nemoto, Y.; Nakayama, M.; Nakayama, Y. Deposition transfection technology using a DNA complex with a thermoresponsive cationic star polymer. J. Control. Release 2007, 123, 239–246. [Google Scholar]
- Langer, R.; Vacanti, J.P. Tissue engineering. Science 1993, 260, 920–926. [Google Scholar]
- Yoon, D.M.; Fisher, J.P. Polymeric Scaffolds for Tissue Engineering Applications. In Tissue Engineering; Fisher, J.P., Ed.; Taylor & Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Kretlow, J.D.; Klouda, L.; Mikos, A.G. Injectable matrices and scaffolds for drug delivery in tissue engineering. Adv. Drug Deliv. Rev. 2007, 59, 263–273. [Google Scholar]
- Reddi, A.H. Growth Factors and Morphogens: Signals for Tissue Engineering. In Tissue Engineering; Fisher, J.P., Ed.; Taylor & Francis: Boca Raton, FL, USA, 2007. [Google Scholar]
- Place, E.S.; George, J.H.; Williams, C.K.; Stevens, M.M. Synthetic polymer scaffolds for tissue engineering. Chem. Soc. Rev. 2009, 38, 1139–1151. [Google Scholar]
- Girgorescu, G.; Hunkelerm, D. Cell Encapsulation: Generalities, Methods, Applications and Bioartificial Pancreas Case Study. In Synthetic Polymers for Biotechnology and Medicine; Freitag, R., Ed.; Eurekah.com/Landes Bioscience: Georgetown, TX, USA, 2003. [Google Scholar]
- Hoerstup, S.P.; Lu, L.; Lysaght, M.J.; Mikos, A.G.; Rein, D.; Schoen, F.J.; Temenoff, J.S.; Tessmar, J.K.; Vacanti, J.P. Tissue Engineering. In Biomaterials Science: An Introduction to Materials in Medicine; Ratner, B.D., Hoffman, A.S., Schoen, F.J., Lemons, J.E., Eds.; Elsevier Academic Press: San Diego, CA, USA, 2004; pp. 709–749. [Google Scholar]
- Matthew, H.W.T. Polymers for Tissue Engineering Scaffolds. In Polymeric Biomaterials; Dumitiu, S., Ed.; Taylor & Francis: Boca Raton, FL, USA, 2001; pp. 167–186. [Google Scholar]
- Vert, M. Polymeric biomaterials: Strategies of the past vs. Strategies of the future. Prog. Polym. Sci. 2007, 32, 755–761. [Google Scholar]
- Salem, A.K.; Leong, K.W. Scaffolds for Delivery and Regenerative Medicine. In Scaffolding in Tissue Engineering; Ma, P.X., Elisseeff, J.H., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2005; pp. 317–334. [Google Scholar]
- Calafiore, R. Bioartificial Pancreas. In Polymeric Biomaterial; Dumitriu, S., Ed.; Taylor & Francis: Boca Raton, FL, USA, 2001; pp. 983–1005. [Google Scholar]
- Cunliffe, D.; Alarcon, C.D.; Peters, V.; Smith, J.R.; Alexander, C. Thermoresponsive surface-grafted poly(N-isopropylacrylamide) copolymers: Effect of phase transitions on protein and bacterial attachment. Langmuir 2003, 19, 2888–2899. [Google Scholar]
- Kumashiro, Y.; Yamato, M.; Okano, T. Cell attachment-detachment control on temperature-responsive thin surfaces for novel tissue engineering. Ann. Biomed. Eng. 2010, 38, 1977–1988. [Google Scholar]
- Varghese, V.M.; Raj, V.; Sreenivasan, K.; Kumary, T.V. In vitro cytocompatibility evaluation of a thermoresponsive nipaam-mma copolymeric surface using l929 cells. J. Mater. Sci.-Mater. Med. 2010, 21, 1631–1639. [Google Scholar]
- Reed, J.A.; Lucero, A.E.; Hu, S.; Ista, L.K.; Bore, M.T.; Lopez, G.P.; Canavan, H.E. A low-cost, rapid deposition method for “smart” films: Applications in mammalian cell release. ACS Appl. Mater. Interfaces 2010, 2, 1048–1051. [Google Scholar]
