
Influence of Monomer Size on CO2 Adsorption and Mechanical Properties in Microporous Cyanate Ester Resins


Abstract
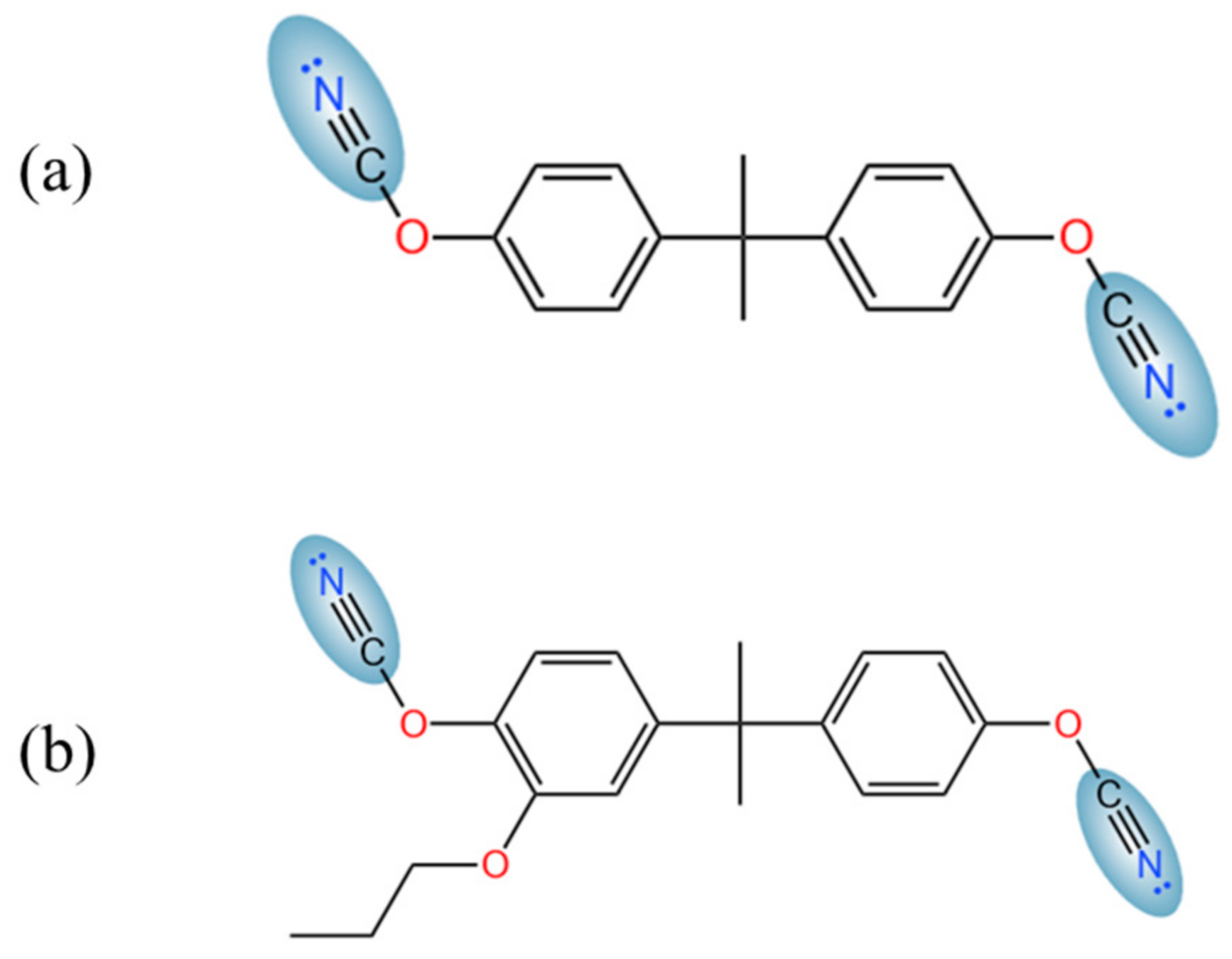
1. Introduction
2. Calculation Methods
2.1. Quantum Calculations Method
2.2. Modelling Framework
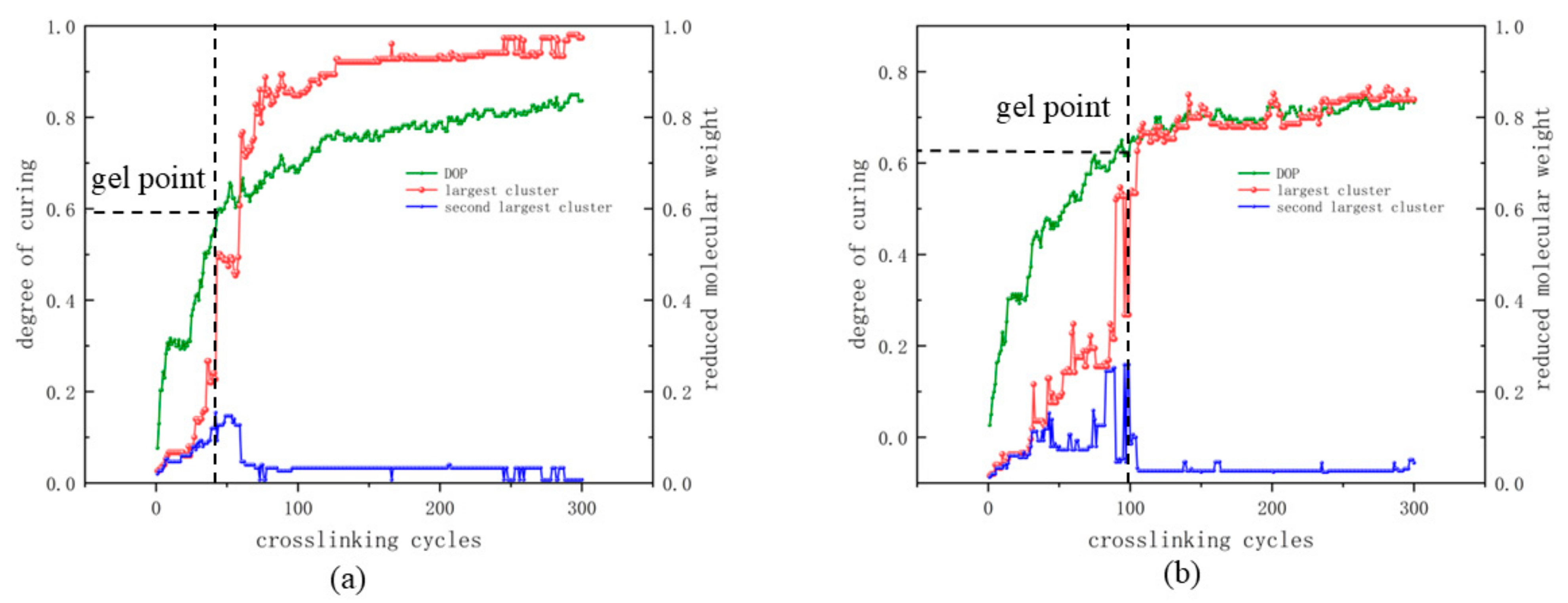
2.3. Crosslinking Method
2.4. Post-Crosslinking Procedure
2.5. Properties Analysis
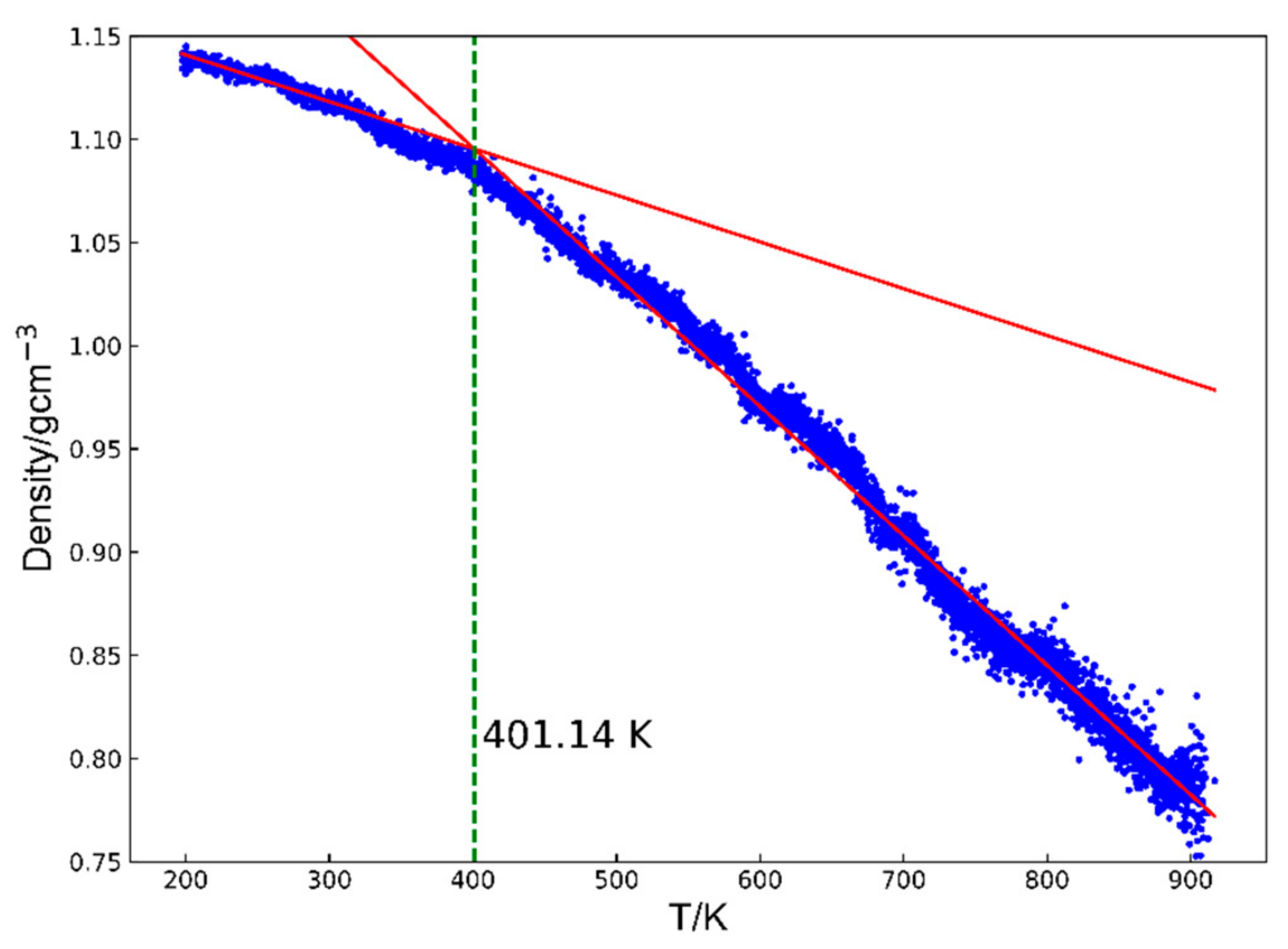
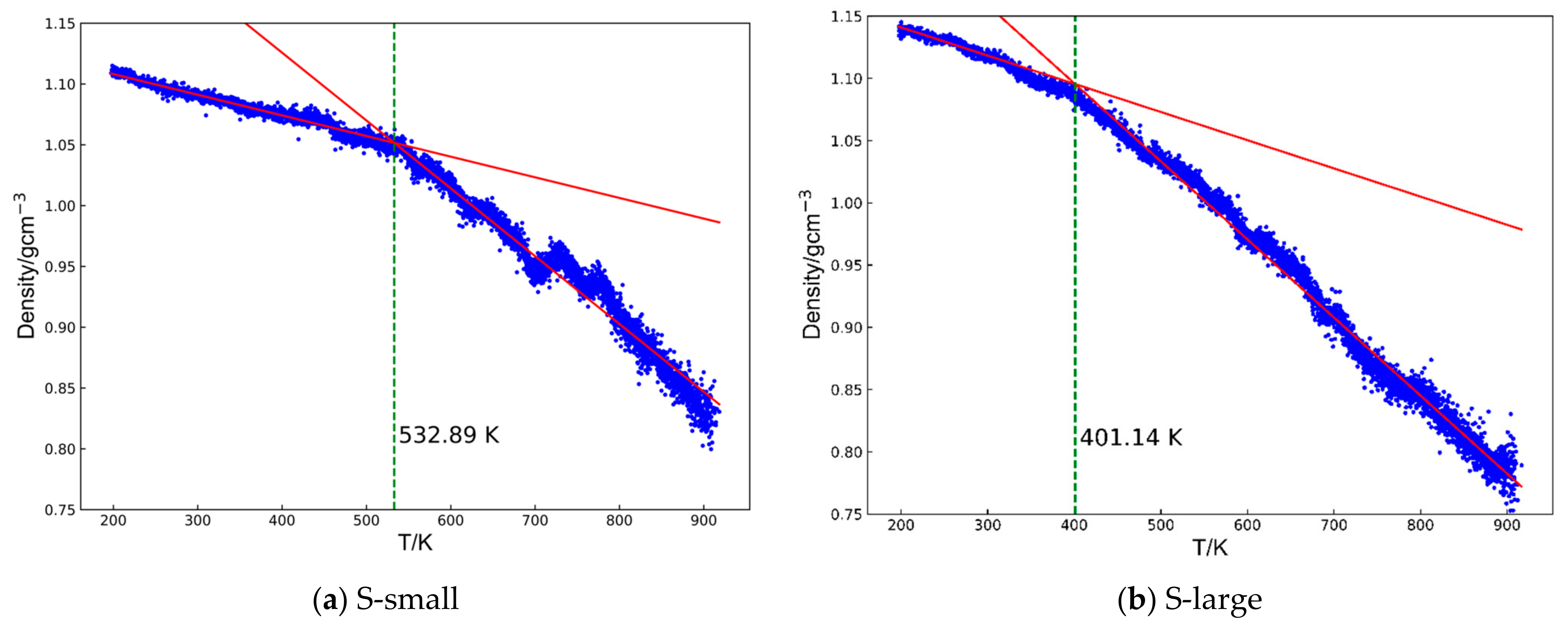
- Glass transition temperature: the glass transition temperature of a cyanate resin, a dynamic property, can be predicted by examining how its density