
Biodegradable and Biocompatible Thermoplastic Poly(Ester-Urethane)s Based on Poly(ε-Caprolactone) and Novel 1,3-Propanediol Bis(4-Isocyanatobenzoate) Diisocyanate: Synthesis and Characterization


 , ,
, ,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
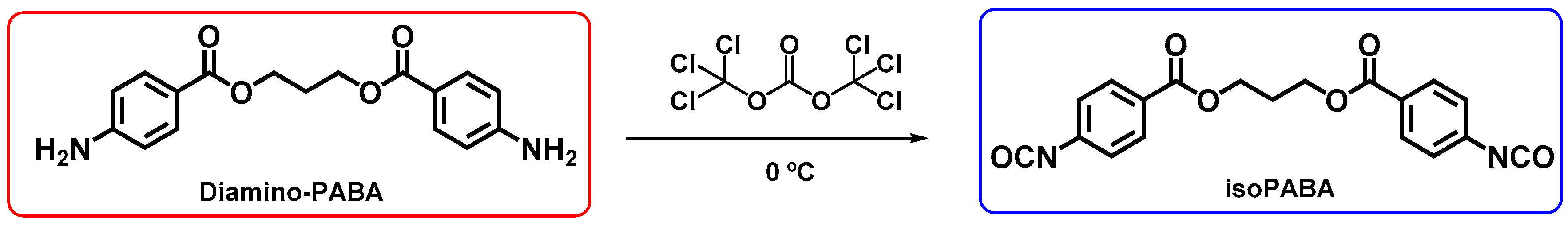
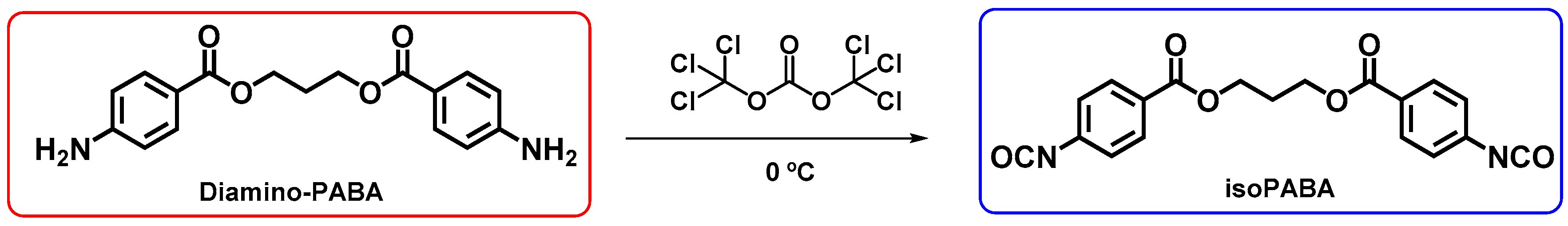
2.3. Synthesis of 1,3-Propanediol Bis(4-Isocyanatobenzoate) (IsoPABA)
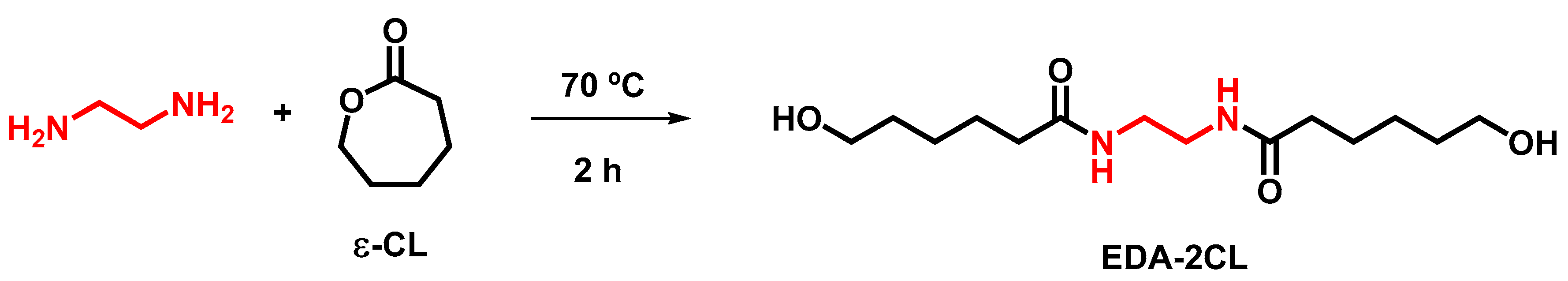
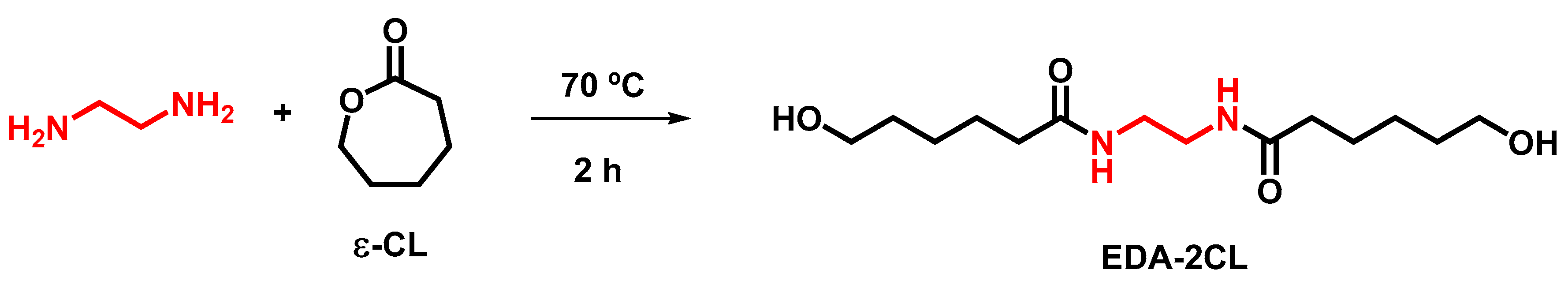
2.4. Synthesis of N, N’-Ethylene-Bis(6-Hydroxycaproamide) (EDA-2CL)
2.5. Synthesis of Hard Segment Models
2.6. Synthesis of Segmented Poly(Ester-Urethane)s
2.7. Hydrolytic and Enzymatic In Vitro Degradation
2.8. Cytotoxicity Test
2.9. Cell Adhesion and Proliferation Assay
3. Results and Discussion
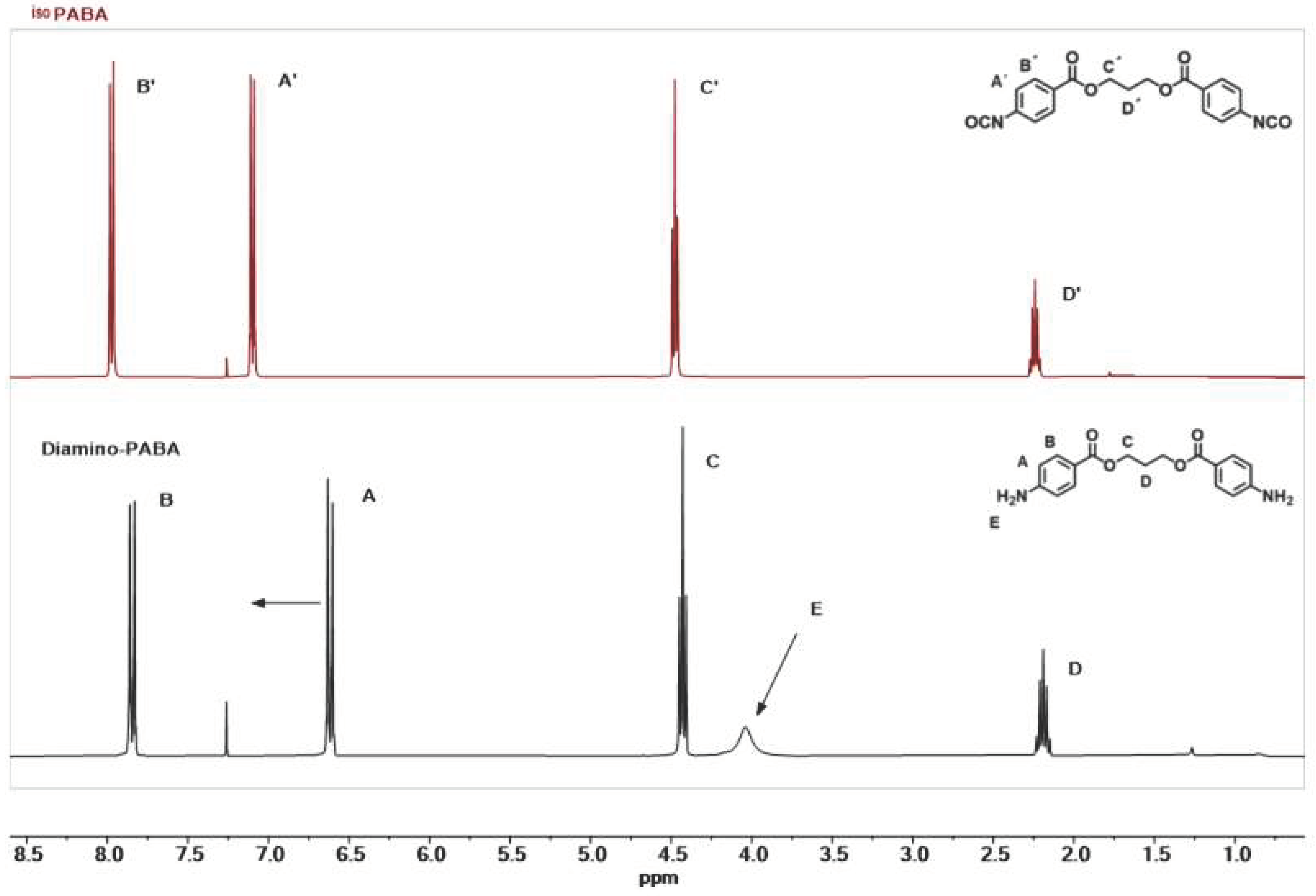
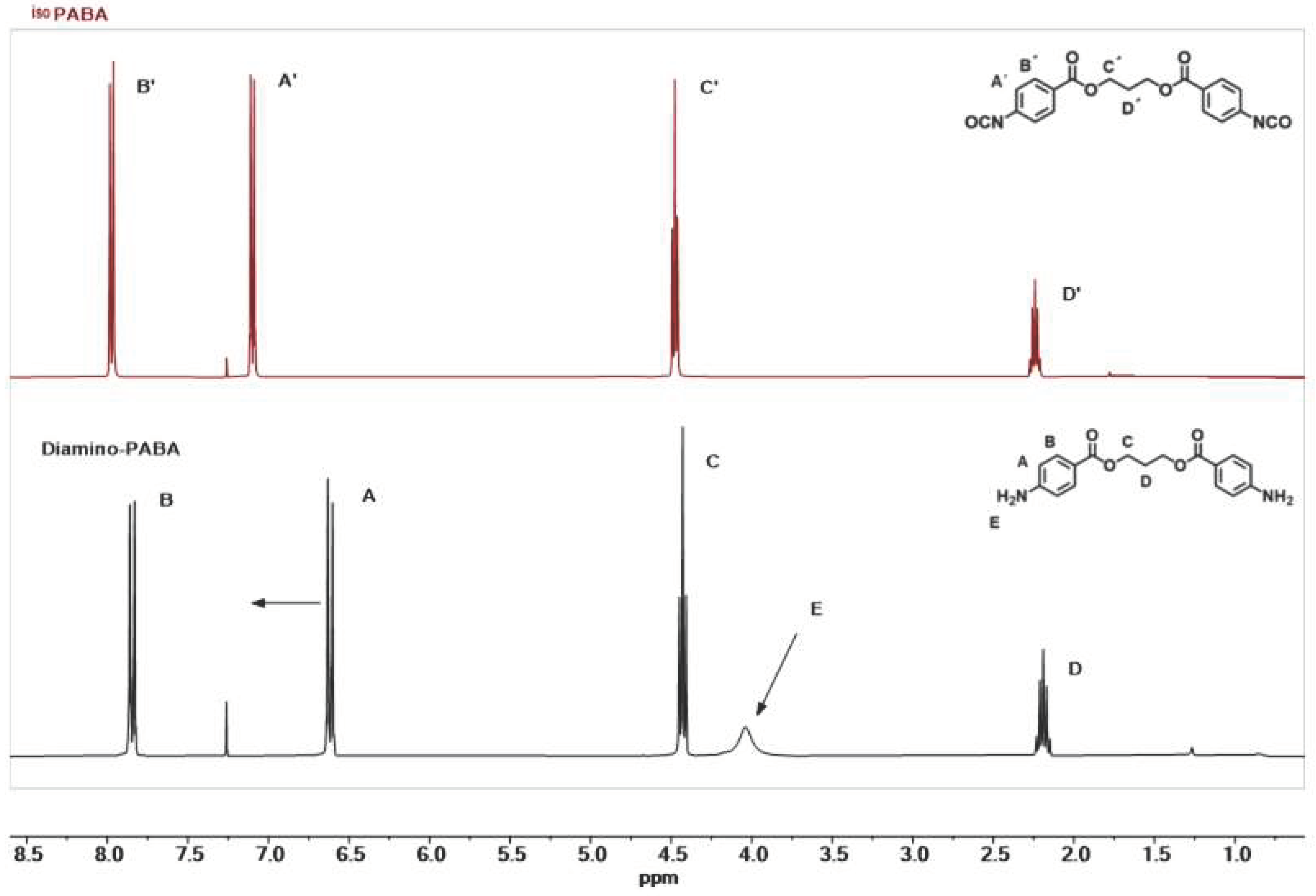
3.1. Synthesis of Aromatic Isocyanate IsoPABA
3.2. Synthesis and Characterization of Hard Segments Models
3.3. Synthesis of Segmented Poly(Ester-Urethane)s
3.4. Characterization of Segmented Poly(ester-Urethane)s
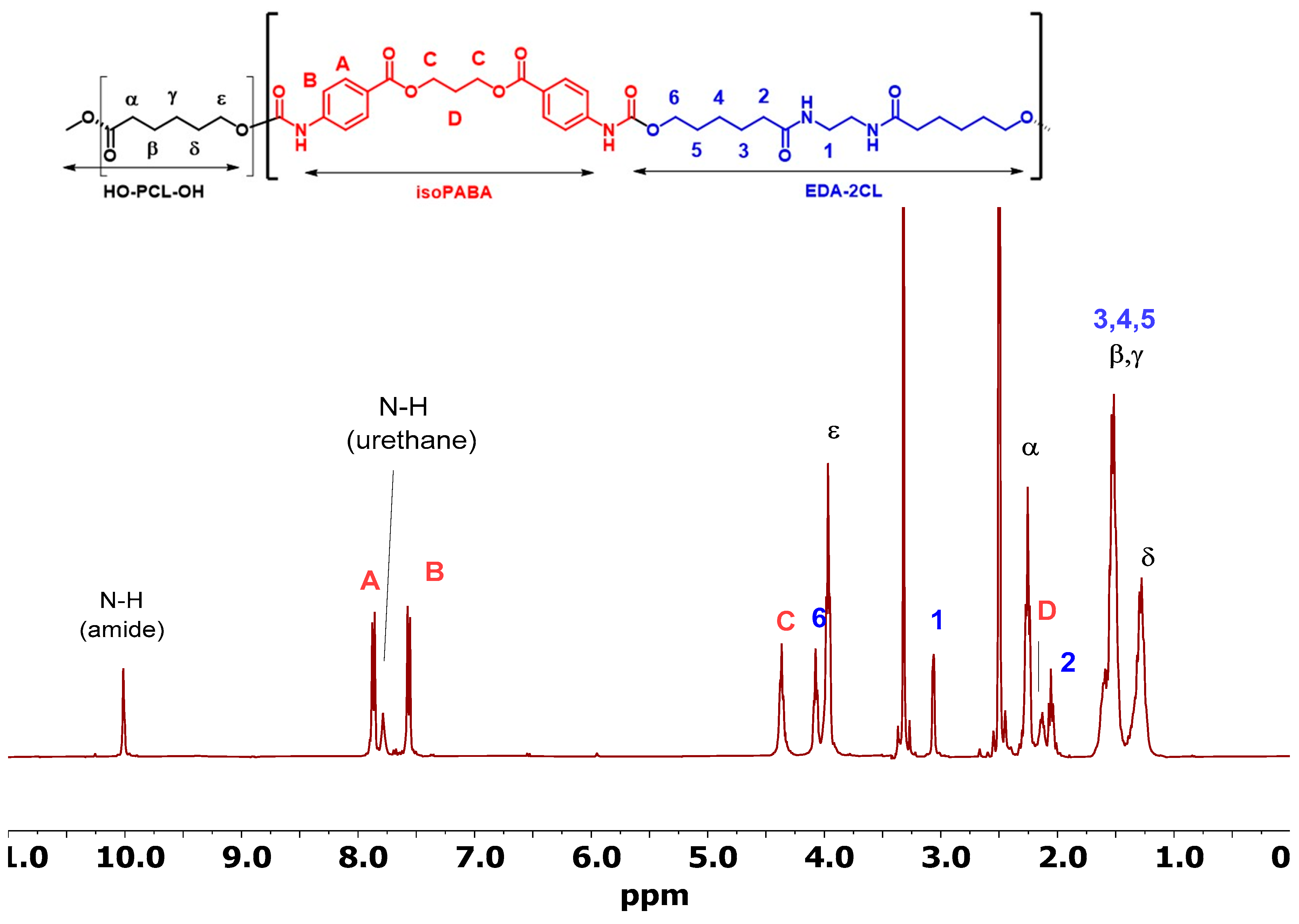
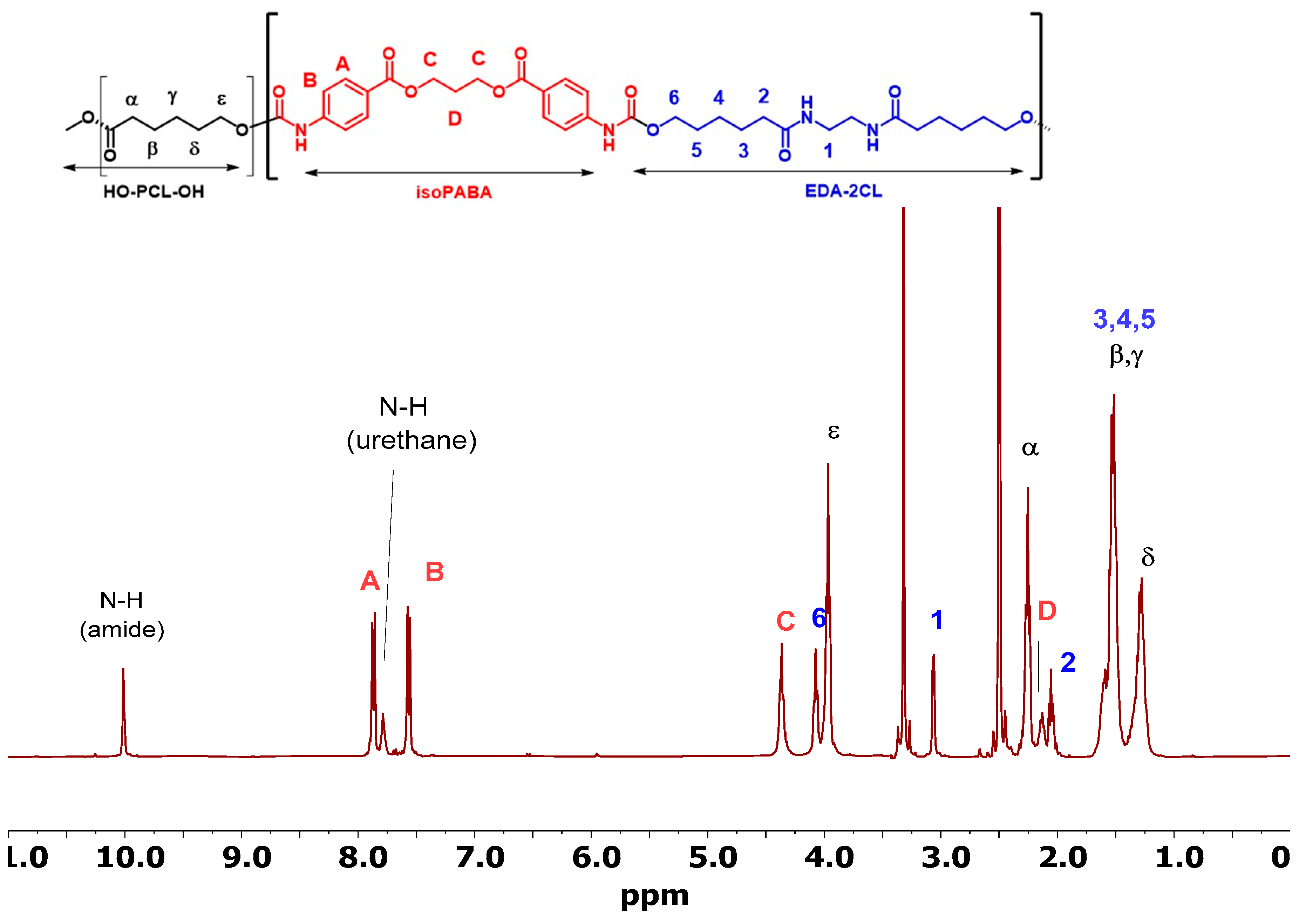
3.4.1. Nuclear Magnetic Resonance (NMR)
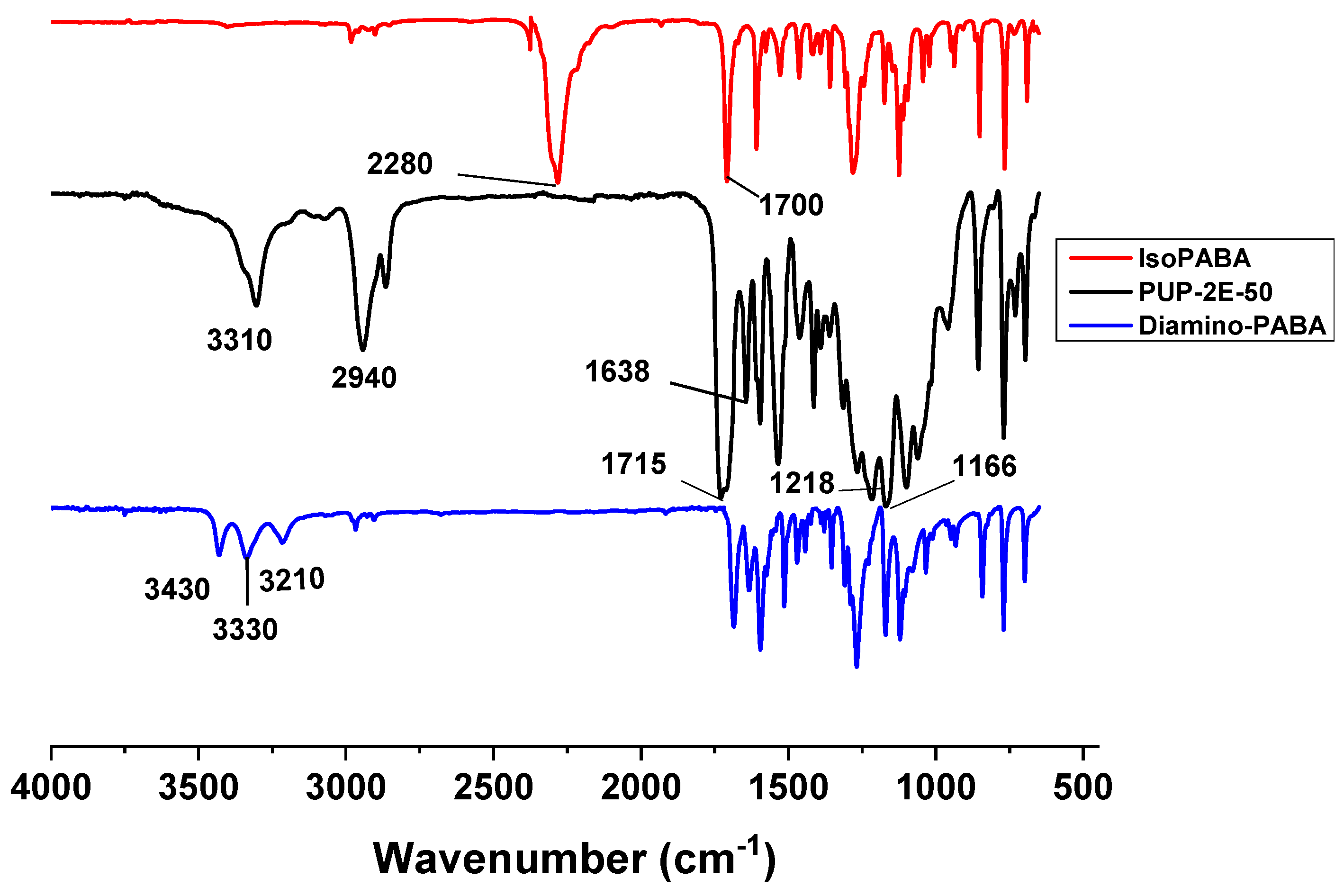
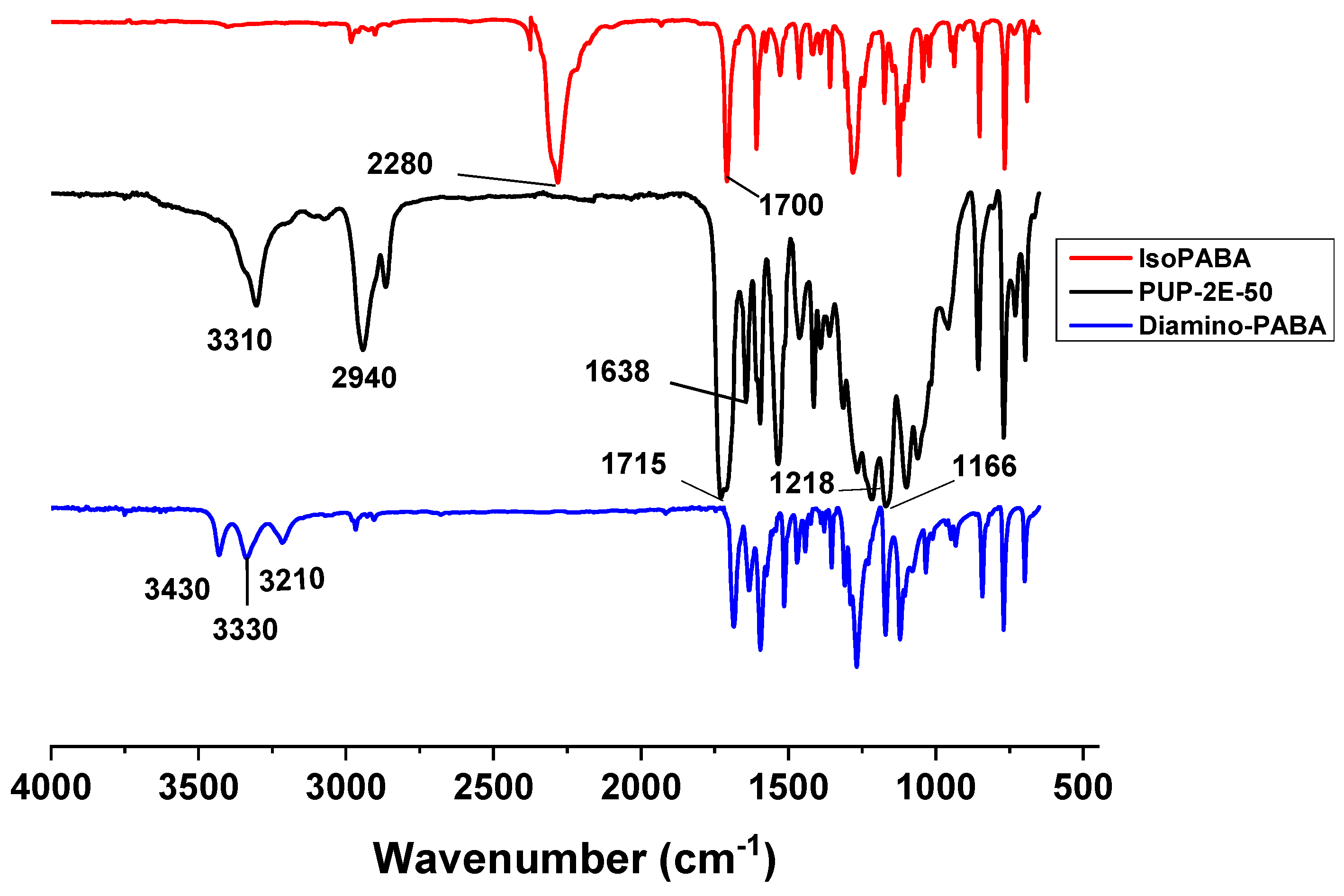
3.4.2. Infrared Analysis (FTIR–ATR)
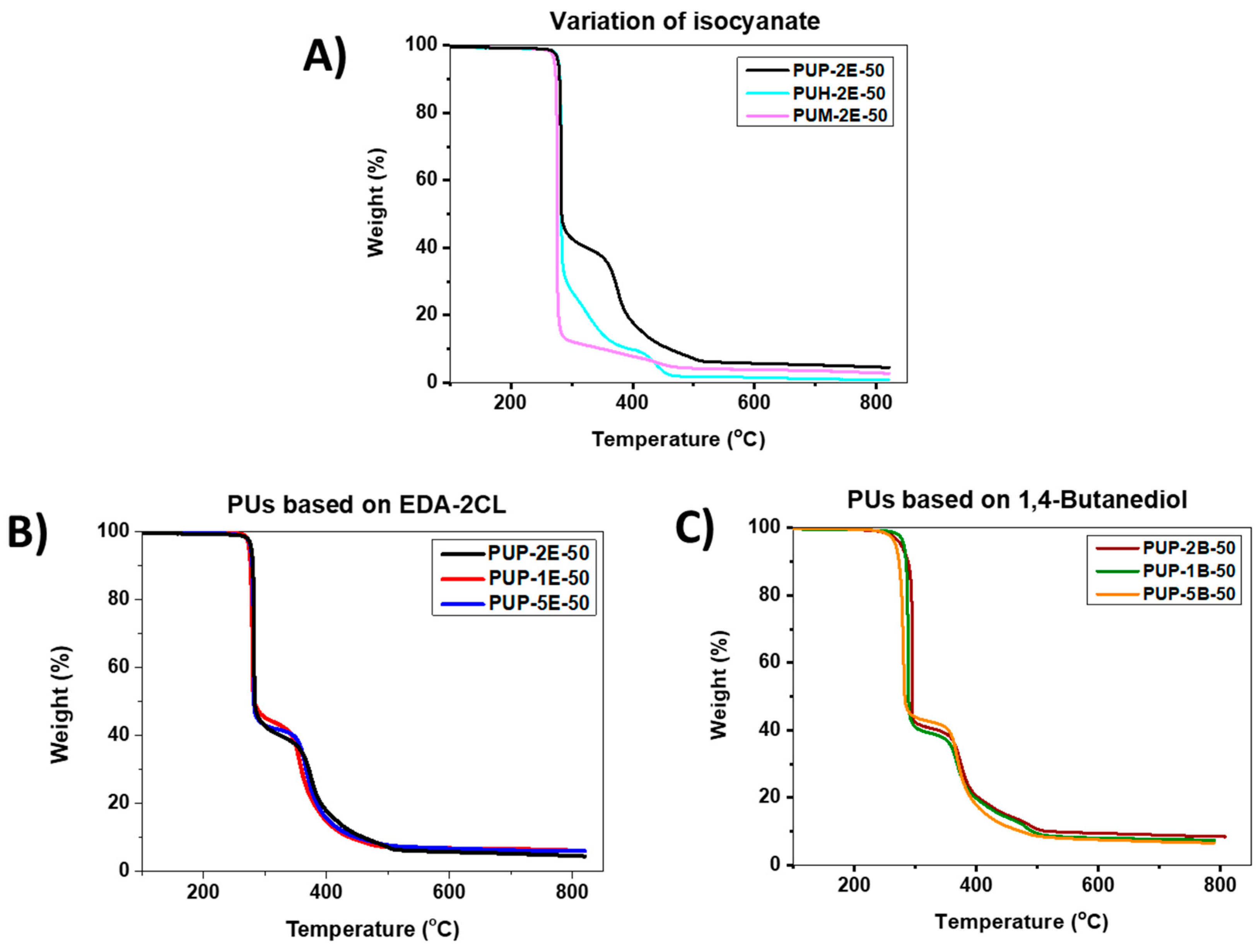
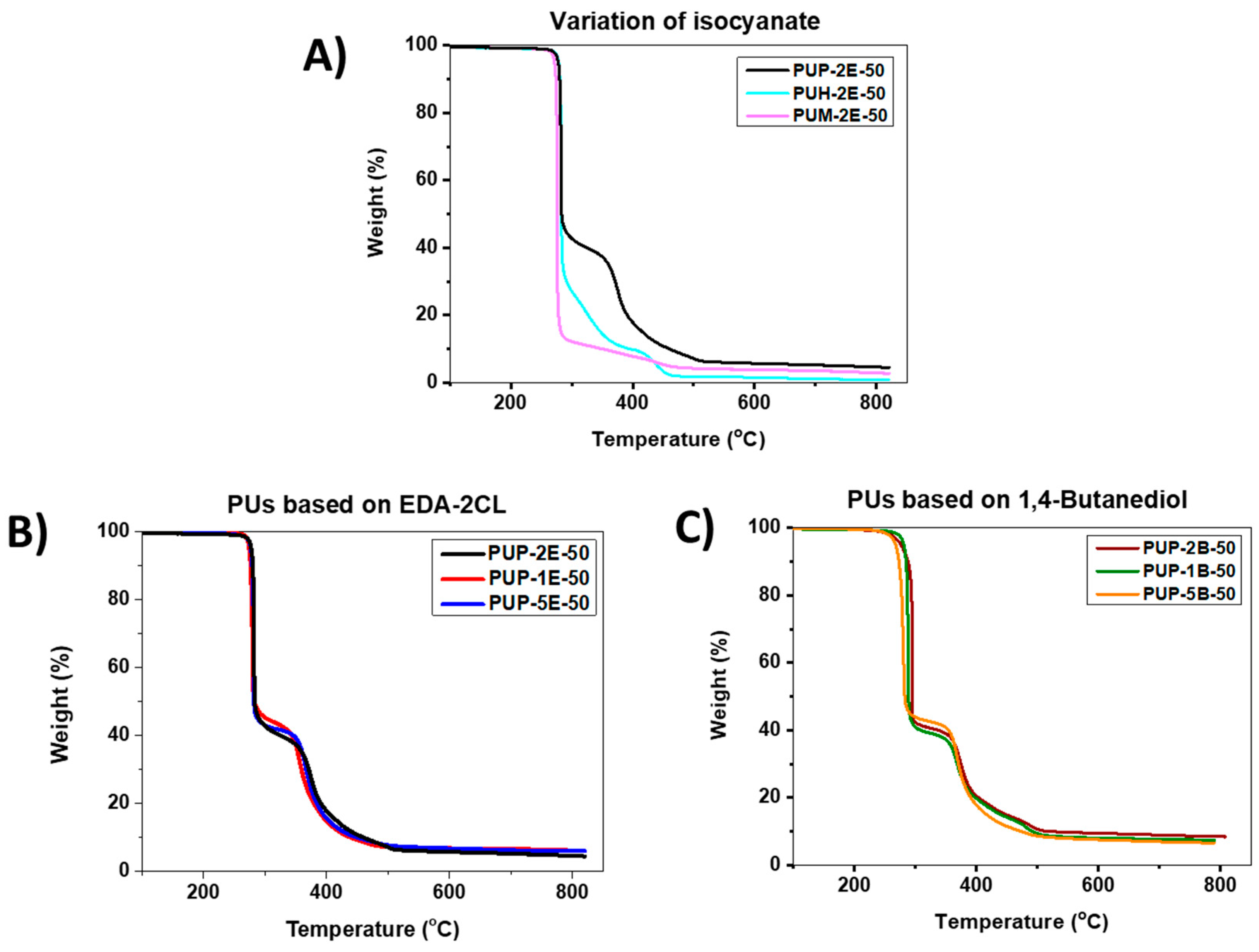
3.4.3. Thermogravimetric Analysis (TGA)
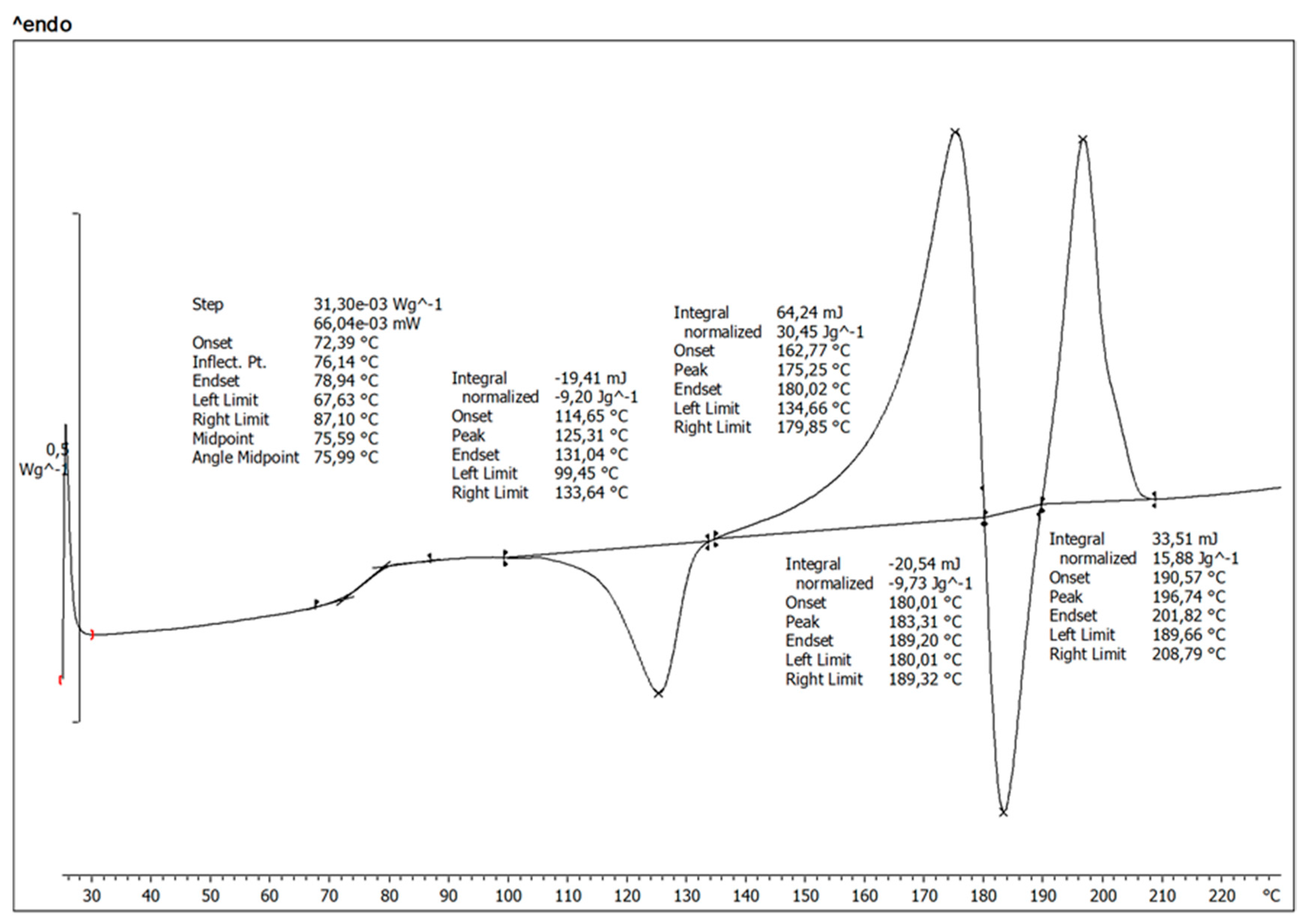
3.4.4. Differential Scanning Calorimetry (DSC)
3.4.5. Mechanical Properties
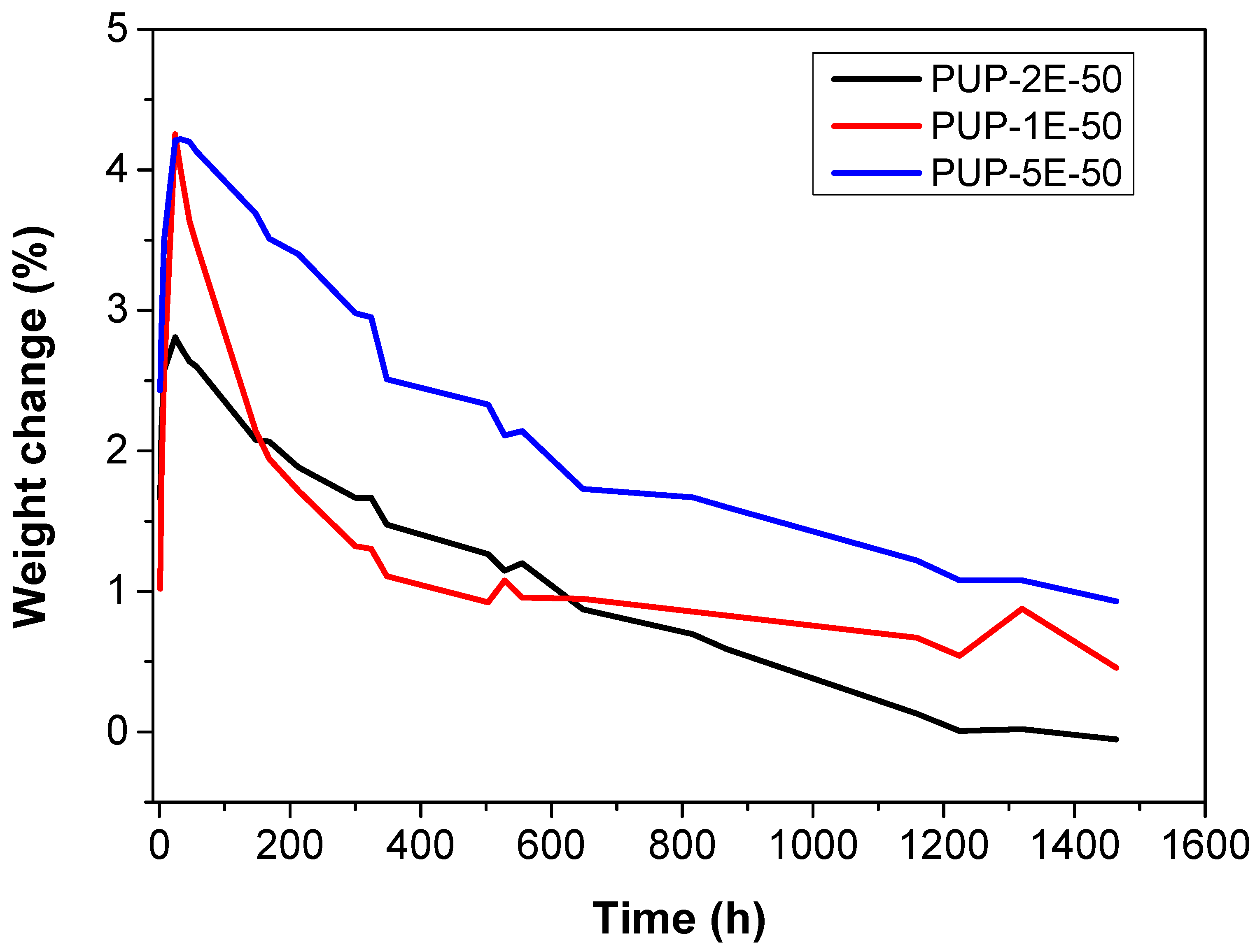
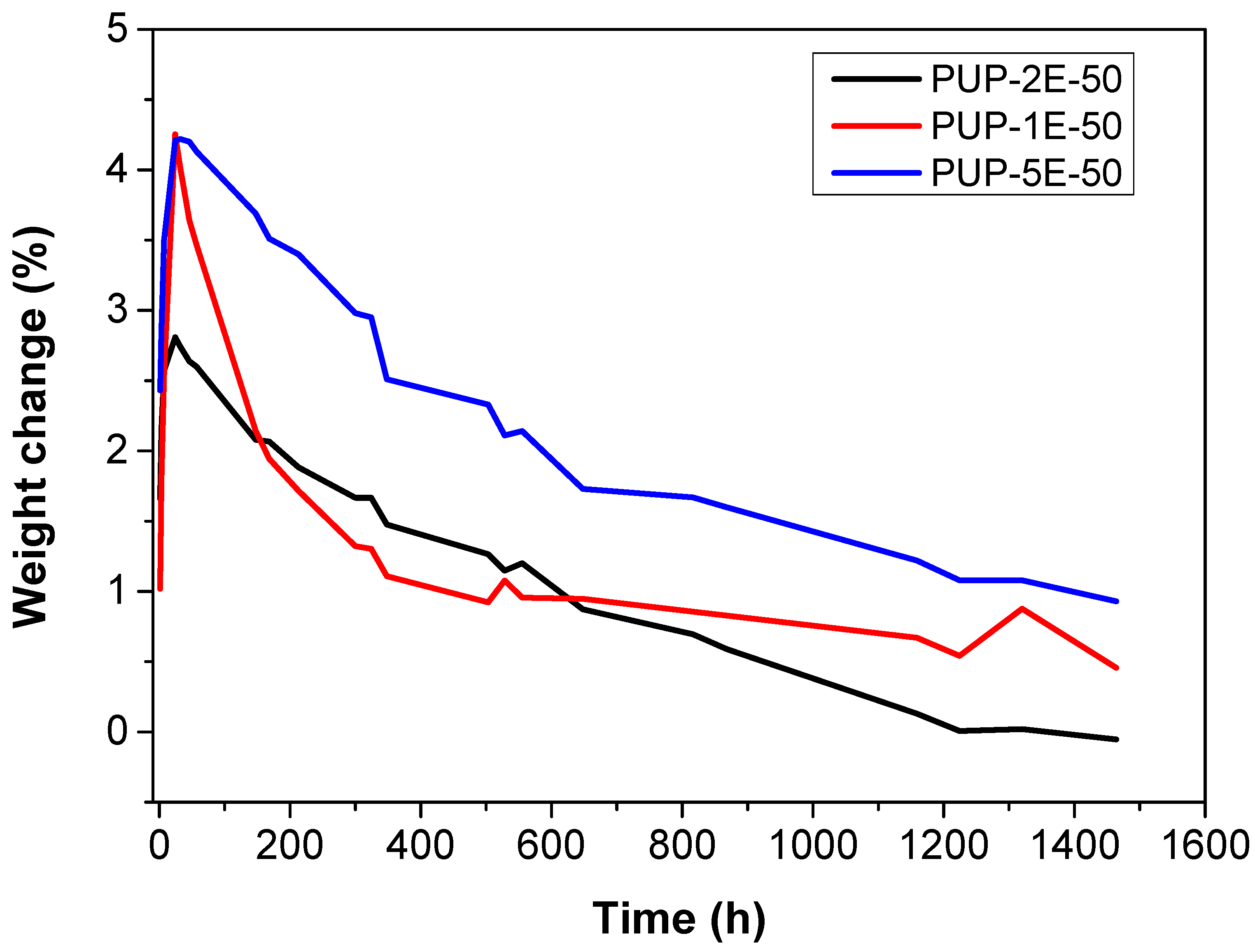
3.4.6. Hydrolytic and Enzymatic In Vitro Degradation
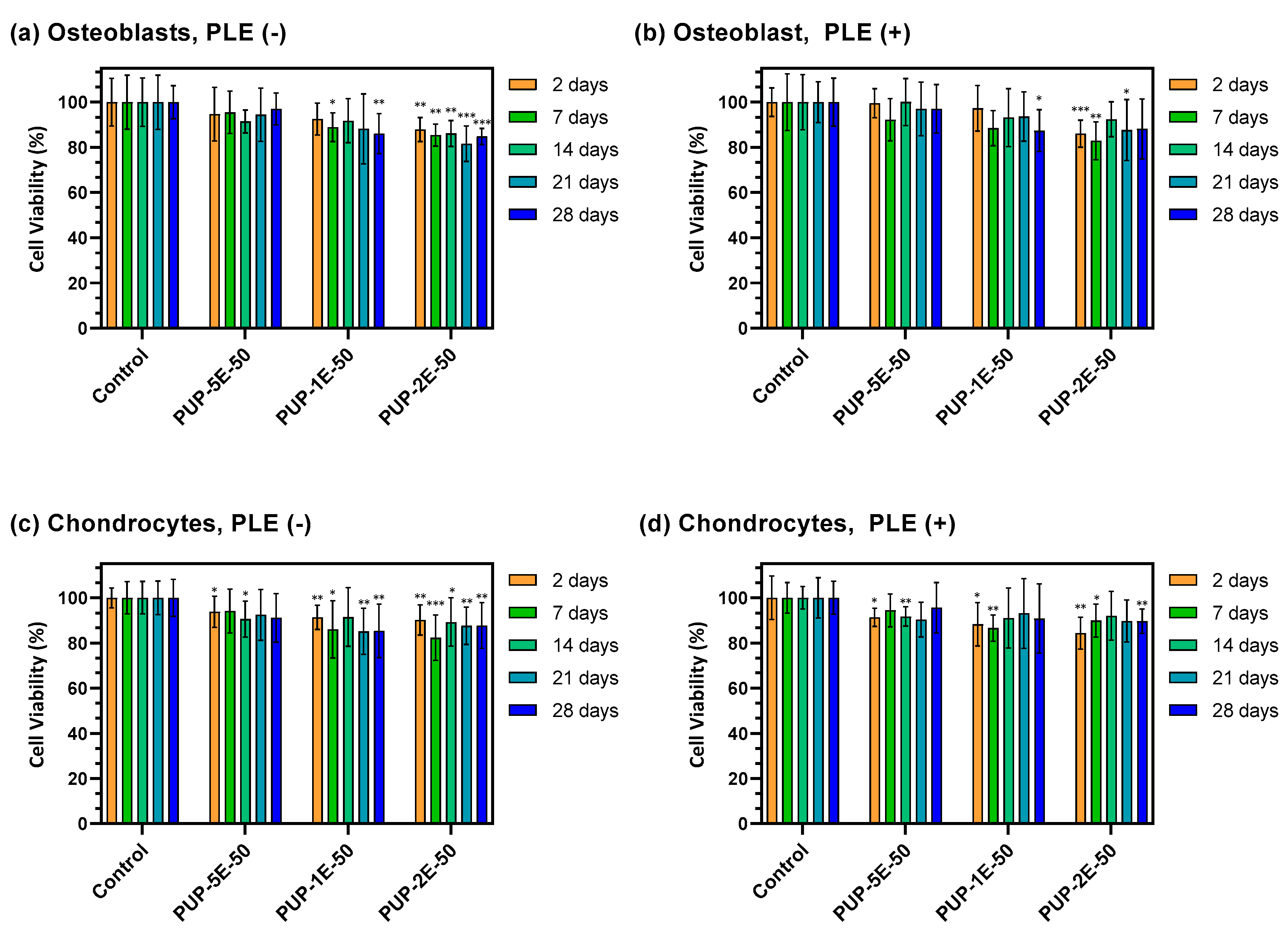
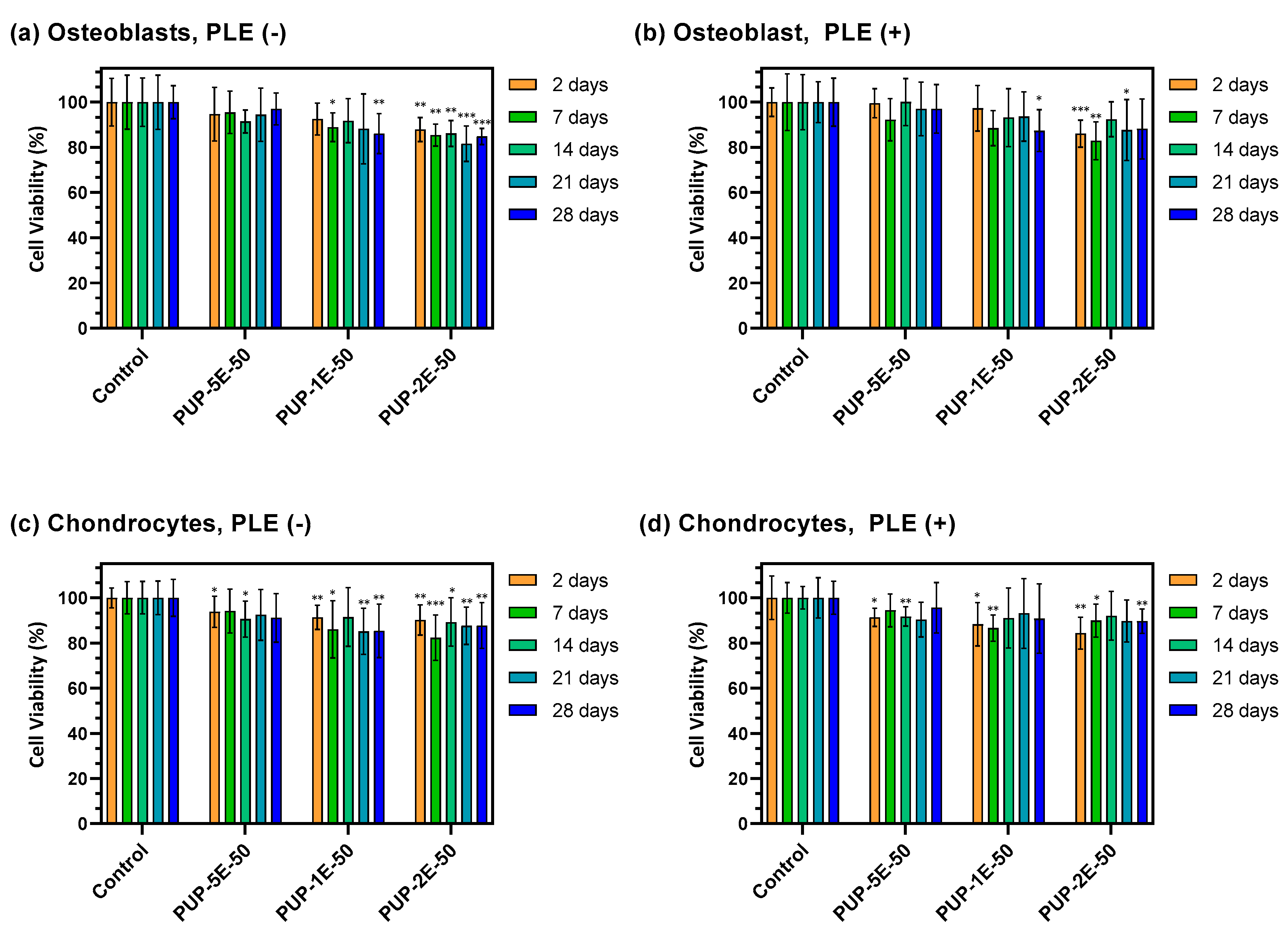
3.4.7. Cytotoxicity Test
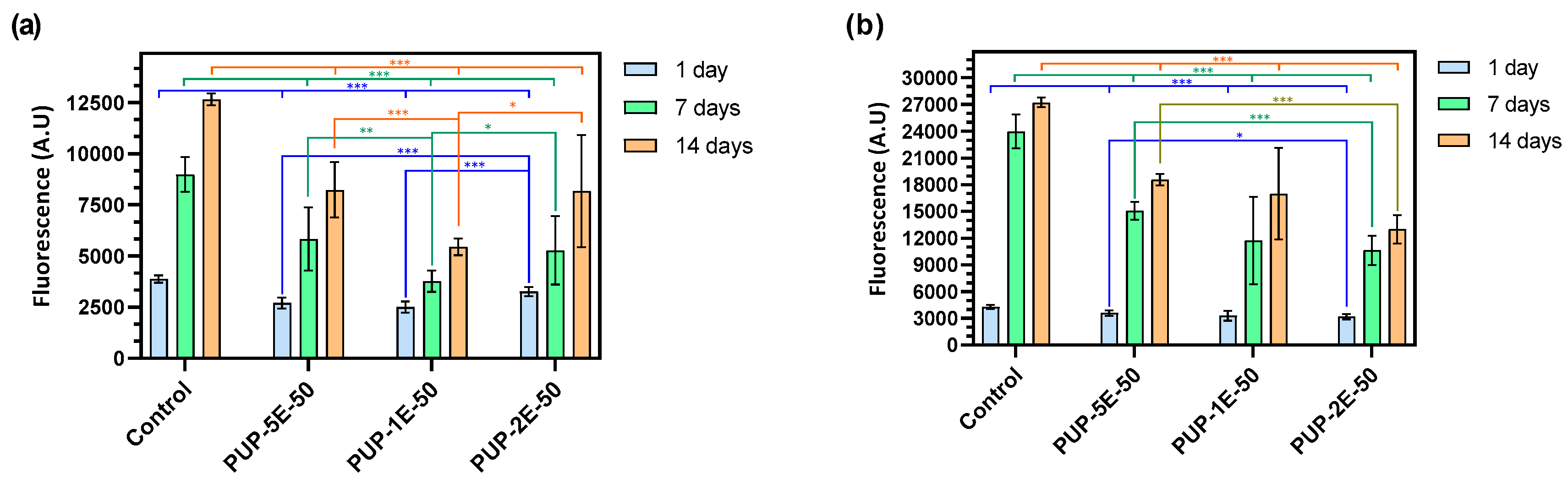
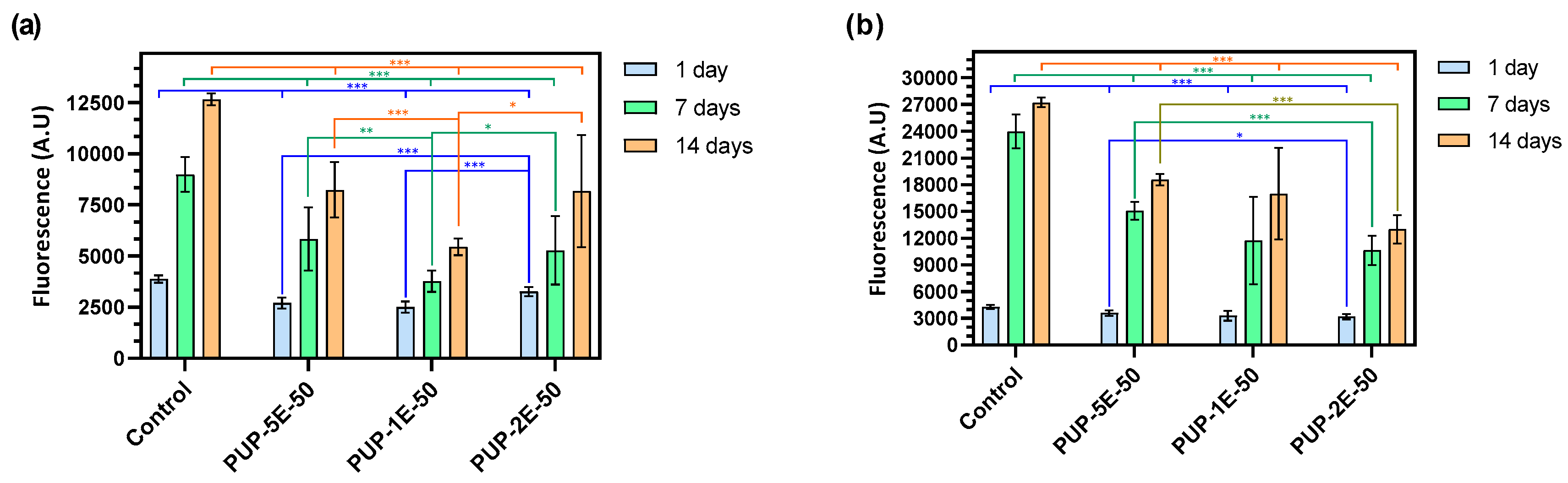