- Nitschke, M.; Gramm, S.; Gotze, T.; Valtink, M.; Drichel, J.; Voit, B.; Engelmann, K.; Werner, C. Thermo-responsive poly(nipaam-co-degma) substrates for gentle harvest of human corneal endothelial cell sheets. J. Biomed. Mater. Res. Part A 2007, 80A, 1003–1010. [Google Scholar]
- Patrickios, C.S.; Georgiou, T.K. Covalent amphiphilic polymer networks. Curr. Opinion Colloid Interface Sci. 2003, 8, 76–85. [Google Scholar]
- Tsitsilianis, C. Responsive reversible hydrogels from associative “smart” macromolecules. Soft Matter 2010, 6, 2372–2388. [Google Scholar]
- Tanaka, T. Gels. Sci. Am. 1981, 244, 124–138. [Google Scholar]
- Okuyama, Y.; Yoshida, R.; Sakai, K.; Okano, T.; Sakurai, Y. Swelling controlled zero-order and sigmoidal drug release from thermoresponsive poly(N-isopropylacrylamide-co-butyl methacrylate) hydrogel. J. Biomater. Sci.-Polym. Ed. 1993, 4, 545–556. [Google Scholar]
- Jones, D.S.; Lorimer, C.P.; McCoy, C.P.; Gorman, S.P. Characterization of the physicochemical, antimicrobial, and drug release properties of thermoresponsive hydrogel copolymers designed for medical device applications. J. Biomed. Mater. Res. Part B 2008, 85B, 417–426. [Google Scholar]
- Jones, D.S.; Lorimer, C.J.; Andrews, G.P.; McCoy, C.P.; Gorman, S.P. An examination of the thermorheological and drug release properties of zinc tetraphenylporphyrin-containing thermoresponsive hydrogels, designed as light activated antimicrobial implants. Chem. Eng. Sci. 2007, 62, 990–999. [Google Scholar]
- Coughlan, D.C.; Corrigan, O.I. Release kinetics of benzoic acid and its sodium salt from a series of poly(N-isopropylacrylamide) matrices with various percentage crosslinking. J. Pharm. Sci. 2008, 97, 318–330. [Google Scholar]
- Coughlan, D.C.; Corrigan, O.I. Drug-polymer interactions and their effect on thermoresponsive poly(N-isopropylacrylamide) drug delivery systems. Int. J. Pharm. 2006, 313, 163–174. [Google Scholar]
- Jhon, Y.K.; Bhat, R.R.; Jeong, C.; Rojas, O.J.; Szleifer, I.; Genzer, J. Salt-induced depression of lower critical solution temperature in a surface-grafted neutral thermoresponsive polymer. Macromol. Rapid Commun. 2006, 27, 697–701. [Google Scholar]
- Martellini, F.; Mei, L.H.I.; Balino, J.L.; Carenza, M. Water and drug transport in radiation-crosslinked poly(2-methoxyethylacrylate-co-dimethyi acrylamide) and poly(2-methoxyethylacrylate-co-acrylamide) hydrogels. Radiat. Phys. Chem. 2003, 66, 155–159. [Google Scholar]
- Kiremitci, A.S.; Ciftci, A.; Ozalp, M.; Gumuesderelioglu, M. Novel chlorhexidine releasing system developed from thermosensitive vinyl ether-based hydrogels. J. Biomed. Mater. Res. Part B 2007, 83B, 609–614. [Google Scholar]
- Yang, H.; Kao, W.Y.J. Thermoresponsive gelatin/monomethoxy poly(ethylene glycol)-poly(D,L-lactide) hydrogels: Formulation, characterization, and antibacterial drug delivery. Pharm. Res. 2006, 23, 205–214. [Google Scholar]
- Yoshida, T.; Aoyagi, T.; Kokufuta, E.; Okano, T. Newly designed hydrogel with both sensitive thermoresponse and biodegradability. J. Polym. Sci. Part A 2003, 41, 779–787. [Google Scholar]
- Bessa, P.C.; Machado, R.; Nurnberger, S.; Dopler, D.; Banerjee, A.; Cunha, A.M.; Rodriguez-Cabello, J.C.; Redl, H.; van Griensven, M.; Reis, R.L.; et al. Thermoresponsive self-assembled elastin-based nanoparticles for delivery of bmps. J. Control. Release 2010, 142, 312–318. [Google Scholar]
