
Advancements in Catalytic Depolymerization Technologies

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract

1. Introduction
2. Catalytic Depolymerization of Cellulose
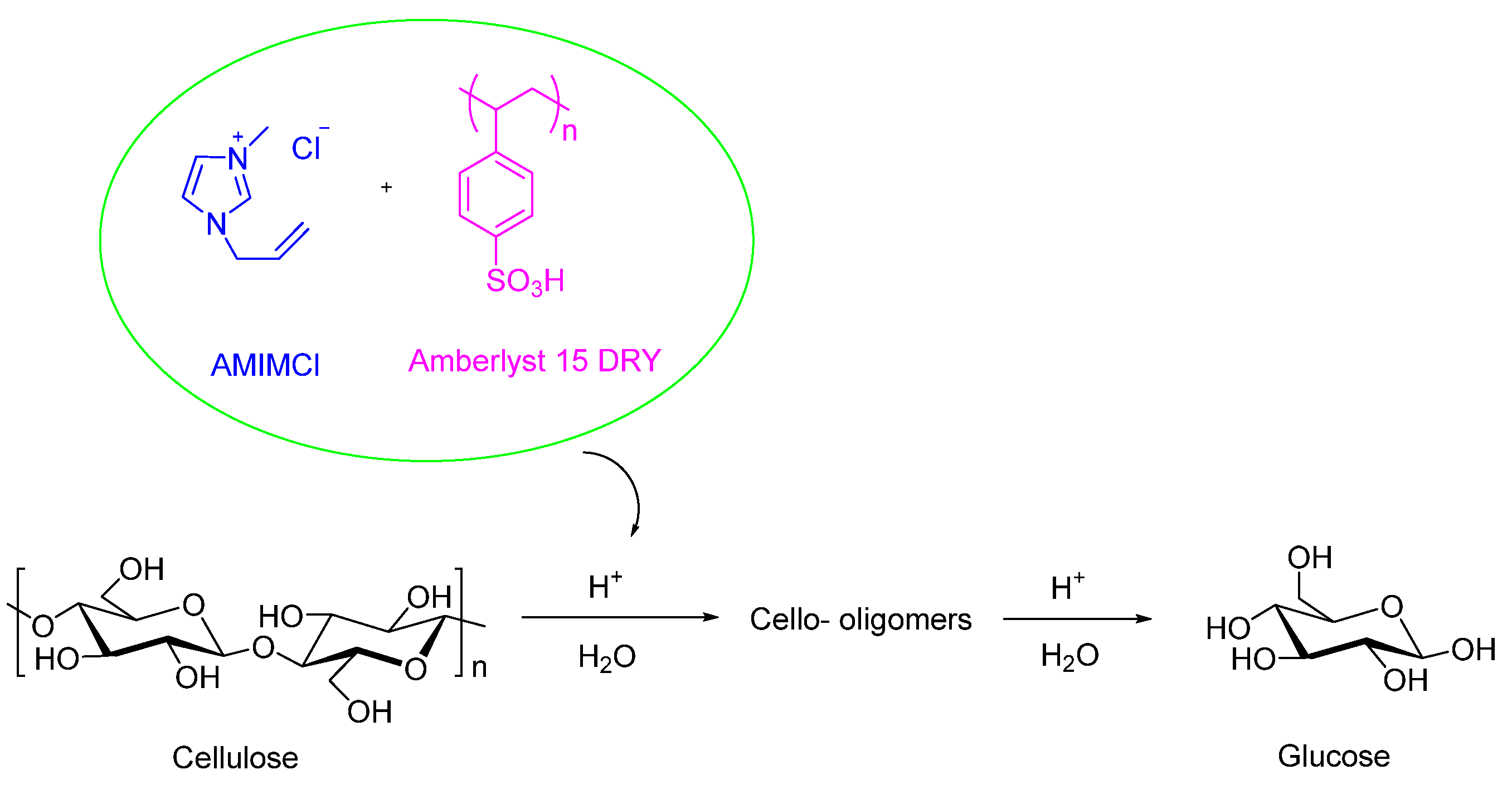
2.1. Solid Acid Catalyst Depolymerization of Cellulose
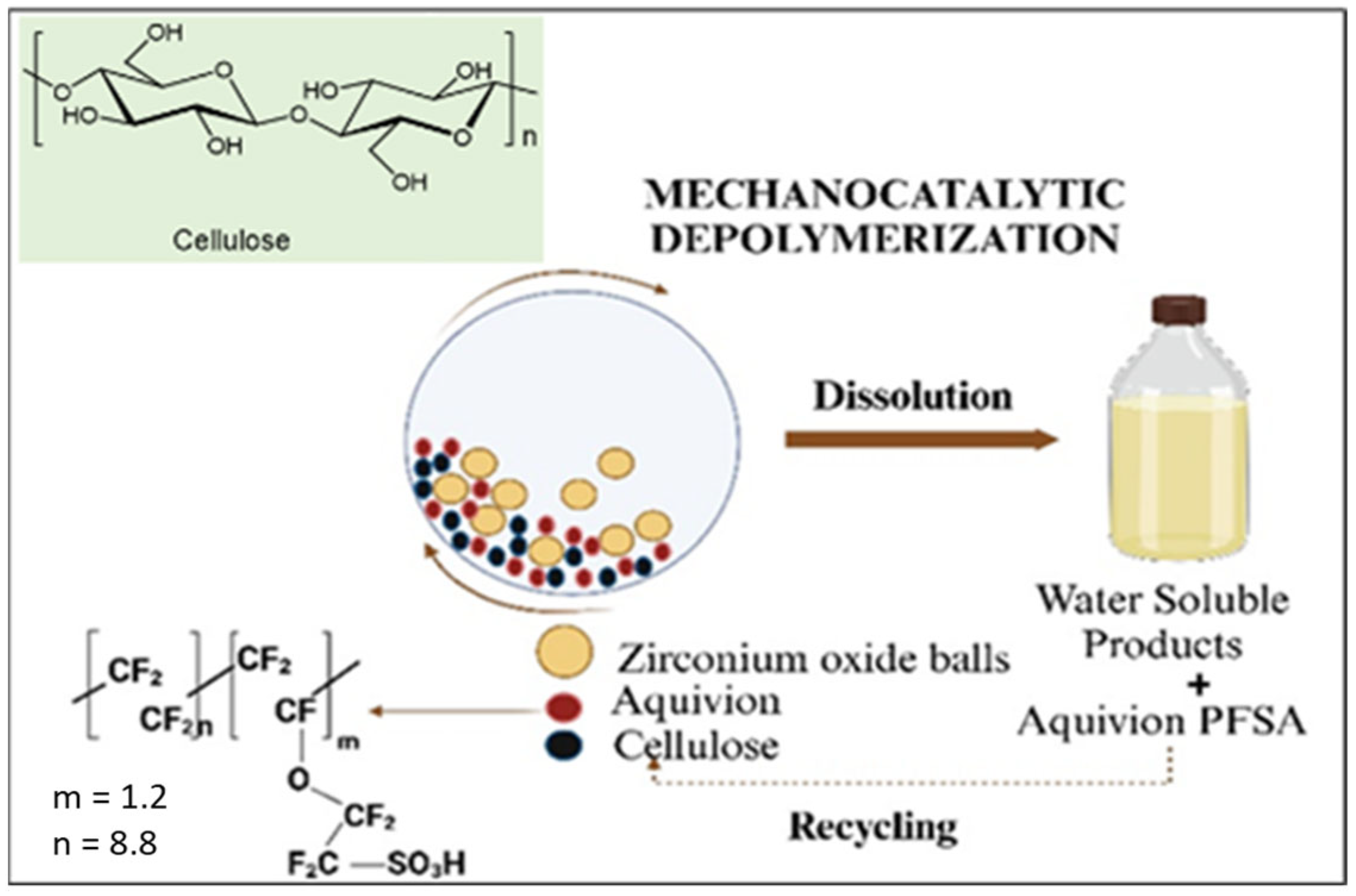
2.2. Mechanocatalytic Depolymerization of Cellulose
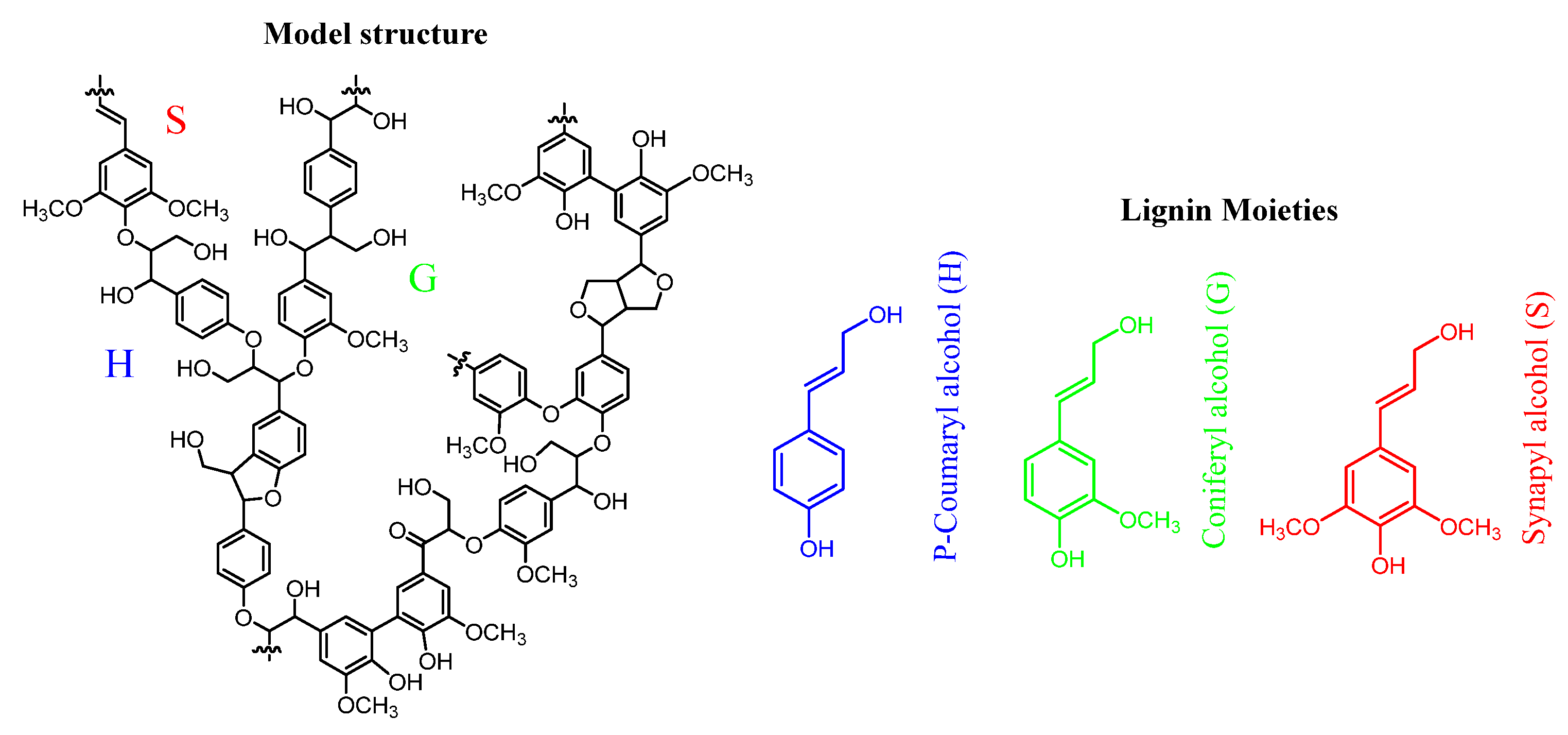
3. Catalytic Depolymerization of Lignin
3.1. Challenges and Opportunities
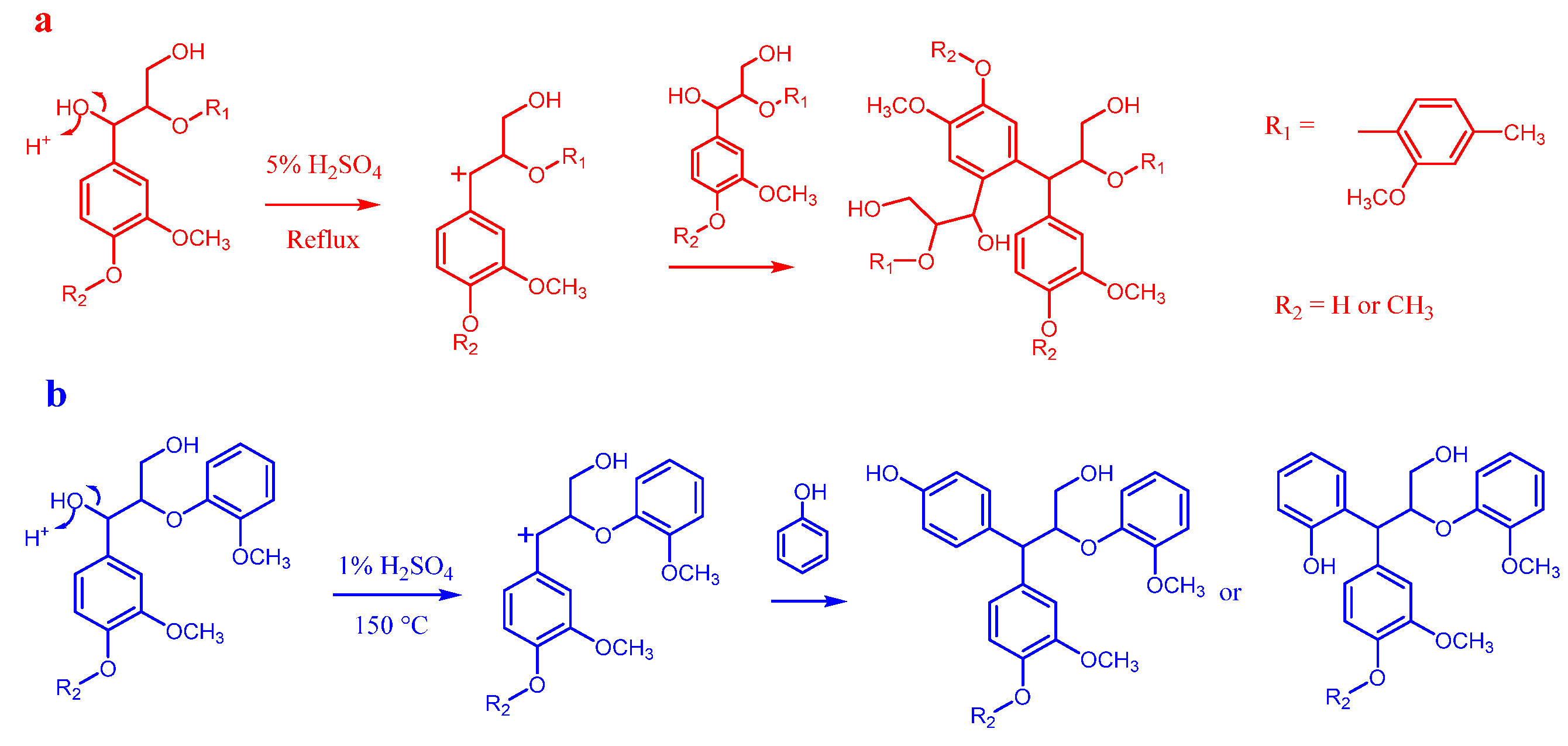
3.1.1. Homogeneous Acid Catalysis
3.1.2. Homogeneous Base Catalysis
Combined Acid-Base Catalysis
3.1.3. Homogeneous Metal Catalysis
- Hydrogenolysis involves the cleavage of bonds by the addition of hydrogen (H2), resulting in the formation of smaller molecules.
- Oxidation entails the removal of electrons from a molecule, leading to the formation of carbonyl groups (C=O) and other oxidized byproducts.
- Solvolysis involves the cleavage of bonds through a reaction with a solvent, producing smaller molecular fragments.
3.1.4. Heterogeneous Solid Acid Catalysis
3.1.5. Heterogeneous Metal-Supported Catalysis
3.1.6. Enzyme Catalysis
- Advantages of Enzyme Catalysis
- Mild Reaction Conditions: Enzymatic depolymerization occurs at ambient temperature and neutral pH, reducing energy requirements.
- Specificity and Selectivity: High specificity minimizes unwanted side reactions, yielding a more controlled product distribution compared to chemical methods.
- Environmental Friendliness: It avoids the use of harsh chemicals and produces minimal waste, supporting sustainable practices [83].
- Challenges and Limitations
- Despite its advantages, enzyme catalysis faces several challenges:
- Sensitivity to Reaction Conditions: Enzymes are sensitive to temperature, pH, and the presence of inhibitors, necessitating careful control of reaction conditions.
- Limited Lignin Accessibility: The complex structure of lignin can hinder enzyme accessibility, reducing depolymerization efficiency.
- Cost and Production Efficiency: The high cost of enzyme production impacts the economic viability of the process [120].
- Strategies for Improvement
- To overcome these challenges, research is focused on the following:
- Engineering more efficient enzymes with enhanced stability and activity.
- Developing cost-effective production methods.
- Implementing pretreatment strategies to improve lignin accessibility [120].
4. Catalytic Depolymerization of Plastics
4.1. Depolymerization of Polyester Plastics
4.2. Depolymerization of Polyamides
4.3. Depolymerization of Polyurethanes
4.4. Depolymerization of Polyethers
5. Summary and Outlook
Future Challenges
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kreps, B.H. The Rising Costs of Fossil-Fuel Extraction: An Energy Crisis That Will Not Go Away. Am. J. Econ. Sociol. 2020, 79, 695–717. [Google Scholar] [CrossRef]
- Ghazouani, T.; Maktouf, S. Impact of natural resources, trade openness, and economic growth on CO2 emissions in oil-exporting countries: A panel autoregressive distributed lag analysis. Nat. Resour. Forum. 2024, 48, 211–231. [Google Scholar] [CrossRef]
- Zibunas, C.; Meys, R.; Kätelhön, A.; Bardow, A. Cost-optimal pathways towards net-zero chemicals and plastics based on a circular carbon economy. Comput. Chem. Eng. 2022, 162, 107798. [Google Scholar] [CrossRef]
- Boulamanti, A.; Moya, J.A. Production costs of the chemical industry in the EU and other countries: Ammonia, methanol and light olefins. Renew. Sustain. Energy Rev. 2017, 68, 1205–1212. [Google Scholar] [CrossRef]
- Axon, S.; James, D. The UN Sustainable Development Goals: How can sustainable chemistry contribute? A view from the chemical industry. Curr. Opin. Green Sustain. Chem. 2018, 13, 140–145. [Google Scholar] [CrossRef]
- Kümmerer, K. Sustainable Chemistry: A Future Guiding Principle. Angew. Chem. Int. Ed. 2017, 56, 16420. [Google Scholar] [CrossRef] [PubMed]
- Zuin, V.G.; Eilks, I.; Elschami, M.; Kümmerer, K. Education in green chemistry and in sustainable chemistry: Perspectives towards sustainability. Green Chem. 2021, 23, 1594–1608. [Google Scholar] [CrossRef]
- Marion, P.; Bernela, B.; Piccirilli, A.; Estrine, B.; Patouillard, N.; Guilbot, J.; Jérôme, C. Sustainable chemistry: How to produce better and more from less? Green Chem. 2017, 19, 4973–4989. [Google Scholar] [CrossRef]
- Fagnani, D.E.; Tami, J.L.; Copley, G.; Clemons, M.N.; Getzler, Y.D.Y.L.; McNeil, A.J. 100th anniversary of macromolecular science viewpoint: Redefining sustainable polymers. ACS Macro Lett. 2021, 10, 41–53. [Google Scholar] [CrossRef]
- Guo, X.; Facchetti, A. The journey of conducting polymers from discovery to application. Nat. Mater. 2020, 19, 922–928. [Google Scholar] [CrossRef]
- Tursi, A.; Olivito, F. Biomass Conversion: General Information, Chemistry, and Processes; Rahimpour, M.R., Kamali, R., Makarem, M.A., Manshadi, M.K.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 3–39. [Google Scholar]
- Wang, R.; Feng, Y.; Li, D.; Li, K.; Yan, Y. Towards the sustainable production of biomass-derived materials with smart functionality: A tutorial review. Green Chem. 2024, 26, 9075–9103. [Google Scholar] [CrossRef]
- Jia, J.; Zang, G.; Paul, M.C. Energy, exergy, and economic (3E) evaluation of a CCHP system with biomass gasifier, solid oxide fuel cells, micro-gas turbine, and absorption chiller. Int. J. Energy Res. 2021, 45, 15182–15199. [Google Scholar] [CrossRef]
- Thakur, V.; Guleria, A.; Kumar, S.; Sharma, S.; Singh, K. Recent advances in nanocellulose processing, functionalization and applications: A review. Mater. Adv. 2021, 2, 1872–1895. [Google Scholar] [CrossRef]
