
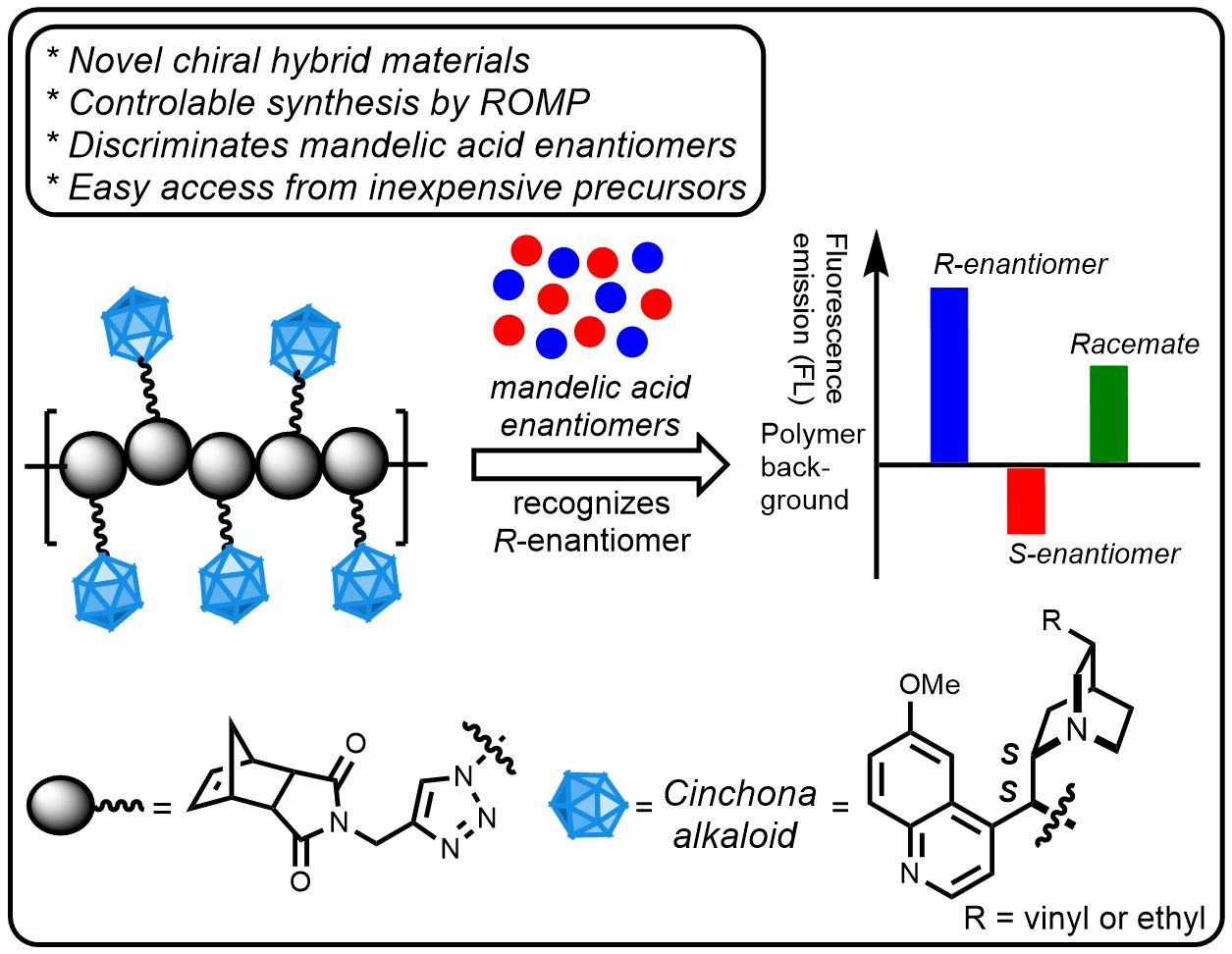
Easy ROMP of Quinine Derivatives Toward Novel Chiral Polymers That Discriminate Mandelic Acid Enantiomers



Abstract

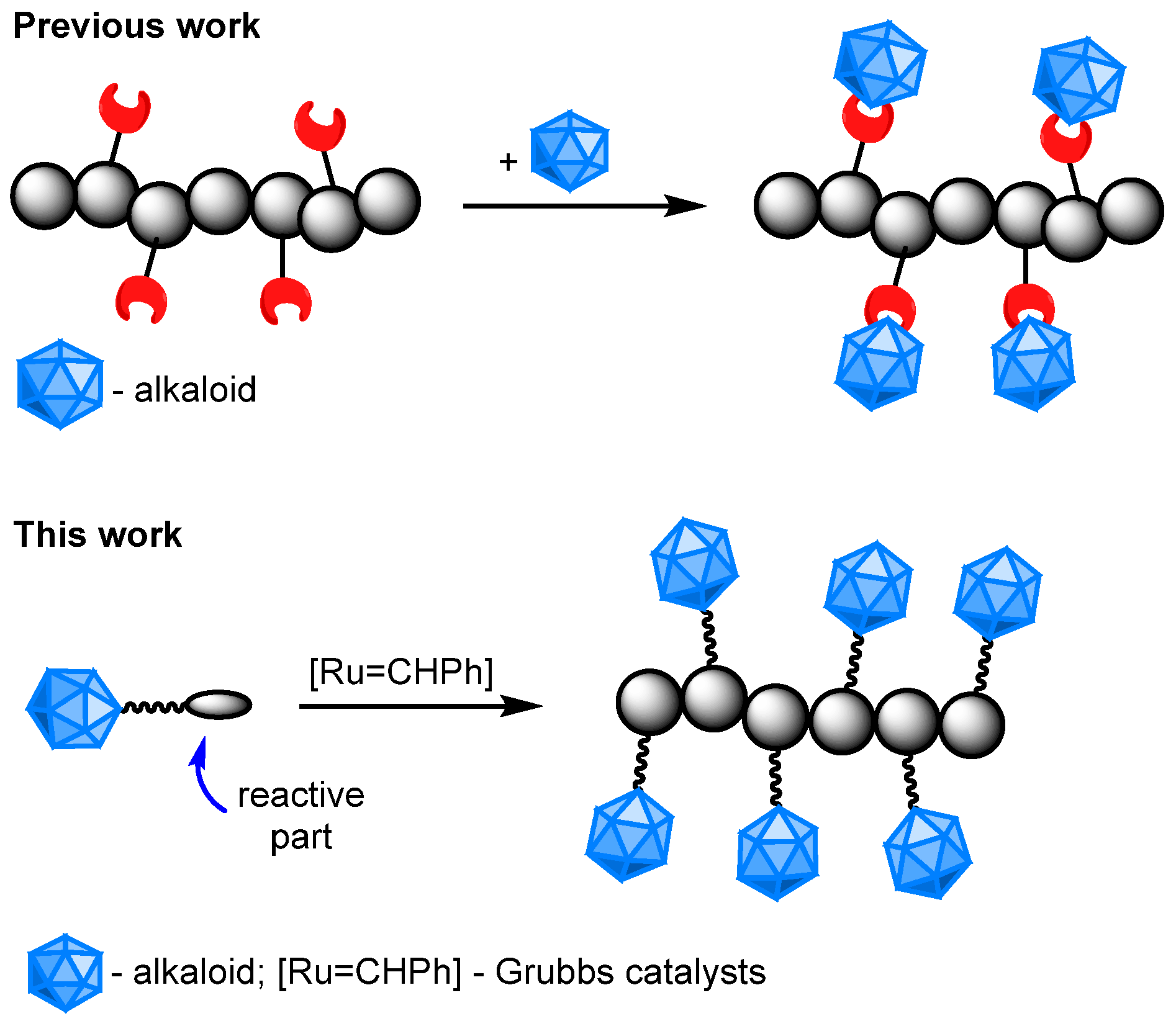
1. Introduction
2. Materials and Methods
2.1. Synthetic Part
2.1.1. General Procedure for Catalytic Screening of ROMP Polymerization
2.1.2. General Procedure for Synthesis of Polymers P1 and P2
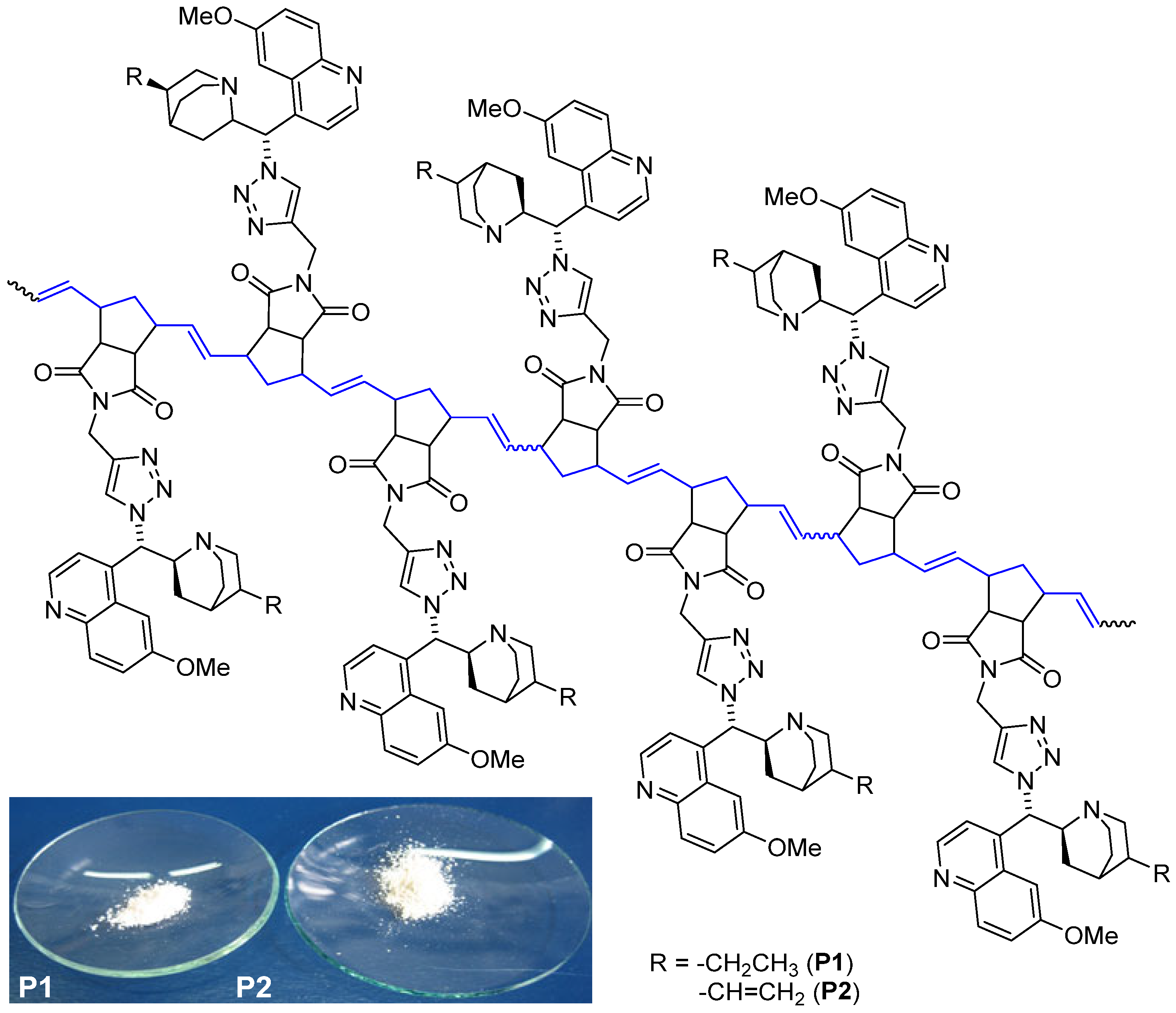
3. Results
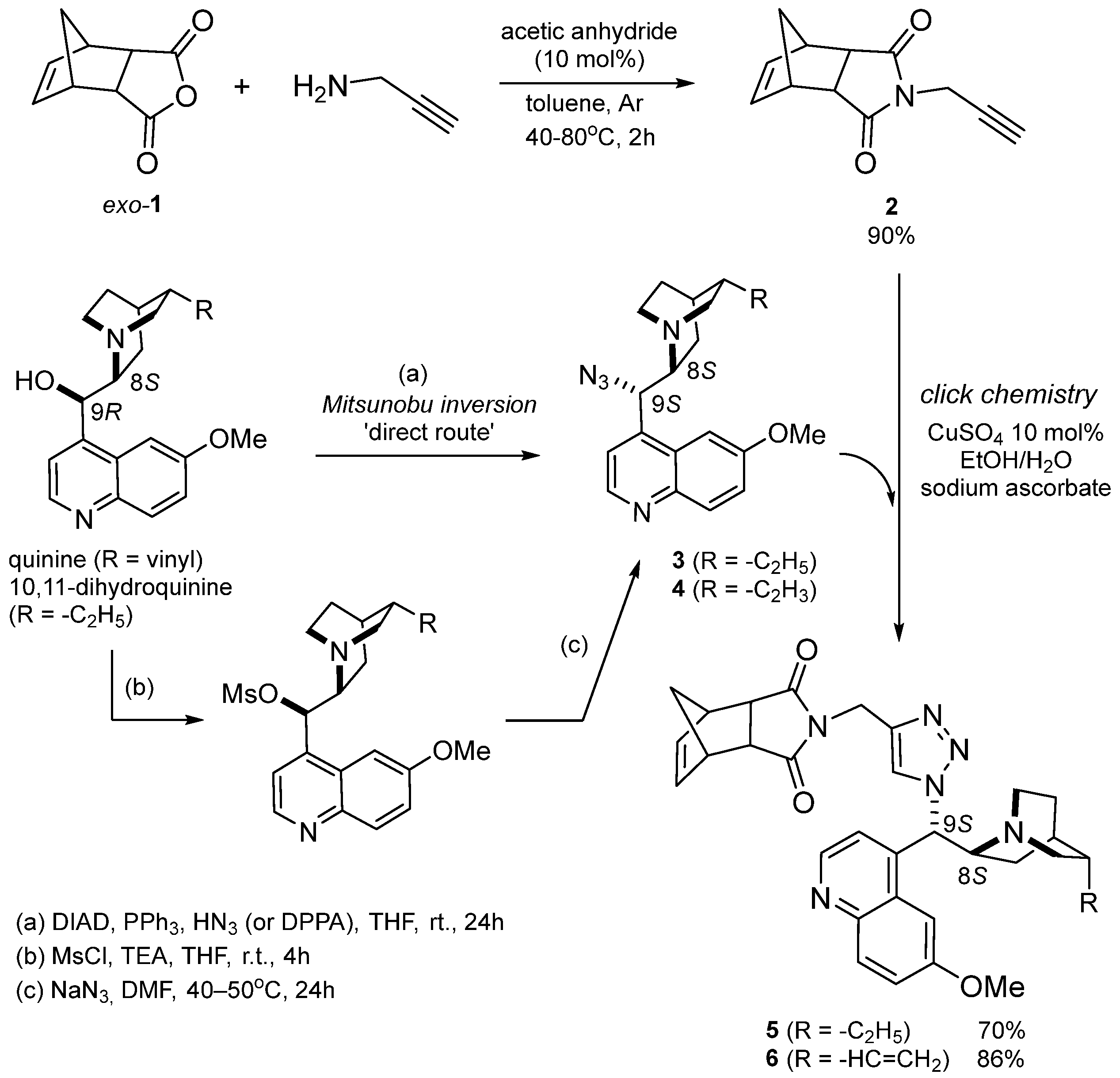
3.1. Synthesis of ROMP-Reactive Exo-Norbornene-Quinine Monomers
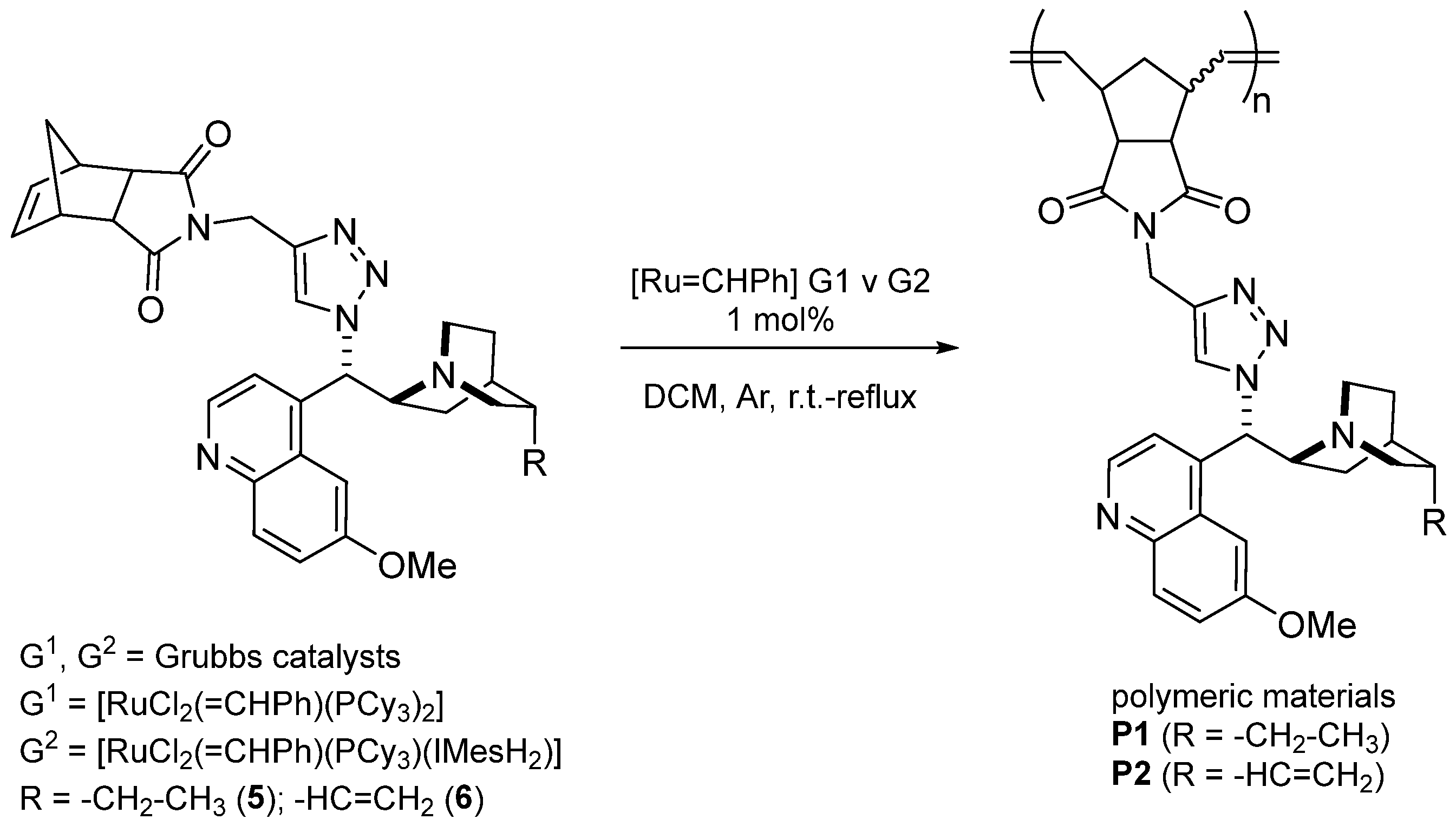
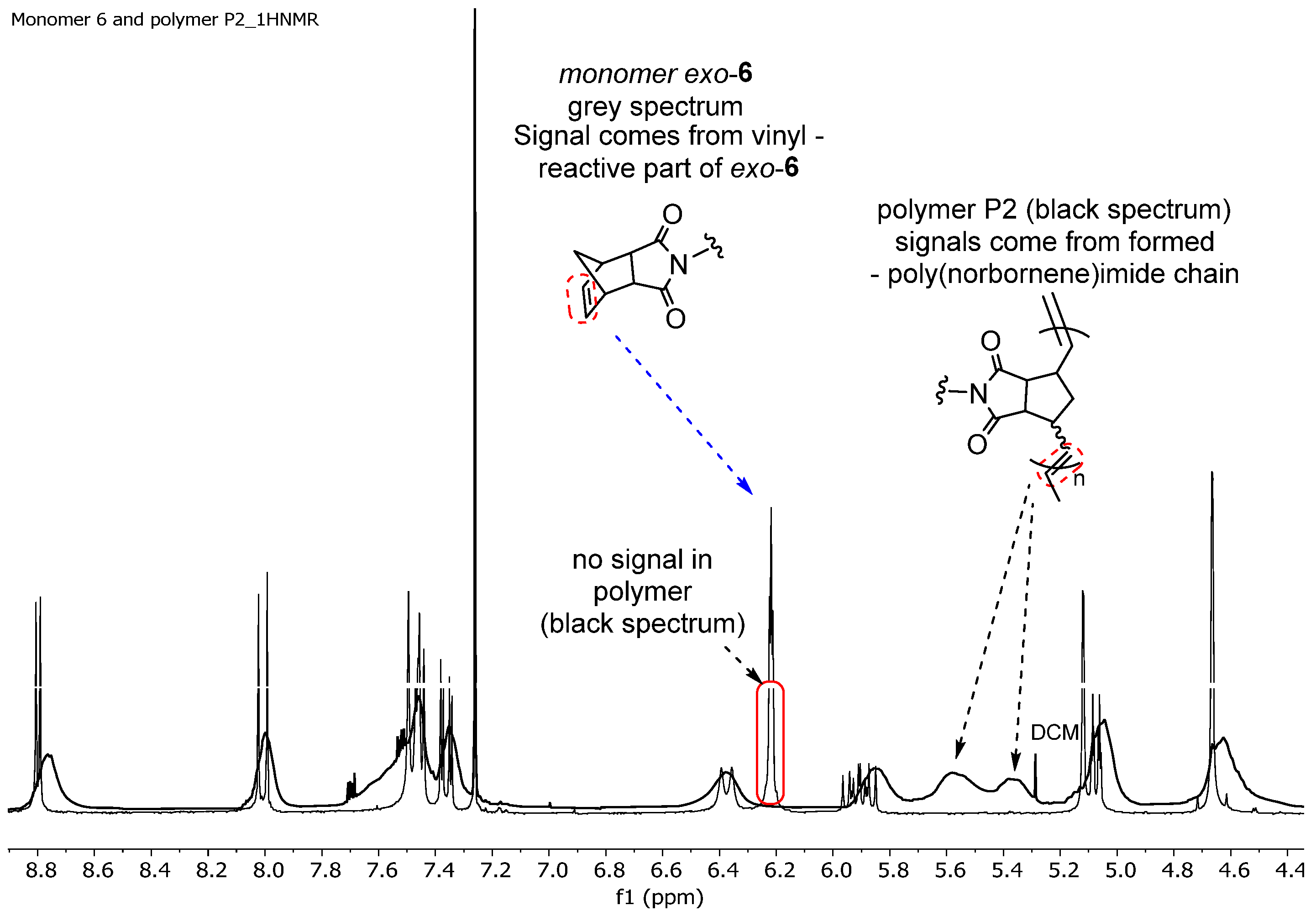
3.2. Polymerization Study
3.3. Characteristics of Polymers—TGA and DSC
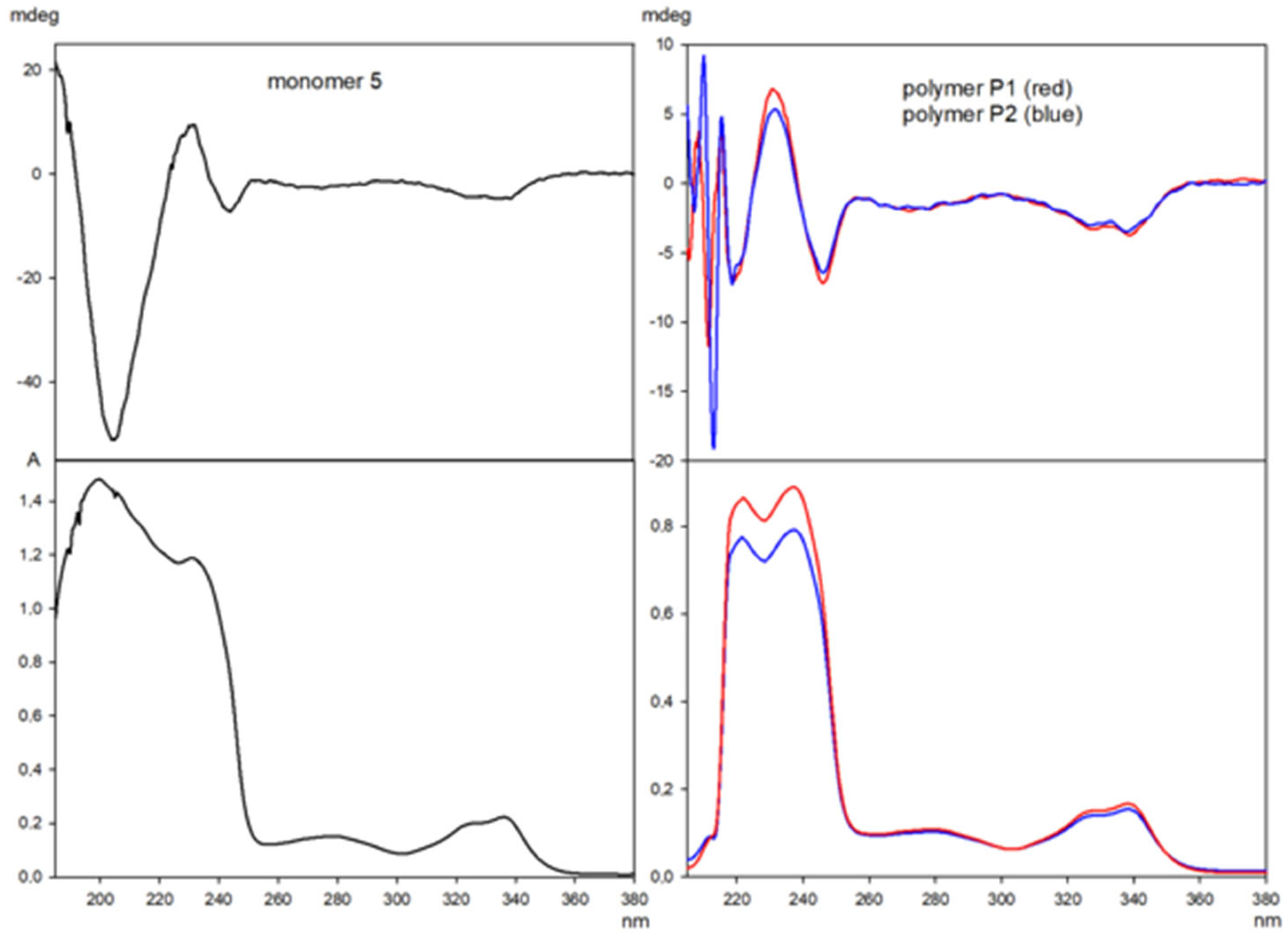
3.4. Characteristics of Polymers—Electronic Circular Dichroism (ECD)
3.5. Chiral Discrimination of Mandelic Acid Enantiomers by Polymer P2
3.6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, C.E. (Ed.) Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Boratyński, P.J.; Zielińska-Błajet, M.; Skarnewski, J. Cinchona Alkaloids-Derivatives and Applications. In Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 82, pp. 29–145. [Google Scholar]
- Ilisz, I.; Bajtai, A.; Lindner, W.; Antal, P. Liquid chromatographic enantiomer separations applying chiral ion-exchangers based on Cinchona alkaloids. J. Pharm. Biomed. Anal. 2018, 1985, 127–152. [Google Scholar] [CrossRef] [PubMed]
