

The Preparation and Characterization of Chitosan/Calcium Phosphate Composite Microspheres for Biomedical Applications


Abstract
1. Introduction
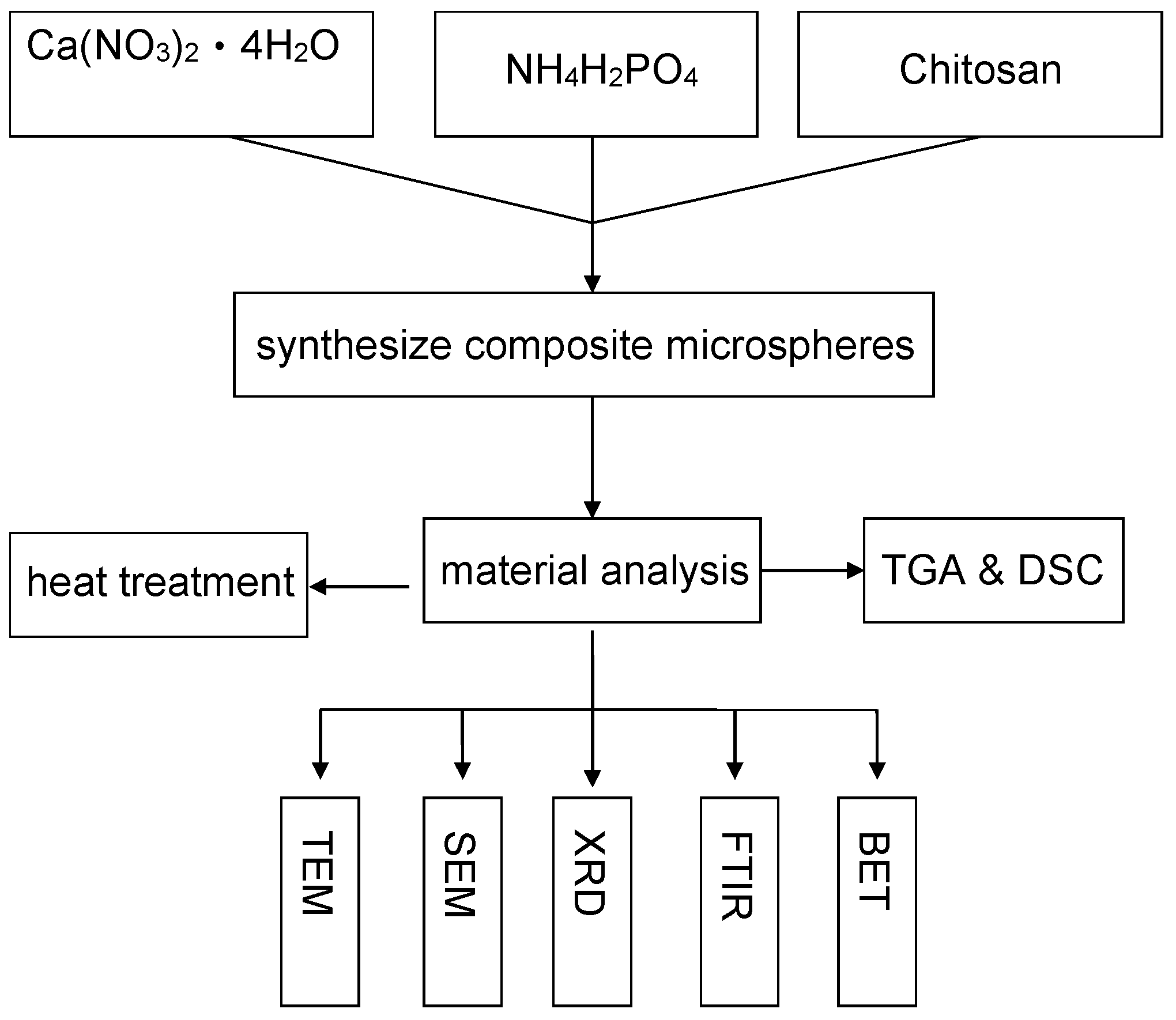
2. Experimental
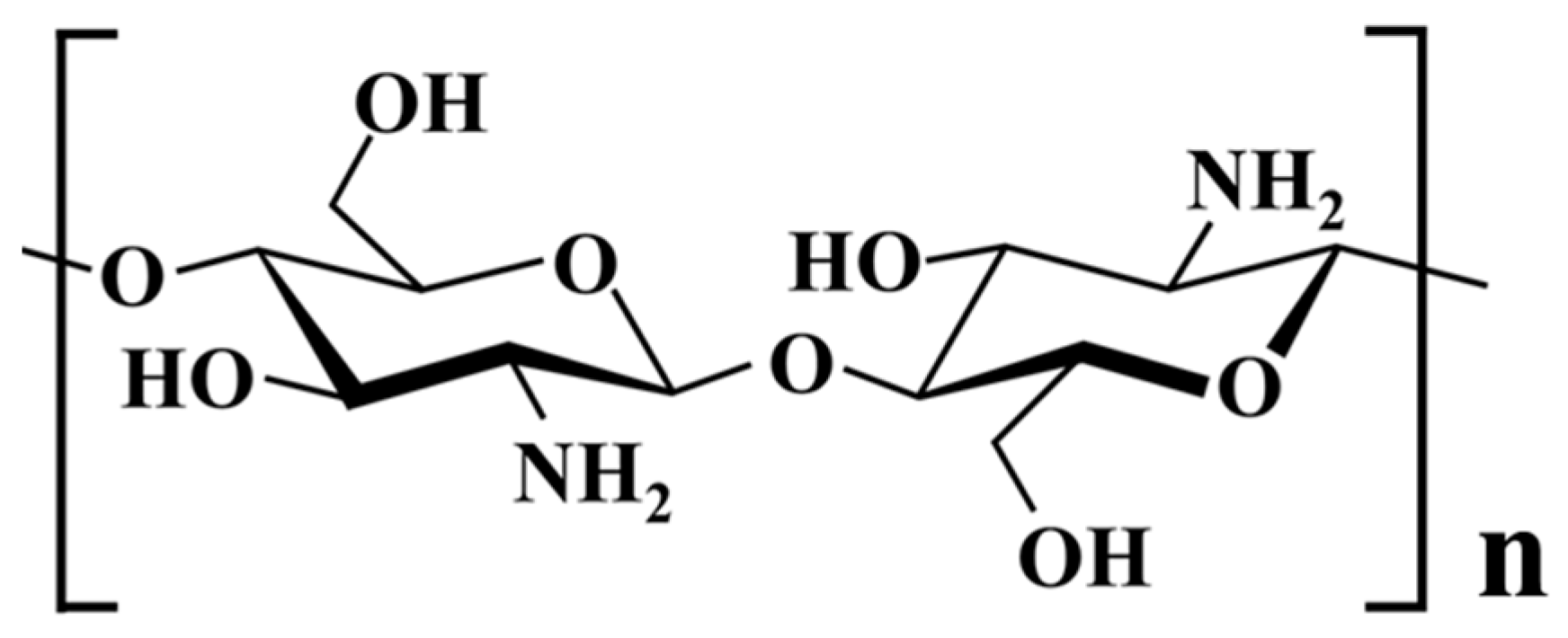
2.1. Materials
2.2. Synthesis of the Porous Chitosan Calcium Phosphate Microsphere (Chi-CaPM)
2.3. Characterization of the Powder
2.3.1. Field Emission Scanning Electron Microscopy (FESEM) Analysis
2.3.2. Fourier Transform Infrared Spectroscopy (FTIR) Analysis
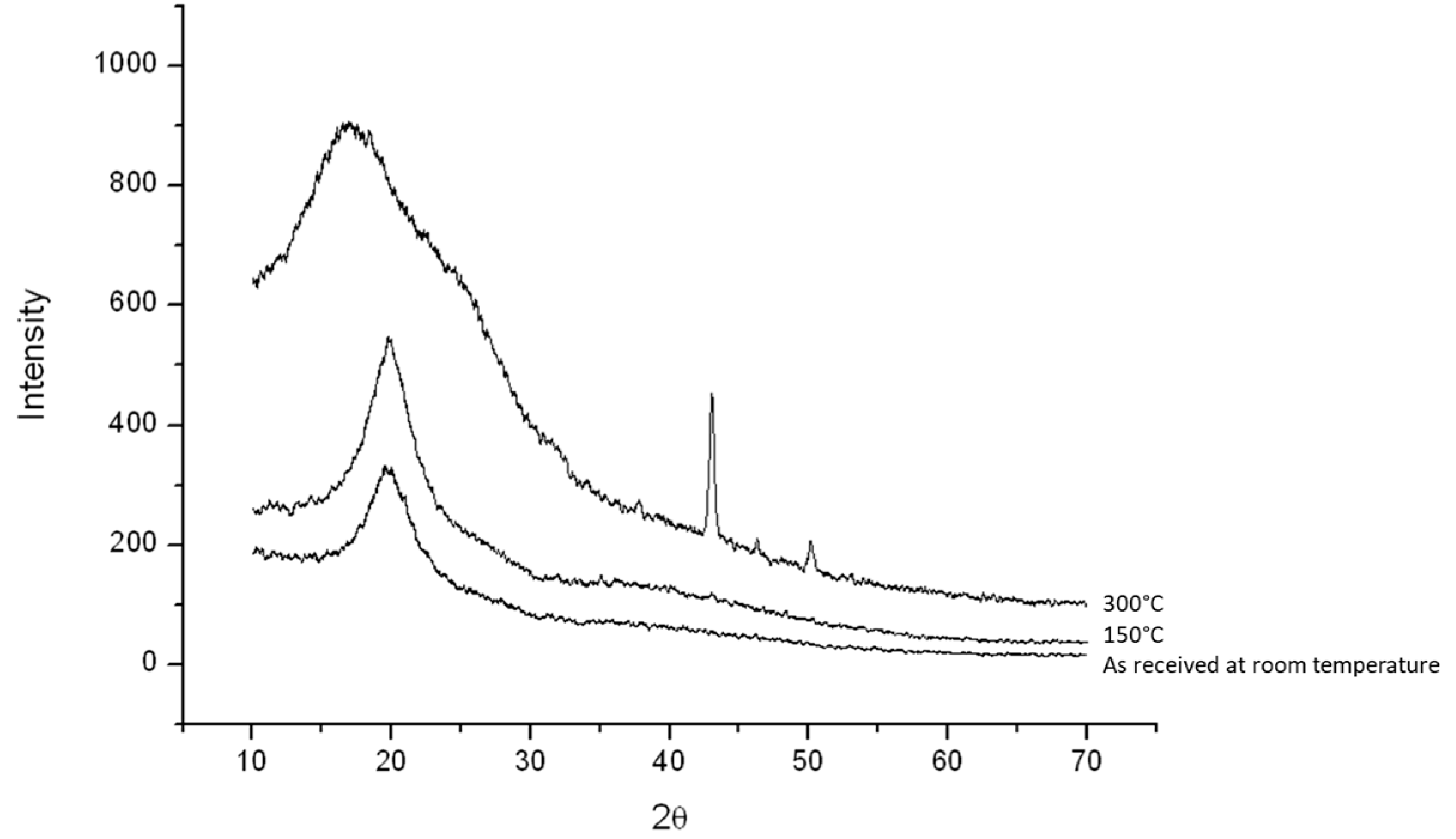
2.3.3. Crystal Structure Analysis
2.3.4. Brunauer–Emmett–Teller (BET) and Barret–Joyner–Halender (BJH) Analysis
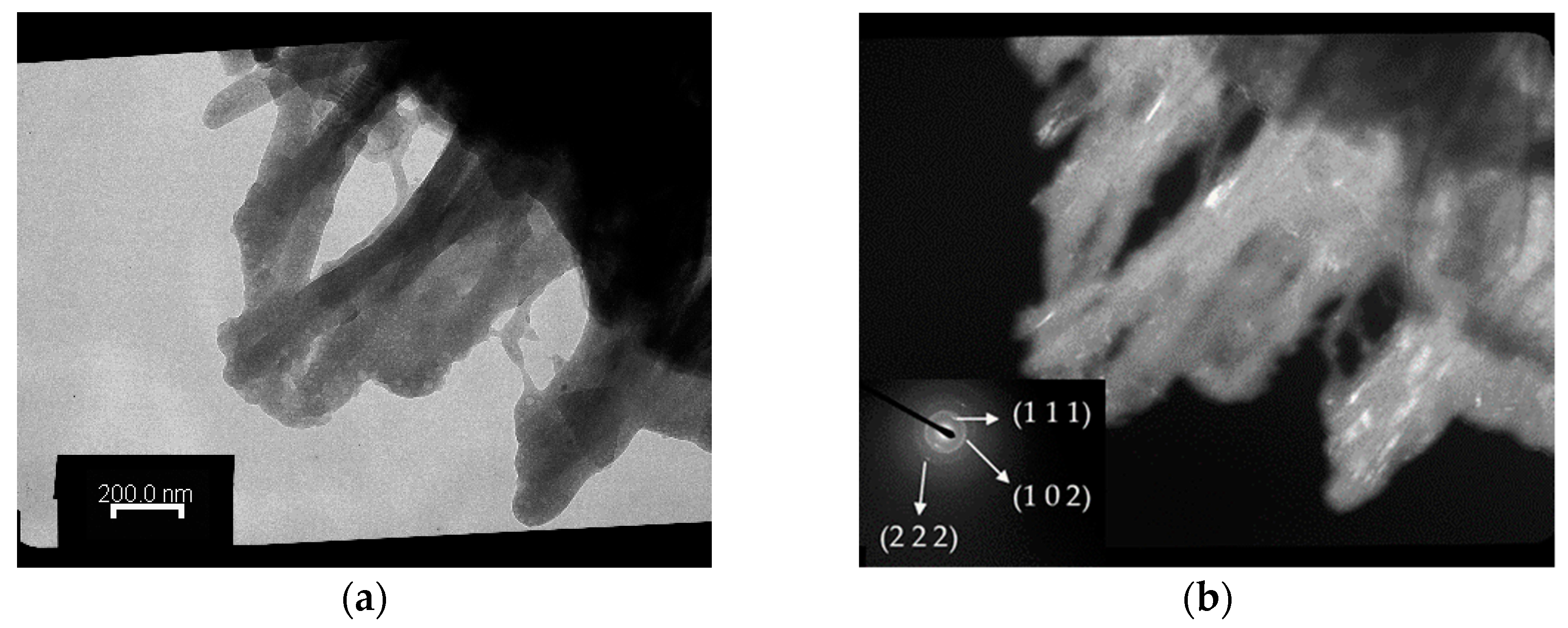
2.3.5. Transmission Electron Microscopy (TEM) Analysis
2.3.6. Inductive Coupled Plasma–Mass Spectrometry (ICP-MS) Analysis
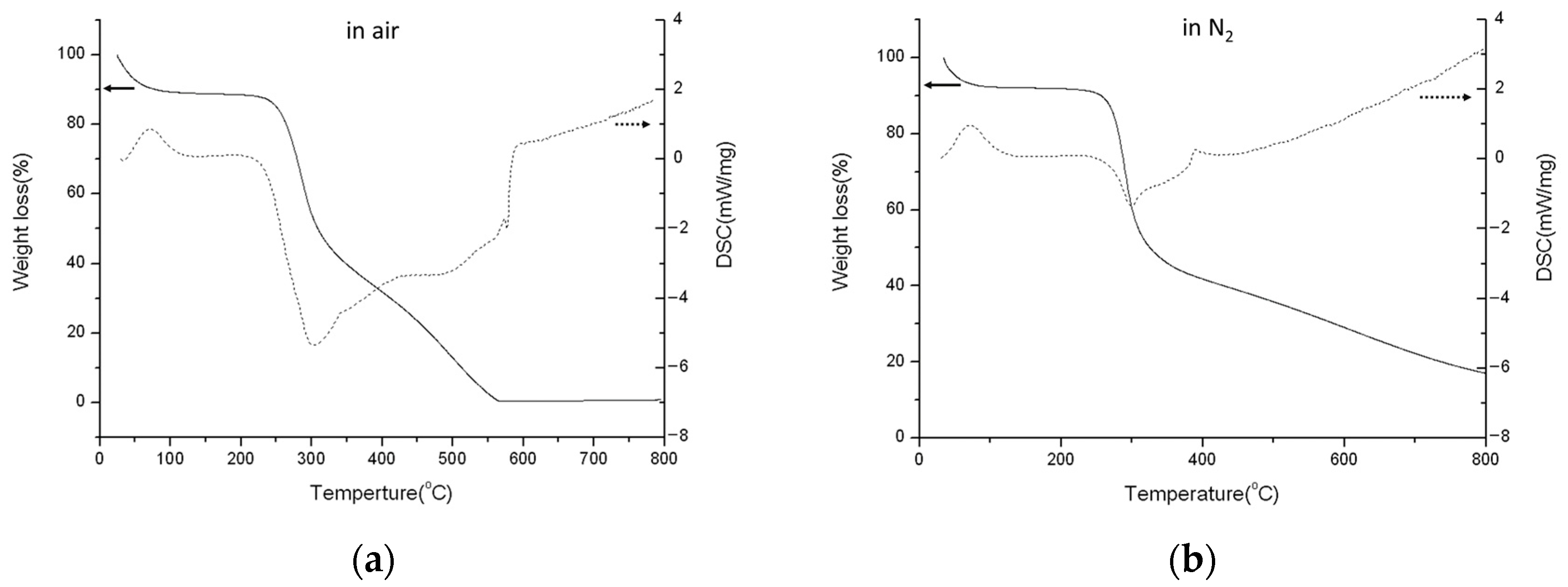
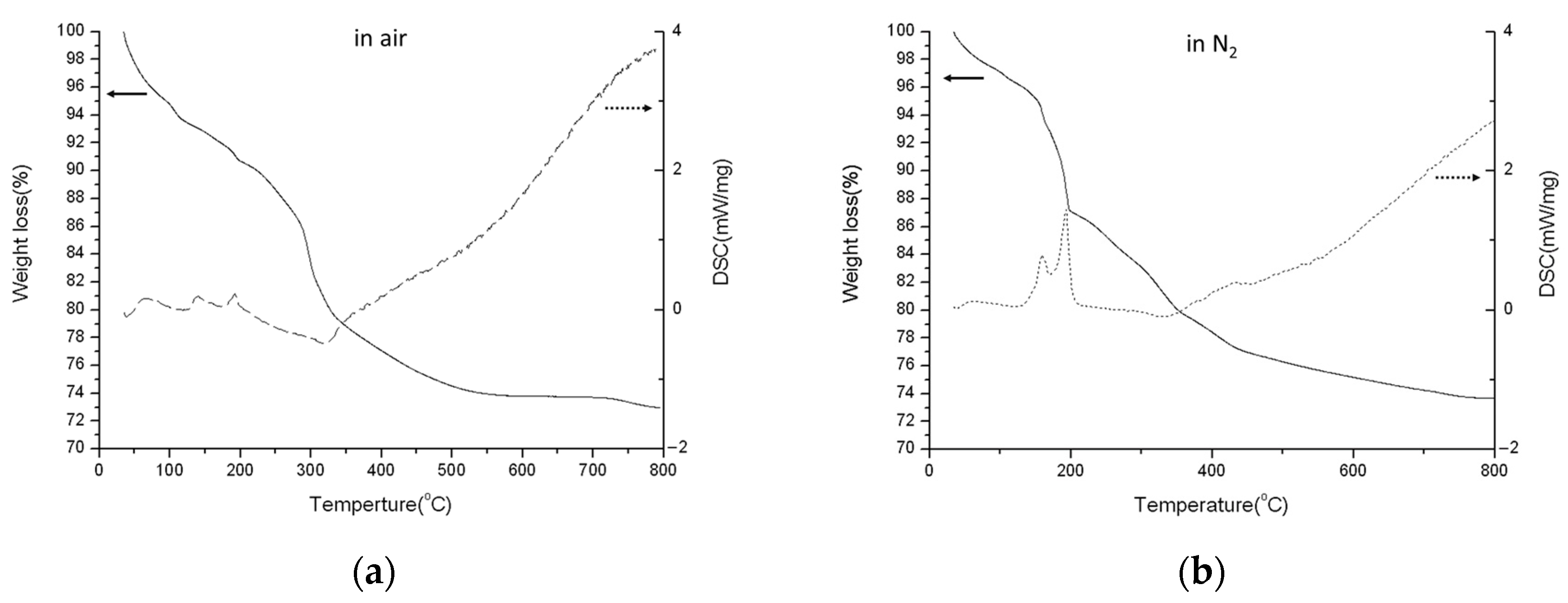
2.4. Thermogravimetric Analysis (TGA)
3. Results and Discussion
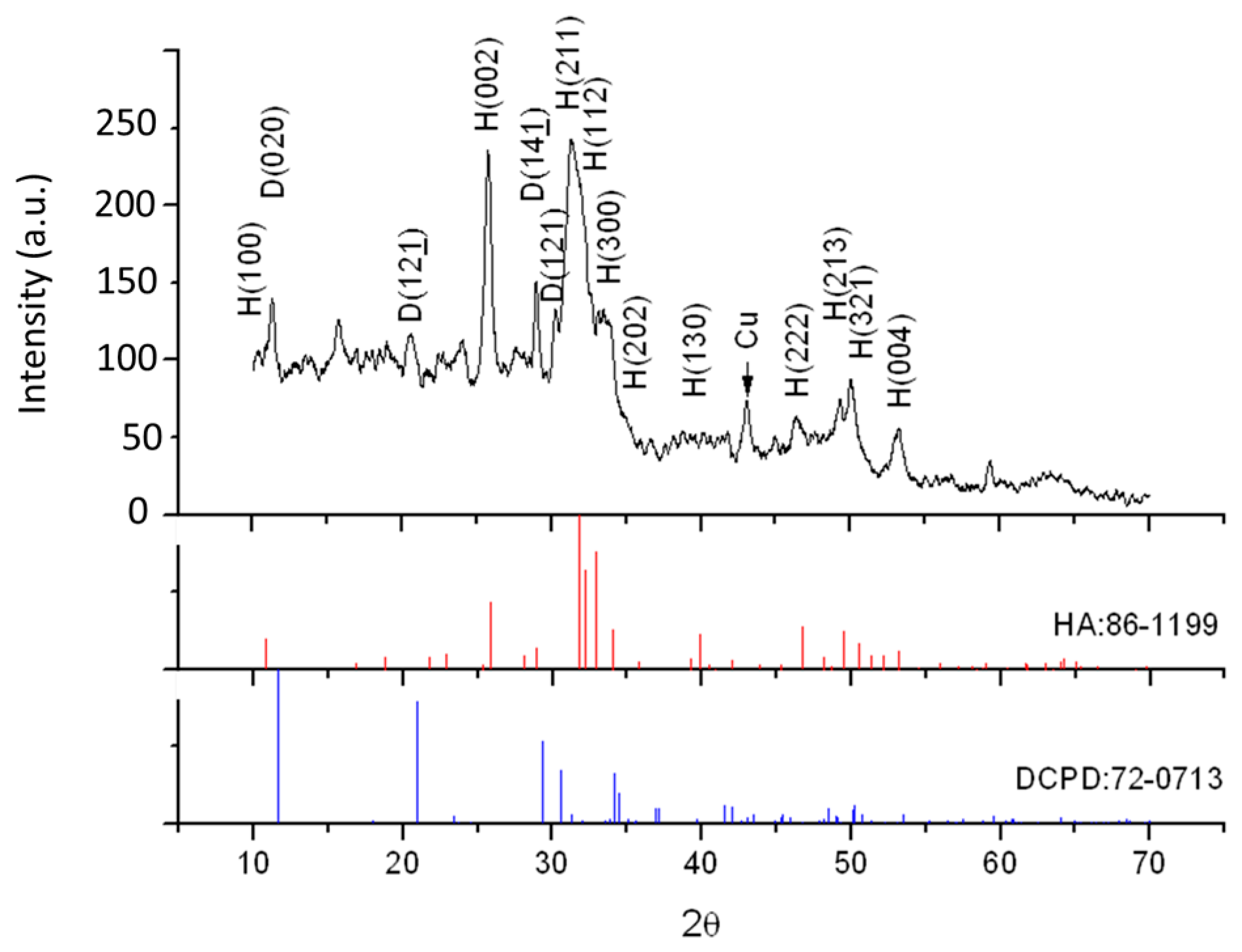
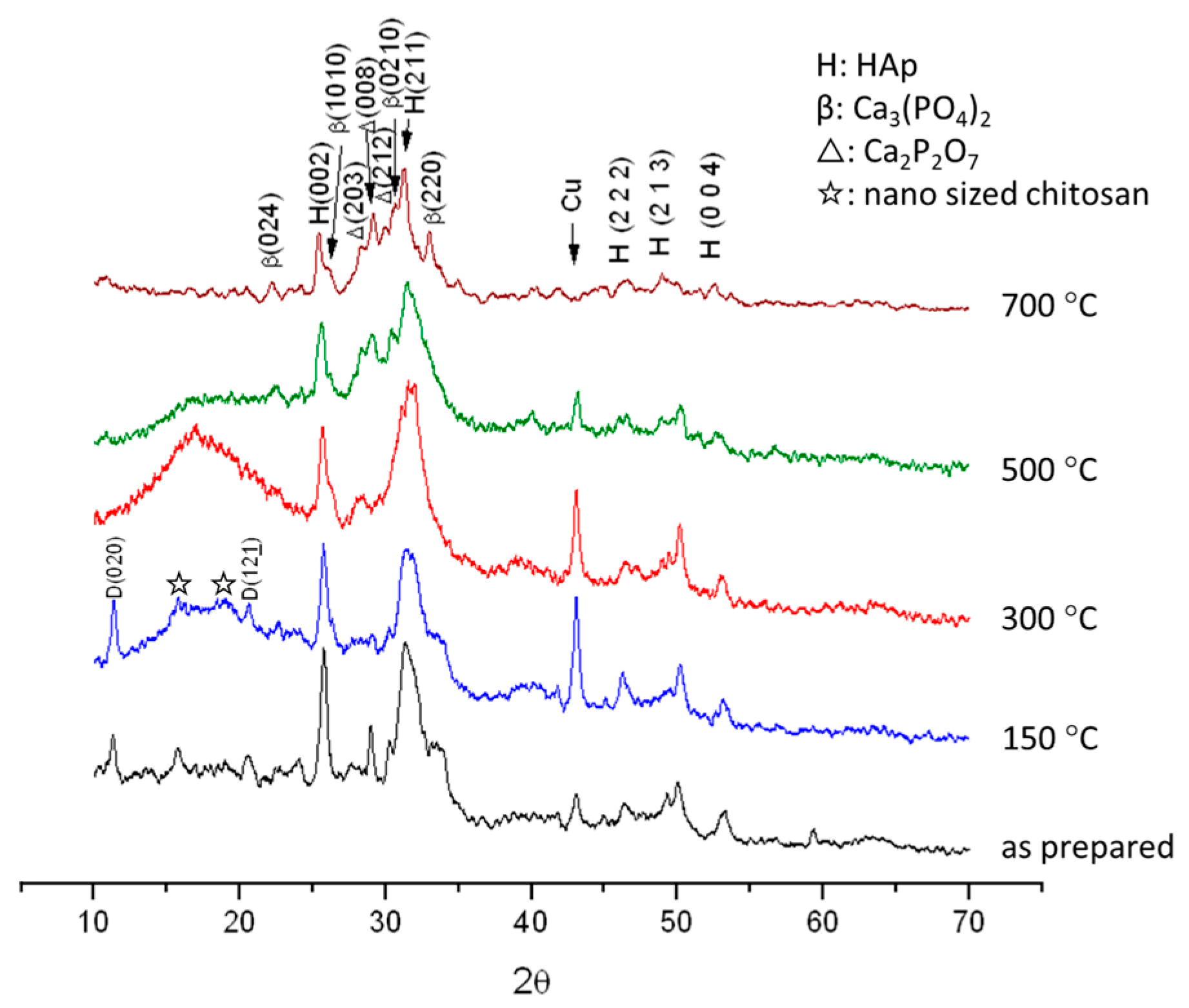
3.1. Crystal Structure and Phase Transformation
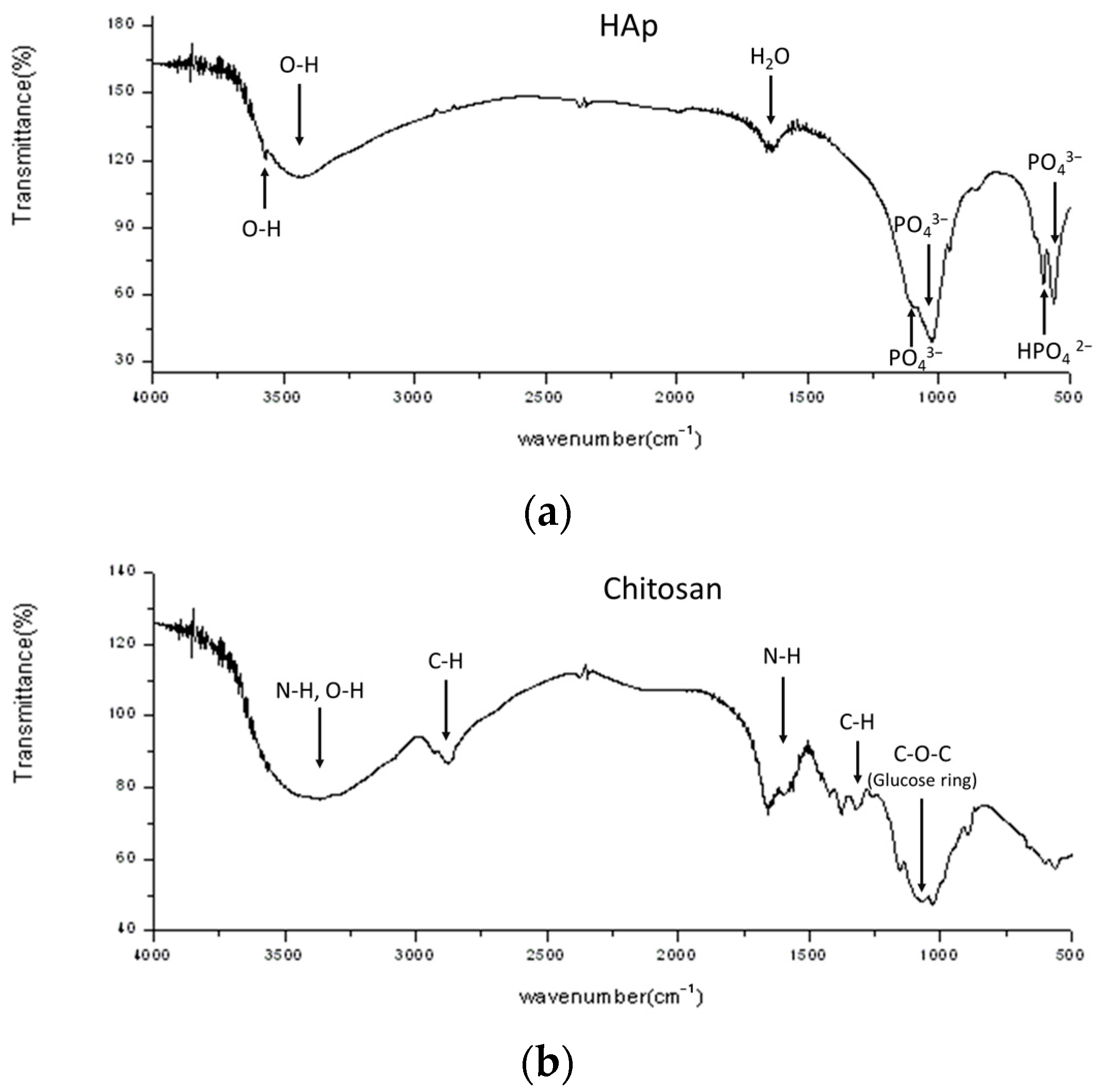
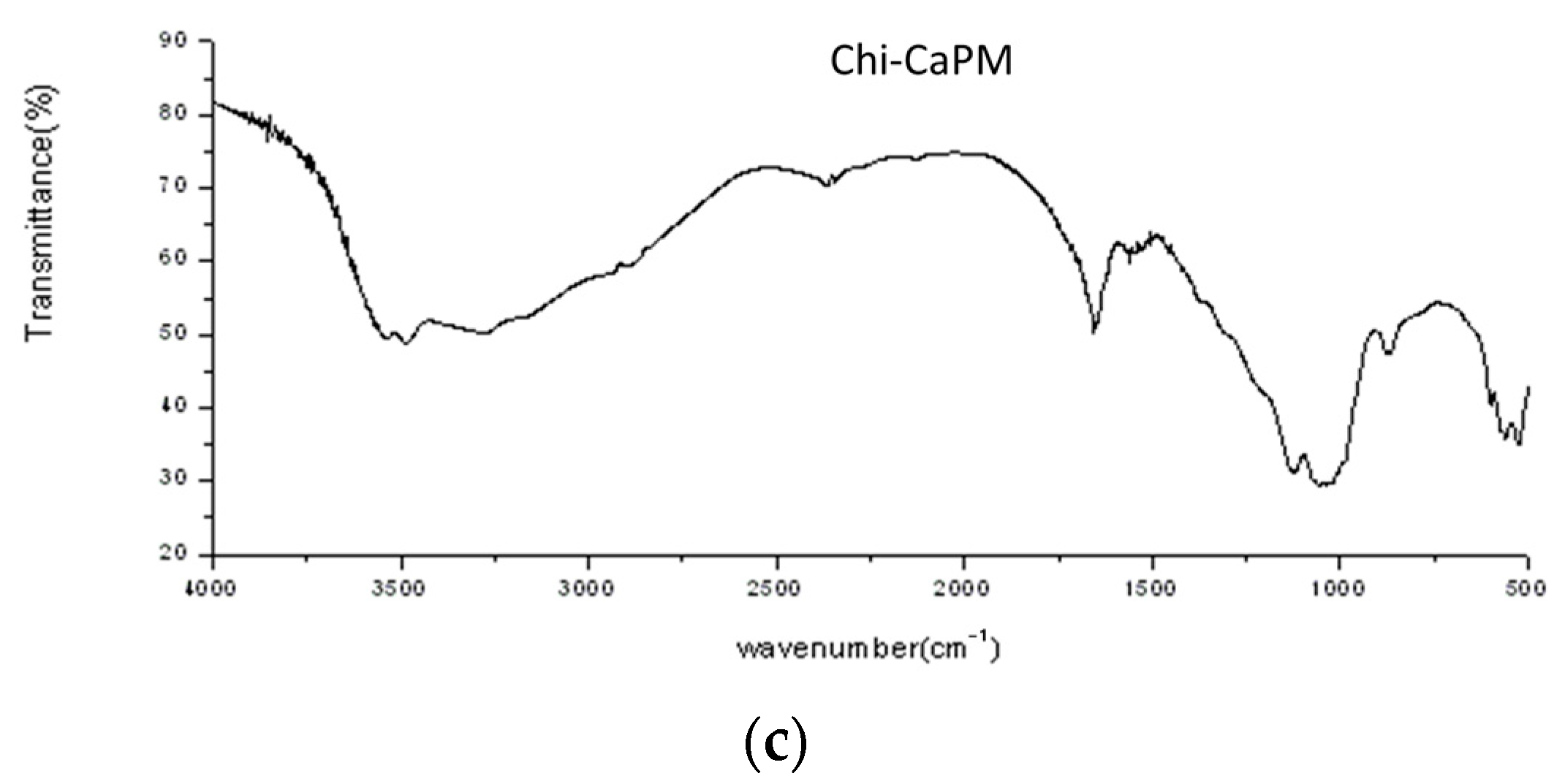
3.2. Chemical Bondings and Compositions
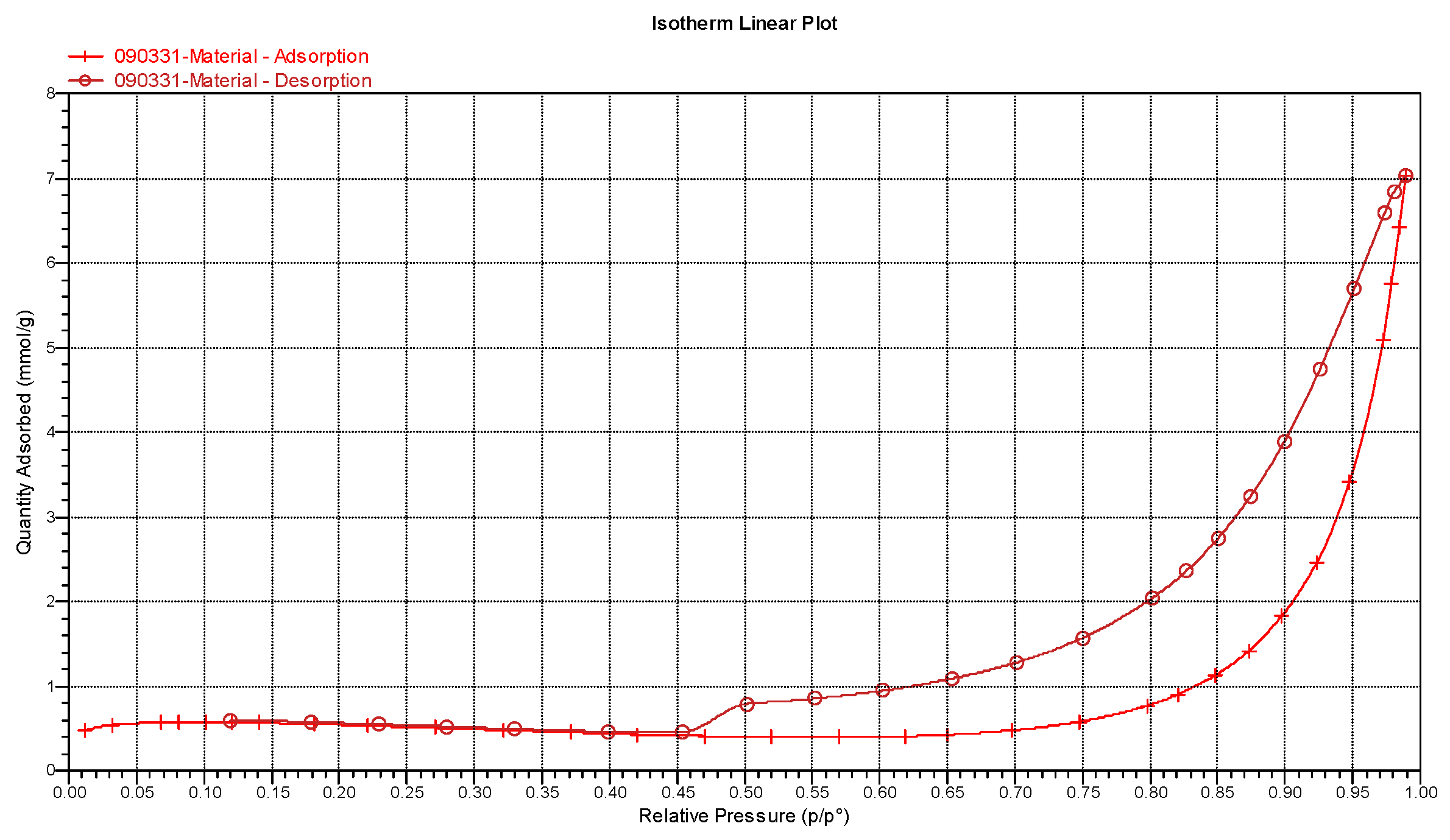
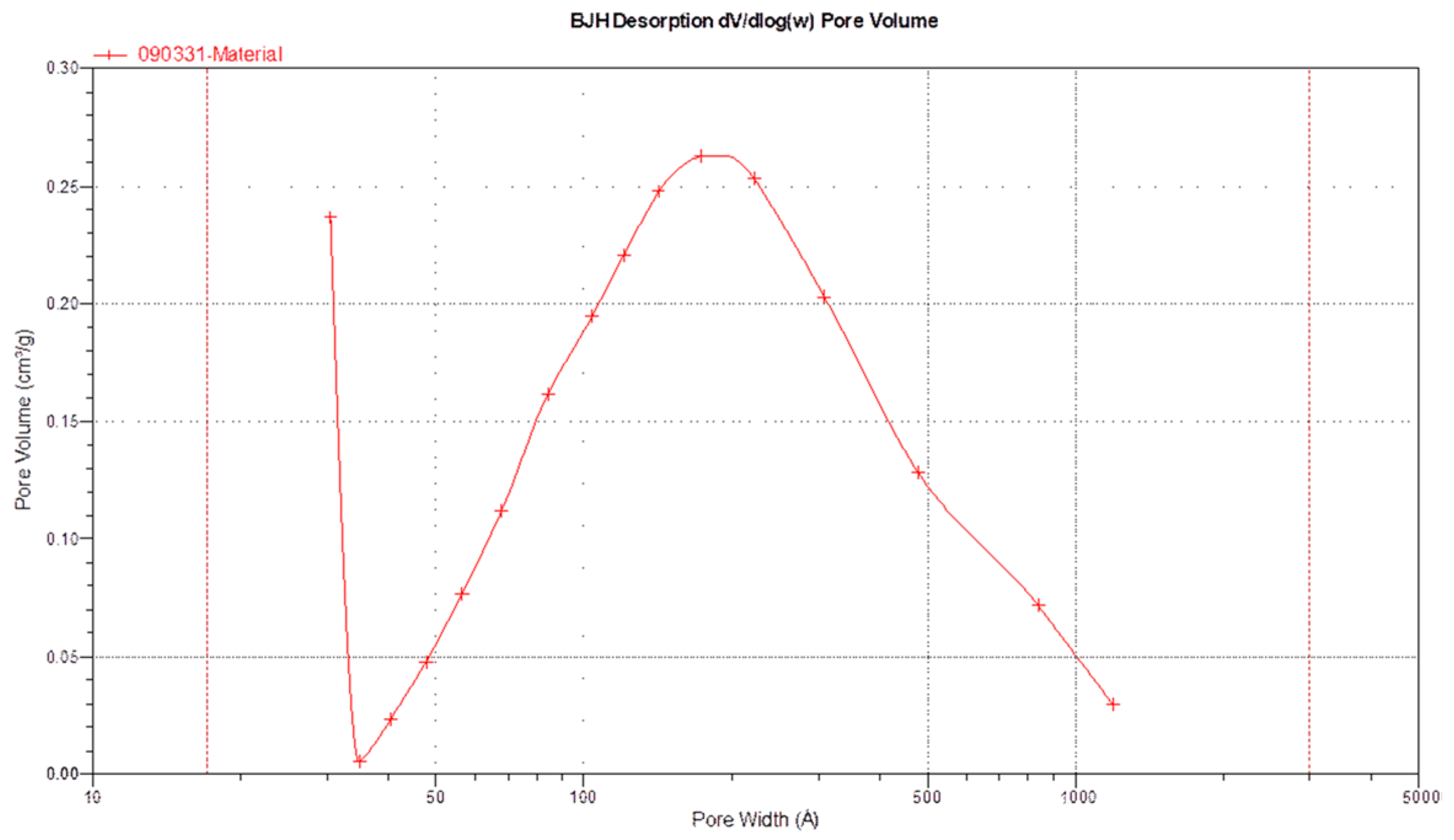
3.3. Surface Morphology, Specific Surface Area, and Pore Volume
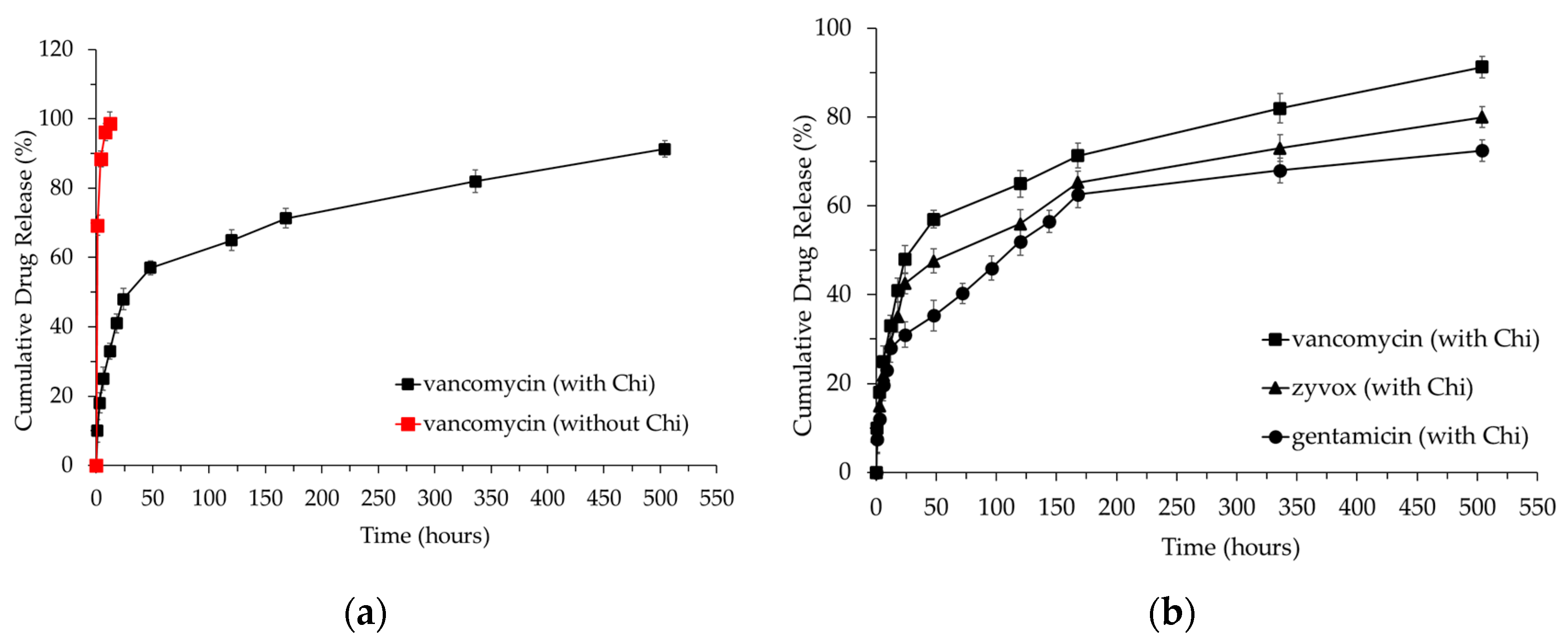
3.4. Antibiotic Loading and Release
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Köse, G.T.; Korkusuz, F.; Özkul, A.; Soysal, Y.; Özdemir, T.; Yildiz, C.; Hasirci, V. Tissue engineered cartilage on collagen and PHBV matrices. Biomaterials 2005, 26, 5187–5197. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Zerankeshi, M.; Alizadeh, R. 3D-printed PLA-Gr-Mg composite scaffolds for bone tissue engineering applications. J. Mater. Res. Technol. 2023, 22, 2440–2446. [Google Scholar] [CrossRef]
- Feng, P.; Shen, S.; Shuai, Y.; Peng, S.; Shuai, C.; Chen, S. PLLA grafting draws GO from PGA phase to the interface in PLLA/PGA bone scaffold owing enhanced interfacial interaction. Sustain. Mater. Technol. 2023, 35, e00566. [Google Scholar] [CrossRef]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Morillo-Bargues, M.J.; Osorno, A.O.; Guerri, C.; Pradas, M.M.; Martínez-Ramos, C. Characterization of Electrospun BDMC-Loaded PLA Nanofibers with Drug Delivery Function and Anti-Inflammatory Activity. Int. J. Mol. Sci. 2023, 24, 10340. [Google Scholar] [CrossRef]