changes differ between the rubbery and glassy states [33,34]. To accurately calculate Tg, the systems were first equilibrated at 200 K for 200 ps and then heated to 900 K at 108 K/s. During this heating process, density changes were recorded. Above Tg, the system enters the rubbery state and its density decreases significantly with increasing temperatures. Below Tg, the system remains in a glassy state, where the density is relatively insensitive to temperature changes. The density–temperature curves were fit using two straight lines corresponding to the glassy and rubbery regions based on the least squares method, following the approach outlined in previous work [35,36] (Figure 2). The calculated root means square errors (RMSEs) for each system are shown in Figure S3.
- Young’s modulus: the models obtained from the NPT equilibration stage were subjected to uniaxial tensile deformation at a strain rate of 107 s−1 along one principal direction. The dimensions in the other two directions were allowed to fluctuate independently under atmospheric pressure conditions (i.e., decoupled boundary conditions). As the model was stretched in one direction, the other directions contracted, and the resulting stress–strain behavior was recorded. From these stress–strain curves, the Young’s modulus was determined by measuring the slope of the linear elastic region up to 5% strain. To obtain a more representative property, the Young’s modulus of each model was averaged over the x, y, and z directions.
- Free volume: the free volume is calculated by the Multiwfn 3.8 software package [37]. A uniform grid was generated inside the box containing the structures, and each grid point lying outside the van der Waals radii of the probe was classified as void. The free volume was then determined using a grid spacing of 0.25 Å.
