
Semi-Crystalline Polyoxymethylene-co-Polyoxyalkylene Multi-Block Telechels as Building Blocks for Polyurethane Applications

 ,
, 
Abstract
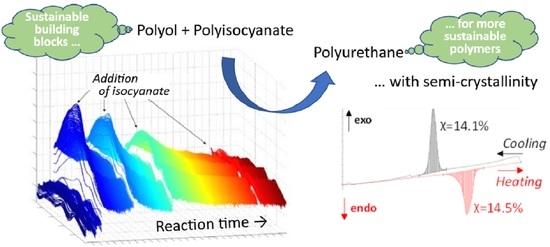
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Synthesis of Copolymer 1 with Diol A
2.1.2. Synthesis of Copolymers 10–13 with Diol A
2.1.3. Synthesis of Copolymers 6–9 with Diol B
2.1.4. Synthesis of Copolymers 2–5 with Diol C
2.1.5. Synthesis of Polyurethane 14
2.2. Methods
2.2.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.2.2. Infra-Red (IR) Spectroscopy
2.2.3. In Situ Infra-Red (IR) Spectroscopy
2.2.4. Gel Permeation Chromatography (GPC)
2.2.5. Differential Scanning Calorimetry (DSC)
2.2.6. Thermogravimetric Analysis (TGA)
2.3. Data Treatment and Kinetics
3. Results and Discussion
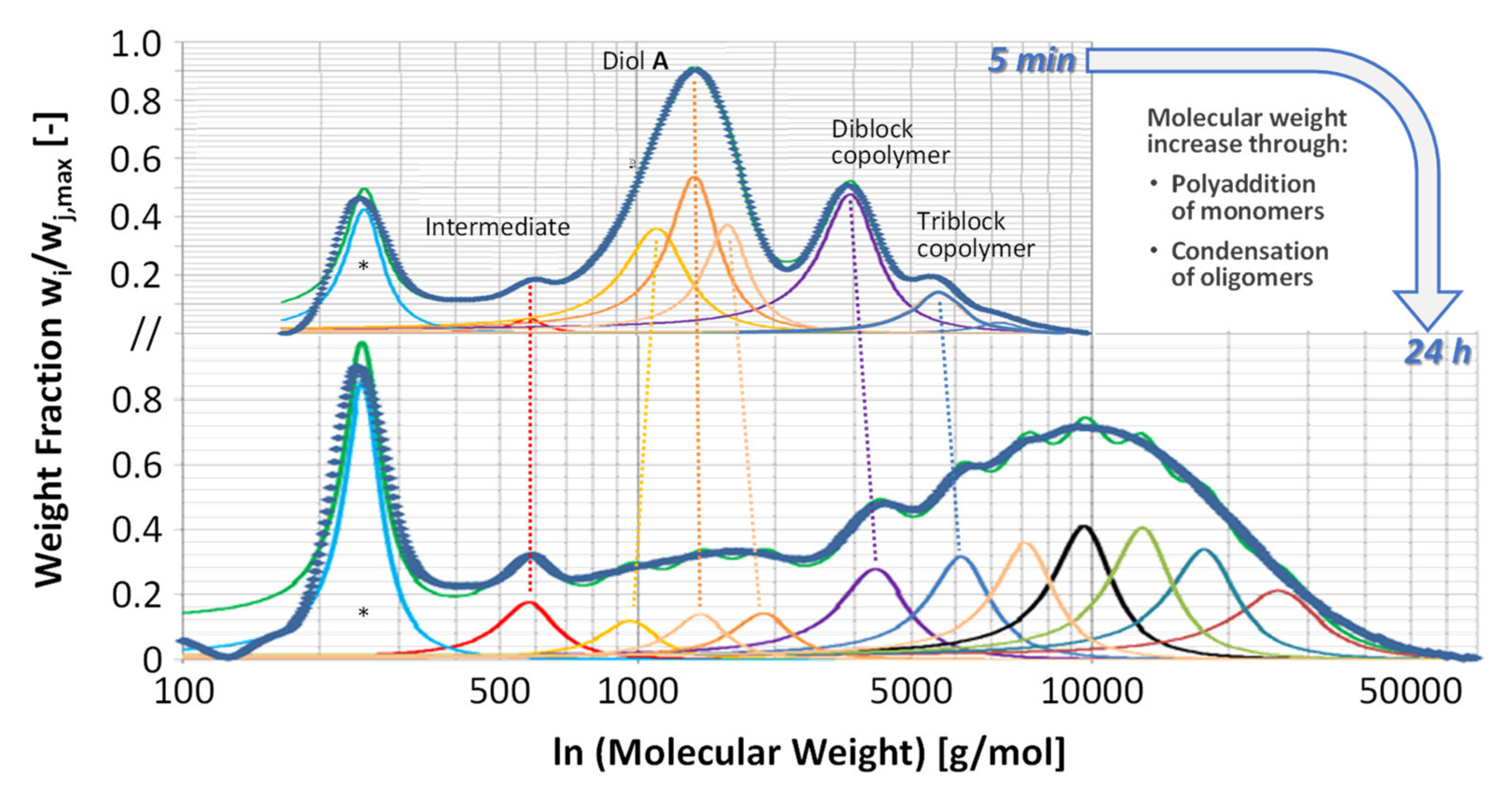
3.1. Synthesis and Characterization of Copolymer 1
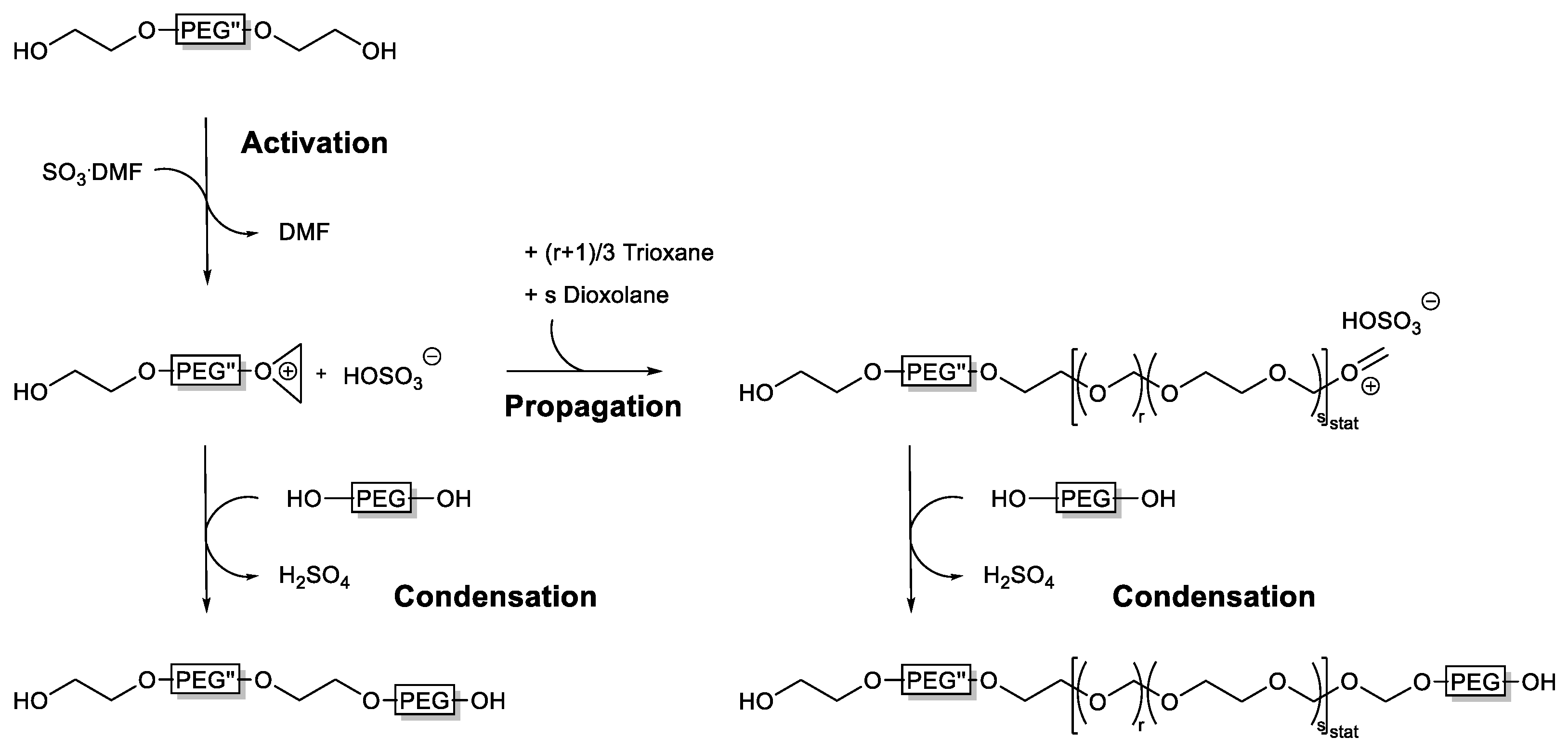
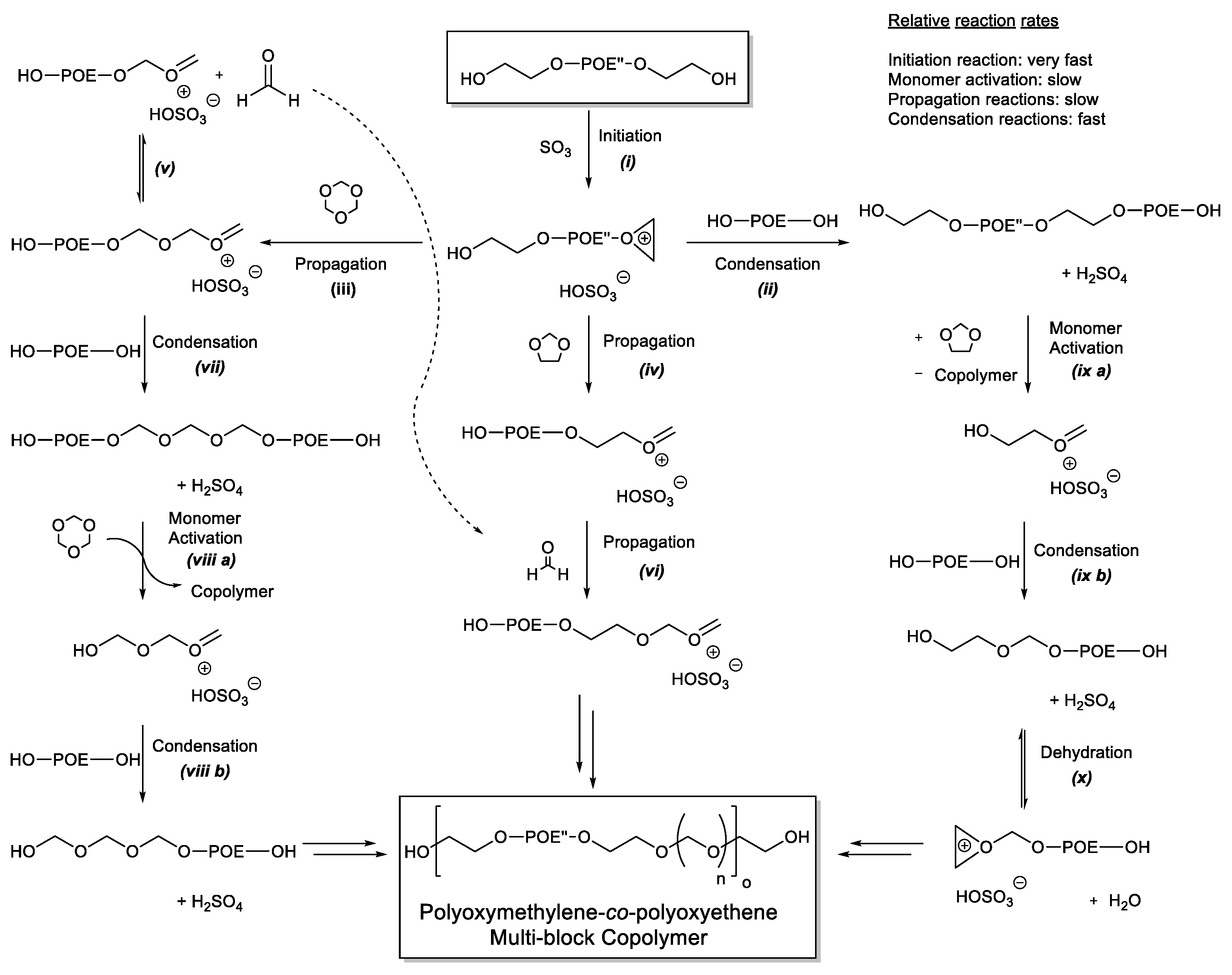
3.2. Reaction Sequence
3.3. Kinetic Model
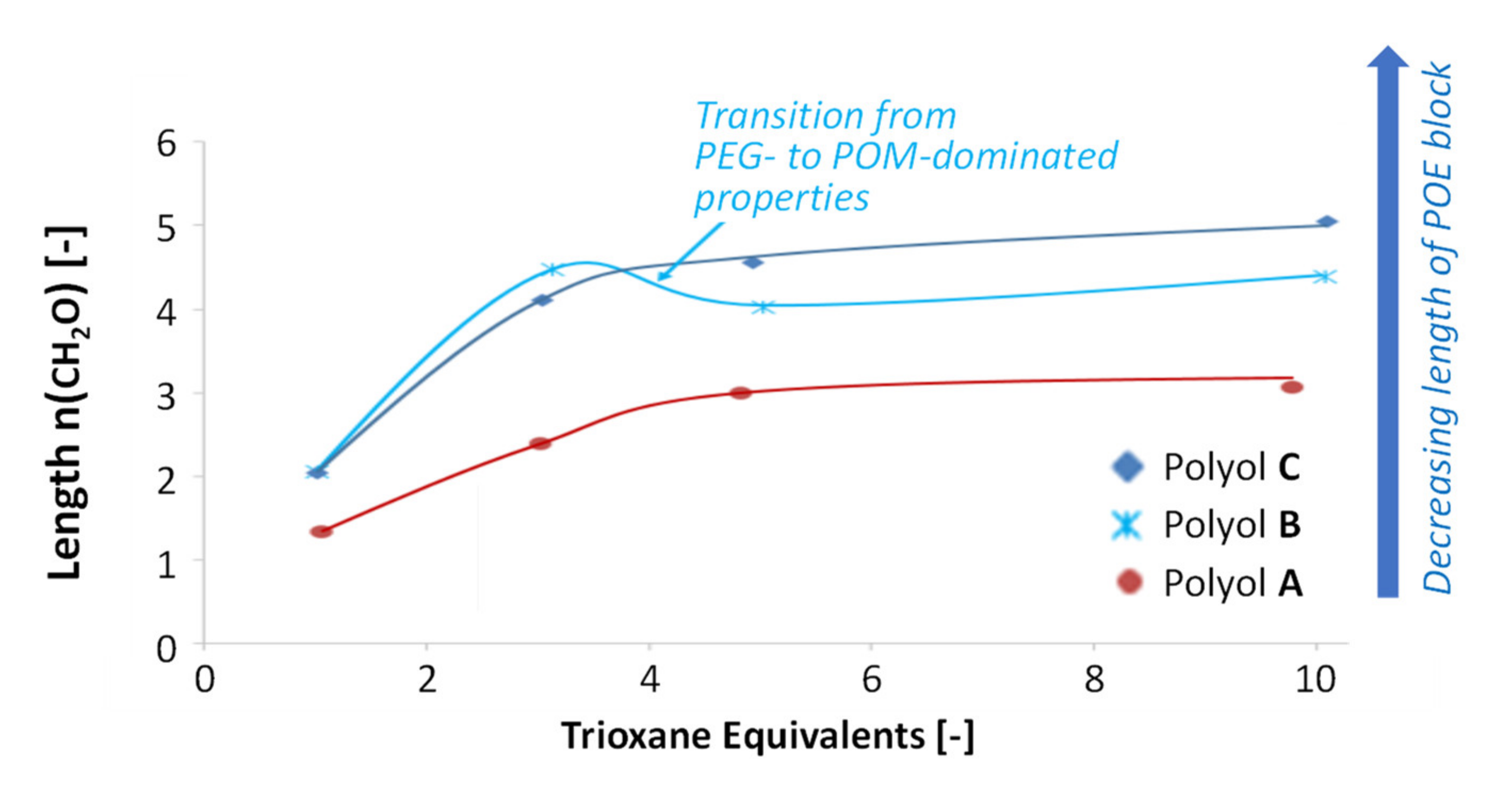
3.4. Variation of Copolymer Composition
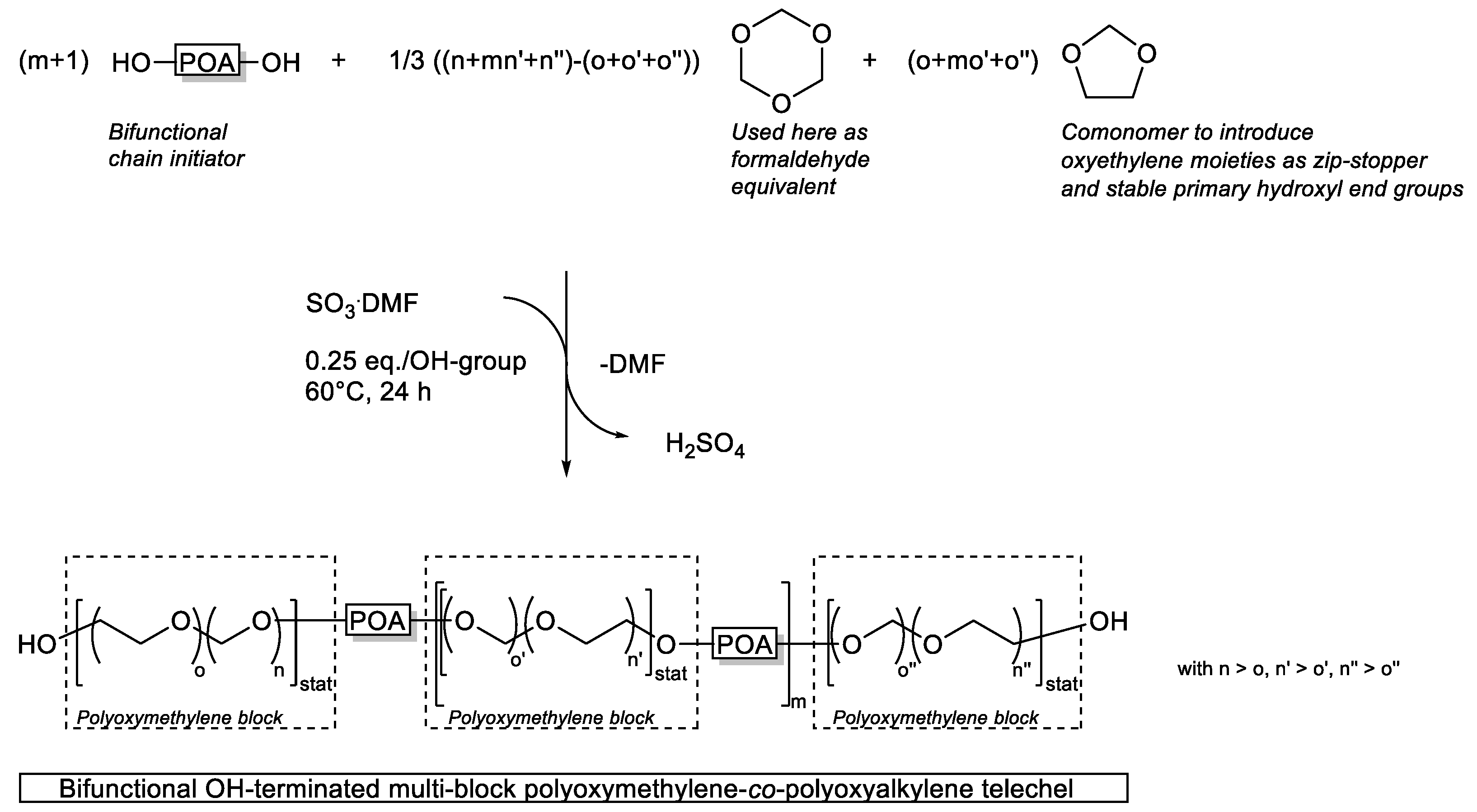
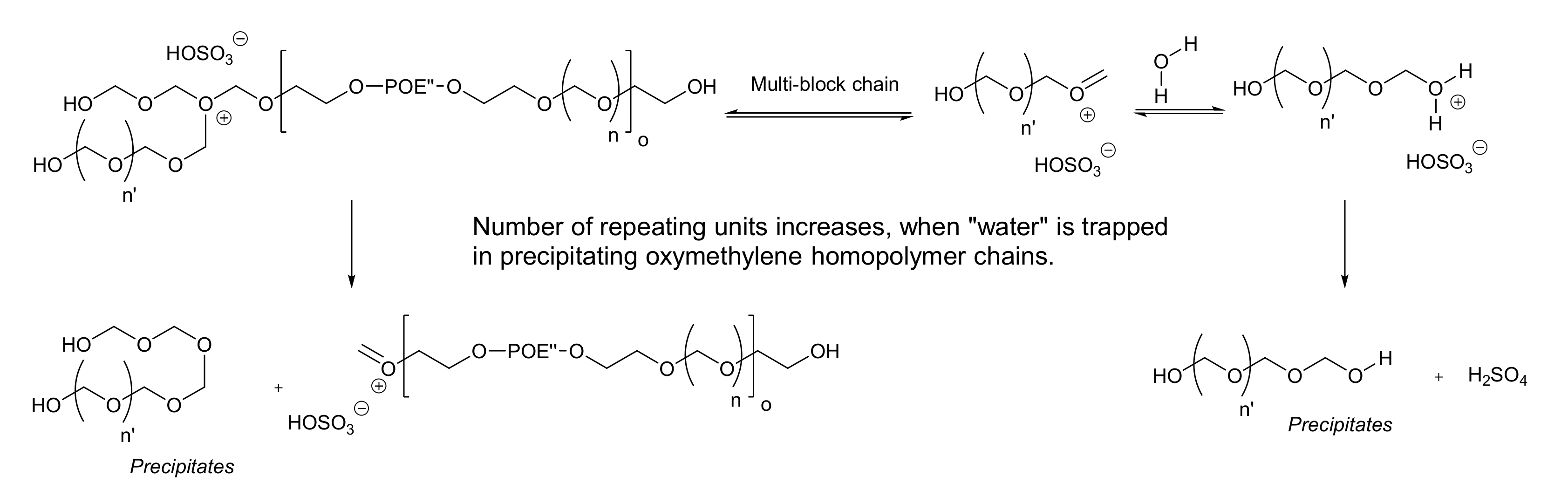
3.5. Mechanism of Multi-Block Copolymer Formation
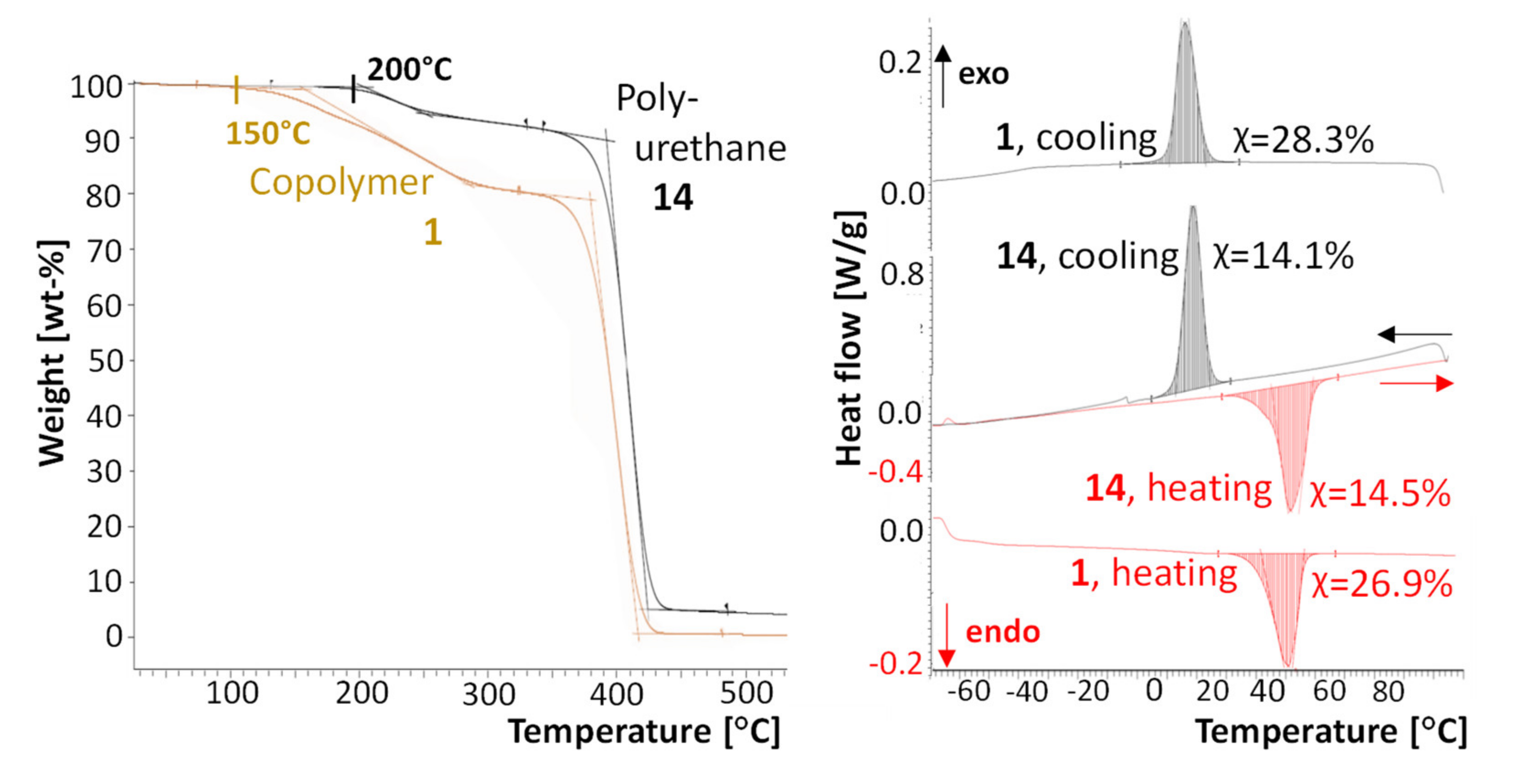
3.6. Polyurethane Formation
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| a.u. | arbitrary units |
| D | Germany |
| DMF | Dimethylformamide |
| DSC | Differential scanning calorimetry |
| e | ecoinvent v3.8 |
| EU | Europe |
| eq. | equivalents |
| OH-functionality | |
| g | GaBi 2021.2 |
| G | Global |
| GPC | Gel permeation chromatography |
| HDPE | High density polyethylene |
| HMBC | Heteronuclear multiple bond coherence |
| HSQC | Heteronuclear single quantum coherence |
| LDPE | Low-density polyethylene |
| LLDPE | Linear low-density polyethylene |
| Me | Methyl |
| mp | market process |
| NMR | Nuclear magnetic resonance |
| PA | Polyamide |
| PC | Polycarbonate |
| PEEK | Polyetheretherketone |
| PEG | Polyethylene glycol |
| PES | Polyethersulfone |
| PET | Polyethyleneterephthalate |
| PMMA | Polymethylmethacrylate |
| POA | Polyoxyalkylene |
| POA′ | Polyoxyalkylene except for a terminal oxyethylene group |
| POA″ | Polyoxyalkylene except for two terminal oxyethylene groups |
| POE | Polyoxyethylene |
| POE′ | Polyoxyalkylene except for a terminal oxyethylene group |
| POE″ | Polyoxyalkylene except for two terminal oxyethylene groups |
| POM | Polyoxymethylene |
| PU | Polyurethane |
| PP | Polypropylene |
| PS | Polystyrene |
| PVC | Polyvinylchloride |
| R | Substituent |
| RoW | Rest of World |
| TGA | Thermal gravimetric analysis |
| TI | Capability by temperature index by Underwriter Laboratories |
| TPU | Thermoplastic polyurethanes |
