
Recent Advances in the Synthesis of Complex Macromolecular Architectures Based on Poly(N-vinyl pyrrolidone) and the RAFT Polymerization Technique

 ,
, 
 and
and 
Abstract
:1. Introduction
- (a)
- (b)
- (c)
- (d)
- (e)
- Suspending agent in two-phase polymerization systems [17].
- (f)
- (g)
- (h)
- (i)
- Environmental protection: removal of heavy metals [15].
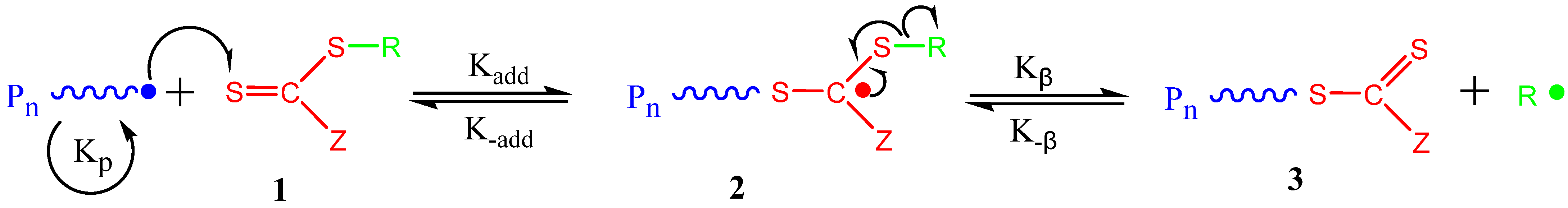
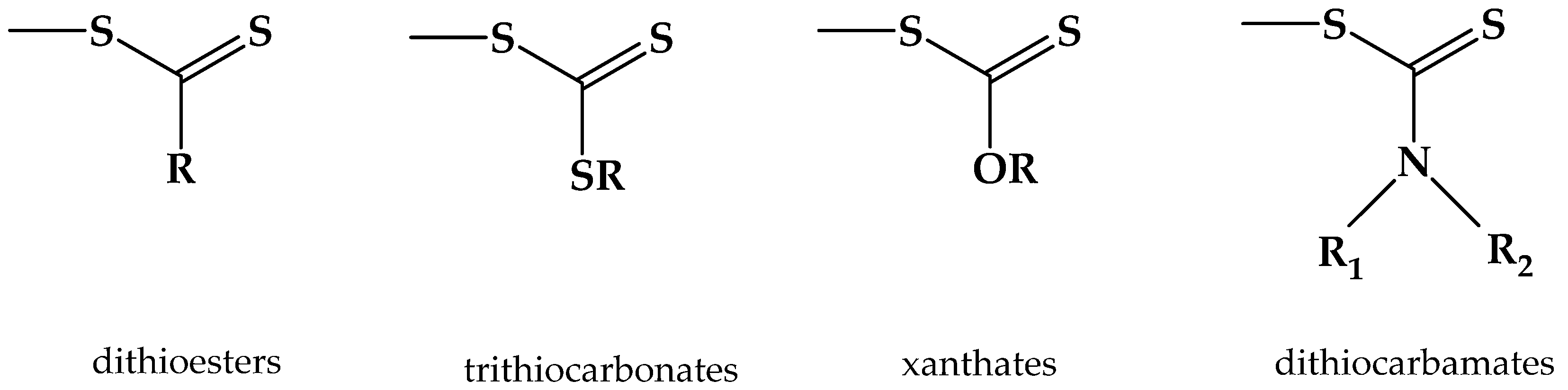
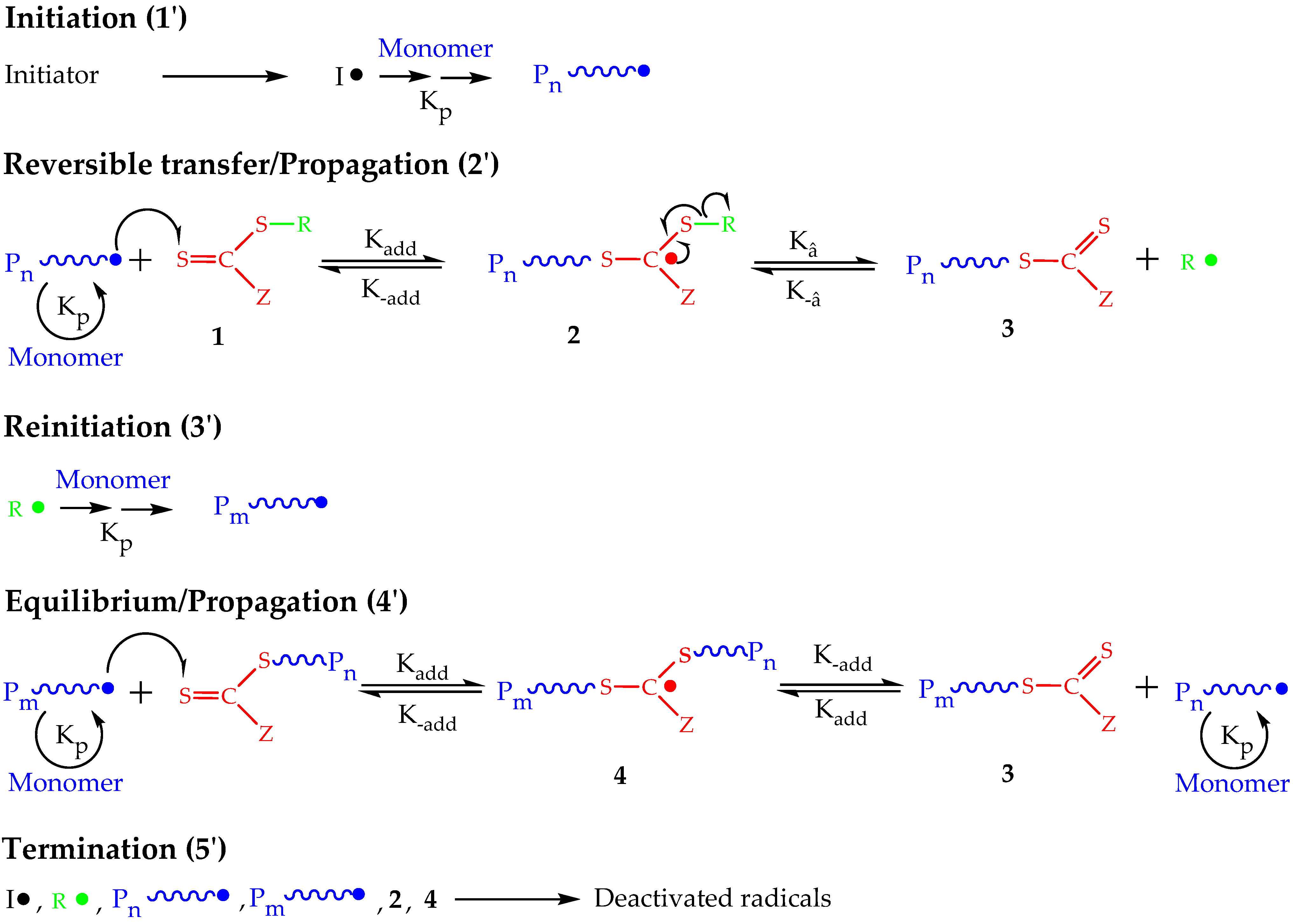
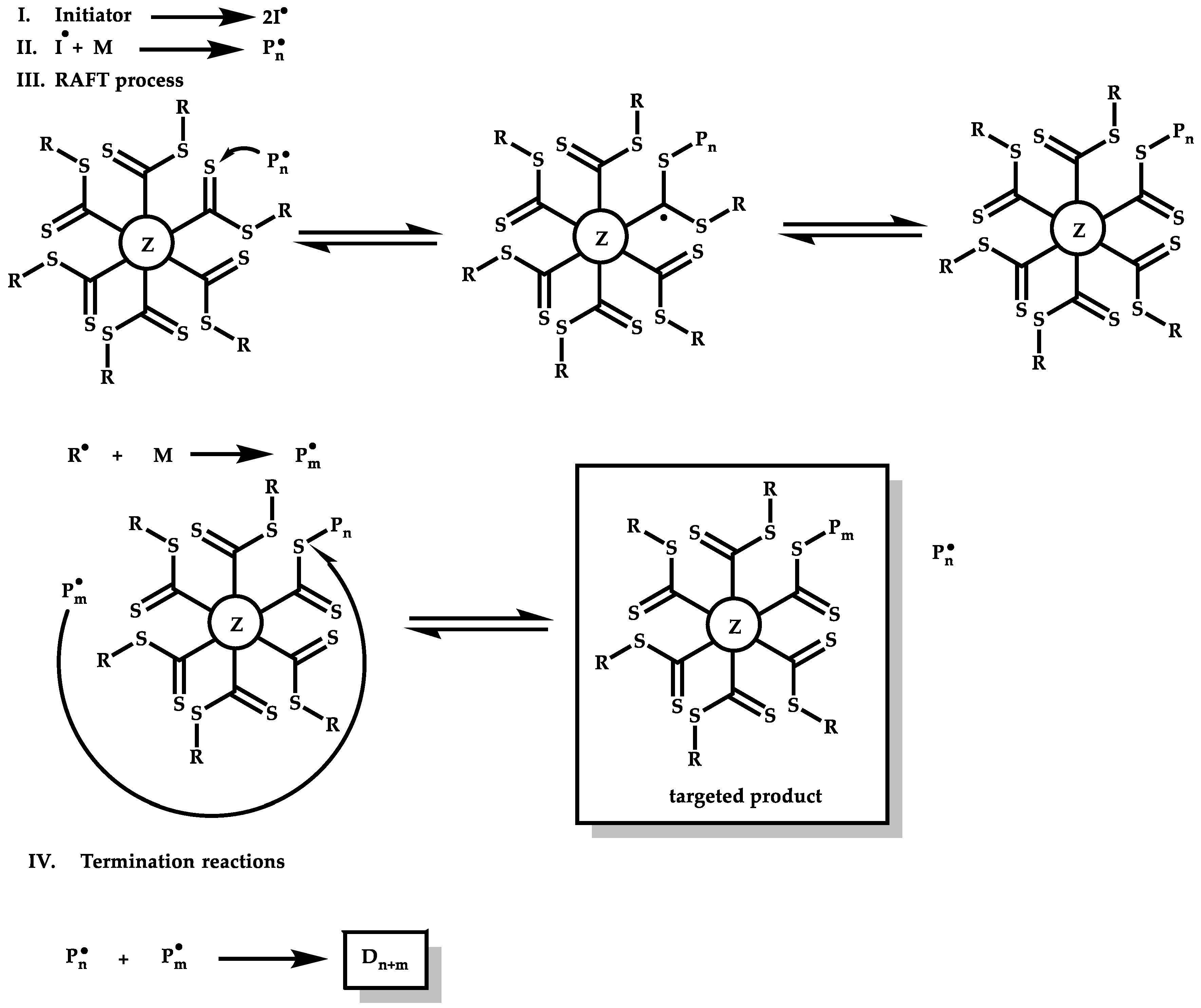
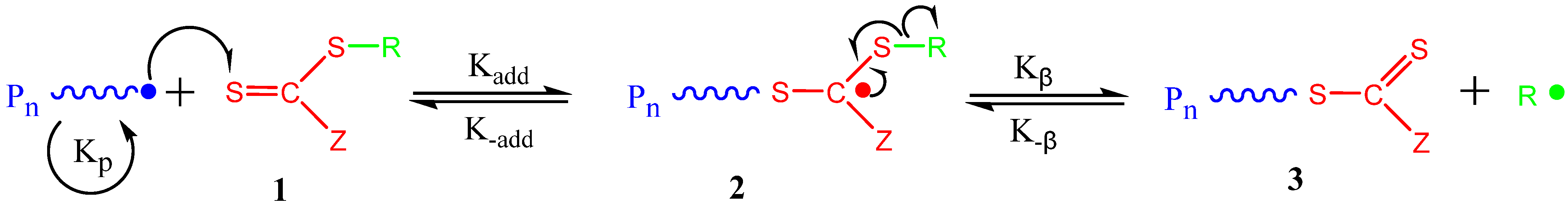
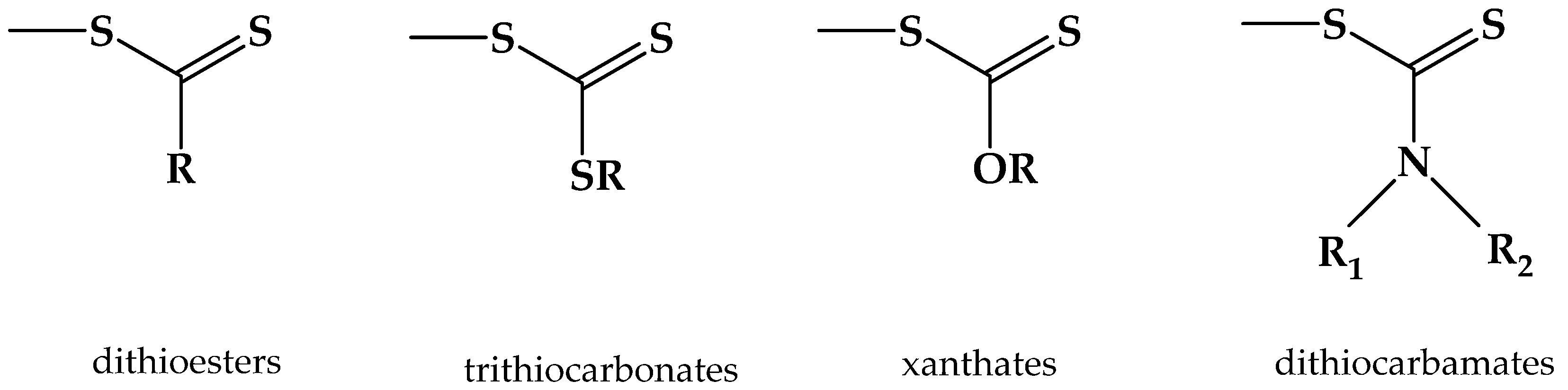
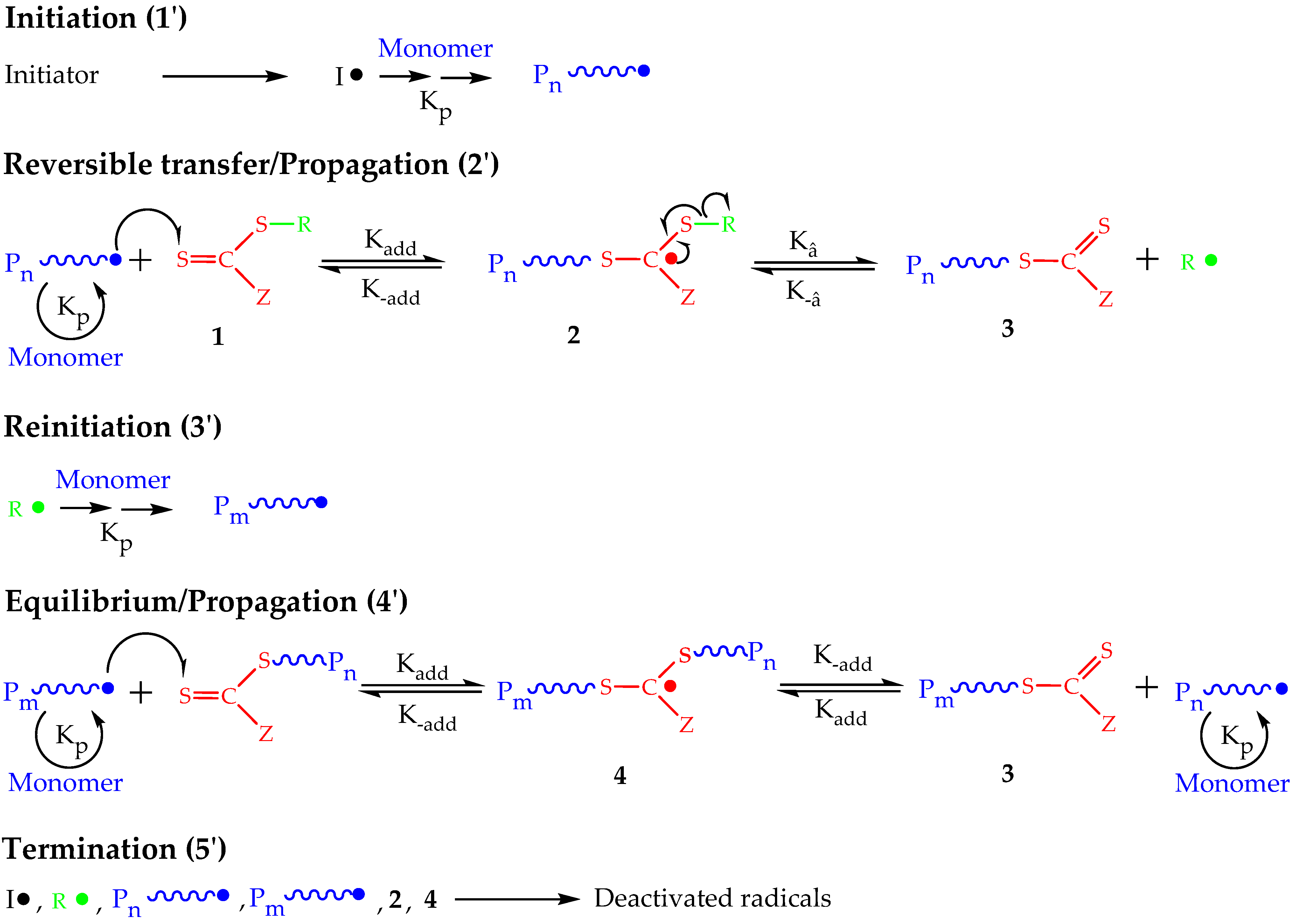