3.4.8. Adhesion and Proliferation Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Das, A.; Mahanwar, P. A brief discussion on advances in polyurethane applications. Adv. Ind. Eng. Polym. Res. 2020, 3, 93–101. [Google Scholar] [CrossRef]
- Lores, N.J.; Hung, X.; Talou, M.H.; Abraham, G.A.; Caracciolo, P.C. Novel three-dimensional printing of poly(ester urethane) scaffolds for biomedical applications. Polym. Adv. Technol. 2021, 32, 3309–3321. [Google Scholar] [CrossRef]
- Mustafov, S.D.; Sen, F.; Seydibeyoglu, M.O. Preparation and characterization of diatomite and hydroxyapatite reinforced porous polyurethane foam biocomposites. Sci. Rep. 2020, 10, 13308. [Google Scholar] [CrossRef] [PubMed]
- Abbasnezhad, N.; Zirak, N.; Shirinbayan, M.; Kouidri, S.; Salahinejad, E.; Tcharkhtchi, A.; Bakir, F. Controlled release from polyurethane films: Drug release mechanisms. J. Appl. Polym. Sci. 2020, 138, 50083. [Google Scholar] [CrossRef]
- Molina, G.A.; Elizalde-Mata, A.; Hernandez-Martinez, A.R.; Fonseca, G.; Cruz Soto, M.; Rodriguez-Morales, A.L.; Estevez, M. Synthesis and Characterization of Inulin-Based Responsive Polyurethanes for Breast Cancer Applications. Polymers 2020, 12, 865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemitson, I.R. Castable Polyurethane Elastomers; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Chan-Chan, L.H.; Tkaczyk, C.; Vargas-Coronado, R.F.; Cervantes-Uc, J.M.; Tabrizian, M.; Cauich-Rodriguez, J.V. Characterization and biocompatibility studies of new degradable poly(urea)urethanes prepared with arginine, glycine or aspartic acid as chain extenders. J. Mater. Sci. Mater. Med. 2013, 24, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Fernández, A.; Abraham, G.A.; Valentín, J.L.; Román, J.S. Synthesis and characterization of biodegradable non-toxic poly(ester-urethane-urea)s based on poly(ε-caprolactone) and amino acid derivatives. Polymer 2006, 47, 785–798. [Google Scholar] [CrossRef]