| up | unit process |
References
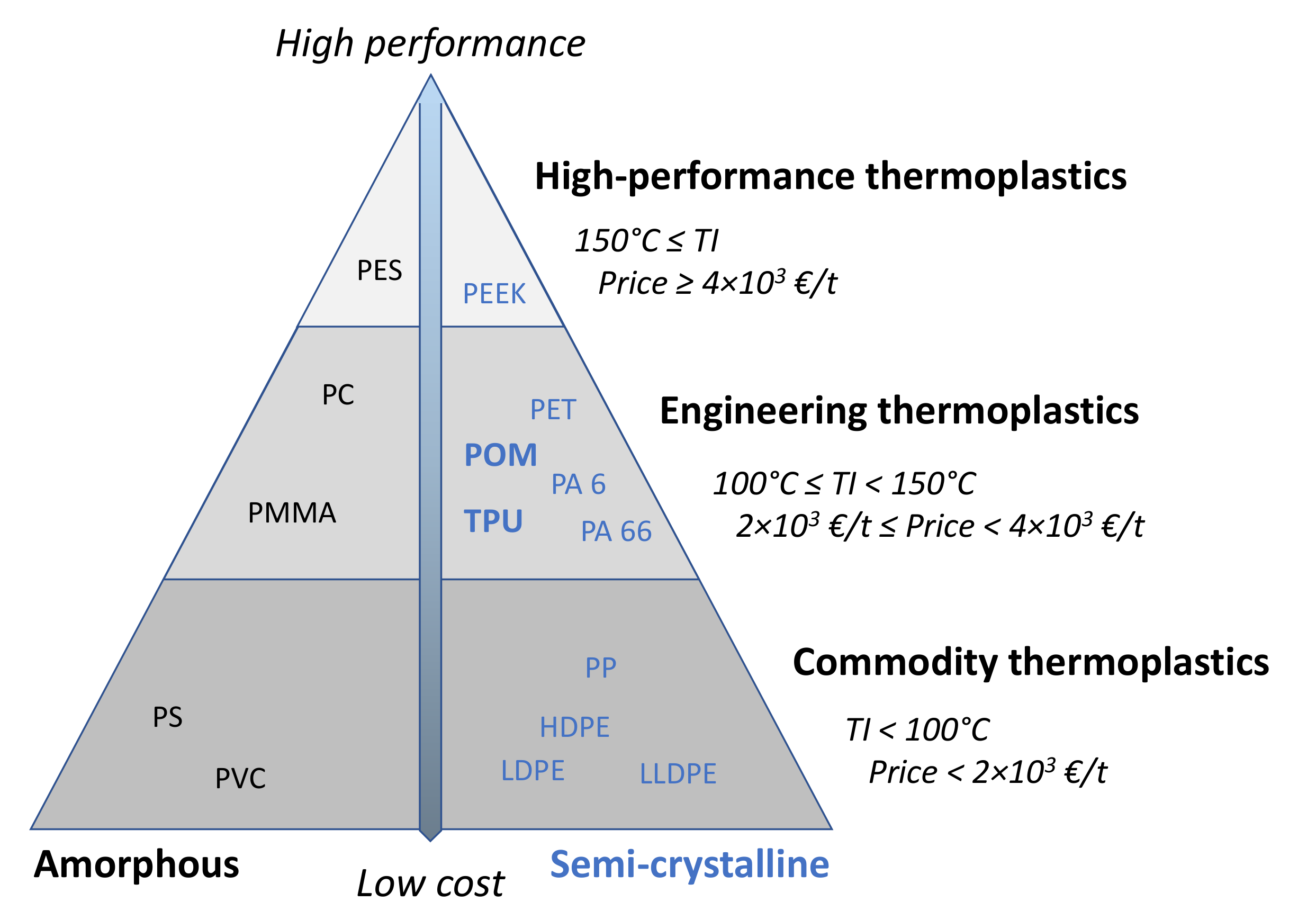
- Peters, E.N. 1—Engineering Thermoplastics—Materials, Properties, Trends. In Applied Plastics Engineering Handbook, 2nd ed.; Kutz, M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2017; pp. 3–26. [Google Scholar] [CrossRef]
- Hong, M.; Chen, E.Y.X. Chemically recyclable polymers: A circular economy approach to sustainability. Green Chem. 2017, 19, 3692–3706. [Google Scholar] [CrossRef]
- Mittal, V. High performance polymers: An overview. In High Performance Polymers and Engineering Plastics; Mittal, V., Ed.; Scrivener Publishing: Salem, MA, USA, 2011; pp. 1–20. [Google Scholar]
- Moha, V.; Cozzula, D.; Hölscher, M.; Leitner, W.; Müller, T.E. A DFT Study on the Co-polymerization of CO2 and Ethylene: Feasibility Analysis for the Direct Synthesis of Polyethylene Esters. ChemSusChem 2016, 9, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Keller, F.; Lee, R.P.; Meyer, B. Life cycle assessment of global warming potential, resource depletion and acidification potential of fossil, renewable and secondary feedstock for olefin production in Germany. J. Clean. Prod. 2020, 250, 119484. [Google Scholar] [CrossRef]
- Reyhanoglu, Y.; Sahmetlioglu, E.; Gokturk, E. Alternative Approach for Synthesizing Polyglycolic Acid Copolymers from C1 Feedstocks and Fatty Ester Epoxides. ACS Sustain. Chem. Eng. 2019, 7, 5103–5110. [Google Scholar] [CrossRef]
- Zhang, D.; del Rio-Chanona, E.A.; Shah, N. Screening Synthesis Pathways for Biomass-Derived Sustainable Polymer Production. ACS Sustain. Chem. Eng. 2017, 5, 4388–4398. [Google Scholar] [CrossRef]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef]
- Joseph, J.; Patel, R.M.; Wenham, A.; Smith, J.R. Biomedical applications of polyurethane materials and coatings. Trans. IMF 2018, 96, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Datta, J.; Kasprzyk, P. Thermoplastic polyurethanes derived from petrochemical or renewable resources: A comprehensive review. Polym. Eng. Sci. 2018, 58 (Suppl. S1), E14–E35. [Google Scholar] [CrossRef] [Green Version]
- Maurya, S.D.; Kurmvanshi, S.K.; Mohanty, S.; Nayak, S.K. A Review on Acrylate-Terminated Urethane Oligomers and Polymers: Synthesis and Applications. Polym.-Plast. Technol. Eng. 2018, 57, 625–656. [Google Scholar] [CrossRef]
- Furtwengler, P.; Avérous, L. Renewable polyols for advanced polyurethane foams from diverse biomass resources. Polym. Chem. 2018, 9, 4258–4287. [Google Scholar] [CrossRef]
- Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M.A.; Müller, T.E.; Leitner, W.; Gürtler, C. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 2014, 16, 1865–1870. [Google Scholar] [CrossRef]