- Kacprzak, K.; Gawroński, J. Resolution of Racemates and Enantioselective Analytics by Cinchona Alkaloids and Their Derivatives. In Cinchona Alkaloids in Synthesis and Catalysis: Ligands, Immobilization and Organocatalysis; Song, C.E., Ed.; Wiley-VCH: Weinheim, Germany, 2009; pp. 421–469. [Google Scholar]
- Singh, R.P.; Deng, L. Science of Synthesis, Asymmetric Organocatalysis 2; List, B., Maruoka, K., Eds.; Thieme: Stuttgart, Germany, 2012; pp. 41–117. ISBN 9783131845313. [Google Scholar] [CrossRef]
- Berrisford, D.J.; Bolm, C.; Sharpless, K.B. Ligandenbeschleunigte Katalyse. Angew. Chem. Int. Ed. Engl. 1995, 34, 1059–1077. [Google Scholar] [CrossRef]
- Yang, T.; Ferrali, A.; Sladojevich, F.; Campbell, L.; Dixon, D.J. Brønsted Base/Lewis Acid Cooperative Catalysis in the Enantioselective Conia-Ene Reaction. J. Am. Chem. Soc. 2009, 131, 9140–9141. [Google Scholar] [CrossRef]
- Jianga, L.; Chen, Y.-C. Recent advances in asymmetric catalysis with cinchona alkaloid-based primary amines. Catal. Sci. Technol. 2011, 1, 354–365. [Google Scholar] [CrossRef]
- Yeboah, E.M.O.; Yeboah, S.O.; Singh, S. Recent applications of Cinchona alkaloids and their derivatives as catalysts in metal-free asymmetric synthesis. Tetrahedron 2011, 67, 1725–1762. [Google Scholar] [CrossRef]
- Ingemann, S.; Hiemstra, H. Comprehensive Enantioselective Organocatalysis, Part II, Non-Amino Acid-Derived Catalysts, Chapter 6: Cinchonas and Cupreidines; Dalko, P.I., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 119–160. [Google Scholar]
