




Thermoresponsive Property of Poly(N,N-bis(2-methoxyethyl)acrylamide) and Its Copolymers with Water-Soluble Poly(N,N-disubstituted acrylamide) Prepared Using Hydrosilylation-Promoted Group Transfer Polymerization

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Measurements
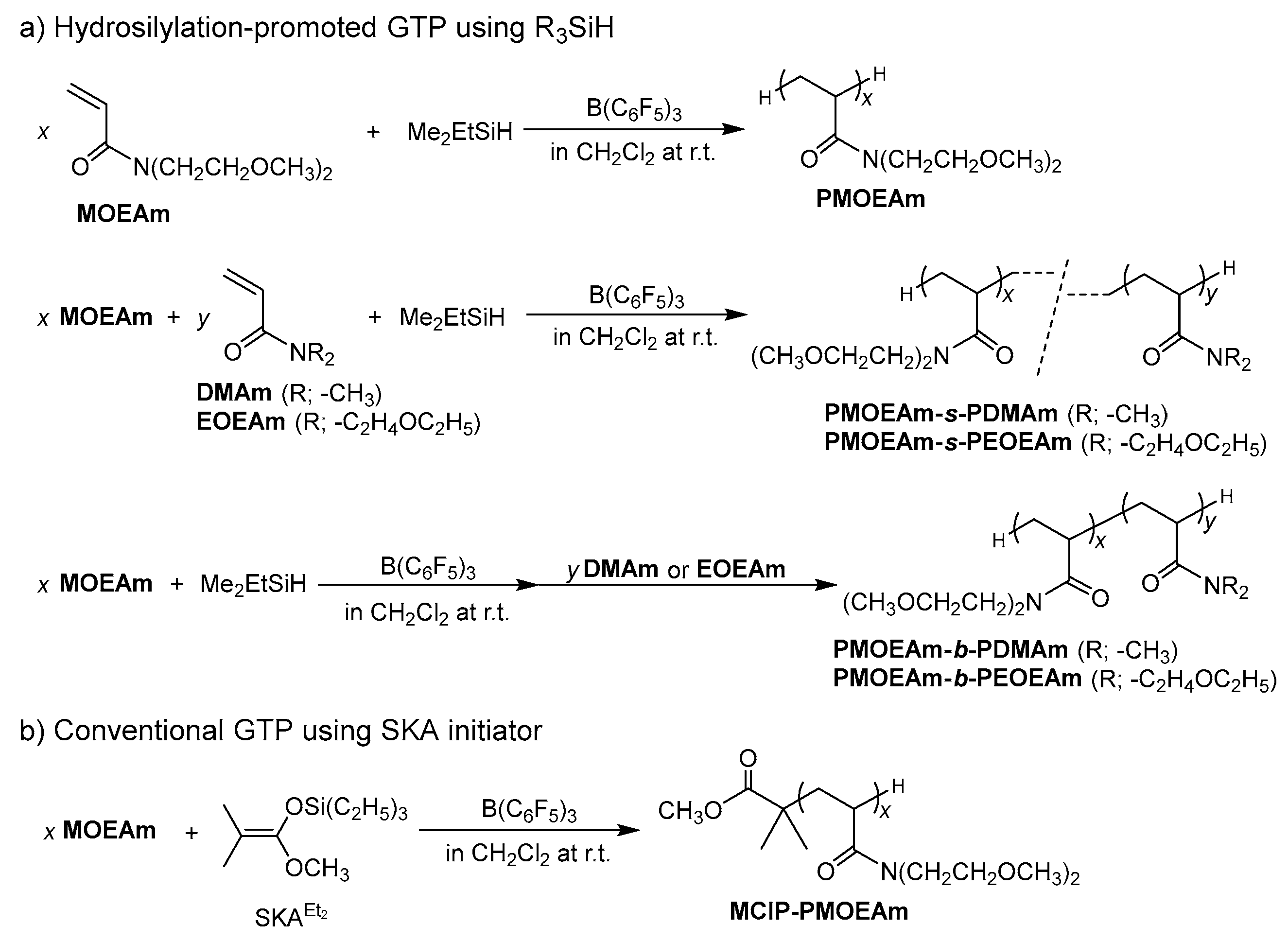
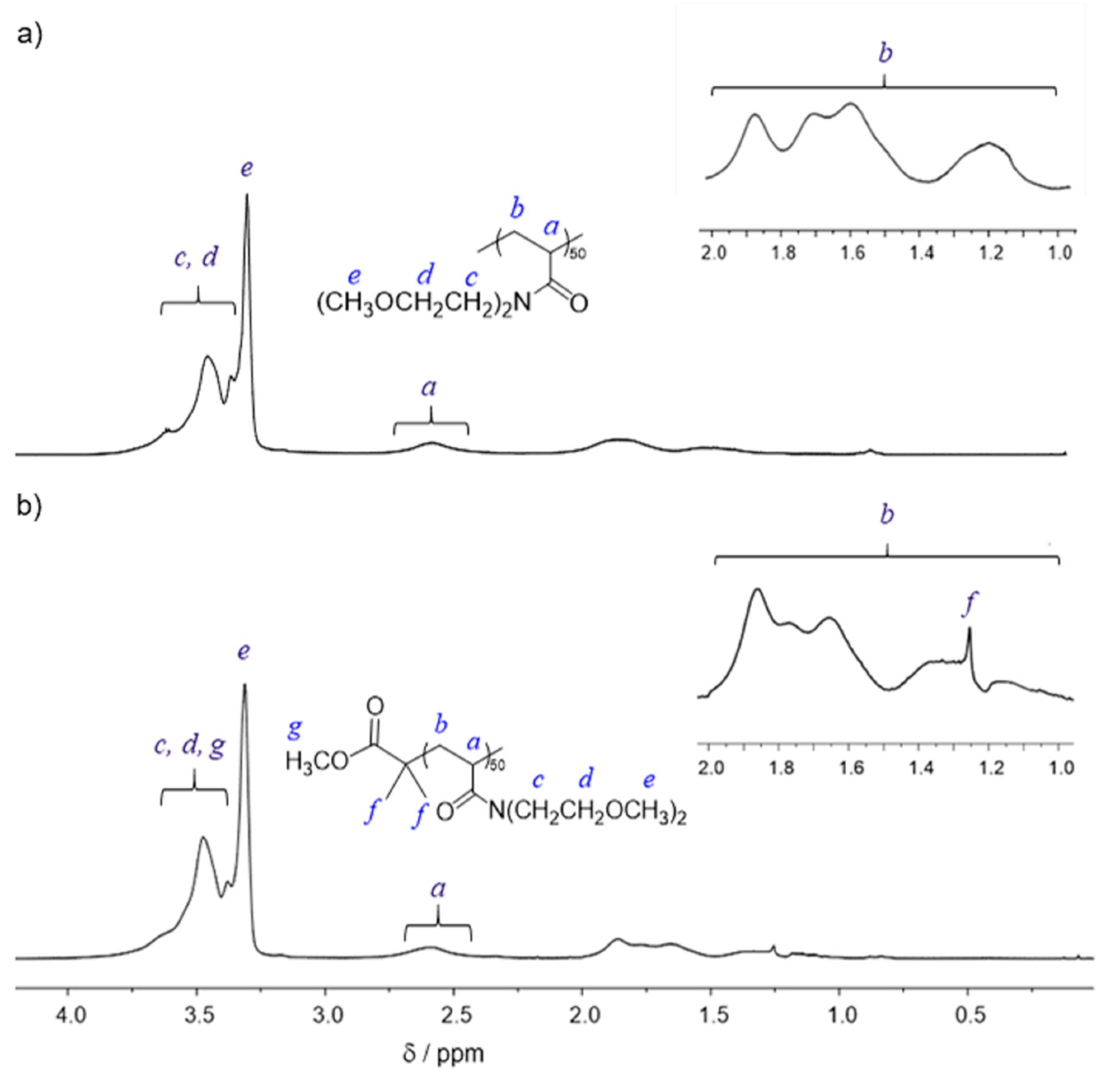
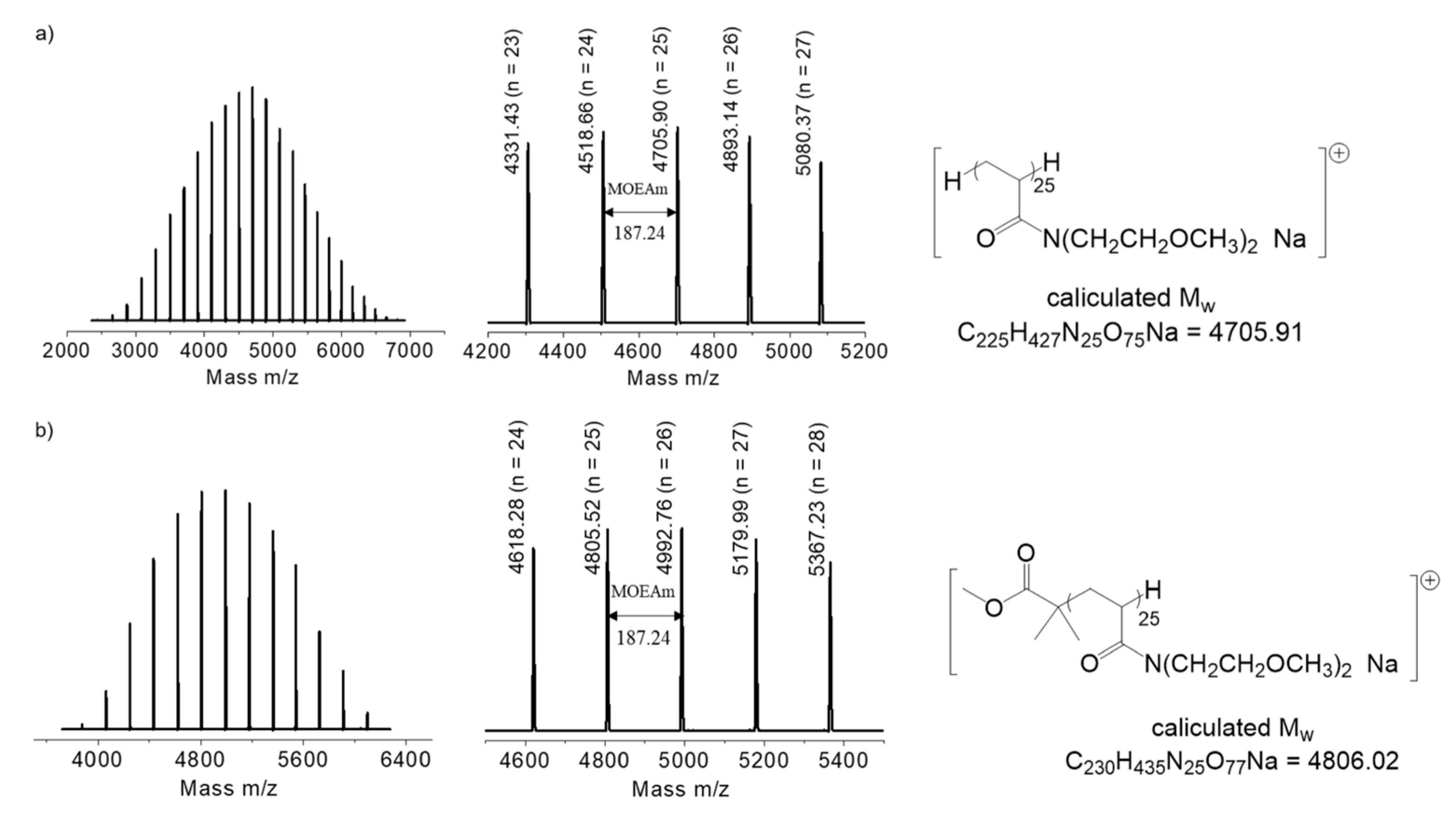
2.3. Synthesis of PMOEAm
2.4. Synthesis of MCIP-PMOEAm
2.5. Synthesis of PMOEAm-s-PDMAm
2.6. Synthesis of PMOEAm-b-PDMAm
3. Results and Discussion
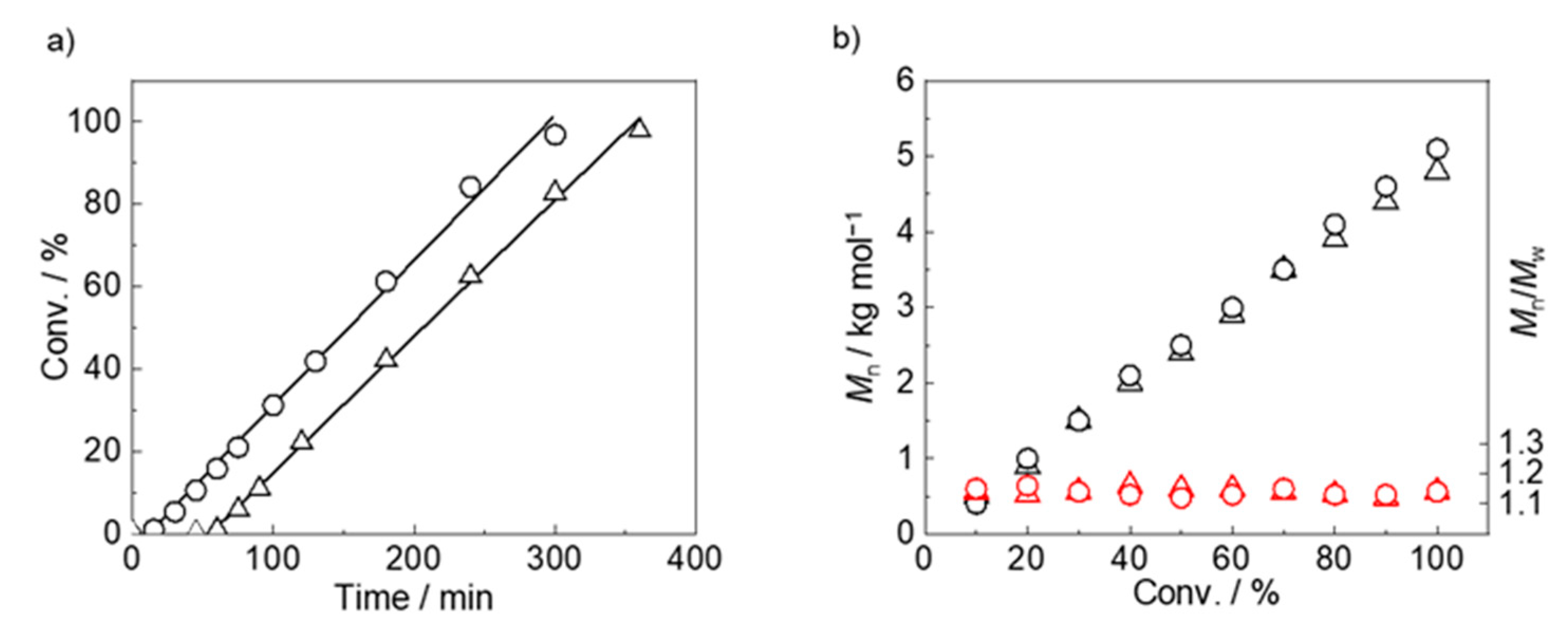
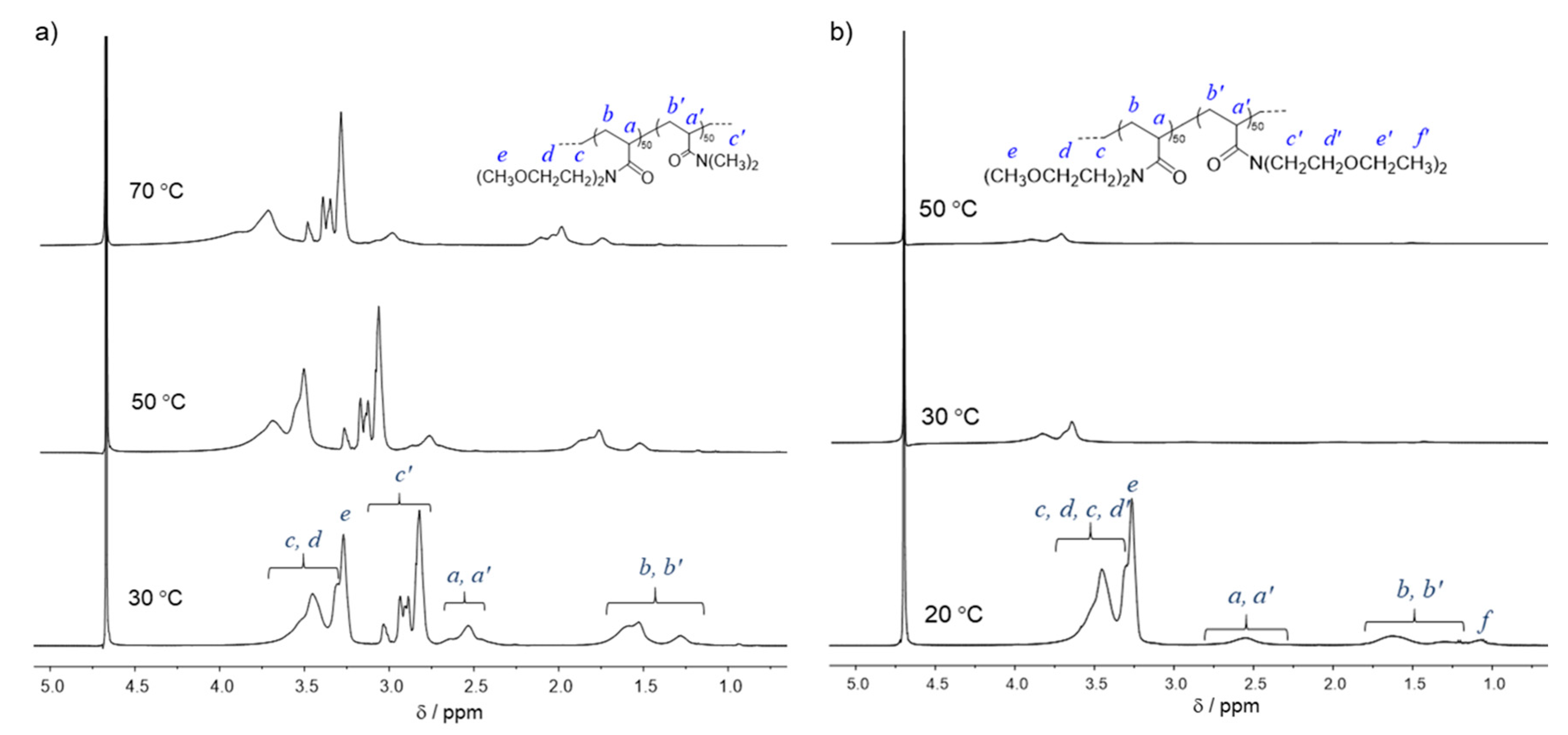
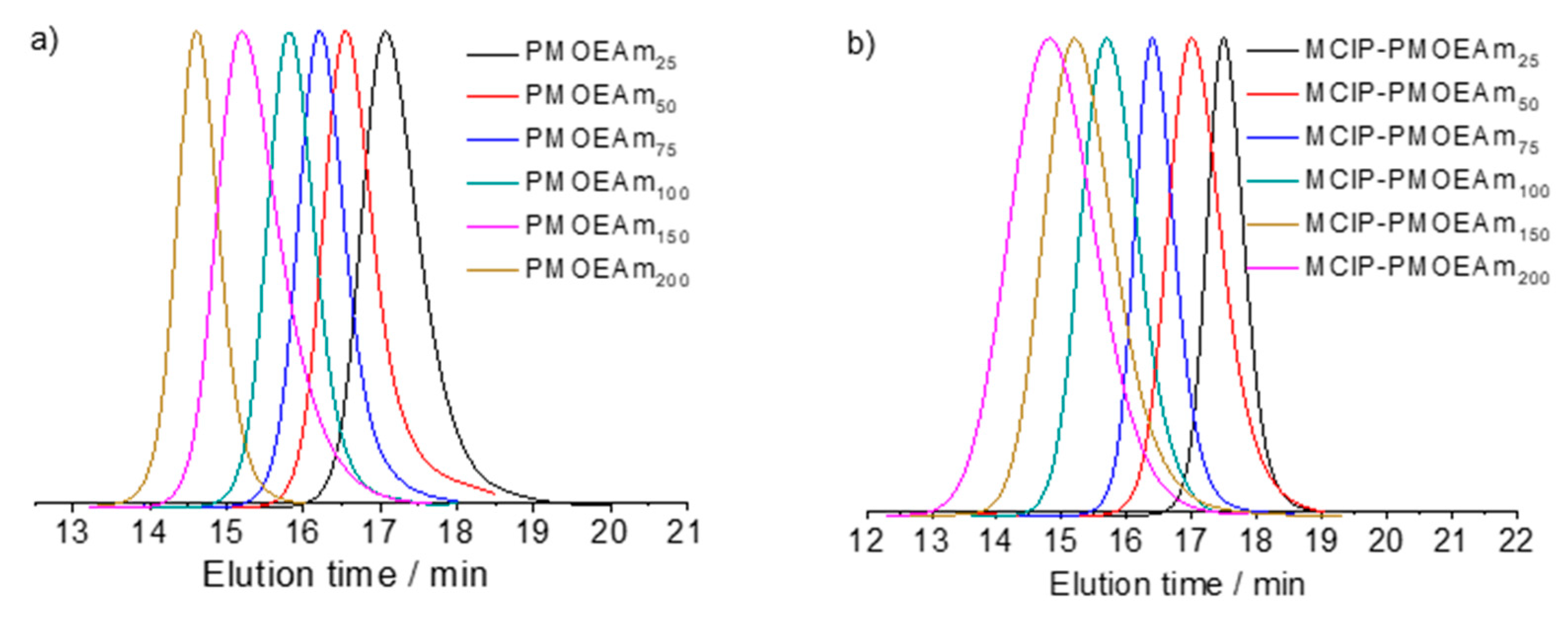
3.1. Synthesis of PMOEAm
3.2. Synthesis of Block and Statistical Copolymers of PMOEAm
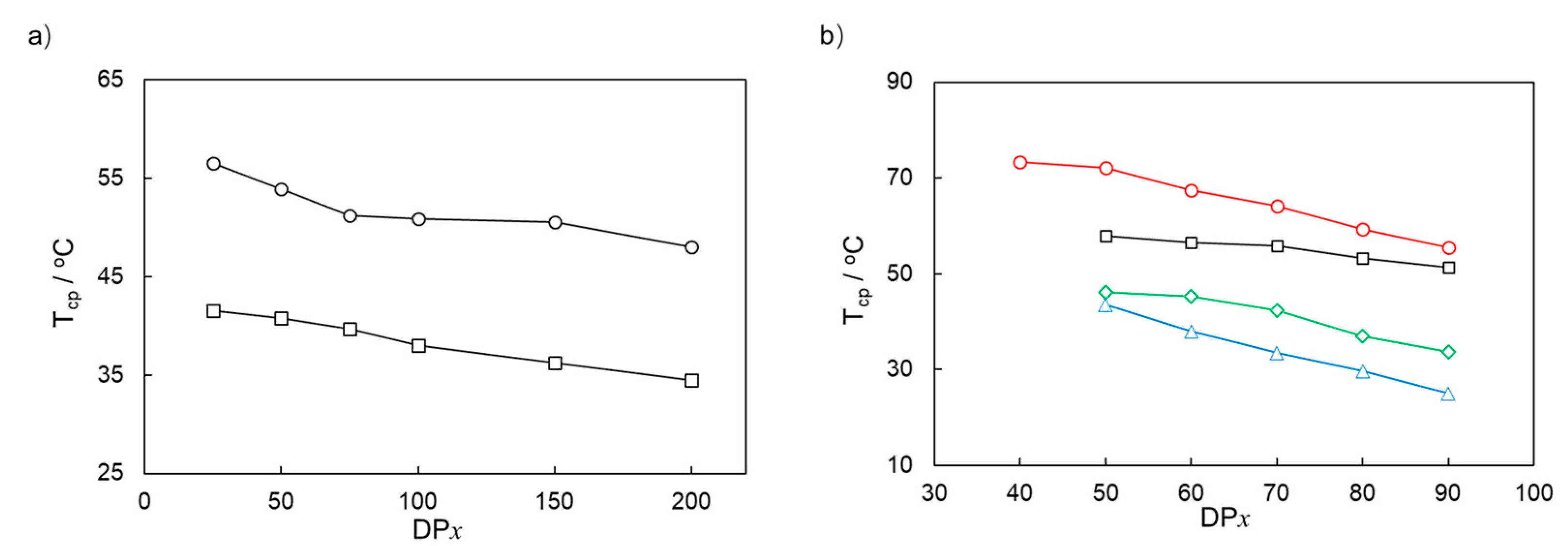
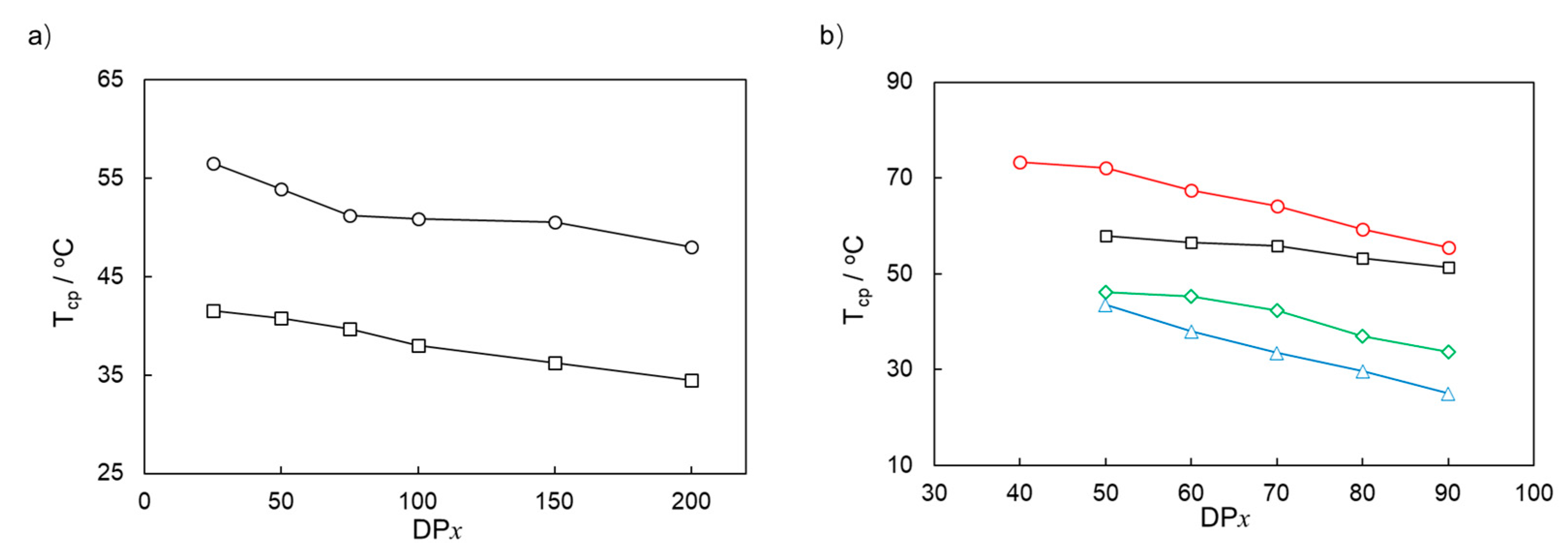
3.3. Thermoresponsive Property of PMOEAm and Its Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Watanabe, E.; Tomoshige, N.; Uyama, H. New Biodegradable and Thermoresponsive Polymers Based on Amphiphilic Poly(asparagine) Derivatives. Macromol. Symp. 2007, 249–250, 509–514. [Google Scholar] [CrossRef]
- Pasparakis, G.; Tsitsilianis, C. LCST Polymers: Thermoresponsive Nanostructured Assemblies towards Bioapplications. Polymer 2020, 211, 123146. [Google Scholar] [CrossRef]
- Lang, X.; Patrick, A.D.; Hammouda, B.; Hore, M.J.A. Chain terminal group leads to distinct thermoresponsive behaviors of linear PNIPAM and polymer analogs. Polymer 2018, 145, 137–147. [Google Scholar] [CrossRef]
- Schroffenegger, M.; Reimhult, E. Thermoresponsive Core-Shell Nanoparticles: Does Core Size Matter? Materials 2018, 11, 1654. [Google Scholar] [CrossRef] [PubMed]
- Nobuyuki, H.; Sho, M.; Shin-nosuke, N.; Tomoyuki, K. Stepwise Thermo-Responsive Amino Acid-Derived Triblock Vinyl Polymers: ATRP Synthesis of Polymers, Aggregation, and Gelation Properties via Flower-Like Micelle Formation. Materials 2018, 11, 424. [Google Scholar]
- Boutris, C.; Chatzi, E.G.; Kiparissides, C. Characterization of the LCST behaviour of aqueous poly(N-isopropylacrylamide) solutions by thermal and cloud point techniques. Polymer 1997, 38, 2567–2570. [Google Scholar] [CrossRef]
- Ni, C.H.; Zhu, X.X.; Wang, Q.L.; Zeng, X.Y. Studies on LCST of poly(N-isopropylacrylamide-co-acrylic acid-co-N-diacetone acrylamide). Chin. Chem. Lett. 2007, 18, 79–80. [Google Scholar] [CrossRef]
- Liu, X.; Song, T.; Chang, M.; Meng, L.; Wang, X.; Sun, R.; Ren, J. Carbon Nanotubes Reinforced Maleic Anhydride-Modified Xylan-g-Poly(N-isopropylacrylamide) Hydrogel with Multifunctional Properties. Materials 2018, 11, 354. [Google Scholar] [CrossRef]
- Saremi, B.; Yao, T.; Yuan, B. Thermo- and pH-sensitive nanoparticles of poly (N-isopropylacrylamide-decenoic acid-1-vinylimidazole) for ultrasound switchable fluorescence imaging. Exp. Biol. Med. 2022, 247, 1005–1012. [Google Scholar] [CrossRef]
