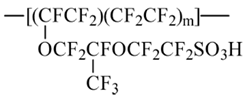4.2. Discussion of the Results of X-ray Diffractometry Experiments
As was shown in the previous section, we studied the growth of crystals with different unit cells of different systems from supersaturated solutions based on natural water and DDW. Let us recall that we have studied potassium chloride with a cubic crystal lattice (
a =
b =
c = 3.63 Å; α = 90°, β = 90°, γ = 90°) [
13]; sodium acetate trihydrate with a monoclinic lattice (
a = 12.28–12.48 Å,
b = 10.41–10.47 Å,
c = 10.36–10.45 Å; α = 90°, β = 111.39–112.65°, γ = 90°) [
14,
15,
16,
17,
18]; and copper sulfate that can crystallize as pentahydrate in a triclinic lattice (
a = 5.97 Å,
b = 6.10 Å,
c = 10.64 Å; α = 77.2°, β = 82.4°, γ = 72.4°) and as trihydrate in a monoclinic lattice (
a = 5.57 Å,
b = 12.98 Å,
c = 7.38 Å; α = 90°, β = 96.5°, γ = 90°) [
19], as well as sucrose C
12H
22O
11 with a monoclinic lattice (
a = 7.72–7.77 Å,
b = 8.68–8.71 Å,
c = 10.82–10.88 Å, α = 90°, β = 102.9°, γ = 90°) [
20,
21].
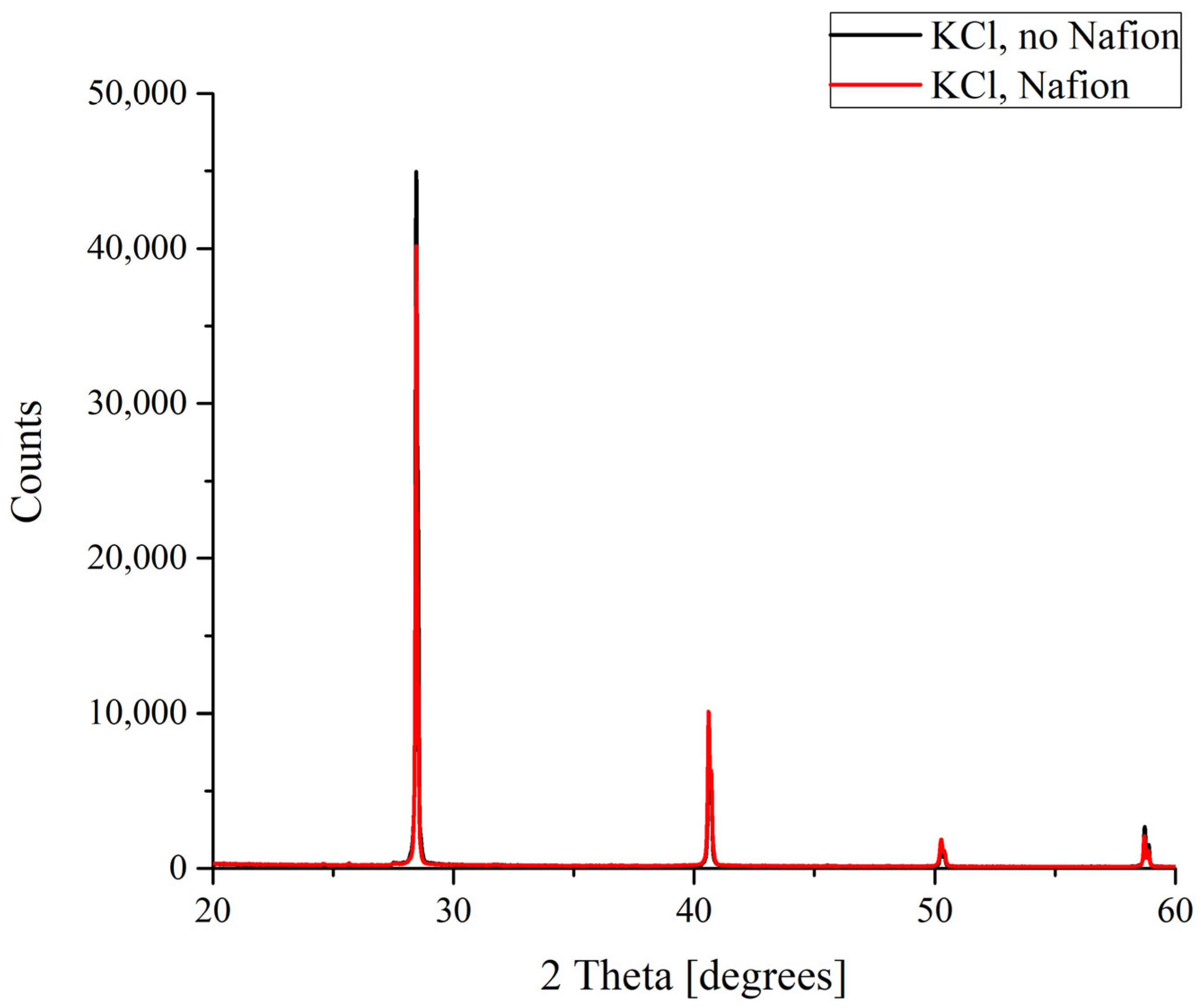
In the case of potassium chloride, which crystallizes to form a cubic lattice solely that does not involve water molecules, no difference was found between the diffraction patterns of the crystals grown on smooth and polymer substrates (
Figure 4).
For sodium acetate, again both diffraction patterns recorded for the deposits formed in the presence and in the absence of a Nafion plate in the Petri dish unambiguously reflected the appearance of a regular trihydrate (
Figure 5). Note also that while the diffraction patterns overlap and nearly coincide in the case of KCl crystallization, in the case of NaCH
3COO·3H
2O we observe a broadening and a slight shift of the profile.
