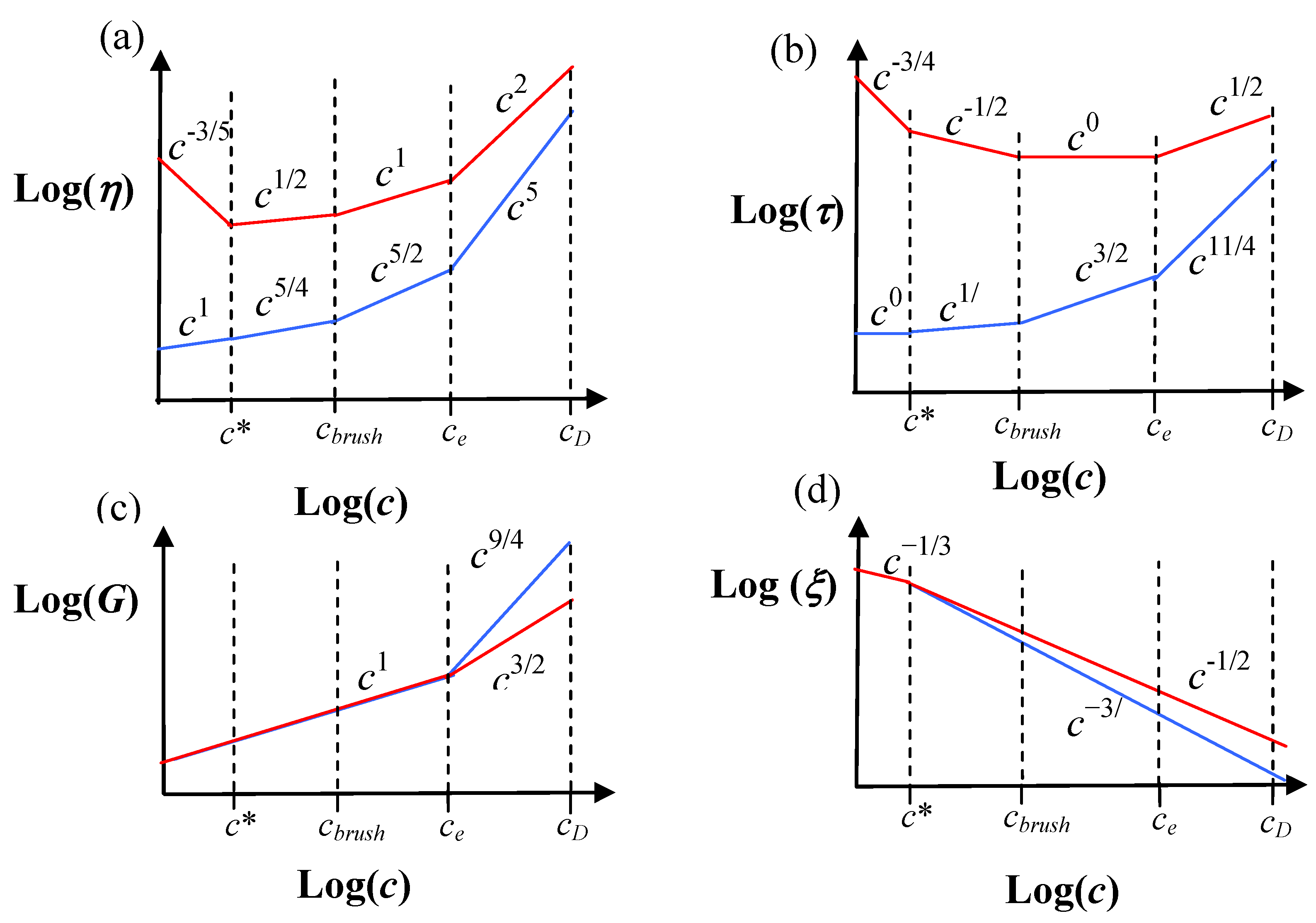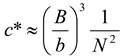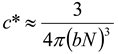1. Structure of Synthetic and Natural Comb Polyelectrolytes
There have been a number of recent review articles on comb polyelectrolytes primarily focused on synthetic materials [
1,
2]. Here we present another short review article, but with the emphasis shifted to biological and biomimetic materials and Manchester centred work in this area.
The comb structural motif is sometimes considered a poor relation of the globular, helical or beta sheet motifs commonly found in biological macromolecules. In most part this is due to the challenges carbohydrates (the predominant side-chain chemistry) present to structural elucidation, which obstructs attempts to make a strong link between the structure and the function of these molecules. Our lack of understanding is not representative of the comb motif’s ubiquity, since the motif is found in a vast range of biomolecular systems. Indeed many molecules from two huge divisions of molecular biology,
glycoproteins [
3,
4,
5] and
proteoglycans [
6,
7], carry the comb motif. These are some of the worst understood biological molecules due to their heterogeneity and the aforementioned problems in structural determination (primarily due to their non-crystallinity).
There are also a few examples of completely proteinaious comb brushes in biology. Neurofilaments play a vital structural role in neurons as intracellular scaffolds and consist of self-assembling protein sub-units with a resultant protein backbone and flexible protein side-chains [
8,
9].
Synthetic biomimetic comb polyelectrolytes can now be made in a wide variety of well defined topologies and provide a new tool for probing the physical properties of combs using well characterised chemistries [
10]. We can now ask fundamental questions, such as what is the function of the side-chains in glycoproteins and proteoglycans, with a reasonable chance of a definitive answer, with theory guided by experiments on well defined synthetic materials [
1].