- Singh, B.; Singh, S.; Gautam, A.; Sutherland, A.; Pal, K. Preparation and characterization of PLA microspheres as drug delivery system for controlled release of Cetirizine with carbon dots as drug carrier. Polym. Bull. 2023, 80, 5741–5757. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, W.; Lu, Y.; Xu, Y.; Wang, C.; Yu, D.-G.; Kim, I. Recent Advances in Poly(α-L-glutamic acid)-Based Nanomaterials for Drug Delivery. Biomolecules 2022, 12, 636. [Google Scholar] [CrossRef] [PubMed]
- Stamatialis, D.F.; Papenburg, B.J.; Gironés, M.; Saiful, S.; Bettahalli, S.N.M.; Schmitmeier, S.; Wessling, M. Medical applications of membranes: Drug delivery, artificial organs and tissue engineering. J. Membr. Sci. 2008, 308, 1–34. [Google Scholar] [CrossRef]
- Murata, Y.; Yutaka, Y.; Hirata, R.; Hidaka, Y.; Hamaji, M.; Yoshizawa, A.; Kishimoto, Y.; Omori, K.; Date, H. Development of novel layered polyglycolic acid sheet for regeneration of critical-size defect in rat trachea. Eur. J. Cardio-Thorac. Surg. 2023, 63. [Google Scholar] [CrossRef]
- Masuko, T.; Iwasaki, N.; Yamane, S.; Funakoshi, T.; Majima, T.; Minami, A.; Ohsuga, N.; Ohta, T.; Nishimura, S.-I. Chitosan–RGDSGGC conjugate as a scaffold material for musculoskeletal tissue engineering. Biomaterials 2005, 26, 5339–5347. [Google Scholar] [CrossRef]
- Hoemann, C.D.; Sun, J.; Légaré, A.; McKee, M.D.; Buschmann, M.D. Tissue engineering of cartilage using an injectable and adhesive chitosan-based cell-delivery vehicle. Osteoarthr. Cartil. 2005, 13, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Riseh, R.S.; Vazvani, M.G.; Kennedy, J.F. The application of chitosan as a carrier for fertilizer: A review. Int. J. Biol. Macromol. 2023, 252, 126483. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhong, S.; Wang, J.; Gao, Y.; Cui, X. Advances in chitosan-based microcapsules and their applications. Carbohydr. Polym. 