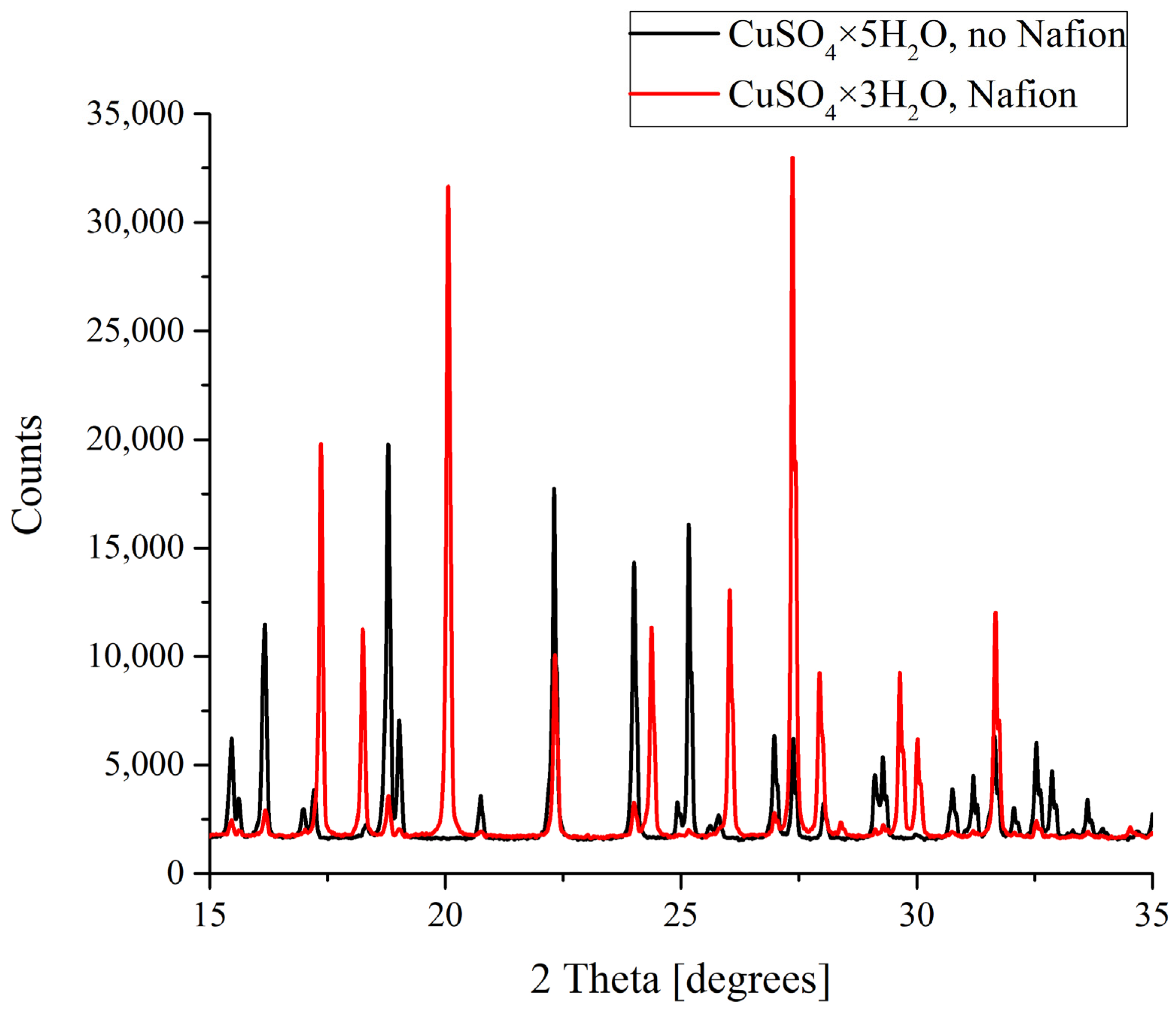
In the case of copper sulfate solutions based on the natural deionized water, a clear difference between the formation of crystals was observed. As was mentioned above, CuSO
4 crystals grown in a supersaturated aqueous solution can be either pentahydrates (CuSO
4·5H
2O) with a triclinic lattice or trihydrates (CuSO
4·3H
2O) with a monoclinic lattice. As follows from the data shown in
Figure 6, it is copper sulfate trihydrate that grows on a Nafion polymeric plate, while a smooth surface promotes the formation of the pentahydrate. Let us stress that in DDW-based solutions a crystalline deposit of copper sulfate pentahydrate with a triclinic lattice was formed irrespectively of whether a Nafion plate was placed in the Petri dish or not.