- Al Nabulsi, A.; Cozzula, D.; Hagen, T.; Leitner, W.; Müller, T.E. Isocyanurate formation during rigid polyurethane foam assembly: A mechanistic study based on in situ IR and NMR spectroscopy. Polym. Chem. 2018, 9, 4891–4899. [Google Scholar] [CrossRef]
- Töpfer, O.; Clauss, M.; Futterer, T.; Schmitt, E. Flame retardants for engineering thermoplastics used in electric and electronic equipment like connectors. In Proceedings of the 2012 Electronics Goes Green 2012+, Berlin, Germany, 9–12 September 2012; pp. 1–7. [Google Scholar]
- Margolis, J.M. Engineering Thermoplastics, Properties and Applications, 1st ed.; CRC Press: New York, NY, USA, 1985; p. 408. [Google Scholar]
- Von der Assen, N.; Bardow, A. Life cycle assessment of polyols for polyurethane production using CO2 as feedstock: Insights from an industrial case study. Green Chem. 2014, 16, 3272–3280. [Google Scholar] [CrossRef] [Green Version]
- Meys, R.; Kätelhön, A.; Bardow, A. Towards sustainable elastomers from CO2: Life cycle assessment of carbon capture and utilization for rubbers. Green Chem. 2019, 21, 3334–3342. [Google Scholar] [CrossRef]
- Von der Assen, N.; Lampe, M.; Müller, L.; Bardow, A. Life-Cycle Assessment Principles for the Integrated Product and Process Design of Polymers from CO2. In Computer Aided Chemical Engineering; Gernaey, K.V., Huusom, J.K., Gani, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 37, pp. 1235–1240. [Google Scholar] [CrossRef]
- Garcia-Garcia, G.; Fernandez, M.C.; Armstrong, K.; Woolass, S.; Styring, P. Analytical Review of Life-Cycle Environmental Impacts of Carbon Capture and Utilization Technologies. ChemSusChem 2021, 14, 995–1015. [Google Scholar] [CrossRef]
- Elmas, S.; Subhani, M.A.; Leitner, W.; Müller, T.E. Anion effect controlling the selectivity in the zinc-catalysed copolymerisation of CO2 and cyclohexene oxide. Beilstein J. Org. Chem. 2015, 11, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Darensbourg, D.J.; Yeung, A.D. A concise review of computational studies of the carbon dioxide–epoxide copolymerization reactions. Polym. Chem. 2014, 5, 3949–3962. [Google Scholar] [CrossRef]
- Kozak, C.M.; Ambrose, K.; Anderson, T.S. Copolymerization of carbon dioxide and epoxides by metal coordination complexes. Coord. Chem. Rev. 2018, 376, 565–587. [Google Scholar] [CrossRef]
- Wang, Y.; Darensbourg, D.J. Carbon dioxide-based functional polycarbonates: Metal catalyzed copolymerization of CO2 and epoxides. Coord. Chem. Rev. 2018, 372, 85–100. [Google Scholar] [CrossRef]
- Romain, C.; Thevenon, A.; Saini, P.K.; Williams, C.K. Dinuclear Metal Complex-Mediated Formation of CO2-Based Polycarbonates. In Carbon Dioxide and Organometallics. Topics in Organometallic Chemistry; Lu, X.B., Ed.; Springer: Cham, Switzerland, 2015; Volume 53. [Google Scholar]