- Li, S.; Song, F.; Sun, C.; Hu, J.; Zhang, Y. Amphiphilic methoxy poly(ethylene glycol)-b-poly(carbonate-selenide) with enhanced ROS responsiveness: Facile synthesis and oxidation process. Eur. Polym. J. 2021, 159, 110752. [Google Scholar] [CrossRef]
- Son, K.; Lee, J. Synthesis and Characterization of Poly(Ethylene Glycol) Based Thermo-Responsive Hydrogels for Cell Sheet Engineering. Materials 2016, 9, 854. [Google Scholar] [CrossRef]
- Gordon, T.N.; Kornmuller, A.; Soni, Y.; Flynn, L.E.; Gillies, E.R. Polyesters based on aspartic acid and poly(ethylene glycol): Functional polymers for hydrogel preparation. Eur. Polym. J. 2021, 152, 110456. [Google Scholar] [CrossRef]
- Kabra, B.G.; Gehrke, S.H.; Hwang, S.T.; Ritschel, W.A. Modification of the dynamic swelling behavior of poly(2-hydroxyethyl methacrylate) in water. J. Appl. Polym. 1991, 42, 2409–2416. [Google Scholar] [CrossRef]
- Moynihan, H.J.; Honey, M.S.; Peppas, N.A. Solute diffusion in swollen membranes. Part V: Solute diffusion in poly(2-hydroxyethyl methacrylate). Polym. Eng. Sci. 1986, 26, 1180–1185. [Google Scholar] [CrossRef]
- Choudhury, N.; Das, S.; Samadder, S.; De, P. Phenylalanine-Tethered pH-Responsive Poly(2-Hydroxyethyl Methacrylate). Chem. Asian J. 2021, 16, 1016–1024. [Google Scholar] [CrossRef]
- Liu, S.; Liu, M. Synthesis and characterization of temperature- and pH-sensitive poly(N,N-diethylacrylamide-co-methacrylic acid). J. Appl. Polym. 2003, 90, 3563–3568. [Google Scholar] [CrossRef]
- Constantin, M.; Bucatariu, S.; Harabagiu, V.; Popescu, I.; Ascenzi, P.; Fundueanu, G. Poly(N-isopropylacrylamide-co-methacrylic acid) pH/thermo-responsive porous hydrogels as self-regulated drug delivery system. Eur. J. Pharm. Sci. 2014, 62, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Noël, M.-H.; van Damme, H.; Hébraud, P. Building of a thermoresponsive cement. Cem. Concr. Res. 2011, 41, 975–980. [Google Scholar] [CrossRef]
- Shi, J.; Wang, X.; Xu, S.; Wu, Q.; Cao, S. Reversible thermal-tunable drug delivery across nano-membranes of hollow PUA/PSS multilayer microcapsules. J. Membr. Sci. 2016, 499, 307–316. [Google Scholar] [CrossRef]
- Bercea, M.; Nita, L.-E.; Eckelt, J.; Wolf, B.A. Polyelectrolyte Complexes: Phase Diagram and Intrinsic Viscosities of the System Water/Poly(2-vinylpyridinium-Br)/Poly(styrene sulfonate-Na). Macromol. Chem. Phys. 2012, 213, 2504–2513. [Google Scholar] [CrossRef]
- An, Q.; Beh, C.; Xiao, H. Preparation and characterization of thermo-sensitive poly(vinyl alcohol)-based hydrogel as drug carrier. J. Appl. Polym. 2013, 131, 39720. [Google Scholar] [CrossRef]
- Petrov, S.; Ivanova, T.; Christova, D.; Ivanova, S. Modification of polyacrylonitrile membranes with temperature sensitive poly(vinylalcohol-co-vinylacetal). J. Membr. Sci. 2005, 261, 1–6. [Google Scholar] [CrossRef]
