




Synthesis and Characterisation of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-b-poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Multi-Block Copolymers Produced Using Diisocyanate Chemistry


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Block Copolymer Synthesis
2.2.1. Drying of Glassware, Solvents and PHBV Reagents
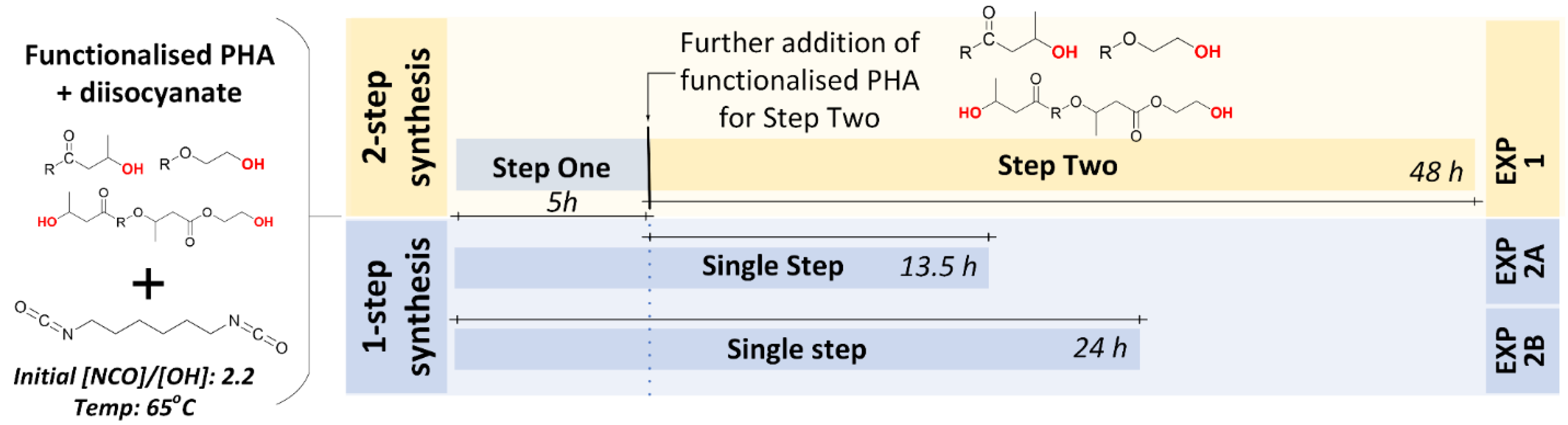
2.2.2. Experiment #1: Two-Step Synthesis of PHBV-b-PHBV Multi-Block Copolymer
2.2.3. Experiment #2A: One-Step Synthesis of PHBV-b-PHBV Multi-Block Copolymers
2.2.4. Experiment #2B: One-Step Synthesis of PHBV-b-PHBV Multi-Block Copolymers with Longer Reaction Time
2.3. Preparation of PHBV Films by Solvent Casting
2.4. Solubility Assessment for As-Produced Block Copolymeric Products
2.5. Material Characterization
2.5.1. Gel Permeation Chromatography (GPC)
2.5.2. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.5.3. Differential Scanning Calorimetry (DSC)
3. Results and Discussion
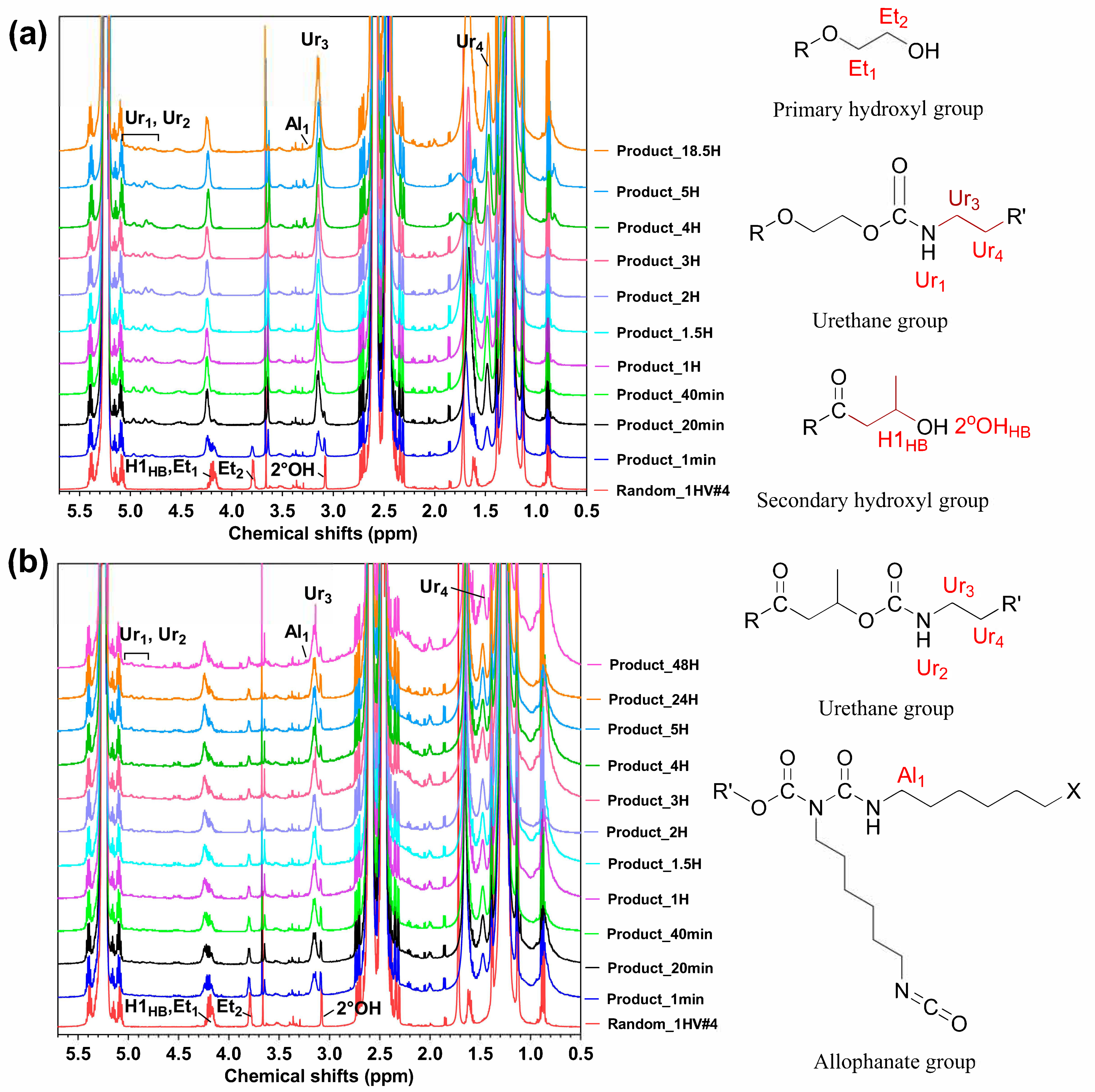
3.1. Molecular Weight and Functional Groups of the Reaction Products
3.1.1. Two-Step Synthesis (Experiment #1)
3.1.2. One-Step Synthesis (Experiment #2A and 2B)
3.2. Thermal Properties of As-Synthesised Block Copolymers
4. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kellerhals, M.B.; Kessler, B.; Witholt, B.; Tchouboukov, A.; Brandl, H. Renewable Long-Chain Fatty Acids for Production of Biodegradable Medium-Chain-Length Polyhydroxyalkanoates (mcl-PHAs) at Laboratory and Pilot Plant Scales. Macromolecules 2000, 33, 4690–4698. [Google Scholar] [CrossRef]
- Chen, G.Q.; Wu, Q. The application of polyhydroxyalkanoates as tissue engineering materials. Biomaterials 2005, 26, 6565–6578. [Google Scholar] [CrossRef]
- Li, Z.B.; Loh, X.J. Water soluble polyhydroxyalkanoates: Future materials for therapeutic applications. Chem. Soc. Rev. 2015, 44, 2865–2879. [Google Scholar] [CrossRef]
- Pan, J.; Li, G.; Chen, Z.; Chen, X.; Zhu, W.; Xu, K. Alternative block polyurethanes based on poly (3-hydroxybutyrate-co-4-hydroxybutyrate) and poly (ethylene glycol). Biomaterials 2009, 30, 2975–2984. [Google Scholar] [CrossRef]
- Bejagam, K.K.; Iverson, C.N.; Marrone, B.L.; Pilania, G. Composition and Configuration Dependence of Glass-Transition Temperature in Binary Copolymers and Blends of Polyhydroxyalkanoate Biopolymers. Macromolecules 2021, 54, 5618–5628. [Google Scholar] [CrossRef]
- Shah, D.T.; Tran, M.; Berger, P.A.; Aggarwal, P.; Asrar, J.; Madden, L.A.; Anderson, A.J. Synthesis and Properties of Hydroxy-Terminated Poly(hydroxyalkanoate)s. Macromolecules 2000, 33, 2875–2880. [Google Scholar] [CrossRef]