- Poland, S.J.; Darensbourg, D.J. A quest for polycarbonates provided via sustainable epoxide/CO2 copolymerization processes. Green Chem. 2017, 19, 4990–5011. [Google Scholar] [CrossRef]
- Ang, R.-R.; Tin Sin, L.; Bee, S.-T.; Tee, T.-T.; Kadhum, A.A.H.; Rahmat, A.R.; Wasmi, B.A. A review of copolymerization of green house gas carbon dioxide and oxiranes to produce polycarbonate. J. Clean. Prod. 2015, 102, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Zhu, Y.; Romain, C.; Brooks, R.; Saini, P.K.; Williams, C.K. Ring-opening copolymerization (ROCOP): Synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51, 6459–6479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taherimehr, M.; Pescarmona, P.P. Green polycarbonates prepared by the copolymerization of CO2 with epoxides. J. Appl. Polym. Sci. 2014, 131, 41141. [Google Scholar] [CrossRef]
- Subhani, M.A.; Gürtler, C.; Leitner, W.; Müller, T.E. Nanoparticulate TiO2-Supported Double Metal Cyanide Catalyst for the Copolymerization of CO2 with Propylene Oxide. Eur. J. Inorg. Chem. 2016, 2016, 1944–1949. [Google Scholar] [CrossRef]
- Pohl, M.; Danieli, E.; Leven, M.; Leitner, W.; Blümich, B.; Müller, T.E. Dynamics of Polyether Polyols and Polyether Carbonate Polyols. Macromolecules 2016, 49, 8995–9003. [Google Scholar] [CrossRef]
- Subhani, M.A.; Köhler, B.; Gürtler, C.; Leitner, W.; Müller, T.E. Transparent Films from CO2-Based Polyunsaturated Poly(ether carbonate)s: A Novel Synthesis Strategy and Fast Curing. Angew. Chem. Int. Ed. 2016, 55, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzarri, C.; Leven, M.; Leitner, W.; Gürtler, C.; Müller, T.E. Method for Production of Polyoxymethylene Block Copolymer Having Increased Proportion of Incorporated Formaldehyde. PCT International Application WO 2018114843 A1, 27 June 2018. [Google Scholar]
- Müller, T.E.; Gürtler, C.; Subhani, M.A.; Ebrahimi, F.; Leitner, W. Method for Production of Polyether Thiocarbonate Polyols. PCT International Application WO 2018114837 A1, 28 June 2018. [Google Scholar]
- Gürtler, C.; Müller, T.E.; Hofmann, J.; Langanke, J.; Leven, M.; Leitner, W. Method for Producing Polymeric Ring-Opening Products. PCT International Application WO 2018029232 A1, 15 February 2018. [Google Scholar]
- Ionescu, M. Chemistry and Technology of Polyols for Polyurethanes; iSmithers Rapra Publishing: Shawbury, UK, 2005; p. 586. [Google Scholar]
- Müller, T.E.; Gürtler, C.; Pohl, M.; Subhani, M.A.; Leitner, W. Method for Producing Polyoxyalkylene Polycarbonate Polyols. European Patent Application EP 3098250 A1, 30 November 2016. [Google Scholar]
- Gürtler, C.; Müller, T.E.; Kermagoret, A.; Dienes, Y.; Barruet, J.; Wolf, A.; Grasser, S. Method for Production of Polyether-Ester-Carbonate Polyols in the Presence of Double Metal Cyanide Catalysts. PCT International Application WO 2013087582 B1, 22 August 2018. [Google Scholar]
