3.1. Influence of Methanol and H2S on the Properties of PP
3.1.1. Effects on the Melt Flow Index (MFI) and MW of the PP
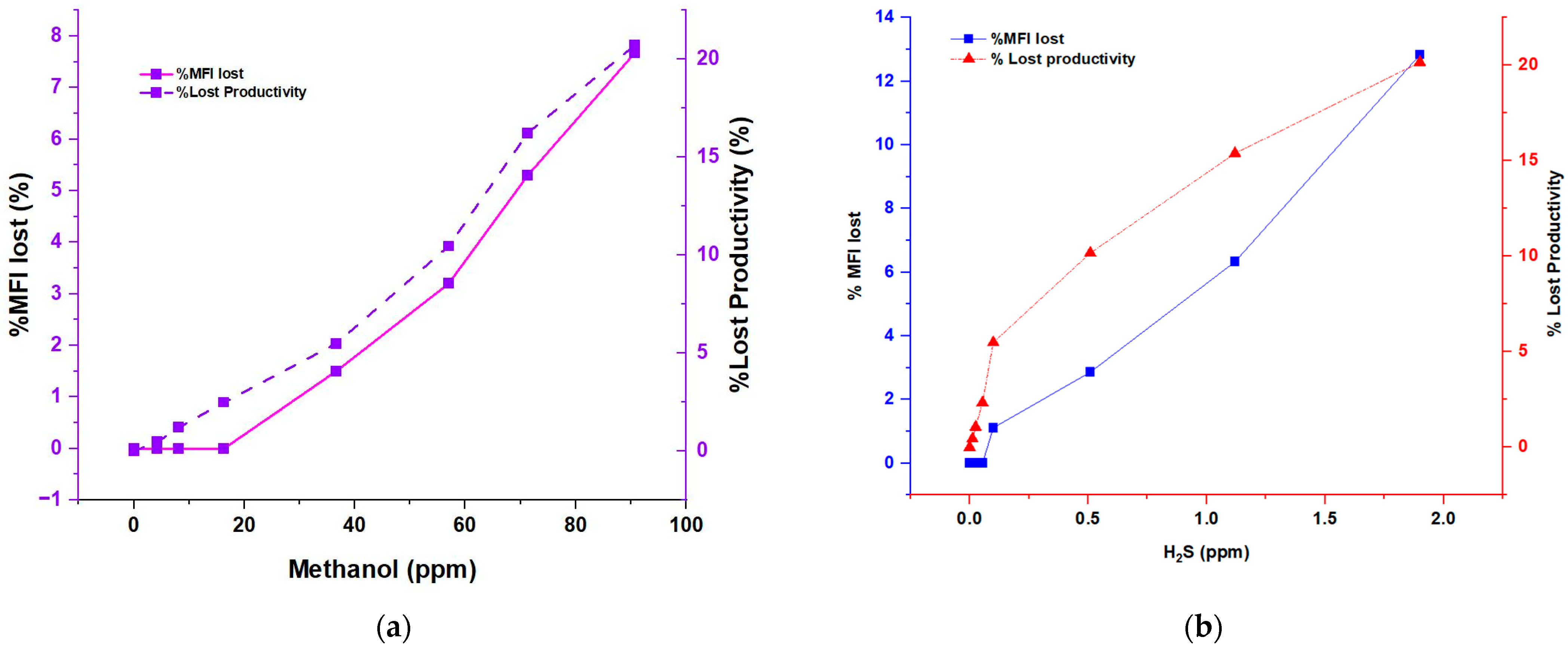
Figure 3 present the results of evaluating the properties of the obtained polymer, which reveals an apparent, direct influence of the presence of H
2S (hydrogen sulfide) and methanol on said properties. The data obtained indicate that as the concentration of the inhibitors (H
2S and methanol) increases, significant changes are produced in the melt index and the average molecular weight of the polymer. Specifically, it was observed that an increase in H
2S concentrations, on average by 0.5 and 1.0 ppm, resulted in a drastic decrease in the molecular weight of the polymer. This implies a shortening in the length of the polymer chains and an increase in the fluidity of the polymer, respectively. On the other hand, it was noted that, in the case of methanol, the change occurs from 60 parts per million, which also causes an abrupt decrease in the molecular weight of the polymer. These findings show a directly proportional relationship between the fluidity index and hydrogen sulfide concentration and methanol, respectively. Furthermore, the concentration of the inhibitors is inversely related to the average molecular weight (Mw), indicating a significant reduction in the resulting polymer chains. Moreover, these results support the negative influence of H
2S and methanol on polymer properties, highlighting the importance of carefully considering and controlling the presence of these compounds during the polymerization process to ensure the desired properties of the final polymer. They also underscore the need for additional studies to better understand the underlying mechanisms and long-term effects of H
2S and methanol exposure on polymers. These other studies may lead to improved formulation and development of polymers with optimized mechanical properties and excellent resistance to environmental conditions. In short, it is crucial to advance in understanding these mechanisms to promote the development of more efficient and sustainable polymers in the chemical industry.
3.1.2. Effects on Lost Melt Flow Index (MFI) and Lost Productivity of Ziegler–Natta Catalyst
Figure 4 presents a detailed analysis of the relationship between the H
2S and CH
3OH concentration as inhibitors and the fundamental parameters in polymerization, such as MFI and Mw. These results provide critical information on the impact of these inhibitors on the properties of the resulting polymer.
A directly proportional correlation was observed between the H2S concentration and both evaluated parameters, indicating that H2S has a significant effect on the physical properties of the polymer. As the H2S concentration increases, further loss of catalyst productivity occurs, affecting the melt flow rate of the polymer. This implies that the presence of H2S causes alterations in the flowability of the polymer and can affect the quality of the final product.
In addition, it was observed that as methanol concentrations increase, the melt flow rate lost in the polymer also increases. This implies that methanol affects the extension of polymer chains, which affects the productivity of the process. These changes in melt flow rate and catalyst productivity demonstrate the need to exercise rigorous control over the presence of H2S and methanol during polymerization to ensure the quality and desired characteristics of the final polymer.
3.1.3. Effects of Inhibitors on the Solubility in Xylene of PP
The analysis illustrated in
Figure 5 examines the impact of methanol concentration as an inhibitor and H
2S on the percentage solubility in xylene. The information on the effects of the concentration of inhibitors such as methanol and hydrogen sulfide on the percent solubility in xylene and the isotacticity of the resulting polymer has important implications for the type of polypropylene (PP) formed.