- Zumbardo-Bacelis, G.A.; Meza-Villegas, L.A.; Pérez-Aranda, C.A.; Vargas-Coronado, R.; Castillo-Cruz, O.; Montaño-Machado, V.; Mantovani, D.; Cauich-Rodríguez, J.V. On arginine-based polyurethane-blends specific to vascular prostheses. J. Appl. Polym. Sci. 2021, 138, 51247. [Google Scholar] [CrossRef]
- Tatai, L.; Moore, T.G.; Adhikari, R.; Malherbe, F.; Jayasekara, R.; Griffiths, I.; Gunatillake, P.A. Thermoplastic biodegradable polyurethanes: The effect of chain extender structure on properties and in-vitro degradation. Biomaterials 2007, 28, 5407–5417. [Google Scholar] [CrossRef]
- Xu, C.; Hong, Y. Rational design of biodegradable thermoplastic polyurethanes for tissue repair. Bioact. Mater. 2021, 15, 250–271. [Google Scholar] [CrossRef]
- Báez, J.E.; Ramírez, D.; Valentín, J.L.; Marcos-Fernández, Á. Biodegradable Poly(ester–urethane–amide)s Based on Poly(ε-caprolactone) and Diamide–Diol Chain Extenders with Crystalline Hard Segments. Synthesis and Characterization. Macromolecules 2012, 45, 6966–6980. [Google Scholar] [CrossRef]
- Caracciolo, P.C.; Buffa, F.; Abraham, G.A. Effect of the hard segment chemistry and structure on the thermal and mechanical properties of novel biomedical segmented poly(esterurethanes). J. Mater. Sci. Mater. Med. 2009, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Acik, G.; Karabulut, H.R.F.; Altinkok, C.; Karatavuk, A.O. Synthesis and characterization of biodegradable polyurethanes made from cholic acid and l-lysine diisocyanate ethyl ester. Polym. Degrad. Stab. 2019, 165, 43–48. [Google Scholar] [CrossRef]
- Hassan, M.K.; Mauritz, K.A.; Storey, R.F.; Wiggins, J.S. Biodegradable aliphatic thermoplastic polyurethane based on poly(ɛ-caprolactone) andL-lysine diisocyanate. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 2990–3000. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, L.; Ding, M.; Tan, H.; Li, J.; Fu, Q. Preparation and rapid degradation of nontoxic biodegradable polyurethanes based on poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) andl-lysine diisocyanate. Polym. Chem. 2011, 2, 601–607. [Google Scholar] [CrossRef]
