
Synthesis of Polyether, Poly(Ether Carbonate) and Poly(Ether Ester) Polyols Using Double Metal Cyanide Catalysts Bearing Organophosphorus Complexing Agents


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of DMC Catalysts Bearing OPCs
2.3. General Procedure for the Semi-Batch ROP of PO
2.4. General Procedure for the Batch ROP of CL
2.5. General Procedure for the Batch Copolymerization of PO with CO2
2.6. Characterization
3. Results and Discussion
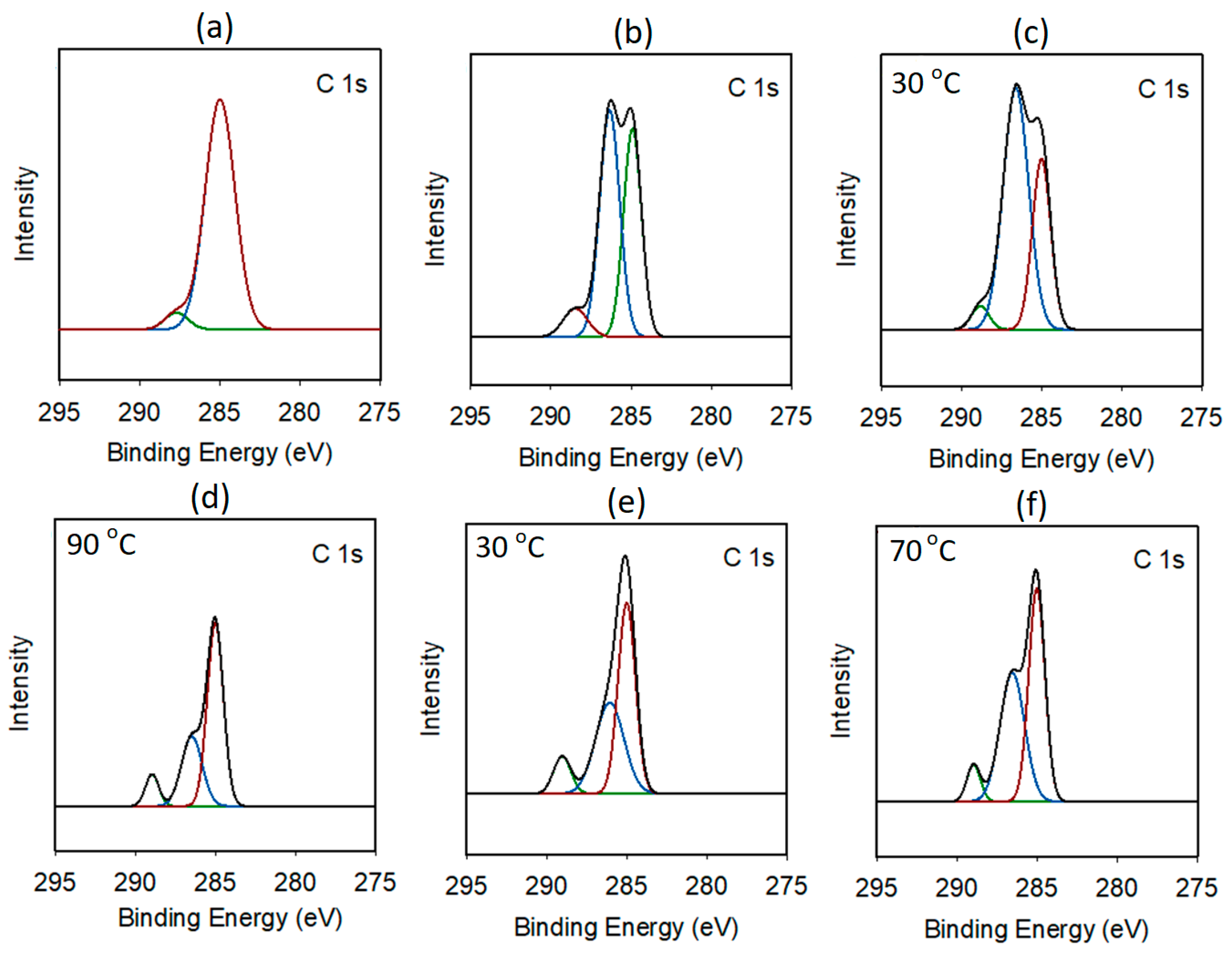
3.1. Preparation and Characterization of DMC Catalysts
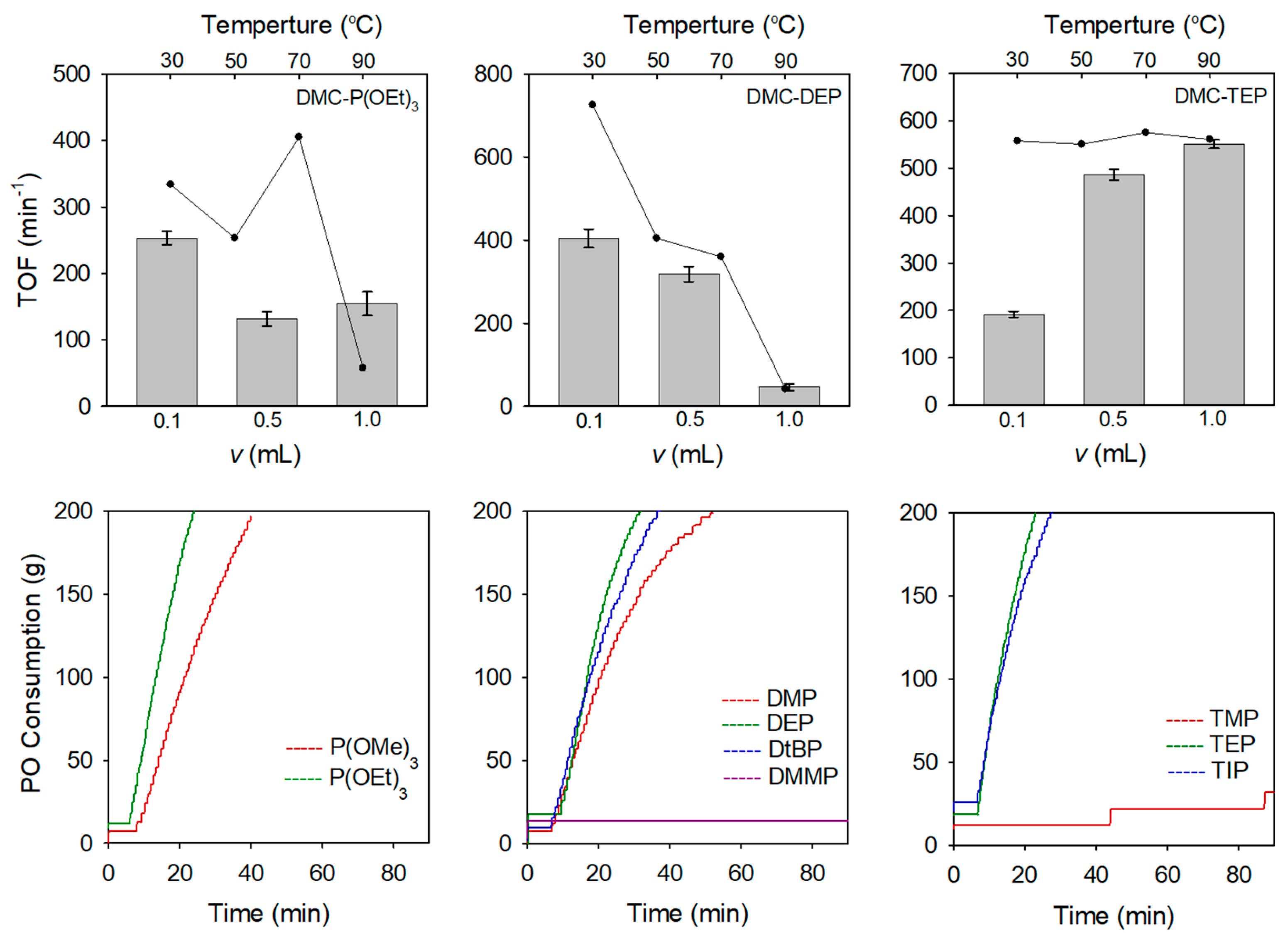
3.2. Catalytic Activities of DMC Catalysts
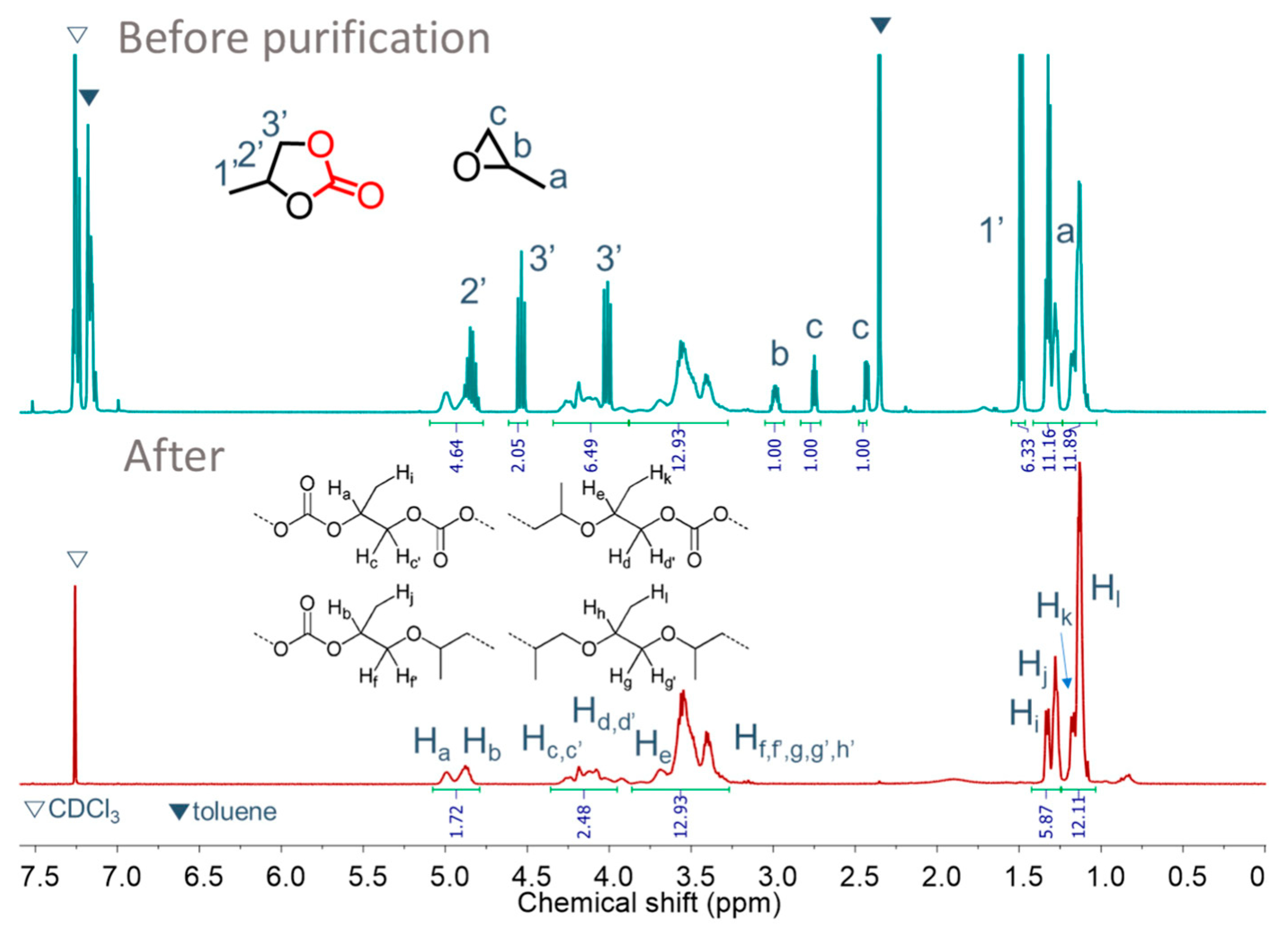
3.3. Copolymerization of PO and CO2 Using DMC-OPCs Catalysts
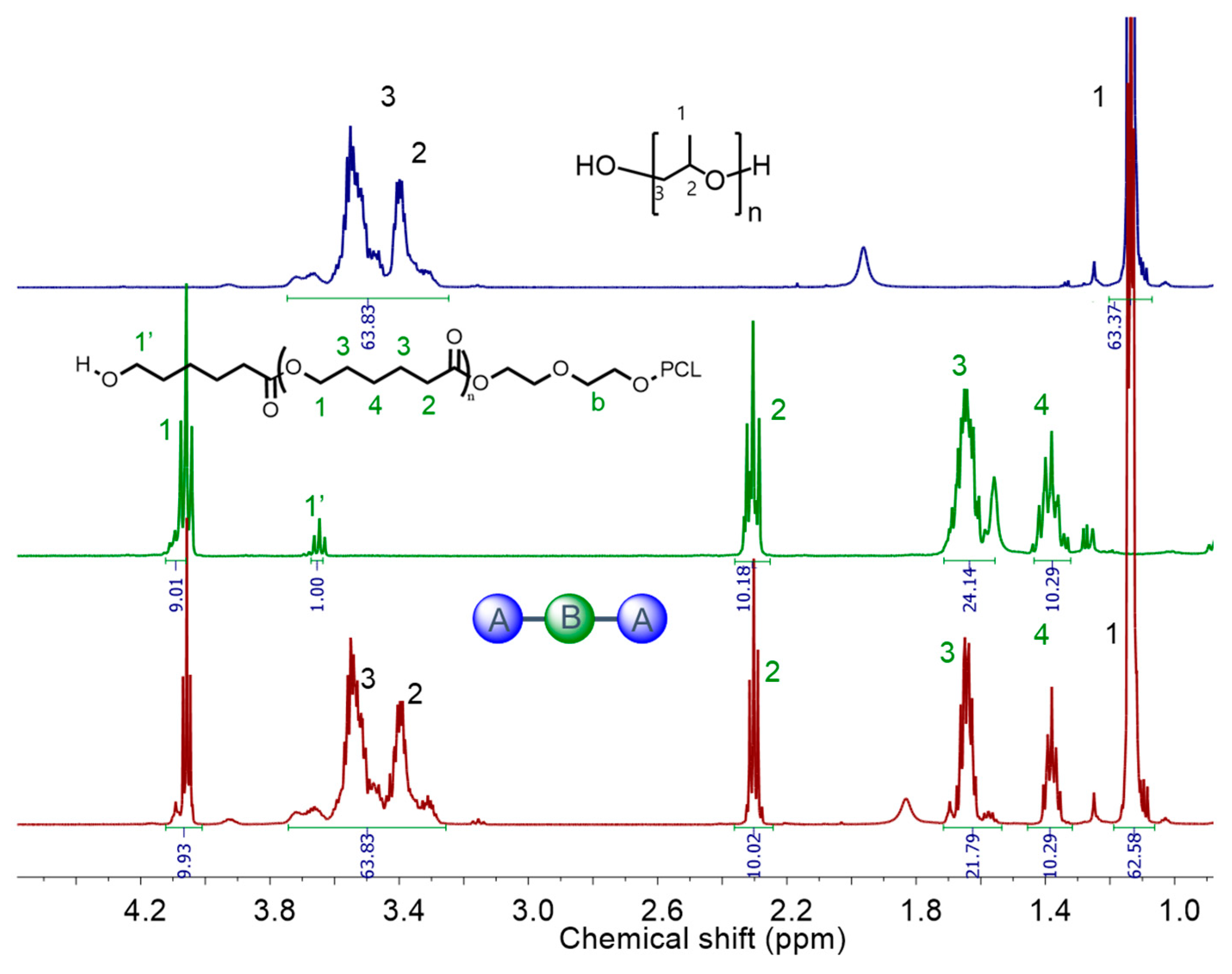
3.4. Synthesis of Triblock Copolymers Using Macroinitiators
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Matos-Peralta, Y.; Antuch, M. Review—Prussian Blue and Its Analogs as Appealing Materials for Electrochemical Sensing and Biosensing. J. Electrochem. Soc. 2019, 167, 037510. [Google Scholar] [CrossRef]
- Liu, Q.; Hu, Z.; Chen, M.; Zou, C.; Jin, H.; Wang, S.; Chou, S.-L.; Liu, Y.; Dou, S.-X. The Cathode Choice for Commercialization of Sodium-Ion Batteries: Layered Transition Metal Oxides versus Prussian Blue Analogs. Adv. Funct. Mater. 2020, 30, 1909530. [Google Scholar] [CrossRef]
- Karpova, E.V.; Karyakin, A.A. Noninvasive monitoring of diabetes and hypoxia by wearable flow-through biosensors. Curr. Opin. Electrochem. 2020, 23, 16–20. [Google Scholar] [CrossRef]