Percent xylene solubility measures the polymer’s ability to dissolve in xylene, a commonly used solvent in the characterization of isotactic polypropylenes. A high percentage of solubility in xylene indicates a greater capacity of the polymer to dissolve, which is related to a greater isotacticity. The more isotactic the polypropylene, the more ordered its structure and the better its mechanical properties. An inversely proportional relationship was observed between the H2S concentration and the percentage of solubility in xylene, which indicates that as the H2S concentration increases, there is a decrease in the solubility of the polymer in xylene. This same relationship is observed in the case of methanol since both inhibitors presented similar behaviors. These findings suggest that both H2S and methanol can affect the polymer’s structure and its ability to dissolve in xylene, decreasing said percentage, which implies a reduction in the isotacticity of the polymer. This suggests that polymerization in the presence of these inhibitors may result in the formation of polypropylene with lower isotacticity, which may have implications for the final properties of the polymer, such as its mechanical strength and stiffness.
It is important to note that the different types of polypropylene (isotactic, atactic, or syndiotactic) have other properties and applications. Isotactic polypropylene is the most desirable in many applications due to its high stiffness, strength, and thermal stability. Therefore, the decrease in the percentage of solubility in xylene and the possible reduction in the isotacticity of the polypropylene formed in the presence of inhibitors can hurt its properties and limit its usefulness in specific applications. In addition, the constant presence of a selective control agent in this study helps to modulate this effect, maintaining a high percentage of solubility in xylene at low concentrations of H
2S and methanol, respectively. This implies that selective control agents can help keep properties and low solubility rates in xylene, which is relevant to maintaining a higher isotacticity in the resulting polymer [
25,
26].
These findings highlight the importance of carefully controlling the concentration of methanol and hydrogen sulfide inhibitors during polymerization. In addition, the importance of considering selective control agents to maintain properties and low percentages of solubility in xylene is emphasized, which contributes to maintaining greater isotacticity in the resulting polymer. These measures can improve the formulation and development of polymers with optimized isotacticity characteristics, which is essential to determine the type of polypropylene produced and its applicability in different industries.
3.1.4. Effects of Inhibitors on Catalyst Productivity and Melt Flow Rate
Below are the graphs that show the results of the productivity of the Ziegler–Natta catalyst expressed in metric tons per kilogram (MT/Kg) about the melt flow index (MFI). These variables were plotted as a function of the concentration of two inhibitors, methanol (
Figure 6a) and hydrogen sulfide (
Figure 6b). By examining these graphs, an inverse relationship between methanol concentration and catalyst productivity can be seen. As the concentration of the inhibitor increases, productivity decreases, which is reflected in an increase in the melt index. This suggests that methanol affects the catalytic activity of the catalyst, resulting in a decrease in polymer production.
Similarly, an inverse relationship between hydrogen sulfide concentration and catalyst productivity is evident. As the H2S concentration increases, productivity decreases, and at the same time, an increase in the flow rate is observed. These effects indicate that hydrogen sulfide also affects the catalytic activity of the catalyst and results in decreased productivity.
It is important to note that these influences are more noticeable in the case of hydrogen sulfide (H2S) than in the case of methanol. For example, at deficient concentrations, such as 2.0 ppm H2S, a melt flow rate of 22.5 g/10 min and a 10 MT/Kg decrease in catalyst productivity are observed. This is similar to the reduction of catalyst productivity when a methanol concentration of 100 ppm is reached.
3.2. Frontier Molecular Orbitals and Global Structure–Activity Descriptors
Density functional theory (DFT) has successfully provided a theoretical foundation for explaining important chemical concepts such as electronegativity, hardness, and softness as well as more specific concepts like the Fukui function and local softness. By using the Koopman’s theorem, we can calculate the ionization potential and electron affinity of certain chemical substances, enabling us to obtain values for electronegativity and hardness.
When the energy of the highest occupied molecular orbital (HOMO) is higher, it signifies that the molecule is more prone to react with substances known as electrophiles. Conversely, a lower energy level in the lowest unoccupied molecular orbital (LUMO) is crucial for the molecule to react with substances known as nucleophiles.
This index measures how willing a chemical substance is to accept electrons. A highly reactive nucleophile will have a low value of ω, while a highly reactive electrophile will have a high value of ω. This new reactivity index helps us quantify how much energy is stabilized when a system acquires additional electrons from its surroundings.
According to Pearson’s theory, we can calculate the number of electrons transferred from the inhibiting molecule to the metal atom. In a reaction between two systems with different electronegativity levels (such as a metal surface and an inhibiting molecule), there is an electron flow from the less electronegative molecule to the more electronegative one until the chemical potentials are equalized.
The inhibitory effect of a compound is generally attributed to the adsorption of the molecule on the metal surface. Adsorption can be physical (physisorption) or chemical (chemisorption) depending on the strength of adsorption. During chemisorption, one of the reactive species acts as an electron pair donor, and the other acts as an electron pair acceptor. The energy of the highest occupied molecular orbital (EHOMO) measures the tendency of a molecule to donate electrons [
26]. High EHOMO values indicate the molecule’s tendency to donate electrons to suitable accepting molecules with vacant and low-energy molecular orbitals. Increasing EHOMO values facilitate adsorption, thereby enhancing inhibition efficiency by influencing the transport process through the adsorbed layer. Therefore, higher EHOMO values indicate a better electron-donating tendency, improving inhibitor adsorption on mild steel and consequently enhancing inhibition efficiency. ELUMO indicates the molecule’s capacity to accept electrons. The inhibitor’s binding ability to the metal surface increases with higher HOMO values and lower LUMO energy values. The frontier molecular orbital diagrams of H
2S and methanol are shown in
Figure 7.
Table 2 represents the total energy and energy levels calculated in (eV) for the HOMO, LUMO, and energy gap of the investigated molecules.
According to the frontier molecular orbital (FMO) theory in chemical reactivity, electron transfer occurs due to the interaction between two types of orbitals: the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of reacting substances [
27]. The EHOMO value is a chemical parameter often associated with a molecule’s electron-donating capacity. A high EHOMO value suggests that the molecule has a tendency to donate electrons to another molecule with a low-energy vacant orbital [
28].
The inhibitor not only can donate electrons to the vacant (LUMO) orbital of the metal ion but can also accept electrons from the metal d orbital, creating a feedback type of bonding. According to our results, the highest EHOMO value, which is −7.1648 (eV) for methanol, indicates a more efficient capacity of the inhibitor to induce Ziegler–Natta catalyst productivity loss.