- CO2 adsorption: CO2 uptake was simulated using a combined GCMC/MD approach at 50 atm and 298 K. The GCMC method allows the insertion and deletion of gas molecules based on energy and chemical potential considerations, thereby identifying energetically favorable sites within the polymer. After each GCMC step, system volume adjustments were made to emulate the real gas uptake process. The CO2 molecules were modeled using parameters derived from the quantum chemistry calculations. For each GCMC/MD cycle, 10,000 GCMC trials were performed, followed by 10,000 NPT MD steps. This procedure was repeated for 40 cycles in this study, ensuring that the systems reached equilibrium and were fully saturated with CO2.
3. Results
3.1. Crosslinking Process
3.2. Glass Transition Temperature (Tg)
3.3. Physical Properties
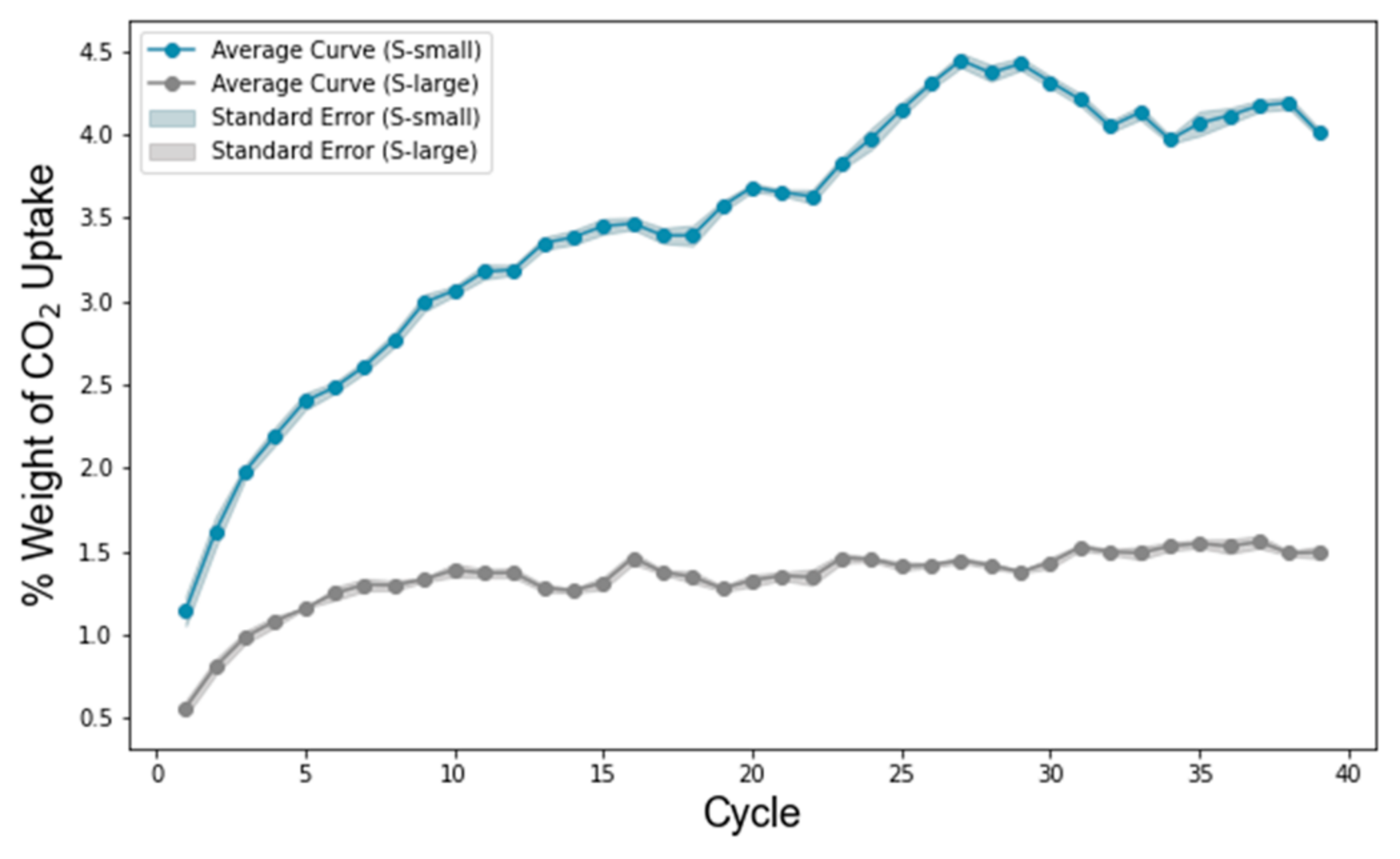
3.4. CO2 Adsorption and Free Volume
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, T.; Shimp, D.A. Polycyanate esters: Science and applications. Prog. Polym. Sci. 1995, 20, 61–118. [Google Scholar] [CrossRef]
- Barroeta Robles, J.; Hubert, P. Material characterization of cyanate ester material for structures with high dimensional stability requirements. Compos. Part A Appl. Sci. Manuf. 2023, 175, 107747. [Google Scholar] [CrossRef]
- Zaman, S.; Favela, S.; Herrera, N.E.; Gandara, A.; Molina, L.; Hassan, M.S.; Gomez, S.G.; Ramirez, J.E.M.; Mahmud, M.S.; Martinez, A.C.; et al. 3D printing of cyanate ester resins with interpenetration networks for enhanced thermal and mechanical properties. J. Appl. Polym. Sci. 2024, 141, e55423. [Google Scholar] [CrossRef]
- Dai, X.; Li, P.; Sui, Y.; Zhang, C. Synthesis and performance of flexible epoxy resin with long alkyl side chains via click reaction. J. Polym. Sci. 2021, 59, 627–637. [Google Scholar] [CrossRef]
- Zuo, P.; Ye, C.; Jiao, Z.; Luo, J.; Fang, J.; Schubert, U.S.; McKeown, N.B.; Liu, T.L.; Yang, Z.; Xu, T. Near-frictionless ion transport within triazine framework membranes. Nature 2023, 617, 299–305. [Google Scholar] [CrossRef]
- Licari, J.J.; Swanson, D.W. Adhesives Technology for Electronic Applications: Materials, Processing, Reliability; William Andrew: Norwich, NY, USA, 2011. [Google Scholar]
- Deng, G.; Wang, Z. Triptycene-Based Microporous Cyanate Resins for Adsorption/Separations of Benzene/Cyclohexane and Carbon Dioxide Gas. ACS Appl. Mater. Interfaces 2017, 9, 41618–41627. [Google Scholar] [CrossRef]
- Moore, L.M.J.; Redeker, N.D.; Browning, A.R.; Sanders, J.M.; Ghiassi, K.B. Polycyanurates via Molecular Dynamics: In Situ Crosslinking from Di(Cyanate Ester) Resins and Model Validation Through Comparison to Experiment. Macromolecules 2021, 54, 6275–6284. [Google Scholar] [CrossRef]