- Etale, A.; Onyianta, A.J.; Turner, S.R.; Eichhorn, S.J. Cellulose: A review of water interactions, applications in composites, and water treatment. Chem. Rev. 2023, 123, 2016–2048. [Google Scholar] [CrossRef]
- Maiuolo, L.; Olivito, F.; Algieri, V.; Costanzo, P.; Jiritano, A.; Tallarida, M.A.; Tursi, A.; Sposato, C.; Feo, A.; De Nino, A. Synthesis, Characterization and Mechanical Properties of Novel Bio-Based Polyurethane Foams Using Cellulose-Derived Polyol for Chain Extension and Cellulose Citrate as a Thickener Additive. Polymers 2021, 13, 2802. [Google Scholar] [CrossRef]
- Romeo, I.; Olivito, F.; Tursi, A.; Algieri, V.; Beneduci, A.; Chidichimo, G.; Maiuolo, L.; Sicilia, E.; De Nino, A. Totally green cellulose conversion into bio-oil and cellulose citrate using molten citric acid in an open system: Synthesis, characterization and computational investigation of reaction mechanisms. RSC Adv. 2020, 10, 34738–34751. [Google Scholar] [CrossRef]
- Chen, P.; Shrotri, A.; Fukuoka, A. Synthesis of cello-oligosaccharides by depolymerization of cellulose: A review. Appl. Catal. 2021, 621, 118177. [Google Scholar] [CrossRef]
- Tyufekchiev, M.; Ralph, K.; Duan, P.; Yuan, S.; Schmidt-Rohr, K.; Timko, M.T. Rapid depolymerization of decrystallized cellulose to soluble products via ethanolysis under mild conditions. ChemSusChem 2020, 13, 2634. [Google Scholar] [CrossRef]
- Clark, J.H.; Deswarte, F.E.I. Introduction to Chemicals from Biomass; John Wiley & Sons: Chichester, UK, 2008. [Google Scholar]
- Farmer, T.J.; Mascal, M. Biomass-derived Platform Chemicals. In Introduction to Chemicals from Biomass, 2nd ed.; Clark, J.H., Deswarte, F.E.I., Eds.; Wiley: Chichester, UK, 2015; pp. 61–92. [Google Scholar]
- Yabushita, M.; Kobayashi, H.; Fukuoka, A. Catalytic transformation of cellulose into platform chemicals. Appl. Catal. B Environ. 2014, 145, 1–9. [Google Scholar] [CrossRef]
- Forsberg, C.W.; Dale, B.E.; Jones, D.S.; Hossain, T.; Morais, A.R.C.; Wendt, L.M. Replacing liquid fossil fuels and hydrocarbon chemical feedstocks with liquid biofuels from large-scale nuclear biorefineries. Appl. Energy 2021, 298, 117225. [Google Scholar] [CrossRef]
- Olivito, F.; Jagdale, P.; Oza, G. Direct Production of Furfural from Fructose Catalyzed by Iron(III) Sulfate Using a Simple Distillation Apparatus. ACS Sustain. Chem. Eng. 2023, 11, 17595–17599. [Google Scholar] [CrossRef]
- Tomishige, K.; Yabushita, M.; Cao, J.; Nakagawa, Y. Hydrodeoxygenation of potential platform chemicals derived from biomass to fuels and chemicals. Green Chem. 2022, 24, 5652–5690. [Google Scholar] [CrossRef]
- Mascal, M. Across the Board: Mark Mascal on the Challenges of Lignin Biorefining. ChemSusChem 2020, 13, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Bai, J.; Innocent, M.T.; Wang, Q.; Xiang, H.; Tang, J.; Zhu, M. Lignin-based carbon fibers: Formation, modification and potential applications. Green Energy Environ. 2022, 7, 578–605. [Google Scholar] [CrossRef]
- Sethupathy, S.; Morales, G.M.; Gao, L.; Wang, H.; Yang, B.; Jiang, J.; Sun, J.; Zhu, D. Lignin valorization: Status, challenges and opportunities. Bioresour. Technol. 2022, 347, 126696. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C. Lignin Source and Structural Characterization. ChemSusChem 2020, 13, 4385–4393. [Google Scholar] [CrossRef]
- Haq, I.; Mazumder, P.; Kalamdhad, A.S. Recent advances in removal of lignin from paper industry wastewater and its industrial applications—A review. Bioresour. Technol. 2020, 312, 123636. [Google Scholar] [CrossRef] [PubMed]
- Mboowa, D. A review of the traditional pulping methods and the recent improvements in the pulping processes. Biomass Convers. Bioref. 2024, 14, 1–12. [Google Scholar] [CrossRef]
- Zhang, B.; Meng, Q.; Liu, H.; Han, B. Catalytic Conversion of Lignin into Valuable Chemicals: Full Utilization of Aromatic Nuclei and Side Chains. Acc. Chem. Res. 2023, 56, 3558–3571. [Google Scholar] [CrossRef]
- Karagoz, P.; Khiawjan, S.; Marques, M.P.; Santzouk, S.; Bugg, T.D.; Lye, G.J. Pharmaceutical applications of lignin-derived chemicals and lignin-based materials: Linking lignin source and processing with clinical indication. Biomass Convers. Bioref. 2024, 14, 26553–26574. [Google Scholar] [CrossRef]
- Salcedo-Puerto, O.; Mendoza-Martinez, C.; Saari, J.; Vakkilainen, E. Hydrothermal carbonization of industrial kraft lignin: Assessment of operational parameters. Fuel 2024, 373, 132389. [Google Scholar] [CrossRef]
- Sun, P.; Wang, Z.; Li, C.; Tang, B.; Peng, C. Catalytic conversion of lignin and its derivatives to alkanes over multifunctional catalysts: A review. Fuel 2024, 361, 130726. [Google Scholar] [CrossRef]
- Diao, X.; Xiong, Y.; Shi, Y.; Ma, L.; Dong, C.; Zhang, S.; Ji, N. Catalytic hydrodeoxygenation and C–C coupling of lignin and its derivatives into renewable jet-fuel-range cycloalkanes. Green Chem. 2024, 26, 11406–11426. [Google Scholar] [CrossRef]
- Rosenboom, J.G.; Langer, R.; Traverso, G. Bioplastics for a circular economy. Nat. Rev. Mater. 2022, 7, 117–137. [Google Scholar] [CrossRef]
- Kumar, A.; Awasthi, M.K.; Sheet, N.; Kharde, T.A.; Singh, S.K. One-pot Upcycling of Waste Plastics for Selective Hydrogen Production at Low-Temperature. ChemCatChem 2023, 15, e202300574. [Google Scholar] [CrossRef]
- Faust, K.; Denifl, P.; Hapke, M. Recent advances in catalytic chemical recycling of polyolefins. ChemCatChem 2023, 15, e202300310. [Google Scholar] [CrossRef]
- Yang, S.; Li, Y.; Nie, M.; Liu, X.; Wang, Q.; Chen, N.; Zhang, C. Lifecycle management for sustainable plastics: Recent progress from synthesis, processing to upcycling. Adv. Mater. 2024, 36, 2404115. [Google Scholar] [CrossRef]