- Rubio Hernandez-Sampelayo, A.; Navarro, R.; Marcos-Fernandez, A. Preparation of High Molecular Weight Poly(urethane-urea)s Bearing Deactivated Diamines. Polymers 2021, 13, 1914. [Google Scholar] [CrossRef]
- Oprea, S.; Potolinca, V.O.; Oprea, V. Synthesis and characterization of novel poly(urethane-urea) elastomers based on 1,3-propanediol bis(4-aminobenzoate) as chain extender. Mater. Today Commun. 2020, 22, 100860. [Google Scholar] [CrossRef]
- Malamed, S.F. Handbook of Local Anesthesia-E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Navarro, R.; García, C.; Rodríguez-Hernández, J.; Elvira, C.; Marcos-Fernández, A.; Gallardo, A.; Reinecke, H. General approach to prepare polymers bearing pendant isocyanate groups. Polym. Chem. 2020, 11, 5140–5146. [Google Scholar] [CrossRef]
- Irusta, L.; Fernandez-Berridi, M. Aromatic poly (ether-urethanes): Effect of the polyol molecular weight on the photochemical behaviour. Polymer 1999, 40, 4821–4831. [Google Scholar] [CrossRef]
- Shor, L.; Güçeri, S.; Wen, X.; Gandhi, M.; Sun, W. Fabrication of three-dimensional polycaprolactone/hydroxyapatite tissue scaffolds and osteoblast-scaffold interactions in vitro. Biomaterials 2007, 28, 5291–5297. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Bera, A.; Nutan, B.; Jewrajka, S.K. Reactive compatibilizer mediated precise synthesis and application of stimuli responsive polysaccharides-polycaprolactone amphiphilic co-network gels. Polymer 2016, 99, 470–479. [Google Scholar] [CrossRef]
- Chandel, A.K.S.; Jewrajka, S.K. Amphiphilic Polymer Co-Networks: Synthesis, Properties, Modelling and Applications; Royal Society of Chemistry: London, UK, 2020. [Google Scholar]
- Nutan, B.; Chandel, A.K.S.; Jewrajka, S.K. Liquid prepolymer-based in situ formation of degradable poly (ethylene glycol)-linked-poly (caprolactone)-linked-poly (2-dimethylaminoethyl) methacrylate amphiphilic conetwork gels showing polarity driven gelation and bioadhesion. ACS Appl. Bio Mater. 2018, 1, 1606–1619. [Google Scholar] [CrossRef] [PubMed]
