
Scaffold Chemical Model Based on Collagen—Methyl Methacrylate Graft Copolymers

 , , ,
, , ,  ,
,  and
and 
Abstract
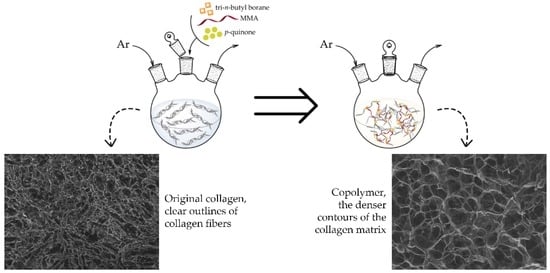
1. Introduction
- Obtain copolymers of methyl methacrylate and fish collagen in the presence of tributyl borane and several p-quinones differing in their structure and reactivity, in addition to the previously obtained data [36];
- Characterize the obtained products using the methods of gel-penetrating chromatography and scanning electron microscopy;
- Evaluate the propensity of copolymers for biodegradation under the action of enzymes in comparison with collagen to understand the prospects of their use as precursors of scaffolds;
- Conduct a cytotoxicity examination;
- Form a chemical model of scaffold construction based on collagen and methyl methacrylate graft-copolymers using tributyl borane and p-quinones.
2. Materials and Methods
2.1. Materials
2.2. Synthesis of TBB
2.3. Polymerization Procedure
2.4. Determination of the Unreacted Monomer
2.5. Enzymatic Hydrolysis
2.6. Size-Exclusion Chromatography
2.7. Scanning Electron Microscopy
2.8. NMR Spectroscopy
2.9. Cytotoxicity Examination via MTT Assay
- RGI 100%—rank 0;
- RGI 99–75%—rank 1 corresponds to the absence of cytotoxicity;
- RGI 74–50%—rank 2 corresponds to a mild degree of cytotoxicity;
- RGI 49–25%—rank 3 corresponds to an average degree of cytotoxicity;
- RGI 24–0%—rank 4 corresponds to high cytotoxicity [40].
3. Results and Discussion
 , p-benzoquinone
, p-benzoquinone  , duroquinone
, duroquinone  , and p-naphthoquinone
, and p-naphthoquinone  .
.4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ozbolat, I.T.; Hospodiuk, M. Current advances and future perspectives in extrusion-based bioprinting. Biomaterials 2016, 76, 321–343. [Google Scholar] [CrossRef]
- Sundar, G.; Joseph, J.; John, A.; Abraham, A. Natural collagen bioscaffolds for skin tissue engineering strategies in burns: A critical review. Int. J. Polym. Mater. 2021, 70, 593–604. [Google Scholar] [CrossRef]
- Puppi, D.; Chiellini, F. Biodegradable polymers for biomedical additive manufacturing. Appl. Mater. Today 2020, 20, 100700. [Google Scholar] [CrossRef]
- Bracaglia, L.G.; Smith, B.T.; Watson, E.; Arumugasaamy, N.; Mikos, A.G.; Fisher, J.P. 3D printing for the design and fabrication of polymer-based gradient scaffolds. Acta Biomater. 2017, 56, 3–13. [Google Scholar] [CrossRef]