2. Principles of RAFT Polymerization Technique
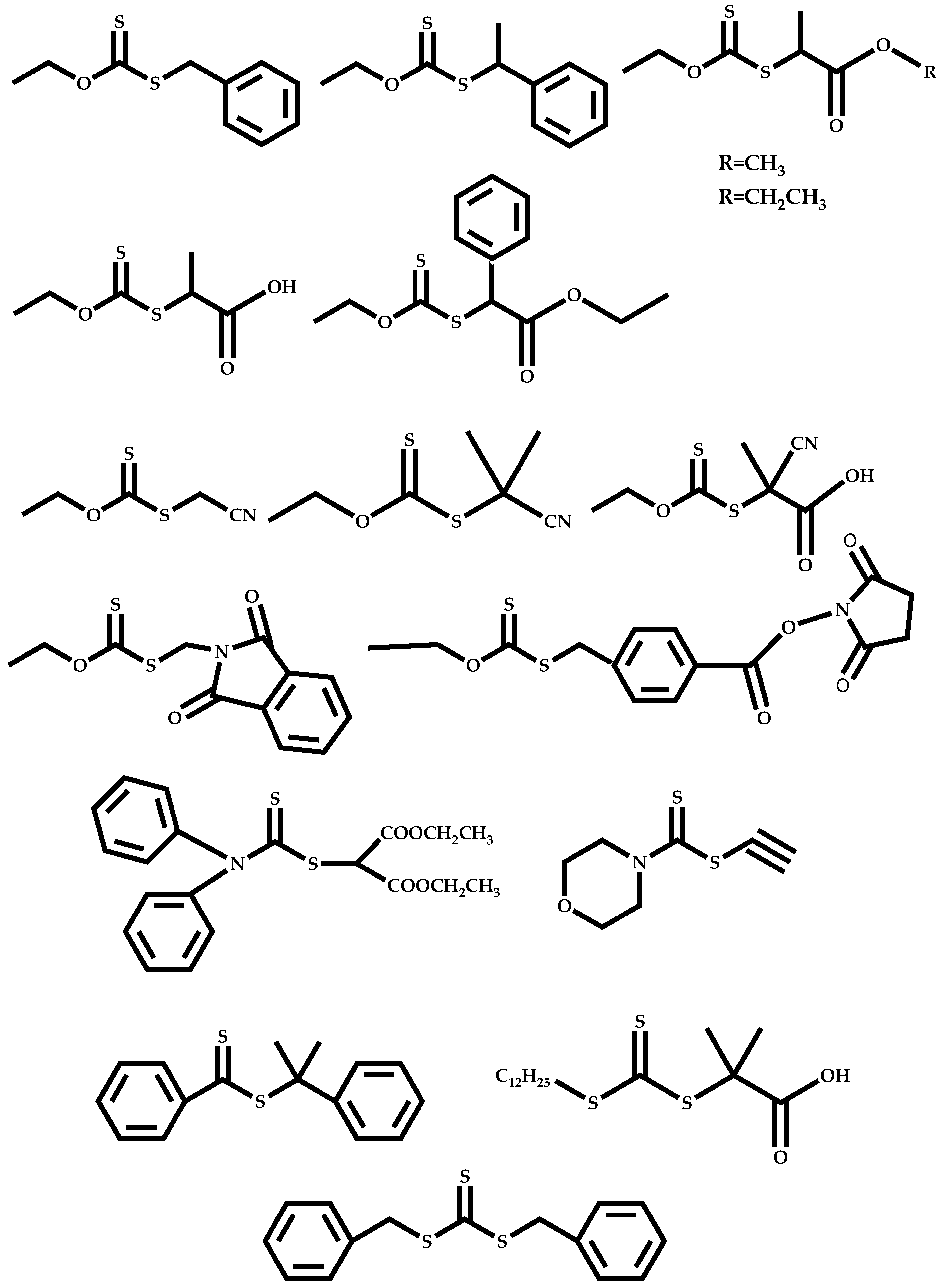
3. Polymerization of NVP by RAFT
4. Synthesis of Complex Macromolecular Architectures
4.1. Statistical Copolymers
4.1.1. Introduction
4.1.2. Evaluation of the Reactivity Ratios
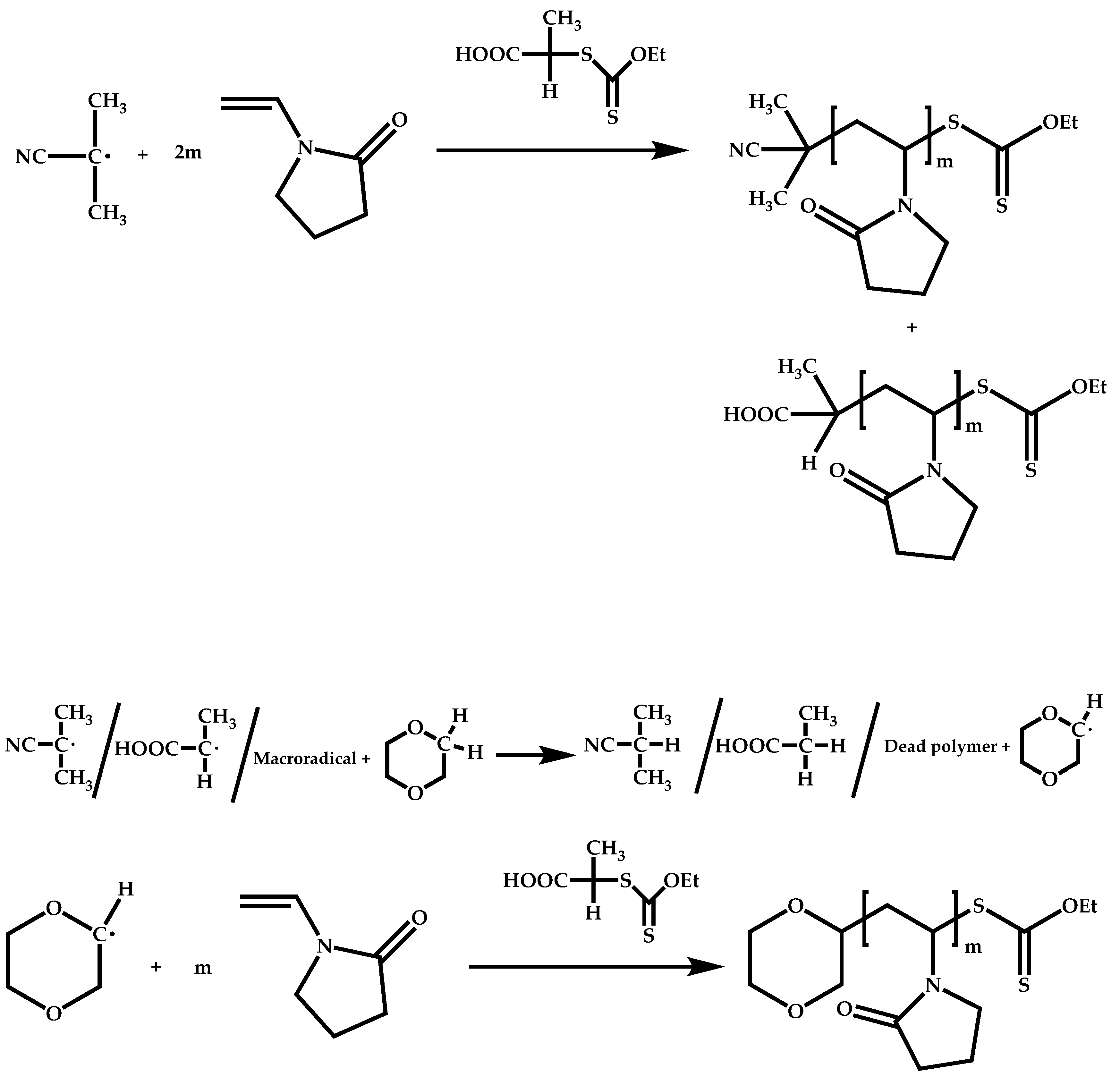
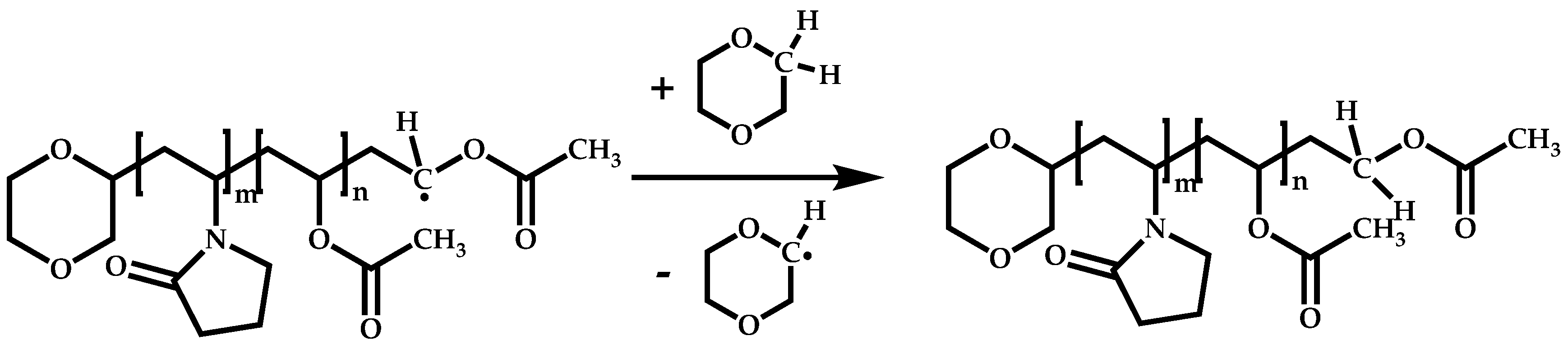