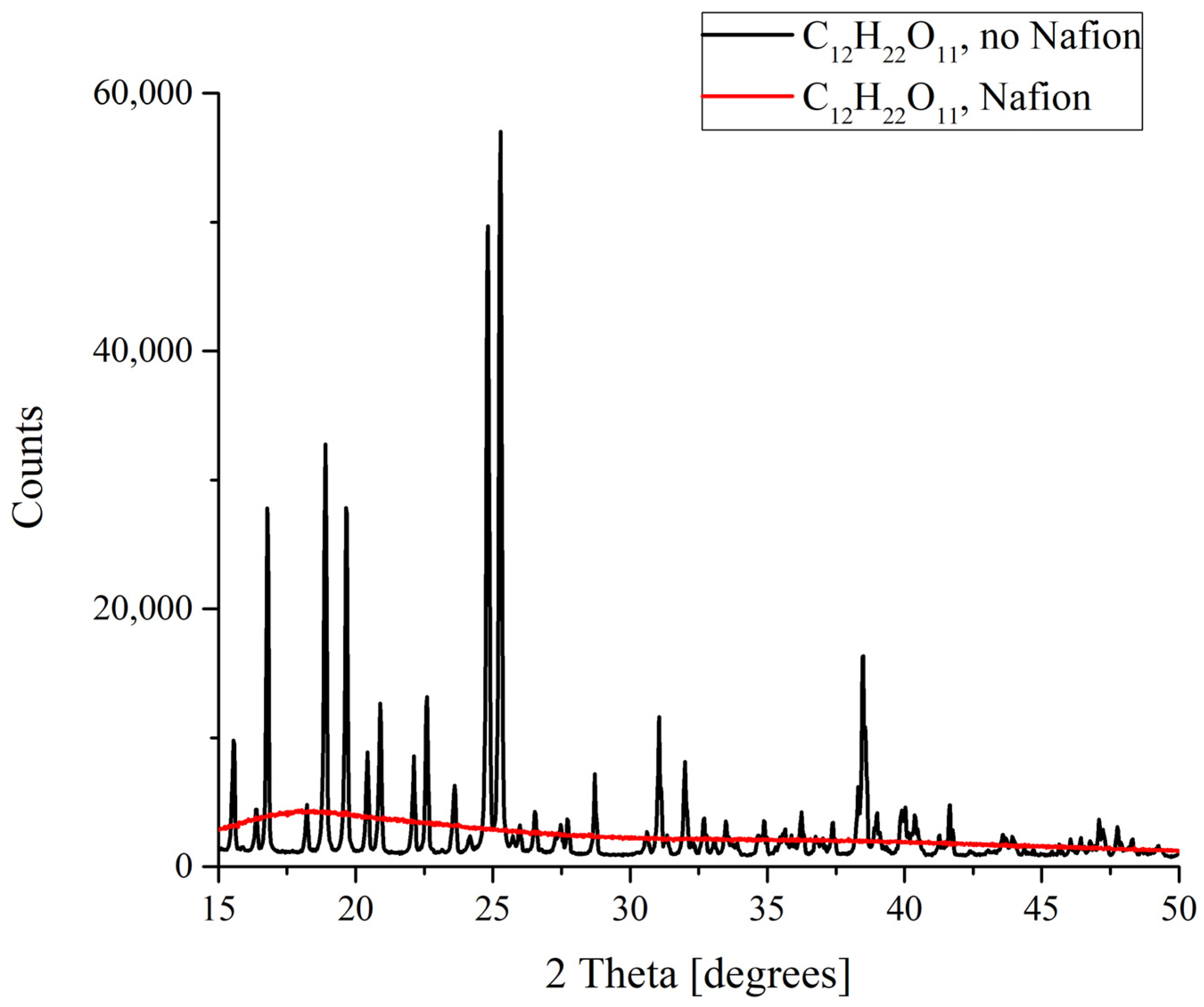
Finally, drastic differences in the formation of deposits from the saturated solutions were discovered for sucrose C
12H
22O
11. It is well known that sucrose exists in the form of monoclinic crystals. When melted sucrose is cooled down, a transparent amorphous bulk named caramel is formed. Judging from the results obtained, it is caramel that forms as a result of the deposition on a Nafion plate (
Figure 7). This means that in this case there is something that prevents an ordered deposition of particles., and this is probably the existence of more or less extended polymer fibers in the nucleation region which promote the strong disordering of crystal lattice cells and the resulting amorphizing.
Despite its relative compactness, the set of compounds we use can be considered as quite representative, since they have principally different syngonies, different molecular sizes, and different kinds of involvement of water molecules. Potassium chloride crystallizes with no lattice water, and eight (absolutely equivalent) K…Cl distances of about 3.15 Å, while Cl…Cl distances are proportional to 3.63 and 5.14 Å depending on the lattice direction. Sodium acetate can be anhydrous, but its typical stable form involves water molecules, each sodium atom having five neighboring molecules at a mean Na…O distance of 2.42 Å; but the local coordination is not highly symmetric, since the totally six oxygen neighbors of sodium (those of five water molecules and one acetate group) form a distorted octahedron with the Na…O distances varying in a range of 2.34 to 2.50 Å, which is quite typical of sodium [
22]. Copper sulfate hydrates are very regular and at the same time involve water molecules. Additionally, the counterion in this case is complementary to the terminal SO
3H groups of Nafion. As to the structural parameters, in trihydrate, the first shell of each copper atom involves three water molecules and three sulfate groups, whereas in pentahydrate, there are four water molecules and two sulfate groups, sulfates opposing each other in the extended octahedral-like configuration. Thus, the latter configuration can be treated as the one with the highest local symmetry among all three salt hydrates, while that of sodium acetate can be treated as the least structured. Finally, sucrose C
12H
22O
11 is a relatively large molecule that involves penta- and hexamolecular cycles with numerous hydroxyl groups, which may cause certain spatial effects.
As shown below, all of the experimental results can be explained based on the nanoscale consideration of the systems. The most interesting result concerns copper sulfate. Because of the different syngony of its tri- and pentahydrates, the initial hypothesis was the following. The presence of a Nafion specimen in a Petri dish probably favors the formation of crystal lattices of a particular kind. Insofar as the key difference between the tri- and pentahydrate is the equality of one lattice angle to 90° in the former case, it was reasonable to assume that the orientation of the deposit growing on a Nafion surface is somehow spatially restricted in the tangential direction. This may be possible if there are some Nafion fibers that are oriented normally to the mean surface. This general peculiarity should be manifested in other situations as well. Two other salts considered in the work were potassium chloride and sodium acetate, which are crystallized (as noticed above) in cubic and monoclinic lattices. Both crystal lattices are complementary to the normal orientation of Nafion fibers with respect to the mean surface of the specimen, which means that the presence of Nafion may only either slightly distort or, by contrast, support the formation of those lattices depending on the complementarity of the parameters of the crystal lattice cells and the actual distances between the Nafion fibers. However, the crystal lattice of sucrose is of the same symmetry as that of the copper sulfate trihydrate. Then, the normal orientation of the fibers is not the sole condition that predetermines the crystal growth, and the arrangement of the fibers may be no less important.