- Fioretta, E.S.; Motta, S.E.; Lintas, V.; Loerakker, S.; Parker, K.K.; Baaijens, F.P.; Volkmar, F.; Simon, P.H.; Emmert, M.Y. Next-generation tissue-engineered heart valves with repair, remodelling and regeneration capacity. Nat. Rev. Cardiol. 2021, 18, 92–116. [Google Scholar] [CrossRef]
- Jung, O.; Smeets, R.; Hartjen, P.; Schnettler, R.; Feyerabend, F.; Klein, M.; Wegner, N.; Walther, F.; Stangier, D.; Henningsen, A. Improved In Vitro Test Procedure for Full Assessment of the Cytocompatibility of Degradable Magnesium Based on ISO 10993-5/-12. Int. J. Mol. Sci. 2019, 20, 255. [Google Scholar] [CrossRef]
- Hussey, G.S.; Dziki, J.L.; Badylak, S.F. Extracellular matrix-based materials for regenerative medicine. Nat. Rev. Mater. 2018, 3, 159–173. [Google Scholar] [CrossRef]
- Rodrigues, M.; Kosaric, N.; Bonham, C.A.; Gurtner, G.C. Wound healing: A cellular perspective. Physiol. Rev. 2019, 99, 665–706. [Google Scholar] [CrossRef]
- Asadian, M.; Chan, K.V.; Norouzi, M.; Grande, S.; Cools, P.; Morent, R.; De Geyter, N. Fabrication and Plasma Modification of Nanofibrous Tissue Engineering Scaffolds. Nanomaterials 2020, 10, 119. [Google Scholar] [CrossRef]
- Spang, M.T.; Christman, K.L. Extracellular matrix hydrogel therapies: In vivo applications and development. Acta Biomater. 2018, 68, 1–14. [Google Scholar] [CrossRef]
- Ambekar, R.; Kandasubramanian, B. Progress in the Advancement of Porous Biopolymer Scaffold: Tissue Engineering Application. Ind. Eng. Chem. Res. 2019, 58, 6163–6194. [Google Scholar] [CrossRef]
- Chen, F.M.; Liu, X. Advancing biomaterials of human origin for tissue engineering. Prog. Polym. Sci. 2016, 53, 86–168. [Google Scholar] [CrossRef]
- Dinescu, S.; Albu Kaya, M.; Chitoiu, L.; Ignat, S.; Kaya, D.A.; Costache, M. Collagen-Based Hydrogels and Their Applications for Tissue Engineering and Regenerative Medicine. In Polymers and Polymeric Composites: A Reference Series; Mondal, M., Ed.; Springer: Cham, Switzerland, 2019; pp. 1643–1664. ISBN 978-3-319-77829-7. [Google Scholar]
- Szymczyk-Ziółkowska, P.; Łabowska, M.B.; Detyna, J.; Michalak, I.; Gruber, P. A review of fabrication polymer scaffolds for biomedical applications using additive manufacturing techniques. Biocybern. Biomed. Eng. 2020, 40, 624–638. [Google Scholar] [CrossRef]