- Liu, J.; Detrembleur, C.; Hurtgen, M.; Debuigne, A.; De Pauw-Gillet, M.-C.; Mornet, S.; Jérôme, C. Thermo-responsive gold/poly(vinyl alcohol)-b-poly(N-vinylcaprolactam) core–corona nanoparticles as a drug delivery system. Polym. Chem. 2014, 5, 5289–5299. [Google Scholar] [CrossRef]
- Cheng, S.; Feng, W.; Pashikin, I.; Yuan, L.; Deng, H.; Zhou, Y. Radiation polymerization of thermo-sensitive poly(N-vinylcaprolactam). Radiat. Phys. Chem. 2002, 63, 517–519. [Google Scholar] [CrossRef]
- Medeiros, S.F.; Barboza, J.C.S.; Giudici, R.; Santos, A.M. Thermally-sensitive and Biocompatible Poly(N-vinylcaprolactam): A Kinetic Study of Free Radical Polymerization in Ethanol. J. Macromol. Sci. A 2013, 50, 763–773. [Google Scholar] [CrossRef]
- Winninger, J.; Iurea, D.M.; Atanase, L.I.; Salhi, S.; Delaite, C.; Riess, G. Micellization of novel biocompatible thermo-sensitive graft copolymers based on poly(ε-caprolactone), poly(N-vinylcaprolactam) and poly(N-vinylpyrrolidone). Eur. Polym. J. 2019, 119, 74–82. [Google Scholar] [CrossRef]
- Zhang, X.; Burton, T.F.; In, M.; Bégu, S.; Aubert-Pouëssel, A.; Robin, J.-J.; Giani, O. Synthesis and behavior of PEG-b-PDEAm block copolymers in aqueous solution. Mater. Today Commun. 2020, 24, 100987. [Google Scholar] [CrossRef]
- Chen, J.; Liu, M.; Jin, S.; Liu, H. Synthesis and characterization of κ-carrageenan/poly(N,N-diethylacrylamide) semi-interpenetrating polymer network hydrogels with rapid response to temperature. Polym. Adv. Technol. 2008, 19, 1656–1663. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Polymers for Biomedical Applications. Polymers 2011, 3, 1215–1242. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, G.; Li, J. Preparation of amphoteric polyacrylamide flocculant and its application in the treatment of tannery wastewater. J. Appl. Polym. Sci. 2010, 120, 518–523. [Google Scholar] [CrossRef]
- Danch, A.; Laggner, P.; Degovics, G.; Sek, D.; Stelzer, F. Thermodynamic and structure investigations of new side-chain liquid crystal polymer. Proc. SPIE-Int. Soc. Opt. Eng. 1998, 3319, 271–275. [Google Scholar]
- Biswas, C.S.; Hazer, B. Synthesis and characterization of stereoregular poly(N-ethylacrylamide) hydrogel by using Y(OTf)3 Lewis acid. Colloid Polym. Sci. 2014, 293, 143–152. [Google Scholar] [CrossRef]
- Liu, C.; Wang, S.; Zhou, H.; Gao, C.; Zhang, W. Thermoresponsive poly(ionic liquid): Controllable RAFT synthesis, thermoresponse, and application in dispersion RAFT polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 54, 945–954. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.; Sun, T.; Du, J.; Yang, X.; Wang, J. Surface-modulated and thermoresponsive polyphosphoester nanoparticles for enhanced intracellular drug delivery. Sci. China Chem. 2014, 57, 579–585. [Google Scholar] [CrossRef]
- Cheng, C.; Wei, H.; Shi, B.-X.; Cheng, H.; Li, C.; Gu, Z.-W.; Zhuo, R.-X.; Zhang, X.-Z.; Zhuo, R.-X. Biotinylated thermoresponsive micelle self-assembled from double-hydrophilic block copolymer for drug delivery and tumor target. Biomaterials 2008, 29, 497–505. [Google Scholar] [CrossRef]