The energy difference, calculated as (ΔE = ELUMO − EHOMO), is a key factor influencing an inhibiting molecule’s ability to interact with the metal surface. When this energy difference decreases, it means the molecule is more reactive, resulting in higher efficiency as an inhibitor since less energy is required to participate in the necessary chemical reactions [
29]. A molecule with a low-energy gap is more adaptable and is usually associated with higher chemical activity but lower long-term stability. The results in
Table 2 show that H
2S (7.8352 eV) has the lowest-energy gap, suggesting it could be more effective as an inhibitor, as supported by experimental results.
Ionization energy is a key indicator of the reactivity of chemical substances, whether they are atoms or molecules. When this energy is high, it signifies that substances are stable and do not readily react. Conversely, when it is low, it indicates high reactivity and a propensity to interact with other substances. In this case, the low ionization energy, which is 0.7929 (eV) for methanol, indicates that this compound is highly effective as an inhibitor (See
Table 3).
Absolute hardness and softness are key properties for assessing the stability and reactivity of a molecule. Chemical hardness essentially tells us how resistant a molecule is to changes in its electron cloud when subjected to small alterations during a chemical reaction. A “hard” molecule has a large energy gap between its components, whereas a “soft” molecule has a small energy gap [
30]. In our study, H
2S exhibits low hardness with a value of 3.9176 (eV) compared to methanol, indicating a small energy gap, as observed in
Table 2. For easier electron transfer, adsorption typically occurs in the part of the molecule where softness (S), which is a local property, has the highest value [
31]. H
2S has the highest softness value at 0.2552, albeit with a difference from methanol of only 0.0327.
According to Sanderson’s electronegativity equalization principle [
32], H
2S, with its high electronegativity (3.3221 eV), tends to quickly equalize its properties, suggesting it is less reactive and therefore has lower efficiency as an inhibitor. In
Table 3, we see that H
2S has a higher electronegativity than methanol. This implies that the electronegativity difference between the metal and the inhibitor is greater in the case of methanol compared to H
2S.
Mulliken population analysis is used to identify the sites in a molecule where inhibitors attach and calculate how charge is distributed throughout the molecule [
33]. The general consensus among experts is that the more negatively charged an atom in the molecule (known as a heteroatom) is, the more easily it can adhere to the metal surface through a donor–acceptor type of interaction [
34]. The charge distribution diagram for the catalyst and methanol and the one for the catalyst and H
2S is shown in
Figure 8, where regions marked in green indicate positive charges within the structure, and negative charges are highlighted in red areas. Brighter shades indicate larger charge magnitudes. It is essential to consider the scenario where a molecule will receive a certain amount of charge at one location and release a certain amount of charge through the same or another location [
35]. Parr and Yang proposed that a higher value of the Fukui function suggests greater reactivity [
36]. Therefore, when the condensed Fukui function has a higher value, it means that a specific center in the molecule is more reactive.
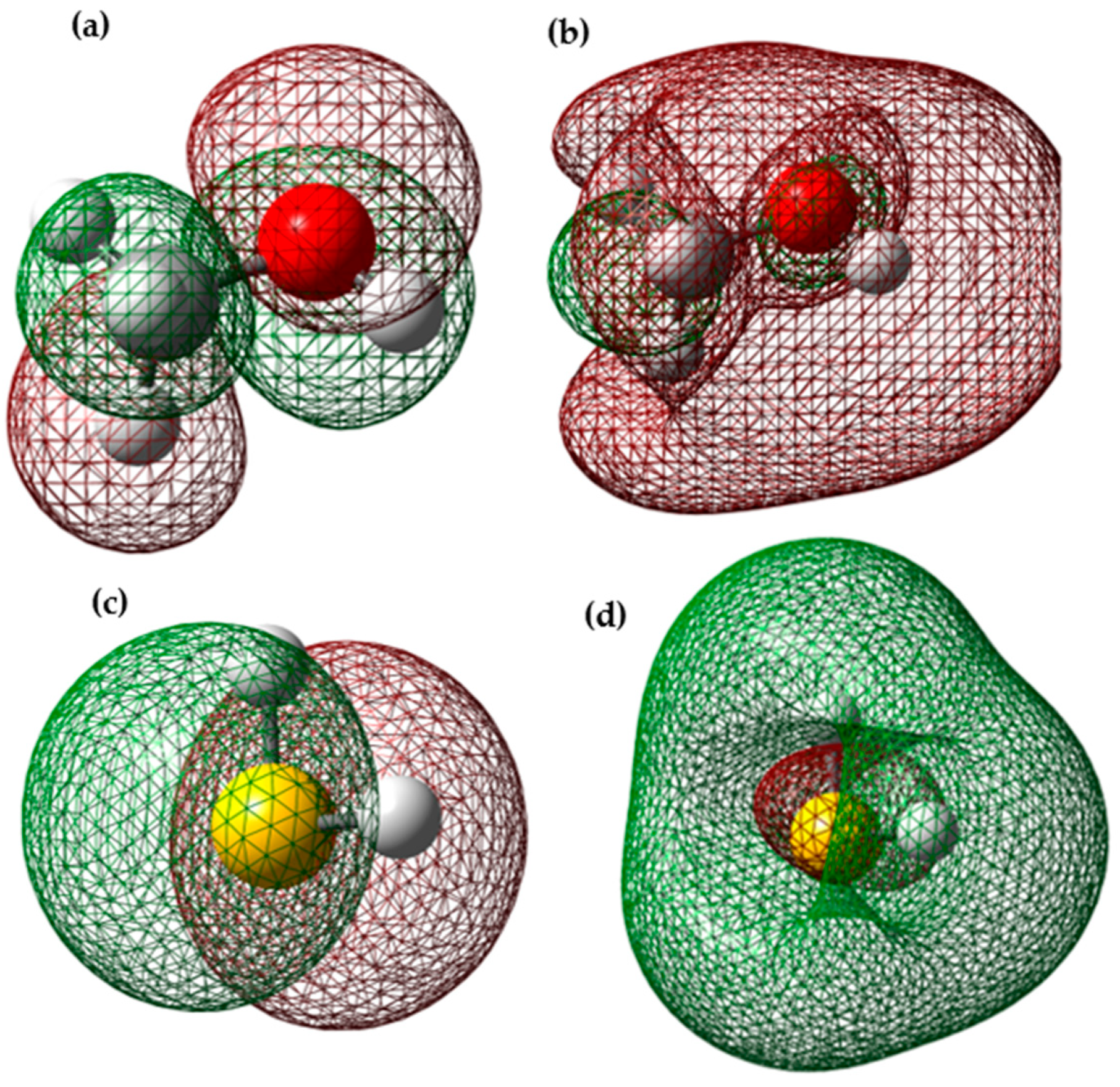
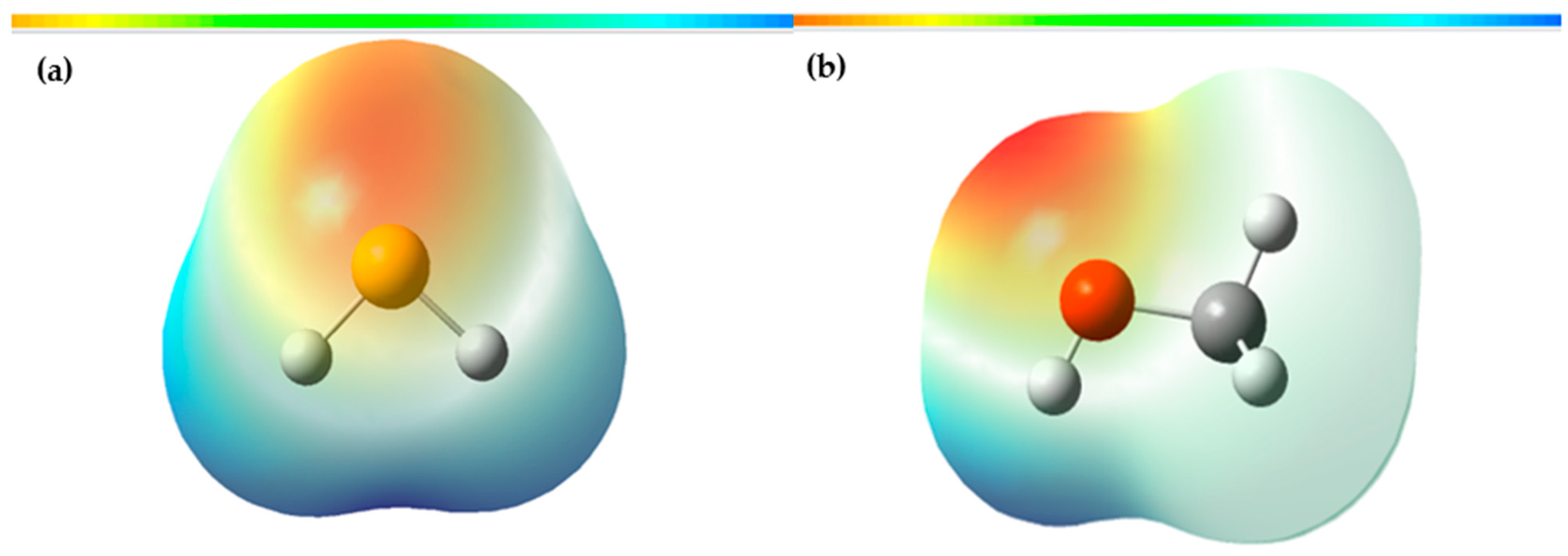
3.3. Molecular Electrostatic Potential of H2S and Methanol