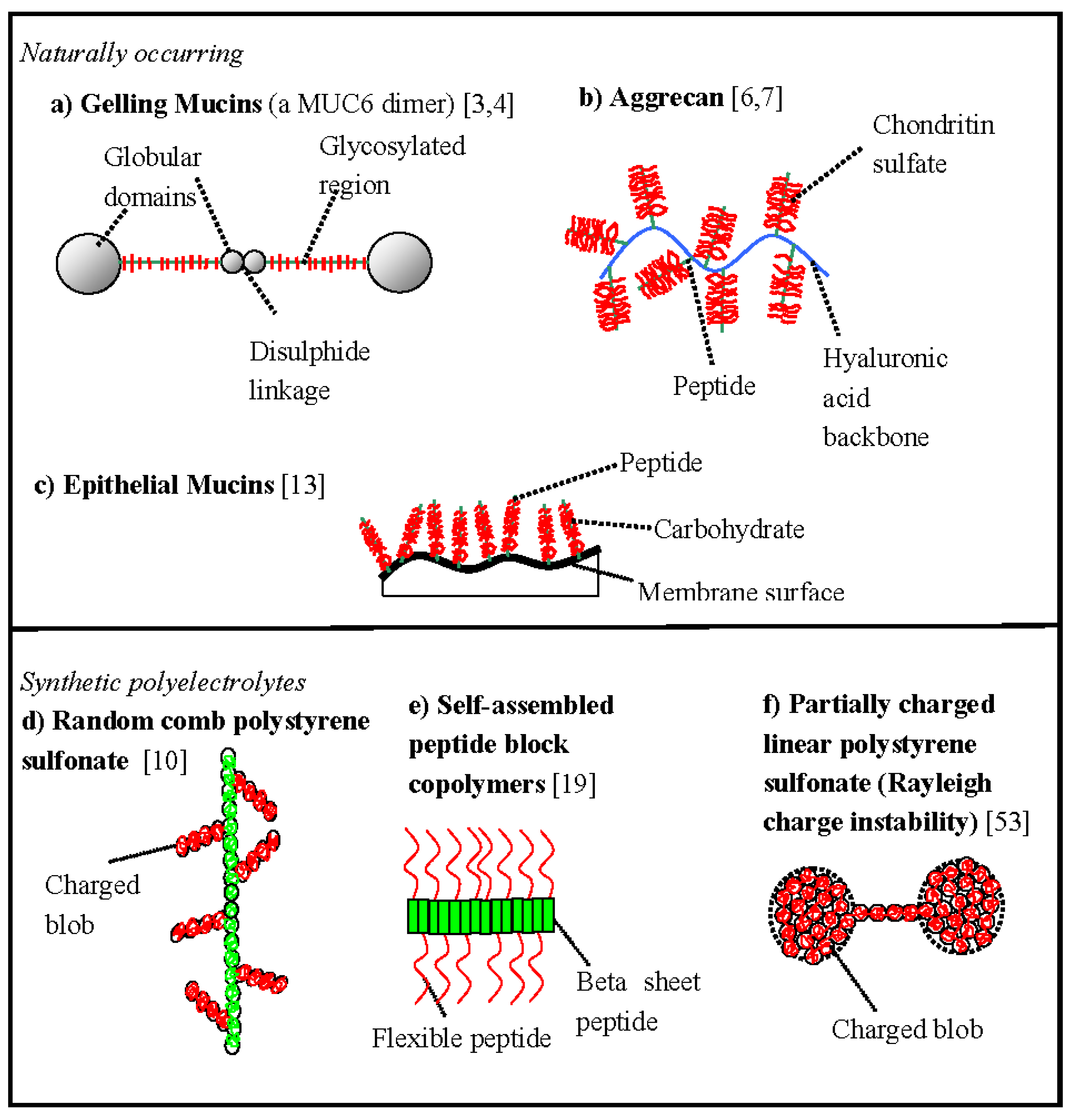
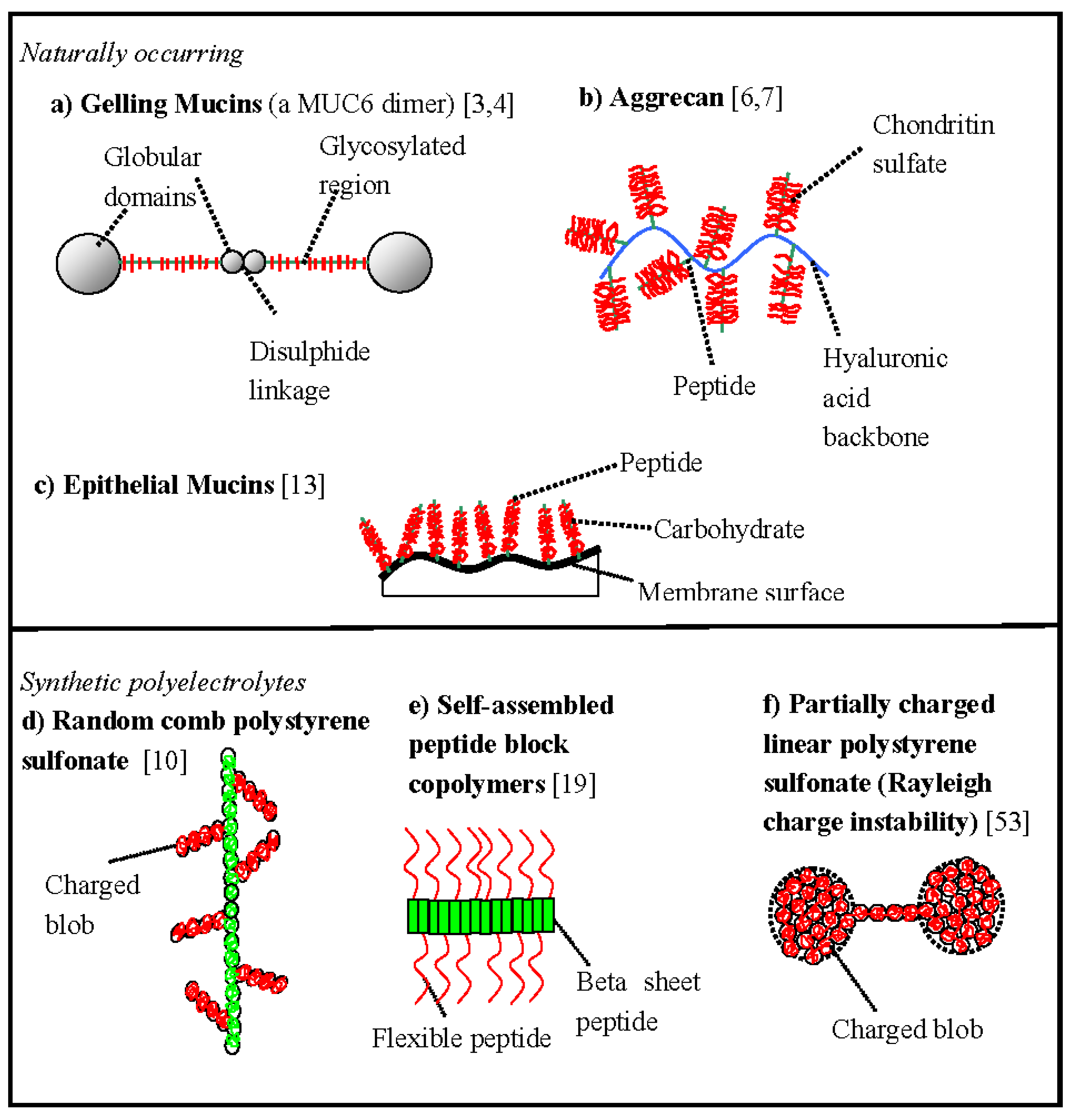
Some schematic comb polyelectrolyte structures and associated biopolymer morphologies are shown in
Figure 1. Although the comb motif is seen in a wide range of biological macromolecules, it is often in combination with other structural effects, e.g., MUC6 mucin has a dumbbell structure [
11] (two globules separated by a comb spacer—a mucin dimer is shown in
Figure 1a) and aggrecan has a double-hierarchical comb structure (a bottle brush of bottle brushes,
Figure 1b) [
6]. The chemistries corresponding to the morphologies shown in
Figure 1 are described in more detail in
Table 1.