2023, 300, 120265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Alsarra, I.A.; Neau, S.H. An in vitro evaluation of a chitosan-containing multiparticulate system for macromolecule delivery to the colon. Int. J. Pharm. 2002, 239, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Shao, H.; Li, X.; Ullah, M.W.; Luo, G.; Xu, Z.; Ma, L.; He, X.; Lei, Z.; Li, Q.; et al. Injectable immunomodulation-based porous chitosan microspheres/HPCH hydrogel composites as a controlled drug delivery system for osteochondral regeneration. Biomaterials 2022, 285, 121530. [Google Scholar] [CrossRef] [PubMed]
- Nsereko, S.; Amiji, M. Localized delivery of paclitaxel in solid tumors from biodegradable chitin microparticle formulations. Biomaterials 2002, 23, 2723–2731. [Google Scholar] [CrossRef] [PubMed]
- Pandey, N.; Bohra, B.S.; Tiwari, H.; Pal, M.; Negi, P.B.; Dandapat, A.; Mehta, S.P.S.; Sahoo, N.G. Development of biodegradable chitosan/graphene oxide nanocomposite via spray drying method for drug loading and delivery application. J. Drug Deliv. Sci. Technol. 2022, 74, 103555. [Google Scholar] [CrossRef]
- Ogunjimi, A.T.; Fiegel, J.; Brogden, N.K. Design and Characterization of Spray-Dried Chitosan-Naltrexone Microspheres for Microneedle-Assisted Transdermal Delivery. Pharmaceutics 2020, 12, 496. [Google Scholar] [CrossRef]
- Shi, X.-Y.; Tan, T.-W. Preparation of chitosan/ethylcellulose complex microcapsule and its application in controlled release of Vitamin D2. Biomaterials 2002, 23, 4469–4473. [Google Scholar] [CrossRef]
- Zhang, S.; Kang, L.; Hu, S.; Hu, J.; Fu, Y.; Hu, Y.; Yang, X. Carboxymethyl chitosan microspheres loaded hyaluronic acid/gelatin hydrogels for controlled drug delivery and the treatment of inflammatory bowel disease. Int. J. Biol. Macromol. 2021, 167, 1598–1612. [Google Scholar] [CrossRef]
- Li, H.; Huo, J.; Zhang, H.; Liu, Y.; Shi, X.; Zhao, Z.; Zhou, J.; Wang, X.; Zhang, C. Eudragit S100-coated halloysite nanotube/chitosan microspheres for colon-targeted release of paeoniflorin. J. Drug Deliv. Sci. Technol. 2021, 61, 102258. [Google Scholar] [CrossRef]
- Tsai, M.L.; Bai, S.W.; Chen, R.H. Cavitation effects versus stretch effects resulted in different size and polydispersity of ionotropic gelation chitosan–sodium tripolyphosphate nanoparticle. Carbohydr. Polym. 2008, 71, 448–457. [Google Scholar] [CrossRef]