- Rincon, A.C.; Molina-Martinez, I.T.; de Las Heras, B.; Alonso, M.; Bailez, C.; Rodriguez-Cabello, J.C.; Herrero-Vanrell, R. Biocompatibility of elastin-like polymer poly(vpavg) microparticles: In vitro and in vivo studies. J. Biomed. Mater. Res. Part A 2006, 78A, 343–351. [Google Scholar]
- Xiao, H.; Nayak, B.R.; Lowe, T.L. Synthesis and characterization of novel thermoresponsive-co-biodegradable hydrogels composed of N-isopropylacrylamide, poly(L-lactic acid), and dextran. J. Polym. Sci. Part A 2004, 42, 5054–5066. [Google Scholar]
- Wu, D.Q.; Qiu, F.; Wang, T.; Jiang, X.J.; Zhang, X.Z.; Zhuo, R.X. Toward the development of partially biodegradable and injectable thermoresponsive hydrogels for potential biomedical applications. ACS Appl. Mater. Interfaces 2009, 1, 319–327. [Google Scholar]
- Brun-Graeppi, A.; Richard, C.; Bessodes, M.; Scherman, D.; Narita, T.; Ducouret, G.; Merten, O.W. Study on the sol-gel transition of xyloglucan hydrogels. Carbohydr. Polym. 2010, 80, 555–562. [Google Scholar]
- Kan, P.; Lin, X.Z.; Hsieh, M.F.; Chang, K.Y. Thermogelling emulsions for vascular embolization and sustained release of drugs. J. Biomed. Mater. Res. Part B 2005, 75B, 185–192. [Google Scholar]
- Meenach, S.A.; Anderson, K.W.; Hilt, J.Z. Synthesis and characterization of thermoresponsive poly(ethylene glycol)-based hydrogels and their magnetic nanocomposites. J. Polym. Sci. Part A 2010, 48, 3229–3235. [Google Scholar]
- Papaphilippou, P.C.; Pourgouris, A.; Marinica, O.; Taculescu, A.; Athanasopoulos, G.I.; Vekas, L.; Krasia-Christoforou, T. Fabrication and characterization of superparamagnetic and thermoresponsive hydrogels based on oleic-acid-coated Fe3O4 nanoparticles, hexa(ethylene glycol) methyl ether methacrylate and 2-(acetoacetoxy)ethyl methacrylate. J. Magn. Magn. Mater. 2011, 323, 557–563. [Google Scholar]
- Wang, N.; Dong, A.; Radosz, M.; Shen, Y.Q. Thermoresponsive degradable poly(ethylene glycol) analogues. J. Biomed. Mater. Res. Part A 2008, 84A, 148–157. [Google Scholar]
- Garty, S.; Kimelman-Bleich, N.; Hayouka, Z.; Cohn, D.; Friedler, A.; Pelled, G.; Gazit, D. Peptide-modified “smart” hydrogels and genetically engineered stem cells for skeletal tissue engineering. Biomacromolecules 2010, 11, 1516–1526. [Google Scholar]
- Borden, B.A.; Yockman, J.; Kim, S.W. Thermoresponsive hydrogel as a delivery scaffold for transfected rat mesenchymal stem cells. Mol. Pharm. 2010, 7, 963–968. [Google Scholar]
- Kwon, I.K.; Matsuda, T. Photo-iniferter-based thermoresponsive block copolymers composed of poly(ethylene glycol) and poly(N-isopropylacrylamide) and chondrocyte immobilization. Biomaterials 2006, 27, 986–995. [Google Scholar]
- Kirkland, S.E.; Hensarling, R.M.; McConaughy, S.D.; Guo, Y.; Jarrett, W.L.; McCormick, C.L. Thermoreversible hydrogels from raft-synthesized bab triblock copolymers: Steps toward biomimetic matrices for tissue regeneration. Biomacromolecules 2008, 9, 481–486. [Google Scholar]
- Ma, Z.W.; Nelson, D.M.; Hong, Y.; Wagner, W.R. Thermally responsive injectable hydrogel incorporating methacrylate-polylactide for hydrolytic lability. Biomacromolecules 2010, 11, 1873–1881. [Google Scholar]
- Stabenfeldt, S.E.; Garcia, A.J.; LaPlaca, M.C. Thermoreversible laminin-functionalized hydrogel for neural tissue engineering. J. Biomed. Mater. Res. Part A 2006, 77A, 718–725. [Google Scholar]