The molecular electrostatic potential (MEP) map is a valuable tool in investigating global molecular structure and reactivity, as it provides detailed information about the charge distribution and the availability of electrons in a molecule. This uses colors to represent the different regions of the molecule according to their electron density. In the MEP, the red areas indicate a higher electron density, suggesting the presence of nucleophilic sites in the molecule, that is, areas where the molecule has a high probability of donating electrons. On the other hand, the blue areas represent an electron deficiency, indicating the presence of electrophilic sites, where the molecule has a higher affinity for accepting electrons. These characteristics are fundamental to understanding the molecule’s chemical reactivity since they allow us to predict the interactions with other chemical species. By identifying the nucleophilic and electrophilic sites in the MEP, we can determine which areas of the molecule are most likely to participate in chemical reactions and how they can interact with other substances.
Figure 9 presents a three-dimensional representation of the electrostatic effect, which covers a range of values from −3.502 × 10
−2 to 3.502 × 10
−2.
According to
Figure 9a, the most prone site for an electrophilic attack in the case of H
2S is located around the sulfur, indicated by the red area. In contrast, the most susceptible site for a nucleophilic attack is the blue areas distributed around the hydrogen atoms. In
Figure 9b, the MEP of methanol is shown in a range that varies from −6.288 × 10
−2 to 6.288 × 10
−2 eV, where the blue, green, and red colors indicate the regions with the most positive electrostatic potential, zero potential, and most negative electrostatic potential, respectively. The red and yellow regions are primarily located on the oxygen atom, which is the most reactive site for an electrophilic attack. On the other hand, the blue regions are found around the hydrogen atoms, which are the most reactive sites for a nucleophilic attack.
3.4. Local Chemical Reactivity Descriptors (Fukui Function)
The Fukui function is a valuable tool for identifying the most reactive sites in a molecule, either for nucleophilic or electrophilic reactions. Proposed by Parr and Yang in 1984, this function provides us with information about the changes in electron density at each site in the molecule.
By calculating the Fukui functions, we can determine which sites are most likely to accept or donate electrons. Sites with higher values are considered favorable sites for nucleophilic attack, meaning they have a more remarkable ability to accept electrons. On the other hand, the sites with higher values of are conducive to an electrophilic attack, indicating that they have a higher affinity to donate electrons. The dual descriptor f(r) is a better way to identify a reactive site in a molecule. It is defined as = , providing an adequate distinction between nucleophilic and electrophilic attack in a precise region according to its sign. When is less than zero (< 0), the site is favorable for electrophilic attack. On the other hand, when is more significant than zero (> 0), the site is good for a nucleophilic attack. This approach allows us to more precisely identify the reactive sites in a molecule and predict what type of chemical reaction is most likely to occur. Calculations were performed, and tables containing the Fukui functions (, , and ) and the dual descriptor () for H2S were created, allowing us to better understand the chemical reactivity of this molecule.
The maximum values of the local descriptors of the Fukui function, such as , , and indicate the sites where electrophilic, nucleophilic, or free radical attack on H2S is most likely to occur. According to the calculations, the sites most susceptible to an electrophilic attack in hydrogen sulfide are the ones in which the sulfide atom shows greater reactivity. On the other hand, the sites most vulnerable to nucleophilic attack are hydrogen atoms. On the other hand, sulfur sites are most susceptible to free radical attack.