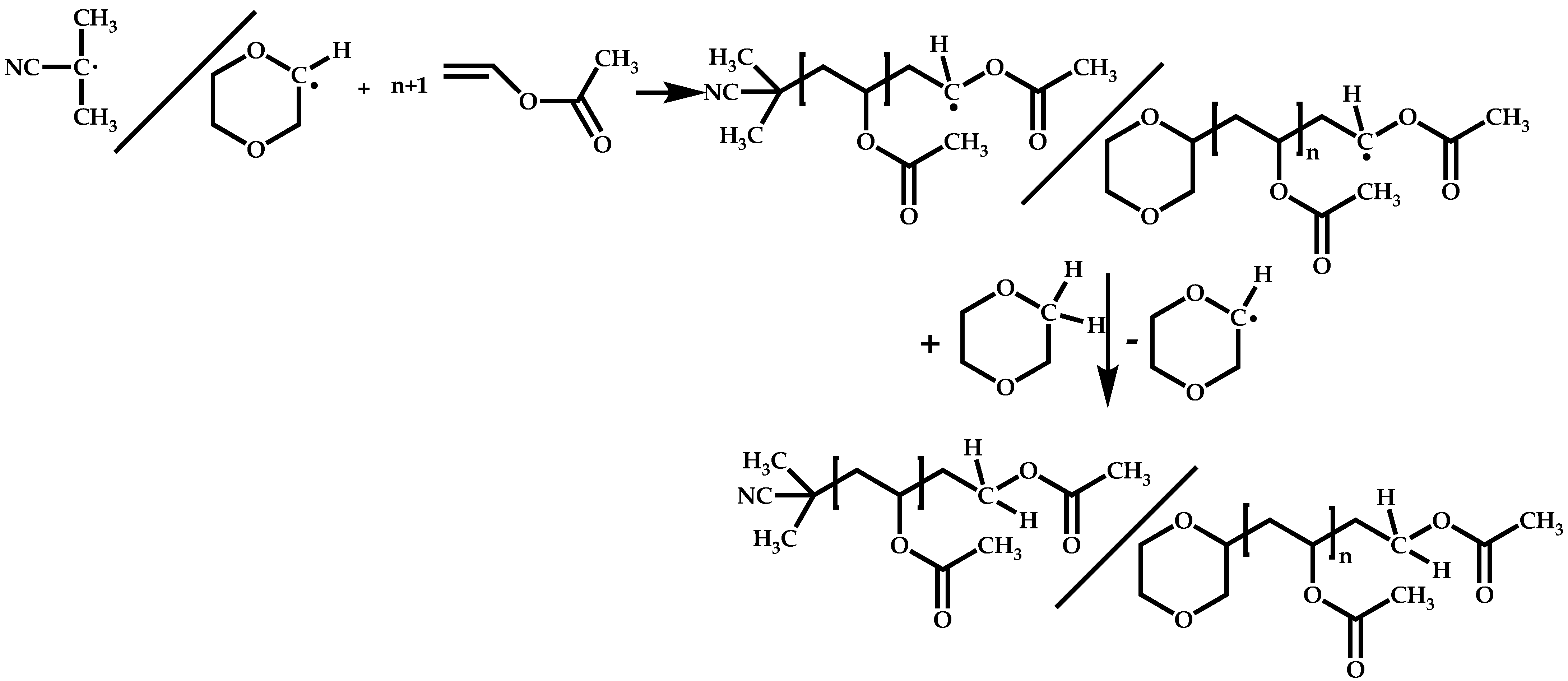
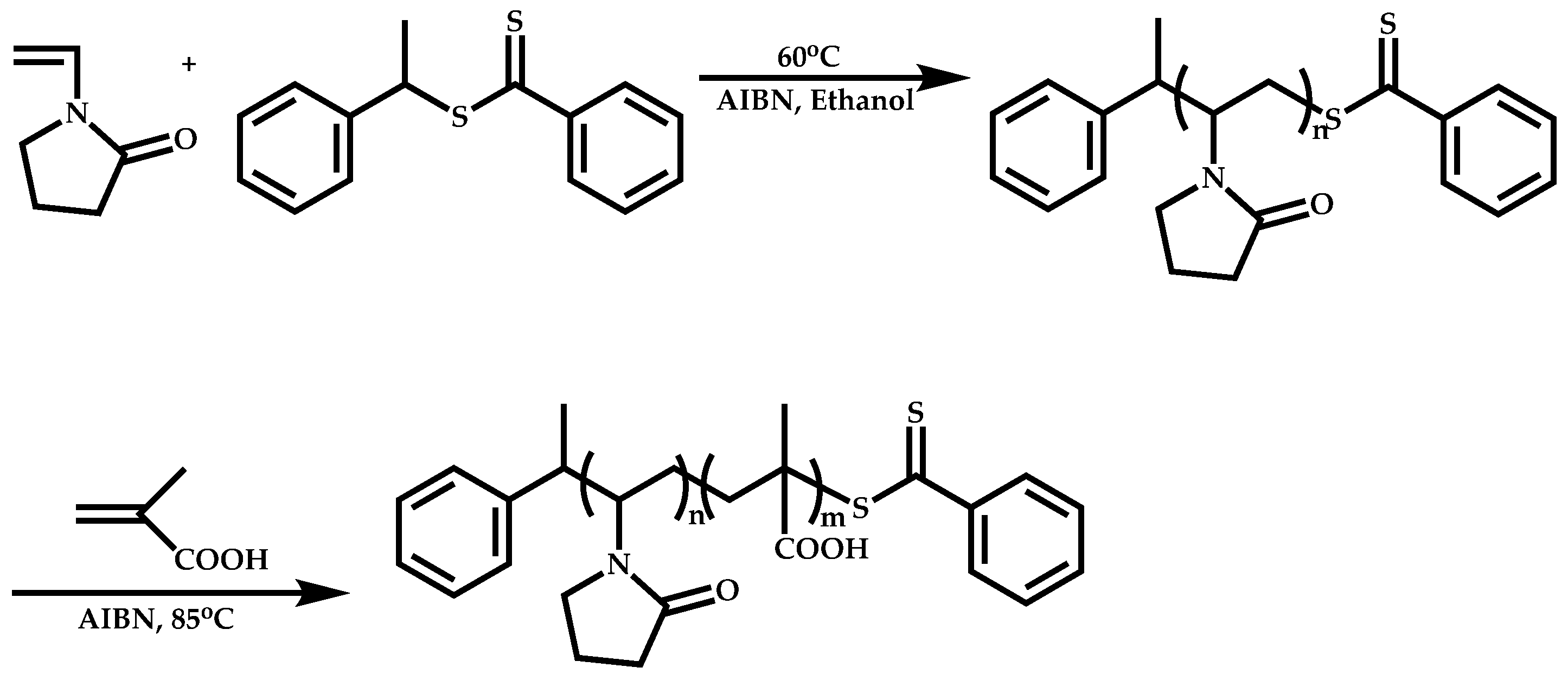
4.1.3. Statistical Copolymers of NVP via RAFT
4.2. Block Copolymers Based on NVP Synthesized via RAFT Polymerization
4.2.1. Introduction
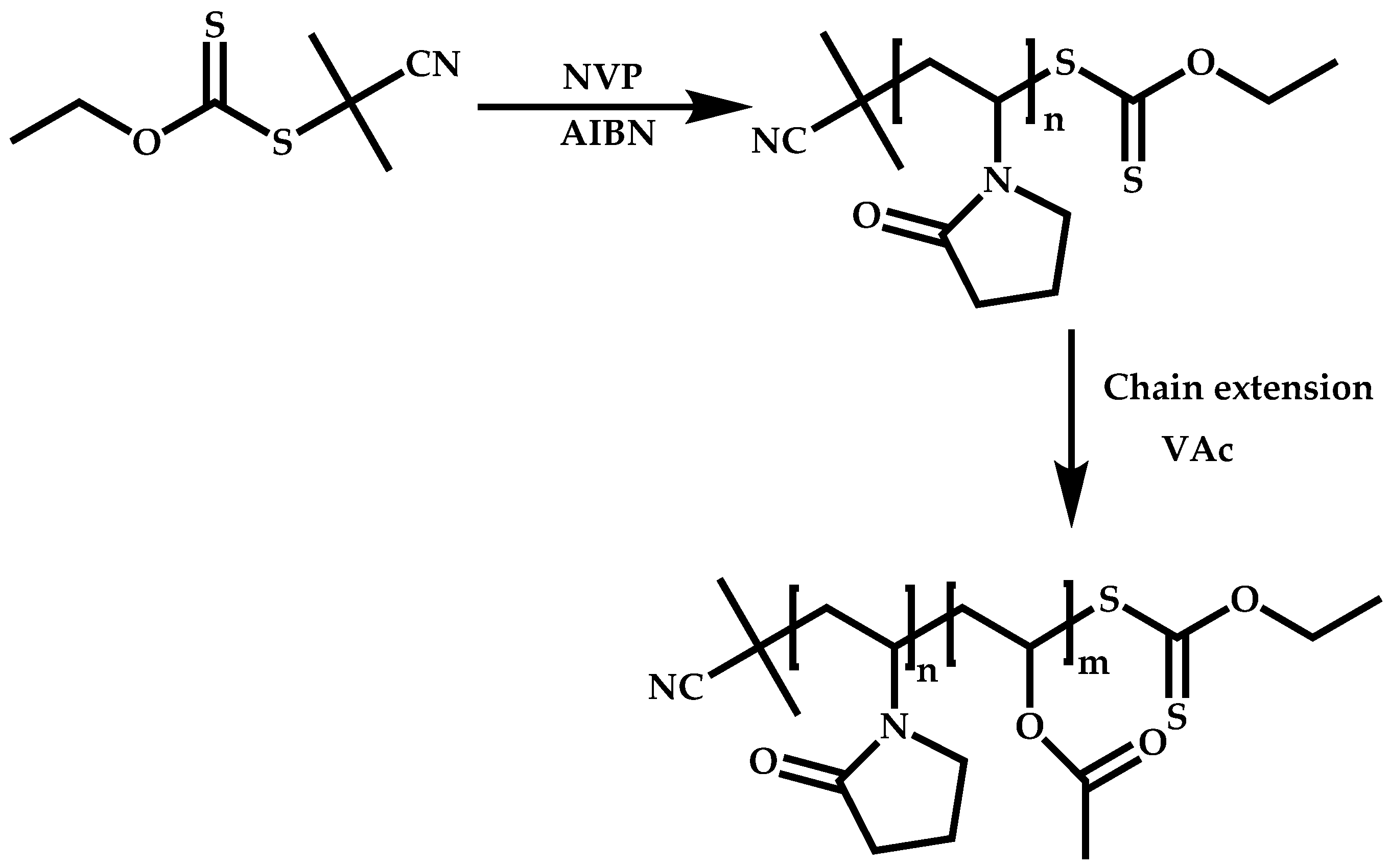
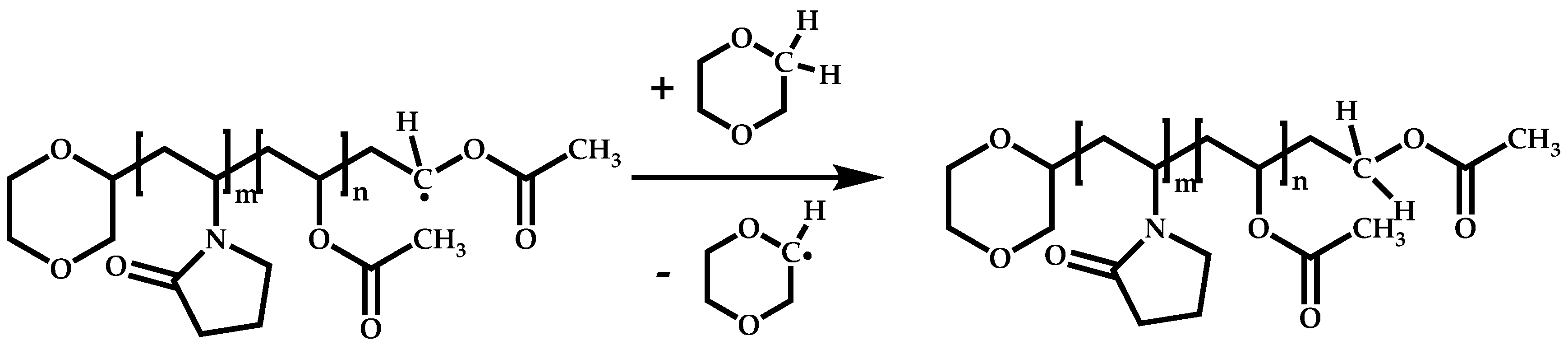
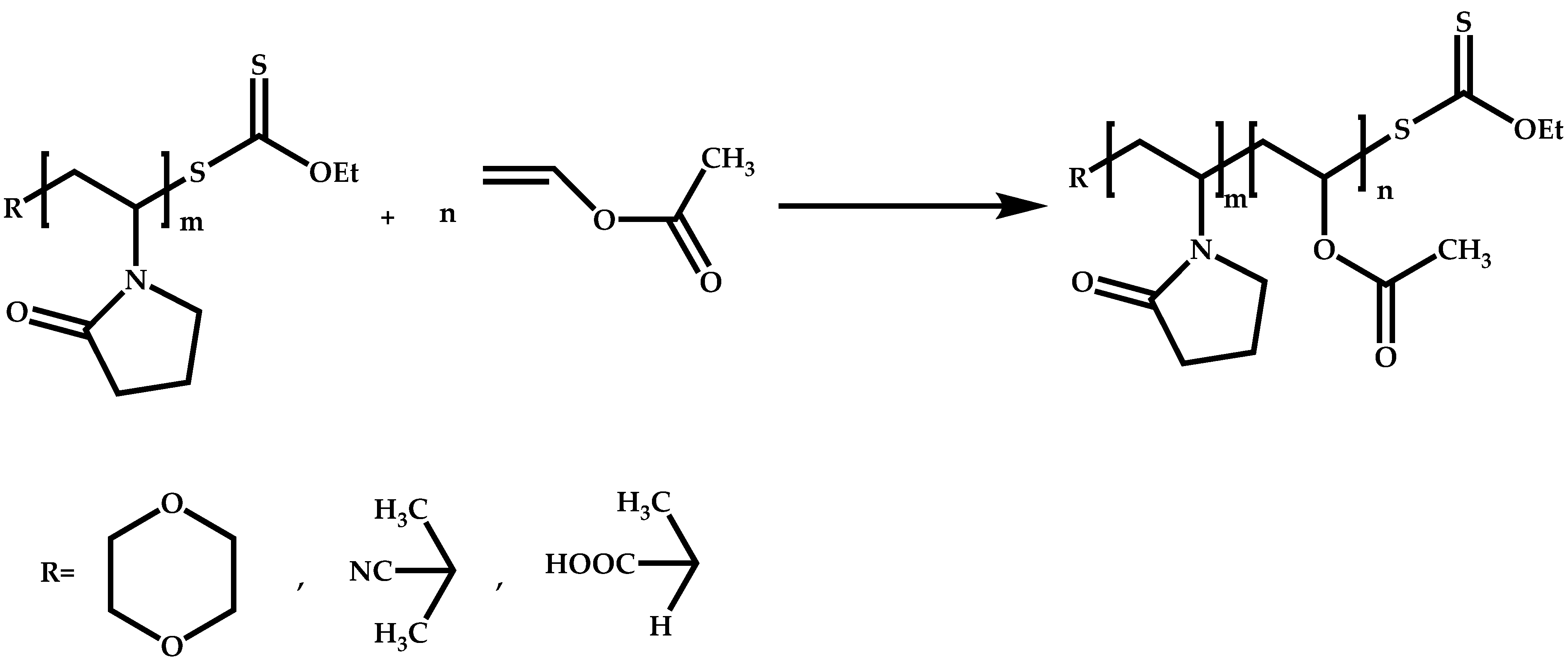