- Spicer, C.D. Hydrogel scaffolds for tissue engineering: The importance of polymer choice. Polym. Chem. 2020, 11, 184–219. [Google Scholar] [CrossRef]
- Do, A.V.; Khorsand, B.; Geary, S.M.; Salem, A.K. 3D printing of scaffolds for tissue regeneration applications. Adv. Healthc. Mater. 2015, 4, 1742–1762. [Google Scholar] [CrossRef]
- Yoon, D.M.; Fisher, J.P. Natural and Synthetic Polymeric Scaffolds. In Biomedical Materials, 2nd ed.; Narayan, R., Ed.; Springer: Cham, Switzerland, 2021; pp. 257–283. ISBN 978-3-030-49205-2. [Google Scholar]
- Reddy, M.S.B.; Ponnamma, D.; Choudhary, R.; Sadasivuni, K.K. A Comparative Review of Natural and Synthetic Biopolymer Composite Scaffolds. Polymers 2021, 13, 1105. [Google Scholar] [CrossRef]
- Vedadghavami, A.; Minooei, F.; Mohammadi, M.H.; Khetani, S.; Rezaei, A.; Mashayekhan, S.; Sanati-Nezhad, A. Manufacturing of hydrogel biomaterials with controlled mechanical properties for tissue engineering applications. Acta Biomater. 2017, 62, 42–63. [Google Scholar] [CrossRef]
- Nistor, M.T.; Vasile, C.; Chiriac, A.P. Hybrid collagen-based hydrogels with embedded montmorillonite nanoparticles. Mater. Sci. Eng. C 2015, 53, 212–221. [Google Scholar] [CrossRef]
- Lohrasbi, S.; Mirzaei, E.; Karimizade, A.; Takallu, S.; Rezaei, A. Collagen/cellulose nanofiber hydrogel scaffold: Physical, mechanical and cell biocompatibility properties. Cellulose 2020, 27, 927–940. [Google Scholar] [CrossRef]
- Semenycheva, L.L.; Egorikhina, M.N.; Chasova, V.O.; Valetova, N.B.; Kuznetsova, Y.L.; Mitin, A.V. Enzymatic Hydrolysis of Marine Collagen and Fibrinogen Proteins in the Presence of Thrombin. Mar. Drugs 2020, 18, 208. [Google Scholar] [CrossRef]
- Goodarzi, H.; Jadidi, K.; Pourmotabed, S.; Sharifi, E.; Aghamollaei, H. Preparation and in vitro characterization of cross-linked collagen–gelatin hydrogel using EDC/NHS for corneal tissue engineering applications. Int. J. Biol. Macromol. 2019, 126, 620–632. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, X.; Fan, D.; Zhu, C.; Deng, J.; Hui, J.; Ma, P. Synthesis and characterization of hyaluronic acid/human-like collagen hydrogels. Mater. Sci. Eng. C 2014, 43, 547–554. [Google Scholar] [CrossRef]