- Galperin, A.; Long, T.J.; Ratner, B.D. Degradable, thermo-sensitive poly(N-isopropyl acrylamide)-based scaffolds with controlled porosity for tissue engineering applications. Biomacromolecules 2010, 11, 2583–2592. [Google Scholar]
- Shin, H.S.; Kim, S.Y.; Lee, Y.M. Indomethacin release behaviors from pH and thermoresponsive poly(vinyl alcohol) and poly(acrylic acid) IPN hydrogels for site-specific drug delivery. J. Appl. Polym. Sci. 1997, 65, 685–693. [Google Scholar]
- Wang, Q.F.; Li, S.M.; Wang, Z.Y.; Liu, H.Z.; Li, C.J. Preparation and characterization of a positive thermoresponsive hydrogel for drug loading and release. J. Appl. Polym. Sci. 2009, 111, 1417–1425. [Google Scholar]
- Mundargi, R.C.; Shelke, N.B.; Babu, V.R.; Patel, P.; Rangaswamy, V.; Aminabhavi, T.M. Novel thermo-responsive semi-interpenetrating network microspheres of gellan gum-poly(N-isopropylacrylamide) for controlled release of atenolol. J. Appl. Polym. Sci. 2010, 116, 1832–1841. [Google Scholar]
- Kim, S.; Chung, E.H.; Gilbert, M.; Healy, K.E. Synthetic mmp-13 degradable ecms based on poly(N-isopropylacrylamide-co-acrylic acid) semi-interpenetrating polymer networks. I. Degradation and cell migration. J. Biomed. Mater. Res. Part A 2005, 75A, 73–88. [Google Scholar]
- Chung, J.E.; Yokoyama, M.; Aoyagi, T.; Sakurai, Y.; Okano, T. Effect of molecular architecture of hydrophobically modified poly(N-isopropylacrylamide) on the formation of thermoresponsive core-shell micellar drug carriers. J. Control. Release 1998, 53, 119–130. [Google Scholar]
- Akimoto, J.; Nakayama, M.; Sakai, K.; Okano, T. Temperature-induced intracellular uptake of thermoresponsive polymeric micelles. Biomacromolecules 2009, 10, 1331–1336. [Google Scholar]
- Hruby, M.; Konak, C.; Kucka, J.; Vetrik, M.; Filippov, S.K.; Vetvicka, D.; Mackova, H.; Karlsson, G.; Edwards, K.; Rihova, B.; et al. Thermoresponsive, hydrolytically degradable polymer micelles intended for radionuclide delivery. Macromol. Biosc. 2009, 9, 1016–1027. [Google Scholar]
- Loh, X.J.; Zhang, Z.X.; Wu, Y.L.; Lee, T.S.; Li, J. Synthesis of novel biodegradable thermoresponsive triblock copolymers based on poly(r)-3-hydroxybutyrate and poly(N-isopropylacrylamide) and their formation of thermoresponsive micelles. Macromolecules 2009, 42, 194–202. [Google Scholar]
- Wei, H.; Zhang, X.Z.; Cheng, H.; Chen, W.Q.; Cheng, S.X.; Zhuo, R.X. Self-assembled thermo- and pH-responsive micelles of poly(10-undecenoic acid-b-N-isopropylacrylamide) for drug delivery. J. Control. Release 2006, 116, 266–274. [Google Scholar]
- Wei, H.; Zhang, X.Z.; Zhou, Y.; Cheng, S.X.; Zhuo, R.X. Self-assembled thermoresponsive micelles of poly(N-isopropylacrylamide-b-methyl methacrylate). Biomaterials 2006, 27, 2028–2034. [Google Scholar]
- Akimoto, J.; Nakayama, M.; Sakai, K.; Okano, T. Thermally controlled intracellular uptake system of polymeric micelles possessing poly(N-isopropylacrylamide)-based outer coronas. Mol. Pharm. 2010, 7, 926–935. [Google Scholar]
- Neradovic, D.; van Nostrum, C.F.; Hennink, W.E. Thermoresponsive polymeric micelles with controlled instability based on hydrolytically sensitive N-isopropylacrylamide copolymers. Macromolecules 2001, 34, 7589–7591. [Google Scholar]