- Zhao, Z.; Xiong, Y.; Cheng, X.; Hou, X.; Yang, Y.; Tian, Y.; You, J.; Xu, L. Adsorptive removal of trace thallium(I) from wastewater: A review and new perspectives. J. Hazard. Mater. 2020, 393, 122378. [Google Scholar] [CrossRef]
- Zhou, A.; Cheng, W.; Wang, W.; Zhao, Q.; Xie, J.; Zhang, W.; Gao, H.; Xue, L.; Li, J. Hexacyanoferrate-Type Prussian Blue Analogs: Principles and Advances Toward High-Performance Sodium and Potassium Ion Batteries. Adv. Energy Mater. 2021, 11, 2000943. [Google Scholar] [CrossRef]
- Estelrich, J.; Busquets, M.A. Prussian Blue: A Safe Pigment with Zeolitic-Like Activity. Int. J. Mol. Sci. 2021, 22, 780. [Google Scholar] [CrossRef]
- Song, W.; Zhang, Y.; Tran, C.H.; Choi, H.K.; Yu, D.-G.; Kim, I. Porous organic polymers with defined morphologies: Synthesis, assembly, and emerging applications. Prog. Polym. Sci. 2023, 142, 101691. [Google Scholar] [CrossRef]
- Jack, M. Method of Making a Polyether Using a Double Metal Cyanide Complex Compound. 1966. Available online: https://pubchem.ncbi.nlm.nih.gov/patent/US-3278458-A (accessed on 15 February 2024).
- Kim, I.; Ahn, J.-T.; Ha, C.S.; Yang, C.S.; Park, I. Polymerization of Propylene Oxide by Using Double Metal Cyanide Catalysts and the Application to Polyurethane Elastomer. Polymer 2003, 44, 3417–3428. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, I.K.; Ha, J.Y.; Jo, J.K.; Park, I.; Ha, C.-S.; Suh, H.; Kim, I. Tuning of the Activity and Induction Period of the Polymerization of Propylene Oxide Catalyzed by Double Metal Cyanide Complexes Bearing β-Alkoxy Alcohols as Complexing Agents. Ind. Eng. Chem. Res. 2010, 49, 4107–4116. [Google Scholar] [CrossRef]
- Gupta, P.O.H. Double-Metal Cyanide Catalysts for Producing Polyether Polyols. U.S. Patent US6586566B1, 1 July 2003. [Google Scholar]
