

Structure-Properties Correlations of PVA-Cellulose Based Nanocomposite Films for Food Packaging Applications

 ,
,  ,
,  , ,
, ,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Analytical Techniques
2.3.1. Scanning Electron Microscopy (SEM)
2.3.2. Attenuated Total Reflection Fourier Transform Infrared Spectroscopy (ATR-FTIR)
2.3.3. X-Ray Diffraction (XRD)
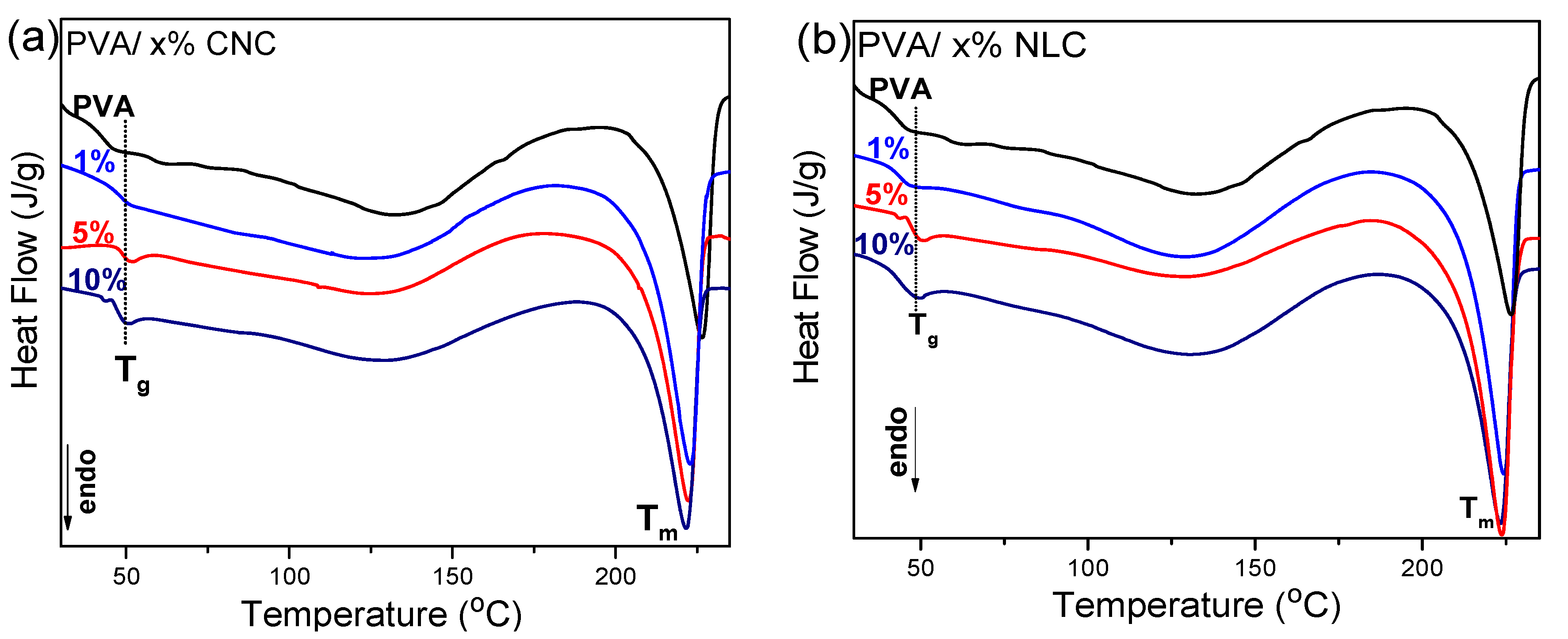
2.3.4. Differential Scanning Calorimetry (DSC)
2.3.5. Gas Permeability Measurements
2.3.6. Water Vapor Transmittance Rate Measurements
2.3.7. Swelling Properties
2.3.8. Mechanical Properties
2.3.9. Raman Scattering
3. Results and Discussion
3.1. Morphology of the Films
3.2. Structure at Molecular Level
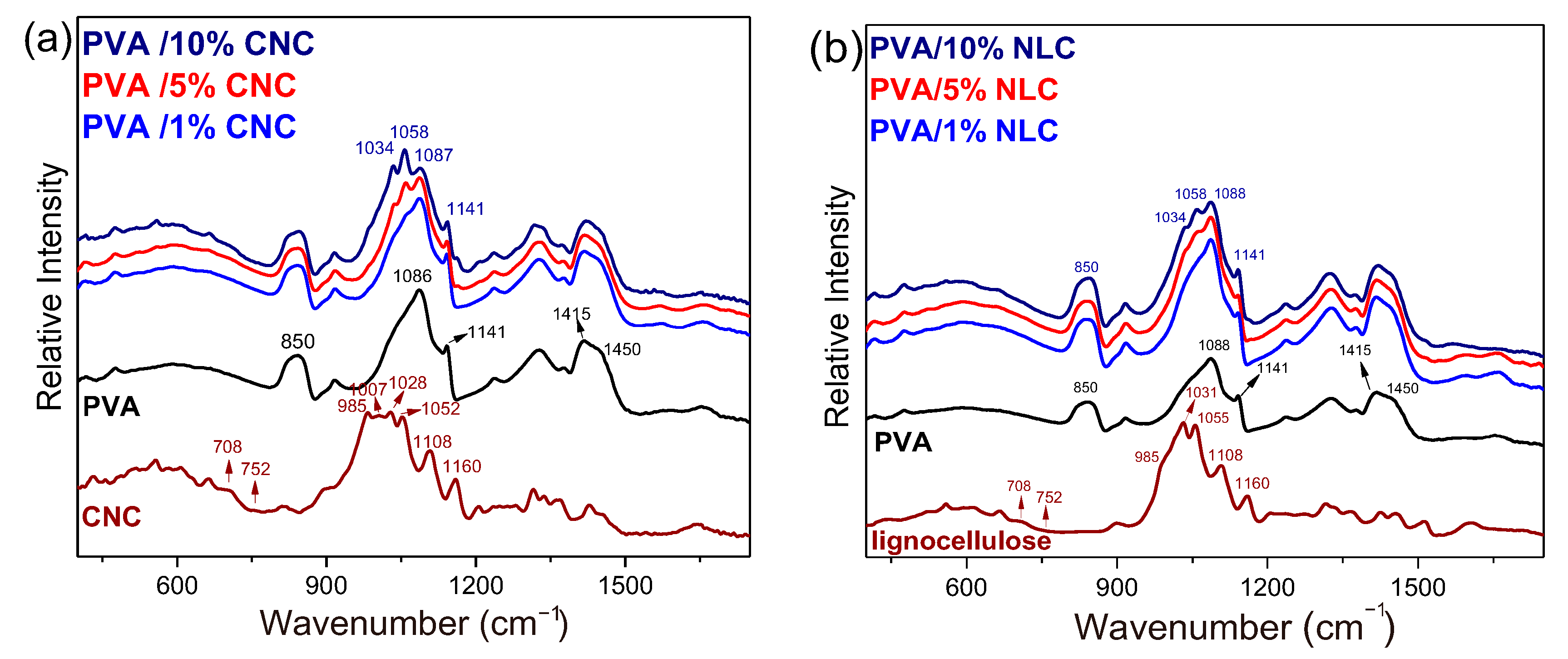
3.2.1. Conformational Alterations and H-Bonds of Cellulose in the Composites
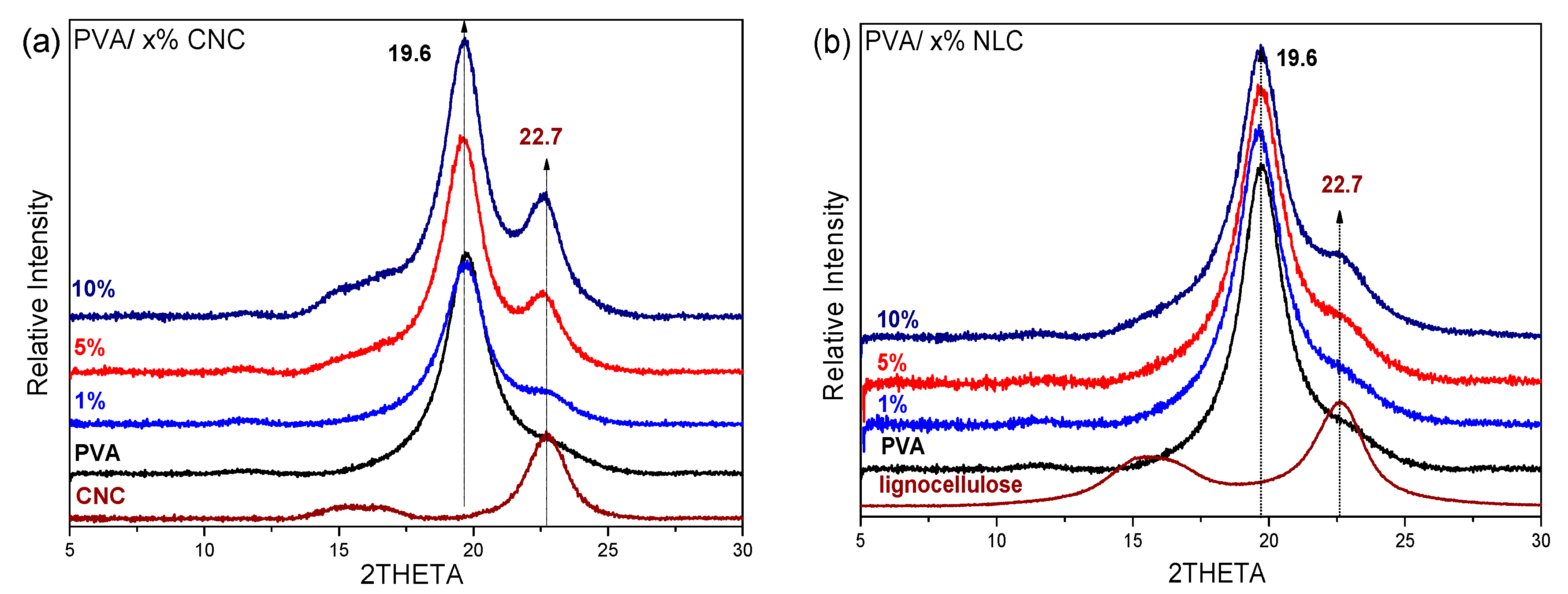
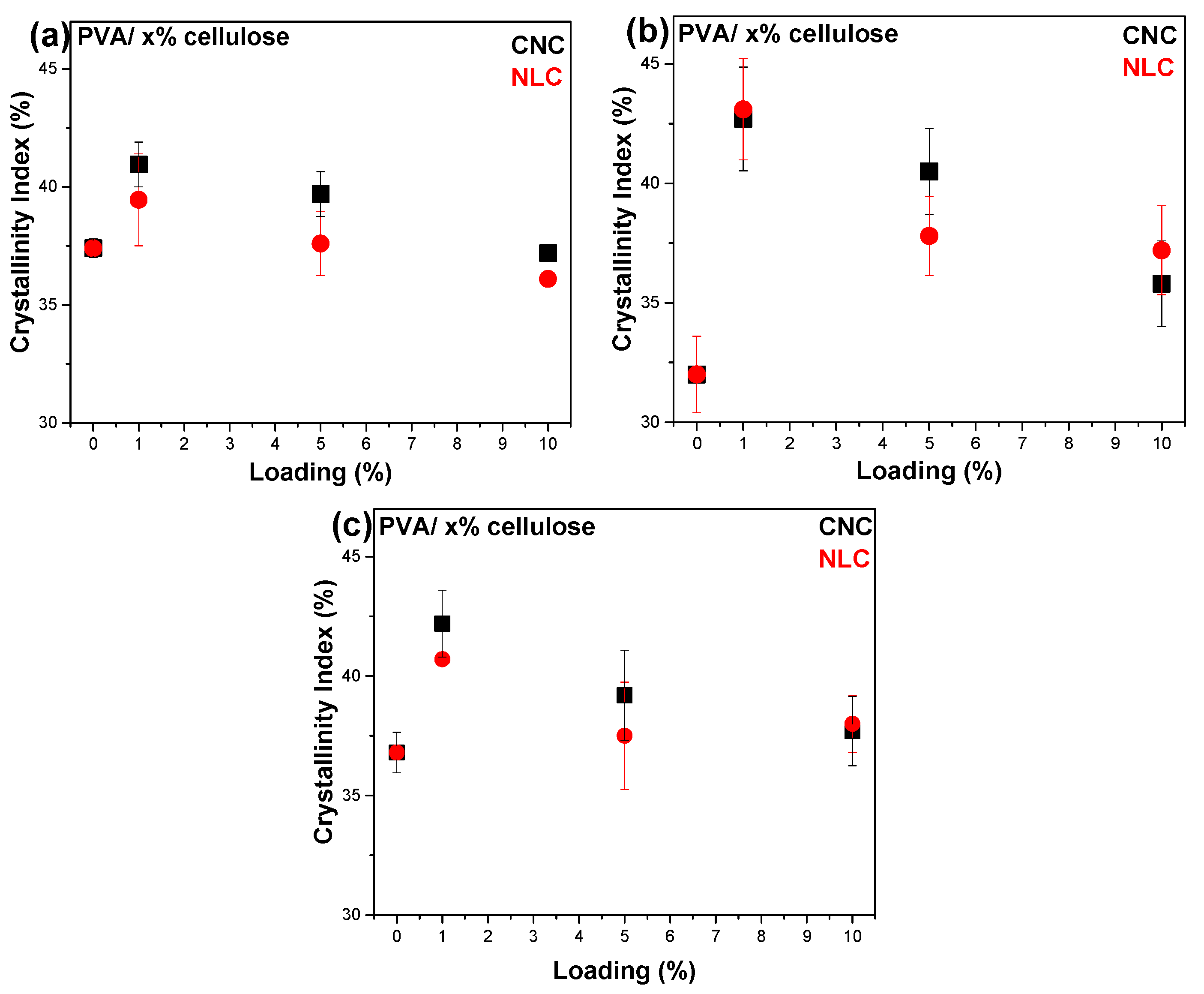
3.2.2. Study of the Crystalline Phases
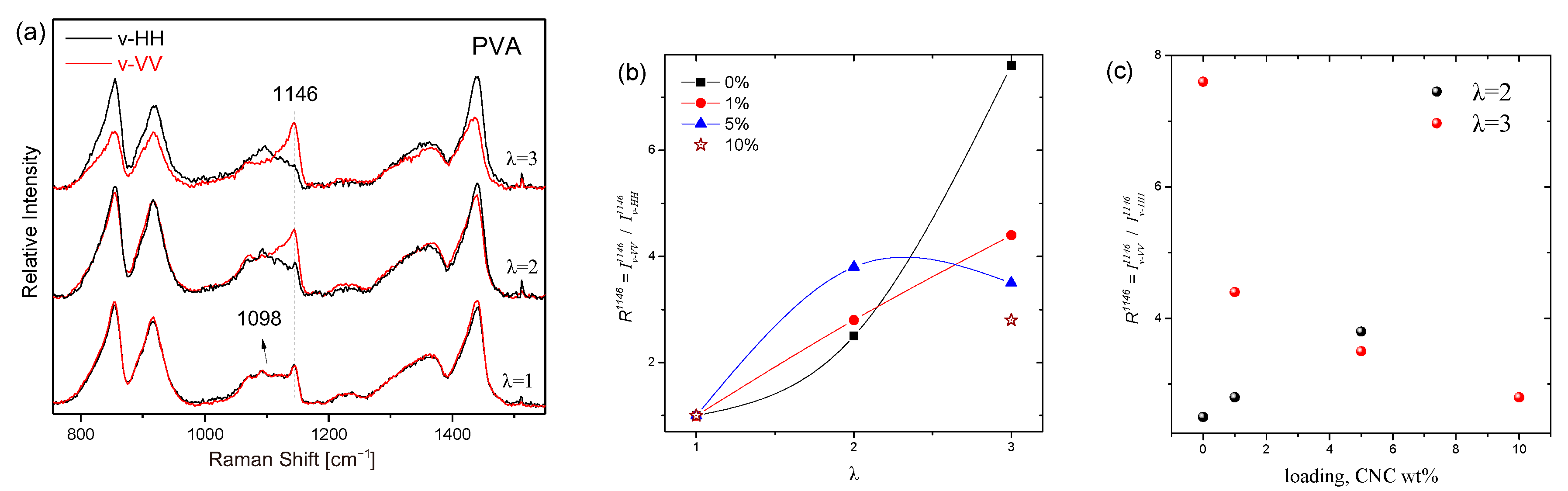
3.2.3. Anisotropy and Macromolecular Orientation
3.3. Macroscopic Properties
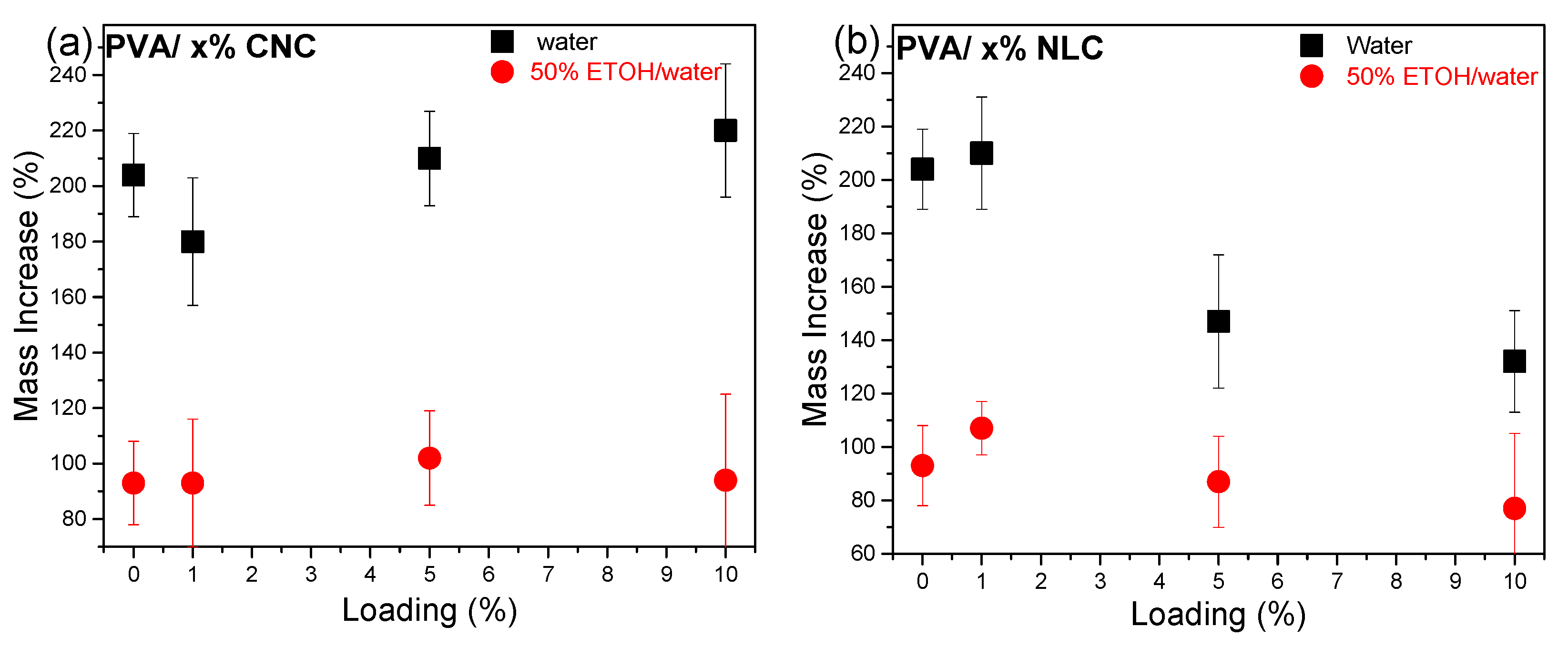
3.3.1. Swelling Properties
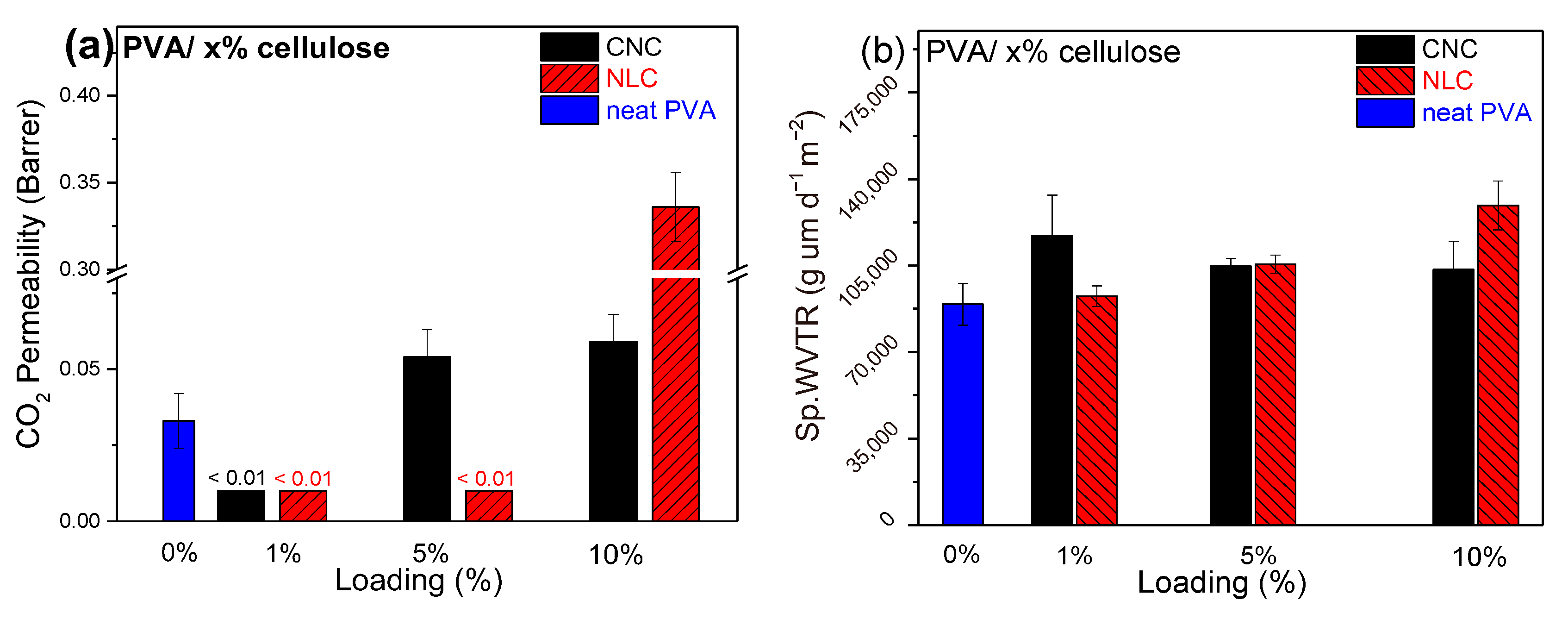
3.3.2. Gas Permeability Measurements
3.3.3. Water Vapor Transmittance Rate
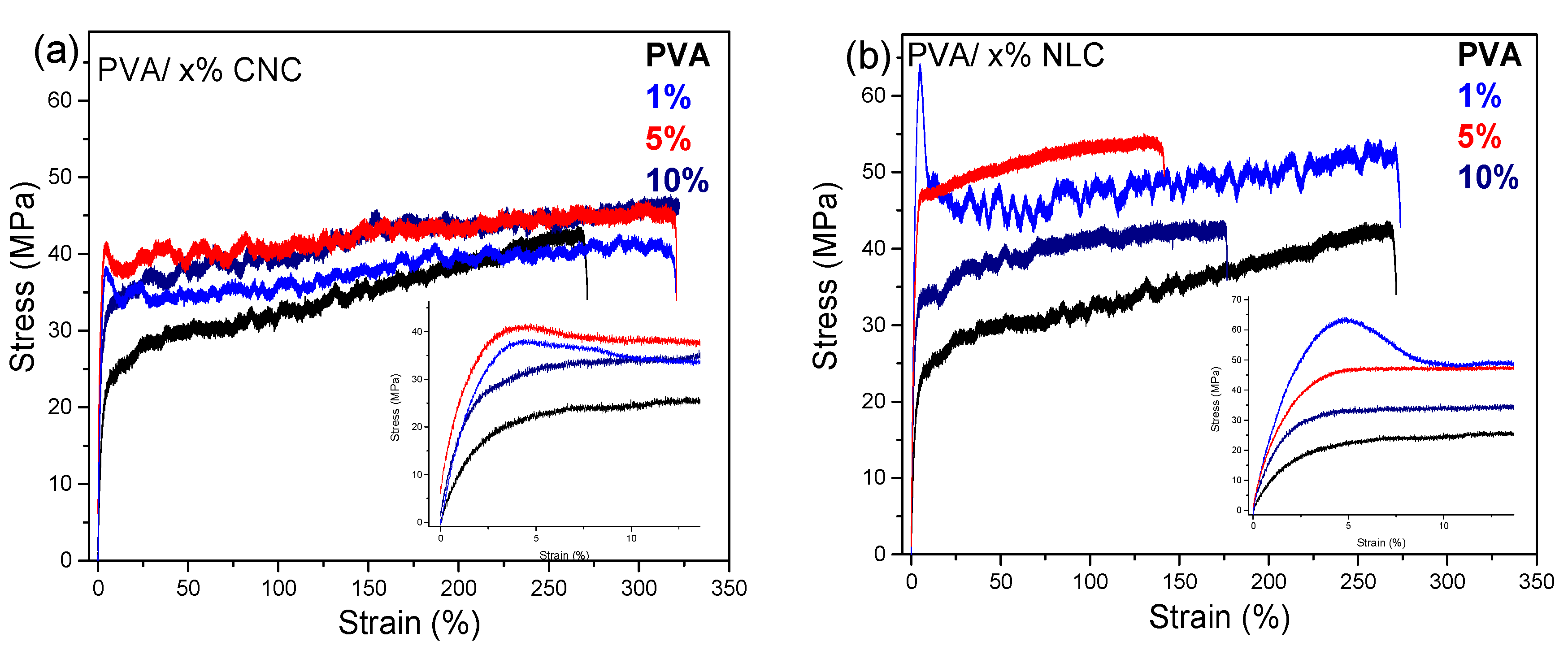
3.3.4. Mechanical Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| wt.% | % weight percentage concentration |
| CNC | Cellulose Nano Crystals |
| NLC | Nano Lignocellulose |
| PVA | PolyVinyl Alcohol |
| ATR-FTIR | Attenuated total reflectance- Fourier transform infrared spectroscopy |
| XRD | X-Ray Diffraction |
| DSC | Differential Scanning Calorimetry |
| WVTR | Water Vapor Transmittance Rate |
| Cr.I% | % Crystallinity Index |
References
- Siracusa, V.; Rocculi, P.; Romani, S.; Rosa, M.D. Biodegradable polymers for food packaging: A review. Trends Food Sci. Technol. 2008, 19, 634–643. [Google Scholar] [CrossRef]
- Rhim, J.-W.; Park, H.-M.; Ha, C.-S. Bio-nanocomposites for food packaging applications. Prog. Polym. Sci. 2013, 38, 1629–1652. [Google Scholar] [CrossRef]