- Bartnikowski, M.; Dargaville, T.R.; Ivanovski, S.; Hutmacher, D.W. Degradation mechanisms of polycaprolactone in the context of chemistry, geometry and environment. Prog. Polym. Sci. 2019, 96, 1–20. [Google Scholar] [CrossRef]
- Navarro, R.; Seoane-Rivero, R.; Cuevas, J.M.; Marcos-Fernandez, Á. A novel strategy to polyurethanes with improved mechanical properties by photoactivation of amidocoumarin moieties. RSC Adv. 2020, 10, 29935–29944. [Google Scholar] [CrossRef]
- Elzein, T.; Nasser-Eddine, M.; Delaite, C.; Bistac, S.; Dumas, P. FTIR study of polycaprolactone chain organization at interfaces. J. Colloid Interface Sci. 2004, 273, 381–387. [Google Scholar] [CrossRef]
- Lyu, J.S.; Lee, J.S.; Han, J. Development of a biodegradable polycaprolactone film incorporated with an antimicrobial agent via an extrusion process. Sci. Rep. 2019, 9, 20236. [Google Scholar] [CrossRef]
- Petrović, Z.S.; Zavargo, Z.; Flyn, J.H.; Macknight, W.J. Thermal degradation of segmented polyurethanes. J. Appl. Polym. Sci. 1994, 51, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Báez, J.E.; Marcos-Fernández, Á.; Galindo-Iranzo, P. On the Effect of Alkyl End Group in Poly(ϵ-caprolactone) Oligomers: Preparation and Characterization. Polym.-Plast. Technol. Eng. 2011, 50, 839–850. [Google Scholar] [CrossRef]
- Báez, J.E.; Marcos-Fernández, Á.; Navarro, R. Similarities between homopolymers and triblock copolymers derived from poly(ε-caprolactone) (PCL) macrodiols (HOPCL–E–PCLOH and HOPCL–PEG–PCLOH) and their poly(ester-ether-urethanes): Synthesis and characterization. Chem. Pap. 2019, 73, 1287–1299. [Google Scholar] [CrossRef]
- Gharibi, R.; Agarwal, S. Polyurethanes from Hydrophobic Elastic Materials to Hydrogels with Potent Nonleaching Biocidal and Antibiofilm Activity. ACS Appl. Polym. Mater. 2021, 3, 4695–4707. [Google Scholar] [CrossRef]