- Dharman, M.M.; Yu, J.-I.; Ahn, J.-Y.; Park, D.-W. Selective production of cyclic carbonate over polycarbonate using a double metal cyanide–quaternary ammonium salt catalyst system. Green Chem. 2009, 11, 1754–1757. [Google Scholar] [CrossRef]
- Marquez, C.; Corbet, M.; Smolders, S.; Marion, P.; De Vos, D. Double metal cyanides as heterogeneous Lewis acid catalysts for nitrile synthesis via acid-nitrile exchange reactions. Chem. Commun. 2019, 55, 12984–12987. [Google Scholar] [CrossRef]
- Sebastian, J.; Srinivas, D. Novel application of a Fe–Zn double-metal cyanide catalyst in the synthesis of biodegradable, hyperbranched polymers. Chem. Commun. 2011, 47, 10449–10451. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Dong, X. Novel application of double metal cyanide in the synthesis of hyperbranched polyether polyols. Des. Monomers Polym. 2013, 16, 72–78. [Google Scholar] [CrossRef]
- Peeters, A.; Valvekens, P.; Ameloot, R.; Sankar, G.; Kirschhock, C.E.A.; De Vos, D.E. Zn–Co Double Metal Cyanides as Heterogeneous Catalysts for Hydroamination: A Structure–Activity Relationship. ACS Catal. 2013, 3, 597–607. [Google Scholar] [CrossRef]
- Tran, C.H.; Lee, M.-W.; Lee, S.-J.; Choi, J.-H.; Lee, E.-G.; Choi, H.-K.; Kim, I. Highly Active Heterogeneous Double Metal Cyanide Catalysts for Ring-Opening Polymerization of Cyclic Monomers. Polymers 2022, 14, 2507. [Google Scholar] [CrossRef]
- Tran, C.H.; Lee, S.J.; Moon, B.-R.; Lee, E.-G.; Choi, H.-K.; Kim, I. Organonitriles as complexing agents for the double metal cyanide-catalyzed synthesis of polyether, polyester, and polycarbonate polyols. Catal. Today 2023, 418, 114125. [Google Scholar] [CrossRef]