To check the idea, non-empirical simulations of the model Nafion fragments either individually or in combination with water molecules and cation-anion salt pairs were carried out. In accordance with the general Formula (1), in which
m = 6.5 for Nafion N117 used in this work, a fragment selected for modeling involved the perfluorinated hydrocarbon skeleton with two side perfluorinated vinyl ether chains terminated by SO
3H groups and two terminal methyl groups, so that the general composition of the fragment was CF
3C(F)(O-CF
2CF(CF
3)OCF
2CF
2SO
3H)(CF
2)
14C(F)(O-CF
2CF(CF
3)OCF
2CF
2SO
3H)CF
3 (
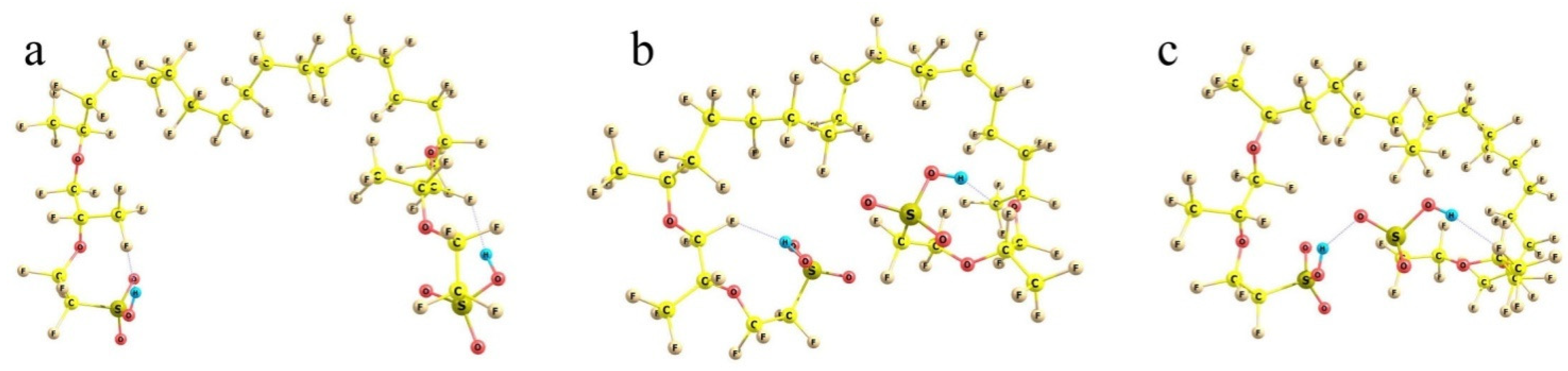
Figure 9). At first, various conformations of the model were considered.
Because of the very large number of the degrees of freedom of the system considered related to the internal rotations around all single bonds, it is nearly impossible to find the absolute energy minimum of the system. However, judging from the known peculiarities of hydrocarbons, it is clear that mutual rotations of the neighboring functional groups, which involve atoms of the same kind (fluorine in our case), result in energetically either equivalent or close minima. The small differences appear due to the interactions with the more distant groups, all of which are of the prevailing dispersion nature. Such interactions provide contributions of no larger than a kcal/mole. At the same time, there are SO
3H-ended chains, which should interact electrostatically; and these effects are much larger. Then, any set of the structures with the principally different arrangement of the side chains can give tentative estimates of the relative stability of those conformations. In any actual system, numerous interactions between the neighboring groups should inevitably distort any such structure; but the general energy trends should be the same. And the first result obtained is reasonable and quite expected. The more folded the branches and the closer the terminal sulfonate groups to each other, the lower the energy of the system. It decreases on going from (a) to (b) and (c) configurations in
Figure 9.
When the SO
3H-ended chains are initially kept at the largest possible distance from each other (taking into account that the structure should be extended in both directions, and any group has neighbors from both sides, the mean angles between the direction of the hydrophobic backbone and its polyether side chain should not exceed 90°) and extended to the utmost degree, the energy of the system is the highest (
Figure 9a). When the branches are initially intorted, they tend to approach each other to minimize the net dipole moment (
Figure 9b); and the electronic energy difference between these two structures is ca. 15 kcal/mol. Finally, if the terminal groups can form an H-bond, the stabilization of the system is maximum. The structure shown in
Figure 9c is lower than the previous one by another 10 kcal/mol, which makes it already definitely favorable compared to the one with extended terminal branches (with an energy difference of ca. −25 kcal/mol). When thermal increments to the energy determined also by the different folding of the backbone (which becomes progressively less extended as the SO
3H-ended chains become involved in the interaction with each other) are taken into account, the energy differences between the structures are decreased, but remain quite substantial: the Δ
Gvib relative Gibbs energies are 0 vs. −11 vs. −19 kcal/mol for the (a), (b), and (c) structures, respectively.
Thus, one can assume that in a dry state, Nafion fibers should be packed in such a way that their SO3H-ended chains are intorted as strongly as possible (in the presence of the neighboring segments), while the sulfonate groups themselves tend to approach each other to form hydrogen bonds, which provide closed segments within the hydrophobic matrix. Naturally, the number of chains (and sulfonate groups) involved in one H-bonded knot can be larger than two, but restricted because of the spatial limitations, and the additional groups can be either those from the adjacent fibers or of the neighboring segments of the same fiber. This makes the whole structure quite strongly entangled.
Now, let us turn to the hydration of a Nafion plate. To mimic the effect produced by water molecules, it is reasonable to restrict the consideration to a single-chain segment of the above Nafion model, i.e., to just one side chain connected to the perfluorinated backbone, and analyze what happens when the number of water molecules is gradually increased. Again, it is absolutely impossible and unnecessary to find all the local arrangements of water molecules around the model fragment. The most important conditions to be met are the reasonably large number of hydrogen bonds formed between water molecules and the smallest possible number of OH groups uninvolved in the bonds. The former condition corresponds to the largest possible contribution of the H-bond energies (each no less than 5 kcal/mol) to the total energy of the system, while the latter is the condition of the overall stability of the system against external perturbations.