- Kee Wong, M.; Lock, S.S.M.; Chan, Y.H.; Yeoh, S.J.; Tan, I.S. Towards sustainable production of bio-based ethylene glycol: Progress, perspective and challenges in catalytic conversion and purification. J. Chem. Eng. 2023, 468, 143699. [Google Scholar] [CrossRef]
- Volanti, M.; Cespi, D.; Passarini, F.; Neri, E.; Cavani, F.; Mizsey, P.; Fozer, D. Terephthalic acid from renewable sources: Early-stage sustainability analysis of a bio-PET precursor. Green Chem. 2019, 21, 885–896. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, Q.; Che, Y.; Niu, Z. Opportunities and challenges for plastic depolymerization by biomimetic catalysis. Chem. Sci. 2024, 15, 6200–6217. [Google Scholar] [CrossRef]
- Liu, Y.; Akula, K.C.; Dandamudi, K.P.R.; Liu, Y.; Xu, M.; Sanchez, A.; Zhu, D.; Deng, S. Effective depolymerization of polyethylene plastic wastes under hydrothermal and solvothermal liquefaction conditions. Chem. Eng. 2022, 446, 137238. [Google Scholar] [CrossRef]
- Cao, J.; Liang, H.; Yang, J.; Zhu, Z.; Deng, J.; Li, X.; Elimelech, M.; Lu, X. Depolymerization mechanisms and closed-loop assessment in polyester waste recycling. Nat. Commun. 2024, 15, 6266. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Tong, D.; Dong, Y.; Ren, L.; Fang, K.; Zhou, C.; Yu, W. Kaolinite: A natural and stable catalyst for depolymerization of cellulose to reducing sugars in water. Appl. Clay Sci. 2020, 188, 105512. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, X.; Xu, J.; Shi, M.; Wang, F.; Chen, C.; Xu, J. Depolymerization of cellulose to glucose by oxidation–hydrolysis. Green Chem. 2015, 17, 1519–1524. [Google Scholar] [CrossRef]
- Zhou, L.; Gao, D.; Ma, Y.; Li, H.; Su, Y.; Yang, X.; Lu, T. Depolymerization of cellulose promoted by lignin via oxidation-hydrolysis route. Ind. Crops Prod. 2021, 174, 114179. [Google Scholar] [CrossRef]
- Pang, S.C.; Voon, L.K.; Chin, S.F. Controlled depolymerization of cellulose fibres isolated from lignocellulosic biomass wastes. Int. J. Polym. Sci. 2018, 2018, 6872893. [Google Scholar] [CrossRef]
- Luo, X.; Wu, H.; Li, C.; Li, Z.; Li, H.; Zhang, H.; Li, Y.; Su, Y.; Yang, S. Heteropoly acid-based catalysts for hydrolytic depolymerization of cellulosic biomass. Front. Chem. 2020, 8, 580146. [Google Scholar] [CrossRef]
- Negi, A.; Kesari, K.K. Light-driven depolymerization of cellulosic biomass into hydrocarbons. Polymers 2023, 15, 3671. [Google Scholar] [CrossRef]
- Wu, T.; Li, N.; Pan, X.; Chen, S.L. Homogenous hydrolysis of cellulose to glucose in an inorganic ionic liquid catalyzed by zeolites. Cellulose 2020, 27, 9201–9215. [Google Scholar] [CrossRef]
- Brandt, A.; Gräsvik, J.; Hallett, J.P.; Welton, T. Deconstruction of lignocellulosic biomass with ionic liquids. Green Chem. 2013, 15, 550. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, Q.; Sabnis, S.; Zheng, W.; Vlachos, D.G.; Fan, W.; Li, W.; Ma, L. Production of high-yield short-chain oligomers from cellulose via selective hydrolysis in molten salt hydrates and separation. Green Chem. 2019, 21, 5030–5038. [Google Scholar] [CrossRef]
- Paiva, M.F.; Albuquerque, E.M.; de Souza, P.M.; Bitter, J.H.; Vanhove, G.; Wojcieszak, R.; Noronha, F.B. Cellulose depolymerization using zinc chloride hydrate and solid acid catalysts. Cellulose 2024, 31, 7953–7972. [Google Scholar] [CrossRef]
- Deng, W.; Kennedy, J.R.; Tsilomelekis, G.; Zheng, W.; Nikolakis, V. Cellulose hydrolysis in acidified LiBr molten salt hydrate media. Ind. Eng. Chem. Res. 2015, 54, 5226–5236. [Google Scholar] [CrossRef]
- Zhang, Y.R.; Song, Y.N.; Chen, C.Z.; Li, M.F.; Zhang, Z.T.; Fan, Y.M. One-vessel synthesis of 5-hydroxymethylfurfural in concentrated zinc chloride solution from lignocellulosic materials. BioResources 2017, 12, 7807–7818. [Google Scholar] [CrossRef]
- Wei, W.; Wu, S. Depolymerization of cellulose into high-value chemicals by using synergy of zinc chloride hydrate and sulfate ion promoted titania catalyst. Bioresour. Technol. 2017, 241, 760–766. [Google Scholar] [CrossRef]
- Liang, C.; Wang, J.; Du, C.; Hu, P.; Wu, X.; Qu, W.; Xu, J. Comparison of corncob-derived solid acids and evaluation of catalytic cellulose hydrolysis performance in LiBr. Biomass Convers. Bioref. 2024, 14, 2019–2031. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, R.; Yi, W.; Xie, Y.; Wang, M.; Wang, Y.; Cui, H. Efficient catalytic conversion of cellulose to levulinic acid in the biphasic system of molten salt hydrate and methyl isobutyl ketone. Green Chem. 2020, 22, 4240–4251. [Google Scholar] [CrossRef]
- Furusato, S.; Takagaki, A.; Hayashi, S.; Miyazato, A.; Kikuchi, R.; Oyama, S.T. Mechanochemical decomposition of crystalline cellulose in the presence of protonated layered niobium molybdate solid acid catalyst. ChemSusChem 2018, 11, 888–896. [Google Scholar] [CrossRef]
- Karam, A.; De Oliveira Vigier, K.; Marinkovic, S.; Estrine, B.; Oldani, C.; Jérôme, F. Acid Catalysts in Green Chemistry. ChemSusChem 2017, 10, 3604. [Google Scholar] [CrossRef]
- Hick, S.M.; Griebel, C.; Restrepo, D.T.; Truitt, J.H.; Buker, E.J.; Bylda, C.; Blair, R.G. Mechanocatalysis for biomass-derived chemicals and fuels. Green Chem. 2010, 12, 468. [Google Scholar] [CrossRef]
- Yu, P.; Yu, H.; Cheng, J.; Nie, J.; Liu, Y.; Niu, Q.; Yang, Q.; Liu, Y.; Ji, G. Enhancing Enzymatic Hydrolysis of Rice Straw by Acid-Assisted Mechanocatalytic Depolymerization Pretreatment. Agronomy 2024, 14, 2550. [Google Scholar] [CrossRef]
- Lignin Market—By Raw Material, Product, Application, and Forecast 2024–2032. Market Research Report. Available online: https://www.gminsights.com/industry-analysis/lignin-market (accessed on 6 June 2025).