- Kwon, I.K.; Matsuda, T. Photo-iniferter-based thermoresponsive block copolymers composed of poly(ethylene glycol) and poly(N-isopropylacrylamide) and chondrocyte immobilization. Biomaterials 2006, 27, 986–995. [Google Scholar] [CrossRef]
- Porjazoska, A.; Dimitrov, P.; Dimitrov, I.; Cvetkovska, M.; Tsvetanov, C.B. Synthesis and aqueous solution properties of functionalized and thermoresponsive poly(D,L-lactide)/polyether block copolymers. Macromol. Symp. 2004, 210, 427–436. [Google Scholar] [CrossRef]
- Peleshanko, S.; Anderson, K.D.; Goodman, M.; Determan, M.D.; Mallapragada, S.K.; Tsukruk, V.V. Thermoresponsive Reversible Behavior of Multistimuli Pluronic-Based Pentablock Copolymer at the Air–Water Interface. Langmuir 2007, 23, 25–30. [Google Scholar] [CrossRef]
- Yoshioka, H.; Mikami, M.; Mori, Y.; Tsuchida, E. Preparation of thermoresponsive surfaces using polyvinylchloride-graft-poly(N-isopropylacrylamide). Polym. Adv. Technol. 1993, 4, 519–521. [Google Scholar] [CrossRef]
- Oh, B.H.L.; Bismarck, A.; Chan-Park, M.B. High Internal Phase Emulsion Templating with Self-Emulsifying and Thermoresponsive Chitosan-graft-PNIPAM-graft-Oligoproline. Biomacromolecules 2014, 15, 1777–1787. [Google Scholar] [CrossRef]
- Wu, Y.; Deng, Y.; Yuan, Q.; Ling, Y.; Tang, H. Thermoresponsive poly(γ-propyl-l-glutamate)-graft-(oligo ethylene glycol)s: Synthesis, characterization, and properties. J. Appl. Polym. 2014, 131, 41022. [Google Scholar] [CrossRef]
- Kelland, M.A. Tuning the thermoresponsive properties of hyperbranched poly(ester amide)s based on diisopropanolamine and cyclic dicarboxylic anhydrides. J. Appl. Polym. 2011, 121, 2282–2290. [Google Scholar] [CrossRef]
- Tong, J.; Wei, Z.; Yang, L.; Yang, Y.; Chen, Y. Study on the phase transition behaviors of thermoresponsive hyperbranched polyampholytes in water. Polymer 2016, 84, 107–116. [Google Scholar] [CrossRef]
- Rezaei, S.J.T.; Nabid, M.R.; Niknejad, H.; Entezami, A.A. Folate-decorated thermoresponsive micelles based on star-shaped amphiphilic block copolymers for efficient intracellular release of anticancer drugs. Int. J. Pharm. 2012, 437, 70–79. [Google Scholar] [CrossRef]
- Ida, S.; Toyama, Y.; Takeshima, S.; Kanaoka, S. Thermoresponsive core cross-linked star-shaped poly(N-isopropylacrylamide) for reversible and controlled aggregation of nanoscale molecular units. Polym. J. 2019, 52, 359–363. [Google Scholar] [CrossRef]
- Ma, C.; Han, T.; Niu, N.; Al-Shok, L.; Efstathiou, S.; Lester, D.W.; Huband, S.; Haddleton, D. Well-defined polyacrylamides with AIE properties via rapid Cu-mediated living radical polymerization in aqueous solution: Thermoresponsive nanoparticles for bioimaging. Polym. Chem. 2021, 13, 58–68. [Google Scholar] [CrossRef]