- Ben Halima, N. Poly(vinyl alcohol): Review of its promising applications and insights into biodegradation. RSC Adv. 2016, 6, 39823–39832. [Google Scholar] [CrossRef]
- Muhammad, F.F.; Aziz, S.B.; Hussein, S.A. Effect of the dopant salt on the optical parameters of PVA:NaNO3 solid polymer electrolyte. J. Mater. Sci. Mater. Electron. 2015, 26, 521–529. [Google Scholar] [CrossRef]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater. Sci. Eng. C 2008, 28, 539–548. [Google Scholar] [CrossRef]
- Hassan, C.M.; Peppas, N.A. Structure and Applications of Poly(vinyl alcohol) Hydrogels Produced by Conventional Crosslinking or by Freezing/Thawing Methods. In Biopolymers PVA Hydrogels, Anionic Polymerisation Nanocomposites; Springer: Berlin/Heidelberg, Germany, 2000; pp. 37–65. [Google Scholar] [CrossRef]
- Oun, A.A.; Shin, G.H.; Rhim, J.-W.; Kim, J.T. Recent advances in polyvinyl alcohol-based composite films and their applications in food packaging. Food Packag. Shelf Life 2022, 34, 100991. [Google Scholar] [CrossRef]
- Aslam, M.; Kalyar, M.A.; Ali Raza, Z. Polyvinyl alcohol: A review of research status and use of polyvinyl alcohol based nanocomposites. Polym. Eng. Sci. 2018, 58, 2119–2132. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.-P.; Sain, M. Biocomposites reinforced with natural fibers: 2000–2010. Prog. Polym. Sci. 2012, 37, 1552–1596. [Google Scholar] [CrossRef]
- Spitalsky, Z.; Tasi, D.; Papgelis, K.; Galioti, C. Carbon nanotube-polymer composites: Chemistry, processing mechanical and electrical properties. Prog. Polym. Sci. 2010, 35, 357–401. [Google Scholar] [CrossRef]
- Wang, F.; Hu, Z.; Ouyang, S.; Wang, S.; Liu, Y.; Li, M.; Wu, Y.; Li, Z.; Qian, J.; Wu, Z.; et al. Application progress of nanocellulose in food packaging: A review. Int. J. Biol. Macromol. 2024, 268, 131936. [Google Scholar] [CrossRef]
- Fortunati, E.; Puglia, D.; Monti, M.; Santulli, C.; Maniruzzaman, M.; Kenny, J.M. Cellulose nanocrystals extracted from okra fibers in PVA nanocomposites. J. Appl. Polym. Sci. 2013, 128, 3220–3230. [Google Scholar] [CrossRef]
- Jahan, Z.; Niazi, M.B.K.; Gregersen, Ø.W. Mechanical, thermal and swelling properties of cellulose nanocrystals/PVA nanocomposites membranes. J. Ind. Eng. Chem. 2018, 57, 113–124. [Google Scholar] [CrossRef]