The proposed double-globule morphology of the
gelling mucins (
Figure 1a) is a new development in the field that appears to be in agreement with a wide range of experiments including atomic force microscopy, transmission electron microscopy and small-angle X-ray scattering [
11]. There is a fascinating analogy between the mucin double-globules and the multi-globules observed in synthetic hydrophobic polyelectrolytes [
12] (
Figure 1f), but it is now believed the mucin globular morphology, although driven by hydrophobicity, is more closely determined by the primary chemical structure than normally expected with synthetic polyelectrolyte Rayleigh charge instabilities for random copolymers. In mucin the comb motif acts as a hydrophilic globule breaker, splitting a single globule into two subglobules [
11]. MUC6 mucin has a block copolymer type primary structure, in the language of synthetic polymer chemistry, two hydrophobic globular blocks separated by a hydrophilic comb block.
Epithelial mucins present a second major structural subclassification in the mucin family (
Figure 1c). These large flexible biopolymer combs are found chemically attached to the surfaces of a wide range of naturally occurring membranes. In particular they are found in combination with the ciliated linings of the lung’s surface and intense research is currently underway to understand their role in respiratory diseases [
13].
Figure 1.
The morphology of some schematic comb biopolymers and related polyelectrolyte structures. (a) Gelling mucins, e.g., porcine MUC6 [
11]. (b) Proteoglycans, e.g., bovine aggrecan [
6,
7]. (c) Epithelial mucins [
13]. (d) Synthetic flexible comb polyelectrolytes, e.g., polystyrene sulfonate [
10]. (e) Synthetic self-assembled peptide block copolymers [
19]. (f) Synthetic hydrophobic linear polyelectrolytes demonstrating the Rayleigh charge instability, e.g., partially charged linear polystyrene sulfonate [
53]. For simplicity the counterions have not been shown. Additional chemical details are given in
Table 1.
Figure 1.
The morphology of some schematic comb biopolymers and related polyelectrolyte structures. (a) Gelling mucins, e.g., porcine MUC6 [
11]. (b) Proteoglycans, e.g., bovine aggrecan [
6,
7]. (c) Epithelial mucins [
13]. (d) Synthetic flexible comb polyelectrolytes, e.g., polystyrene sulfonate [
10]. (e) Synthetic self-assembled peptide block copolymers [
19]. (f) Synthetic hydrophobic linear polyelectrolytes demonstrating the Rayleigh charge instability, e.g., partially charged linear polystyrene sulfonate [
53]. For simplicity the counterions have not been shown. Additional chemical details are given in
Table 1.
Table 1.
Additional chemical details corresponding to the morphologies shown in
Figure 1.
Table 1.
Additional chemical details corresponding to the morphologies shown in Figure 1.
(a)Gelling mucin [3,4]
Main chain—unique complex protein sequence (hydrophobic globules at ends), flexible moderately charged.
Side-chains—polymorphic complex hydrophilic carbohydrate sequences (both charged anionic and uncharged isoforms exist), short and relatively rigid. | (b) Aggrecan [6,7]
Main chain—hyaluronic acid, hydrophilic, flexible, highly charged, anionic, polyelectrolyte.
Main side-chains—unique flexible complex protein sequence (hydrophilic).
Side-chains—flexible, polymorphic complex carbohydrate sequences (hydrophilic, anionic, strongly charged). | (c) Epithelial mucin [13]
Main chain—unique flexible complex protein sequence (hydrophilic).
Side-chains—flexible, polymorphic complex carbohydrate sequences (hydrophilic, anionic, strongly charged). |
(d) Comb fully charged [10] polystyrene sulfonate
Main chain—hydrophilic, highly charged, anionic, flexible.
Side-chains—hydrophilic, highly charged, anionic, flexible. | (e) Diblock peptide [19]
Main chain—semi-flexible self-assembled hydrophobic blocks of valine.
Side-chains—flexible hydrophobic cationic charged polylysine chains. | (f) Linear partially charged polystyrene sulfonate [53]
Main chain—hydrophobic partially charged flexible, anionic
(~66% sulfonation). |
A detailed analysis of the physical properties of epithelial mucins could provide inspiration for the development of low frictional or low wear coatings for nano-motors. Such charged comb tethered surfaces would not only provide steric stabilisation (a repulsive interaction), but also a hover craft of counterions [
14] causing an additional repulsive interaction and thus a further reduction in friction. Another important emergent application is found in tissue engineering where comb polyelectrolytes could be used to protect fragile engineered tissues from damage, a particular problem with skin grafts (replacement proteoglycans).
More generally comb polyelectrolytes are found in two main physical roles in biology; they reduce wear at surfaces (protective coatings that are either physically or chemically adsorbed, the
gelling mucins versus the
epithelial mucins) or they increase the viscosity in hydrated composites as dissipative fillers (many
proteoglycans perform this task,
Figure 1b).
The physical properties of charged polymers are much more challenging than their neutral counterparts, and many fundamental unresolved questions still exist in this field [
15]. However, scaling predictions for the viscosity, modulus, relaxation time, correlation length and diffusion coefficient for linear flexible polyelectrolytes have been relatively successful [
15]. The technical challenge is provided by the long range nature of the charge interaction of these polymers combined with complex monomer/solvent and excluded volume interactions. New scaling predictions have recently been made for neutral and charged combs in solution, but they have not yet been tested in detail [
6]. Critical for these new predictions is the insight that the introduction of an additional well controlled relaxation time in the system (the side-chain motions) can introduce a huge increase in the relaxation time of the chain backbone motion. Comb polyelectrolytes are thus ideal viscosity modifiers (and thus dissipative fillers) and the viscosity can be simply modified by the addition of extra side-chains or by changing the length of the side-chains. In common with linear flexible polyelectrolytes [
16], comb polyelectrolytes have an unusual polymer concentration dependence of the relaxation time,
i.e., counter intuitively the relaxation time can speed up with increased polymer concentration in the unentangled regime due to charge screening effects on the chain conformations.
2. Synthetic Polyelectrolytes
Both standard polymer synthesis techniques and more advanced peptide self-assembly methods have been recently used to create synthetic polyelectrolyte combs [
17,
18,
19,
20] (
Figure 1d,e). Similarly the self-assembly of surfactants on to polypeptide backbones has also been employed as a synthesis route [
21].