- Golmohammadian Tehrani, A.; Makki, H.; Ghaffarian Anbaran, S.R.; Vakili, H.; Ghermezcheshme, H.; Zandi, N. Superior anti-biofouling properties of mPEG-modified polyurethane networks via incorporation of a hydrophobic dangling chain. Prog. Org. Coatings 2021, 158, 106358. [Google Scholar] [CrossRef]
- Nakajima-Kambe, T.; Shigeno-Akutsu, Y.; Nomura, N.; Onuma, F.; Nakahara, T. Microbial degradation of polyurethane, polyester polyurethanes and polyether polyurethanes. Appl. Microbiol. Biotechnol. 1999, 51, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, V. Microbial Degradation of Synthetic Biopolymers Waste. Polymers 2019, 11, 1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javaid, M.A.; Zia, K.M.; Khera, R.A.; Jabeen, S.; Mumtaz, I.; Younis, M.A.; Shoaib, M.; Bhatti, I.A. Evaluation of cytotoxicity, hemocompatibility and spectral studies of chitosan assisted polyurethanes prepared with various diisocyanates. Int. J. Biol. Macromol. 2019, 129, 116–126. [Google Scholar] [CrossRef]
- Portan, D.V.; Deligianni, D.D.; Deligianni, K.; Kroustalli, A.A.; Tyllianakis, M.; Papanicolaou, G.C. Modeling of the interaction between osteoblasts and biocompatible substrates as a function of adhesion strength. J. Biomed. Mater. Res. Part A 2018, 106, 621–628. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, D.M.; Marcos-Fernandez, A.; Rodriguez-Lorenzo, L.M.; Jimenez-Gallegos, R.; Vargas-Becerril, N.; Tellez-Jurado, L. Synthesis and in Vitro Cytocompatibility of Segmented Poly(Ester-Urethane)s and Poly(Ester-Urea-Urethane)s for Bone Tissue Engineering. Polymers 2018, 10, 991. [Google Scholar] [CrossRef] [Green Version]
- Naureen, B.; Haseeb, A.; Basirun, W.J.; Muhamad, F. Recent advances in tissue engineering scaffolds based on polyurethane and modified polyurethane. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 118, 111228. [Google Scholar] [CrossRef]