- O’Donnell, M.J. The Enantioselective Synthesis of r-Amino Acids by Phase-Transfer Catalysis with Achiral Schiff Base Esters. Acc. Chem. Res. 2004, 37, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Chhanda, S.A.; Itsuno, S. ADMET Polymerization of Dimeric Cinchona Squaramides for the Preparation of a Highly Enantioselective Polymeric Organocatalyst. Catalysts 2020, 10, 591. [Google Scholar] [CrossRef]
- Kristensen, T.E.; Hansen, T. Polymer-Supported Chiral Organocatalysts: Synthetic Strategies for the Road Towards Affordable Polymeric Immobilization. Eur. J. Org. Chem. 2010, 2010, 3179–3204. [Google Scholar] [CrossRef]
- Kristensen, T.E.; Hansen, T. Synthesis of Chiral Catalysts Supported on Organic Polymers. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques and Applications; Gruttadauria, M., Giacalone, F., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 209–256. [Google Scholar]
- Jumde, R.P.; Di Pietro, A.; Manariti, A.; Mandoli, A. New Polymer-Supported Mono- and Bis-Cinchona Alkaloid Derivatives: Synthesis and Use in Asymmetric Organocatalyzed Reactions. Chem. Asian J. 2015, 102, 397–404. [Google Scholar] [CrossRef]
- Kacprzak, K.; Maier, N.M.; Lindner, W. Triazolo-linked cinchona alkaloid carbamate anion exchange-type chiral stationary phases: Synthesis by click chemistry and evaluation. J. Chromatogr. A 2011, 1218, 1452–1460. [Google Scholar] [CrossRef]