A challenge is to extract structural information about the conformations of the side-chains in dilute solution [
22,
23]. A possible approach is to use a variable density cylindrical model with small-angle X-ray scattering, small-angle neutron scattering, and static light scattering data, but this is probably only rigorously applicable to densely grafted combs that closely conform to a cylindrical morphology [
10]. An alternative method can be used with less densely grafted combs, but depends on a decoupling approximation between the side-chain and main-chain conformations [
23], and questions exist on the handling of the electrostatic interaction.
In semi-dilute solutions polyelectrolyte combs overlap and a universal scaling of the correlation length (the average length between neighbouring chains,
ξ) on the polymer concentration (
ξ~
c−1/2) was found in a recent study on polystyrene sulfonates [
10]. The conformation of the overlapping chain sections is relatively independent of the topology, it only depends on the polymer concentration.
Some generic predictions for the viscosity (
η), relaxation time (
τ), shear modulus (G) and correlation length (
ξ) for both polyelectrolyte and neutral flexible combs are shown in
Figure 2. The polymer concentration (
c) dependences are shown, since this is an easily accessible experimental parameter with solution state macromolecules. These scaling calculations for the relaxation time, specific viscosity, correlation length and modulus explain a number of phenomena with comb polystyrene sulfonate, mucin and aggrecan solutions, but still need to be tested in more detail [
24]. Predictions for the diffusion coefficient are also readily made using the same scaling model.
The predictions on
Figure 2 are given for unentangled side-chains. Entanglements in the side-chains give rise to an additional exponential factor slowing the relaxation time and increasing the viscosity [
6]. Although the concentration dependence of the viscosity is stronger for neutral combs on
Figure 2, charged combs typically have higher viscosities due to their more extended conformations (larger chain sizes give a larger prefactor).
Figure 2.
Collection of scaling results for both neutral and charged flexible combs with unentangled side-chains. The predictions for neutral combs are in blue, and charged combs are in red [
6]. (a) Specific viscosity (
η). (b) Relaxation time (
τ). (c) Modulus (G). (d) Correlation length (
ξ). The quantities are shown as a function of polymer concentration (c).
c* is the semi-dilute overlap concentration, c
brush is the brush overlap concentration,
ce is the entanglement concentration and
cD is the electrostatic blob overlap concentration. Note that
c*,
cbrush and
ce are not expected to coincide for both neutral and charged comb solutions, and are just drawn in this manner for convenience (see
Table 2 for their accurate scaling dependences), e.g., the c* for the charged combs (
c*
c) will be much smaller than that for the neutral combs (
c*
n) for an equivalent polymer chemistry,
i.e.,
c*
c <<
c*
n. We have assumed
cbrush <
ce, which may not be true with some small side chain architecture, e.g., see
Table 2 for rigid polymers.
Figure 2.
Collection of scaling results for both neutral and charged flexible combs with unentangled side-chains. The predictions for neutral combs are in blue, and charged combs are in red [
6]. (a) Specific viscosity (
η). (b) Relaxation time (
τ). (c) Modulus (G). (d) Correlation length (
ξ). The quantities are shown as a function of polymer concentration (c).
c* is the semi-dilute overlap concentration, c
brush is the brush overlap concentration,
ce is the entanglement concentration and
cD is the electrostatic blob overlap concentration. Note that
c*,
cbrush and
ce are not expected to coincide for both neutral and charged comb solutions, and are just drawn in this manner for convenience (see
Table 2 for their accurate scaling dependences), e.g., the c* for the charged combs (
c*
c) will be much smaller than that for the neutral combs (
c*
n) for an equivalent polymer chemistry,
i.e.,
c*
c <<
c*
n. We have assumed
cbrush <