When the number of molecules is small, they gather around the hydrophilic SO
3H group (
Figure 10a) and form a kind of a hat with an almost planar abat-jour visually parallel to the nearest contour of fluorine atoms. In the case of 16 molecules, which is sufficient for the formation of the first hydration shell around the SO
3H group, the group dissociates (to form a hydronium ion and a negatively charged –SO
3− residue), and the corresponding energy decrease equals 49 kcal/mol, although the formal number of hydrogen bonds stabilizing the system is increased only by one, from 25 in the relaxed water cluster to 26 in the cluster bound to a hydrophilic head of the model Nafion fragment. Note that here the reference water cluster is the one that formed upon the relaxation of the aqueous coat upon removal of the model Nafion structure from it. If we would consider the lowest-energy water cluster composed of the same number of molecules, the energy gain upon the hydration of a model Nafion fragment is smaller, but insubstantially, by 5 kcal/mol, because the total number of hydrogen bonds that stabilize the cluster is larger only by one. The above figures mean that the H-bonds themselves are strengthened upon the reorganization of water molecules around the appeared proton and the negatively charged –SO
3− residue, and the mean solvation energy per one terminal group at a low water content can be estimated as ca. 45 kcal/mol.
When the number of water molecules is increased nearly two-fold (
Figure 10b) and they are initially randomly arranged around the model Nafion fragment, the number of molecules in the close neighborhood of the –SO
3− residue is not substantially increased. The crown of its hat becomes thicker, and concurrently a water shell forms over the fluorocarbon chain separated from it by about 2.5 Å on the average. Water molecules are oriented in such a way that only some of the peripheral ones have dangling OH groups, while all the protons of the so-to-speak inner-structure molecules are involved in hydrogen bonds. This extension of the water shell corresponds to the formal total hydration energy of the model Nafion structure of 24.5 kcal/mol if the reference water cluster corresponds to a compact fragment of the bulk H-bond network. At the same time, if we would take the water cluster obtained upon the relaxation of the hydration shell of a model Nafion structure as the reference, the estimated energy gain would be much larger, about 50.4 kcal/mol. It means that when water molecules are rearranged around fluorocarbon chains, much energy is spent on the distortion of the original H-bond network of water. If it is compensated by the efficient hydration of hydronium ions and –SO
3− residues, the process is thermodynamically possible, but it gradually becomes less favorable as the length of fluorocarbon chains to be coated is increased. This trend becomes ultimately clear at the larger amount of water.
When the number of water molecules is increased to half a hundred (
Figure 10c) and they are initially randomly distributed around a model Nafion fragment, they can already form a closed monomolecular layer with the –SO
3− residue naturally built in this layer and the fluorocarbon chain residing in a kind of a tunnel; and a continuous network of 85 H-bonds is observed. However, this variant is not energetically favorable. Formation of such a water coat requires a dramatic distortion of the hydrogen-bond network of water. Instead of a compact ensemble of water molecules stabilized by a three-dimensional network of intermolecular bonds (
Figure 11a), a strongly expanded two-dimensional bubble (
Figure 11b) should appear, which is not additionally stabilized by the interactions with the structure it covers except for the coordination to an –SO
3− residue. Therefore, the energy difference between the hydrated Nafion fragment (
Figure 10c) and a combination of the individual Nafion fragment and the relaxed water cluster (
Figure 11a) equals 48 kcal/mol. Formally, this difference can be explained by the smaller number of hydrogen bonds the 2D bubble is stabilized with, namely 85 vs. 94 in the 3D ensemble, which accounts for about 5.3 kcal/mol per one additional bond. This is not an individual bond energy because of the collective effects typical of hydrogen-bond networks. Nevertheless, this is a reasonable estimate of the energy that should be supplied to the water system in one way or another to provide the necessary reorganization of hydrogen bonds. Note that it is nearly equal to the energy liberated upon the hydration of the –SO
3− residue, which means that the extension of hydration shells over the fluorocarbon chains is thermodynamically possible only at the cost of the hydration of hydrophilic segments, and the most energetically favorable variant is the one when only –SO
3− residues and their close neighbors are hydrated.
An additional argument in favor of such a conclusion is that even at the smallest considered number of water molecules, the SO
3H group is dissociated (see above), and an H
3O
+ fragment separated from the –SO
3− residue with one water molecule appears. Insofar as all water molecules in the above systems are spent on the formation of a hydrating monolayer, there is no driving force for the migration of a detached proton to a larger distance from the –SO
3− residue. Only when the number of water molecules increases to 60, and some of the molecules act as nucleating sites of the second hydration shell, do there appear migration paths for the proton, and its departure from the –SO
3− residue becomes possible (
Figure 10d); and in the optimum structure it resides close to the end of the fluorocarbon chain.
These results show that the hydration of side chains of a Nafion fiber is energetically favorable when water molecules do not penetrate deep and close to the perfluorinated Teflon backbone, and the energy necessary for the unfolding and extension of the fibers can easily be compensated by the hydration energy, especially taking into account that in this situation water molecules can be arranged at a larger distance from the perfluorinated carbon skeleton, which is also energetically favorable.
Thus, based on these model simulations, we can state that hydration of a Nafion plate should result in the unfolding of the side chains involving terminal SO3H groups, and the orientation of the chains should minimize the electrostatic repulsion of the residues and balance all the ionic interactions, which is best at their normal orientation to the Teflon backbone. Based on this conclusion, we can analyze the possible variants of the deposit crystal growth on such a surface.