- Szczepanczyk, P.; Szlachta, M.; Zlocista-Szewczyk, N.; Chlopek, J.; Pielichowska, K. Recent Developments in Polyurethane-Based Materials for Bone Tissue Engineering. Polymers 2021, 13, 946. [Google Scholar] [CrossRef]
- Shaabani, A.; Sedghi, R.; Motasadizadeh, H.; Dinarvand, R. Self-healable conductive polyurethane with the body temperature-responsive shape memory for bone tissue engineering. Chem. Eng. J. 2021, 411, 128449. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | PU | CE | Mn (KDa) | Mw (KDa) | Ð |
|---|---|---|---|---|---|
| 1 | PUP-2E-50 | EDA-2CL | 74.4 | 114.4 | 1.54 |
| 2 | PUP-1E-50 | EDA-2CL | 40.2 | 61.6 | 1.53 |
| 3 | PUP-5E-50 | EDA-2CL | 94.2 | 142.4 | 1.51 |
| 4 | PUM-2E-50 | EDA-2CL | 51.9 | 109.5 | 2.11 |
| 5 | PUH-2E-50 | EDA-2CL | 68.6 | 148.3 | 2.16 |
| 6 | PUP-2B-50 | BD | 32.3 | 47.9 | 1.48 |
| 7 | PUP-1B-50 | BD | 36.0 | 61.9 | 1.72 |
| 8 | PUP-5B-50 | BD | 56.7 | 85.7 | 1.51 |
| Entry | PU | CE | Tg (°C) | Tm1 (°C) | ΔH1 (J/g) | Xi (PCL) (%) | Tm2 (°C) | ΔH2 (J/g) | ΔH2 (J/gHS) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | PUP-2E-50 | EDA-2CL | −46 | 31 | 3.1 | 4.6 | 167/188 | 5.4 + 5 | 20.8 |
| 2 | PUP-1E-50 | EDA-2CL | −20 | 142 | 6.8 | 13.68 | |||
| 3 | PUP-5E-50 | EDA-2CL | 5 | 142 | 4.3 | 8.6 | |||
| 4 | PUM-2E-50 | EDA-2CL | −52 | 32 | 6.5 | 9.6 | 109 | 2.43 | 4.9 |
| 5 | PUH-2E-50 | EDA-2CL | −50 | 35 | 4.7 | 6.9 | 186 | 13.7 | 27.4 |
| 6 | PUP-2B-50 | BD | −41 | 35 | 0.2 | 0.3 | 143 | 1 | 2 |
| 7 | PUP-1B-50 | BD | −22 | 188 | 8.1 | 16.2 | |||
| 8 | PUP-5B-50 | BD | 8 | 134/167 | 0.9 + 1.18 | 4.2 |
| Entry | PU | CE | Stress at Break (MPa) | Strain at Break (%) | Modulus |
|---|---|---|---|---|---|
| 1 | PUP-2E-50 | EDA-2CL | 24 ± 2 | 1060 ± 110 | 55 ± 4 |
| 2 | PUP-1E-50 | EDA-2CL | 6.8 ± 0.2 | 220 ± 30 | 34 ± 2 |
| 3 | PUP-5E-50 | EDA-2CL | 2.3 ± 1.1 | 2800 ± 500 | 4.0 ± 0.8 |
| 4 | PUM-2E-50 | EDA-2CL | 18 ± 5 | 440 ± 60 | 65 ± 15 |
| 5 | PUH-2E-50 | EDA-2CL | 23 ± 2 | 810 ± 120 | 64 ± 12 |
| 6 | PUP-2B-50 | BD | 6.7 ± 0.3 | 120 ± 18 | 43 ± 5 |
| 7 | PUP-1B-50 | BD | 5.7 ± 0.5 | 130 ± 5 | 28 ± 3 |