- Akaraonye, E.; Keshavarz, T.; Roy, I. Production of polyhydroxyalkanoates: The future green materials of choice. J. Chem. Technol. Biotechnol. 2010, 85, 732–743. [Google Scholar]
- Ferre-Guell, A.; Winterburn, J. Biosynthesis and Characterization of Polyhydroxyalkanoates with Controlled Composition and Microstructure. Biomacromolecules 2018, 19, 996–1005. [Google Scholar] [CrossRef]
- Arcos-Hernández, M.V.; Laycock, B.; Donose, B.C.; Pratt, S.; Halley, P.; Al-Luaibi, S.; Werker, A.; Lant, P.A. Physicochemical and mechanical properties of mixed culture polyhydroxyalkanoate (PHBV). Eur. Polym. J. 2013, 49, 904–913. [Google Scholar] [CrossRef]
- Li, S.Y.; Dong, C.L.; Wang, S.Y.; Ye, H.M.; Chen, G.Q. Microbial production of polyhydroxyalkanoate block copolymer by recombinant Pseudomonas putida. Appl. Microbiol. Biotechnol. 2011, 90, 659–669. [Google Scholar] [CrossRef]
- Tripathi, L.; Wu, L.P.; Chen, J.C.; Chen, G.Q. Synthesis of Diblock copolymer poly-3-hydroxybutyrate -block-poly-3-hydroxyhexanoate PHB-b-PHHx by a beta-oxidation weakened Pseudomonas putida KT2442. Microb. Cell. Fact. 2012, 11, 11. [Google Scholar] [CrossRef]
- Westlie, A.H.; Chen, E.Y.X. Catalyzed Chemical Synthesis of Unnatural Aromatic Polyhydroxyalkanoate and Aromatic–Aliphatic PHAs with Record-High Glass-Transition and Decomposition Temperatures. Macromolecules 2020, 53, 9906–9915. [Google Scholar] [CrossRef]
- Lin, Y.; Böker, A.; He, J.; Sill, K.; Xiang, H.; Abetz, C.; Li, X.; Wang, J.; Emrick, T.; Long, S.; et al. Self-directed self-assembly of nanoparticle/copolymer mixtures. Nature 2005, 434, 55–59. [Google Scholar] [CrossRef]
- Hu, D.; Chung, A.-L.; Wu, L.-P.; Zhang, X.; Wu, Q.; Chen, J.-C.; Chen, G.-Q. Biosynthesis and Characterization of Polyhydroxyalkanoate Block Copolymer P3HB-b-P4HB. Biomacromolecules 2011, 12, 3166–3173. [Google Scholar] [CrossRef]
- Oliveira, F.C.; Dias, M.L.; Castilho, L.R.; Freire, D.M.G. Characterization of poly(3-hydroxybutyrate) produced by Cupriavidus necator in solid-state fermentation. Bioresour. Technol. 2007, 98, 633–638. [Google Scholar] [CrossRef]
- Luo, S.; Grubb, D.T.; Netravali, A.N. The effect of molecular weight on the lamellar structure, thermal and mechanical properties of poly(hydroxybutyrate-co-hydroxyvalerates). Polymer 2002, 43, 4159–4166. [Google Scholar] [CrossRef]
- Yu, J.; Chen, L.X.L. Cost-Effective Recovery and Purification of Polyhydroxyalkanoates by Selective Dissolution of Cell Mass. Biotechnol. Prog. 2006, 22, 547–553. [Google Scholar] [CrossRef]
- Tanadchangsaeng, N.; Yu, J. Microbial synthesis of polyhydroxybutyrate from glycerol: Gluconeogenesis, molecular weight and material properties of biopolyester. Biotechnol. Bioeng. 2012, 109, 2808–2818. [Google Scholar] [CrossRef]