In the case of potassium chloride, the regular cubic lattice implies the basic Cl…Cl and K…K distances equal to 3.63 Å at K…Cl contacts of 3.15 Å (
Figure 12a). This means that the lattice segment that can be formed between two Nafion fibers should be characterized by a boundary distance proportional to 3.63 Å. It is worth noting that there are –SO
3− residues, which play the role of coordinating sites at the ends of side Nafion chains, where potassium cations can be bound. Thus, formally –SO
3− residues should substitute a chloride ion at some boundary lattice points. Potassium sulfate is known to crystallize in an orthorhombic lattice with the following cell parameters:
a = 7.46 Å,
b = 10.08 Å,
c = 5.78 Å, α = 90°, β = 90°, γ = 90° [
23]. The orientation of the crystal axes is the same as in a cubic lattice of KCl; but the internuclear distances differ, falling in a range of 3.80 to 4.06 Å in the case of K…K and of 5.00 to 10.08 Å in the case of S…S, while the K…O contacts are all 2.9 to 3.1 Å (
Figure 12b). Thus, the K…Cl and K…O distances in the potassium chloride and sulfate crystals are sufficiently close, which formally means that the replacement of one K…Cl contact at the fiber boundary with a K…O–S(O
2) should cause a minor perturbation rather than a substantial distortion of the crystal lattice. Furthermore, Cl…Cl distances in a potassium chloride lattice are proportional to 3.63 Å, while the distance between the oxygen atoms of the terminal –SO
3− residues of the two neighboring perfluorinated ether chains of Nafion (when the SO
3H groups have symmetrically equivalent orientation) is about 14.6 Å (
Figure 12c), which is almost exactly a four-fold Cl…Cl distance. This means that a fragment of KCl lattice can almost exactly fit in between the two neighboring fibers with minor perturbation compared to the individual lattice of the substance. Then, the growth of a cubic KCl crystal on a Nafion plate should be straightforward, and the side Nafion chains act just as coordinating and slightly armoring peripheral inclusions.
In the case of sodium acetate, the situation is more complex. Here, the counterion of the salt involves a CH
3 group in addition to the carboxyl fragment. Those CH
3 groups face each other in the crystal lattice being separated by a C…C distance of 3.56 Å (
Figure 13a); and this is a strong limitation imposed on the flexibility of the particle arrangement. As a result, the orientations of the acetic groups are alternating in two orthogonal directions in the lattice. Taking into account that methyl groups cannot be located close to the Nafion fibers, the distances between the carbon atoms of those acetic groups, which have opposite orientations, are 6.23, 6.48, and 9.16 Å. No direction coincides with any lattice vector (
Figure 13b), which means that the direction of crystal growth can by no means be driven by nearly parallel Nafion chains if they extend to the maximum possible degree and form a symmetric repeated brush on a Nafion surface. However, for the two acetic groups of the same orientation, the distance between the carbon atoms of their carboxyl groups is 5.34 Å, while the distances between the oxygen atoms of the groups are 5.24 and 5.93 Å. The latter values if increased threefold provide inter-nuclear distances of 15.72 and 17.79 Å as boundaries. In Nafion model conformations where both SO
3H groups have the same spatial orientation, the O…O distances fall in a range of 15 to 16 Å (
Figure 13c). Taking into account that the Nafion fibers are sufficiently flexible due to the possibility of numerous internal rotations within any perfluorinated chain, and their terminal groups can also change their positions especially under the effect of solvating water molecules, the distances between the terminal –SO