- Kanaoka, S.; Grubbs, R.H. Synthesis of Block Copolymers of Silicon-Containing Norbornene Derivatives via Living Ring-Opening Metathesis Polymerization Catalyzed by a Ruthenium Carbene Complex. Macromolecules 1995, 28, 4707–4713. [Google Scholar] [CrossRef]
- Bielawski, C.W.; Grubbs, R.H. Highly Efficient Ring-Opening Metathesis Polymerization (ROMP) Using New Ruthenium Catalysts Containing N-Heterocyclic Carbene Ligands. Angew. Chem. 2000, 39, 3025–3028. [Google Scholar] [CrossRef]
- Maynard, H.D.; Okada, S.Y.; Grubbs, R.H. Synthesis of Norbornenyl Polymers with Bioactive Oligopeptides by Ring-Opening Metathesis Polymerization. Macromolecules 2000, 33, 6239–6248. [Google Scholar] [CrossRef]
- Joseph, D.R.; Jeffrey, S.M. ROMP Reactivity of endo- and exo-Dicyclopentadiene. Macromolecules 2002, 35, 7878–7882. [Google Scholar]
- Pollino, J.M.; Stubbs, L.P.; Weck, M. Living ROMP of exo-Norbornene Esters Possessing PdII SCS Pincer Complexes or Diaminopyridines. Macromolecules 2003, 36, 2230–2234. [Google Scholar] [CrossRef]
- Majchrzak, M.; Hine, P.J.; Khosravi, E. An autonomous self-healing system based on ROMP of norbornene dicarboximide monomers. Polymer 2012, 53, 5251–5257. [Google Scholar] [CrossRef]
- Roper, S.; Franz, M.H.; Wartchow, R.; Hoffmann, H.M. Fused Triazoles via Tandem Reactions of Activated Cinchona Alkaloids with Azide Ion. Second Cinchona Rearrangement Exemplified. Org. Lett. 2003, 5, 2773–2776. [Google Scholar] [CrossRef]