- McChalicher, C.W.J.; Srienc, F. Investigating the structure–property relationship of bacterial PHA block copolymers. J. Biotechnol. 2007, 132, 296–302. [Google Scholar] [CrossRef]
- Pederson, E.N.; McChalicher, C.W.J.; Srienc, F. Bacterial Synthesis of PHA Block Copolymers. Biomacromolecules 2006, 7, 1904–1911. [Google Scholar] [CrossRef]
- Tripathi, L.; Wu, L.-P.; Meng, D.; Chen, J.; Chen, G.-Q. Biosynthesis and Characterization of Diblock Copolymer of P(3-Hydroxypropionate)-block-P(4-hydroxybutyrate) from Recombinant Escherichia coli. Biomacromolecules 2013, 14, 862–870. [Google Scholar] [CrossRef]
- Kageyama, Y.; Tomita, H.; Isono, T.; Satoh, T.; Matsumoto, K. Artificial polyhydroxyalkanoate poly [2-hydroxybutyrate-block-3-hydroxybutyrate] elastomer-like material. Sci. Rep. 2021, 11, 22446. [Google Scholar] [CrossRef]
- Nakaoki, T.; Yasui, J.; Komaeda, T. Biosynthesis of P3HBV-b-P3HB-b-P3HBV Triblock Copolymer by Ralstonia eutropha. J. Polym. Environ. 2019, 27, 2720–2727. [Google Scholar] [CrossRef]
- Li, Z.B.; Yang, J.; Loh, X.J. Polyhydroxyalkanoates: Opening doors for a sustainable future. NPG Asia Mater. 2016, 8, 20. [Google Scholar] [CrossRef]
- Meng, D.C.; Shen, R.; Yao, H.; Chen, J.C.; Wu, Q.; Chen, G.Q. Engineering the diversity of polyesters. Curr. Opin. Biotechnol. 2014, 29, 24–33. [Google Scholar] [CrossRef]
- Müller, A.J.; Arnal, M.L.; Balsamo, V. Crystallization in Block Copolymers with More than One Crystallizable Block. In Progress in Understanding of Polymer Crystallization; Reiter, G., Strobl, G.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 229–259. [Google Scholar]
- Samui, A.B.; Kanai, T. Polyhydroxyalkanoates based copolymers. Int. J. Biol. Macromol. 2019, 140, 522–537. [Google Scholar] [CrossRef]
- Adamus, G.; Sikorska, W.; Janeczek, H.; Kwiecień, M.; Sobota, M.; Kowalczuk, M. Novel block copolymers of atactic PHB with natural PHA for cardiovascular engineering: Synthesis and characterization. Eur. Polym. J. 2012, 48, 621–631. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Ni, X.; Leong, K.W. Synthesis and Characterization of New Biodegradable Amphiphilic Poly(ethylene oxide)-b-poly[(R)-3-hydroxy butyrate]-b-poly(ethylene oxide) Triblock Copolymers. Macromolecules 2003, 36, 2661–2667. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Ni, X.; Wang, X.; Li, H.; Leong, K.W. Self-assembled supramolecular hydrogels formed by biodegradable PEO–PHB–PEO triblock copolymers and α-cyclodextrin for controlled drug delivery. Biomaterials 2006, 27, 4132–4140. [Google Scholar] [CrossRef]
- Loh, X.J.; Goh, S.H.; Li, J. New Biodegradable Thermogelling Copolymers Having Very Low Gelation Concentrations. Biomacromolecules 2007, 8, 585–593. [Google Scholar] [CrossRef]