3− residues and the growing crystal can, so to speak, adapt to each other. However, if the orientations of the –SO
3− residues of the neighboring chains do not comply with the above requirement, the reorganization may require additional time and energy. As a result, the growing bottom part of the crystal (at least two cells in thickness) may be defective. It is this defectiveness that probably predetermines the aforementioned broadening and small shifts of the signals in the XRD patterns of the sodium acetate deposit grown on a Nafion substrate compared to that formed on a glass surface. Thus, in this situation, the fibers can by no means act as armoring elements but rather as superficial inclusions, which do not produce a noticeable distortion.
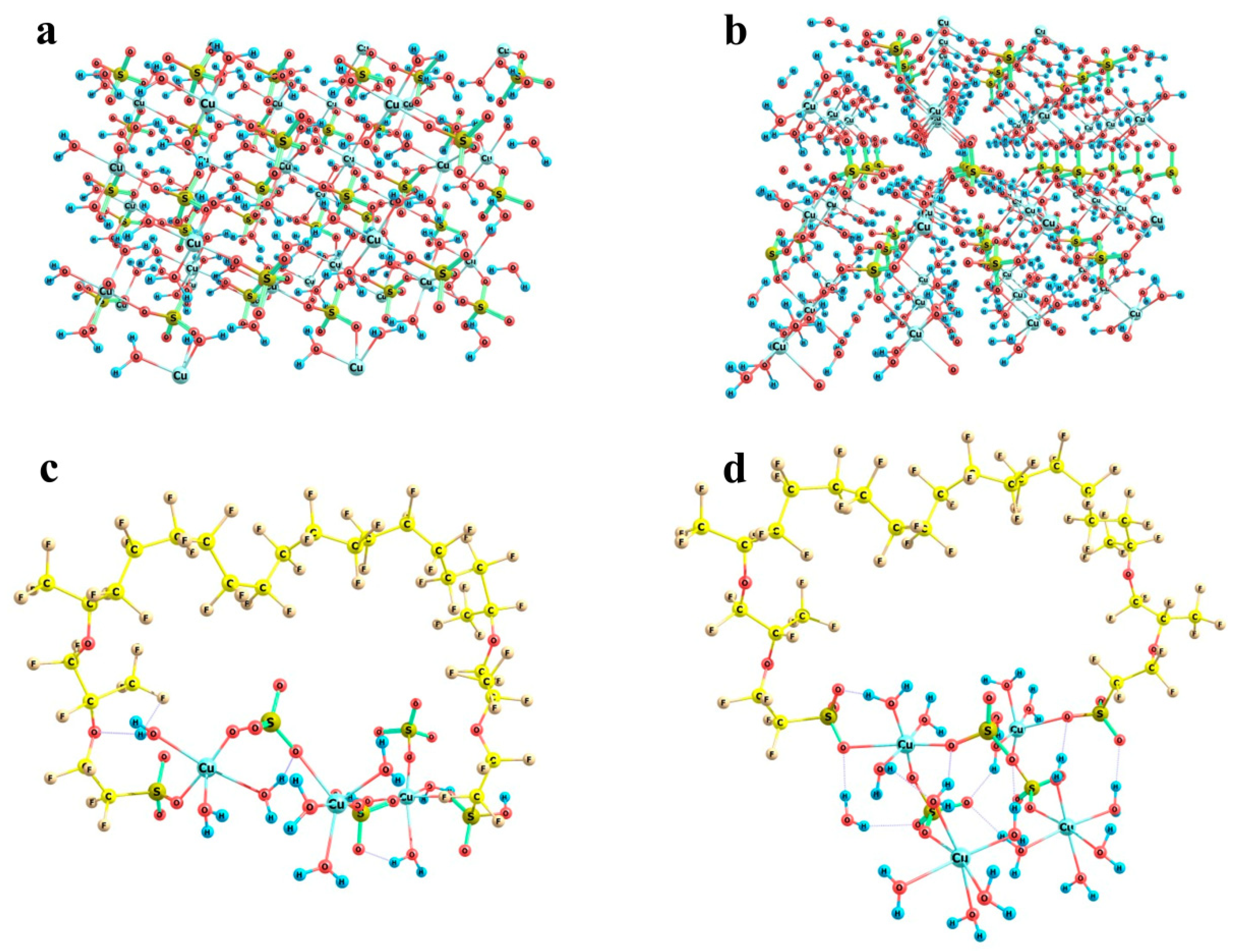
Now, let us turn to the most interesting example of copper sulfate salt. As was mentioned above, it can crystallize to form two different hydrates, namely trihydrate with a monoclinic lattice and pentahydrate with a triclinic lattice. Fragments of both crystal structures are shown in
Figure 14a,b respectively. In trihydrate, the first shell of any copper atom involves three water molecules and three sulfate groups, so that the Cu…O distances are not equal (falling in ranges of 1.96 to 1.98 Å in the case of water oxygens and 1.96 to 2.45 Å in the case of sulfate oxygens), but the directions of bonds are quite regular in view of the replicated cells. In pentahydrate, the situation is quite similar, namely, the first shell of copper involves four water molecules and two sulfate groups, and the sulfates are arranged along a nearly C
4 local symmetry axis (if only oxygens rather than whole water molecules are taken into account). As a result, here the local O-symmetry of a copper site corresponds to an elongated octahedron: two Cu…O distances to sulfates are equal to 2.41 Å, and Cu…O distances to water oxygens are equal in opposing pairs with a slight difference between the pairs (1.97 and 1.94 Å). The higher local symmetry of pentahydrate imposes stronger restrictions on the possible boundaries that may be imposed by Nafion chains, whereas the generally less symmetric trihydrate structure with the variations in the sulfate group orientations makes the problem of fitting of a crystal structure fragment to the space between Nafion chains more easily solvable.
Figure 14c,d illustrates the almost exact inclusion of such fragments. Two variants were considered, namely a (Cu)
3(SO
4)
3(H
2O)
9 cluster (
Figure 14c) and a (Cu)
4(SO
4)
3(H
2O)
16 cluster (
Figure 14d). The initial mutual arrangement of copper ions, sulfate groups, and water molecules in both combined model systems corresponded to the copper sulfate pentahydrate lattice. Then, the structures were optimized at the imposed restrictions on the mutual positions of copper and sulfur atoms of the sulfate groups, as well as the distances between copper atoms and water oxygens in order to prevent the agglomeration of water molecules and the approach of the distant oppositely charged ions, both of which are pronounced at such small relative numbers of water molecules in small ensembles of particles. The two systems of different size were considered in order to illustrate that rotations of the copper sulfate hydrate ensemble with respect to the Nafion chains can provide a compensation for the changes in the distances between the Nafion –SO
3− residues. The corresponding S…S distances in the combined model systems were 12.7 and 11.5 Å, respectively. This means that here Nafion chains can probably be built in the crystal deposit in the most efficient way.