- Franz, M.H.; Röper, S.; Wartchow, R.; Hoffmann, H.M.R. The First and Second Cinchona Rearrangement. Two Fundamental Transformations of Alkaloid Chemistry. J. Org. Chem. 2004, 69, 2983–2991. [Google Scholar] [CrossRef]
- Brunner, H.; Bügler, J.; Nuber, B. Preparation of 9-Amino(9-deoxy)cinchona Alkaloids. Tetrahedron Asymmetry 1995, 6, 1699–1702. [Google Scholar] [CrossRef]
- Kacprzak, K.; Gierczyk, B. Clickable 9-azido-(9-deoxy)-Cinchona alkaloids: Synthesis and conformation. Tetrahedron Asymmetry 2010, 21, 2740–2745. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.R.; Williams, J.E.; Blanchard, K.R. The Evaluation of Strain in Hydrocarbons. The Strain in Adamantane and Its Origin. J. Am. Chem. Soc. 1970, 92, 2377–2386. [Google Scholar] [CrossRef]
- Berova, N.; Nakanishi, K.; Woody, R.W. (Eds.) Circular Dichroism: Principles and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2000; ISBN 0-471-33003-5. [Google Scholar]
- Gawroński, J.; Brzostowska, M.; Koput, J. A Circular Dichroism Study of Molecular Association of Cinchona Alkaloids and Carboxylic Acids. Croat. Chem. Acta 1989, 62, 97–107. [Google Scholar]
- Tsangaris, J.M.; Kabanos, T.A. Circular Dichroism of the Complexes of Quinine with Copper(II), Nickel(II), Cobalt(II), Chromium(III) and Palladium(II) Chlorides. Monat. Chem. 1982, 113, 1393–1398. [Google Scholar] [CrossRef]