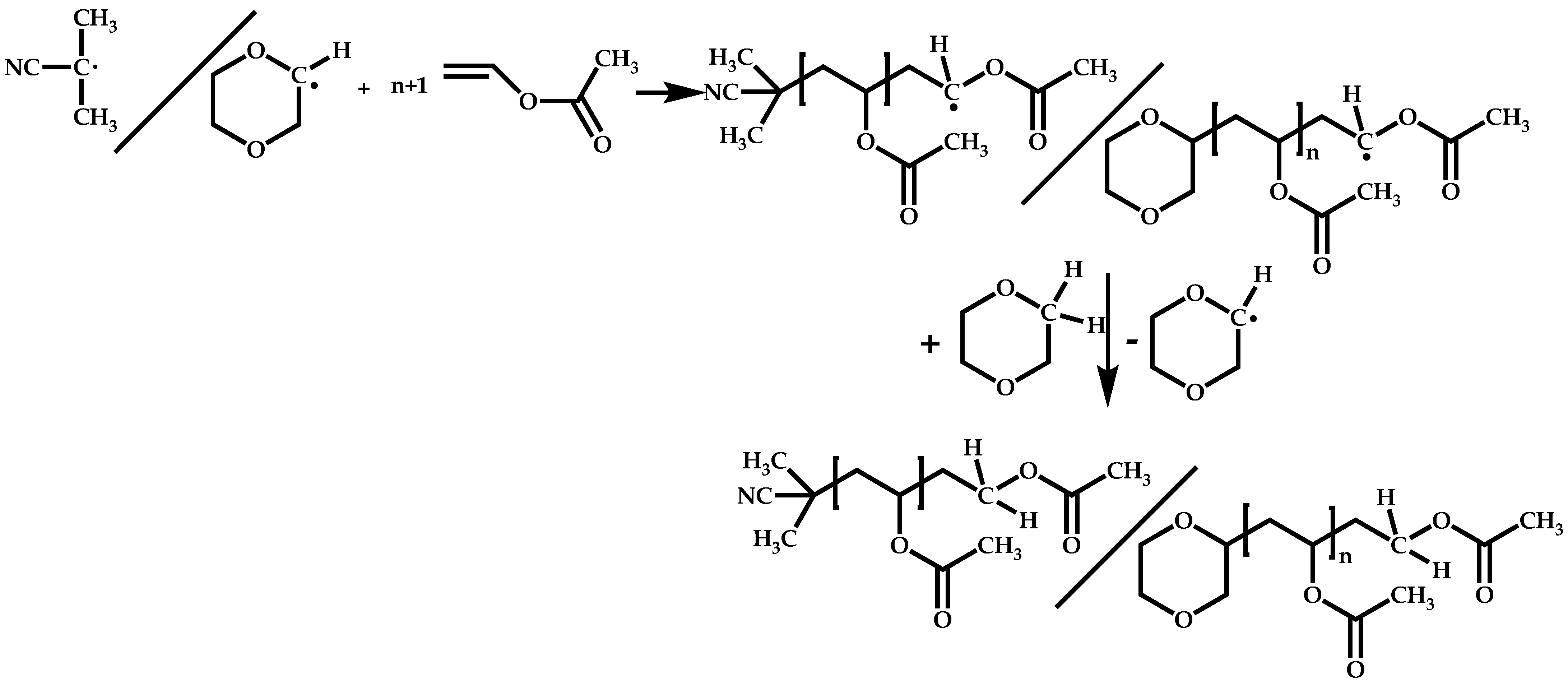
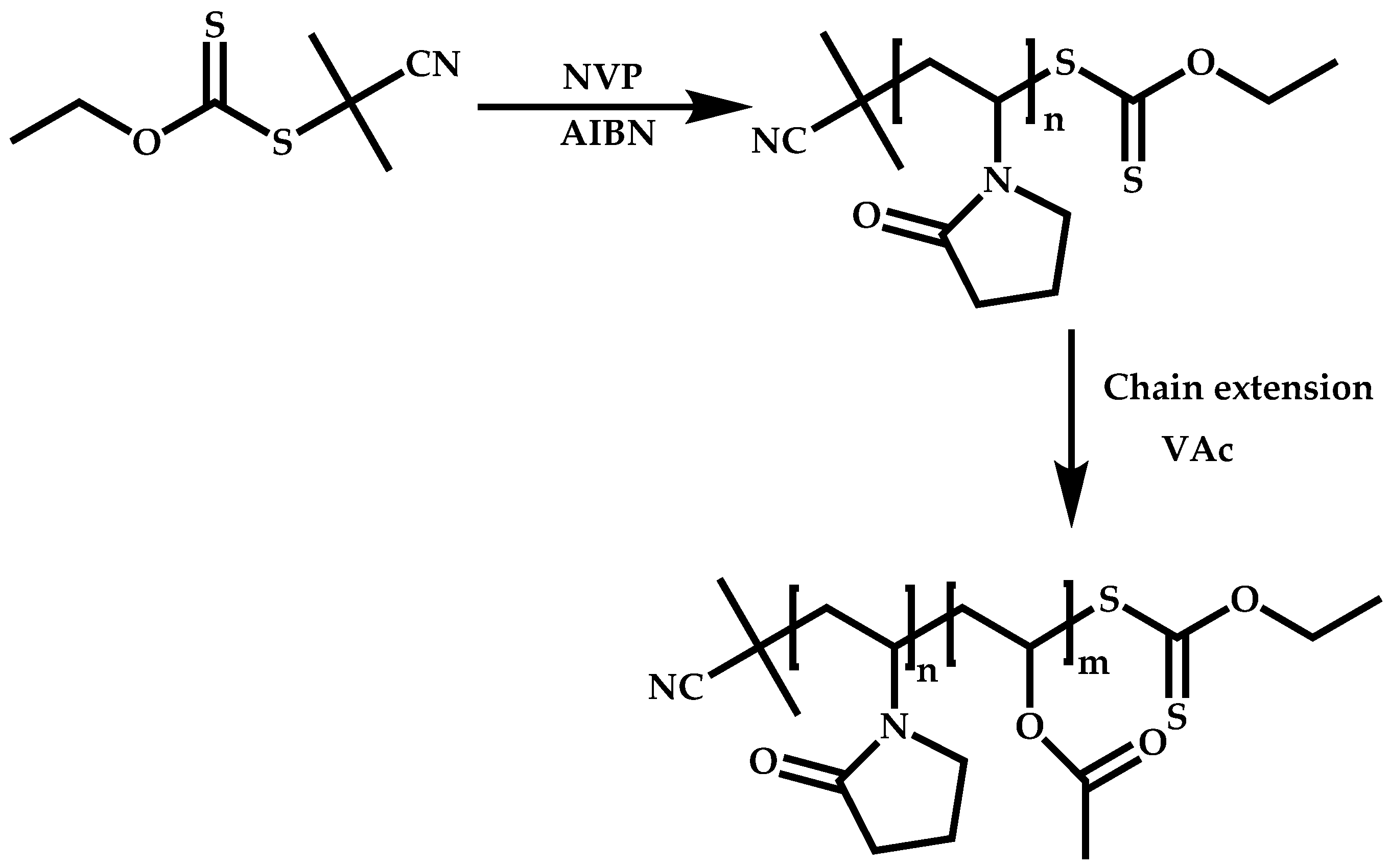
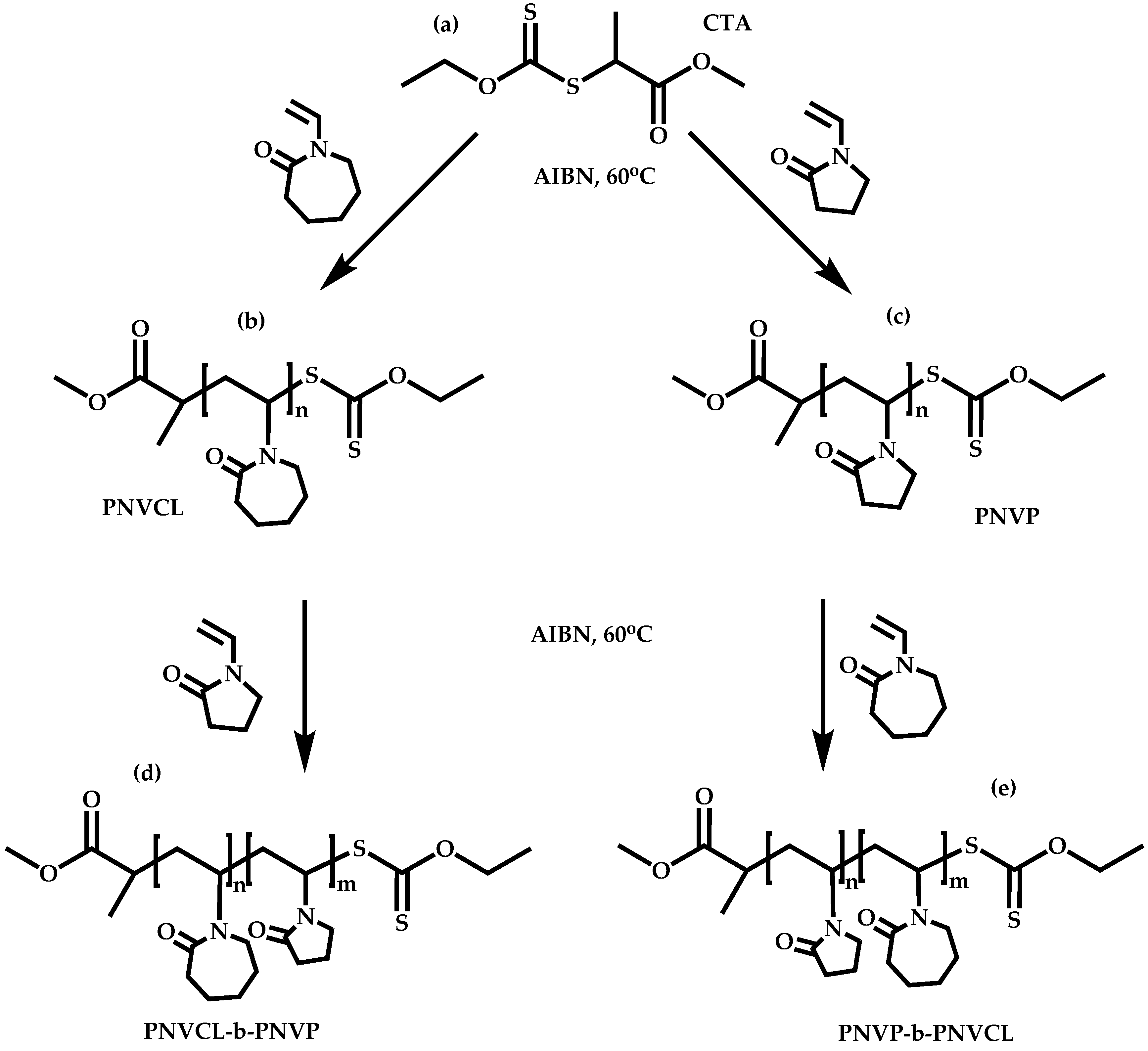
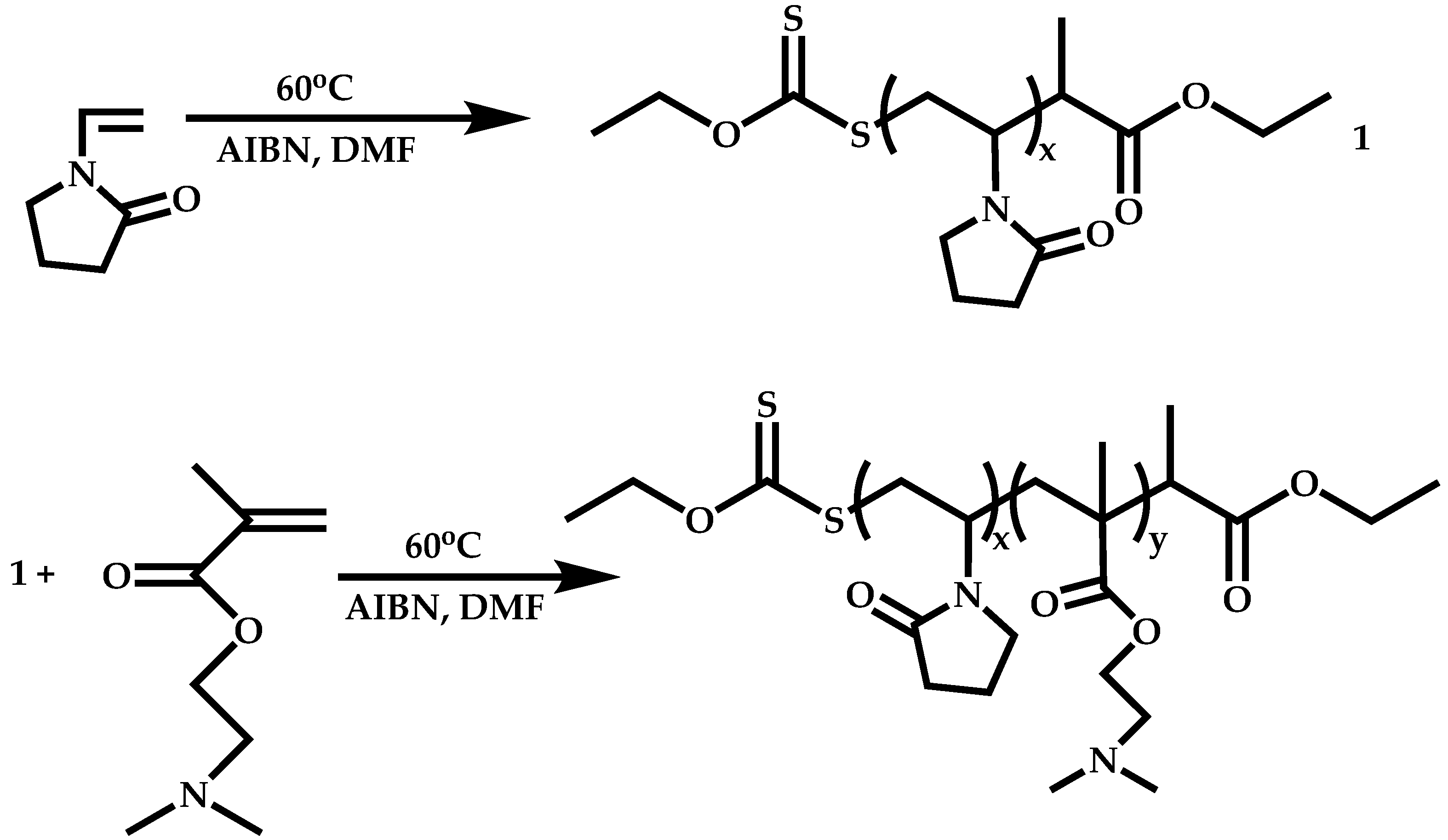
4.2.2. Sequential Addition of Monomers
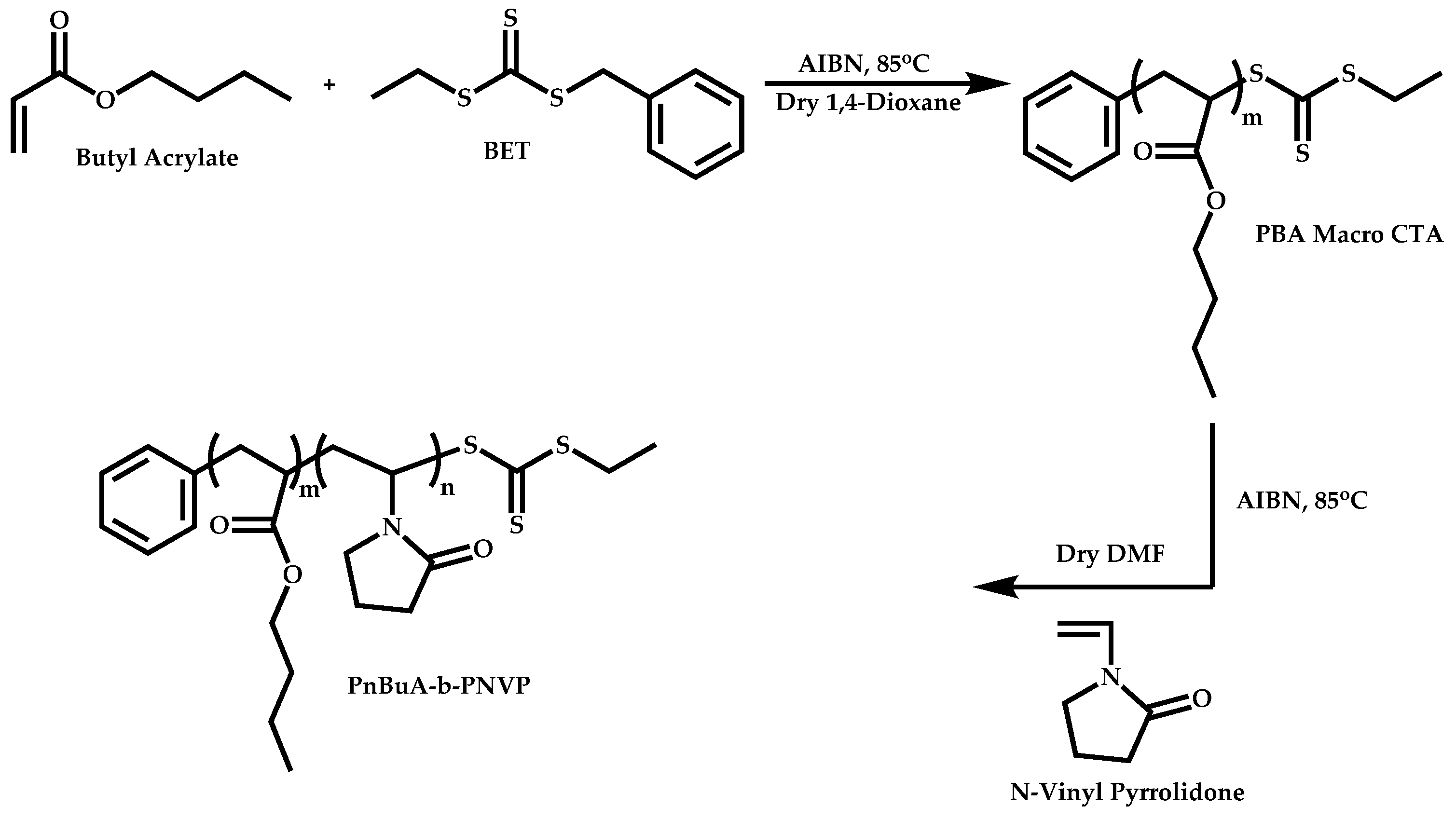
Block Copolymers with Low Activated Monomers (LAMs)
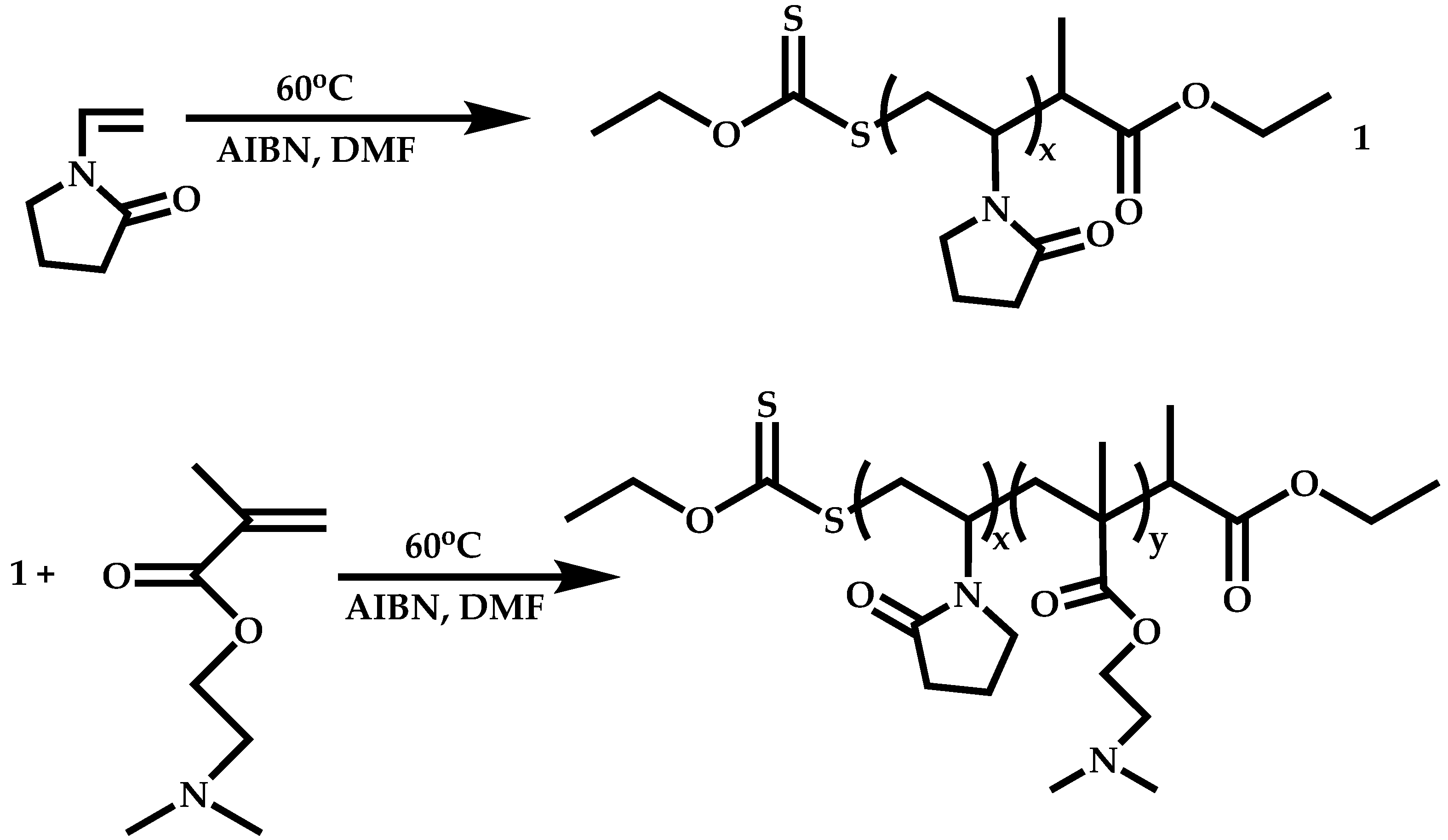