- Müller, T.E.; Gürtler, C.; Wohak, M.; Hofmann, J.; Subhani, M.A.; Leitner, W.; Peckermann, I.; Wolf, A. Polyether Carbonate Polyol Production by Three Stage Process with Suspending Agent and Last Step Continuous Addition of H-Functional Starter. PCT International Application WO 2014033070 A1, 6 March 2014. [Google Scholar]
- Gürtler, C.; Müller, T.E.; Bizzarri, C.; Leven, M.; Leitner, W.; Winkelhaus, D. Method for Production of Polyoxymethylene-Containing Prepolymer and Method for Production of Polyurethane. European Patent Application EP 3498743 A1, 19 June 2019. [Google Scholar]
- Gürtler, C.; Müller, T.E.; Bizzarri, C.; Leven, M.; Leitner, W. Method for the Production of Polyurethane Polymers. European Patent Application EP 3498746 A1, 19 June 2019. [Google Scholar]
- Gürtler, C.; Marker, V.; Müller, T.E.; Leven, M.; Bizzari, C.; Leitner, W. Method for Producing Polyurethanes with Reduced Heat of Combustion. PCT International Application WO 2018037040 A1, 1 March 2018. [Google Scholar]
- Müller, T.E.; Gürtler, C.; Langstein, G.; Bizzarri, C.; Leitner, W. Method for Production of Degradable Polyurethanes by Using Oxymethylene Polyols. European Patent Application EP 3287475 A1, 28 February 2018. [Google Scholar]
- Hoffmann, M.; Bizzarri, C.; Leitner, W.; Müller, T.E. Reaction pathways at the initial steps of trioxane polymerisation. Catal. Sci. Technol. 2018, 8, 5594–5603. [Google Scholar] [CrossRef]
- Bizzarri, C.; Vogt, H.; Baráth, G.; Gürtler, C.; Leitner, W.; Müller, T.E. Oligomeric poly(acetalcarbonates) obtained by repolymerisation of paraformaldehyde. Green Chem. 2016, 18, 5160–5168. [Google Scholar] [CrossRef]
- Von der Assen, N.; Sternberg, A.; Kätelhön, A.; Bardow, A. Environmental potential of carbon dioxide utilization in the polyurethane supply chain. Faraday Discuss. 2015, 183, 291–307. [Google Scholar] [CrossRef] [Green Version]
- Ionescu, M.; Sinharoy, S.; Petrović, Z.S. Polyacetal Polyols for Polyurethanes. J. Polym. Environ. 2009, 17, 123. [Google Scholar] [CrossRef]
- Lüftl, S.; Visakh, P.M.; Chandran, S. Polyoxymethylene Handbook, Structure, Properties, Applications and Their Nanocomposites, 1st ed.; Wiley & Sons Ltd.: Chichester, UK, 2014. [Google Scholar]
- Hajek, J.; Zidek, R. Production of formaldehyde by methanol oxidation. Chem. Prum. 1975, 25, 298. [Google Scholar]