- Skopinska-Wisniewska, J.; Kuderko, J.; Bajek, A.; Maj, M.; Sionkowska, A.; Ziegler-Borowska, M. Collagen/elastin hydrogels cross-linked by squaric acid. Mater. Sci. Eng. C 2016, 60, 100–108. [Google Scholar] [CrossRef]
- Moxon, S.R.; Corbett, N.J.; Fisher, K.; Potjewyd, G.; Domingos, M.; Hooper, N.M. Blended alginate/collagen hydrogels promote neurogenesis and neuronal maturation. Mater. Sci. Eng. C 2019, 104, 109904. [Google Scholar] [CrossRef]
- Yang, K.; Sun, J.; Guo, Z.; Yang, J.; Wei, D.; Tan, Y.; Guo, L.; Luo, H.; Fan, H.; Zhang, X. Methacrylamide-modified collagen hydrogel with improved anti-actin-mediated matrix contraction behavior. J. Mater. Chem. B 2018, 6, 7543–7555. [Google Scholar] [CrossRef]
- Lan, W.; Xu, M.; Zhang, X.; Zhao, L.; Huang, D.; Wei, X.; Chen, W. Biomimetic polyvinyl alcohol/type II collagen hydrogels for cartilage tissue engineering. J. Biomater. Sci. Polym. Ed. 2020, 31, 1179–1198. [Google Scholar] [CrossRef]
- Ji, Q.; Zhang, H.; Zhang, X.; Ma, Q.; Teng, L.; Qiu, L. Hydrosoluble collagen based biodegradable hybrid hydrogel for biomedical scaffold. J. Biomater. Sci. Polym. Ed. 2020, 31, 2199–2219. [Google Scholar] [CrossRef]
- Bao, Z.; Gao, M.; Fan, X.; Cui, Y.; Yang, J.; Peng, X.; Nian, R. Development and characterization of a photo-cross-linked functionalized type-I collagen (Oreochromis niloticus) and polyethylene glycol diacrylate hydrogel. Int. J. Biol. Macromol. 2020, 155, 163–173. [Google Scholar] [CrossRef]
- Ullah, K.; Khan, S.A.; Murtaza, G.; Sohail, M.; Manan, A.; Afzal, A. Gelatin-based hydrogels as potential biomaterials for colonic delivery of oxaliplatin. Int. J. Pharm. 2019, 556, 236–245. [Google Scholar] [CrossRef]
- Kojima, K.; Iguchi, S.; Kajima, Y.; Yoshikuni, M. Grafting of Methyl Methacrylate onto Collagen Initiated by Tributylborane. J. Appl. Polym. Sci. 1983, 28, 87–95. [Google Scholar] [CrossRef]
- Kuznetsova, Y.L.; Morozova, E.A.; Sustaeva, K.S.; Markin, A.V.; Mitin, A.V.; Batenykin, M.A.; Salomatina, E.V.; Shurygina, M.P.; Gushchina, K.S.; Pryazhnikova, M.I.; et al. Tributylborane in the synthesis of graft copolymers of collagen and polymethyl methacrylate. Russ. Chem. Bull. 2022, 71, 389–398. [Google Scholar] [CrossRef]