- Bai, Y.; Kikugawa, G.; Xi, Y.; Kishimoto, N. Development of a multistep-GRRM/MC/MD simulation method for the formation of crosslinked network structures via multistep reversible reaction pathways. Polymer 2024, 292, 126606. [Google Scholar] [CrossRef]
- Xi, Y.; Fukuzawa, H.; Kikugawa, G.; Zhao, Y.; Kawagoe, Y.; Okabe, T.; Kishi, H.; Kishimoto, N. Enhancing epoxy resin curing: Investigating the catalytic role of water as a trace impurity in dense crosslinked network formation using an advanced cat-GRRM/MC/MD Method1. Polymer 2024, 313, 127675. [Google Scholar] [CrossRef]
- Li, G.; Wang, Z. Microporous Polyimides with Uniform Pores for Adsorption and Separation of CO2 Gas and Organic Vapors. Macromolecules 2013, 46, 3058–3066. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, Z. Building ultramicropores within organic polymers based on a thermosetting cyanate ester resin. Chem. Commun. 2009, 33, 5027–5029. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Feng, Y.; Wang, J.; Liang, B.; Li, Y.; Song, Z.; Itkis, D.M.; Song, J. A robust, highly stretchable ion-conducive skin for stable lithium metal batteries. Chem. Eng. J. 2020, 396, 125254. [Google Scholar] [CrossRef]
- Crawford, A.O.; Howlin, B.J.; Cavalli, G.; Hamerton, I. Examining the thermo-mechanical properties of novel cyanate ester blends through empirical measurement and simulation. React. Funct. Polym. 2012, 72, 596–605. [Google Scholar] [CrossRef]
- Song, C.; Hu, F.; Meng, Z.; Li, S.; Shao, W.; Zhang, T.; Liu, S.; Jian, X. Atomistic structure generation of covalent triazine-based polymers by molecular simulation. RSC Adv. 2020, 10, 4258–4263. [Google Scholar] [CrossRef]
- Okabe, T.; Takehara, T.; Inose, K.; Hirano, N.; Nishikawa, M.; Uehara, T. Curing reaction of epoxy resin composed of mixed base resin and curing agent: Experiments and molecular simulation. Polymer 2013, 54, 4660–4668. [Google Scholar] [CrossRef]
- Xi, Y.; Fukuzawa, H.; Fukunaga, S.; Kikugawa, G.; Zhao, Y.; Kawagoe, Y.; Okabe, T.; Kishimoto, N. Development of cat-GRRM/MC/MD method for the simulation of cross-linked network structure formation with molecular autocatalysis. Mol. Catal. 2024, 552, 113680. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Saita, K.; Suzuki, K.; Ichino, T.; Sumiya, Y.; Sugiyama, K.; Ono, Y. Implementation and performance of the artificial force induced reaction method in the GRRM17 program. J. Comput. Chem. 2018, 39, 233–251. [Google Scholar] [CrossRef]