Table 4 shows the variation of
depending on the atoms. The results reveal that, from the calculated values of
, the sulfide atom (1) appears to be the site prone to an electrophilic attack since the value of
= −0.2929. Meanwhile, hydrogen atoms (2–3) present positive
values, indicating they are the most favorable sites for a nucleophilic attack, with
values of 0.1436 and ∆f of 0.1482, respectively.
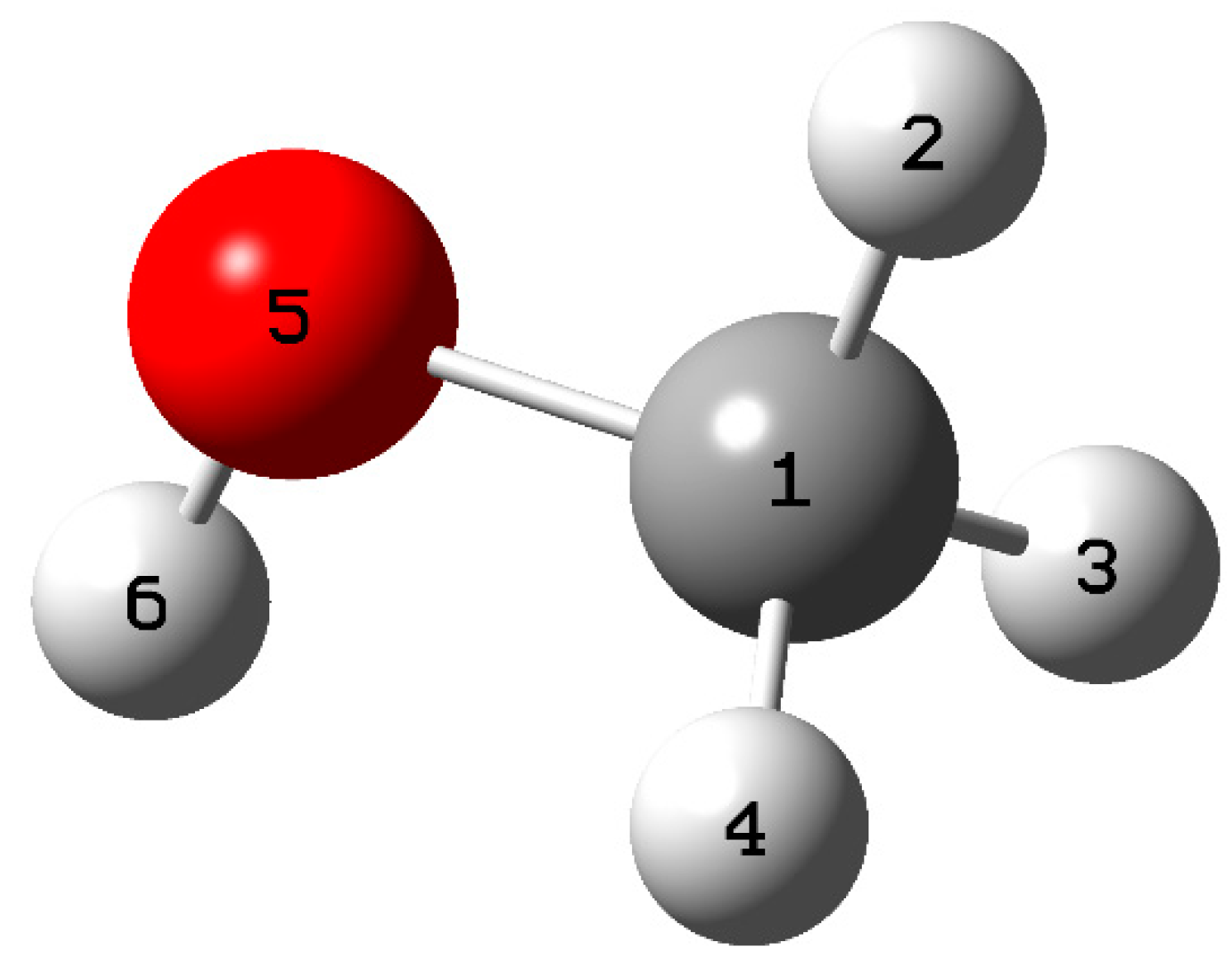
Table 5 shows the specific values of the Fukui functions and the dual descriptor for each site in the methanol molecule.
The calculations identified the sites most prone to electrophilic, nucleophilic, and free radical attacks in methanol. According to the results, the oxygen atom at position 5 is more susceptible to electrophilic and nucleophilic attack. Furthermore, it was found that the carbon at position one and the oxygen at position 5 are the sites most vulnerable to free radical attack (
Figure 10). These findings are supported by
Table 5, which shows the variations in the dual descriptor (
) about atoms. The results indicate that the oxygen atom at position 5 has a negative value of ∆f (−0.2800), indicating its higher propensity to electrophilic attack. On the other hand, the carbon at position 1 has a positive value of
(0.2166), indicating that it is a more favorable site for a nucleophilic attack.