- Hiroki, I.; Zhenglin, T.; Yashima, E. Synthesis and Bifunctional Asymmetric Organocatalysis of Helical Poly(phenylacetylene)s Bearing Cinchona Alkaloid Pendants via a Sulfonamide Linkage. J. Pol. Chem. Part A Pol. Chem. 2013, 51, 2869–2879. [Google Scholar]
- Amol, M.; Kendhale, A.M.; Poniman, L.; Dong, Z.; Laxmi-Reddy, K.; Kauffmann, B.; Ferrand, Y.; Huc, I. Absolute Control of Helical Handedness in Quinoline Oligoamides. J. Org. Chem. 2011, 76, 195–200. [Google Scholar]
- Poloński, T.; Milewska, M.J.; Gdaniec, M.; Gilski, M. Molecular Geometry and Circular Dichroism Spectra of Bicyclo [2.2.1]heptane-2,3-dicarboxylic Anhydrides and Imides. J. Org. Chem. 1993, 58, 3134–3139. [Google Scholar] [CrossRef]
- Tejera, S.; Dorta, R.L.; Vázquez, J.T. Study of aryl triazoles for absolute configuration determination. Tetrahedron Asymmetry 2016, 27, 896–909. [Google Scholar] [CrossRef]
- Larsen, S.; de Diego, H.L.; Kozma, D. Optical Resolution Through Diastereomeric Salt Formation: The Crystal Structures of Cinchoninium (R)-Mandelate and Cinchoninium (S)-Mandelate at Low Temperature, Acta Crystallogr. Sect. B Struct. Sci. 1993, 49, 310–316. [Google Scholar] [CrossRef]
- Gjerlov, A.; Larsen, S. A Study of Diastereomeric Mandelate Salts of Cinchonidine and the Relation to Their Quasidiastereomeric Analogues, Acta Crystallogr. Sect. B Struct. Sci. 1997, 53, 708–718. [Google Scholar]