- Herzberger, J.; Niederer, K.; Pohlit, H.; Seiwert, J.; Worm, M.; Wurm, F.R.; Frey, H. Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: Synthesis, novel polymer architectures, and bioconjugation. Chem. Rev. 2016, 116, 2170–2243. [Google Scholar] [CrossRef] [PubMed]
- Pitet, L.M.; Hait, S.B.; Lanyk, T.J.; Knauss, D.M. Linear and Branched Architectures from the Polymerization of Lactide with Glycidol. Macromolecules 2007, 40, 2327–2334. [Google Scholar] [CrossRef]
- Lee, K.W.; Chung, J.W.; Kwak, S.-Y. Highly Branched Polycaprolactone/Glycidol Copolymeric Green Plasticizer by One-Pot Solvent-Free Polymerization. ACS Sustain. Chem. Eng. 2018, 6, 9006–9017. [Google Scholar] [CrossRef]
- Song, W.; Tang, Y.; Moon, B.Y.; Liao, Q.; Xu, H.; Hou, Q.; Zhang, H.; Yu, D.-G.; Liao, Y.; Kim, I. Green synthesis of hypercrosslinked polymers for CO2 capture and conversion: Recent advances, opportunities, and challenges. Green Chem. 2024, 26, 2476–2504. [Google Scholar] [CrossRef]
- Chen, S.; Wang, H.; Li, Z.; Wei, F.; Zhu, H.; Xu, S.; Xu, J.; Liu, J.; Gebru, H.; Guo, K. Metallic organophosphate catalyzed bulk ring-opening polymerization. Polym. Chem. 2018, 9, 732–742. [Google Scholar] [CrossRef]
- Gao, C.-Y.; Ai, J.; Tian, H.-R.; Wu, D.; Sun, Z.-M. An ultrastable zirconium-phosphonate framework as bifunctional catalyst for highly active CO2 chemical transformation. Chem. Commun. 2017, 53, 1293–1296. [Google Scholar] [CrossRef]
- ASTM D4671-05; Standard Test Method for Polyurethane Raw Materials Determination of Unsaturation of Polyols. iTeh Standards: Newark, DE, USA, 2005.