- Popescu, M.-C. Structure and sorption properties of CNC reinforced PVA films. Int. J. Biol. Macromol. 2017, 101, 783–790. [Google Scholar] [CrossRef]
- Cheng, Q.; Wang, S.; Rials, T.G. Poly(vinyl alcohol) nanocomposites reinforced with cellulose fibrils isolated by high intensity ultrasonication. Compos. Part A: Appl. Sci. Manuf. 2009, 40, 218–224. [Google Scholar] [CrossRef]
- Liu, D.; Sun, X.; Tian, H.; Maiti, S.; Ma, Z. Effects of cellulose nanofibrils on the structure and properties on PVA nanocomposites. Cellulose 2013, 20, 2981–2989. [Google Scholar] [CrossRef]
- Fortunati, E.; Puglia, D.; Luzi, F.; Santulli, C.; Kenny, J.; Torre, L. Binary PVA bio-nanocomposites containing cellulose nanocrystals extracted from different natural sources: Part I. Carbohydr. Polym. 2013, 97, 825–836. [Google Scholar] [CrossRef]
- Du, J.; Guo, J.; Zhu, Q.; Guo, J.; Gu, J.; Wu, Y.; Ren, L.; Yang, S.; Jiang, J. Enhancement of Polyvinyl Alcohol-Based Films by Chemically Modified Lignocellulosic Nanofibers Derived from Bamboo Shoot Shells. Polymers 2025, 17, 1571. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Z.; Li, C.; Zheng, Y.; Xu, J. A strong, excellent water resistance, and anti-ultraviolet poly(vinyl alcohol)/lignocellulose/poly(butylene adipate-co-terephthalate) composite with “sandwich” structure. Int. J. Biol. Macromol. 2025, 289, 138779. [Google Scholar] [CrossRef]
- Lv, C.; Liu, D.; Tian, H.; Xiang, A. Non-isothermal crystallization kinetics of polyvinyl alcohol plasticized with glycerol and pentaerythritol. J. Polym. Res. 2020, 27, 66. [Google Scholar] [CrossRef]
- ASTM E96-95; Standard Test Methods for Water Vapor Transmission of Materials. ASTM International: West Conshohocken, PA, USA, 1995.
- Bounos, G.; Andrikopoulos, K.; Moschopoulou, H.; Lainioti, G.; Roilo, D.; Checchetto, R.; Ioannides, T.; Kallitsis, J.; Voyiatzis, G. Enhancing water vapor permeability in mixed matrix polypropylene membranes through carbon nanotubes dispersion. J. Membr. Sci. 2017, 524, 576–584. [Google Scholar] [CrossRef]