- Güney, A.; Gardiner, C.; McCormack, A.; Malda, J.; Grijpma, D.W. Thermoplastic PCL-b-PEG-b-PCL and HDI Polyurethanes for Extrusion-Based 3D-Printing of Tough Hydrogels. Bioengineering 2018, 5, 99. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Wu, L.; Chen, Z.; Lin, Y.; Xu, K.; Chen, G.Q. Synthesis, characterization and biocompatibility of biodegradable elastomeric poly(ether-ester urethane)s Based on Poly(3-hydroxybutyrate-co-3-hydroxyhexanoate) and Poly(ethylene glycol) via melting polymerization. J. Biomater. Sci. Polym. Ed. 2009, 20, 1179–1202. [Google Scholar] [CrossRef]
- Ou, W.; Qiu, H.; Chen, Z.; Xu, K. Biodegradable block poly(ester-urethane)s based on poly(3-hydroxybutyrate-co-4-hydroxybutyrate) copolymers. Biomaterials 2011, 32, 3178–3188. [Google Scholar] [CrossRef]
- Ramier, J.; Renard, E.; Grande, D. Microwave-assisted synthesis and characterization of biodegradable block copolyesters based on poly(3-hydroxyalkanoate)s and poly(D,L-lactide). J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1445–1455. [Google Scholar] [CrossRef]
- Velankar, S.; Cooper, S.L. Microphase separation and rheological properties of polyurethane melts. 1. Effect of block length. Macromolecules 1998, 31, 9181–9192. [Google Scholar] [CrossRef]
- Andrade, A.P.; Neuenschwander, P.; Hany, R.; Egli, T.; Witholt, B.; Li, Z. Synthesis and characterization of novel copoly (ester− urethane) containing blocks of poly-[(R)-3-hydroxyoctanoate] and poly-[(R)-3-hydroxybutyrate]. Macromolecules 2002, 35, 4946–4950. [Google Scholar] [CrossRef]
- Shen, Y.; Deng, J.J.; Luo, X.; Zhang, X.; Zeng, X.; Feng, M.; Pan, S.R. Synthesis and Characterization of a Sterically Stabilized Polyelectrolyte Using Isophorone Diisocyanate as the Coupling Reagent. J. Biomater. Sci.-Polym. Ed. 2009, 20, 1217–1233. [Google Scholar] [CrossRef]
- Chen, Z.F.; Cheng, S.T.; Xu, K.T. Block poly(ester-urethane)s based on poly(3-hydroxybutyrate-co-4-hydroxybutyrate) and poly(3-hydroxyhexanoate-co-3-hydroxyoctanoate). Biomaterials 2009, 30, 2219–2230. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, M.; Chen, Y. Synthesis and characterization of multi-block copolymers containing poly [(3-hydroxybutyrate)-co-(3-hydroxyvalerate)] and poly(ethylene glycol). Polym. Int. 2010, 59, 842–850. [Google Scholar] [CrossRef]
- Kai, D.; Loh, X.J. Polyhydroxyalkanoates: Chemical Modifications Toward Biomedical Applications. ACS Sustain. Chem. Eng. 2014, 2, 106–119. [Google Scholar] [CrossRef]
- Li, X.; Loh, X.J.; Wang, K.; He, C.; Li, J. Poly(ester urethane)s Consisting of Poly[(R)-3-hydroxybutyrate] and Poly(ethylene glycol) as Candidate Biomaterials: Characterization and Mechanical Property Study. Biomacromolecules 2005, 6, 2740–2747. [Google Scholar] [CrossRef]