- ASTM E1899-97; Standard Test Method for Hydroxyl Groups Using Reaction with p-Toluenesulfonyl Isocyanate (TSI) and Potentiometric Titration with Tetrabutylammonium Hydroxide. iTeh Standards: Newark, DE, USA, 1997.
- Tran, C.H.; Jang, H.B.; Moon, B.-r.; Lee, E.-g.; Choi, H.-k.; Kim, I. Organic carbonates as green and sustainable complexing agents for double metal cyanide catalysts for the synthesis of polyether and poly(ether-carbonate) polyols. Catal. Today 2024, 425, 114319. [Google Scholar] [CrossRef]
- Tran, C.H.; Lee, M.W.; Park, S.W.; Jeong, J.E.; Lee, S.J.; Song, W.; Huh, P.; Kim, I. Heterogeneous Double Metal Cyanide Catalyzed Synthesis of Poly(ε-caprolactone) Polyols for the Preparation of Thermoplastic Elastomers. Catalysts 2021, 11, 1033. [Google Scholar] [CrossRef]
- ASTM D274-5; Standard Test Method for Relative Tinting Strength of White Pigments by Reflectance Measurements. American Society for Testing and Materials: West Conshohocken, PA, USA, 2017.
- Ahmed, A.A.; Gypser, S.; Leinweber, P.; Freese, D.; Kühn, O. Infrared spectroscopic characterization of phosphate binding at the goethite–water interface. Phys. Chem. Chem. Phys. 2019, 21, 4421–4434. [Google Scholar] [CrossRef] [PubMed]
- Kloß, S.; Selent, D.; Spannenberg, A.; Franke, R.; Börner, A.; Sharif, M. Effects of Substitution Pattern in Phosphite Ligands Used in Rhodium-Catalyzed Hydroformylation on Reactivity and Hydrolysis Stability. Catalysts 2019, 9, 1036. [Google Scholar] [CrossRef]
- Siow, K.S.; Britcher, L.; Kumar, S.; Griesser, H.J. Deposition and XPS and FTIR Analysis of Plasma Polymer Coatings Containing Phosphorus. Plasma Process. Polym. 2014, 11, 133–141. [Google Scholar] [CrossRef]
- Zhong, Q.; Yang, Y.; Chen, L.; Li, Q.; Xu, B.; Jiang, T. Intensification Behavior of Mercury Ions on Gold Cyanide Leaching. Metals 2018, 8, 80. [Google Scholar] [CrossRef]