- Shenbagamuthuraman, V.; Patel, A.; Khanna, S.; Banerjee, E.; Parekh, S.; Karthick, C.; Ashok, B.; Velvizhi, G.; Nanthagopal, K.; Ong, H.C. State of art of valorising of diverse potential feedstocks for the production of alcohols and ethers: Current changes and perspectives. Chemosphere 2022, 286, 131587. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, D.; Calvo-Serrano, R.; Martín-Gamboa, M.; Galán-Martín, A.; Guillén-Gosálbez, G. Social life cycle assessment of green methanol and benchmarking against conventional fossil methanol. Sci. Total Environ. 2022, 824, 153840. [Google Scholar] [CrossRef]
- Sha, F.; Han, Z.; Tang, S.; Wang, J.; Li, C. Hydrogenation of Carbon Dioxide to Methanol over Non-Cu-based Heterogeneous Catalysts. ChemSusChem 2020, 13, 6160–6181. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Jaén, S.; Virginie, M.; Bonin, J.; Robert, M.; Wojcieszak, R.; Khodakov, A.Y. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Z.; Liu, X.; Hua, K.; Shao, Z.; Wei, B.; Huang, C.; Wang, H.; Sun, Y. A Short Review of Recent Advances in Direct CO2 Hydrogenation to Alcohols. Top. Catal. 2021, 64, 371–394. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Zhang, S.; Jing, X.; Wang, Y.; Li, F. Towards Carbon-Neutral Methanol Production from Carbon Dioxide Electroreduction. ChemNanoMat 2021, 7, 728–736. [Google Scholar] [CrossRef]
- Boutin, E.; Robert, M. Molecular Electrochemical Reduction of CO2 beyond Two Electrons. Trends Chem. 2021, 3, 359–372. [Google Scholar] [CrossRef]
- Kinzel, N.W.; Werlé, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. [Google Scholar] [CrossRef]
- Dhiman, S.S.; Shrestha, N.; David, A.; Basotra, N.; Johnson, G.R.; Chadha, B.S.; Gadhamshetty, V.; Sani, R.K. Producing methane, methanol and electricity from organic waste of fermentation reaction using novel microbes. Bioresour. Technol. 2018, 258, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hernández Villamor, D.; Peng, H.; Chen, J.; Liu, L.; Haritos, V.; Ledesma-Amaro, R. Metabolic engineering strategies to enable microbial utilization of C1 feedstocks. Nat. Chem. Biol. 2021, 17, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Sharma, T.; Nguyen, D.D.; Cheng, C.K.; Lam, S.S.; Xia, C.; Nadda, A.K. Integrated catalytic insights into methanol production: Sustainable framework for CO2 conversion. J. Environ. Manag. 2021, 289, 112468. [Google Scholar] [CrossRef] [PubMed]
- Thenert, K.; Beydoun, K.; Wiesenthal, J.; Leitner, W.; Klankermayer, J. Ruthenium-Catalyzed Synthesis of Dialkoxymethane Ethers Utilizing Carbon Dioxide and Molecular Hydrogen. Angew. Chem. Int. Ed. 2016, 55, 12266–12269. [Google Scholar] [CrossRef] [PubMed]
- Tomkins, P.; Müller, T.E. Evaluating the carbon inventory, carbon fluxes and carbon cycles for a long-term sustainable world. Green Chem. 2019, 21, 3994–4013. [Google Scholar] [CrossRef]
- Wyndorps, J.; Röver, L. Analyse der Kopernikus P2X-Wertschöpfungskette zu Herstellung von PME Polymeren. In Roadmap des Kopernikus-Projektes P2X; Ausfelder, F., Dura, H., Eds.; Dechema: Frankfurt am Main, Germany, 2021; pp. 73–78. [Google Scholar]