| 8 | PUP-5B-50 | BD | 0.8 ± 0.2 | 330 ± 40 | 3 ± 1 |
| Weight Change (7 days) | ||
|---|---|---|
| PLE (−) | PLE (+) | |
| PUP-2E-50 | 1.67% | −1.90% |
| PUP-1E-50 | 2.13% | −3.42% |
| PUP-5E-50 | 2.94% | −4.37% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubio Hernández-Sampelayo, A.; Navarro, R.; González-García, D.M.; García-Fernández, L.; Ramírez-Jiménez, R.A.; Aguilar, M.R.; Marcos-Fernández, Á. Biodegradable and Biocompatible Thermoplastic Poly(Ester-Urethane)s Based on Poly(ε-Caprolactone) and Novel 1,3-Propanediol Bis(4-Isocyanatobenzoate) Diisocyanate: Synthesis and Characterization. Polymers 2022, 14, 1288. https://doi.org/10.3390/polym14071288
Rubio Hernández-Sampelayo A, Navarro R, González-García DM, García-Fernández L, Ramírez-Jiménez RA, Aguilar MR, Marcos-Fernández Á. Biodegradable and Biocompatible Thermoplastic Poly(Ester-Urethane)s Based on Poly(ε-Caprolactone) and Novel 1,3-Propanediol Bis(4-Isocyanatobenzoate) Diisocyanate: Synthesis and Characterization. Polymers. 2022; 14(7):1288. https://doi.org/10.3390/polym14071288
Chicago/Turabian StyleRubio Hernández-Sampelayo, Alejandra, Rodrigo Navarro, Dulce María González-García, Luis García-Fernández, Rosa Ana Ramírez-Jiménez, María Rosa Aguilar, and Ángel Marcos-Fernández. 2022. "Biodegradable and Biocompatible Thermoplastic Poly(Ester-Urethane)s Based on Poly(ε-Caprolactone) and Novel 1,3-Propanediol Bis(4-Isocyanatobenzoate) Diisocyanate: Synthesis and Characterization" Polymers 14, no. 7: 1288. https://doi.org/10.3390/polym14071288
APA StyleRubio Hernández-Sampelayo, A., Navarro, R., González-García, D. M., García-Fernández, L., Ramírez-Jiménez, R. A., Aguilar, M. R., & Marcos-Fernández, Á. (2022). Biodegradable and Biocompatible Thermoplastic Poly(Ester-Urethane)s Based on Poly(ε-Caprolactone) and Novel 1,3-Propanediol Bis(4-Isocyanatobenzoate) Diisocyanate: Synthesis and Characterization. Polymers, 14(7), 1288. https://doi.org/10.3390/polym14071288