- Fernández-Rodríguez, J.; Erdocia, X.; Sánchez, C.; González Alriols, M.; Labidi, J. Lignin depolymerization for phenolic monomers production by sustainable processes. J. Energy Chem. 2017, 26, 622–631. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef]
- Roy, R.; Jadhav, B.; Rahman, M.S.; Raynie, D.E. Characterization of residue from catalytic hydrothermal depolymerization of lignin. Curr. Res. Green Sustain. Chem. 2021, 4, 100052. [Google Scholar] [CrossRef]
- Rath, S.; Pradhan, D.; Du, H.; Mohapatra, S.; Thatoi, H. Transforming lignin into value-added products: Perspectives on lignin chemistry, lignin-based biocomposites, and pathways for augmenting ligninolytic enzyme production. Adv. Compos. Hybrid Mater. 2024, 7, 27. [Google Scholar] [CrossRef]
- Roy, R.; Rahman, M.S.; Amit, T.A.; Jadhav, B. Recent advances in lignin depolymerization techniques: A comparative overview of traditional and greener approaches. Biomass 2022, 2, 130–154. [Google Scholar] [CrossRef]
- Patel, R.; Dhar, P.; Babaei-Ghazvini, A.; Nikkhah Dafchahi, M.; Acharya, B.B. Transforming lignin into renewable fuels, chemicals, and materials: A review. Bioresour. Technol. Rep. 2023, 22, 101463. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, F. Catalytic Cleavage of Lignin C–O and C–C Bonds. In Advances in Inorganic Chemistry; Ford, P.C., van Eldik, R., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 77, pp. 175–218. ISBN 9780323850582. [Google Scholar]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, F. Catalytic Lignin Depolymerization to Aromatic Chemicals. Acc. Chem. Res. 2020, 53, 470–484. [Google Scholar] [CrossRef]
- Ventura, M.; Domine, M.E.; Chávez-Sifontes, M. Catalytic Processes For Lignin Valorization into Fuels and Chemicals (Aromatics). Curr. Catal. 2019, 8, 45–55. [Google Scholar] [CrossRef]
- Luo, J.; Zhang, X.; Lu, J.; Zhang, J. Fine Tuning the Redox Potentials of Carbazolic Porous Organic Frameworks for Visible-Light Photoredox Catalytic Degradation of Lignin β-O-4 Models. ACS Catal. 2017, 7, 5062–5070. [Google Scholar] [CrossRef]
- Nguyen, S.T.; Murray, P.R.D.; Knowles, R.R. Light-Driven Depolymerization of Native Lignin Enabled by Proton-Coupled Electron Transfer. ACS Catal. 2020, 10, 800–805. [Google Scholar] [CrossRef]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C.C. Hydrolytic depolymerization of hydrolysis lignin: Effects of catalysts and solvents. Bioresour. Technol. 2015, 190, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Wanmolee, W.; Daorattanachai, P.; Laosiripojana, N. Depolymerization of organosolv lignin to valuable chemicals over homogeneous and heterogeneous acid catalysts. Energy Procedia 2016, 100, 173–177. [Google Scholar] [CrossRef]
- Yong, K.J.; Wu, T.Y. Recent advances in the application of alcohols in extracting lignin with preserved β-O-4 content from lignocellulosic biomass. Bioresour. Technol. 2023, 384, 129238. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, L.; Hou, Q.; Mo, Z.; Liu, X.; Li, W. A review on catalytic depolymerization of lignin towards high-value chemicals: Solvent and catalyst. Fermentation 2023, 9, 386. [Google Scholar] [CrossRef]
- Purushothaman, R.K.P.; van Erven, G.; van Es, D.S.; Rohrbach, L.; Frissen, A.E.; van Haveren, J.; Gosselink, R.J.A. New insights into the base catalyzed depolymerization of technical lignins: A systematic comparison. RSC Adv. 2023, 13, 4898. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Qin, J.; Sun, S.; Gao, D.; Fang, Y.; Chen, G.; Tian, C.; Bao, C.; Zhang, S. Progress in developing methods for lignin depolymerization and elucidating the associated mechanisms. Eur. Polym. J. 2024, 210, 112995. [Google Scholar] [CrossRef]
- Li, C.; Jiang, D.; Cheng, X.; Li, H.; He, S.; Mu, M.; Cao, B.; Esakkimuthu, S.; Wang, S. Elucidating reaction mechanisms in the oxidative depolymerization of sodium lignosulfonate for enhancing vanillin production: A Density Functional Theory study. J. Anal. Appl. Pyrolysis 2024, 179, 106499. [Google Scholar] [CrossRef]
- Paananen, H.; Eronen, E.; Mäkinen, M.; Jänis, J.; Suvanto, M.; Pakkanen, T.T. Base-catalyzed oxidative depolymerization of softwood kraft lignin. Ind. Crops Prod. 2020, 152, 112473. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, Y.I.; Um, B.H. Base-catalyzed depolymerization of organosolv lignin into monoaromatic phenolic compounds. Waste Biomass Valor. 2025, 16, 257–270. [Google Scholar] [CrossRef]
- Biswas, B.; Kumar, A.; Krishna, B.B.; Bhaskar, T. Effects of solid base catalysts on depolymerization of alkali lignin for the production of phenolic monomer compounds. Renew. Energy 2021, 175, 270–280. [Google Scholar] [CrossRef]
- Zhang, X.; Li, W.; Wang, J.; Zhang, B.; Guo, G.; Shen, C.; Jiang, Y. Depolymerization of Kraft lignin into liquid fuels over a WO3 modified acid-base coupled hydrogenation catalyst. Fuel 2022, 323, 124428. [Google Scholar] [CrossRef]