- Sanders, J.; Estridge, C.E.; Jackson, M.B.; Mustard, T.J.; Tucker, S.J.; Giesen, D.J.; Christensen, S.; Browning, A.; Halls, M.D. Computational method for simulating thermoset polymer curing and prediction of thermophysical properties. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Garg, M.; Apostolopoulou-Kalkavoura, V.; Linares, M.; Kaldéus, T.; Malmström, E.; Bergström, L.; Zozoulenko, I. Moisture uptake in nanocellulose: The effects of relative humidity, temperature and degree of crystallinity. Cellulose 2021, 28, 9007–9021. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Banerjee, A.; Adams, N.; Simons, J.; Shepard, R. Search for stationary points on surfaces. J. Phys. Chem. 1985, 89, 52–57. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K.; Morokuma, K. Systematic exploration of the mechanism of chemical reactions: The global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. [Google Scholar] [CrossRef]
- Senda, N. Development of molecular calculation support system “Winmostar”. Idemitsu Tech. Rep. 2006, 49, 106–111. [Google Scholar]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Kawagoe, Y.; Kikugawa, G.; Shirasu, K.; Okabe, T. Thermoset resin curing simulation using quantum-chemical reaction path calculation and dissipative particle dynamics. Soft Matter 2021, 17, 6707–6717. [Google Scholar] [CrossRef]
- Du, X.; Jasim, D.J.; Sajadi, S.M.; Hekmatifar, M.; Salahshour, S.; Sabetvand, R.; Arefpour, A.; Toghraie, D. The molecular dynamics description of Polycaprolactone coating effect on mechanical behavior of Polycaprolactone/BG-AK bio-nanocomposites. Int. J. Adhes. Adhes. 2024, 129, 103577. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- Buneman, O. Computer Simulation Using Particles (R. W. Hockney and J. W. Eastwood). SIAM Rev. 1983, 25, 425–426. [Google Scholar] [CrossRef]
- Demir, B.; Perli, G.; Chan, K.-Y.; Duchet-Rumeau, J.; Livi, S. Molecular-Level Investigation of Cycloaliphatic Epoxidised Ionic Liquids as a New Generation of Monomers for Versatile Poly(Ionic Liquids). Polymers 2021, 13, 1512. [Google Scholar] [CrossRef] [PubMed]
- Vu-Bac, N.; Bessa, M.A.; Rabczuk, T.; Liu, W.K. A Multiscale Model for the Quasi-Static Thermo-Plastic Behavior of Highly Cross-Linked Glassy Polymers. Macromolecules 2015, 48, 6713–6723. [Google Scholar] [CrossRef]