- Rijcken, C.J.F.; Veldhuis, T.F.J.; Ramzi, A.; Meeldijk, J.D.; van Nostrum, C.F.; Hennink, W.E. Novel fast degradable thermosensitive polymeric micelles based on peg-block-poly(N-(2-hydroxyethyl)methacrylamide-oligolactates). Biomacromolecules 2005, 6, 2343–2351. [Google Scholar]
- Huang, X.N.; Du, F.S.; Cheng, J.; Dong, Y.Q.; Liang, D.H.; Ji, S.P.; Lin, S.S.; Li, Z.C. Acid-sensitive polymeric micelles based on thermoresponsive block copolymers with pendent cyclic orthoester groups. Macromolecules 2009, 42, 783–790. [Google Scholar]
- Hruby, M.; Filippov, S.K.; Panek, J.; Novakova, M.; Mackova, H.; Kucka, J.; Vetvicka, D.; Ulbrich, K. Polyoxazoline thermoresponsive micelles as radionuclide delivery systems. Macromol. Biosci. 2010, 10, 916–924. [Google Scholar]
- Wang, Y.-C.; Tang, L.-Y.; Li, Y.; Wang, J. Thermoresponsive block copolymers of poly(ethylene glycol) and polyphosphoester: Thermo-induced self-assembly, biocompatibility, and hydrolytic degradation. Biomacromolecules 2008, 10, 66–73. [Google Scholar]
- Gotzamanis, G.; Tsitsilianis, C. Design of responsive double-hydrophilic A-b-(B-co-C) diblock terpolymers with tunable thermosensitivity. Polymer 2007, 48, 6226–6233. [Google Scholar]
- Wei, H.; Zhang, X.Z.; Chen, W.Q.; Cheng, S.X.; Zhuo, R.X. Self-assembled thermosensitive micelles based on poly(L-lactide-star block-N-isopropylacrylamide) for drug delivery. J. Biomed. Mater. Res. Part A 2007, 83A, 980–989. [Google Scholar]
- Read, E.S.; Armes, S.P. Recent advances in shell cross-linked micelles. Chem. Commun. 2007, 3021–3035. [Google Scholar]
- Wei, H.; Cheng, C.; Chang, C.; Chen, W.Q.; Cheng, S.; Zhang, X.Z.; Zhuo, R.X. Synthesis and applications of shell cross-linked thermoresponsive hybrid micelles based on poly(N-isopropylacrylamide-co-3-(trimethoxysilyl)propyl methacrylate)-b-poly(methyl methacrylate). Langmuir 2008, 24, 4564–4570. [Google Scholar]
- Wei, H.; Quan, C.Y.; Chang, C.; Zhang, X.Z.; Zhuo, R.X. Preparation of novel ferrocene-based shell cross-linked thermoresponsive hybrid micelles with antitumor efficacy. J. Phys. Chem. B 2010, 114, 5309–5314. [Google Scholar]
- Cohn, D.; Sagiv, H.; Benyamin, A.; Lando, G. Engineering thermoresponsive polymeric nanoshells. Biomaterials 2009, 30, 3289–3296. [Google Scholar]
- Meng, F.; Zhong, Z.; Feijen, J. Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules 2009, 10, 197–209. [Google Scholar]
- Du, J.Z.; O'Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544–3561. [Google Scholar]
- Kim, K.T.; Meeuwissen, S.A.; Nolte, R.J.M.; van Hest, J.C.M. Smart nanocontainers and nanoreactors. Nanoscale 2010, 2, 844–858. [Google Scholar]
- Qin, S.H.; Geng, Y.; Discher, D.E.; Yang, S. Temperature-controlled assembly and release from polymer vesicles of poly(ethylene oxide)-block-poly(N-isopropylacrylamide). Adv. Mater. 2006, 18, 2905–2909. [Google Scholar]
- Li, Y.; Lokitz, B.S.; McCormick, C.L. Thermally responsive vesicles and their structural “locking” through polyelectrolyte complex formation. Angew. Chem., Int. Ed. 2006, 45, 5792–5795. [Google Scholar]
- Qin, S.; Geng, Y.; Discher, D.E.; Yang, S. Temperature-controlled assembly and release from polymer vesicles of poly(ethylene oxide)-block-poly(N-isopropylacrylamide). Adv. Mater. 2006, 18, 2905–2909. [Google Scholar]