- Algharib, S.A.; Dawood, A.; Zhou, K.; Chen, D.; Li, C.; Meng, K.; Zhang, A.; Luo, W.; Ahmed, S.; Huang, L.; et al. Preparation of chitosan nanoparticles by ionotropic gelation technique: Effects of formulation parameters and in vitro characterization. J. Mol. Struct. 2022, 1252, 132129. [Google Scholar] [CrossRef]
- Abbas, A.; Alhamdany, A. Floating Microspheres of Enalapril Maleate as a Developed Controlled Release Dosage Form: Investigation of the Effect of an Ionotropic Gelation Technique. Turk. J. Pharm. Sci. 2020, 17, 159–171. [Google Scholar] [CrossRef]
- Bayomi, M.A.; Al-Suwayeh, S.A.; El-Helw, A.M.; Mesnad, A.F. Preparation of casein–chitosan microspheres containing diltiazem hydrochloride by an aqueous coacervation technique. Pharm. Acta Helv. 1998, 73, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Çelik, Ö.; Akbuğa, J. Preparation of superoxide dismutase loaded chitosan microspheres: Characterization and release studies. Eur. J. Pharm. Biopharm. 2007, 66, 42–47. [Google Scholar] [CrossRef]
- Wei, W.; Wang, L.-Y.; Yuan, L.; Yang, X.-D.; Su, Z.-G.; Ma, G.-H. Bioprocess of uniform-sized crosslinked chitosan microspheres in rats following oral administration. Eur. J. Pharm. Biopharm. 2008, 69, 878–886. [Google Scholar] [CrossRef]
- Teterina, A.Y.; Minaychev, V.V.; Smirnova, P.V.; Kobiakova, M.I.; Smirnov, I.V.; Fadeev, R.S.; Egorov, A.A.; Ashmarin, A.A.; Pyatina, K.V.; Senotov, A.S.; et al. Injectable Hydrated Calcium Phosphate Bone-like Paste: Synthesis, In Vitro, and In Vivo Biocompatibility Assessment. Technologies 2023, 11, 77. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, T.; Xu, Y.; Liang, C.; Li, G.; Guo, Y.; Zhang, Z.; Lian, J.; Ren, L. Double-layer calcium phosphate sandwiched siloxane composite coating to enhance corrosion resistance and biocompatibility of magnesium alloys for bone tissue engineering. Prog. Org. Coat. 2023, 177, 107417. [Google Scholar] [CrossRef]
- Habraken, W.J.E.M.; Wolke, J.G.C.; Jansen, J.A. Ceramic composites as matrices and scaffolds for drug delivery in tissue engineering. Adv. Drug Deliv. Rev. 2007, 59, 234–248. [Google Scholar] [CrossRef]