- Kumaran, M. Interlaboratory Comparison of the ASTM Standard Test Methods for Water Vapor Transmission of Materials (E 96-95). J. Test. Eval. 1998, 26, 83–88. [Google Scholar] [CrossRef]
- ASTM D882-18; Standard Test Method for Tensile Properties of Thin Plastic Sheeting. ASTM International: West Conshohocken, PA, USA, 2018.
- Roohani, M.; Habibi, Y.; Belgacem, N.M.; Ebrahim, G.; Karimi, A.N.; Dufresne, A. Cellulose whiskers reinforced polyvinyl alcohol copolymers nanocomposites. Eur. Polym. J. 2008, 44, 2489–2498. [Google Scholar] [CrossRef]
- Tretinnikov, O.N.; Zagorskaya, S.A. Determination of the degree of crystallinity of poly(vinyl alcohol) by FTIR spectroscopy. J. Appl. Spectrosc. 2012, 79, 521–526. [Google Scholar] [CrossRef]
- Maréchal, Y.; Chanzy, H. The hydrogen bond network in Iβ cellulose as observed by infrared spectrometry. J. Mol. Struct. 2000, 523, 183–196. [Google Scholar] [CrossRef]
- Gohil, J.M.; Bhattacharya, A.; Ray, P. Studies on the Crosslinking of Poly (Vinyl Alcohol). J. Polym. Res. 2006, 13, 161–169. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies; John Wiley and Sons, Ltd.: New York, NY, USA, 2001; ISBN 978-0-470-09307-8. [Google Scholar]
- Larkin, P.J. Infrared and Raman Spectroscopy Principles and Spectral Interpretation; Elsevier: Amsterdam, The Netherlands, 2011; ISBN 978-0-12-804162-8. [Google Scholar]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Kim, H.C.; Kim, H.Y.; Chung, Y.S.; Park, W.H.; Youk, J.H. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef]
- French, A.D. Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 2014, 21, 885–896. [Google Scholar] [CrossRef]
- Zhang, W.; He, X.; Li, C.; Zhang, X.; Lu, C.; Zhang, X.; Deng, Y. High performance poly (vinyl alcohol)/cellulose nanocrystals nanocomposites manufactured by injection molding. Cellulose 2014, 21, 485–494. [Google Scholar] [CrossRef]
- Ricciardi, R.; Auriemma, F.; De Rosa, C.; Lauprêtre, F. X-ray Diffraction Analysis of Poly(vinyl alcohol) Hydrogels, Obtained by Freezing and Thawing Techniques. Macromolecules 2004, 37, 1921–1927. [Google Scholar] [CrossRef]