- Díaz-Urrutia, C.; Chen, W.; Crites, C.; Daccache, J.; Korobkov, I.; Baker, R.T. Towards lignin valorisation: Comparing homogeneous catalysts for the aerobic oxidation and depolymerisation of organosolv lignin. RSC Adv. 2015, 5, 70502. [Google Scholar] [CrossRef]
- Wang, S.; Li, X.; Ma, R.; Song, G. Catalytic Hydrogenolysis of Lignin into Serviceable Products. Acc. Chem. Res. 2025, 58, 529–542. [Google Scholar] [CrossRef]
- Kaur, P.; Singh, G.; Arya, S.K. Tandem catalytic approaches for lignin depolymerization: A review. Biomass Convers. Biorefin. 2024, 14, 6143–6154. [Google Scholar] [CrossRef]
- Ji, D.; Wang, Y.; Peng, J.; Yuan, D.; Li, Z.; Ji, D.; Wu, H. Recent Advances in Depolymerization and High-Value Utilization of Lignin: A Review. Ind. Eng. Chem. Res. 2024, 63, 19916–19935. [Google Scholar] [CrossRef]
- Khan, A.; Evans, L.W.; Martin, D. Visible light-driven ligand-to-metal charge transfer-mediated selective cleavage of β-O-4 lignin model compounds: A greener route to lignin valorization. Green Chem. 2025, 27, 4664. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, H.; Niu, M.; Guo, Y.; Li, H. Research progress on vanillin synthesis by catalytic oxidation of lignin: A review. Ind. Crops Prod. 2024, 214, 118443. [Google Scholar] [CrossRef]
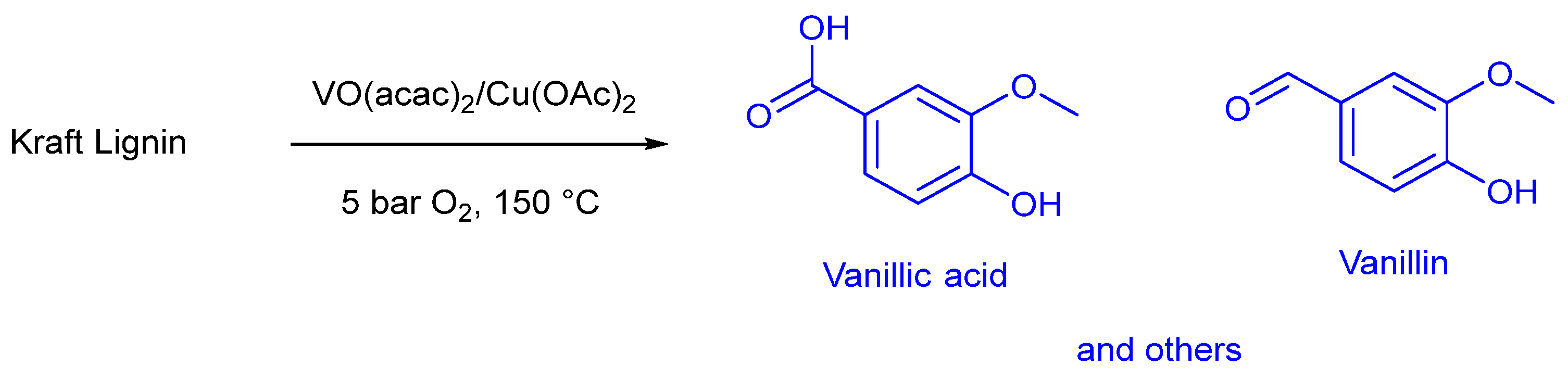
- Walch, F.; Abdelaziz, O.Y.; Meier, S.; Bjelić, S.; Hulteberg, C.P.; Riisager, A. Oxidative depolymerization of Kraft lignin to high-value aromatics using a homogeneous vanadium–copper catalyst. Catal. Sci. Technol. 2021, 11, 1843–1853. [Google Scholar] [CrossRef]
- Abdelaziz, O.Y.; Clemmensen, I.; Meier, S.; Walch, F.; Bjelić, S.; Riisager, A. Oxidative Depolymerization of Kraft Lignin to Aromatics Over Bimetallic V–Cu/ZrO2 Catalysts. Top. Catal. 2023, 66, 1369–1380. [Google Scholar] [CrossRef]
- Martinez, D.V.; Rodriguez, A.; Juarros, M.A.; Martinez, E.J.; Alam, T.M.; Simmons, B.A.; Sale, K.L.; Singer, S.W.; Kent, M.S. Depolymerization of lignin for biological conversion through sulfonation and a chelator-mediated Fenton reaction. Green Chem. 2022, 24, 1627–1640. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, L.; Jiang, Y.; Liu, X.; Wang, J.; Tang, Z. Recent advances in the acid-catalyzed conversion of lignin. Biomass Convers. Biorefin. 2023, 13, 519–539. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, C.; Chee, P.L.; Qu, C.; Fok, A.Z.; Yong, F.H.; Ong, Z.L.; Kai, D. Strategies for lignin depolymerization and reconstruction towards functional polymers. Sustain. Energy Fuels 2023, 7, 2953–2964. [Google Scholar] [CrossRef]
- Zhang, B.; Li, W.; Li, X. The selectivity depolymerization of corn stover lignin via nickel-doped tin phosphate catalyst in the absence of hydrogen. Ind. Crops Prod. 2021, 174, 114211. [Google Scholar] [CrossRef]
- Zhang, C.; Shen, X.; Jin, Y.; Cheng, J.; Cai, C.; Wang, F. Catalytic strategies and mechanism analysis orbiting the center of critical intermediates in lignin depolymerization. Chem. Rev. 2023, 123, 4510–4601. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Tsang, C.W.; Lin, C.S.K.; Len, C.; Hu, H.; Liang, C. A review on high catalytic efficiency of solid acid catalysts for lignin valorization. Bioresour. Technol. 2020, 298, 122432. [Google Scholar] [CrossRef] [PubMed]
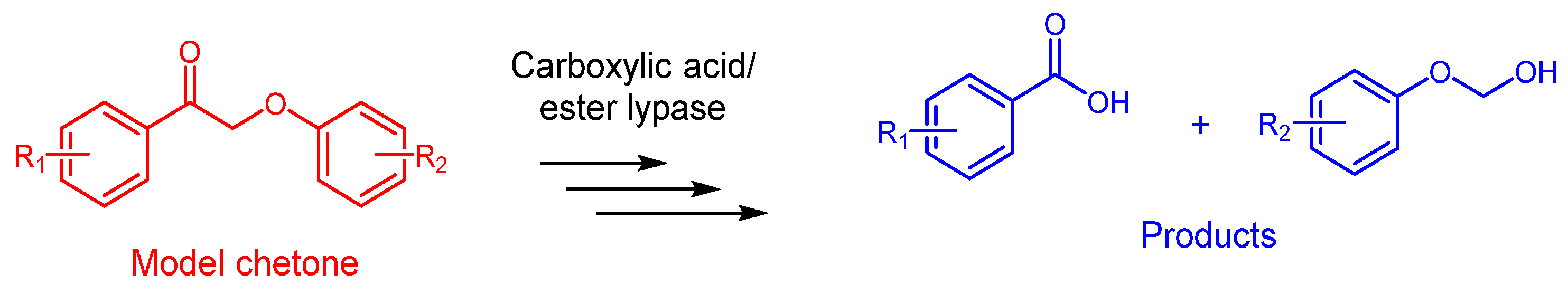
- Zeng, J.; Mills, M.J.L.; Simmons, B.A.; Kent, M.S.; Sale, K.L. Understanding factors controlling depolymerization and polymerization in catalytic degradation of β-ether linked model lignin compounds by versatile peroxidase. Green Chem. 2017, 19, 2145–2151. [Google Scholar] [CrossRef]
- Ma, H.; Li, H.; Zhao, W.; Li, L.; Liu, S.; Long, J.; Li, X. Selective depolymerization of lignin catalyzed by nickel supported on zirconium phosphate. Green Chem. 2019, 21, 658–667. [Google Scholar] [CrossRef]
- Li, Z.; Bai, X.; Wei, X.Y.; Dilixiati, Y.; Fan, Z.C.; Kong, Q.Q.; Li, L.; Li, J.H.; Lu, K.L.; Zhao, J.; et al. A solid acid-catalyzed depolymerization of pine lignin to obtain guaiacol using a hydrogen-free strategy. Fuel Process. Technol. 2023, 249, 107843. [Google Scholar] [CrossRef]
- Knill, C.J.; Kennedy, J.F. Degradation of cellulose under alkaline conditions. Carbohydr. Polym. 2003, 51, 281–300. [Google Scholar] [CrossRef]
- Chen, R.; Huang, Y.; Rao, C.; Su, H.; Pang, Y.; Lou, H.; Yang, D.; Qiu, X. Enhanced Photocatalytic Degradation of Lignin by In2s3 with Hydrophobic Surface and Metal Defects. Appl. Surf. Sci. 2022, 600, 154110. [Google Scholar] [CrossRef]
- Pei, Z.; Liu, X.; Chen, J.; Wang, H.; Li, H. Research Progress on Lignin Depolymerization Strategies: A Review. Polymers 2024, 16, 2388. [Google Scholar] [CrossRef]
- Shen, Z.; Shi, C.; Liu, F.; Wang, W.; Ai, M.; Huang, Z.; Zhang, X.; Pan, L.; Zou, J.J. Advances in heterogeneous catalysts for lignin hydrogenolysis. Adv. Sci. 2024, 11, 2306693. [Google Scholar] [CrossRef]