- Zhang, C.; Maric, M. Fluorescent, thermoresponsive copolymers via nitroxide-mediated polymerization: Synthesis and effect of fluorescent groups on phase transitions in an ionic liquid. J. Polym. Sci. A Polym. Chem. 2013, 51, 4702–4715. [Google Scholar] [CrossRef]
- Kwan, S.; Marić, M. Thermoresponsive polymers with tunable cloud point temperatures grafted from chitosan via nitroxide mediated polymerization. Polymer 2016, 86, 69–82. [Google Scholar] [CrossRef]
- Kim, D.J.; Kang, S.M.; Kong, B.; Kim, W.-J.; Paik, H.; Choi, H.; Choi, I.S. Formation of Thermoresponsive Gold Nanoparticle/PNIPAAm Hybrids by Surface-Initiated, Atom Transfer Radical Polymerization in Aqueous Media. Macromol. Chem. Phys. 2005, 206, 1941–1946. [Google Scholar] [CrossRef]
- Zhang, K.; Ma, J.; Zhang, B.; Zhao, S.; Li, Y.; Xu, Y.; Wang, J. Synthesis of thermoresponsive silica nanoparticle/PNIPAM hybrids by aqueous surface-initiated atom transfer radical polymerization. Mater. Lett. 2007, 61, 949–952. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kadla, J.F. Preparation of a Thermoresponsive Lignin-Based Biomaterial through Atom Transfer Radical Polymerization. Biomacromolecules 2010, 11, 981–988. [Google Scholar] [CrossRef]
- Kessel, S.; Truong, N.P.; Jia, Z.; Monteiro, M.J. Aqueous reversible addition-fragmentation chain transfer dispersion polymerization of thermoresponsive diblock copolymer assemblies: Temperature directed morphology transformations. J. Polym. Sci. A Polym. Chem. 2012, 50, 4879–4887. [Google Scholar] [CrossRef]
- Pascual, S.; Monteiro, M.J. Shell-crosslinked nanoparticles through self-assembly of thermoresponsive block copolymers by RAFT polymerization. Eur. Polym. J. 2009, 45, 2513–2519. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.; Luo, X.; Kong, N.; Cui, L.; Liu, J. Preparation of biodegradable and thermoresponsive enzyme–polymer conjugates with controllable bioactivity via RAFT polymerization. Eur. Polym. J. 2013, 49, 2949–2960. [Google Scholar] [CrossRef]
- Webster, O.W. Molecular architecture control in acrylic polymers by group transfer polymerization. Macromol. Symp. 1993, 70–71, 75–81. [Google Scholar] [CrossRef]
- Raynaud, J.; Ciolino, A.; Baceiredo, A.; Destarac, M.; Bonnette, F.; Kato, T.; Taton, D. Harnessing the Potential of N-Heterocyclic Carbenes for the Rejuvenation of Group-Transfer Polymerization of (Meth)Acrylics. Angew. Chem. Int. Ed. 2008, 47, 5390–5393. [Google Scholar] [CrossRef]
- Scholten, M.D.; Hedrick, J.L.; Waymouth, R.M. Group Transfer Polymerization of Acrylates Catalyzed by N-Heterocyclic Carbenes. Macromolecules 2008, 41, 7399–7404. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Chen, Y. Organocatalyzed Group Transfer Polymerization of Alkyl Sorbate: Polymer Synthesis, Postpolymerization Modification, and Thermal Properties. Macromolecules 2021, 54, 9039–9052. [Google Scholar] [CrossRef]