- Wei, H.; Zhang, X.-Z.; Chen, W.-Q.; Cheng, S.-X.; Zhuo, R.-X. Self-assembled thermosensitive micelles based on poly(L-lactide-star block-N-isopropylacrylamide) for drug delivery. J. Biomed. Mater. Res. Part A 2007, 83A, 980–989. [Google Scholar]
- Zhou, Y.; Yan, D.; Dong, W.; Tian, Y. Temperature-responsive phase transition of polymer vesicles: Real-time morphology observation and molecular mechanism. J. Phys. Chem. B 2007, 111, 1262–1270. [Google Scholar]
- Agut, W.; Brûlet, A.; Schatz, C.; Taton, D.; Lecommandoux, S.B. pH and temperature responsive polymeric micelles and polymersomes by self-assembly of poly[2-(dimethylamino)ethyl methacrylate]-b-poly(glutamic acid) double hydrophilic block copolymers. Langmuir 2010, 26, 10546–10554. [Google Scholar]
- Wilson, S.J.; Gorelov, A.V.; Rochev, Y.A.; McGillicuddy, F.; Dawson, K.A.; Gallagher, W.M.; Keenan, A.K. Extended delivery of the antimitotic agent colchicine from thermoresponsive N-isopropylacrylamide-based copolymer films to human vascular smooth muscle cells. J. Biomed. Mater. Res. Part A 2003, 67A, 667–673. [Google Scholar]
- Dinarvand, R.; Ansari, M. The use of thermoresponsive hydrogel membrane as modulated drug delivery system. Daru 2002, 10, 105–110. [Google Scholar]
- Zakharchenko, S.; Puretskiy, N.; Stoychev, G.; Stamm, M.; Ionov, L. Temperature controlled encapsulation and release using partially biodegradable thermo-magneto-sensitive self-rolling tubes. Soft Matter 2010, 6, 2633–2636. [Google Scholar]
- Zhu, Z.C.; Sukhishvili, S.A. Temperature-induced swelling and small molecule release with hydrogen-bonded multilayers of block copolymer micelles. ACS Nano 2009, 3, 3595–3605. [Google Scholar]
- Schmaljohann, D. Thermo-responsive polymers and hydrogels in tissue engineering. E-Polymers 2005, no. 21. [Google Scholar]
- Li, L.; Yang, X.M.; Liu, F.J.; Shang, J.Q.; Yan, G.P.; Li, W. Thermosensitive poly(N-isopropylacrylamide)-b-polycaprolacton-b-poly(N-isopropylacrylamide) triblock copolymers prepared via atom transfer radical polymerization for control of cell adhesion and detachment. J. Chil. Chem. Soc. 2009, 54, 397–400. [Google Scholar]
- Abraham, T.N.; Raj, V.; Prasad, T.; Kumar, P.R.A.; Sreenivasan, K.; Kumary, T.V. A novel thermoresponsive graft copolymer containing phosphorylated hema for generating detachable cell layers. J. Appl. Polym. Sci. 2010, 115, 52–62. [Google Scholar]
- Chen, G.P.; Ito, Y.; Imanishi, Y. Regulation of growth and adhesion of cultured cells by insulin conjugated with thermoresponsive polymers. Biotechnol. Bioeng. 1997, 53, 339–344. [Google Scholar]
- Hatakeyama, H.; Kikuchi, A.; Yamato, M.; Okano, T. Influence of insulin immobilization to thermoresponsive culture surfaces on cell proliferation and thermally induced cell detachment. Biomaterials 2005, 26, 5167–5176. [Google Scholar]
- to, Y.; Chen, G.P.; Guan, Y.Q.; Imanishi, Y. Patterned immobilization of thermoresponsive polymer. Langmuir 1997, 13, 2756–2759. [Google Scholar]
- Liu, H.C.; Ito, Y. Cell attachment and detachment on micropattern-immobilized poly(N-isopropylacrylamide) with gelatin. Lab Chip 2002, 2, 175–178. [Google Scholar]
- Tsuda, Y.; Kikuchi, A.; Yamato, M.; Nakao, A.; Sakurai, Y.; Umezu, M.; Okano, T. The use of patterned dual thermoresponsive surfaces for the collective recovery as co-cultured cell sheets. Biomaterials 2005, 26, 1885–1893. [Google Scholar]