- Mallapragada, S.K.; Peppas, N.A. Dissolution mechanism of semicrystalline poly(vinyl alcohol) in water. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 1339–1346. [Google Scholar] [CrossRef]
- Karimi, K.; Taherzadeh, M.J. A critical review of analytical methods in pretreatment of lignocelluloses: Composition, imaging, and crystallinity. Bioresour. Technol. 2016, 200, 1008–1018. [Google Scholar] [CrossRef]
- Voronova, M.I.; Surov, O.V.; Guseinov, S.S.; Barannikov, V.P.; Zakharov, A.G. Thermal stability of polyvinyl alcohol/nanocrystalline cellulose composites. Carbohydr. Polym. 2015, 130, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W. Thermal Analysis of Polymeric Materials; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar] [CrossRef]
- Menczel, J.D.; Prime, R.B. Thermal Analysis of Polymers: Fundamentals and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Ning, N.; Fu, S.; Zhang, W.; Chen, F.; Wang, K.; Deng, H.; Zhang, Q.; Fu, Q. Realizing the enhancement of interfacial interaction in semicrystalline polymer/filler composites via interfacial crystallization. Prog. Polym. Sci. 2012, 37, 1425–1455. [Google Scholar] [CrossRef]
- Peng, J.; Ellingham, T.; Sabo, R.; Turng, L.-S.; Clemons, C.M. Short cellulose nanofibrils as reinforcement in polyvinyl alcohol fiber. Cellulose 2014, 21, 4287–4298. [Google Scholar] [CrossRef]
- Shrestha, S.; Montes, F.; Schueneman, G.T.; Snyder, J.F.; Youngblood, J.P. Effects of aspect ratio and crystal orientation of cellulose nanocrystals on properties of poly(vinyl alcohol) composite fibers. Compos. Sci. Technol. 2018, 167, 482–488. [Google Scholar] [CrossRef]
- Zhang, N.; Pang, Y.; Li, Z.; Yang, C.; Zong, L.; Yang, H.; Wu, H.; Duan, Y.; Zhang, J. Rubber-like and biodegradable poly (vinyl alcohol) composites with triple networks for high-efficiency solvent barrier. Compos. Sci. Technol. 2023, 231, 109801. [Google Scholar] [CrossRef]
- Hossain, K.M.Z.; Ahmed, I.; Parsons, A.J.; Scotchford, C.A.; Walker, G.S.; Thielemans, W.; Rudd, C.D. Physico-chemical and mechanical properties of nanocomposites prepared using cellulose nanowhiskers and poly(lactic acid). J. Mater. Sci. 2012, 47, 2675–2686. [Google Scholar] [CrossRef]