- Ute, K.; Tarao, T.; Nakao, S.; Kitayama, T. Preparation and properties of disyndiotactic poly(alkylcrotonate)s. Polymer 2003, 44, 7869–7874. [Google Scholar] [CrossRef]
- Raynaud, J.; Liu, N.; Fèvre, M.; Gnanou, Y.; Taton, D. No matter the order of monomer addition for the synthesis of well-defined block copolymers by sequential group transfer polymerization using N-heterocyclic carbenes as catalysts. Polym. Chem. 2011, 2, 1706. [Google Scholar] [CrossRef]
- Kikuchi, S.; Chen, Y.; Ichinohe, E.; Kitano, K.; Sato, S.; Duan, Q.; Shen, X.; Kakuchi, T. Synthesis and Thermoresponsive Property of Linear, Cyclic, and Star-Shaped Poly(N,N-diethylacrylamide)s Using B(C6F5)3-Catalyzed Group Transfer Polymerization as Facile End-Functionalization Method. Macromolecules 2016, 49, 4828–4838. [Google Scholar] [CrossRef]
- Chen, Y.; Kitano, K.; Tsuchida, S.; Kikuchi, S.; Takada, K.; Satoh, T.; Kakuchi, T. B(C6F5)3-catalyzed group transfer polymerization of alkyl methacrylates with dimethylphenylsilane through in situ formation of silyl ketene acetal by B(C6F5)3-catalyzed 1,4-hydrosilylation of methacrylate monomer. Polym. Chem. 2015, 6, 3502–3511. [Google Scholar] [CrossRef]
- Xu, T.; Chen, E.Y.-X. Silylium dual catalysis in living polymerization of methacrylates viaIn situhydrosilylation of monomer. J. Polym. Sci. A Polym. Chem. 2015, 53, 1895–1903. [Google Scholar] [CrossRef]
- Li, J.; Mizutani, S.; Sato, S.; Narumi, A.; Haba, O.; Kawaguchi, S.; Kawaguchi, S.; Kakuchi, T.; Shen, X. Thermoresponsive property of well-defined poly(N-methyl-N-n-propylacrylamide) and its copolymer architectures prepared by hydrosilylation-promoted group transfer polymerization. Polymer 2020, 122678. [Google Scholar] [CrossRef]
- Hidaka, T.; Sugihara, S.; Maeda, Y. Infrared spectroscopic study on LCST behavior of poly(N,N-bis(2-methoxyethyl)acrylamide). Eur. Polym. J. 2013, 49, 675–681. [Google Scholar] [CrossRef]
- Song, J.M.; Winnik, F.M.; Brash, J.L. Synthesis and Solution Properties of Fluorescently Labeled Amphiphilic (N-alkylacrylamide) Oligomers. Macromolecules 1998, 31, 109–115. [Google Scholar] [CrossRef]
- Koyama, M.; Hirano, T.; Ohno, K.; Katsumoto, Y. Molecular Understanding of the UCST-Type Phase Separation Behavior of a Stereocontrolled Poly(N-isopropylacrylamide) in Bis(2-methoxyethyl) Ether. J. Phys. Chem. B 2008, 112, 10854–10860. [Google Scholar] [CrossRef]
- Yamazaki, A.; Winnik, F.M.; Cornelius, R.M.; Brash, J.L. Modification of liposomes with N-substituted polyacrylamides: Identification of proteins adsorbed from plasma. Biochim. Biophys. Acta Biomembr. 1999, 1421, 103–115. [Google Scholar] [CrossRef]
- Hechenbichler, M.; Laschewsky, A.; Gradzielski, M. Poly(N,N-bis(2-methoxyethyl)acrylamide), a thermoresponsive non-ionic polymer combining the amide and the ethyleneglycolether motifs. Colloid Polym. Sci. 2020, 299, 205–219. [Google Scholar] [CrossRef]