- Báthori, N.B.; Nassimbeni, L.R.; Oliver, C.L. Quininium mandelates—A systematic study of chiral discrimination in crystals of diastereomeric salts. Chem. Commun. 2011, 479, 2670–2672. [Google Scholar]
- Mo, M.-Y.; Wang, X.-J.; Shen, R.-Z.; Hu, C.-Y.; Li, X.-C.; Li, G.-W.; Liu, L.-T. Enantiospecific Analysis of Carboxylic Acids Using Cinchona Alkaloid Dimers as Chiral Solvating Agents. Anal. Chem. 2024, 96, 7487–7496. [Google Scholar] [CrossRef] [PubMed]
- Kacprzak, K.; Maier, N.; Lindner, W. Unexpected enantioseparation of mandelic acids and their derivatives on 1,2,3-triazololinked quinine tert-butyl carbamate anion exchange-type chiral stationary phase. J. Sep. Sci. 2010, 33, 2590–2598. [Google Scholar] [CrossRef]
- Pu, L. Fluorescence of Organic Molecules in Chiral Recognition. Chem. Rev. 2004, 104, 1687–1716. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer (Catalyst Used) | Mn a (g/mol) ×104 | Mw a (g/mol) ×104 | ĐM (Mw/Mn) | Yield (%) b |
|---|---|---|---|---|
| P1 (G2) | 2.2365 c | 2.9020 c | 1.32 | 96 |
| P2 (G2) | 2.2618 c | 3.0494 c | 1.35 | 89 |
| Polymer (Catalyst Used) | Td5 (°C) a | Td10 (°C) a | 1st Step of Thermal Decomp. (Weight Resid. (%)) Top Temp. a (°C) | 2nd Step of Thermal Decomp. (Weight Resid. (%)) Top Temp. a (°C) | 3th Step of Thermal Decomp. (Weight Resid. (%)) Top Temp. a (°C) | Total Weight Residue (%) |
|---|---|---|---|---|---|---|
| P1 (G1) (Figure S14 ESI) | 242 | 285 | 200–390 (38) Max. 306 | 390–550 (22) Max. 431 | ---- | 75 |