- Farid, O.; Mansour, F.; Habib, M.; Robinson, J.; Tarleton, S. Investigating the sorption influence of poly(vinyl alcohol) (PVA) at different crosslinking content. J. Environ. Chem. Eng. 2016, 4, 293–298. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Liu, Q.L.; Chen, Y.; Wu, J.Y.; Zhu, A.M. Microstructure dependent diffusion of water–ethanol in swollen poly(vinyl alcohol): A molecular dynamics simulation study. Chem. Eng. Sci. 2009, 64, 334–340. [Google Scholar] [CrossRef]
- Klepić, M.; Setničková, K.; Lanč, M.; Žák, M.; Izák, P.; Dendisová, M.; Fuoco, A.; Jansen, J.C.; Friess, K. Permeation and sorption properties of CO2-selective blend membranes based on polyvinyl alcohol (PVA) and 1-ethyl-3-methylimidazolium dicyanamide ([EMIM][DCA]) ionic liquid for effective CO2/H2 separation. J. Membr. Sci. 2020, 597, 117623. [Google Scholar] [CrossRef]
- Idris, A.; Muntean, A.; Mesic, B.; Lestelius, M.; Javed, A. Oxygen Barrier Performance of Poly(vinyl alcohol) Coating Films with Different Induced Crystallinity and Model Predictions. Coatings 2021, 11, 1253. [Google Scholar] [CrossRef]
- Idris, A.; Muntean, A.; Mesic, B. A review on predictive tortuosity models for composite films in gas barrier applications. J. Coat. Technol. Res. 2022, 19, 699–716. [Google Scholar] [CrossRef]
- Abdullah, Z.W.; Dong, Y.; Han, N.; Liu, S. Water and gas barrier properties of polyvinyl alcohol (PVA)/starch (ST)/glycerol (GL)/halloysite nanotube (HNT) bionanocomposite films: Experimental characterisation and modelling approach. Compos. Part B Eng. 2019, 174, 107033. [Google Scholar] [CrossRef]
- Gonzalez, J.S.; Ludueña, L.N.; Ponce, A.; Alvarez, V.A. Poly(vinyl alcohol)/cellulose nanowhiskers nanocomposite hydrogels for potential wound dressings. Mater. Sci. Eng. C Mater. Biol. Appl. 2014, 34, 54–61. [Google Scholar] [CrossRef]
- Ma, R.; Xiong, D.; Miao, F.; Zhang, J.; Peng, Y. Novel PVP/PVA hydrogels for articular cartilage replacement. Mater. Sci. Eng. C 2009, 29, 1979–1983. [Google Scholar] [CrossRef]