- Clark, E.S. Delrin material characteristics. J. Heart Valve Dis. 1996, 5 (Suppl. S2), S184–S189. [Google Scholar]
- Majer, J.; Hainová, O. Bestimmung des Kristallinitätsgrades von Polyoxymethylen und von einem Mischpolymeren Trioxan-zyklischer Äther durch Ultrarotspektroskopie. Kolloid-Z. Z. Polym. 1965, 201, 23–26. [Google Scholar] [CrossRef]
- Engel, D. PEAXACT 3.0.7-Software for Quantitative Spectroscopy and Chromatography; S•PACT GmbH: Aachen, Germany, 2013. [Google Scholar]
- Crivello, J.V.; Varlemann, U. Structure and Reactivity Relationships in the Photoinitiated Cationic Polymerization of 3,4-Epoxycyclohexylmethyl-3′,4′-epoxycyclohexane Carboxylate. In Photopolymerization; American Chemical Society: Washington, DC, USA, 1997; Volume 673, pp. 82–94. [Google Scholar] [CrossRef]
- Crivello, J.V.; Varlemann, U. Mechanistic study of the reactivity of 3,4-epoxycyclohexylmethyl 3′,4′-epoxycyclohexancarboxylate in photoinitiated cationic polymerizations. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 2473–2486. [Google Scholar] [CrossRef]
- Hopkinson, A.C.; Lien, M.H.; Csizmadia, I.G.; Yates, K. Quantum chemical studies on electrophilic addition. Theor. Chim. Acta 1978, 47, 97–109. [Google Scholar] [CrossRef]
- Sharavanan, K.; Ortega, E.; Moreau, M.; Lorthioir, C.; Lauprêtre, F.; Desbois, P.; Klatt, M.; Vairon, J.-P. Cationic Copolymerization of 1,3,5-Trioxane with 1,3-Dioxepane: A Comprehensive Approach to the Polyacetal Process. Macromolecules 2009, 42, 8702–8710. [Google Scholar] [CrossRef]
- Noppel, M.; Vehkamäki, H.; Kulmala, M. An improved model for hydrate formation in sulfuric acid–water nucleation. J. Chem. Phys. 2001, 116, 218–228. [Google Scholar] [CrossRef]
- Heist, R.H.; Reiss, H. Hydrates in supersaturated binary sulfuric acid-water vapor. J. Chem. Phys. 1974, 61, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Kern, W.; Cherdon, H.; Jaacks, V. Polyoxymethylene. Angew. Chem. 1961, 73, 177. [Google Scholar] [CrossRef]
- Kern, W.; Jaacks, V. Some kinetic effects in the polymerization of 1,3,5-trioxane. J. Polym. Sci. 1960, 48, 399–404. [Google Scholar] [CrossRef]
- Weissermel, K.; Fischer, E.; Gutweiler, K.; Hermann, H.D.; Cherdron, H. Polymerization of Trioxane. Angew. Chem. Int. Ed. Engl. 1967, 6, 526–533. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level | Compound | GWI [kg CO2-eq./kg] | Source 1 |
|---|---|---|---|
| Poly- urethane | Rigid foam | 4.59 | g-mp-EU |
| Flexible foam, TDI-based | 3.67–3.28 | g-mp-EU | |
| Flexible foam, MDI-based | 3.04 | g-mp-EU | |
| Iso- cyanate | TDI | 4.76–3.22 | g-mp-EU |
| MDI | 3.82–2.85 | g-mp-EU | |
| Polyol | Production mix | 4.11–4.01 | e-mp/up-EU/RoW |