ce, which may not be true with some small side chain architecture, e.g., see
Table 2 for rigid polymers.
![Polymers 02 00057 g002]()
The backbone and side-chains of comb polymers can be flexible, semi-flexible or rigid. Furthermore charged varieties of each of the six permutations are possible providing a wide variety of possible ways to tailor their subsequent performance. A few implications of the flexibility mechanisms for comb polymer dynamics have been recently studied [
6], but much more detailed research is required.
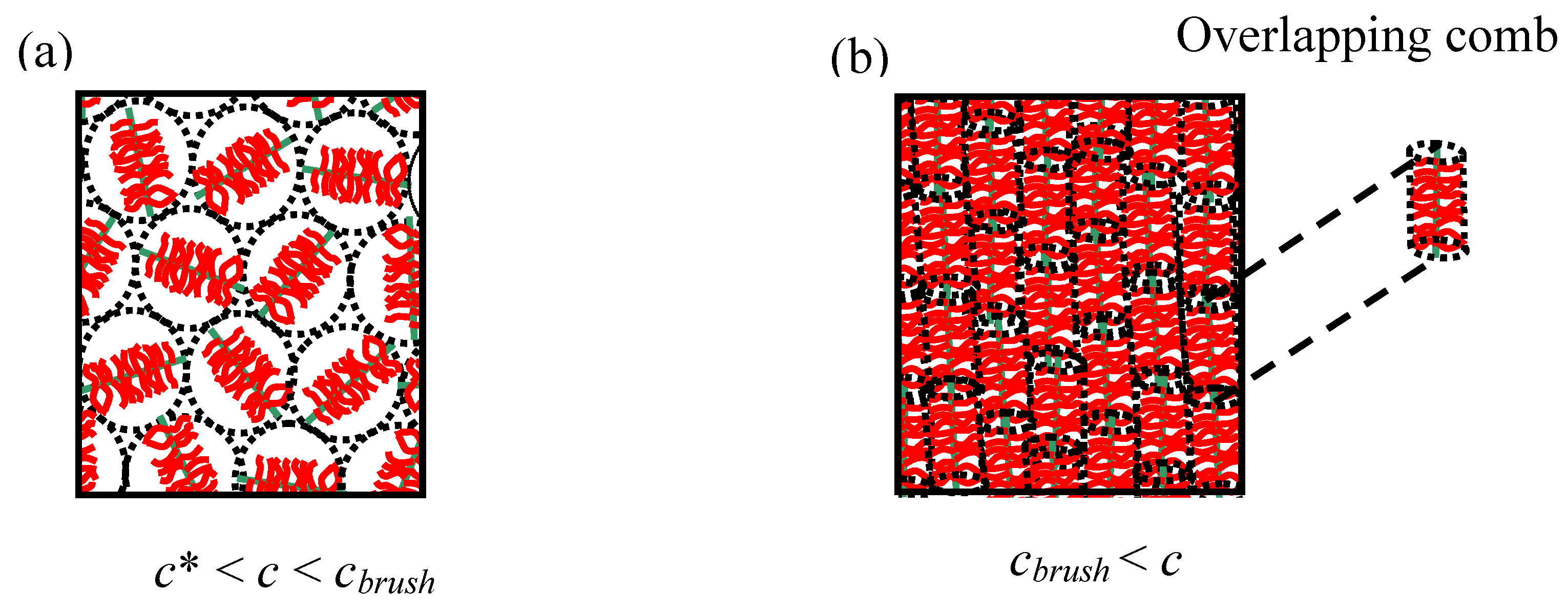
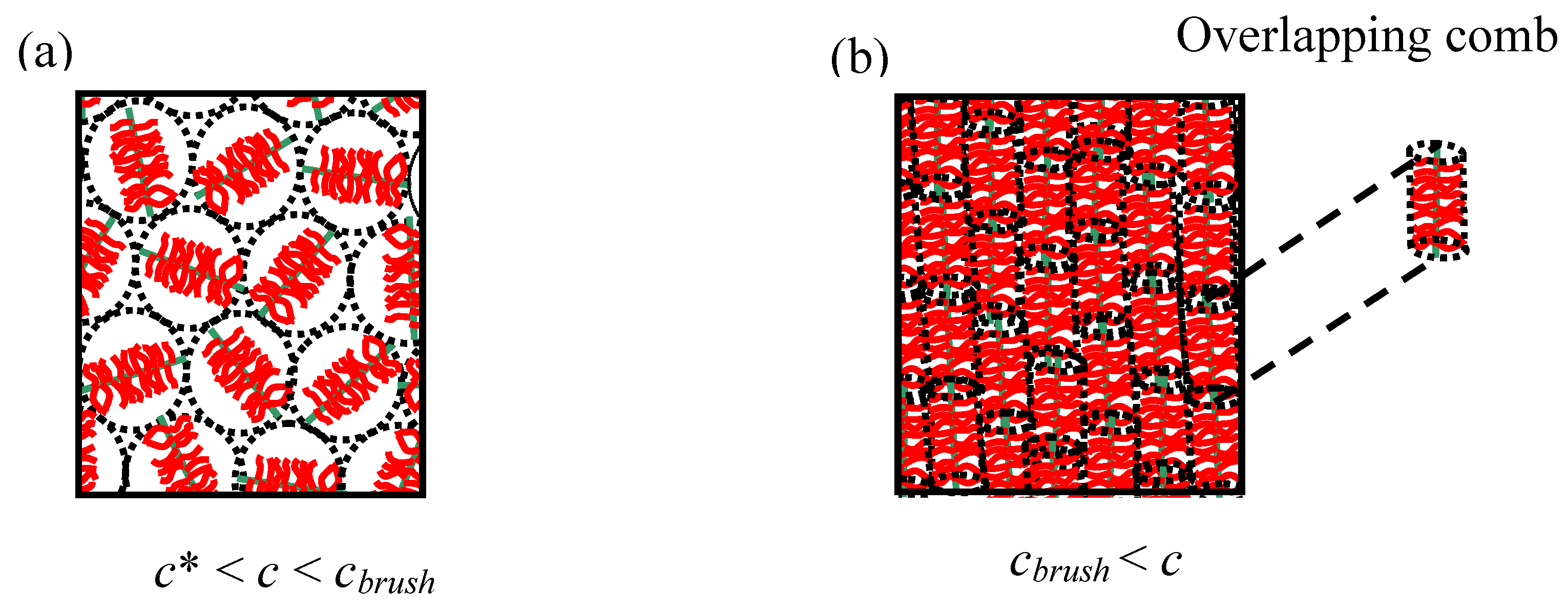
An unusual feature of comb polyelectrolytes is that, unlike many other polymeric topologies (linear, dendrimer, hyperbranched polymer,
etc.), they can have two well-defined overlap concentrations (
c* and
cbrush) if both the backbone and side-chains are reasonably monodisperse. Additional phenomena are associated with the existence of the second overlap concentration (
cbrush) with semi-dilute comb solutions (
Figure 3). The overlap concentrations can be calculated relatively simply using expressions from
Table 2. Simply put
cbrush occurs for flexible comb architectures when the side-chain length equals the mesh size (
ξ) of the semi-dilute polymer solution. For example a densely grafted polyelectrolyte comb which has side-chains that are half the backbone length will have a
cbrush at a concentration four times larger than
c*,
i.e.,
cbrush = 4
c*. This makes the brush overlap concentration relatively easy to investigate experimentally.
Table 2.
Scaling expressions can be used to calculate the critical concentrations (in units of the number of monomers per unit volume) in comb solutions: semi-dilute overlap concentration (
c*), brush overlap concentration (
cbrush) and the entanglement concentration (
ce).
n is the number of overlapping chains required for an entanglement, B is the ratio of the contour length to the end to end length,
b is the monomer length,
N is the number of monomers in the backbone and
K is the number of monomers in a side-chain [
5,
6,
24,
54,
55].
Table 2.
Scaling expressions can be used to calculate the critical concentrations (in units of the number of monomers per unit volume) in comb solutions: semi-dilute overlap concentration (c*), brush overlap concentration (cbrush) and the entanglement concentration (ce). n is the number of overlapping chains required for an entanglement, B is the ratio of the contour length to the end to end length, b is the monomer length, N is the number of monomers in the backbone and K is the number of monomers in a side-chain [5,6,24,54,55].
| Type of Comb | Semi-dilute overlap
concentration (c*) | Brush overlap
concentration (cbrush) | Entanglement
concentration (ce) |
|---|
| Flexible neutral [6,55] | ![Polymers 02 00057 i001]() | ![Polymers 02 00057 i002]() | ![Polymers 02 00057 i003]() |
| Flexible polyelectrolyte [24] | ![Polymers 02 00057 i004]() | ![Polymers 02 00057 i005]() | ![Polymers 02 00057 i006]() |
| Rigid [5,54] | ![Polymers 02 00057 i007]() | ![Polymers 02 00057 i008]() | ![Polymers 02 00057 i009]() |
With
rigid comb polymers the brush overlap concentration is expected to be lower than the isotropic-nematic transition (
cbrush ≤
cnematic), which is dependent on the flexibility of the side-chain and the backbone. Above the brush overlap concentration (
cbrush) comb molecules often exhibit liquid crystalline phases [
5,
25,
26,
27], and there is a switch in the viscoelasticity of the solutions due to screening of the hydrodynamics of the side-chains. Furthermore the inter-comb potential should have a marked change at distances corresponding to the brush overlap concentration (
cbrush) and a step change is also expected in the osmotic compressibility.
Figure 3.
Two overlap concentrations are observed in comb polymer solutions (rigid combs morphologies are shown). (a) The standard semi-dilute overlap concentration (c*). (b) The brush overlap concentration (
cbrush). The flexibility mechanism tends to destroy the long range nematic ordering in flexible polyelectrolyte comb systems, but nematics have been observed in some glycoprotein systems [
26].
Figure 3.