- Uromicheva, M.A.; Kuznetsova, Y.L.; Valetova, N.B.; Mitin, A.V.; Semenycheva, L.L.; Smirnova, O.N. Synthesis of grafted polybutyl acrylate copolymer on fish collagen. Proceed. Uni. Appl. Biochem. Biotechnol. 2021, 11, 16–25. [Google Scholar] [CrossRef]
- Ludin, D.V.; Zaitsev, S.D.; Kuznetsova, Y.L.; Markin, A.V.; Mochalova, A.E.; Salomatina, E.V. Starch-graft-poly(methyl acrylate) copolymer: The new approach to synthesis and copolymer characterization. J. Polym. Res. 2017, 24, 1–9. [Google Scholar] [CrossRef]
- Kuznetsova, Y.; Gushchina, K.; Sustaeva, K.; Mitin, A.; Egorikhina, M.; Chasova, V.; Semenycheva, L. Grafting of Methyl Methacrylate onto Gelatin Initiated by Tri-Butylborane—2,5-Di-Tert-Butyl-p-Benzoquinone System. Polymers 2022, 14, 3290. [Google Scholar] [CrossRef]
- Semenycheva, L.L.; Astanina, M.V.; Kuznetsova, J.L.; Valetova, N.B.; Geras’kina, E.V.; Tarankova, O.A. Method for Producing of Acetic Dispersion of High Molecular Fish Collagen. WO2016056949A1, 14 April 2016. [Google Scholar]
- Armarego, W.L.E.; Chai, C.L.L. Purification of Laboratory Chemicals, 7th ed.; Elsevier: Oxford, UK, 2013; p. 1014. ISBN 978-0-12-382161-4. [Google Scholar]
- Kuznetsova, Y.L.; Sustaeva, K.S.; Mitin, A.V.; Zakharychev, E.A.; Egorikhina, M.N.; Chasova, V.O.; Farafontova, E.A.; Kobyakova, I.I.; Semenycheva, L.L. Graft Polymerization of Acrylamide in an Aqueous Dispersion of Collagen in the Presence of Tributylborane. Polymers 2022, 14, 4900. [Google Scholar] [CrossRef]
- Shanmugam, S.; Gopal, B. Antimicrobial and cytotoxicity evaluation of aliovalent substituted hydroxyapatite. Appl. Surf. Sci. 2014, 303, 277–281. [Google Scholar] [CrossRef]
- Schrieber, R.; Gareis, H. Gelatin Handbook. Theory and Industrial Practice; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; p. 347. ISBN 978-3-527-61096-9. [Google Scholar]
- Arif, M.M.; Khan, S.M.; Gull, N.; Tab, T.A.; Zia, S.; Khan, R.U.; Awais, S.M.; Butt, M.A. Polymer-based biomaterials for chronic wound management: Promises and challenges. Int. J. Pharm. 2021, 598, 120270. [Google Scholar] [CrossRef]
- Shah, B.M.; Palakurthi, S.S.; Khare, T.; Khare, S.; Palakurthi, S. Natural proteins and polysaccharides in the development of micro/nano delivery systems for the treatment of inflammatory bowel disease. Int. J. Biol. Macromol. 2020, 165, 722. [Google Scholar] [CrossRef]