| P1 (G2) (Figure S15 ESI) | 286 | 299 | 257–367 (22) Max. 301.0 | 351–509 (21) Max. 447 | 504–769 (48) Max. 594 | 92 |
| P2 (G1) (Figure S16 ESI) | 219 | 287 | 231.7–379.2 (45) Max. 315.5 | 375–531 (30) Max. 423 | ---- | 80 |
| P2 (G2) (Figure S17 ESI) | 297 | 310 | 271–379 (22) Max. 312 | 382–542 (33) Max. 450 | ---- | 76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majchrzak, M.; Kacprzak, K.; Piętka, M.; Garbarek, J.; Taras-Goślińska, K. Easy ROMP of Quinine Derivatives Toward Novel Chiral Polymers That Discriminate Mandelic Acid Enantiomers. Polymers 2025, 17, 1661. https://doi.org/10.3390/polym17121661
Majchrzak M, Kacprzak K, Piętka M, Garbarek J, Taras-Goślińska K. Easy ROMP of Quinine Derivatives Toward Novel Chiral Polymers That Discriminate Mandelic Acid Enantiomers. Polymers. 2025; 17(12):1661. https://doi.org/10.3390/polym17121661
Chicago/Turabian StyleMajchrzak, Mariusz, Karol Kacprzak, Marta Piętka, Jerzy Garbarek, and Katarzyna Taras-Goślińska. 2025. "Easy ROMP of Quinine Derivatives Toward Novel Chiral Polymers That Discriminate Mandelic Acid Enantiomers" Polymers 17, no. 12: 1661. https://doi.org/10.3390/polym17121661
APA StyleMajchrzak, M., Kacprzak, K., Piętka, M., Garbarek, J., & Taras-Goślińska, K. (2025). Easy ROMP of Quinine Derivatives Toward Novel Chiral Polymers That Discriminate Mandelic Acid Enantiomers. Polymers, 17(12), 1661. https://doi.org/10.3390/polym17121661