- Alferov, K.; Wang, S.; Li, T.; Xiao, M.; Guan, S.; Meng, Y. Co-Ni Cyanide Bi-Metal Catalysts: Copolymerization of Carbon Dioxide with Propylene Oxide and Chain Transfer Agents. Catalysts 2019, 9, 632. [Google Scholar] [CrossRef]
- Troev, K.D. 1—Poly(alkylene H-phosphonate)s. In Polyphosphoesters; Troev, K.D., Ed.; Elsevier: Oxford, UK, 2012; pp. 1–127. [Google Scholar] [CrossRef]
- Harsági, N.; Keglevich, G. The Hydrolysis of Phosphinates and Phosphonates: A Review. Molecules 2021, 26, 2840. [Google Scholar] [CrossRef]
- Shaarani, F.W.; Bou, J.J. Synthesis of vegetable-oil based polymer by terpolymerization of epoxidized soybean oil, propylene oxide and carbon dioxide. Sci. Total Environ. 2017, 598, 931–936. [Google Scholar] [CrossRef]
- Tran, C.H.; Pham, L.T.T.; Lee, Y.; Jang, H.B.; Kim, S.; Kim, I. Mechanistic Insights on Zn(II)−Co(III) Double Metal Cyanide-Catalyzed Ring-Opening Polymerization of Epoxides. J. Catal. 2019, 372, 86–102. [Google Scholar] [CrossRef]
- Bao, Y.-F.; Wang, Y.-J.; Wang, Y.-C.; Liu, D.-H. Homogeneous base catalyst with high activity and stability for synthesis of dimethyl carbonate by transesterification. RSC Adv. 2023, 13, 9347–9352. [Google Scholar] [CrossRef]
- Vyas, C.K.; Rao, C.V.S.B.; Jayalakshmi, S.; Joshirao, P.M.; Manchanda, V.K. Phosphonates as alternative to tributyl phosphate for the separation of actinides from fission products. Radiochim. Acta 2015, 103, 277–285. [Google Scholar] [CrossRef]
- Rajani, P.; Gopakumar, G.; Nagarajan, S.; Brahmmananda Rao, C.V.S. Does the basicity of phosphoryl oxygen change with alkyl chain length in phosphate ligands? Chem. Phys. Lett. 2021, 775, 138641. [Google Scholar] [CrossRef]