- Yang, J.; An, X.; Liu, L.; Tang, S.; Cao, H.; Xu, Q.; Liu, H. Cellulose, hemicellulose, lignin, and their derivatives as multi-components of bio-based feedstocks for 3D printing. Carbohydr. Polym. 2020, 250, 116881. [Google Scholar] [CrossRef]
- Schweizer, T.A.; Shambat, S.M.; Haunreiter, V.D.; Mestres, C.A.; Weber, A.; Maisano, F.; Zinkernagel, A.S.; Hasse, B. Polyester Vascular Graft Material and Risk for Intracavitary Thoracic Vascular Graft Infection. Emerg. Infect. Dis. 2020, 26, 2448–2452. [Google Scholar] [CrossRef]
- Chasova, V.; Semenycheva, L.; Egorikhina, M.; Charykova, I.; Linkova, D.; Rubtsova, Y.; Fukina, D.; Koryagin, A.; Valetova, N.; Suleimanov, E. Cod Gelatin as an Alternative to Cod Collagen in Hybrid Materials for Regenerative Medicine. Macromol. Res. 2022, 30, 212–222. [Google Scholar] [CrossRef]
- Kuznetsova, Y.L.; Chesnokov, S.A.; Zaitsev, S.D.; Dodonov, V.A. Photopolymerization of methyl methacrylate in the presence of the tri-n-butylborane–p-quinone system. Vysokomol. Soed. Ser. B 2010, 52, 498–505. (In Russian) [Google Scholar]
- Semenycheva, L.L.; Egorikhina, M.N.; Chasova, V.O.; Valetova, N.B.; Podguzkova, M.V.; Astanina, M.V.; Kuznetsova, Y.L. Enzymatic hydrolysis of collagen by pancreatin and thrombin as a step in the formation of scaffolds. Russ. Chem. Bull. 2020, 69, 164–168. [Google Scholar] [CrossRef]
- Laurens, N.; Engelse, M.A.; Jungerius, C.; Löwik, C.W.; Van Hinsbergh, V.W.M.; Koolwijk, P. Single and combined effects of αvβ3- and α5β1-integrins on capillary tube formation in a human fibrinous matrix. Angiogenesis 2009, 12, 275–285. [Google Scholar] [CrossRef]
- Liu, S.; Zheng, Z.; Li, M.; Wang, X. Effect of oxidation progress of tributylborane on the grafting of polyolefins. J. Appl. Polym. Sci. 2012, 125, 3335–3344. [Google Scholar] [CrossRef]
- Simándi, T.L.; Tüdös, F. Kinetics of radical polymerization—XLV. Steric effects in the radical reactivity of quinones. Eur. Polym. J. 1985, 21, 865. [Google Scholar] [CrossRef]
- Sukhikh, S.; Noskova, S.; Ivanova, S.; Ulrikh, E.; Izgaryshev, A.; Larichev, T.; Kozlova, O.; Prosekov, A.; Babich, O. Comparative Analysis of Collagen-Containing Waste Biodegradation, Amino Acid, Peptide and Carbohydrate Composition of Hydrolysis Products. Appl. Sci. 2021, 11, 11511. [Google Scholar] [CrossRef]















{kind=link}
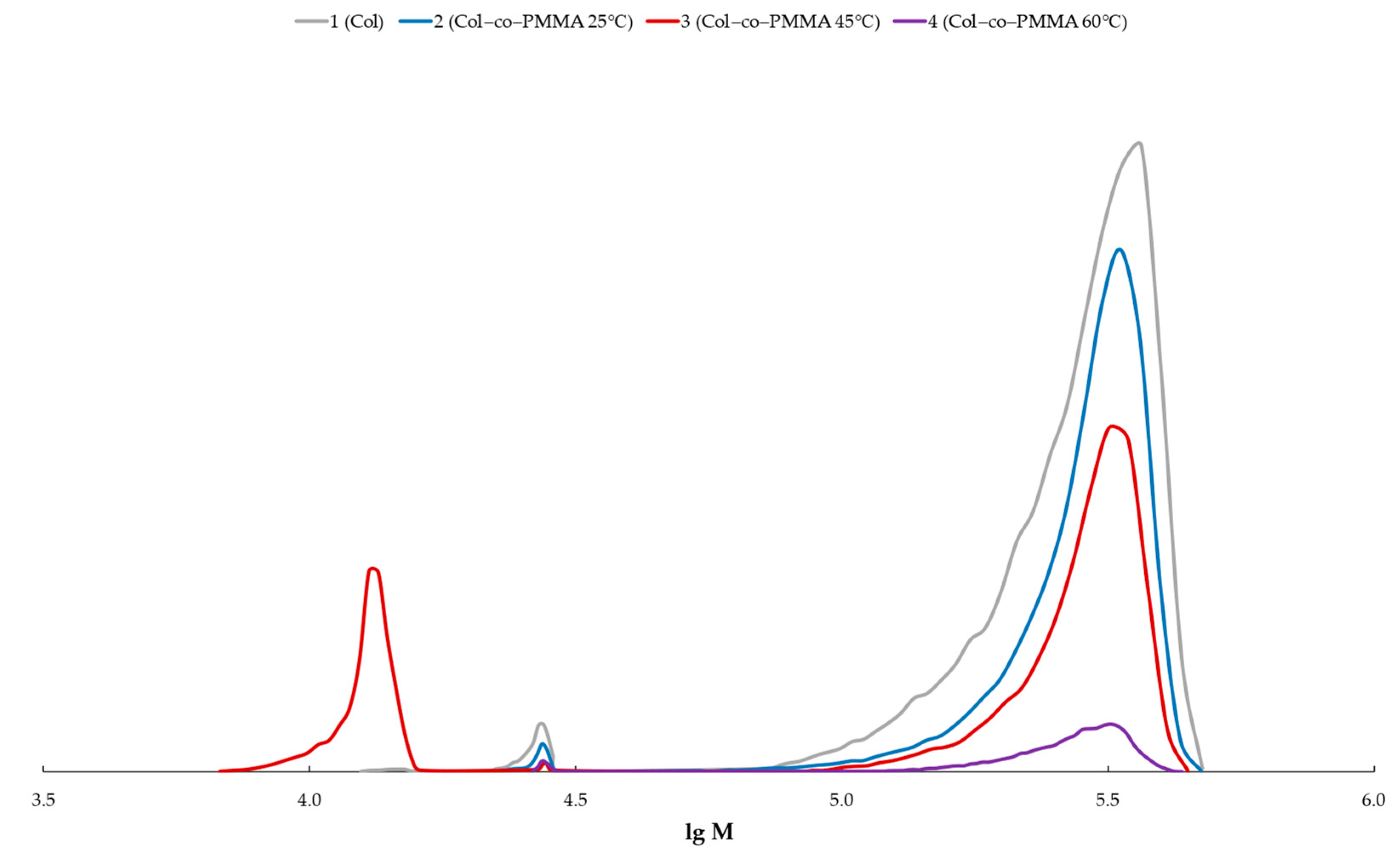{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