- Ito, Y. Surface micropatterning to regulate cell functions. Biomaterials 1999, 20, 2333–2342. [Google Scholar]
- von Recum, H.; Okano, T.; Kim, S.W. Growth factor release from thermally reversible tissue culture substrates. J. Control. Release 1998, 55, 121–130. [Google Scholar]
- Kikuchi, A.; Okano, T. Nanostructured designs of biomedical materials: Applications of cell sheet engineering to functional regenerative tissues and organs. J. Control. Release 2005, 101, 69–84. [Google Scholar]
- Da Silva, R.M.P.; Mano, J.F.; Reis, R.L. Smart thermoresponsive coatings and surfaces for tissue engineering: Switching cell-material boundaries. Trends Biotechnol. 2007, 25, 577–583. [Google Scholar]
- Mizutani, A.; Kikuchi, A.; Yamato, M.; Kanazawa, H.; Okano, T. Preparation of thermoresponsive polymer brush surfaces and their interaction with cells. Biomaterials 2008, 29, 2073–2081. [Google Scholar]
- Moran, M.T.; Carroll, W.M.; Gorelov, A.; Rochev, Y. Intact endothelial cell sheet harvesting from thermoresponsive surfaces coated with cell adhesion promoters. J. R. Soc. Interface 2007, 4, 1151–1157. [Google Scholar]
- Cheng, X.H.; Wang, Y.B.; Hanein, Y.; Bohringer, K.F.; Ratner, B.D. Novel cell patterning using microheater-controlled thermoresponsive plasma films. J. Biomed. Mater. Res. Part A 2004, 70A, 159–168. [Google Scholar]
- Poon, Y.F.; Cao, Y.; Liu, Y.X.; Chan, V.; Chan-Park, M.B. Hydrogels based on dual curable chitosan-graft-polyethylene glycol-graft-methacrylate: Application to layer-by-layer cell encapsulation. ACS Appl. Mater. Interfaces 2010, 2, 2012–2025. [Google Scholar]
- Li, L.H.; Jiang, X.L.; Zhuo, R.X. Synthesis and characterization of thermoresponsive polymers containing reduction-sensitive disulfide linkage. J. Polym. Sci. Part A 2009, 47, 5989–5997. [Google Scholar]
- Neradovic, D.; Soga, O.; van Nostrum, C.F.; Hennink, W.E. The effect of the processing and formulation parameters on the size of nanoparticles based on block copolymers of poly(ethylene glycol) and poly(N-isopropylacrylamide) with and without hydrolytically sensitive groups. Biomaterials 2004, 25, 2409–2418. [Google Scholar]
- Zhang, S.M.; Zhang, L.N.; He, B.F.; Wu, Z.S. Preparation and characterization of thermosensitive pnipaa-coated iron oxide nanoparticles. Nanotechnology 2008, 19, 325608. [Google Scholar]
- Purushotham, S.; Chang, P.E.J.; Rumpel, H.; Kee, I.H.C.; Ng, R.T.H.; Chow, P.K.H.; Tan, C.K.; Ramanujan, R.V. Thermoresponsive core-shell magnetic nanoparticles for combined modalities of cancer therapy. Nanotechnology 2009, 20, 305101. [Google Scholar]
- Purushotham, S.; Ramanujan, R.V. Modeling the performance of magnetic nanoparticles in multimodal cancer therapy. J. Appl. Phys. 2010, 107, 114701. [Google Scholar]
- Liu, C.Y.; Guo, J.; Yang, W.L.; Hu, J.H.; Wang, C.C.; Fu, S.K. Magnetic mesoporous silica microspheres with thermo-sensitive polymer shell for controlled drug release. J. Mater. Chem. 2009, 19, 4764–4770. [Google Scholar]
- Zhu, Y.F.; Kaskel, S.; Ikoma, T.; Hanagata, N. Magnetic SBA-15/poly(N-isopropylacrylamide) composite: Preparation, characterization and temperature-responsive drug release property. Microporous Mesoporous Mater. 2009, 123, 107–112. [Google Scholar]
- Lutz, J.-F.; Stiller, S.; Hoth, A.; Kaufner, L.; Pison, U.; Cartier, R. One-pot synthesis of pegylated ultrasmall iron-oxide nanoparticles and their in vivo evaluation as magnetic resonance imaging contrast agents. Biomacromolecules 2006, 7, 3132–3138. [Google Scholar]