- Yu, Z.; Kong, W.; Liang, W.; Guo, Y.; Cui, J.; Hu, Y.; Sun, Z.; Elangovan, S.; Xu, F. Heterogeneously catalyzed reductive depolymerization of lignin to value-added chemicals. ChemSusChem 2025, 18, e202401399. [Google Scholar] [CrossRef]
- Kumi, D.O.; Phaahlamohlaka, T.N.; Dlamini, M.W.; Mangezvo, I.T.; Mhlanga, S.D.; Scurrell, M.S.; Coville, N.J. Effect of a titania covering on CNTS as support for the Ru catalysed selective CO methanation. Appl. Catal. B 2018, 232, 492–500. [Google Scholar] [CrossRef]
- Biswas, B.; Kumar, A.; Krishna, B.B.; Baltrusaitis, J.; Adhikari, S.; Bhaskar, T. Catalytic depolymerization of lignin for the selective production of phenolic monomers over cobalt-supported calcium catalysts. Energy Fuels 2023, 37, 3813–3824. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, Y.; Song, W.; He, Z.; Yao, R.; Deng, Z. Metal supported hydrotalcite catalytic depolymerization of lignin: Effects of reaction parameter on compounds distribution. Fuel 2023, 340, 127559. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Barta, K.; Beckham, G.T.; Luterbacher, J.S.; Ralph, J.; Rinaldi, R.; Román-Leshkov, Y.; Samec, J.S.M.; Sels, B.F.; Wang, F. Guidelines for performing lignin-first biorefining. Energy Environ. Sci. 2021, 14, 262–292. [Google Scholar] [CrossRef]
- Su, S.; Xiao, L.P.; Chen, X.; Wang, S.; Chen, X.H.; Guo, Y.; Zhai, S.R. Lignin-first depolymerization of lignocellulose into monophenols over carbon nanotube-supported ruthenium: Impact of lignin sources. ChemSusChem 2022, 15, e202200365. [Google Scholar] [CrossRef]
- Hatakka, A. Lignin-modifying enzymes from selected white-rot fungi: Production and role from in lignin degradation. FEMS Microbiol. Rev. 1994, 13, 125–135. [Google Scholar] [CrossRef]
- Hofrichter, M. lignin conversion by manganese peroxidase (MnP). Enzym. Microb. Technol. 2002, 30, 454–466. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, R.; Xin, Y.; Li, Y.; Shi, G.; Zhu, R.; Zhang, L. Enhancing lignin model compound depolymerization using mediator-enzyme catalysis: A sustainable approach to C–C bond cleavage. ACS Sustain. Chem. Eng. 2024, 12, 5842–5849. [Google Scholar] [CrossRef]
- Dillies, J.; Vivien, C.; Chevalier, M.; Rulence, A.; Châtaigné, G.; Flahaut, C.; Senez, V.; Froidevaux, R. Enzymatic depolymerization of industrial lignins by laccase-mediator systems in 1, 4-dioxane/water. Biotechnol. Appl. Biochem. 2020, 67, 774–782. [Google Scholar] [CrossRef]
- Shorey, R.; Salaghi, A.; Fatehi, P.; Mekonnen, T.H. Valorization of lignin for advanced material applications: A review. RSC Sustain. 2024, 2, 804. [Google Scholar] [CrossRef]
- Kim, D.H.; Han, D.O.; Shim, K.I.; Kim, J.K.; Pelton, J.G.; Ryu, M.H.; Joo, J.C.; Han, J.W.; Kim, H.T.; Kim, K.H. One-pot chemo-bioprocess of PET depolymerization and recycling enabled by a biocompatible catalyst, betaine. ACS Catal. 2021, 11, 3996–4008. [Google Scholar] [CrossRef]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.L.; Texier, H.; Gavalda, S.; et al. An engineered PET depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Rorrer, N.A.; Nicholson, S.R.; Erickson, E.; DesVeaux, J.S.; Avelino, A.F.T.; Lamers, P.; Bhatt, A.; Zhang, Y.; Avery, G.; et al. Techno-economic, life-cycle, and socioeconomic impact analysis of enzymatic recycling of poly (ethylene terephthalate). Joule 2021, 5, 2479–2503. [Google Scholar] [CrossRef]
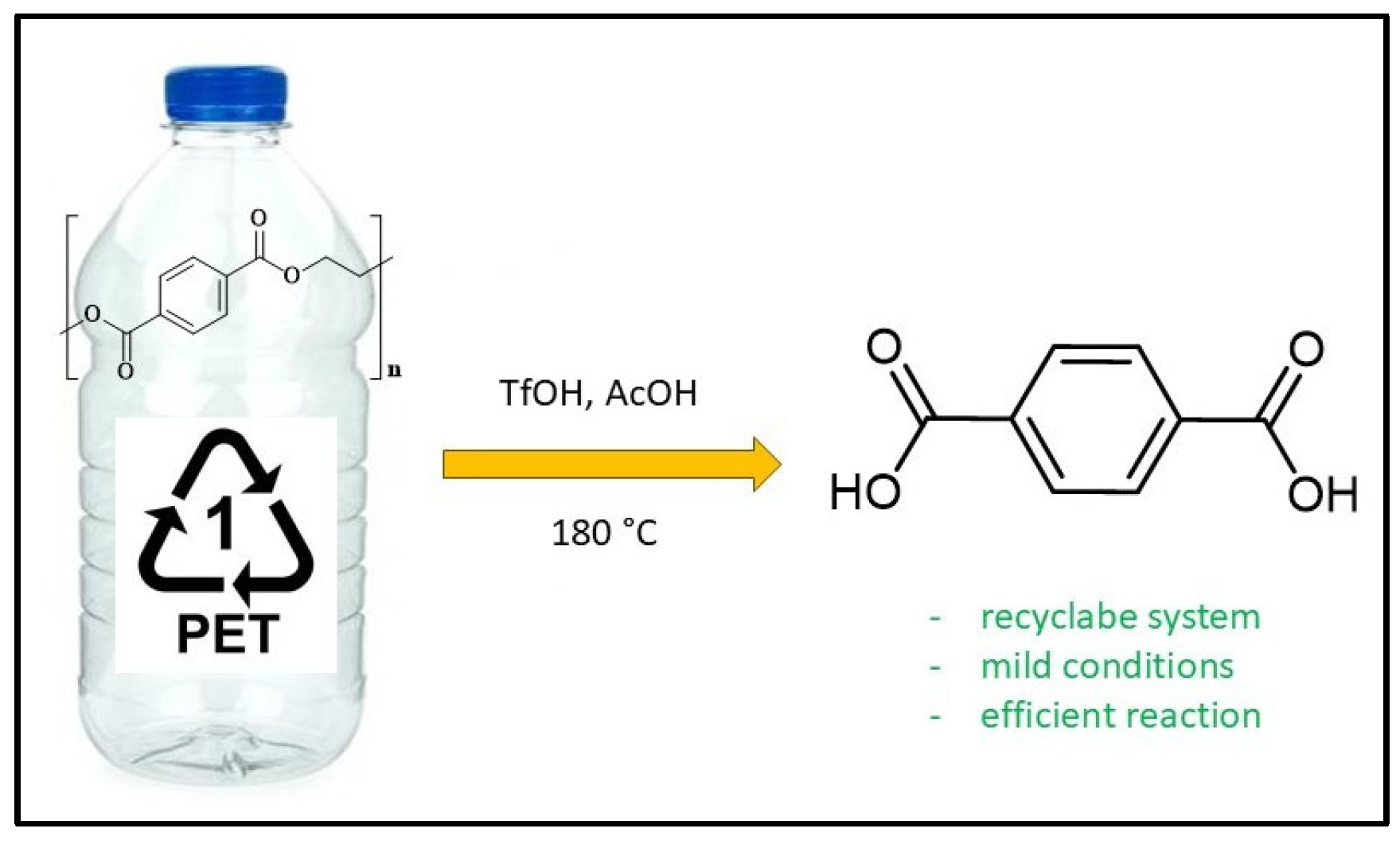
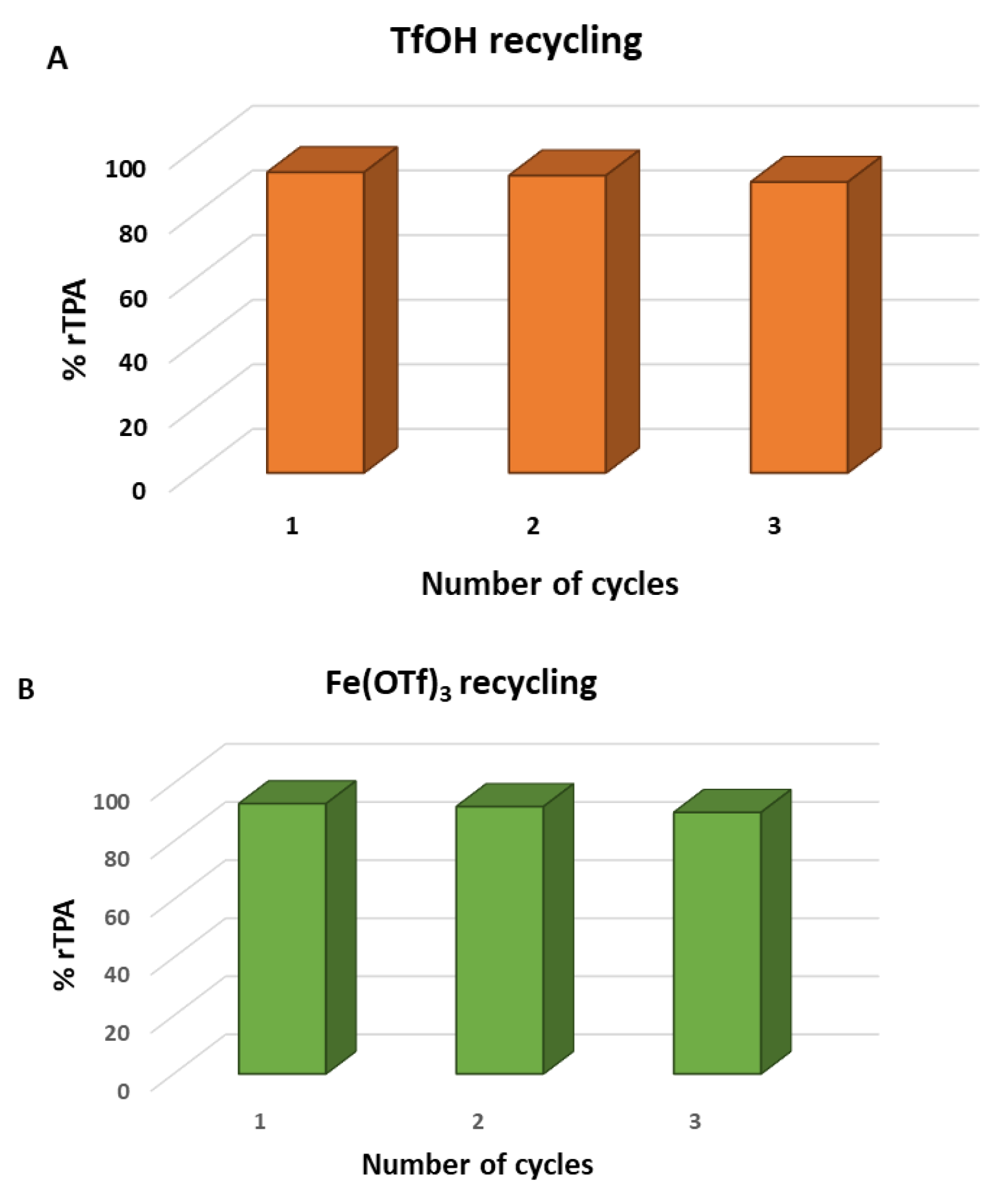
- Luo, Y.; Sun, J.; Li, Z. Rapid chemical recycling of waste polyester plastics catalyzed by recyclable catalyst. Green Chem. Eng. 2024, 5, 257–265. [Google Scholar] [CrossRef]
- Tanaka, S.; Koga, M.; Kuragano, T.; Ogawa, A.; Ogiwara, H.; Sato, K.; Nakajima, Y. Depolymerization of polyester fibers with dimethyl carbonate-aided methanolysis. ACS Mater. Au 2024, 4, 335–345. [Google Scholar] [CrossRef]
- Li, H.; Aguirre-Villegas, H.A.; Allen, R.D.; Bai, X.; Benson, C.H.; Beckham, G.T.; Bradshaw, S.L.; Brown, J.L.; Brown, R.C.; Cecon, V.S.; et al. Expanding plastics recycling technologies: Chemical aspects, technology status and challenges. Green Chem. 2022, 24, 8899–9002. [Google Scholar] [CrossRef]
- Majumdar, A.; Shukla, S.; Singh, A.A.; Arora, S. Circular fashion: Properties of fabrics made from mechanically recycled poly-ethylene terephthalate (PET) bottles. Resour. Conserv. Recycl. 2020, 161, 104915. [Google Scholar] [CrossRef]
- Tanaka, S.; Sato, J.; Nakajima, Y. Capturing ethylene glycol with dimethyl carbonate towards depolymerisation of polyethylene terephthalate at ambient temperature. Green Chem. 2021, 23, 9412–9416. [Google Scholar] [CrossRef]
- Bai, X.; Aireddy, D.R.; Roy, A.; Ding, K. Solvent-Free Depolymerization of Plastic Waste Enabled by Plastic-Catalyst Interfacial Engineering. Angew. Chem. Int. Ed. 2023, 62, e202309949. [Google Scholar] [CrossRef]
- Gordy, W.; Thomas, W.J.O. Electronegativities of the elements. J. Chem. Phys. 1956, 24, 439–444. [Google Scholar] [CrossRef]