- Mai, J.J.; Chan, C.M.; Colwell, J.; Pratt, S.; Laycock, B. Characterisation of end groups of hydroxy-functionalised scl-PHAs prepared by transesterification using ethylene glycol. Polym. Degrad. Stab. 2022, 205, 110123. [Google Scholar] [CrossRef]
- Mai, J.; Chan, C.M.; Laycock, B.; Pratt, S. Understanding the Reaction of Hydroxy-Terminated Poly (3-hydroxybutyrate-co-3-hydroxyvalerate)(PHBV) Random Copolymers with a Monoisocyanate. Macromolecules 2023, 56, 2328–2338. [Google Scholar] [CrossRef]
- Li, G.; Li, D.; Niu, Y.; He, T.; Chen, K.C.; Xu, K. Alternating block polyurethanes based on PCL and PEG as potential nerve regeneration materials. J. Biomed. Mater. Res. Part A 2014, 102, 685–697. [Google Scholar] [CrossRef]
- Kinard, L.A.; Kasper, F.K.; Mikos, A.G. Synthesis of oligo(poly(ethylene glycol) fumarate). Nat. Protoc. 2012, 7, 1219–1227. [Google Scholar] [CrossRef]
- Marchessault, R.; Okamura, K.; Su, C. Physical properties of poly (β-hydroxy butyrate). II. Conformational aspects in solution. Macromolecules 1970, 3, 735–740. [Google Scholar] [CrossRef]
- Sekkar, V.; Bhagawan, S.S.; Prabhakaran, N.; Rama Rao, M.; Ninan, K.N. Polyurethanes based on hydroxyl terminated polybutadiene: Modelling of network parameters and correlation with mechanical properties. Polymer 2000, 41, 6773–6786. [Google Scholar] [CrossRef]
- Petrović, Z.S.; Javni, I.; Divjaković, V. Structure and physical properties of segmented polyurethane elastomers containing chemical crosslinks in the hard segment. J. Polym. Sci. Part B Polym. Phys. 1998, 36, 221–235. [Google Scholar] [CrossRef]
- Ishida, M.; Yoshinaga, K.; Horii, F. Solid-State 13C NMR Analyses of the Microphase-Separated Structure of Polyurethane Elastomer. Macromolecules 1996, 29, 8824–8829. [Google Scholar] [CrossRef]
- Stern, T. Hierarchical fractal-structured allophanate-derived network formation in bulk polyurethane synthesis. Polym. Adv. Technol. 2018, 29, 746–757. [Google Scholar] [CrossRef]
- Li, G.; Li, P.; Qiu, H.; Li, D.; Su, M.; Xu, K. Synthesis, characterizations and biocompatibility of alternating block polyurethanes based on P3/4HB and PPG-PEG-PPG. J. Biomed. Mater. Res. Part A 2011, 98, 88–99. [Google Scholar] [CrossRef]
- Desai, S.; Thakore, I.M.; Sarawade, B.D.; Devi, S. Effect of polyols and diisocyanates on thermo-mechanical and morphological properties of polyurethanes. Eur. Polym. J. 2000, 36, 711–725. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp’t No. | Hydroxy-functionalised PHBV Starting Materials | (kDa) | (kDa) | Đ | 3HV (mol%) | 1 °OH and 2 °OH Used in Reaction (mmoles/ g PHA) | Description | Experimental Conditions |