3.5. Study of the Adsorption of Methanol and H2S on the Active Ti Center of the Ziegler–Natta Catalyst
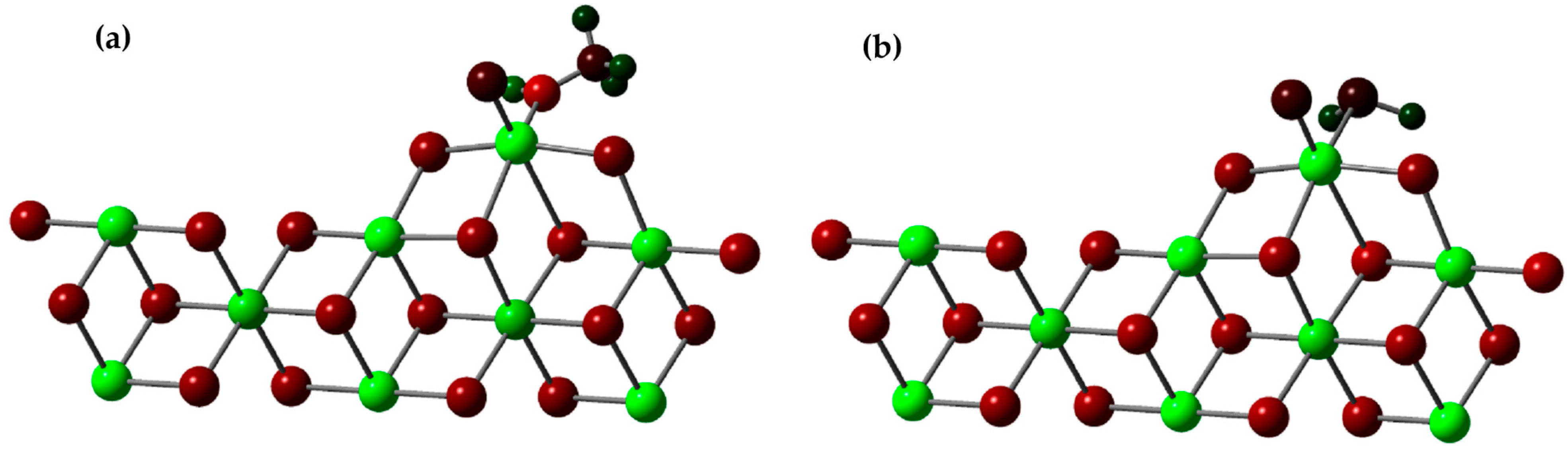
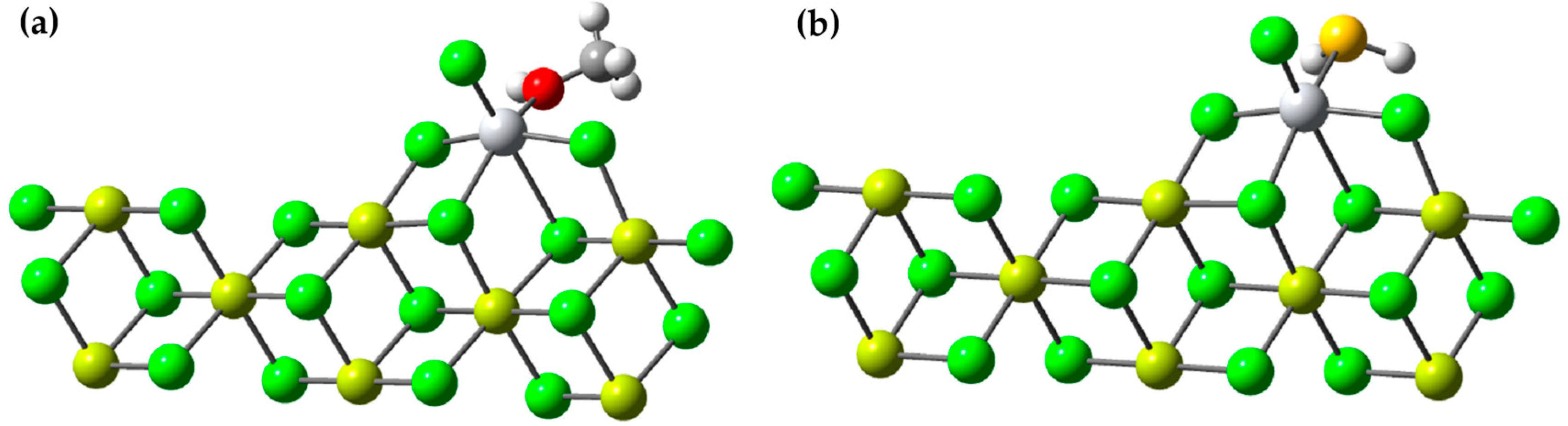
In this study, we investigated the role of methanol and H2S as inhibitors of the active titanium center in a scientifically relevant context. Our analysis focused on the adsorption energy (Ead) of these inhibitors and their ability to stabilize the active titanium center on the 110 surface of MgCl2 through DFT (density functional theory) calculations. To conduct the study, we drew upon both previous experimental and theoretical research, and we selected the β-MgCl2 surface, which has been shown to best explain the experimental observations. We constructed a three-dimensional (3D) model of the crystallographic structure that matched the standard database in the Materials Project. The crystal in the β phase had a trigonal shape and belonged to the P-3M1 space group, with dimensions a = b = 3.641 Å and c = 5.927 Å. We created layered models, which were three-dimensional (3D) objects repeating in two dimensions, arranged parallel to the specific crystallographic plane defined by its indices (h k l).
The adsorption of inhibitors is a key factor and has a significant impact on the productivity of the Ziegler–Natta catalyst, affecting the selectivity of the catalytic process and the form of the polypropylene (PP) that is produced. In this context, various studies were conducted to understand how these two inhibitors affect the Ziegler–Natta catalyst, evaluating how the interaction of inhibitors with the active titanium sites influences catalytic function.
We investigated the adsorption energy (Ead) of these substances using Equation (2).
In this equation,
represents the total energy of the system, which includes a molecule of methanol or H
2S bound to the active center of the catalyst (as seen in
Figure 11).
refers to the energy of the catalyst without any molecule of methanol or H
2S, and the energy of an individual molecule of methanol is
.
n is simply the quantity of methanol or H
2S molecules that have been adsorbed on the surface.
The results revealed that the Ead (adsorption energy) for methanol was −39.92 kcal/mol, while for H2S, it was −14.82 kcal/mol. These values are remarkable on their own, but what makes our findings even more intriguing is the comparison with previous results by Bahri, who reported an Ead of −30.6 kcal/mol for methanol in a similar study.
The significant difference in
Ead values between our study and Bahri’s suggests that methanol may have an even greater capacity to adsorb onto the active titanium center than previously considered [
37]. This finding could have profound implications in catalysis and catalyst design. The choice of effective inhibitors is crucial for controlling chemical reactions and improving the efficiency of catalytic processes. Our results support the idea that methanol could be a promising option as an inhibitor in specific applications involving the active titanium center, potentially opening new avenues for catalyst optimization in a variety of chemistry and industry fields. These findings are highly relevant and make a significant contribution to current knowledge in the field of catalysis, underscoring the importance of our work in the context of high-impact scientific research.