Two overlap concentrations are observed in comb polymer solutions (rigid combs morphologies are shown). (a) The standard semi-dilute overlap concentration (c*). (b) The brush overlap concentration (
cbrush). The flexibility mechanism tends to destroy the long range nematic ordering in flexible polyelectrolyte comb systems, but nematics have been observed in some glycoprotein systems [
26].
Molecular dynamics simulations of comb polyelectrolytes in solvents that are poor for the backbone of the polymer indicate that globular instabilities are possible in synthetic comb architectures. These are reminiscent of the gelling mucin double-globule structures [
28,
29] that will be discussed in the next section with glycoproteins (
Figure 1a,f).
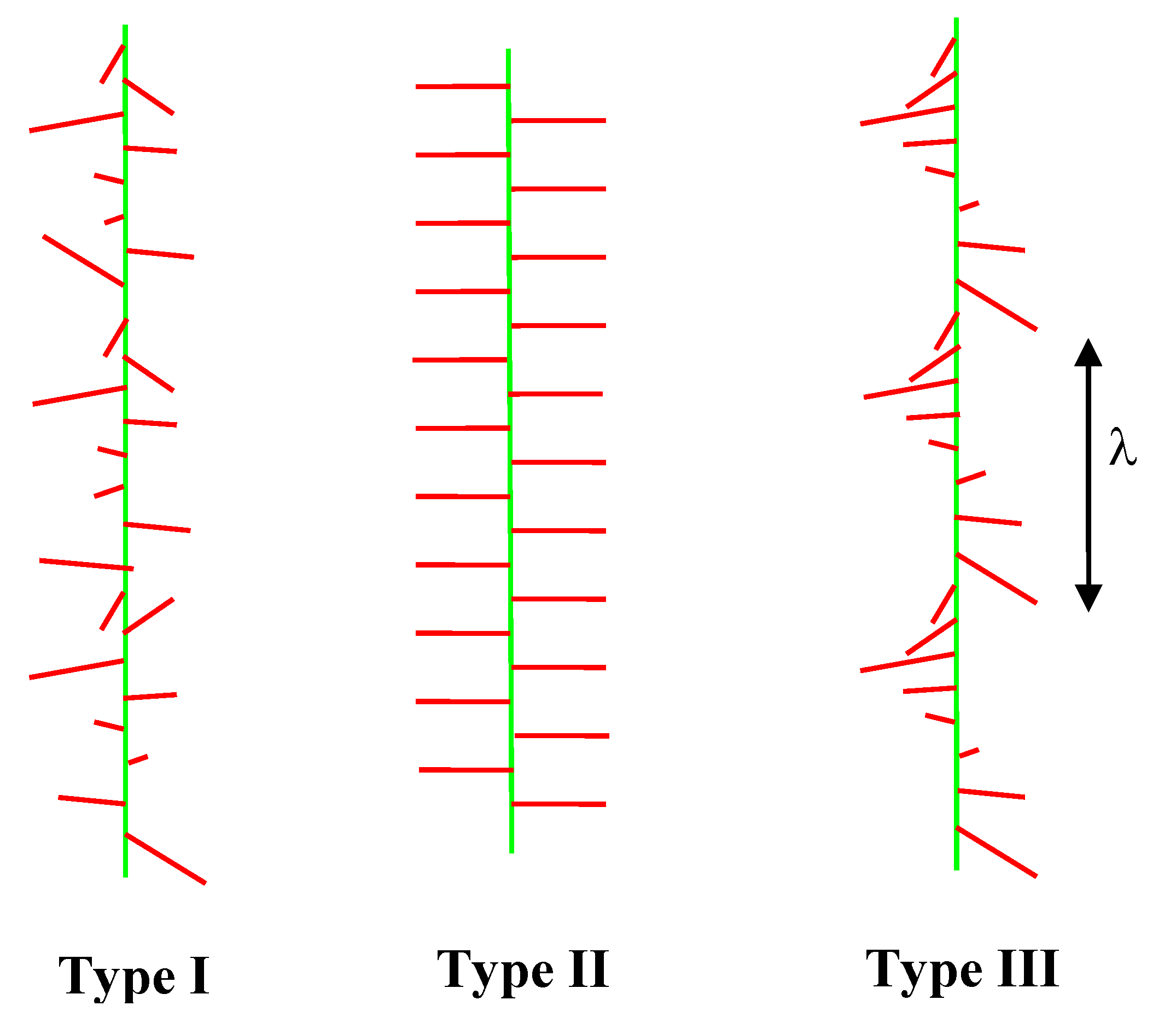
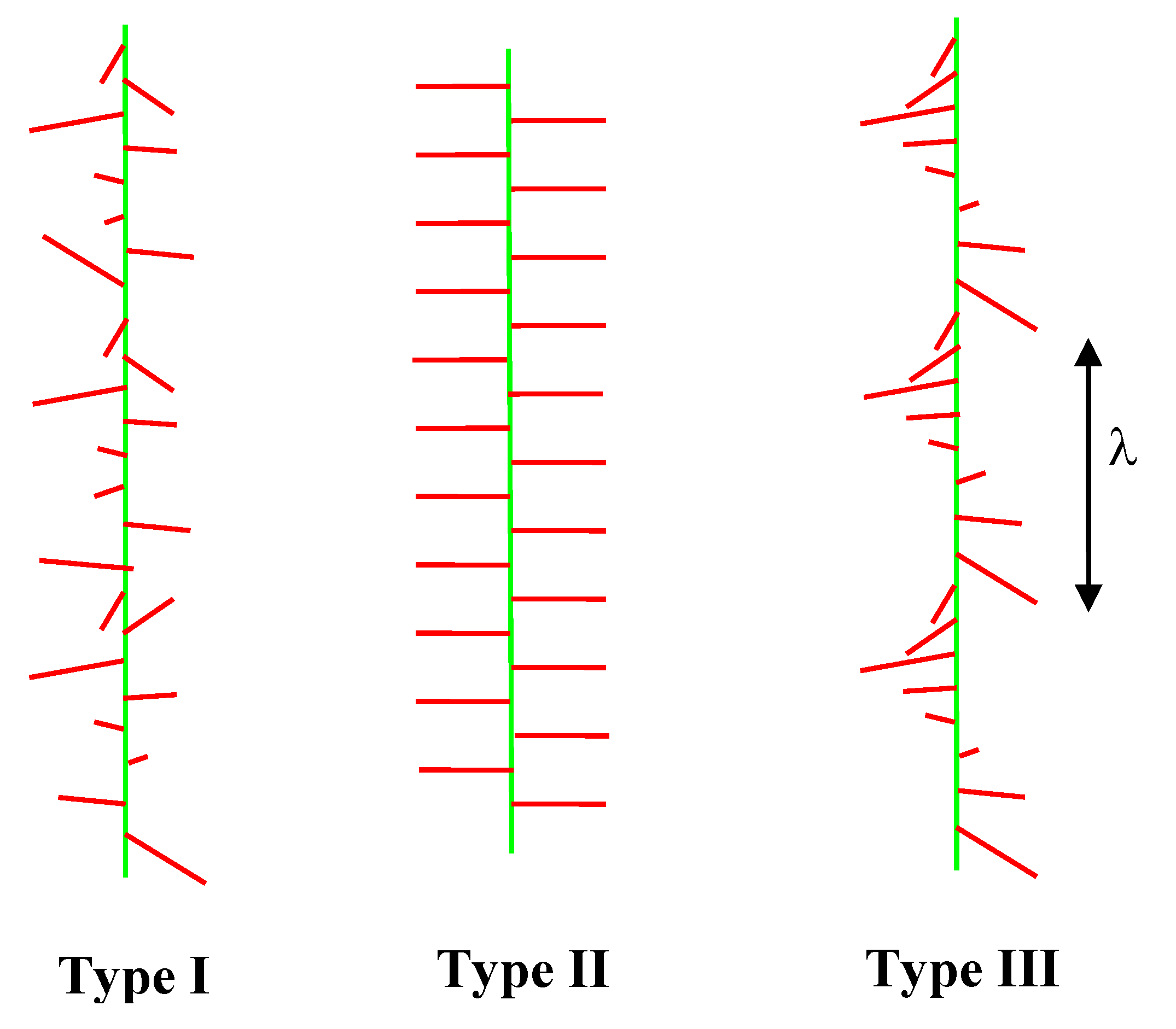
A further fascinating phenomenon that is associated with the long range electrostatic interaction of the comb side-chains is the propensity for the helical state (
Figure 4), which has only recently been demonstrated experimentally. With the long range electrostatic interaction an alternating or random structure of the pendant side-chains can be unfavourable and the helical morphology is stabilised. To date the helical state has only been observed in AFM measurements with comb polyelectrolytes when an attractive interaction is introduced using multivalent counterions [
18,
30]. This situation may well be due to the experimental challenges presented by rapidly fluctuating helical side-chain morphologies in solution,
i.e., helicity is easily disrupted in microscopy experiments if a quenched morphology due to multivalent counterions is not introduced. Helical instabilities have been predicted theoretically in solutions of rod-like polyelectrolytes [
31] and further travelling wave helical solitons might be expected at short time scales (leading to unusual dispersion curves for comb polyelectrolytes in spectroscopy experiments) [
32].
Figure 4.
Electrostatic modulation of the morphologies of the side-chains of charged comb polyelectrolytes. λ is the wavelength of the helical modulation (the pitch). Relatively rigid side-chains are shown, but similar instabilities are possible with flexible polyelectrolyte combs. Three types of side-chain morphology are shown, Type I (random), Type II (alternating) and Type III (helical).
Figure 4.
Electrostatic modulation of the morphologies of the side-chains of charged comb polyelectrolytes. λ is the wavelength of the helical modulation (the pitch). Relatively rigid side-chains are shown, but similar instabilities are possible with flexible polyelectrolyte combs. Three types of side-chain morphology are shown, Type I (random), Type II (alternating) and Type III (helical).
3. Glycoproteins
Technically the difference in classification between glycoproteins and proteoglycans is a little vague, but glycoproteins are typically defined as macromolecules with short carbohydrate chains combined with proteins, e.g., the mucins [
33]. Not all glycoproteins have the comb motif (some are sparsely grafted with only a few short carbohydrate side-chains such as fibrinogen), but many do. A sub-classification of the glycoproteins is presented by the
mucins. There are currently seventeen mucin encoding gene types that have been identified. The mucins can predominantly be arranged into two broad
gelling and
epithelial subdivisions (with the exception of two non-polymeric mucins MUC7, MUC8). Each variety of mucin is vital to many living processes within organisms, but huge gaps still exist in our understanding of them. Little is known about the physical properties of the epithelial mucins, but they are very widely distributed [