- Dorozhkin, S.V.; Epple, M. Biological and Medical Significance of Calcium Phosphates. Angew. Chem. Int. Ed. 2002, 41, 3130–3146. [Google Scholar] [CrossRef]
- Raymond, Y.; Bonany, M.; Lehmann, C.; Thorel, E.; Benítez, R.; Franch, J.; Espanol, M.; Solé-Martí, X.; Manzanares, M.-C.; Canal, C.; et al. Hydrothermal processing of 3D-printed calcium phosphate scaffolds enhances bone formation in vivo: A comparison with biomimetic treatment. Acta Biomater. 2021, 135, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Syukkalova, E.A.; Sadetskaya, A.V.; Demidova, N.D.; Bobrysheva, N.P.; Osmolowsky, M.G.; Voznesenskiy, M.A.; Osmolovskaya, O.M. The effect of reaction medium and hydrothermal synthesis conditions on morphological parameters and thermal behavior of calcium phosphate nanoparticles. Ceram. Int. 2021, 47, 2809–2821. [Google Scholar] [CrossRef]
- Hattori, T.; Lwadate, Y. Hydrothermal preparation of calcium hydroxyapatite powders. J. Am. Ceram. Soc. 1990, 73, 1803–1805. [Google Scholar] [CrossRef]
- Laonapakul, T.; Sutthi, R.; Chaikool, P.; Talangkun, S.; Boonma, A.; Chindaprasirt, P. Calcium phosphate powders synthesized from CaCO3 and CaO of natural origin using mechanical activation in different media combined with solid-state interaction. Mater. Sci. Eng. C 2021, 118, 111333. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, S.; Agarwal, A.K.; Rai, K.N.; Garg, A. Development of high strength hydroxyapatite by solid-state-sintering process. Ceram. Int. 2007, 33, 419–426. [Google Scholar] [CrossRef]
- Fonseca, R.L.M.; de Souza Gonçalves, B.; Balarini, J.C.; Nunes, E.H.M.; Houmard, M. Deliquescent behavior of calcium phosphate materials synthesized by sol–gel technique. J. Sol-Gel Sci. Technol. 2021, 97, 404–413. [Google Scholar] [CrossRef]
- Wang, F.; Liu, M.; Lu, Y.; Qi, Y. A simple sol–gel technique for preparing hydroxyapatite nanopowders. Mater. Lett. 2005, 59, 916–919. [Google Scholar]
- Gutiérrez-Arenas, D.A.; Cuca-García, M.; Méndez-Rojas, M.A.; Pro-Martínez, A.; Becerril-Pérez, C.M.; Mendoza-Álvarez, M.E.; Ávila-Ramos, F.; Ramírez-Bribiesca, J.E. Designing Calcium Phosphate Nanoparticles with the Co-Precipitation Technique to Improve Phosphorous Availability in Broiler Chicks. Animals 2021, 11, 2773. [Google Scholar] [CrossRef]