- Bakkour, Y.; Darcos, V.; Li, S.; Coudane, J. Diffusion ordered spectroscopy (DOSY) as a powerful tool for amphiphilic block copolymer characterization and for critical micelle concentration (CMC) determination. Polym. Chem. 2012, 3, 2006–2010. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]




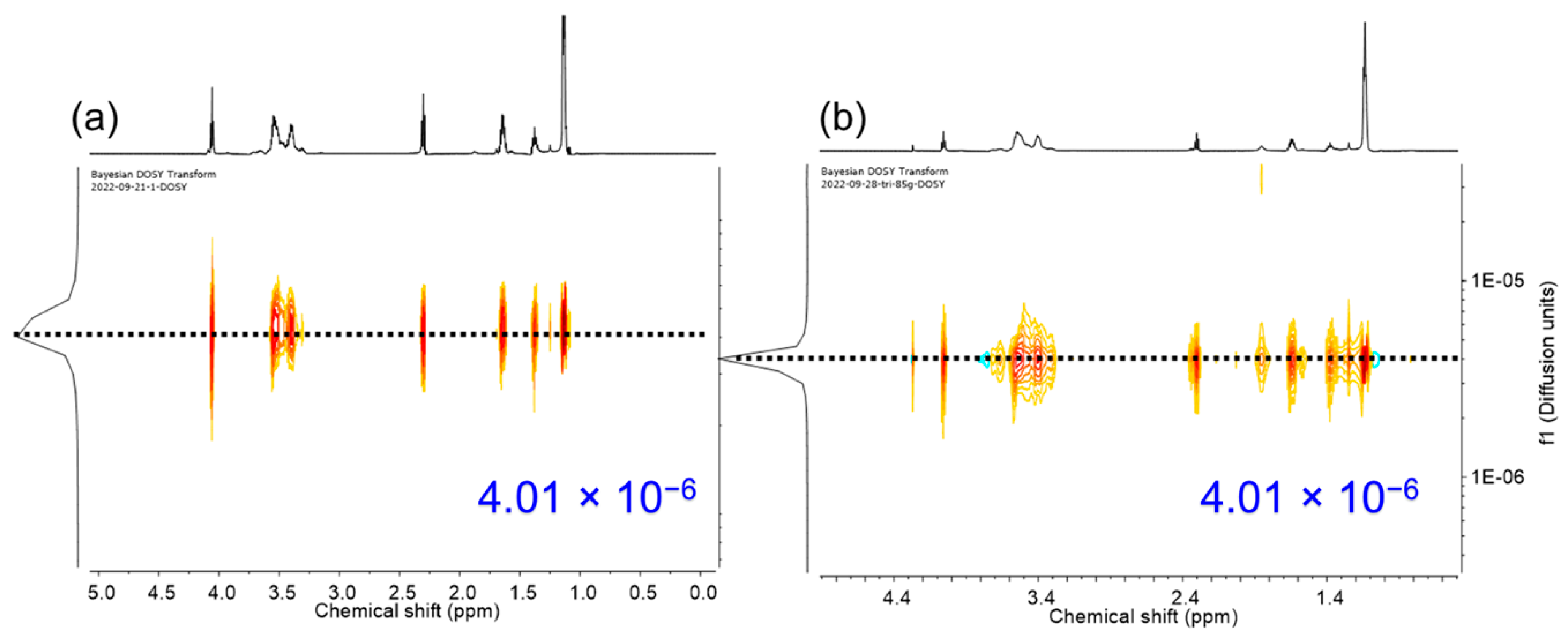

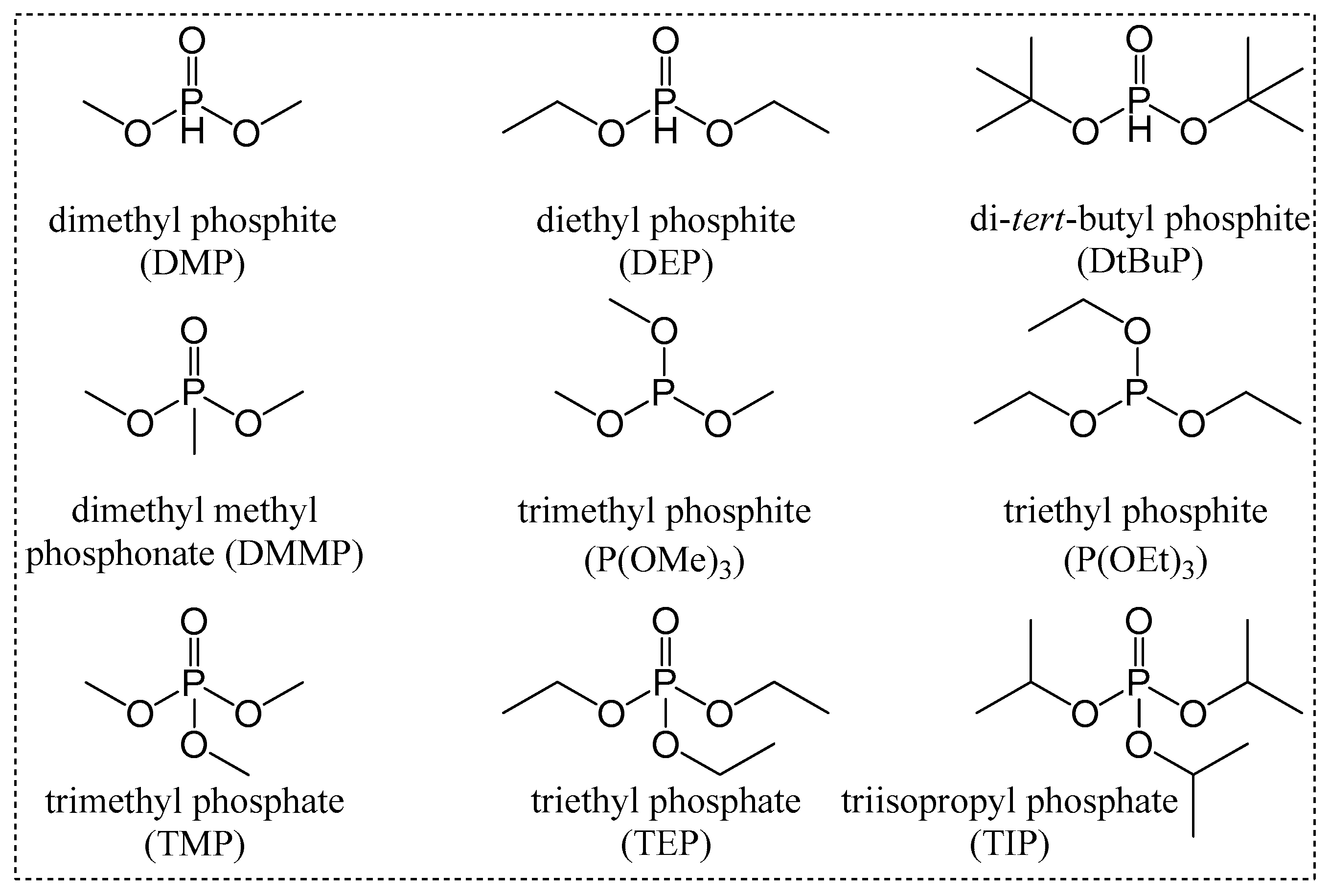{kind=link}
{kind=link}
{kind=link}
{kind=link}
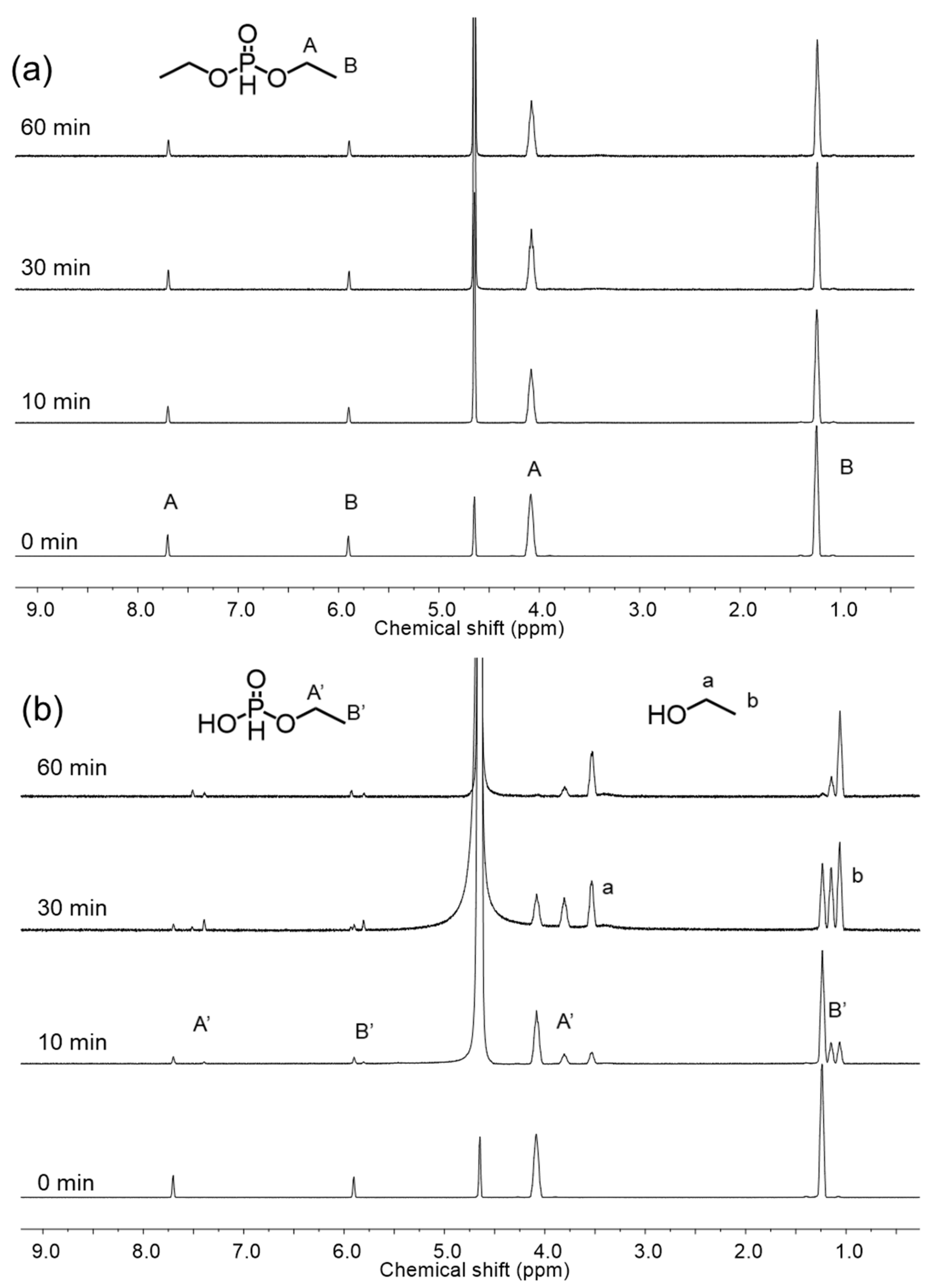{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | ICP-Mass (wt%) | Elemental Analysis (wt%) | TGA (wt%) | Estimated Catalyst Formulation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Zn | Co | P | C | H | N | CA | H2O | P123 | ||
| DMC-pure | 27.6 | 16.6 | − | 20.2 | 1.3 | 23.6 | − | 12.0 | − | Zn1.5Co(CN)6.0·2.37H2O |
| DMC-DEP | 19.7 | 9.0 | 3.9 | 28.4 | 3.4 | 12.2 | 13.2 | 1.4 | 13.8 | Zn1.97Co(CN)5.71·0.82DEP·0.02P123·0.51H2O |
| DMC-P(OEt)3 | 23.0 | 11.2 | 3.5 | 29.0 | 3.2 | 13.3 | 10.2 | 0.5 | 13.7 | Zn1.85Co(CN)4.98·0.60P(OEt)3·0.01P123·0.14H2O |
| DMC-TEP | 16.9 | 8.3 | 6.1 | 30.2 | 3.9 | 11.1 | 21.7 | 0.9 | 14.5 | Zn1.84Co(CN)5.63·1.40TEP·0.02P123·0.37H2O |
| DMC-tBuOH | 23.9 | 10.7 | − | 29.3 | 3.3 | 16.3 | 7.2 | 1.9 | 24.1 | Zn2.0Co(CN)6.4·0.53tBuOH· 0.02P123·0.58H2O |
| Entry | Catalyst | Reaction Condition | Polyol Properties | |||||
|---|---|---|---|---|---|---|---|---|
| nI (mmol) | (bar) | Conv. (%) | Yield of PPC (%) | a (%) | Mn (g mol−1) | Ð | ||
| 1 | DMC-DEP | 0.25 | 30 | 99.9 | 61 | 37 | 4100 | 4.36 |
| 2 | DMC-DEP | 2.5 | 10 | 90.1 | 76.5 | 20 | 3400 | 4.33 |
| 3 | DMC-DEP | 2.5 | 20 | 99.4 | 68.1 | 31 | 4200 | 3.81 |
| 4 | DMC-DEP | 2.5 | 30 | 99.7 | 63 | 33 | 2000 | 4.23 |
| 5 | DMC-DEP | 12.5 | 30 | 99.6 | 60 | 20 | 1200 | 2.09 |
| 6 | DMC-P(OEt)3 | 2.5 | 30 | 97.8 | 56 | 33 | 2400 | 3.47 |
| 7 | DMC-TEP | 2.5 | 30 | 97.3 | 57 | 34 | 1900 | 2.64 |