- Zhao, Y.; Kikugawa, G.; Kawagoe, Y.; Shirasu, K.; Kishimoto, N.; Xi, Y.; Okabe, T. Uncovering the Mechanism of Size Effect on the Thermomechanical Properties of Highly Cross-Linked Epoxy Resins. J. Phys. Chem. B 2022, 126, 2593–2607. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Medvedev, G.A.; Lee, E.-W.; Kim, J.; Caruthers, J.M.; Strachan, A. Molecular dynamics simulations and experimental studies of the thermomechanical response of an epoxy thermoset polymer. Polymer 2012, 53, 4222–4230. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Varshney, V.; Patnaik, S.S.; Roy, A.K.; Farmer, B.L. A Molecular Dynamics Study of Epoxy-Based Networks: Cross-Linking Procedure and Prediction of Molecular and Material Properties. Macromolecules 2008, 41, 6837–6842. [Google Scholar] [CrossRef]
- Guenthner, A.J.; Lamison, K.R.; Vij, V.; Reams, J.T.; Yandek, G.R.; Mabry, J.M. New Insights into Structure–Property Relationships in Thermosetting Polymers from Studies of Cocured Polycyanurate Networks. Macromolecules 2012, 45, 211–220. [Google Scholar] [CrossRef]
- Guenthner, A.J.; Reams, J.T.; Lamison, K.R.; Ramirez, S.M.; Swanson, D.D.; Yandek, G.R.; Sahagun, C.M.; Davis, M.C.; Mabry, J.M. Synergistic physical properties of cocured networks formed from di- and tricyanate esters. ACS Appl. Mater. Interfaces 2013, 5, 8772–8783. [Google Scholar] [CrossRef]
- Yu, H.; Shen, C.; Tian, M.; Qu, J.; Wang, Z. Microporous Cyanate Resins: Synthesis, Porous Structure, and Correlations with Gas and Vapor Adsorptions. Macromolecules 2012, 45, 5140–5150. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S-Small | S-Large | |
|---|---|---|
| Density [g cm–3] | 1.102 ± 0.004 | 1.112 ± 0.004 |
| DOP [%] | 83.8 ± 0.3 | 76.7 ± 1.4 |
| Unreacted functional groups in the largest cluster [%] | 13.7 ± 1.1 | 19.9 ± 0.76 |
| Gel point [%] | 65.2 ± 1.7 | 62.4 ± 2.6 |
| Tg [K] | 522.0 ± 8.4 | 420.7 ± 18.5 |
| Young’s modulus [GPa] | 2.32 ± 0.23 | 1.77 ± 0.18 |
| Free volume [%] | 0.88 ± 0.01 | 0.52 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Kikugawa, G.; Kishimoto, N. Influence of Monomer Size on CO2 Adsorption and Mechanical Properties in Microporous Cyanate Ester Resins. Polymers 2025, 17, 148. https://doi.org/10.3390/polym17020148
Bai Y, Kikugawa G, Kishimoto N. Influence of Monomer Size on CO2 Adsorption and Mechanical Properties in Microporous Cyanate Ester Resins. Polymers. 2025; 17(2):148. https://doi.org/10.3390/polym17020148
Chicago/Turabian StyleBai, Yukun, Gota Kikugawa, and Naoki Kishimoto. 2025. "Influence of Monomer Size on CO2 Adsorption and Mechanical Properties in Microporous Cyanate Ester Resins" Polymers 17, no. 2: 148. https://doi.org/10.3390/polym17020148
APA StyleBai, Y., Kikugawa, G., & Kishimoto, N. (2025). Influence of Monomer Size on CO2 Adsorption and Mechanical Properties in Microporous Cyanate Ester Resins. Polymers, 17(2), 148. https://doi.org/10.3390/polym17020148