- Chang, T.; Wang, Y.; Li, Z.; Hu, Y.; Shi, N.; Zou, X.; Li, C.; Xu, Y.; Li, N.; Guo, K. Double metal cyanide complex as a heterogeneous catalyst for depolymerization of polyesters. Catal. Today 2025, 452, 115241. [Google Scholar] [CrossRef]
- Rodríguez, M.P.; Vázquez-Vélez, E.; Galván-Hernández, A.; Martinez, H.; Torres, A. Surface modification of the Nylon 6, 6 and wasted glass fiber-Nylon 6.6 coatings using atmospheric plasma treatment. J. Appl. Polym. Sci. 2023, 140, e53763. [Google Scholar] [CrossRef]
- Shakiba, M.; Rezvani Ghomi, E.; Khosravi, F.; Jouybar, S.; Bigham, A.; Zare, M.; Abdouss, M.; Moaref, R.; Ramakrishna, S. Nylon—A material introduction and overview for biomedical applications. Polym. Adv. Technol. 2021, 32, 3368–3383. [Google Scholar] [CrossRef]
- Castanha, N.; Balardin, F.; Dantas, H.; Marangoni Júnior, L. Mechanical recycling of pol yamides: A review. Trends Food Sci. Technol. 2025, 162, 105107. [Google Scholar] [CrossRef]
- Zhou, W.; Neumann, P.; Al Batal, M.; Rominger, F.; Hashmi, A.S.K.; Schaub, T. Depolymerization of Technical-Grade Polyamide 66 and Polyurethane Materials through Hydrogenation. ChemSusChem 2021, 14, 4176–4183. [Google Scholar] [CrossRef]
- Coeck, R.; De Vos, D.E. Effective and sustainable depolymerization of Nylon 66–a transamidation for the complete recycling of polyamides. Chem. Commun. 2024, 60, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, G.L.; Short, R.D. Biological effects of acetamide, formamide, and their monomethyl and dimethyl derivatives. CRC Crit. Rev. Toxicol. 1986, 17, 129–182. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Masunaga, H.; Numata, K. Chemoenzymatic Polymerization/Depolymerization of Semiaromatic Polyamides for Chemical Recycling. ACS Sustain. Chem. Eng. 2025, 13, 3994–4004. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, G.; Zhu, X.; Zhang, Y.; Liang, C.; Chen, Y. Sulfonated Fe-MOF Solid Acid Catalyst for High-Efficient Depolymerization of Polyamide 6. J. Polym. Sci. 2025, 63, 1641–1649. [Google Scholar] [CrossRef]
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane types, synthesis and applications–a review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef]
- Maiuolo, L.; Olivito, F.; Ponte, F.; Algieri, V.; Tallarida, M.A.; Tursi, A.; Chidichimo, G.; Sicilia, E.; De Nino, A. A novel catalytic two-step process for the preparation of rigid polyurethane foams: Synthesis, mechanism and computational studies. React. Chem. Eng. 2021, 6, 1238–1245. [Google Scholar] [CrossRef]
- Olivito, F.; Algieri, V.; Jiritano, A.; Tallarida, M.A.; Costanzo, P.; Maiuolo, L.; De Nino, A. Bio-based polyurethane foams for the removal of petroleum-derived pollutants: Sorption in batch and in continuous-flow. Polymers 2023, 15, 1785. [Google Scholar] [CrossRef]
- Olivito, F.; Jagdale, P.; Oza, G. Synthesis and Biodegradation Test of a New Polyether Polyurethane Foam Produced from PEG 400, L-Lysine Ethyl Ester Diisocyanate (L-LDI) and Bis-hydroxymethyl Furan (BHMF). Toxics 2023, 11, 698. [Google Scholar] [CrossRef]
- Mouren, A.; Avérous, L. Aromatic thermoplastic polyurethanes synthesized from different potential sustainable resources. Eur. Polym. J. 2023, 197, 112338. [Google Scholar] [CrossRef]
- Corapi, A.; Gallo, L.; Lucadamo, L.; Tursi, A.; Chidichimo, G. Evaluation of the ecotoxicity of new polyurethane composites on target organisms for aquatic and atmospheric environments. Environ. Toxicol. Chem. 2023, 42, 421–436. [Google Scholar] [CrossRef]
- Zubar, V.; Haedler, A.T.; Schütte, M.; Hashmi, A.S.K.; Schaub, T. Hydrogenative depolymerization of polyurethanes catalyzed by a manganese pincer complex. ChemSusChem 2022, 15, e202101606. [Google Scholar] [CrossRef] [PubMed]
- Luleburgaz, S.; Tunca, U.; Durmaz, H. Polyether Synthesis through Reductive Etherification Reaction Strategy. Macromol. Chem. Phys. 2023, 224, 2300063. [Google Scholar] [CrossRef]
- Li, X.; Yu, Z.; Zhang, L. Synthesis of a green reactive flame-retardant polyether polyol and its application. J. Appl. Polym. Sci. 2021, 138, e50154. [Google Scholar] [CrossRef]
- Maeno, Z.; Yamada, S.; Mitsudome, T.; Mizugaki, T.; Jitsukawa, K. Effective management of polyethers through depolymerization to symmetric and unsymmetric glycol diesters using a proton-exchanged montmorillonite catalyst. Green Chem. 2017, 19, 2612–2619. [Google Scholar] [CrossRef]
- Ansmann, N.; Thorwart, T.; Greb, L. Silicon Catalyzed C− O Bond Ring Closing Metathesis of Polyethers. Angew. Chem. Int. Ed. 2022, 61, e202210132. [Google Scholar] [CrossRef]
- Tschernuth, F.S.; Bichlmaier, L.; Inoue, S. Catalytic degradation of aliphatic ethers using the Lewis superacidic bis (perfluoropinacolato) silane. ChemCatChem 2023, 15, e202300281. [Google Scholar] [CrossRef]
- Ansmann, N.; Johann, K.; Favresse, P.; Johann, T.; Fiedel, M.; Greb, L. Silicon-Catalyzed Depolymerization of Polyethers: Pushing Scope, Practicability and Mechanistic Understanding. ChemCatChem 2024, 16, e202301615. [Google Scholar] [CrossRef]