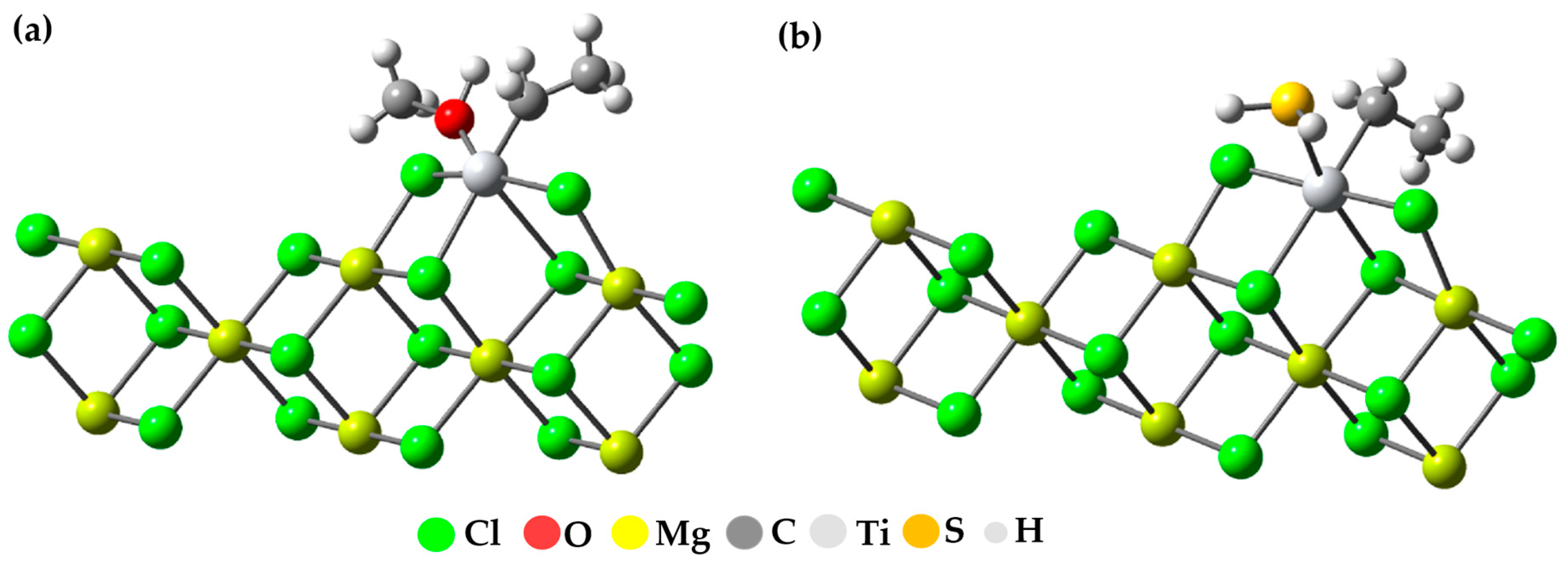
After investigating how the two inhibitors adhere to the active center of the catalyst, we decided to examine how these molecules adhere to the active center when there is an active TiCl
2Et compound present. To do this, we used the same group of atoms that we mentioned in the previous section (an active TiIII compound on the (110) surface). This is because it has been suggested that active titanium atoms may be organized on that plane or have a similar environment to what is assumed after they coordinate on that specific plane. A diagram of the model showing how the inhibiting molecules bond to the set containing the active titanium can be seen in
Figure 12.
Previous research has established that the bond between inhibitory substances and the titanium center is exceptionally strong compared to other interactions. In a previous study conducted by Bahri, using TiCl
2Me as the active center of the Ziegler–Natta catalyst, energy values of −27.2, −15.1, −8.4, −13.1, and −30.6 kcal/mol were obtained for interactions with Ti-H
2O, H
2S, CO
2, O
2, and CH
3OH, respectively. It is important to note that the interaction with the active titanium proved to be the most favorable among all evaluated [
37]. In our study, we focused our attention on interactions with Ti-CH
3OH and H
2S using TiCl
2Et as the active center, and we found energy values of −62.7 and −30.7 kcal/mol, respectively. These results further reinforce the remarkable strength of these interactions.
Our study reveals significantly improved results compared to previous research like Bahri’s. While Bahri obtained energy values of −15.1 and −30.6 kcal/mol for interactions with Ti-H2S and CH3OH, respectively, our findings show substantially lower values, recording −62.7 and −30.7 kcal/mol for interactions with Ti-CH3OH and H2S. These results indicate greater stability in the interactions studied in our approach, suggesting a significant advancement in the understanding of these molecular-level reactions.
In
Tables S1–S7, we present the cartesian coordinates of our calculations in B3LYP-D3 for Ziegler–Natta catalyst, Methanol, MgCl
2-TiCl
4-Methanol interaction, H
2S, MgCl
2-TiCl
4- interaction H
2S, MgCl
2/TiCl
4-Methanol-CH
3 interaction and MgCl
2/TiCl
4-H
2S-CH
3 respectively.