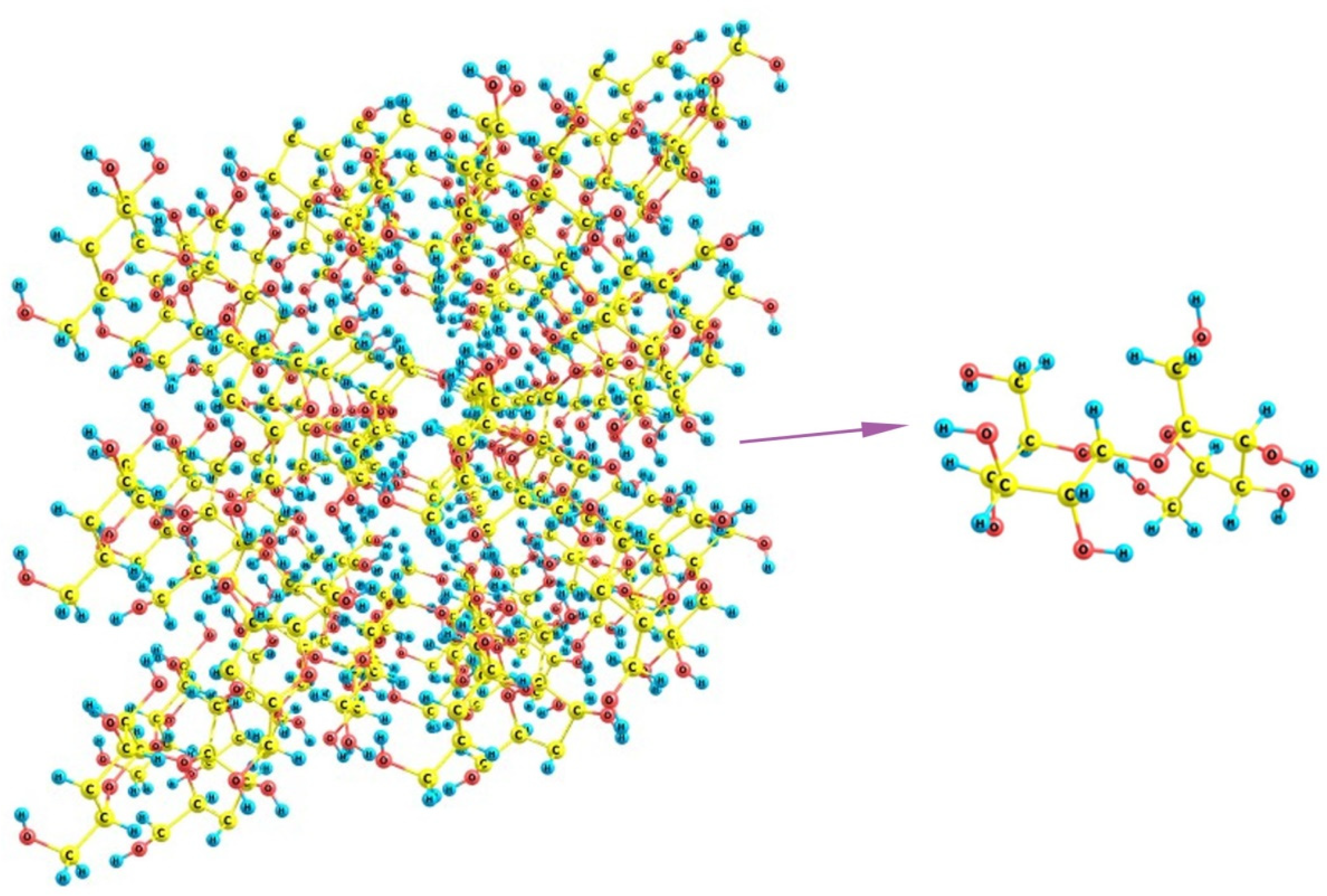
Finally, if we turn to sucrose, it can be noticed that its crystals involve staircase brushes of alternating methylene groups and hydroxyls (
Figure 15) where no distinct regions of predominantly hydrophilic or hydrophobic nature can be distinguished. Additionally, because of the large size of the molecules, which are nevertheless smaller than the above distances between the unfolded SO
3-terminated chains of Nafion, there is no possibility for a regular fitting of sucrose molecule between the fibers. The sole variant is the wavy changes when some two neighboring chains are at a larger distance (about 16 Å between their terminal groups) and, thus, can accommodate two sucrose molecules in between, whereas the distances to their next left- and right-hand-side neighbors are much smaller, so that only one molecule can be located there. This variant is possible only when the orientation of Nafion fibers is not normal to the mean polymer surface, and these are inclined at varying angles. However, such dangling fibers are no longer a regular brush, which could stabilize some cubic, orthorhombic, or monoclinic lattice, but rather an undressed hair that could only distort any kind of molecular organization. Thus, the discrepancy between both individual sucrose molecules and their crystal packing, on one hand, and the possible regular arrangements of Nafion chains, on the other, should make the nucleation of the sucrose deposit strongly unordered and disturbed, which favors the appearance of an amorphous material instead of a crystal.
Summing up, Nafion differs from most coiled or folded polymers susceptible to swelling. Typically, an increase in the volume of the polymer matrix as a result of swelling causes certain mechanochemical or physical changes determined by the acquired inner strains. Usually, such behavior is demonstrated by cross-linked polymers, whose surfaces are not substantially affected by soaking. Nafion is not a cross-linked polymer, but rather a strongly branched one with a drastic difference with respect to hydration between the backbone and side chains. As shown at a nanometer level, these chains should be unfolded upon immersing the polymer in an aqueous solution. Their resulting extension is not as large, about 9 Å, of which only 6 Å segments are reasonably hydrophilic, and the behavior of the segments in water apparently plays a substantial role in the character of the surface processes.
Terminal SO3H groups are shown to be dissociated even in the presence of a small number of water molecules, which means that in actual solutions, the dissociation should always take place. The separation between the side chains, which is about 12 to 14 Å depending on the conformation of the backbone, makes the dissociation of nearly all the groups which are brought in contact with water highly probable. The dissociation in turn promotes further unfolding of other chains and their as regular as possible arrangement over the surface due to the governing balance of electrostatic forces. As a result, –SO3− residues bonded to but separated from the hydrophobic backbone can act as a regular coordinating brush or grid. Their negative charges make them efficient coordinating sites for cations, and when the compound dissolved can crystallize in a lattice, an integer number of whose unit cells can fit in between the extended Nafion chains, crystallization is promoted, and Nafion acts as a true template.
By contrast to typical nearly planar templates, the Nafion template is characterized by a certain variability in the armoring parameters due to the flexibility of the chains. As a result, compounds with different unit cell parameters can be deposited on the Nafion surface. The sole, seemingly principal, restriction is that the syngony should be no lower than monoclinic because of the optimum normal orientation of the chains with respect to the backbone. This is illustrated by the examples of the formation of cubic potassium chloride and monoclinic sodium acetate trihydrate crystal deposits. In the case when the compound can be crystallized in triclinic and monoclinic syngonies, like copper sulfate hydrates, the above feature of Nafion makes the crystallization of monoclinic deposits preferable and selective. At the same time, if the unit cell parameters prevent a stable building of an integer number of the cells in between the Nafion chains and the chemical composition of the molecules is by no means complementary to the hydrophilic segments of the polymer fibers, the deposit becomes amorphous.



 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}