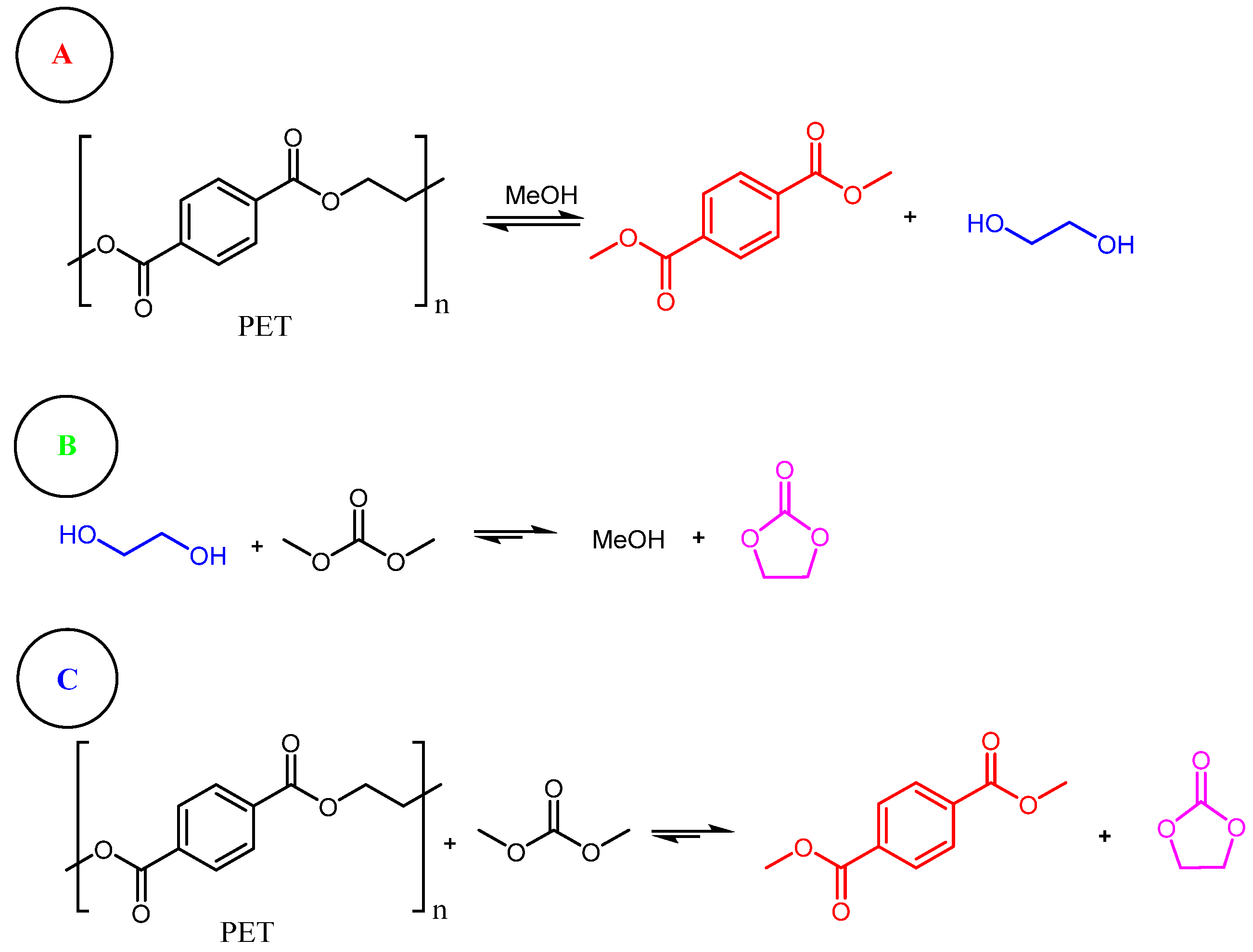

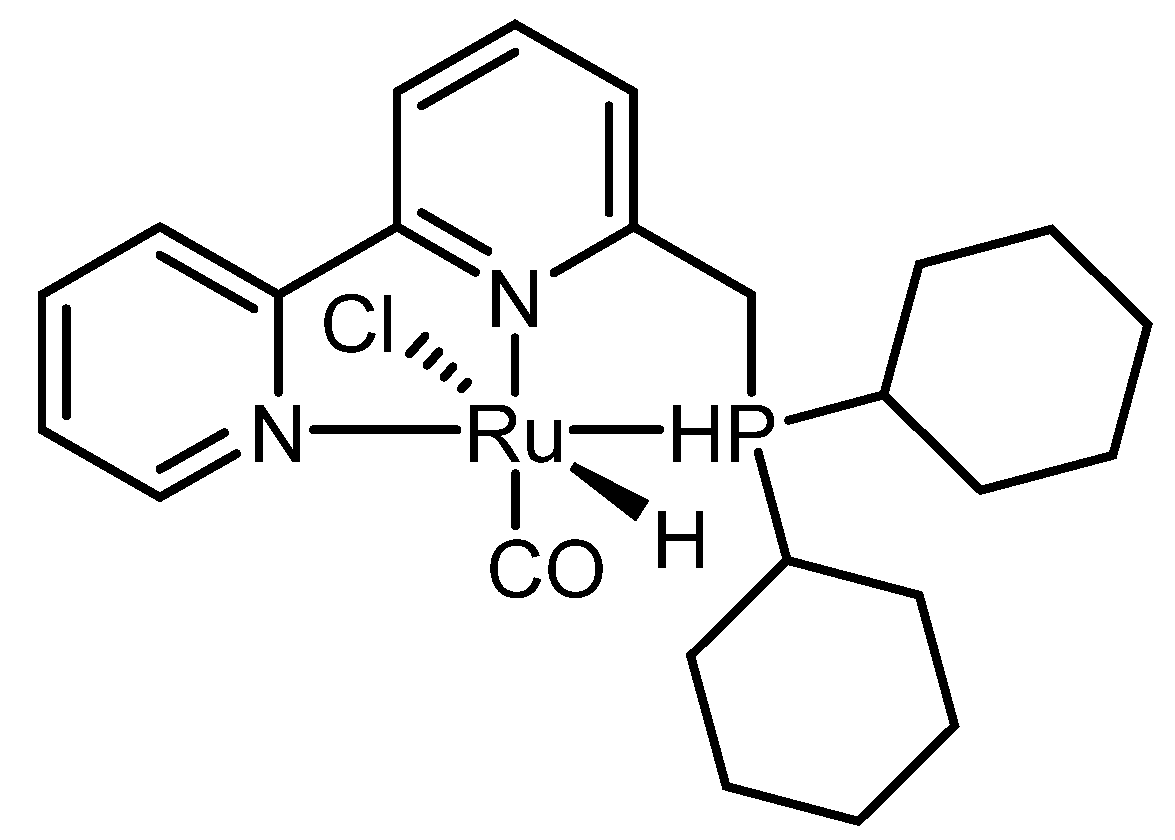
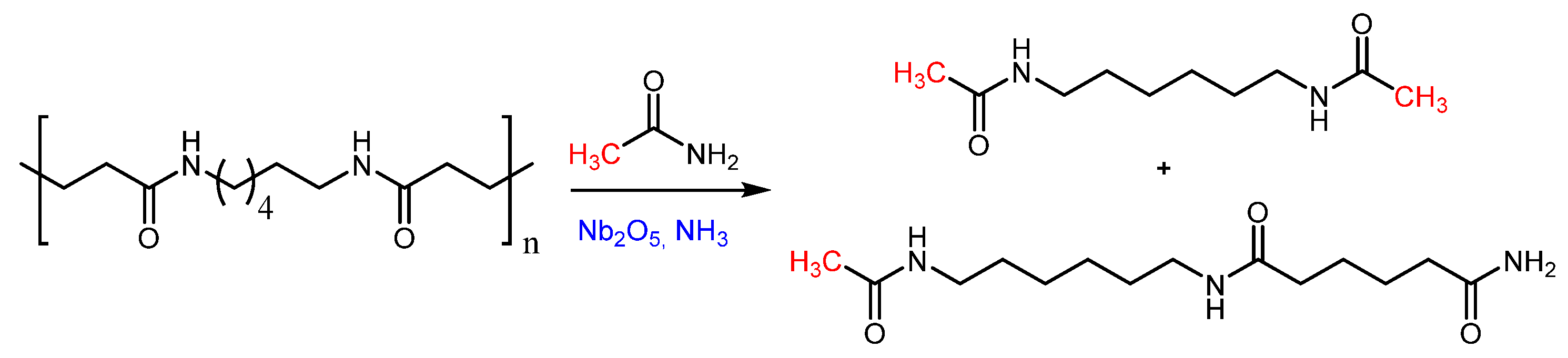
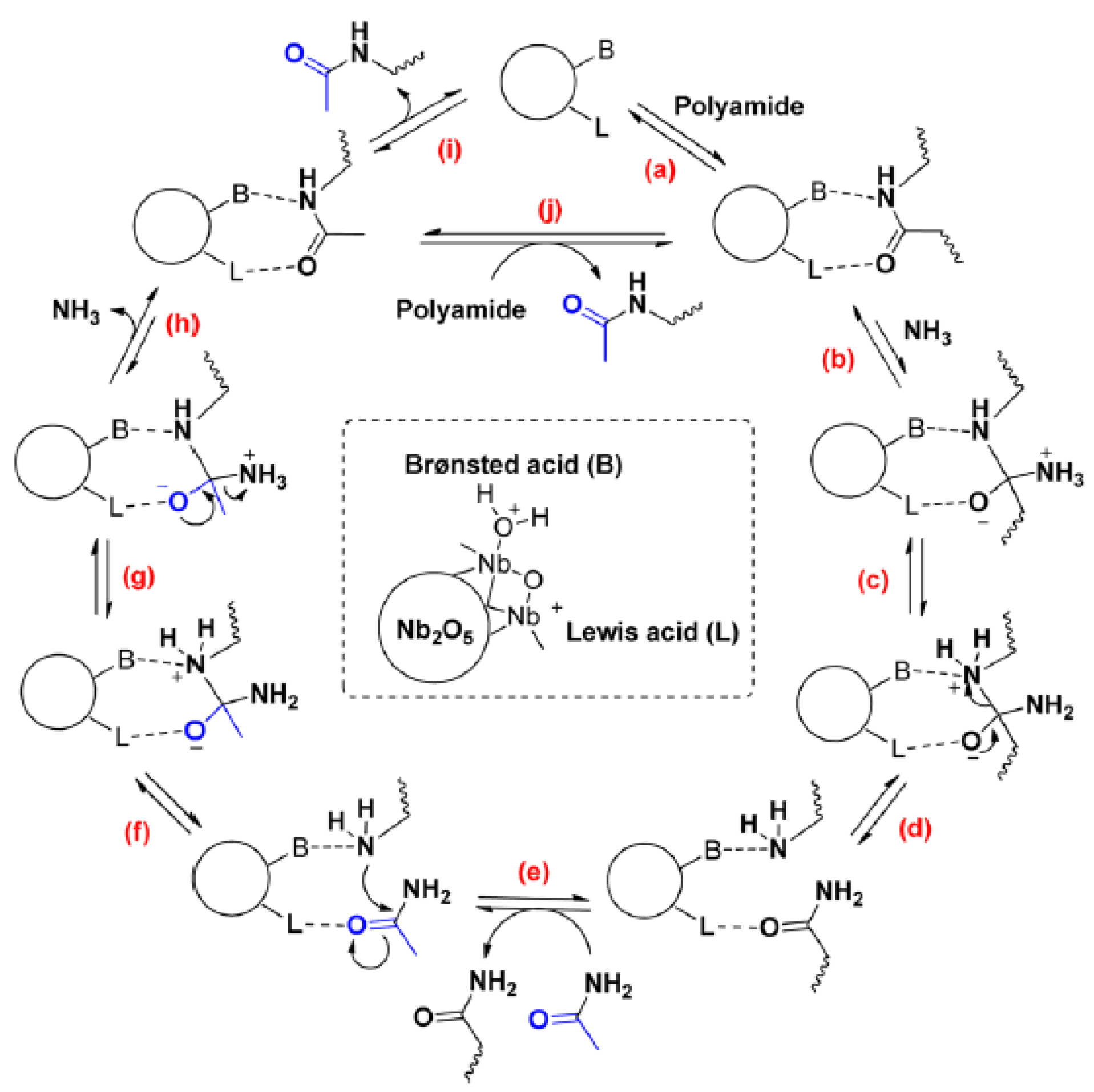

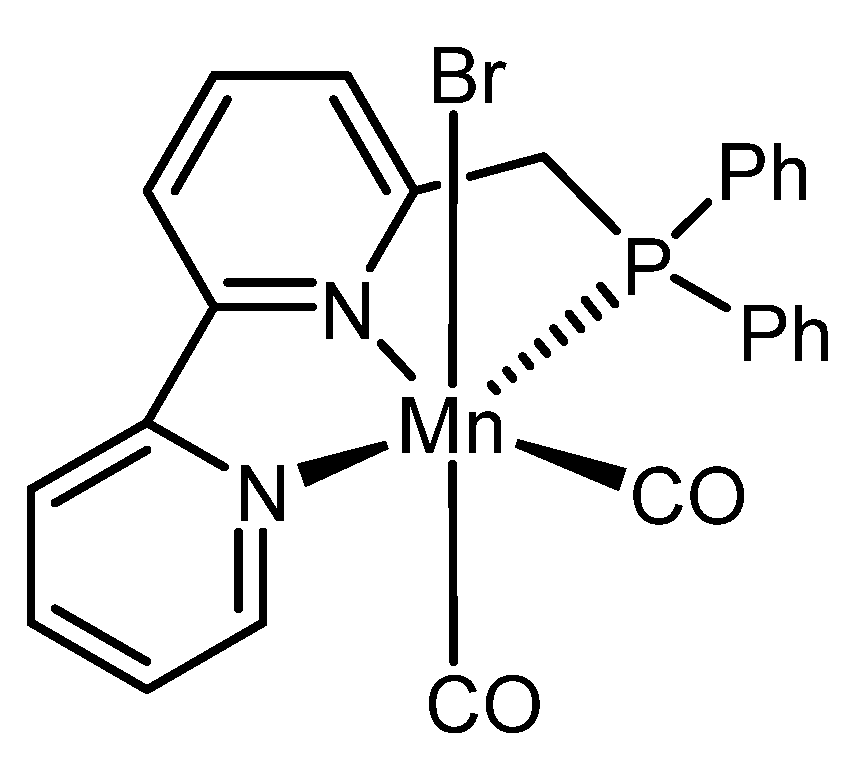


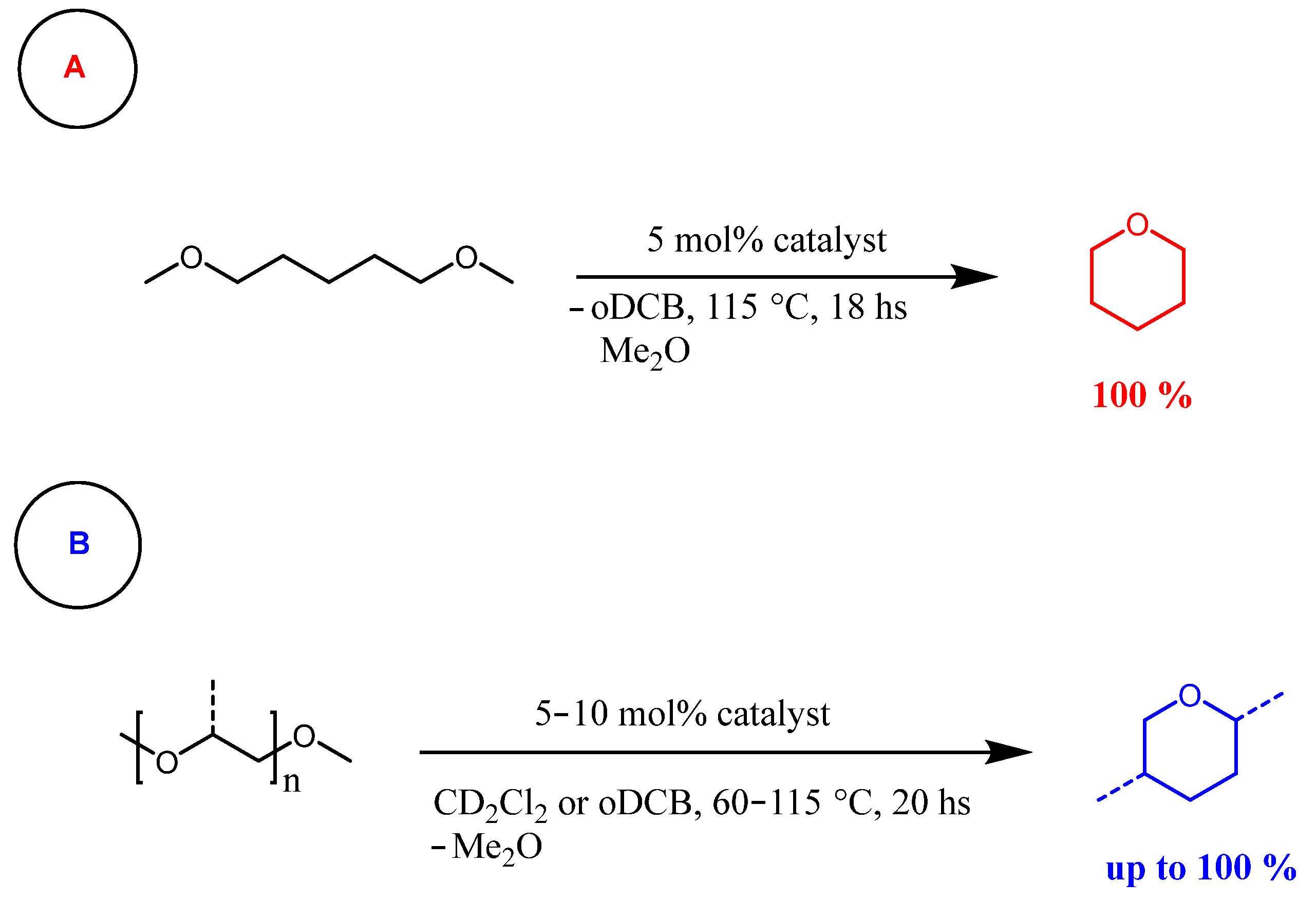
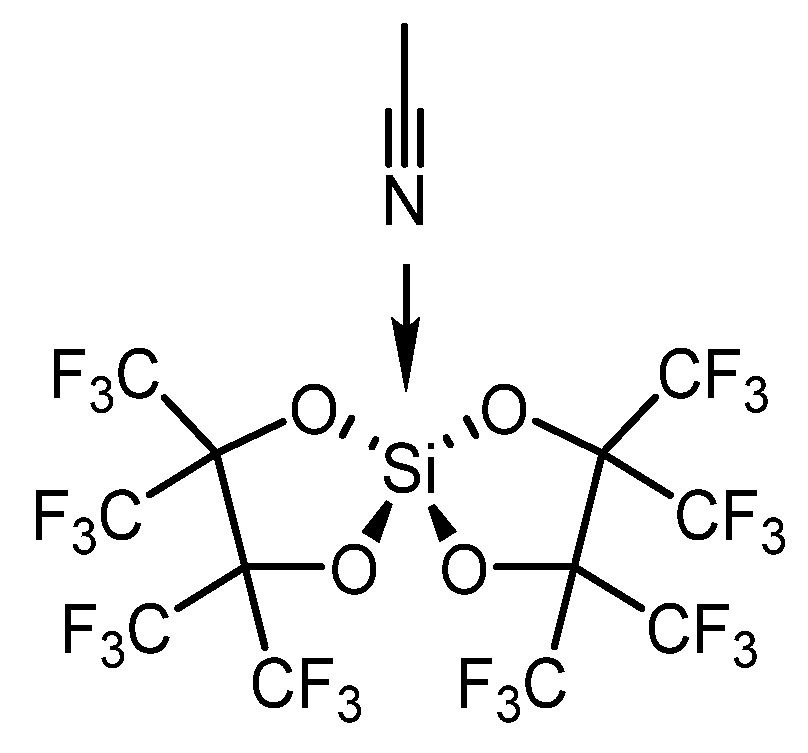

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Cellulose | Mean DPw Peak 0 min and 120 min | Percentage Change of DPw ΔDPw (%) | |
|---|---|---|---|
| Sawdust | 351 | 72 | 79.5 |
| Sago pith wastes | 564 | 30 | 94.7 |
| Corn Cob | 329 | 88 | 78.1 |
| Sugarcane bagasse | 571 | 37 | 93.5 |
| Substrate | MSH/Catalyst | Main Products (%Yield) | Ref. |
|---|---|---|---|
| MCC | Beta and ZSM-5 zeolites with SiO2/Al2O3 = 30 and 50 | Gluco oligomers (54.4%), glucose (28.3%) & HMF (0.2%) | [52] |
| MCC | ZnCl2 (72 wt%)/HCl (0.2 M) | HMF (69.5%) | [57] |
| Cellulose | ZnCl2·3H2O/SO4/TiO2 | Gluco oligomers (9.4%), glucose (50.5%), HMF (3.4%), fructose (5.9%), & LA (5.1%) | [58] |
| MCC | LiBr (55 wt%) Activated/Activated carbon | Glucose (80%) & LA (4%) | [59] |
| Catalyst | H+ Exchange Capacity (mmol/g) | Solubility (%) |
|---|---|---|
| Blank | - | <5 |
| Aquivion PW98 | 1.0 | 90 |
| SBA–SO3H | 0.2 | 60 |
| CMK-3-SO3H | 0.7 | 87 |
| Kaolinite | - | 50 |
| Aquivion PW66 | 1.45 | 99 |
| Aquivion PW79 | 1.26 | 32 |
| Aquivion PW87 | 1.15 | 80 |
| Material | Diameter (mm) | Density (g/cm−3) |
|---|---|---|
| Zirconia | 3 | 5.68 |
| Stainless steel | 2 | 7.8 |
| Stainless steel | 4 | 7.8 |
| Tungsten carbide | 3 | 15.63 |
| Catalyst | Products Yield (%) |
|---|---|
| HNbMoO6 | 14 |
| kaolinite | 4 |
| NiO | 0.3 |
| SnO2 | 0.6 |
| TiO2 | 0.5 |
| Nb2O5 | 0.9 |
| H-Montmorillonite | 3 |
| USY zeolite | 3 |
| Mg–Al HT | 0 |
| Catalysis Type | Advantages | Limitations | References |
|---|---|---|---|
| Homogeneous Acid Catalysis | High reactivity, low cost | Low selectivity, harsh conditions, byproduct formation, corrosivity, toxic reagents | [78,79,80,81] |
| Homogeneous Base Catalysis | High efficiency at moderate temperatures, potential for vanillin production | Solvent dependency, limited performance in aqueous media | [87] |
| Combined Acid–Base Catalysis | Effective stabilization of intermediates, dual activation | Requires careful optimization of multi-functional catalysts | [88] |
| Homogeneous Metal Catalysis | High activity and selectivity, tunable ligands, reusable on supports | High cost, elevated T/P needed, sensitivity to reaction conditions | [95,96] |
| Catalysis Type | Advantages | Disadvantages | References |
|---|---|---|---|
| Heterogeneous–Solid Acid | Regenerable, high conversion, environmentally sustainable | Complex selectivity; possible char formation | [97,98,99,100,101,102,103,104,105] |
| Heterogeneous–Metal-Supported | High activity, large surface area, good selectivity, reusable | Sensitive to metal type and support; potential for deactivation | [106,107,108,109,110,111,112,113,114,115] |
 | ||||
|---|---|---|---|---|
| Catalyst | T (°C) | Ccat (M) | CH20 (M) | Yield (%) |
| Hf(OTf)4 | 180 | 0.25 | 0 | 60 |
| Hf(OTf)4 | 180 | 0.25 | 0.5 | 72 |
| Fe(OTf)3 | 180 | 0.25 | 0.5 | 64 |
| Al(OTf)3 | 180 | 0.25 | 0.5 | 60 |
| TfOH | 180 | 0.25 | 0.5 | 98 |
| Tf2NH | 180 | 0.25 | 0.5 | 58 |
| Catalyst | DMC (mL) | MeOH (mL) | DMT (%) | EC (%) |
|---|---|---|---|---|
| LiOMe | 1.5 | 0.2 | 83 | 70 |
| KOMe | 1.5 | 0.2 | 95 | 93 |
| NaOMe | 1.5 | 0.2 | 95 | 86 |
| NaOMe [b] | 1.5 | 0.2 | 93 | 92 |
| NaOMe | 1 | 0.13 | 98 | 67 |
| NaOMe | 0.5 | 0.065 | 93 | 60 |
 | |||
|---|---|---|---|
| T (°C) | P (H2) Bar | Yield (%) Diamine | Yield (%) Diol |
| 150 | 70 | 12 | <5 |
| 180 | 100 | 60 | 35 |
| 200 | 100 | 78 | 62 |
| 200 | 80 | 70 | 47 |
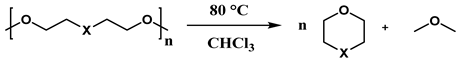 | |||
|---|---|---|---|
| Substrate | Catalyst (%mol) | Time (h) | Yield (%) |
| 1,5 DMP | 1–5 | 1–20 | 96–97 |
| Diglyme | 1–5 | 2.5–96 | 96–99 |
| 18-crown-6 | 30 | 30 | 88 |
| PEG-DME | 225 | 18 | 87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oza, G.; Olivito, F.; Rohokale, A.; Nardi, M.; Procopio, A.; Wan-Mohtar, W.A.A.Q.I.; Jagdale, P. Advancements in Catalytic Depolymerization Technologies. Polymers 2025, 17, 1614. https://doi.org/10.3390/polym17121614
Oza G, Olivito F, Rohokale A, Nardi M, Procopio A, Wan-Mohtar WAAQI, Jagdale P. Advancements in Catalytic Depolymerization Technologies. Polymers. 2025; 17(12):1614. https://doi.org/10.3390/polym17121614
Chicago/Turabian StyleOza, Goldie, Fabrizio Olivito, Apurva Rohokale, Monica Nardi, Antonio Procopio, Wan Abd Al Qadr Imad Wan-Mohtar, and Pravin Jagdale. 2025. "Advancements in Catalytic Depolymerization Technologies" Polymers 17, no. 12: 1614. https://doi.org/10.3390/polym17121614
APA StyleOza, G., Olivito, F., Rohokale, A., Nardi, M., Procopio, A., Wan-Mohtar, W. A. A. Q. I., & Jagdale, P. (2025). Advancements in Catalytic Depolymerization Technologies. Polymers, 17(12), 1614. https://doi.org/10.3390/polym17121614