- Bernard, L.; Freche, M.; Lacout, J.L.; Biscans, B. Preparation of hydroxyapatite by neutralization at low temperature—Influence of purity of the raw material. Powder Technol. 1999, 103, 19–25. [Google Scholar] [CrossRef]
- Nagata, F.; Miyajima, T.; Yokogawa, Y. A method to fabricate hydroxyapatite/poly(lactic acid) microspheres intended for biomedical application. J. Eur. Ceram. Soc. 2006, 26, 533–535. [Google Scholar] [CrossRef]
- Radwan, N.H.; Nasr, M.; Ishak, R.A.H.; Abdeltawab, N.F.; Awad, G.A.S. Chitosan-calcium phosphate composite scaffolds for control of post-operative osteomyelitis: Fabrication, characterization, and in vitro–in vivo evaluation. Carbohydr. Polym. 2020, 244, 116482. [Google Scholar] [CrossRef] [PubMed]
- Ressler, A.; Ohlsbom, R.; Žužić, A.; Gebraad, A.; Frankberg, E.J.; Pakarinen, T.-K.; Ivanković, H.; Miettinen, S.; Ivanković, M. Chitosan/collagen/Mg, Se, Sr, Zn-substituted calcium phosphate scaffolds for bone tissue engineering applications: A growth factor free approach. Eur. Polym. J. 2023, 194, 112129. [Google Scholar] [CrossRef]
- Sinha, A.; Mishra, T.; Ravishankar, N. Polymer assisted hydroxyapatite microspheres suitable for biomedical application. J. Mater. Sci. Mater. Med. 2008, 19, 2009–2013. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.-Q.; Chu, X.; Huang, N.-P.; Wang, T.; Wang, Y.; Shi, X.; Ding, Y.; Gu, Z.-Z. The effect of nanofibrous galactosylated chitosan scaffolds on the formation of rat primary hepatocyte aggregates and the maintenance of liver function. Biomaterials 2009, 30, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Li, B.; Wang, M.; Shen, J. Preparation and characterization of biodegradable chitosan/hydroxyapatite nanocomposite rods via in situ hybridization: A potential material as internal fixation of bone fracture. Biomaterials 2004, 25, 779–785. [Google Scholar] [CrossRef]
- Iqbal, N.; Braxton, T.M.; Anastasiou, A.; Raif, E.M.; Chung, C.K.; Kumar, S.; Giannoudis, P.V.; Jha, A. Dicalcium Phosphate Dihydrate Mineral Loaded Freeze-Dried Scaffolds for Potential Synthetic Bone Applications. Materials 2022, 15, 6245. [Google Scholar] [CrossRef]