13,
34].
Porcine stomach mucins (MUC6) tend to be regarded as the model gelling mucin system, since they are relatively easy to extract in reasonable quantity and are important in a number of intestinal diseases and for drug delivery [
35,
36]. MUC6 mucin can be extracted in associating or non-associating forms dependent on the extraction procedure (disulphide bond disruption). Non-associating MUC6 acts as a viscous fluid up to high frequencies (10
5 Hz). Below these high frequencies the viscoelasticity follows the predictions of linear flexible polyelectrolytes (the side-chains are too small to have a dynamic signature at slow time scales) with a
η~
c1/2 to
η~
c3/2 cross over of the specific viscosity (
η) versus polymer concentration (c) scaling [
3]. Dynamic light scattering, atomic force microscopy, zeta potential, static light scattering and X-ray scattering measurements all point to a double-globular model for the structure of MUC6 (
Figure 1a) [
4]. Comparison of genetically determined protein sequences implies that the double-globular morphology may be conserved over a wide range of the mucin/glycoprotein family, e.g., von Wille de Brand’s cofactor, MUC6, MUC5,
etc. [
11]. Although non-associating mucins conform well to scaling models, a future challenge will be to extend them to naturally gelling specimens [
3,
4,
5,
37]. Thick viscoelastic MUC5 mucus gels are typical of cystic fibrosis conditions and careful analysis of their physical properties is required to develop therapeutic strategies with these materials [
38].
Little is known about the epithelial mucins, but their physical function, attached directly to surfaces, is in complete contrast to the gelling mucins. It is likely that they reduce friction and wear on the delicate membranes.
Many glycoproteins, e.g., fibrinogen, do not conform to the comb morphology (fibrinogen only has 4 short carbohydrate side-chains) and have a diverse range of functions often based on a double-globular motif, e.g., fibrinogen self-assembles into semi-flexible fibres to stem blood flow [
39].
4. Proteoglycans
Proteoglycans in contrast to the glycoproteins, consist of long carbohydrate chains combined with proteins (
Figure 1b). Although the relevant data has been available for thirty years [
40] only recently have the self-assembling properties of proteoglycans been properly recognized [
6]. This process of self-assembly has a clear role
in vivo in the construction of cartilaginous materials, where aggrecans dissipate energy in these nanocomposite materials when combined with collagens.
Aggrecans are some of the most studied proteoglycans due to their importance in osteoarthritis conditions, although their exact physical properties are still being elucidated. A crucial role for aggrecan is as a dissipative filler in viscoelastic composites when combined with collagen in cartilage. Aggrecan is a fascinating system in which to study viscoelasticity due to its modular architecture and the recent availability of microrheology techniques that allow its viscoelasticity to be mapped over an unprecedently wide frequency range. A recent combination of diffusing wave spectroscopy and fast particle tracking measurements provided a range of 0.1–10
6 Hz for the complex shear moduli [
6] as a function of polymer concentration. At high frequencies evidence is found for flexible polymeric Rouse modes in aggrecan in agreement with dynamic light scattering studies [
7].