| Block Copolymer | Reaction Condition a | Polyol Properties | |||||
|---|---|---|---|---|---|---|---|
| Initiator | ΣnPO (mol) | t (min) | Mn (g mol−1) | Ð | |||
| Type | Mn b (g mol−1) | Ð | |||||
| BCP-1 | PCL | 2000 | 1.65 | 0.45 | 27 | 4600 | 1.26 |
| BCP-2 | PCL | 1700 | 1.49 | 0.35 | 18.5 | 3700 | 1.37 |
| BCP-3 | PCL | 1700 | 1.49 | 0.85 | 18 | 6600 | 1.29 |
| BCP-4 | PTMG | 1400 | 1.8 | 0.1 | 10 | 1800 | 1.23 |
| BCP-5 | PTMG | 1400 | 1.8 | 0.4 | 12 | 3600 | 1.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.-G.; Tran, C.-H.; Heo, J.-Y.; Kim, S.-Y.; Choi, H.-K.; Moon, B.-R.; Kim, I. Synthesis of Polyether, Poly(Ether Carbonate) and Poly(Ether Ester) Polyols Using Double Metal Cyanide Catalysts Bearing Organophosphorus Complexing Agents. Polymers 2024, 16, 818. https://doi.org/10.3390/polym16060818
Lee E-G, Tran C-H, Heo J-Y, Kim S-Y, Choi H-K, Moon B-R, Kim I. Synthesis of Polyether, Poly(Ether Carbonate) and Poly(Ether Ester) Polyols Using Double Metal Cyanide Catalysts Bearing Organophosphorus Complexing Agents. Polymers. 2024; 16(6):818. https://doi.org/10.3390/polym16060818
Chicago/Turabian StyleLee, Eun-Gyeong, Chinh-Hoang Tran, Ju-Yeong Heo, So-Young Kim, Ha-Kyung Choi, Byeong-Ryeol Moon, and Il Kim. 2024. "Synthesis of Polyether, Poly(Ether Carbonate) and Poly(Ether Ester) Polyols Using Double Metal Cyanide Catalysts Bearing Organophosphorus Complexing Agents" Polymers 16, no. 6: 818. https://doi.org/10.3390/polym16060818
APA StyleLee, E.-G., Tran, C.-H., Heo, J.-Y., Kim, S.-Y., Choi, H.-K., Moon, B.-R., & Kim, I. (2024). Synthesis of Polyether, Poly(Ether Carbonate) and Poly(Ether Ester) Polyols Using Double Metal Cyanide Catalysts Bearing Organophosphorus Complexing Agents. Polymers, 16(6), 818. https://doi.org/10.3390/polym16060818