- Kong, L.; Gao, Y.; Cao, W.; Gong, Y.; Zhao, N.; Zhang, X. Preparation and characterization of nano-hydroxyapatite/chitosan composite scaffolds. J. Biomed. Mater. Res. Part A 2005, 75A, 275–282. [Google Scholar] [CrossRef]
- Feng, X.; Ma, L.; Lei, J.; Ouyang, Q.; Zeng, Y.; Luo, Y.; Zhang, X.; Song, Y.; Li, G.; Tan, L.; et al. Piezo-Augmented Sonosensitizer with Strong Ultrasound-Propelling Ability for Efficient Treatment of Osteomyelitis. ACS Nano 2022, 16, 2546–2557. [Google Scholar] [CrossRef]
- Meroni, G.; Tsikopoulos, A.; Tsikopoulos, K.; Allemanno, F.; Martino, P.A.; Soares Filipe, J.F. A Journey into Animal Models of Human Osteomyelitis: A Review. Microorganisms 2022, 10, 1135. [Google Scholar] [CrossRef]
- Cheng, Y.; Zhang, Y.; Zhao, Z.; Li, G.; Li, J.; Li, A.; Xue, Y.; Zhu, B.; Wu, Z.; Zhang, X. Guanidinium-Decorated Nanostructure for Precision Sonodynamic-Catalytic Therapy of MRSA-Infected Osteomyelitis. Adv. Mater. 2022, 34, 2206646. [Google Scholar] [CrossRef] [PubMed]
- Zelmer, A.R.; Nelson, R.; Richter, K.; Atkins, G.J. Can intracellular Staphylococcus aureus in osteomyelitis be treated using current antibiotics? A systematic review and narrative synthesis. Bone Res. 2022, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Zhang, Z.; Zhuang, Y.; Yang, H.; Han, Y.; Wu, Q.; Jia, X.; Yin, Y.; Qu, X.; Zheng, Y.; et al. High-strength biodegradable zinc alloy implants with antibacterial and osteogenic properties for the treatment of MRSA-induced rat osteomyelitis. Biomaterials 2022, 287, 121663. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Shao, J.; Ma, J.; Zou, Q.; Li, J.; Zuo, Y.; Yang, F.; Li, Y. Development of ciprofloxacin and nano-hydroxyapatite dual-loaded polyurethane scaffolds for simultaneous treatment of bone defects and osteomyelitis. Mater. Lett. 2019, 253, 86–89. [Google Scholar] [CrossRef]
- Zhong, C.; Wu, Y.; Lin, H.; Liu, R. Advances in the antimicrobial treatment of osteomyelitis. Compos. Part B Eng. 2023, 249, 110428. [Google Scholar] [CrossRef]
- ASTM F1185-03; Standard Specification for Composition of Hydroxylapatite for Surgical Implants. Available online: https://webstore.ansi.org/standards/astm/astmf118503 (accessed on 4 January 2024).