- Millon, L.E.; Oates, C.J.; Wan, W. Compression properties of polyvinyl alcohol-bacterial cellulose nanocomposite. J. Biomed. Mater. Res. Part B Appl. Biomater. 2009, 90B, 922–929. [Google Scholar] [CrossRef]
- Zimmermann, T.; Pöhler, E.; Geiger, T. Cellulose Fibrils for Polymer Reinforcement. Adv. Eng. Mater. 2004, 6, 754–761. [Google Scholar] [CrossRef]
- Lin, S.; Liu, X.; Liu, J.; Yuk, H.; Loh, H.-C.; Parada, G.A.; Settens, C.; Song, J.; Masic, A.; McKinley, G.H.; et al. Anti-fatigue-fracture hydrogels. Sci. Adv. 2019, 5, eaau8528. [Google Scholar] [CrossRef]
- Alvarado, M.C. Recent progress in polyvinyl alcohol (PVA)/nanocellulose composite films for packaging applications: A comprehensive review of the impact on physico-mechanical properties. Food Bioeng. 2024, 3, 189–209. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Description | Characterization Techniques | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SEM | FTIR | XRD | DSC | Gas Perm. | WVTR | Swelling Prop. | Mechanical | Polarized Raman | |
| PVA pure | 1 | 5 | 3 | 3 | 3 | 2 | 3 | 8 | 2 |
| PVA 1% CNC | 1 | 5 | 3 | 3 | 3 | 2 | 3 | 8 | 2 |
| PVA 5% CNC | 1 | 5 | 3 | 3 | 3 | 2 | 3 | 8 | 2 |
| PVA 10% CNC | 1 | 5 | 3 | 3 | 3 | 2 | 3 | 8 | 2 |
| PVA 1% NLC | 1 | 5 | 3 | 3 | 3 | 2 | 3 | 8 | 2 |
| PVA 5% NLC | 1 | 5 | 3 | 3 | 3 | 2 | 3 | 8 | 2 |
| PVA 10% NLC | 1 | 5 | 3 | 3 | 3 | 2 | 3 | 8 | 2 |
| Sample Description | Crystal Size (nm) XRD | Cr.I% XRD | Cr.I% ATR/FTIR | Cr.I% DSC | Tm (°C) DSC |
|---|---|---|---|---|---|
| PVA pure | 4.81 | 32.0 | 37.4 | 36.8 | 226.0 |
| PVA 1% CNC | 4.76 | 42.7 | 40.9 | 42.2 | 222.6 |
| PVA 5% CNC | 5.13 | 40.5 | 39.7 | 39.2 | 221.9 |
| PVA 10% CNC | 5.32 | 35.8 | 37.2 | 37.7 | 221.3 |
| PVA 1% NLC | 4.65 | 43.1 | 39.5 | 40.7 | 223.6 |
| PVA 5% NLC | 4.80 | 37.8 | 37.6 | 37.5 | 222.8 |
| PVA 10% NLC | 4.77 | 37.2 | 36.1 | 38 | 222.3 |
| Sample Description | Young Modulus (MPa) | Elongation at Break (%) | Tensile Strength at 100% Elongation (MPa) |
|---|---|---|---|
| PVA pure | 1002 ± 86 | 270 ± 25 | 33 ± 2 |
| PVA 1% CNC | 1380 ± 130 | 330 ± 9 | 37 ± 4 |
| PVA 5% CNC | 1550 ± 130 | 320 ± 22 | 40 ± 4 |
| PVA 10% CNC | 1600 ± 98 | 300 ± 19 | 39 ± 5 |