| Polyether polyol | 3.91–3.03 | g-mp-EU | |
| Co- monomer | Ethylene oxide | 2.44–1.41 | g/e-mp/up-EU/RoW |
| Propylene oxide, oxirane process | 1.79 | g-mp-D | |
| Formaldehyde | 1.04–0.94 | g/e-mp/up-EU/RoW | |
| Pre- cursor | Methanol, steam reforming | 0.70–0.66 | e-mp/up-RoW |
| Methanol, biomass gasification | 0.69–0.67 | e-mp/up-RoW |
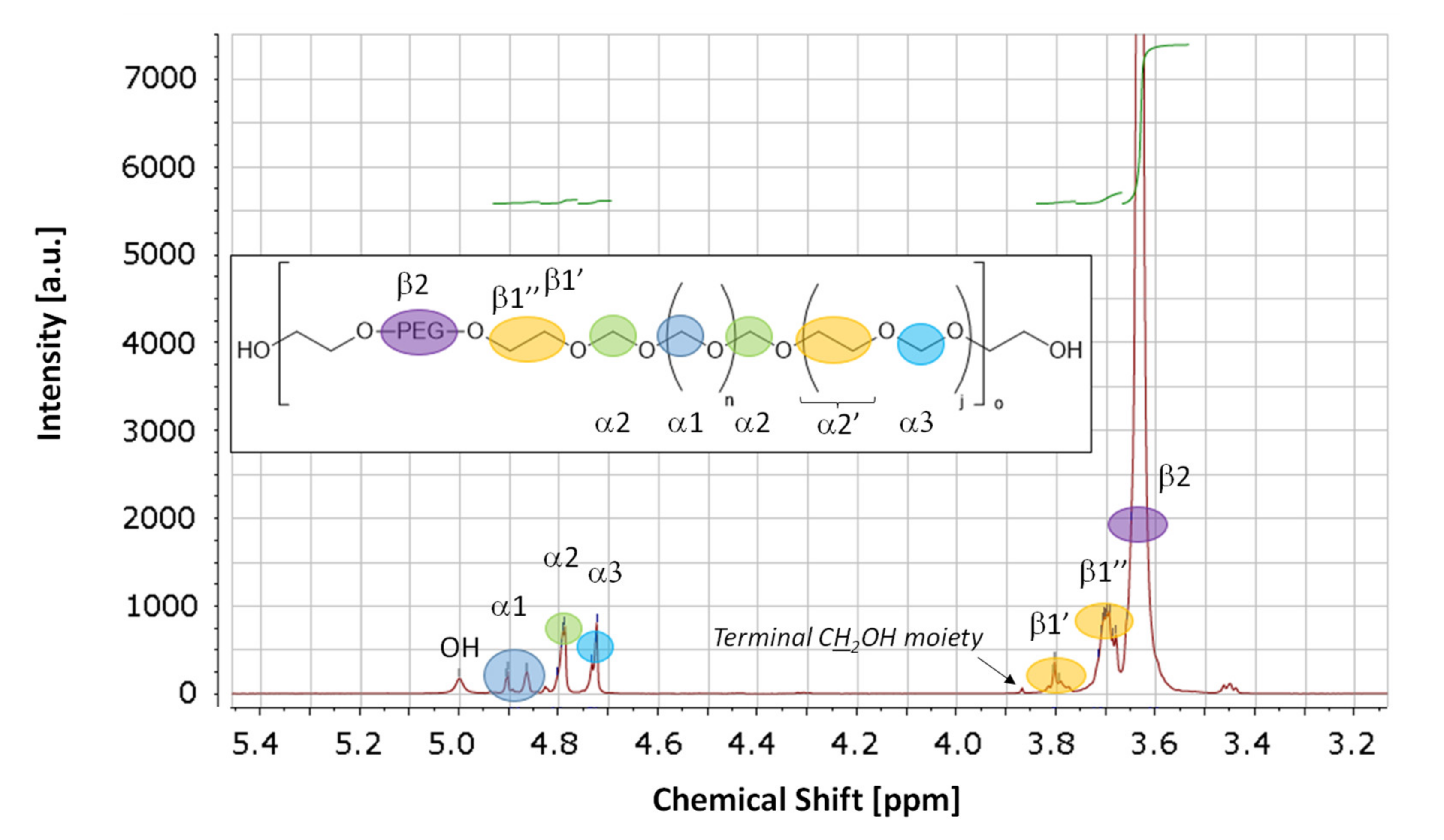
| Signal | Assignment | δ(1H) | δ(13C) |
|---|---|---|---|
| α1 | Inner oxymethylene moieties bound only to oxymethylene moieties | 4.91–4.84 | 89.2–88.8 |
| α2 | Oxymethylene moieties connected to an oxyethylene and oxymethylene moiety | 4.82–4.77 | 92.5–92.1 |
| α3 | Isolated oxymethylene moieties with neighboring oxyethylene moieties | 4.75–4.71 | 95.6 |
| β1′, β1″ | Marginal oxyethylene moieties with a neighboring oxymethylene moieties | 3.82–3.78, 3.74–3.66 | 67.7–67.5, 67.0–66.8 |
| β2 | Oxyethylene moieties within POE block | 3.66–3.57 | 70.7 |
| Co- Polymer | Diol a [g/mol] | Eq.Tx b [-] | Mn c [g/mol] | CH2Od [mol-%] | CH2Oe [-] | avgf [-] |
|---|---|---|---|---|---|---|
| 10 | A | 1.1 | 3.9 × 103 | 3.5 | 2.4 | 1.3 |
| 11 | A | 3.0 | 4.0 × 103 | 13.5 | 5.5 | 2.4 |
| 12 | A | 4.8 | 4.6 × 103 | 20.7 | 8.0 | 3.0 |
| 13 | A | 9.8 | 4.5 × 103 | 10.2 | 7.5 | 3.1 |
| 6 | B | 1.0 | 2.5 × 103 | 13.5 | 4.1 | 2.1 |
| 7 | B | 3.1 | 3.3 × 103 | 34.7 | 6.8 | 4.5 |
| 8 | B | 5.0 | 3.3 × 103 | 36.5 | 7.9 | 4.0 |
| 9 | B | 10.1 | 3.5 × 103 | 36.8 | 7.8 | 4.4 |
| 2 | C | 1.0 | 1.8 × 103 | 19.7 | 4.5 | 2.0 |
| 3 | C | 3.0 | 2.2 × 103 | 45.6 | 7.0 | 4.1 |
| 4 | C | 4.9 | 2.4 × 103 | 52.0 | 7.9 | 4.6 |
| 5 | C | 10.1 | 2.7 × 103 | 49.0 | 7.3 | 5.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, M.; Hermesmann, M.; Leven, M.; Leitner, W.; Müller, T.E. Semi-Crystalline Polyoxymethylene-co-Polyoxyalkylene Multi-Block Telechels as Building Blocks for Polyurethane Applications. Polymers 2022, 14, 882. https://doi.org/10.3390/polym14050882
Hoffmann M, Hermesmann M, Leven M, Leitner W, Müller TE. Semi-Crystalline Polyoxymethylene-co-Polyoxyalkylene Multi-Block Telechels as Building Blocks for Polyurethane Applications. Polymers. 2022; 14(5):882. https://doi.org/10.3390/polym14050882
Chicago/Turabian StyleHoffmann, Matthias, Matthias Hermesmann, Matthias Leven, Walter Leitner, and Thomas Ernst Müller. 2022. "Semi-Crystalline Polyoxymethylene-co-Polyoxyalkylene Multi-Block Telechels as Building Blocks for Polyurethane Applications" Polymers 14, no. 5: 882. https://doi.org/10.3390/polym14050882
APA StyleHoffmann, M., Hermesmann, M., Leven, M., Leitner, W., & Müller, T. E. (2022). Semi-Crystalline Polyoxymethylene-co-Polyoxyalkylene Multi-Block Telechels as Building Blocks for Polyurethane Applications. Polymers, 14(5), 882. https://doi.org/10.3390/polym14050882