- Morsy, M.; Mostafa, K.; Amyn, H.; El-Ebissy, A.A.-h.; Salah, A.M.; Youssef, M.A. Synthesis and characterization of freeze dryer chitosan nano particles as multi functional eco-friendly finish for fabricating easy care and antibacterial cotton textiles. Egypt. J. Chem. 2019, 62, 1277–1293. [Google Scholar] [CrossRef]
- Gheisari, H.; Karamian, E.; Abdellahi, M. A novel hydroxyapatite–Hardystonite nanocomposite ceramic. Ceram. Int. 2015, 41, 5967–5975. [Google Scholar] [CrossRef]
- Sing, K.S. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Wang, Y.; Moo, Y.X.; Chen, C.; Gunawan, P.; Xu, R. Fast precipitation of uniform CaCO3 nanospheres and their transformation to hollow hydroxyapatite nanospheres. J. Colloid Interface Sci. 2010, 352, 393–400. [Google Scholar] [CrossRef]
- Jiang, S.-D.; Yao, Q.-Z.; Zhou, G.-T.; Fu, S.-Q. Fabrication of Hydroxyapatite Hierarchical Hollow Microspheres and Potential Application in Water Treatment. J. Phys. Chem. C 2012, 116, 4484–4492. [Google Scholar] [CrossRef]
- Daryan, S.H.; Khavandi, A.; Javadpour, J. Surface engineered hollow hydroxyapatite microspheres: Hydrothermal synthesis and growth mechanisms. Solid State Sci. 2020, 106, 106301. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Liang, Y.-H.; Yen, S.-K. Effects of Chitosan on Loading and Releasing for Doxorubicin Loaded Porous Hydroxyapatite–Gelatin Composite Microspheres. Polymers 2022, 14, 4276. [Google Scholar] [PubMed]
- Ordikhani, F.; Tamjid, E.; Simchi, A. Characterization and antibacterial performance of electrodeposited chitosan–vancomycin composite coatings for prevention of implant-associated infections. Mater. Sci. Eng. C 2014, 41, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-C.; Lin, C.-C.; Liao, J.-W.; Yen, S.-K. Vancomycin–chitosan composite deposited on post porous hydroxyapatite coated Ti6Al4V implant for drug controlled release. Mater. Sci. Eng. C 2013, 33, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- López-Iglesias, C.; Barros, J.; Ardao, I.; Monteiro, F.J.; Alvarez-Lorenzo, C.; Gómez-Amoza, J.L.; García-González, C.A. Vancomycin-loaded chitosan aerogel particles for chronic wound applications. Carbohydr. Polym. 2019, 204, 223–231. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Wang, J.; Qu, Y.; Liu, G. In vivo drug release and antibacterial properties of vancomycin loaded hydroxyapatite/chitosan composite. Drug Deliv. 2012, 19, 264–269. [Google Scholar] [CrossRef]
- Chang, S.-H.; Lin, H.-T.V.; Wu, G.-J.; Tsai, G.J. pH Effects on solubility, zeta potential, and correlation between antibacterial activity and molecular weight of chitosan. Carbohydr. Polym. 2015, 134, 74–81. [Google Scholar] [CrossRef]
- Maleki Dizaj, S.; Lotfipour, F.; Barzegar-Jalali, M.; Zarrintan, M.-H.; Adibkia, K. Physicochemical characterization and antimicrobial evaluation of gentamicin-loaded CaCO3 nanoparticles prepared via microemulsion method. J. Drug Deliv. Sci. Technol. 2016, 35, 16–23. [Google Scholar] [CrossRef]
- Yang, X.; Shi, G.; Guo, J.; Wang, C.; He, Y. Exosome-encapsulated antibiotic against intracellular infections of methicillin-resistant Staphylococcus aureus. Int. J. Nanomed. 2018, 13, 8095–8104. [Google Scholar] [CrossRef]
- Babaei, M.; Ghaee, A.; Nourmohammadi, J. Poly (sodium 4-styrene sulfonate)-modified hydroxyapatite nanoparticles in zein-based scaffold as a drug carrier for vancomycin. Mater. Sci. Eng. C 2019, 100, 874–885. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | wt.% | at.% |
|---|---|---|
| C | 15.28 | 25.17 |
| O | 41.52 | 51.33 |
| P | 15.11 | 9.64 |
| Ca | 28.09 | 13.86 |
| Totals | 100.00 | 100.00 |
| (a) 5% HCl | (b) Chi-CaPM Dissolved in 5% HCl | (c) Pure Chi-CaPM | |
|---|---|---|---|
| P | 0.0295 | 109 | 108.97 |
| Ca | 0.015 | 160 | 159.99 |
| As | 0.0773 | 0.1553 | 0.0780 |
| Cd | 0.00093 | 0.01233 | 0.0114 |
| Hg | 0.00126 | 0.02308 | 0.0218 |
| Pb | 0.0257 | 0.1451 | 0.1194 |
| BET Adsorption Theory | BJH | ||
|---|---|---|---|
| Adsorption | Desorption | ||
| Surface area | 38.16 m2/g | 33.32 m2/g | 76.29 m2/g |
| Pore volume | 0.244 cm3/g | 0.244 cm3/g | |
| Pore size | 18.49 nm | 29.29 nm | 12.80 nm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-Y.; Huang, S.-W.; Kao, I.-F.; Yen, S.-K. The Preparation and Characterization of Chitosan/Calcium Phosphate Composite Microspheres for Biomedical Applications. Polymers 2024, 16, 167. https://doi.org/10.3390/polym16020167
Wu M-Y, Huang S-W, Kao I-F, Yen S-K. The Preparation and Characterization of Chitosan/Calcium Phosphate Composite Microspheres for Biomedical Applications. Polymers. 2024; 16(2):167. https://doi.org/10.3390/polym16020167
Chicago/Turabian StyleWu, Meng-Ying, Shih-Wei Huang, I-Fang Kao, and Shiow-Kang Yen. 2024. "The Preparation and Characterization of Chitosan/Calcium Phosphate Composite Microspheres for Biomedical Applications" Polymers 16, no. 2: 167. https://doi.org/10.3390/polym16020167
APA StyleWu, M.-Y., Huang, S.-W., Kao, I.-F., & Yen, S.-K. (2024). The Preparation and Characterization of Chitosan/Calcium Phosphate Composite Microspheres for Biomedical Applications. Polymers, 16(2), 167. https://doi.org/10.3390/polym16020167