| PVA 1% NLC | 2620 ± 170 | 270 ± 25 | 46 ± 3 |
| PVA 5% NLC | 2705 ± 170 | 127 ± 27 | 51 ± 3 |
| PVA 10% NLC | 1725 ± 130 | 170 ± 18 | 42 ± 2 |
| Sample Description | Properties | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| WVTR | Barrier CO2 | Swelling H2O | Swelling EtOH | Young Modulus | Elongation at Break | Tensile Strength | Best PVA Orientation | ||
| λ = 2 | λ = 3 | ||||||||
| PVA pure | ●● | ●●●● | ●● | ●●●●● | ●● | ●●●● | ●● | ● | ●●●●● |
| PVA 1% CNC | ● | ●●●●● | ●●● | ●●●●● | ●●● | ●●●●● | ●●● | ●● | ●●● |
| PVA 5% CNC | ●● | ●●● | ●● | ●●●●● | ●●●● | ●●●●● | ●●●● | ●●● | ●● |
| PVA 10% CNC | ●● | ●●● | ● | ●●●●● | ●●●● | ●●●●● | ●●●● | - | ● |
| PVA 1% NLC | ●● | ●●●●● | ●● | ●●●● | ●●●●● | ●●●● | ●●●● | - | - |
| PVA 5% NLC | ●● | ●●●●● | ●●●● | ●●●●● | ●●●●● | ●● | ●●●●● | - | - |
| PVA 10% NLC | ● | ●● | ●●●● | ●●●●● | ●●●● | ●●● | ●●●● | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papapetros, K.; Mathioudakis, G.N.; Vroulias, D.; Koutroumanis, N.; Voyiatzis, G.A.; Andrikopoulos, K.S. Structure-Properties Correlations of PVA-Cellulose Based Nanocomposite Films for Food Packaging Applications. Polymers 2025, 17, 1911. https://doi.org/10.3390/polym17141911
Papapetros K, Mathioudakis GN, Vroulias D, Koutroumanis N, Voyiatzis GA, Andrikopoulos KS. Structure-Properties Correlations of PVA-Cellulose Based Nanocomposite Films for Food Packaging Applications. Polymers. 2025; 17(14):1911. https://doi.org/10.3390/polym17141911
Chicago/Turabian StylePapapetros, Konstantinos, Georgios N. Mathioudakis, Dionysios Vroulias, Nikolaos Koutroumanis, George A. Voyiatzis, and Konstantinos S. Andrikopoulos. 2025. "Structure-Properties Correlations of PVA-Cellulose Based Nanocomposite Films for Food Packaging Applications" Polymers 17, no. 14: 1911. https://doi.org/10.3390/polym17141911
APA StylePapapetros, K., Mathioudakis, G. N., Vroulias, D., Koutroumanis, N., Voyiatzis, G. A., & Andrikopoulos, K. S. (2025). Structure-Properties Correlations of PVA-Cellulose Based Nanocomposite Films for Food Packaging Applications. Polymers, 17(14), 1911. https://doi.org/10.3390/polym17141911