- El-Ejmi, S.; Huglin, B. Thermoreversible behaviour in water of chemically crosslinked poly(2-methoxyethylacrylate-co-N,N-dimethylacrylamide). Polym. Int. 1997, 44, 277–282. [Google Scholar] [CrossRef]
- Fu, X.; Wang, Y.; Xu, L.; Narumi, A.; Sato, S.; Yang, X.; Shen, X.; Kakuchi, T. Thermoresponsive Property of Poly(N,N-bis(2-ethoxyethyl)acrylamide) and Its Multiblock Copolymers with Poly(N,N-dimethylacrylamide) Prepared by Hydrosilylation-Promoted Group Transfer Polymerization. Polym. Chem. 2023, 14, 5060–5070. [Google Scholar] [CrossRef]
- Kikuchi, S.; Chen, Y.; Kitano, K.; Takada, K.; Satoh, T.; Kakuchi, T. Organic acids as efficient catalysts for group transfer polymerization of N,N-disubstituted acrylamide with silyl ketene acetal: Polymerization mechanism and synthesis of diblock copolymers. Polym. Chem. 2015, 6, 6845–6856. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Polymer | [MOEAm]0/[Me2EtSiH]0/[B(C6F5)3]0 | Time h | Mn,calcd b kg mol−1 | Mn,SEC (Đ) c kg mol−1 | Tcp d °C |
|---|---|---|---|---|---|---|
| 1 | PMOEAm25 | 25/1/0.1 | 6 | 4.7 | 4.5 (1.13) | 56.5 |
| 2 | PMOEAm50 | 50/1/0.1 | 6 | 9.4 | 9.6 (1.16) | 53.9 |
| 3 | PMOEAm75 | 75/1/0.1 | 12 | 14.0 | 13.8 (1.12) | 51.2 |
| 4 | PMOEAm100 | 100/1/0.1 | 12 | 18.7 | 19.0 (1.11) | 50.9 |
| 5 | PMOEAm150 | 150/1/0.2 | 12 | 28.1 | 28.1 (1.12) | 50.5 |
| 6 | PMOEAm200 | 200/1/0.2 | 18 | 37.4 | 37.1 (1.17) | 48.0 |
| Run | Polymer | 1st GTP b | 2nd GTP c | Tcp e °C | ||||
|---|---|---|---|---|---|---|---|---|
| [MOEAm]0/[Me2EtSiH]0 | Mn,calcd /kg mol−1 | Mn,SEC (Đ) d /kg mol−1 | [DMAm or EOEAm]0 | Mn,calcd /kg mol−1 | Mn,SEC (Đ) d /kg mol−1 | |||
| 7 | PMOEAm30-b-PDMAm70 | 30/1 | 5.6 | 5.5 (1.09) | 70 | 12.5 | 12.5 (1.08) | - f |
| 8 | PMOEAm40-b-PDMAm60 | 40/1 | 7.5 | 7.5 (1.10) | 60 | 13.4 | 13.4 (1.10) | - f |
| 9 | PMOEAm50-b-PDMAm50 | 50/1 | 9.4 | 9.6 (1.13) | 50 | 14.3 | 14.4 (1.13) | 58.0 |
| 10 | PMOEAm60-b-PDMAm40 | 60/1 | 11.2 | 11.1 (1.10) | 40 | 15.2 | 15.1 (1.09) | 56.5 |
| 11 | PMOEAm70-b-PDMAm30 | 70/1 | 13.1 | 12.9 (1.11) | 30 | 16.1 | 16.0 (1.10) | 55.8 |
| 12 | PMOEAm80-b-PDMAm20 | 80/1 | 15.0 | 15.1 (1.12) | 20 | 16.9 | 16.8 (1.10) | 53.3 |
| 13 | PMOEAm90-b-PDMAm10 | 90/1 | 16.8 | 16.7 (1.13) | 10 | 17.8 | 17.6 (1.15) | 51.4 |
| 14 | PMOEAm30-b-PEOEAm70 | 30/1 | 5.6 | 5.3 (1.09) | 70 | 20.7 | 21.0 (1.10) | - f |
| 15 | PMOEAm40-b-PEOEAm60 | 40/1 | 7.5 | 7.2 (1.08) | 60 | 20.4 | 20.5 (1.09) | - f |
| 16 | PMOEAm50-b-PEOEAm50 | 50/1 | 9.4 | 9.1 (1.12) | 50 | 20.1 | 20.2 (1.11) | 43.5 |
| 17 | PMOEAm60-b-PEOEAm40 | 60/1 | 11.2 | 10.9 (1.10) | 40 | 19.8 | 19.4 (1.12) | 38.0 |
| 18 | PMOEAm70-b-PEOEAm30 | 70/1 | 13.1 | 12.8 (1.11) | 30 | 19.6 | 19.1 (1.10) | 33.5 |
| 19 | PMOEAm80-b-PEOEAm20 | 80/1 | 15.0 | 15.0 (1.11) | 20 | 19.3 | 19.0 (1.09) | 29.7 |
| 20 | PMOEAm90-b-PEOEAm10 | 90/1 | 16.8 | 16.4 (1.10) | 10 | 19.0 | 18.8 (1.11) | 25.0 |
| Run | Polymer | [MOEAm + DMAm or EOEAm]0/[Me2EtSiH]0 | Mn,calcd kg mol−1 | Mn,SEC (Đ) b kg mol−1 | Tcp c °C |