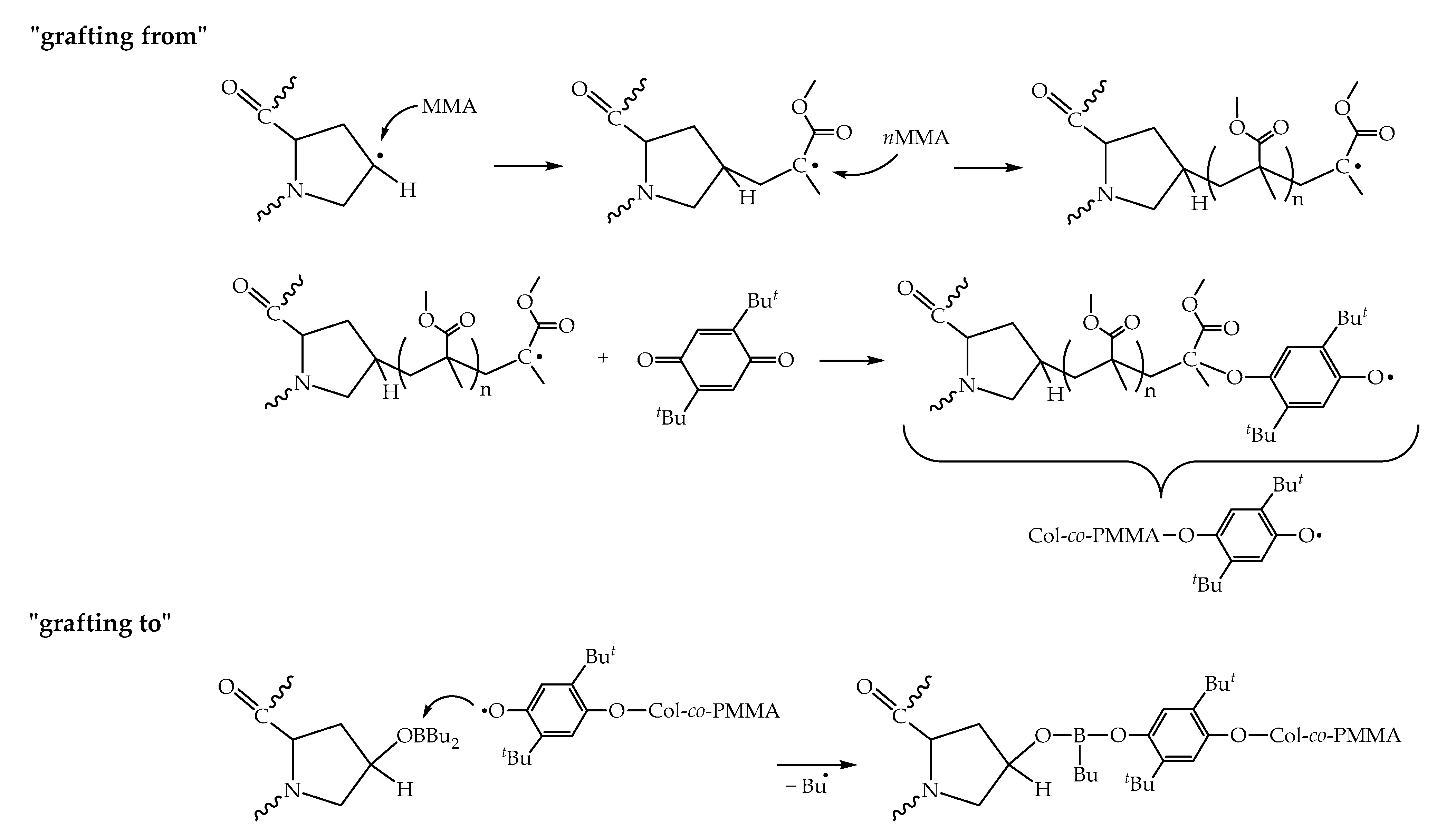{kind=link}
| Monomer | T, °C | Unreacted Monomer, % | MMA Conversion, % | The Yield of PMMA Homopolymer, % | The Proportion of Grafted PMMA in the Copolymer, % |
|---|---|---|---|---|---|
| MMA | 25 | 38 | 62 | 8 | 35 |
| 45 | 38 | 62 | 11 | 31 | |
| 60 | 17 | 83 | 14 | 41 |
| Sample I | Sample II | Sample III | |||||||
|---|---|---|---|---|---|---|---|---|---|
| OD | RGI, % | Rank | OD | RGI, % | Rank | OD | RGI, % | Rank | |
| Control 0:1 | 0.340 ± 0.006 | 100.000 | 0 | 0.500 ± 0.014 | 100.000 | 0 | 0.415 ± 0.009 | 100.000 | 0 |
| Extract 1:0 | 0.291 ± 0.008 | 85.662 | 1 | 0.384 ± 0.023 | 76.664 | 1 | 0.255 ± 0.006 | 61.351 | 2 |
| Extract 1:1 | 0.289 ± 0.006 | 84.861 | 1 | 0.448 ± 0.029 | 89.425 | 1 | 0.280 ± 0.005 | 67.409 | 2 |
| Extract 1:2 | 0.266 ± 0.002 | 78.237 | 1 | 0.334 ± 0.028 | 66.789 | 2 | 0.277 ± 0.008 | 66.843 | 2 |
| Extract 1:4 | 0.275 ± 0.005 | 80.926 | 1 | 0.357 ± 0.027 | 71.373 | 2 | 0.319 ± 0.016 | 76.858 | 1 |
| Extract 1:8 | 0.281 ± 0.004 | 82.689 | 1 | 0.331 ± 0.029 | 66.165 | 2 | 0.342 ± 0.011 | 82.378 | 1 |
| The Concentration of p-Quinone, mol.% | Unreacted Monomer, % | The Yield of PMMA Homopolymer, % | The Proportion of Grafted PMMA in the Copolymer, % |
|---|---|---|---|
| 0.15 | 16 | 6 | 44 |
| 0.25 | 17 | 14 | 41 |
| 0.35 | 43 | 8 | 33 |
| 2.50 | 21 | 52 | 15 |
| Sample IV | Sample V | Sample VI | |||||||
|---|---|---|---|---|---|---|---|---|---|
| OD | RGI, % | Rank | OD | RGI, % | Rank | OD | RGI, % | Rank | |
| Control 0:1 | 0.417 ± 0.012 | 100.000 | 0 | 0.308 ± 0.009 | 100.000 | 0 | 0.269 ± 0.014 | 100.000 | 0 |
| Extract 1:0 | 0.317 ± 0.008 | 76.019 | 1 | 0.232 ± 0.014 | 75.132 | 1 | 0.119 ± 0.001 | 44.249 | 3 |
| Extract 1:1 | 0.350 ± 0.025 | 83.933 | 1 | 0.270 ± 0.018 | 87.627 | 1 | 0.116 ± 0.002 | 43.043 | 3 |
| Extract 1:2 | 0.450 ± 0.044 | 108.004 | 0 | 0.293 ± 0.009 | 94.929 | 1 | 0.194 ± 0.008 | 72.124 | 2 |
| Extract 1:4 | 0.356 ± 0.019 | 85.462 | 1 | 0.313 ± 0.015 | 101.623 | 0 | 0.239 ± 0.007 | 88.868 | 1 |
| Extract 1:8 | 0.584 ± 0.044 | 139.958 | 0 | 0.297 ± 0.012 | 96.430 | 1 | 0.232 ± 0.015 | 86.132 | 1 |
| p-Quinone | Unreacted Monomer, % | The Yield of PMMA Homopolymer, % | The Proportion of Grafted PMMA in the Copolymer, % |