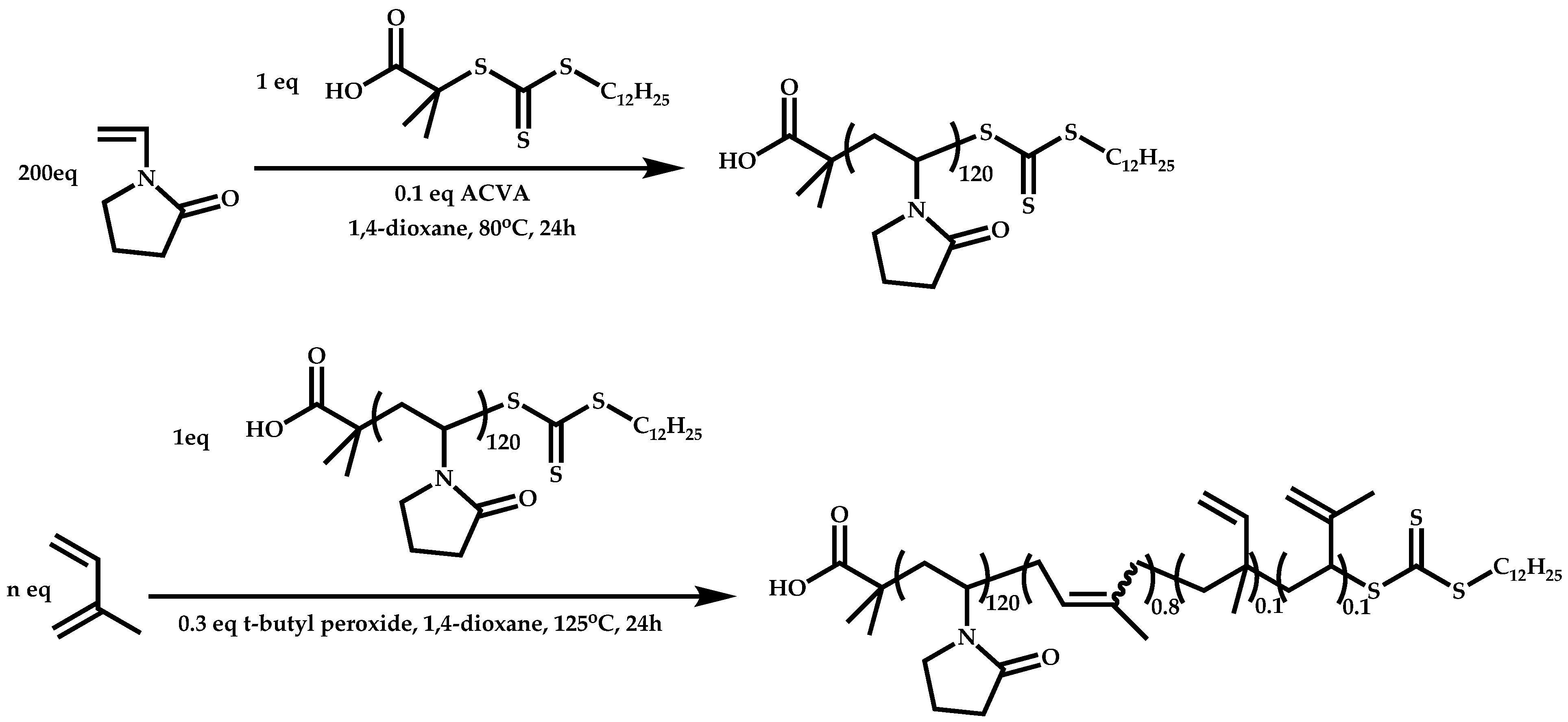
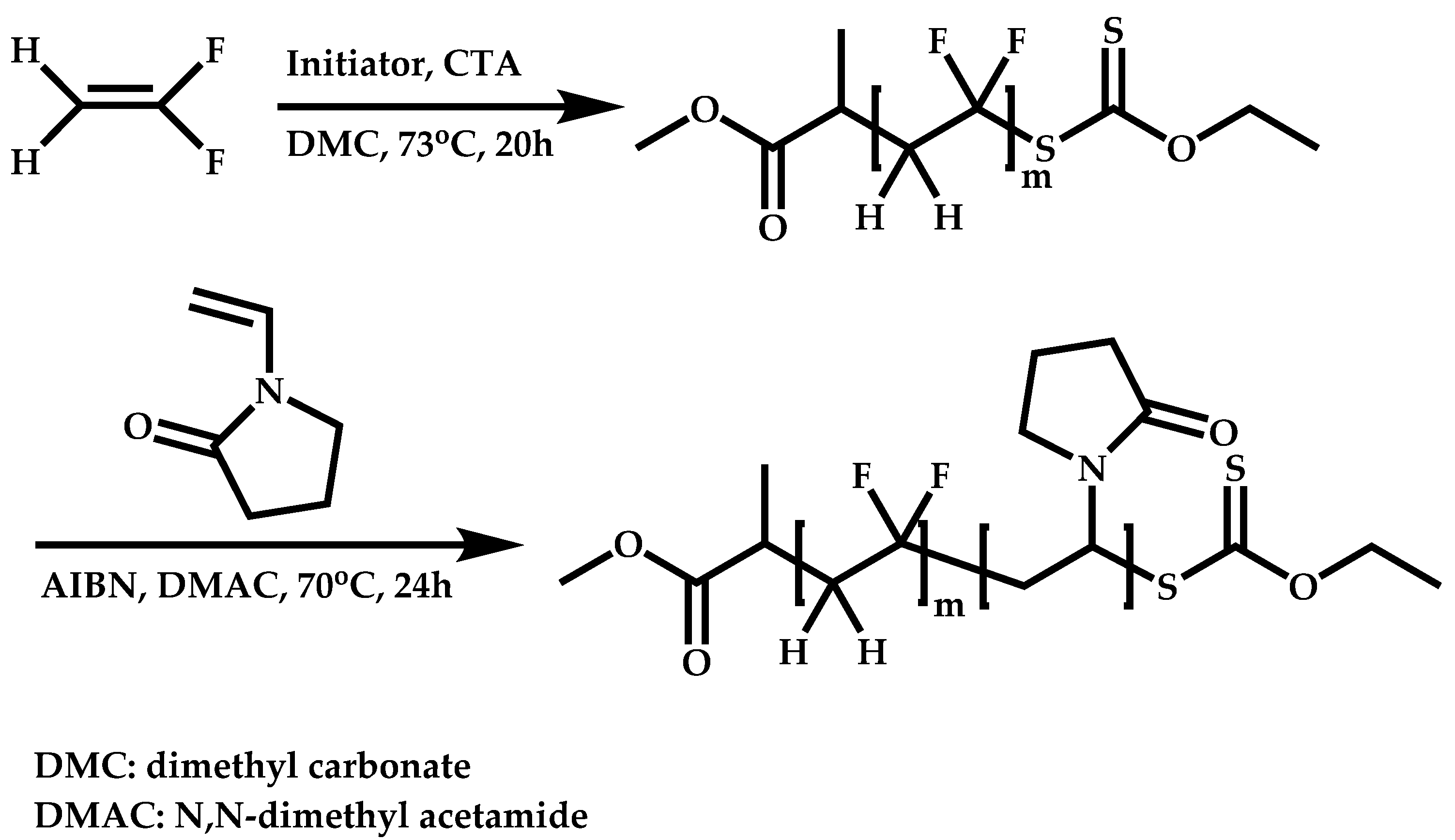
Block Copolymers with More-Activated Monomers (MAMs)
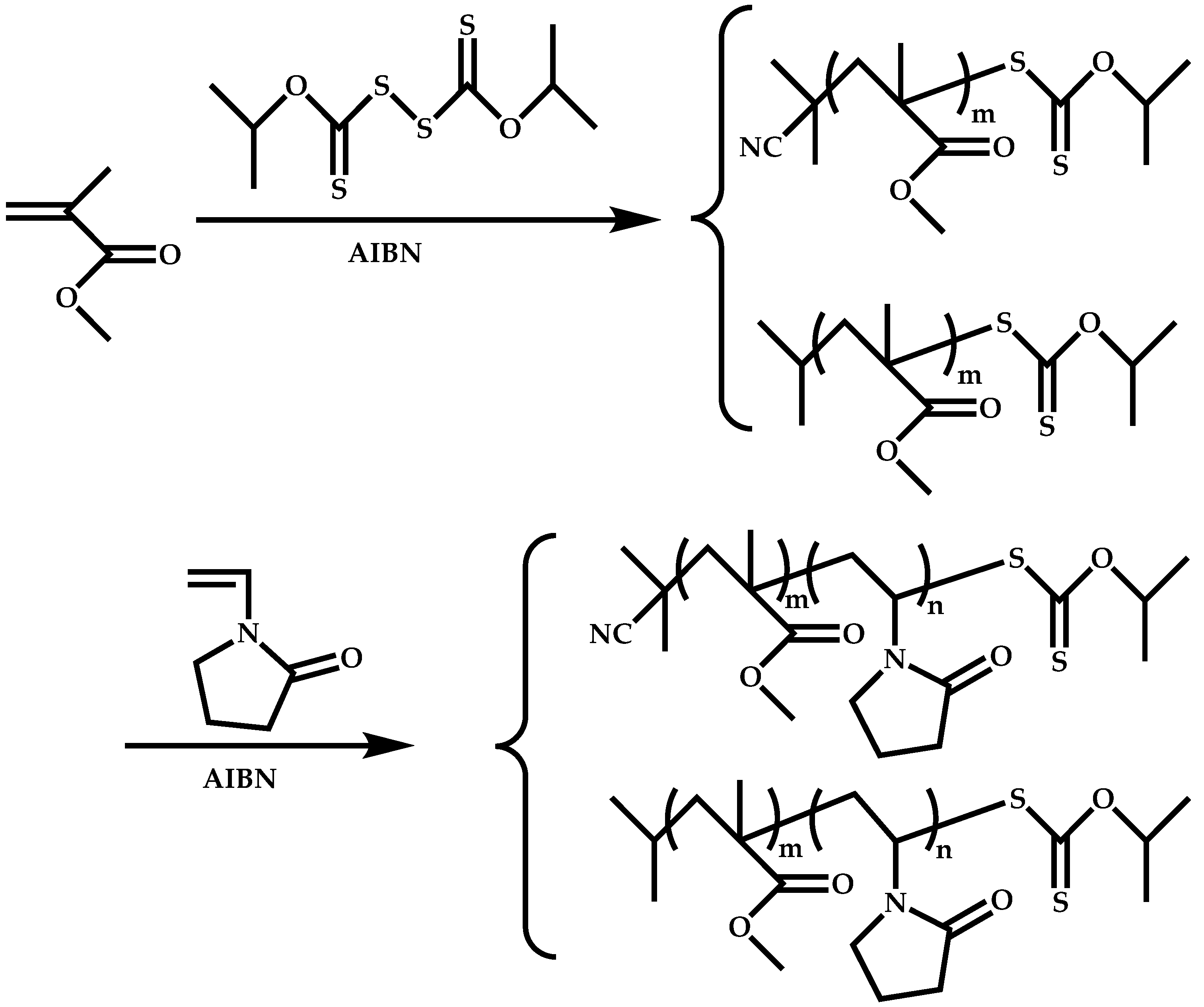
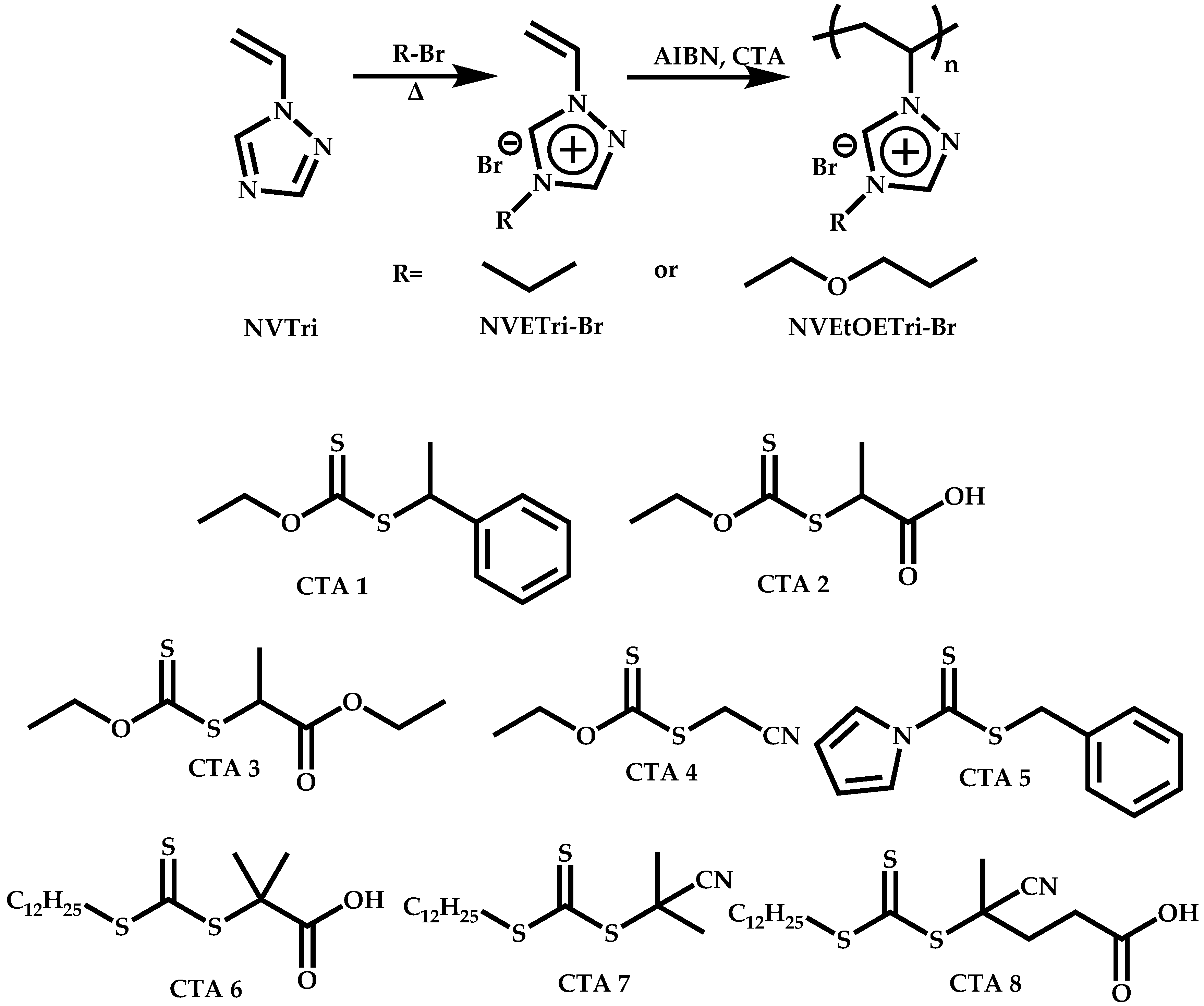
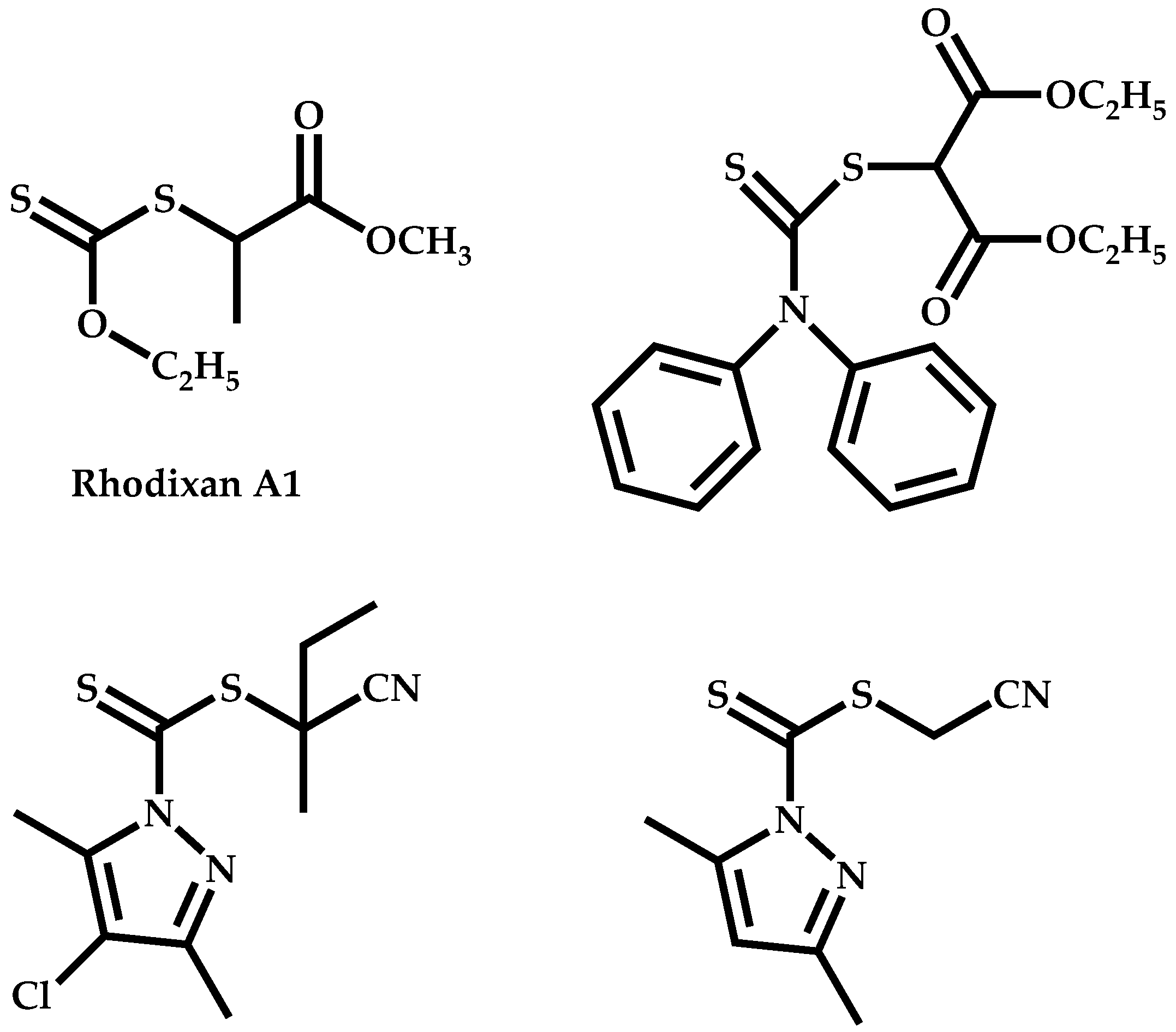
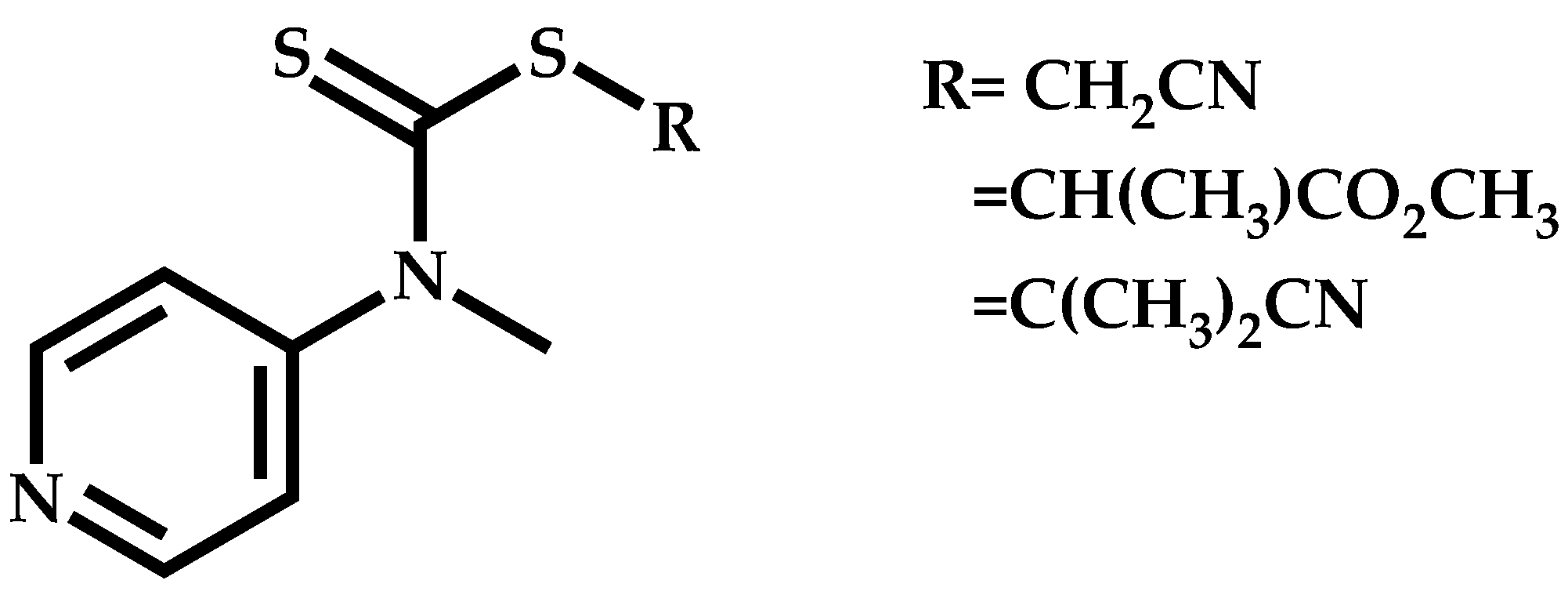