|---|---|---|---|---|---|---|---|---|
| 1 | Random_1HV#4 | 28 | 51 | 1.8 | 1 | 0.084 | First and second steps of the two-step synthesis | [NCO]/[OH] for step 1 = 2.2; Temp = 65 °C; Time (step 1) = 5 h; Theoretical [NCO]/[OH] for step 2 = 0.5 *; Temp = 65 °C; Time (step 2) = 48 hOverall [NCO]/[OH] = 2.2:3 |
| 2A | Random_1HV#4 | 28 | 51 | 1.8 | 1 | 0.084 | One-step synthesis (first step of the two-step synthesis) | [NCO]/[OH] for step 1 = 2.2; Temp = 65 °C: Time = 18.5 h |
| 2B | Random_1HV#5 | 25 | 44 | 1.7 | 1 | 0.103 | One-step synthesis extended | [NCO]/[OH] = 2.2; Temp = 65 °C: Time = 24 h |
| Experiment No. | Final Reaction Products | Insoluble Component (wt.%) |
|---|---|---|
| 1 | Product of two-step synthesis | 2.7 |
| 2A | Block_1HV#1 | 4.4 |
| 2B | Block_1HV#2 | 19.5 |
| PHBV Material | (kDa) | (kDa) | Đ | (°C) | (J/g) | (°C) | (J/g) | (°C) | |
|---|---|---|---|---|---|---|---|---|---|
| random | Random_1HV#1 | 86 | 186 | 2.2 | 157.6/ 174.0 | 102.3 | 98.3 | 87.1 | 2.6 |
| Random_1HV#2 | 117 | 273 | 2.3 | 160.0/ 175.0 | 102.5 | 103.4 | 89.2 | 2.6 | |
| Random_1HV#3 | 194 | 455 | 2.3 | 159.5/ 170.0 | 106.0 | 104.7 | 87.3 | 4.0 | |
| block | Block_1HV#1 | 94 | 159 | 1.7 | 153.1/ 168.5 | 94.0 | 104.9 | 78.9 | 6.3 |
| Block_1HV#2 | 107 | 221 | 2.1 | 152.2/ 167.3 | 89.2 | 110.0 | 83.7 | 8.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mai, J.; Pratt, S.; Laycock, B.; Chan, C.M. Synthesis and Characterisation of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-b-poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Multi-Block Copolymers Produced Using Diisocyanate Chemistry. Polymers 2023, 15, 3257. https://doi.org/10.3390/polym15153257
Mai J, Pratt S, Laycock B, Chan CM. Synthesis and Characterisation of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-b-poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Multi-Block Copolymers Produced Using Diisocyanate Chemistry. Polymers. 2023; 15(15):3257. https://doi.org/10.3390/polym15153257
Chicago/Turabian StyleMai, Jingjing, Steven Pratt, Bronwyn Laycock, and Clement Matthew Chan. 2023. "Synthesis and Characterisation of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-b-poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Multi-Block Copolymers Produced Using Diisocyanate Chemistry" Polymers 15, no. 15: 3257. https://doi.org/10.3390/polym15153257
APA StyleMai, J., Pratt, S., Laycock, B., & Chan, C. M. (2023). Synthesis and Characterisation of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-b-poly(3-hydroxybutyrate-co-3-hydroxyvalerate) Multi-Block Copolymers Produced Using Diisocyanate Chemistry. Polymers, 15(15), 3257. https://doi.org/10.3390/polym15153257