Aggrecan chains do not gel in their native state, providing their solutions with high viscosity and low elasticity. This is a primary requirement for their dissipative filler properties in composite materials. Entanglements between neighboring aggrecan subunits on a single comb level have been demonstrated using atomic force microscopy [
41]. These entanglements lead to high viscosities for aggrecan solutions.
Versican is a much more sparsely grafted aggrecan-type polymer [
42,
43]. It is considered an anti-adhesive molecule important in cell adhesion, migration and proliferation. Also in this family of giant flexible aggrecan-type proteoglycans there are brevican and nuercan which are found in the mammalian brain [
44] and help to maintain the extracellular environment in this organ. Little research has been performed on the physical properties of such materials.
Other examples of proteoglycans include a large variety found in the eye. These proteoglycans contain a range of carbohydrate moieties including chrondroitin sulfate, dermatan sulfate, keratin sulfate and heparan sulfate. A large number of eye based proteoglycans have been catalogued including the lumicans, decorins, biglycans, and fibromodulins [
45]. Here their physical properties are not yet clearly defined, but a common theme is that they bind to collagen fibrils converting them into giant semi-flexible comb polyelectrolytes.
5. Surface Properties of Comb Polyelectrolytes
Good evidence now exists that surface grafted polyelectrolyte and polyzwitterionic brushes have very low frictional properties when in contact with aqueous solutions [
46]. These systems present the synthetic equivalent of epithelial mucins. Low frictional properties have also been observed with synthetic tethered comb polyelectrolyte surfaces [
47]. However it is currently not clear the advantage they have over tethered linear brushes. Ultra-low frictional coefficients have also been measured for zwitterionic polymer chains adsorbed to mica surfaces [
46], although systematic studies [
14] do predict yet lower frictional coefficients are possible if additional repulsive electrostatic interactions are included (polyelectrolyte architectures as opposed to zwitterionic structures).
Comb polyelectrolytes are thought to act as effective ionic strength insensitive (salt stable) dispersants for nanoparticle suspensions [
48]. In this respect they can perform better than the equivalent linear topologies. Polymer brushes have been examined as anti-bacterial coatings [
49]. It would be useful to investigate how comb brushes compare. Similarly surface attached brushes have useful applications in polymer electronics [
50].
Simulations highlight the interplay between electrostatic and steric forces for physical adsorption of comb polyelectrolytes, which sometimes can act in opposition of each other [
51]. Gelling mucins physically adsorb to surfaces to form a barrier to reduce wear. Such mucins are thought to cause this reduction in wear by moving the momentum transfer between two moving surfaces away from the surfaces [
52].


{kind=link}
{kind=link}
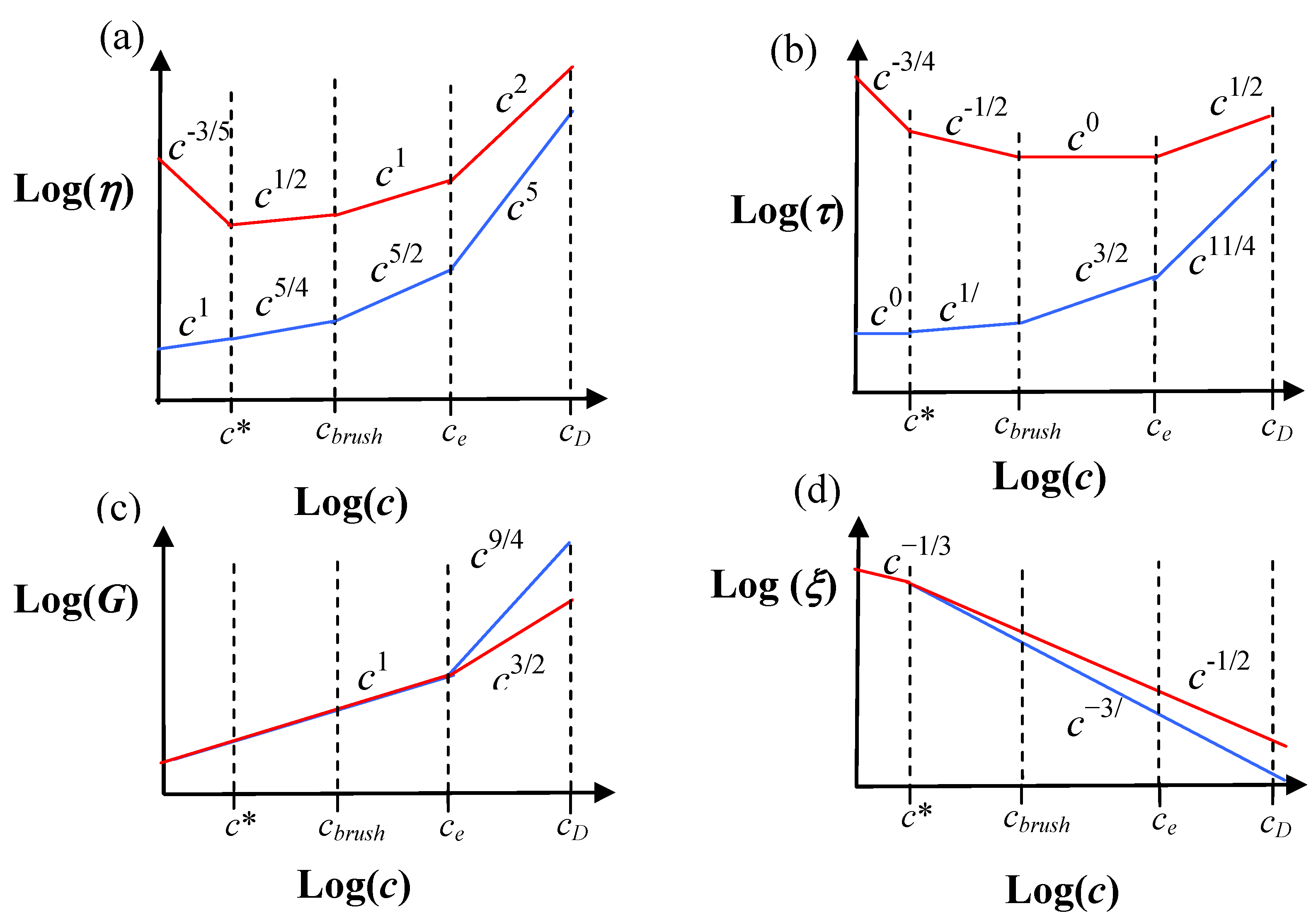{kind=link}
{kind=link}
{kind=link}