|---|---|---|---|---|---|
| 21 | PMOEAm30-s-PDMAm70 | (30 + 70)/1 | 12.5 | 12.5 (1.09) | - d |
| 22 | PMOEAm40-s-PDMAm60 | (40 + 60)/1 | 13.4 | 13.6 (1.12) | 73.4 |
| 23 | PMOEAm50-s-PDMAm50 | (50 + 50)/1 | 14.3 | 14.5 (1.14) | 72.1 |
| 24 | PMOEAm60-s-PDMAm40 | (60 + 40)/1 | 15.2 | 15.3 (1.11) | 67.5 |
| 25 | PMOEAm70-s-PDMAm30 | (70 + 30)/1 | 16.1 | 16.4 (1.15) | 64.2 |
| 26 | PMOEAm80-s-PDMAm20 | (80 + 20)/1 | 16.9 | 16.6 (1.07) | 59.3 |
| 27 | PMOEAm90-s-PDMAm10 | (90 + 10)/1 | 17.8 | 17.7 (1.11) | 55.5 |
| 28 | PMOEAm30-s-PEOEAm70 | (30 + 70)/1 | 20.7 | 20.9 (1.15) | - e |
| 29 | PMOEAm40-s-PEOEAm60 | (40 + 60)/1 | 20.4 | 20.6 (1.14) | - e |
| 30 | PMOEAm50-s-PEOEAm50 | (50 + 50)/1 | 20.1 | 20.5 (1.13) | 46.2 |
| 31 | PMOEAm60-s-PEOEAm40 | (60 + 40)/1 | 19.8 | 19.3 (1.09) | 45.3 |
| 32 | PMOEAm70-s-PEOEAm30 | (70 + 30)/1 | 19.6 | 19.0 (1.10) | 42.4 |
| 33 | PMOEAm80-s-PEOEAm20 | (80 + 20)/1 | 19.3 | 18.9 (1.12) | 37.0 |
| 34 | PMOEAm90-s-PEOEAm10 | (90 + 10)/1 | 19.0 | 18.6 (1.12) | 33.7 |
| Polymer Code | Tcp a | Rh b/nm | |
|---|---|---|---|
| 20 °C | 60 °C | ||
| PMOEAm50 | 53.9 | 15.2 | 425.6 |
| MCIP-PMOEAm50 | 40.8 | 16.1 | 457.2 |
| PMOEAm50-b-PDMAm50 | 58.0 | 16.7 | 402.8 |
| PMOEAm50-s-PDMAm50 | 72.1 | 14.5 | 349.3 c |
| PMOEAm50-b-PEOEAm50 | 25.0 | 18.9 | 551.7 |
| PMOEAm50-s-PEOEAm50 | 33.7 | 18.3 | 482.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, X.; Wang, Y.; Xu, L.; Narumi, A.; Sato, S.-i.; Yang, X.; Shen, X.; Kakuchi, T. Thermoresponsive Property of Poly(N,N-bis(2-methoxyethyl)acrylamide) and Its Copolymers with Water-Soluble Poly(N,N-disubstituted acrylamide) Prepared Using Hydrosilylation-Promoted Group Transfer Polymerization. Polymers 2023, 15, 4681. https://doi.org/10.3390/polym15244681
Fu X, Wang Y, Xu L, Narumi A, Sato S-i, Yang X, Shen X, Kakuchi T. Thermoresponsive Property of Poly(N,N-bis(2-methoxyethyl)acrylamide) and Its Copolymers with Water-Soluble Poly(N,N-disubstituted acrylamide) Prepared Using Hydrosilylation-Promoted Group Transfer Polymerization. Polymers. 2023; 15(24):4681. https://doi.org/10.3390/polym15244681
Chicago/Turabian StyleFu, Xiangming, Yanqiu Wang, Liang Xu, Atsushi Narumi, Shin-ichiro Sato, Xiaoran Yang, Xiande Shen, and Toyoji Kakuchi. 2023. "Thermoresponsive Property of Poly(N,N-bis(2-methoxyethyl)acrylamide) and Its Copolymers with Water-Soluble Poly(N,N-disubstituted acrylamide) Prepared Using Hydrosilylation-Promoted Group Transfer Polymerization" Polymers 15, no. 24: 4681. https://doi.org/10.3390/polym15244681
APA StyleFu, X., Wang, Y., Xu, L., Narumi, A., Sato, S.-i., Yang, X., Shen, X., & Kakuchi, T. (2023). Thermoresponsive Property of Poly(N,N-bis(2-methoxyethyl)acrylamide) and Its Copolymers with Water-Soluble Poly(N,N-disubstituted acrylamide) Prepared Using Hydrosilylation-Promoted Group Transfer Polymerization. Polymers, 15(24), 4681. https://doi.org/10.3390/polym15244681