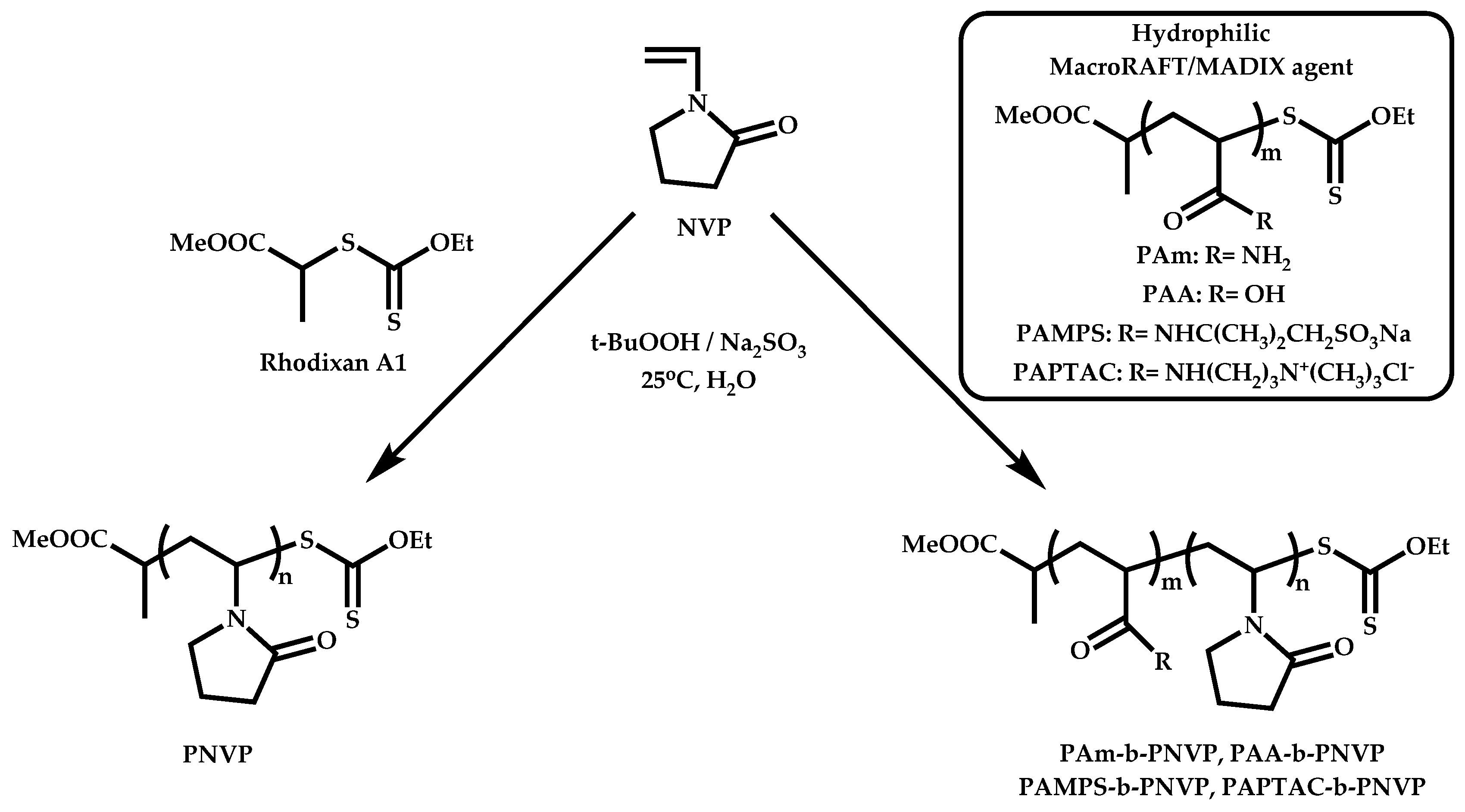
Employment of Universal CTAs
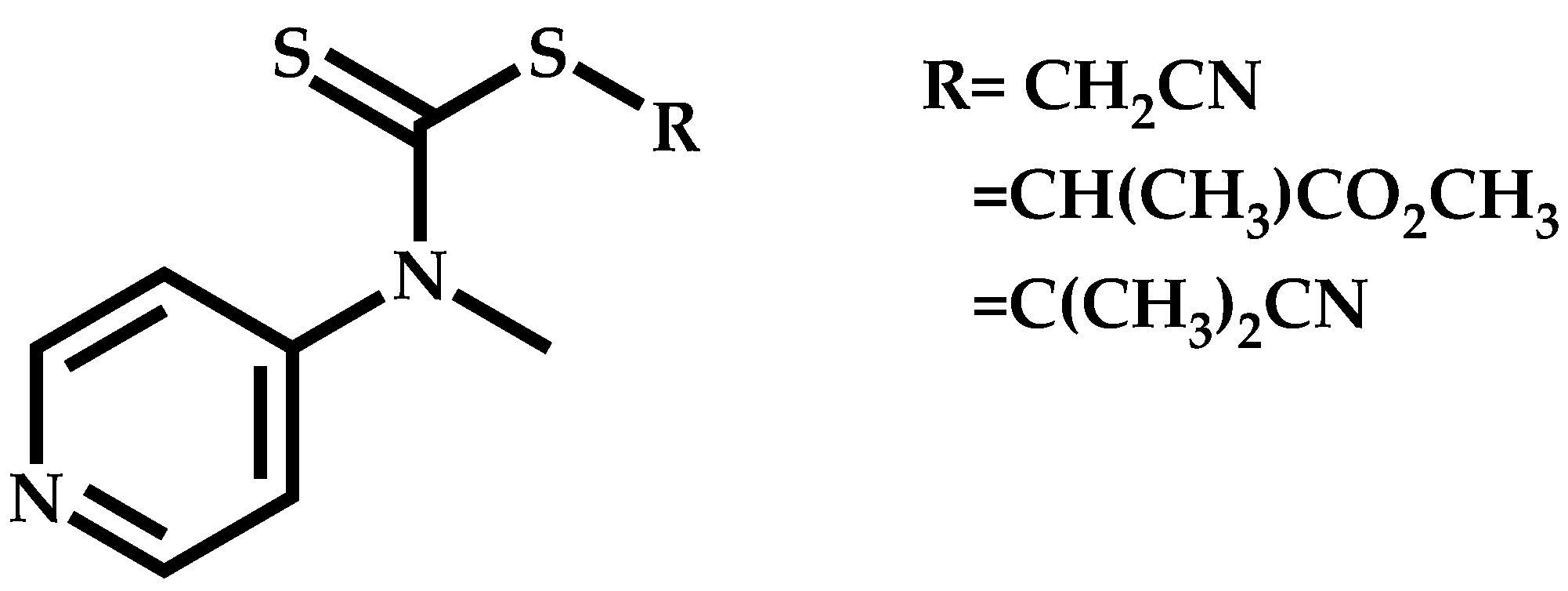
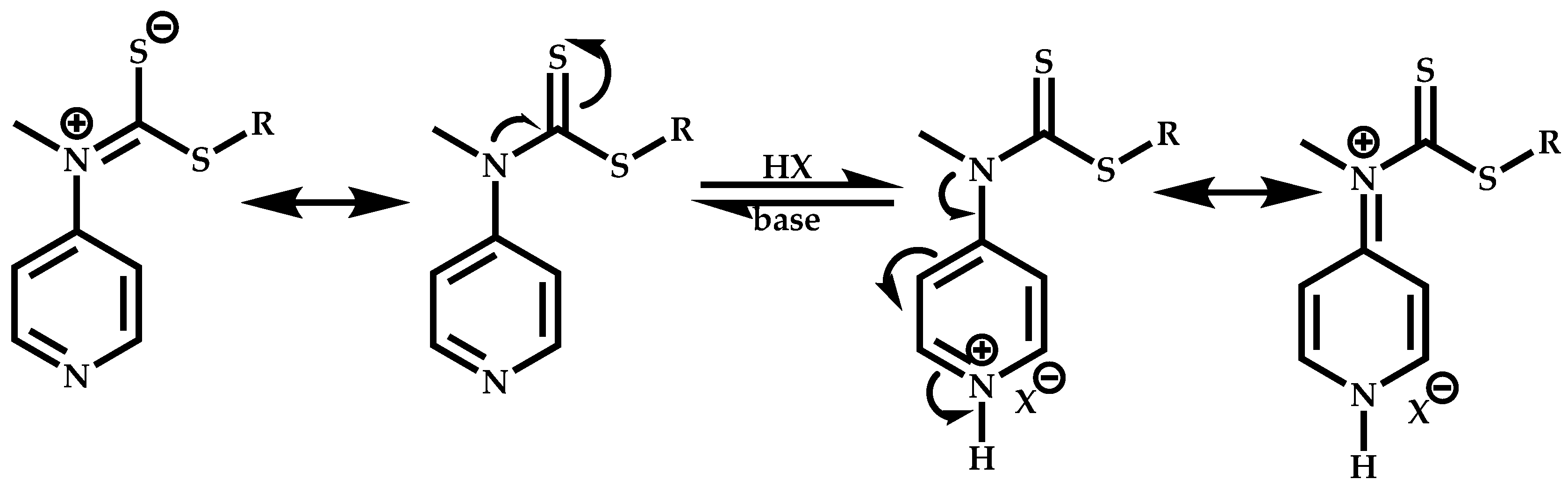
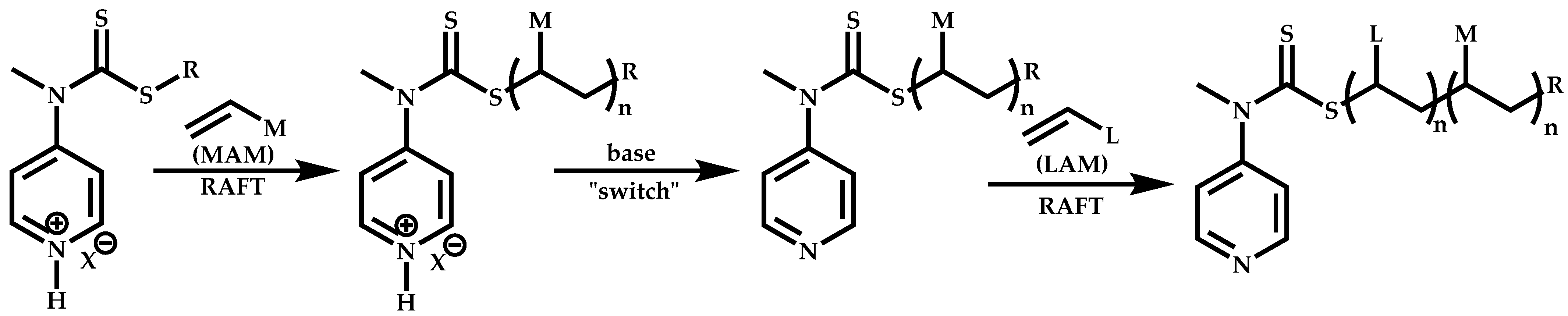
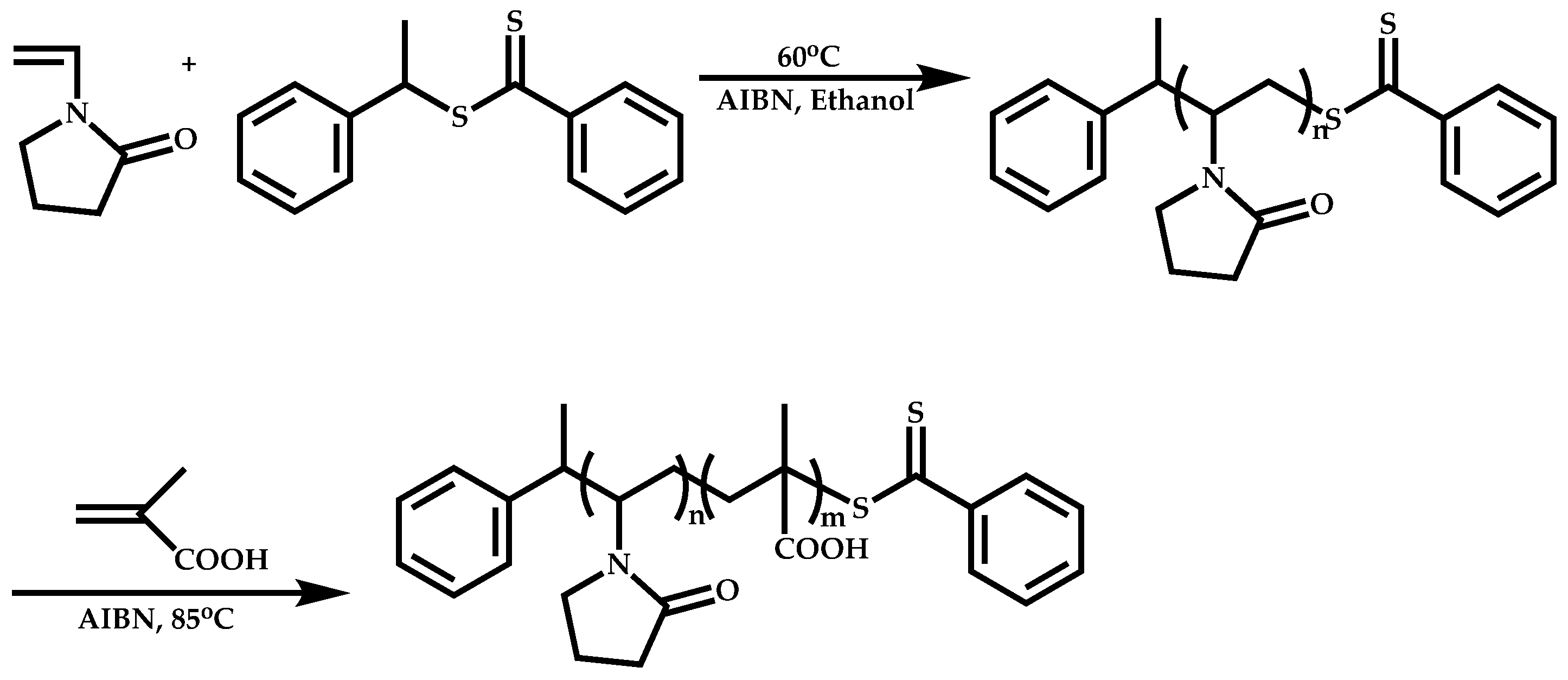
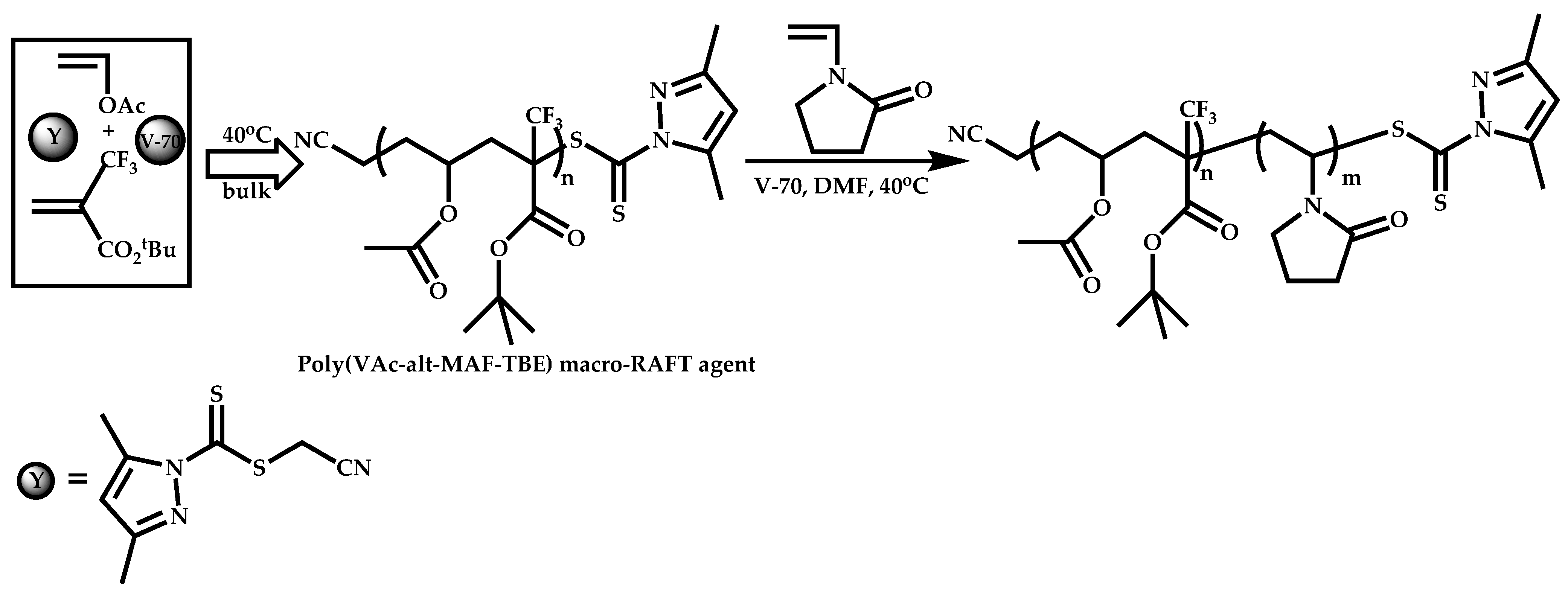
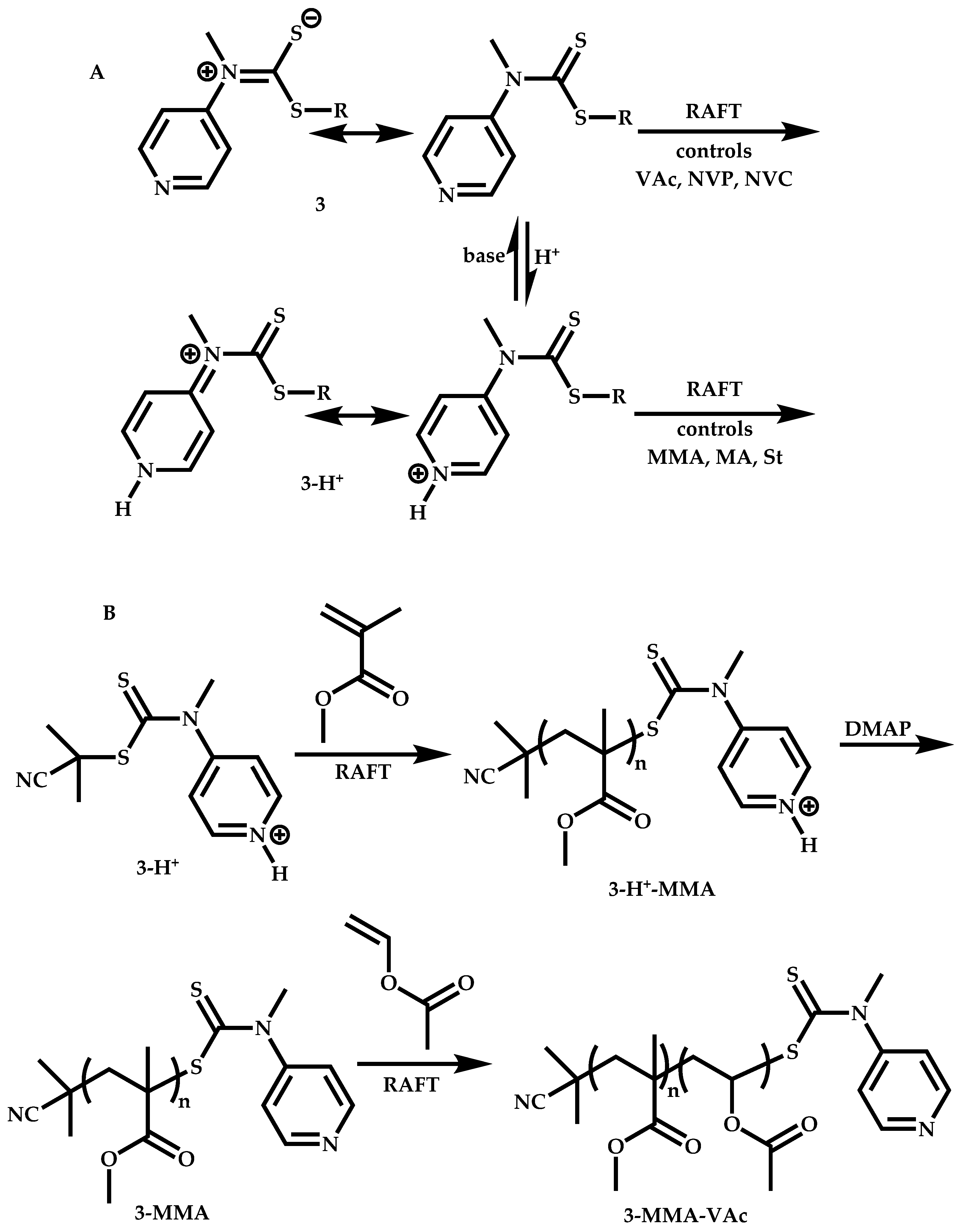
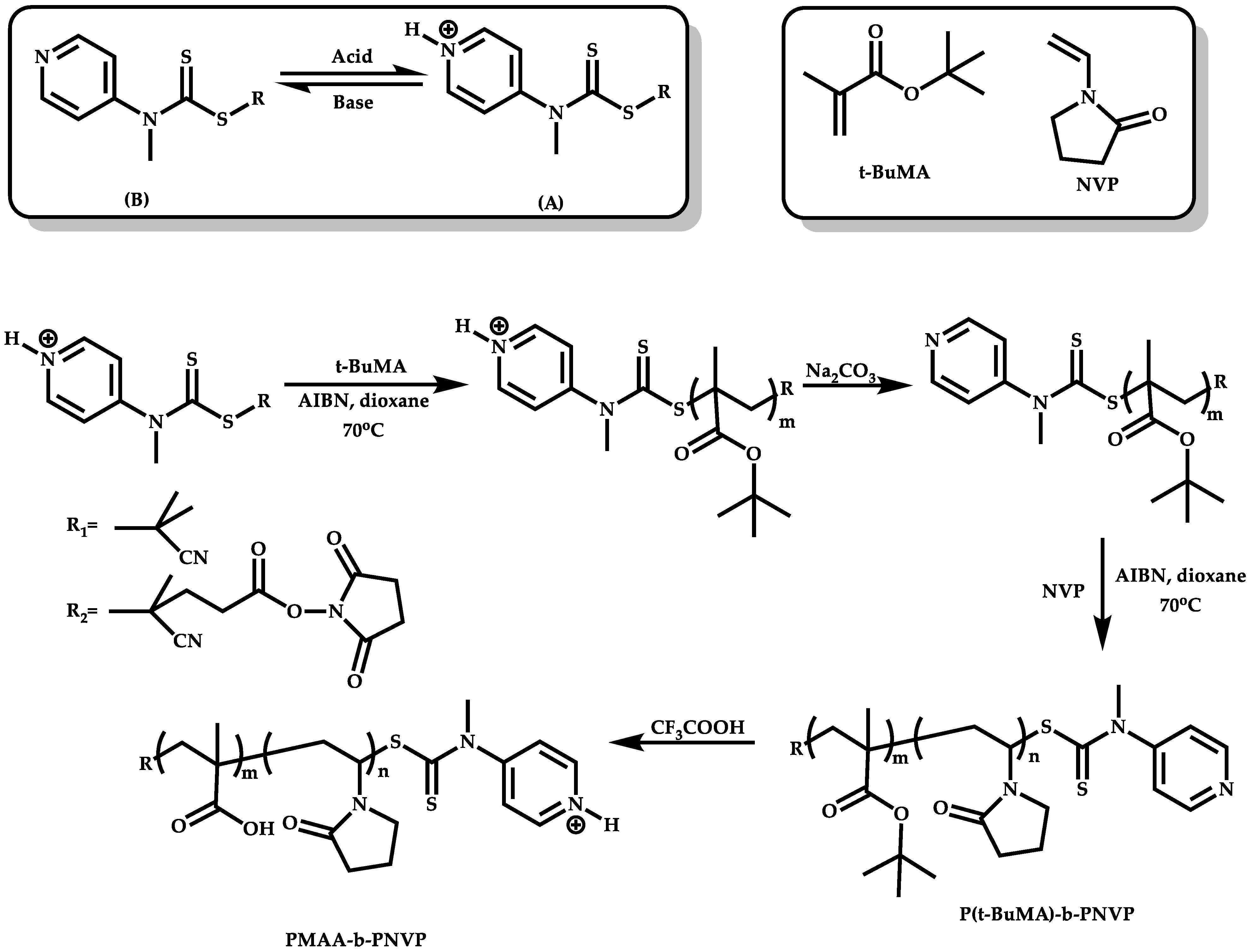