|---|---|---|---|
| 2,5-DTBQ | 17 | 14 | 41 |
| BQ | 0 | 3 | 49 |
| DQ | 33 | 3 | 39 |
| NQ | 65 | 10 | 20 |
| Sample VII | Sample VIII | Sample IX | |||||||
|---|---|---|---|---|---|---|---|---|---|
| OD | RGI, % | Rank | OD | RGI, % | Rank | OD | RGI, % | Rank | |
| Control 0:1 | 0.386 ± 0.009 | 100.000 | 0 | 0.309 ± 0.013 | 100.000 | 0 | 0.269 ± 0.005 | 100.000 | 0 |
| Extract 1:0 | 0.110 ± 0.002 | 28.562 | 3 | 0.184 ± 0.007 | 59.521 | 2 | 0.144 ± 0.003 | 53.671 | 2 |
| Extract 1:1 | 0.098 ± 0.001 | 25.324 | 3 | 0.294 ± 0.013 | 95.526 | 1 | 0.114 ± 0.003 | 42.658 | 3 |
| Extract 1:2 | 0.089 ± 0.002 | 22.959 | 4 | 0.313 ± 0.016 | 101.334 | 0 | 0.267 ± 0.022 | 99.395 | 0 |
| Extract 1:4 | 0.106 ± 0.002 | 27.364 | 3 | 0.274 ± 0.009 | 88.637 | 1 | 0.374 ± 0.019 | 138.987 | 0 |
| Extract 1:8 | 0.223 ± 0.009 | 57.869 | 2 | 0.292 ± 0.012 | 94.379 | 1 | 0.319 ± 0.015 | 118.587 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsova, Y.L.; Gushchina, K.S.; Lobanova, K.S.; Chasova, V.O.; Egorikhina, M.N.; Grigoreva, A.O.; Malysheva, Y.B.; Kuzmina, D.A.; Farafontova, E.A.; Linkova, D.D.; et al. Scaffold Chemical Model Based on Collagen—Methyl Methacrylate Graft Copolymers. Polymers 2023, 15, 2618. https://doi.org/10.3390/polym15122618
Kuznetsova YL, Gushchina KS, Lobanova KS, Chasova VO, Egorikhina MN, Grigoreva AO, Malysheva YB, Kuzmina DA, Farafontova EA, Linkova DD, et al. Scaffold Chemical Model Based on Collagen—Methyl Methacrylate Graft Copolymers. Polymers. 2023; 15(12):2618. https://doi.org/10.3390/polym15122618
Chicago/Turabian StyleKuznetsova, Yulia L., Ksenya S. Gushchina, Karina S. Lobanova, Victoria O. Chasova, Marfa N. Egorikhina, Alexandra O. Grigoreva, Yulia B. Malysheva, Daria A. Kuzmina, Ekaterina A. Farafontova, Daria D. Linkova, and et al. 2023. "Scaffold Chemical Model Based on Collagen—Methyl Methacrylate Graft Copolymers" Polymers 15, no. 12: 2618. https://doi.org/10.3390/polym15122618
APA StyleKuznetsova, Y. L., Gushchina, K. S., Lobanova, K. S., Chasova, V. O., Egorikhina, M. N., Grigoreva, A. O., Malysheva, Y. B., Kuzmina, D. A., Farafontova, E. A., Linkova, D. D., Rubtsova, Y. P., & Semenycheva, L. L. (2023). Scaffold Chemical Model Based on Collagen—Methyl Methacrylate Graft Copolymers. Polymers, 15(12), 2618. https://doi.org/10.3390/polym15122618