3.6. Reaction Mechanisms about Methanol and H2S as Inhibitors of the Z-N Catalyst
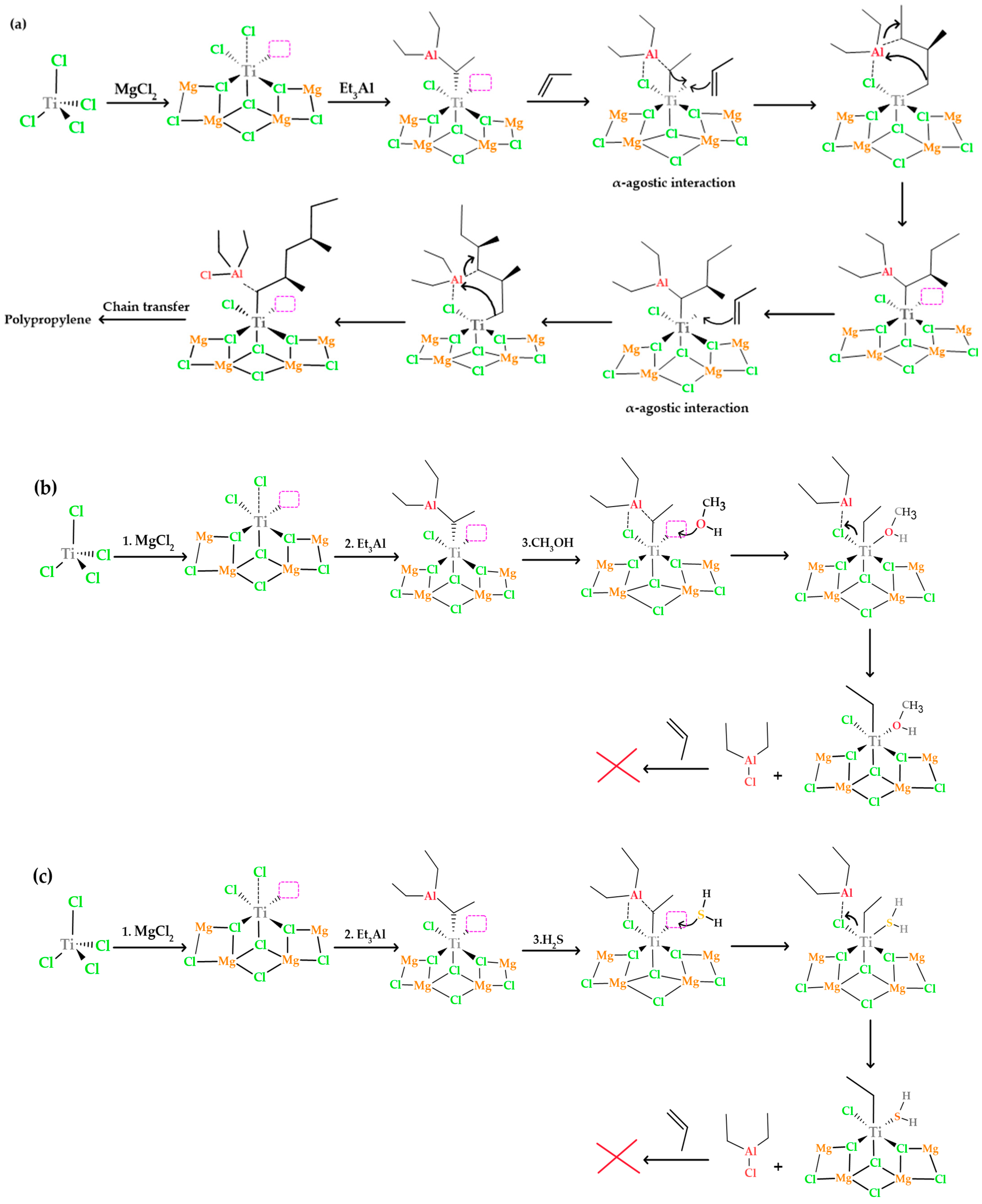
In
Figure 13a, the process of polypropylene formation in the absence of inhibitors is explained. In the first step, the monomer is introduced into the π-complex formed between titanium and ethyl. This insertion process occurs through an intermediate state that has a ring structure and leads to the creation of a propyl cation, via an α-agostic interaction. This interaction involves weak bonding between an atom in the monomer molecule and the titanium and ethyl atoms in the complex, partially sharing their electrons. This agostic interaction contributes to the stability of the structure and is a fundamental part of the process that allows the monomer to join the π-complex and form the propyl cation. During this insertion process, the chemical bond between titanium and the ethyl group is broken, allowing titanium to form a new bond with one of the carbon atoms of propylene, while the other carbon atom of propylene establishes a new bond with the ethyl group [
27,
38].
In a polymerization process, the titanium catalyst (Ti
3+) in its active state, represented as Ti-CH
2CH
3, is essential for the activation of the monomer and its subsequent incorporation into the polymer chain. However, when an inhibitor such as methanol (CH
3OH) or hydrogen sulfide (H
2S) is introduced, interactions occur that significantly affect the course of the reaction (see
Figure 13b,c). In the case of methanol, the oxygen in methanol is highly chemically attractive and capable of donating a pair of electrons to the titanium atom in the catalyst. This results in the formation of a coordinative bond between the oxygen of methanol and titanium, leaving methanol strongly bound to the active center of the catalyst, as observed in the adsorption energies. This interaction effectively inhibits the arrival of the monomer at the active site, as the oxygen of methanol occupies the space that would normally be available for the polymerization reaction. In the case of H
2S, sulfur can also coordinate with the titanium atom, forming a coordinative bond similar to that described above. This blocks the active site of the catalyst and prevents the monomer from approaching and binding to the titanium center, halting the polymerization process.
3.7. Electronic Properties of the Ziegler–Natta Catalyst
Since titanium in the Ti
4+ state has no electrons in its d orbitals, in the molecular orbital diagram in
Figure 14b, we mainly observe the transfer of electrons from the π orbitals of chlorine to the vacant d orbitals of titanium. This transfer is due to the effect of the crystalline field, which divides the d orbitals into dt2g and deg levels. In a simplified model of a Ti
4+ surrounded by six chlorine ligands in an octahedral geometry, dt2g orbitals are shown to influence the molecular orbital (MO) diagram significantly. These orbitals mix with the MOs centered on the metal, giving rise to linear combination orbitals adapted to the system’s symmetry. The lowest-energy electronic transitions correspond to the transfer of charge from the π orbitals of chlorine to the dt2g orbitals of titanium (transition A) and the deg orbitals (transition B). However, the experimental spectra do not simply consist of two bands separated by crystal field separation. The d orbitals of Ti
4+ have a similar symmetry to the π orbitals of the ligands. The π orbitals have lower energy than the d orbitals of the metal, leading to the formation of ligand-centered MO bonds and the occupancy of the bonding t2g MO. In contrast, the antibonding t2g* π orbitals remain vacated.
As a consequence of the interaction between the chlorine ligands and the titanium d orbitals, the metal’s dt2g orbitals experience an increase in energy, leading to a reduction in the crystal field separation (ΔCF) compared to a σ bond. In this framework, the lowest-energy electronic transitions involve the transfer of electrons from the chlorine’s π orbitals to the titanium’s dt2g orbitals (transition A) and the degenerate orbitals (transition B). The computational spectrum derived from the Ziegler–Natta (ZN) catalyst’s structure used in this study (
Figure 14a) is consistent with the spectrum of the Ziegler–Natta catalyst modified by the electron donor (ZNC(DBP)) conducted by Piovano et al. [
27]. This consistency reveals the presence of the characteristic peak corresponding to the first Cl(π) → Ti(dt2g) transition with a minimum absorption wavelength (band A’, 378 nm). However, it is noteworthy that, in the presence of both inhibitors, a shift in the absorption wavelength is observed, shifting from 377 nm to 399 nm. This phenomenon can be attributed to the interaction of these impurities with the catalyst, thereby influencing its chemical environment. Consequently, a modification in the energy associated with the Cl(π) → Ti(dt2g) transition occurs, resulting in a variation in the absorption wavelength recorded in the UV–vis spectrum. This phenomenon is of significance in industrial applications where precise control of catalytic activity is sought after, necessitating a detailed characterization of the molecular interactions involved.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}