- Papaphilippou, P.; Loizou, L.; Popa, N.C.; Han, A.; Vekas, L.; Odysseos, A.; Krasia-Christoforou, T. Superparamagnetic hybrid micelles, based on iron oxide nanoparticles and well-defined diblock copolymers possessing β-ketoester functionalities. Biomacromolecules 2009, 10, 2662–2671. [Google Scholar]
- Zhang, J.; Misra, R.D.K. Magnetic drug-targeting carrier encapsulated with thermosensitive smart polymer: Core-shell nanoparticle carrier and drug release response. Acta Biomater. 2007, 3, 838–850. [Google Scholar]
- Yuan, Q.; Venkatasubramanian, R.; Hein, S.; Misra, R.D.K. A stimulus-responsive magnetic nanoparticle drug carrier: Magnetite encapsulated by chitosan-grafted-copolymer. Acta Biomater. 2008, 4, 1024–1037. [Google Scholar]
- Pich, A.; Bhattacharya, S.; Lu, Y.; Boyko, V.; Adler, H.-J.P. Temperature-sensitive hybrid microgels with magnetic properties. Langmuir 2004, 20, 10706–10711. [Google Scholar]
- Liu, T.Y.; Hu, S.H.; Liu, D.M.; Chen, S.Y.; Chen, I.W. Biomedical nanoparticle carriers with combined thermal and magnetic responses. Nano Today 2009, 4, 52–65. [Google Scholar]
- Das, M.; Sanson, N.; Fava, D.; Kumacheva, E. Microgels loaded with gold nanorods: Photothermally triggered volume transitions under physiological conditions. Langmuir 2007, 23, 196–201. [Google Scholar]
- Chakraborty, S.; Bishnoi, S.W.; Perez-Luna, V.H. Gold nanoparticles with poly(N-isopropylacrylamide) formed via surface initiated atom transfer free radical polymerization exhibit unusually slow aggregation kinetics. J. Phys. Chem. C 2010, 114, 5947–5955. [Google Scholar]
- Fundueanu, G.; Constantin, M.; Ascenzi, P. Poly(N-isopropylacrylamide-co-acrylamide) cross-linked thermoresponsive microspheres obtained from preformed polymers: Influence of the physico-chemical characteristics of drugs on their release profiles. Acta Biomater. 2009, 5, 363–373. [Google Scholar]
- Fundueanu, G.; Constantin, M.; Ascenzi, P. Fast-responsive porous thermoresponsive microspheres for controlled delivery of macromolecules. Int. J. Pharm. 2009, 379, 9–17. [Google Scholar]
- Wang, Y.C.; Li, Y.; Yang, X.Z.; Yuan, Y.Y.; Yan, L.F.; Wang, J. Tunable thermosensitivity of biodegradable polymer micelles of poly(epsilon-caprolactone) and polyphosphoester block copolymers. Macromolecules 2009, 42, 3026–3032. [Google Scholar]
- Wang, Y.C.; Yuan, Y.Y.; Du, J.Z.; Yang, X.Z.; Wang, J. Recent progress in polyphosphoesters: From controlled synthesis to biomedical applications. Macromol. Biosci. 2009, 9, 1154–1164. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ward, M.A.; Georgiou, T.K. Thermoresponsive Polymers for Biomedical Applications. Polymers 2011, 3, 1215-1242. https://doi.org/10.3390/polym3031215
Ward MA, Georgiou TK. Thermoresponsive Polymers for Biomedical Applications. Polymers. 2011; 3(3):1215-1242. https://doi.org/10.3390/polym3031215
Chicago/Turabian StyleWard, Mark A., and Theoni K. Georgiou. 2011. "Thermoresponsive Polymers for Biomedical Applications" Polymers 3, no. 3: 1215-1242. https://doi.org/10.3390/polym3031215
APA StyleWard, M. A., & Georgiou, T. K. (2011). Thermoresponsive Polymers for Biomedical Applications. Polymers, 3(3), 1215-1242. https://doi.org/10.3390/polym3031215