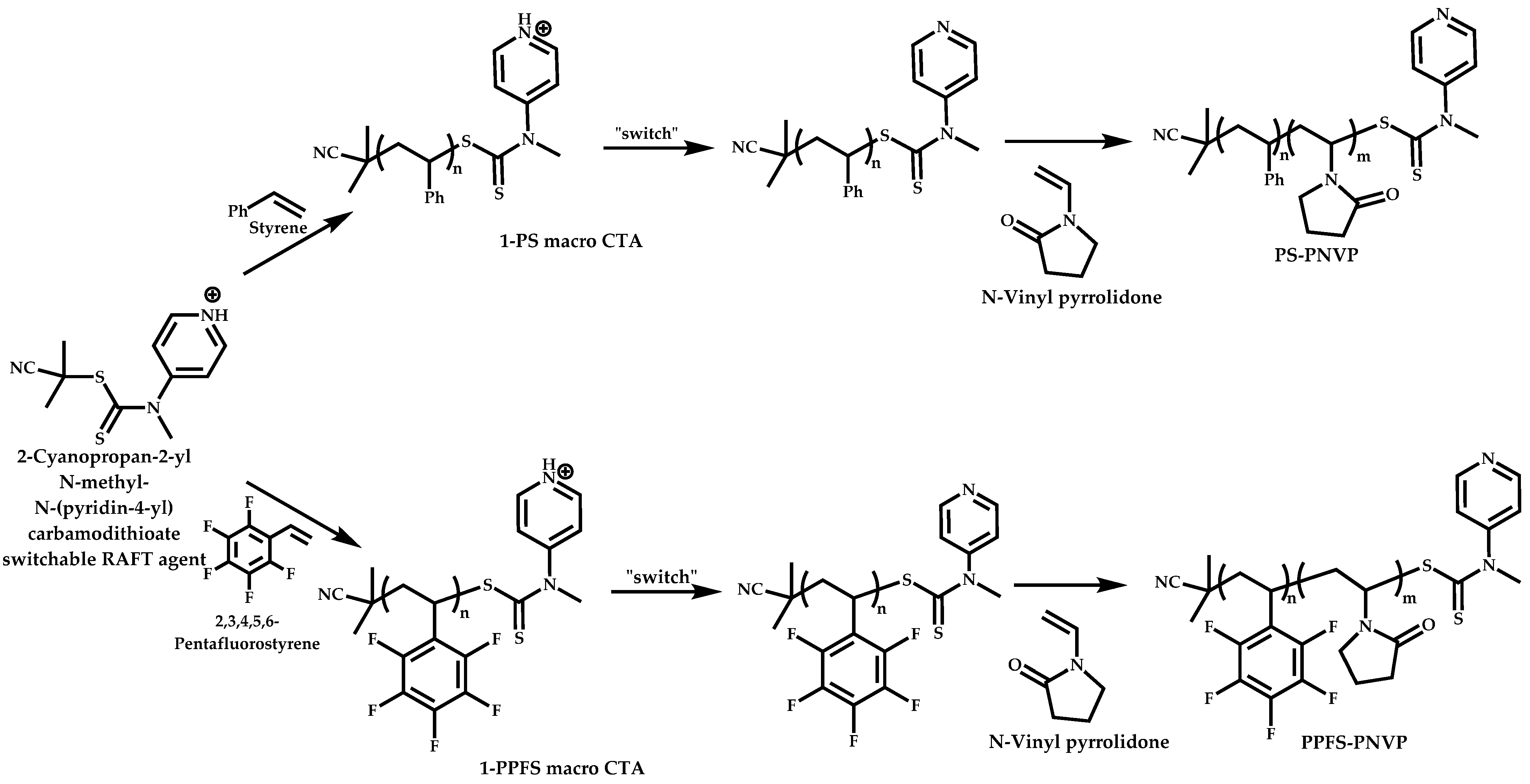
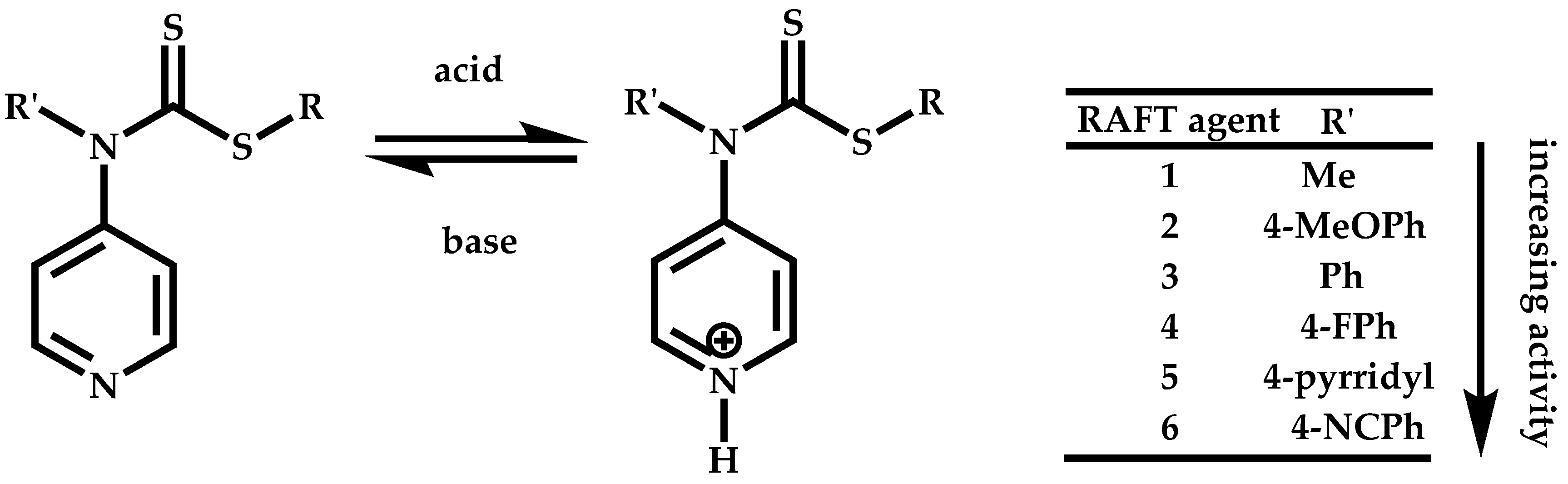
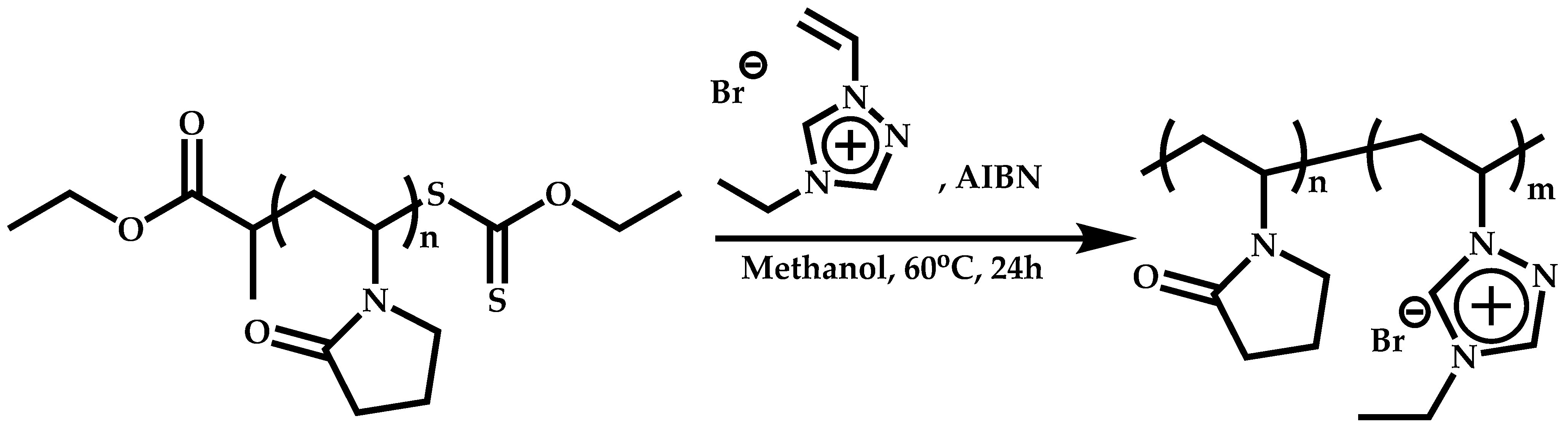
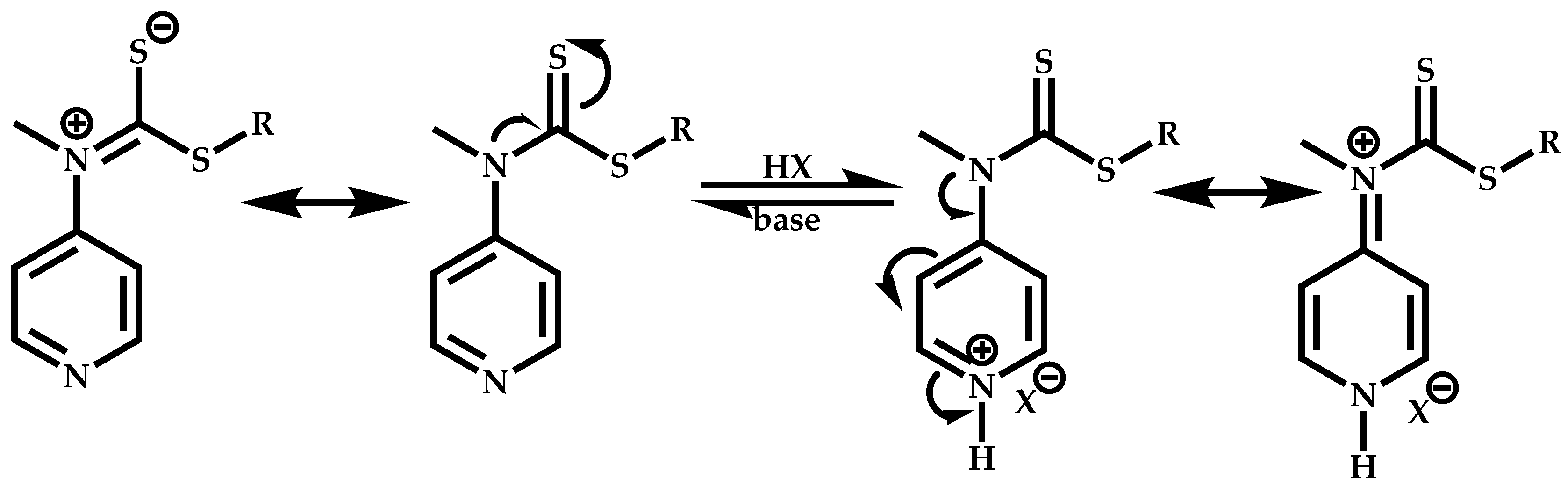
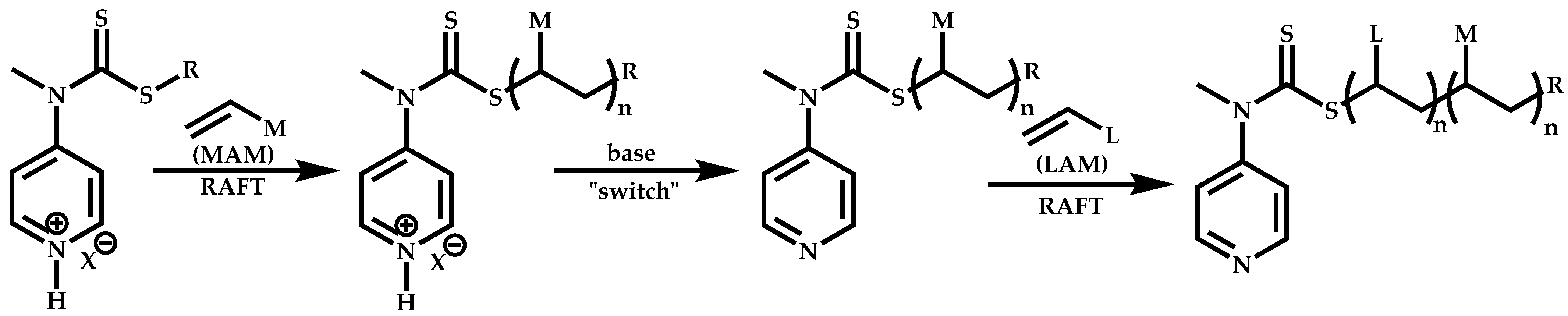
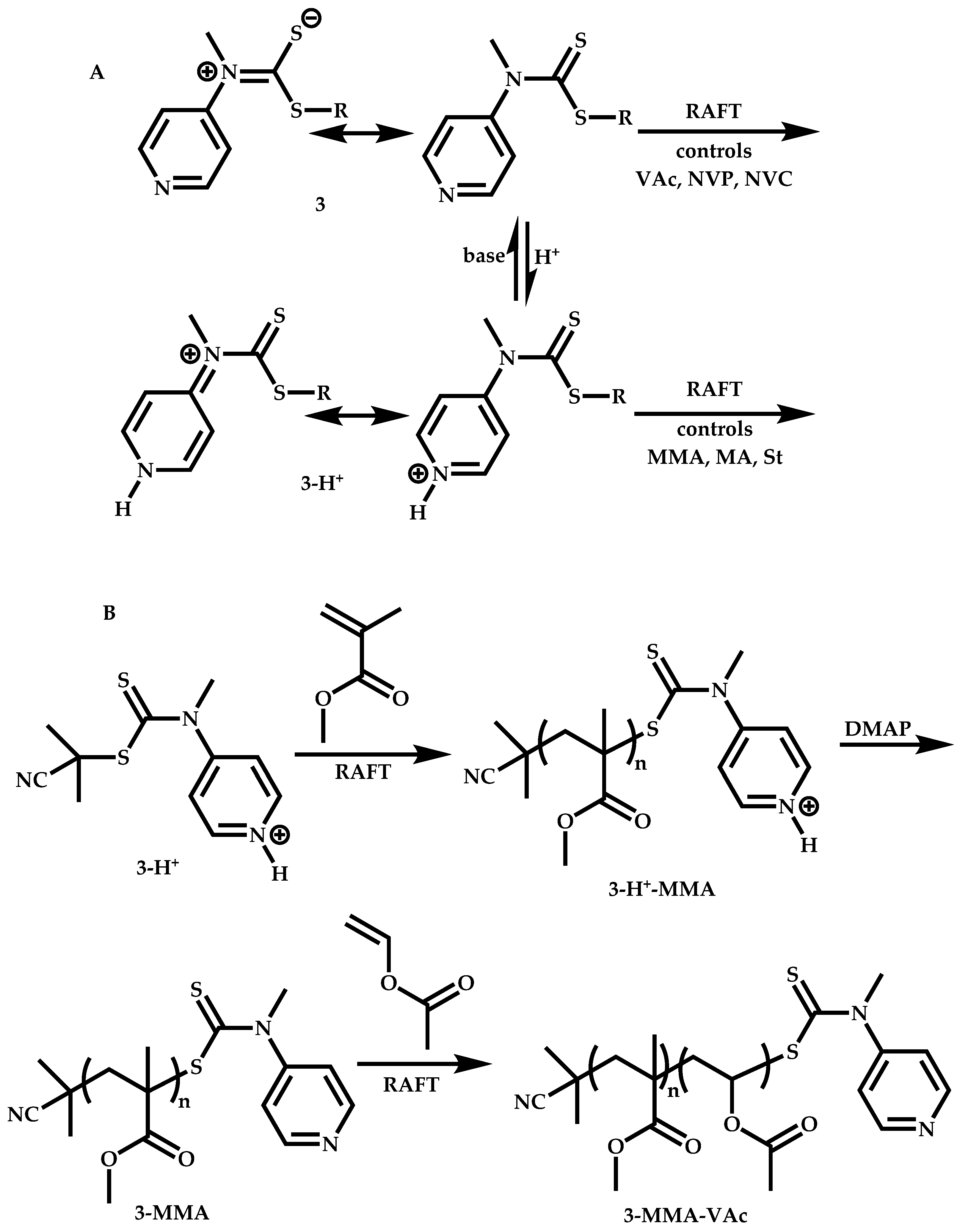
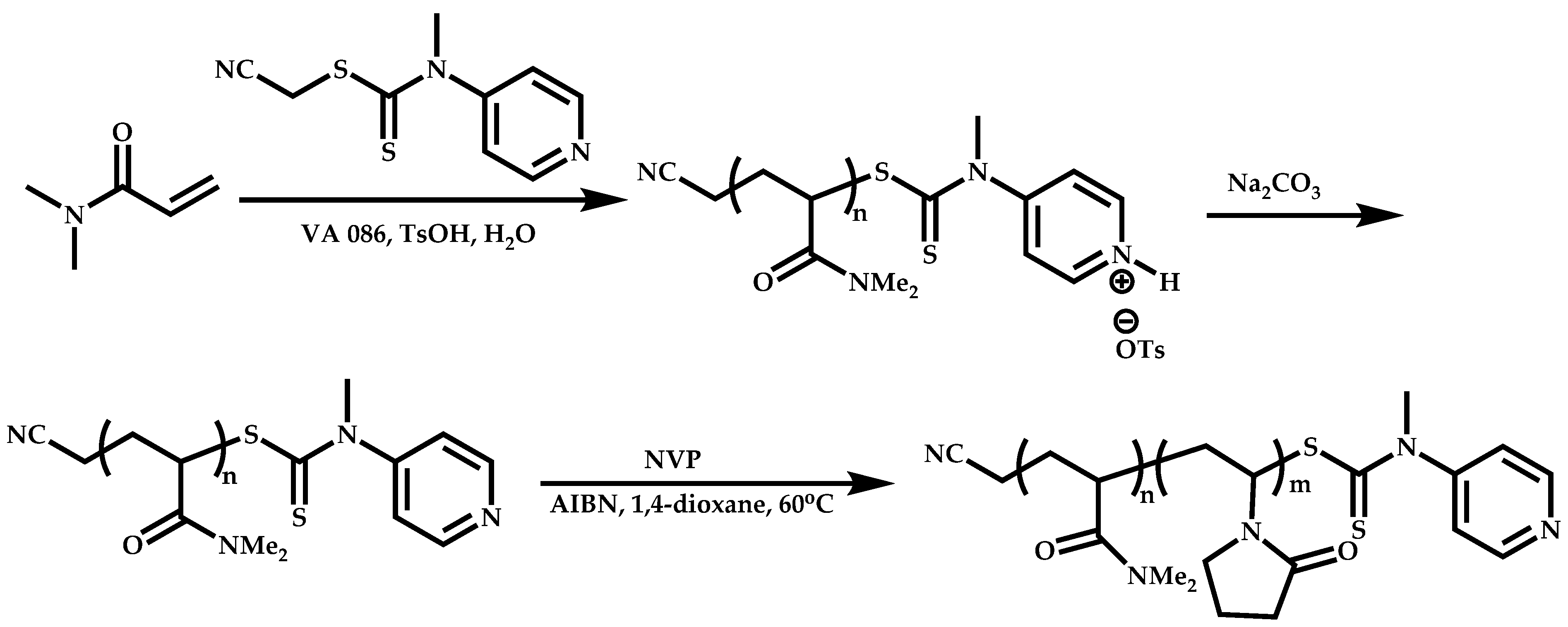
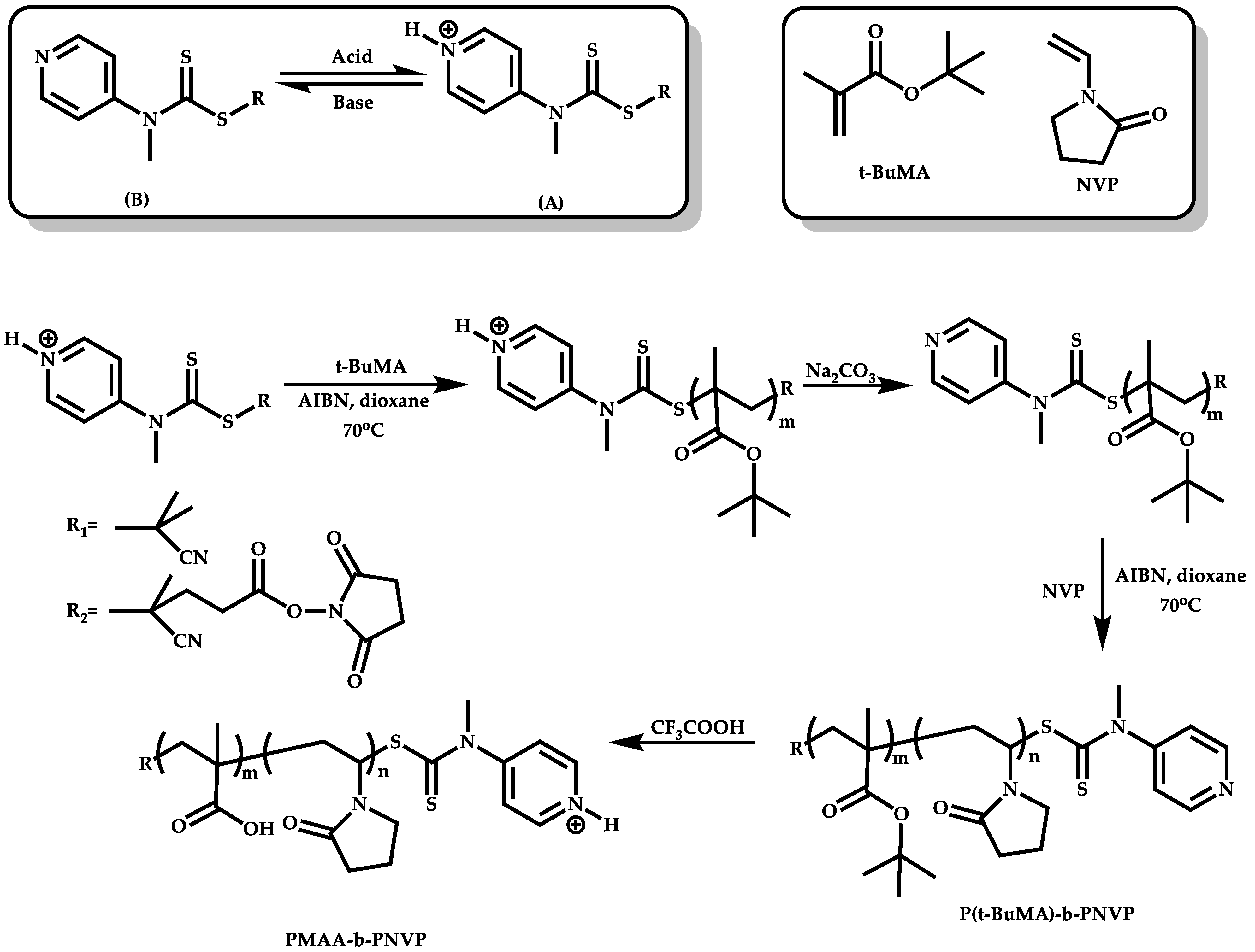
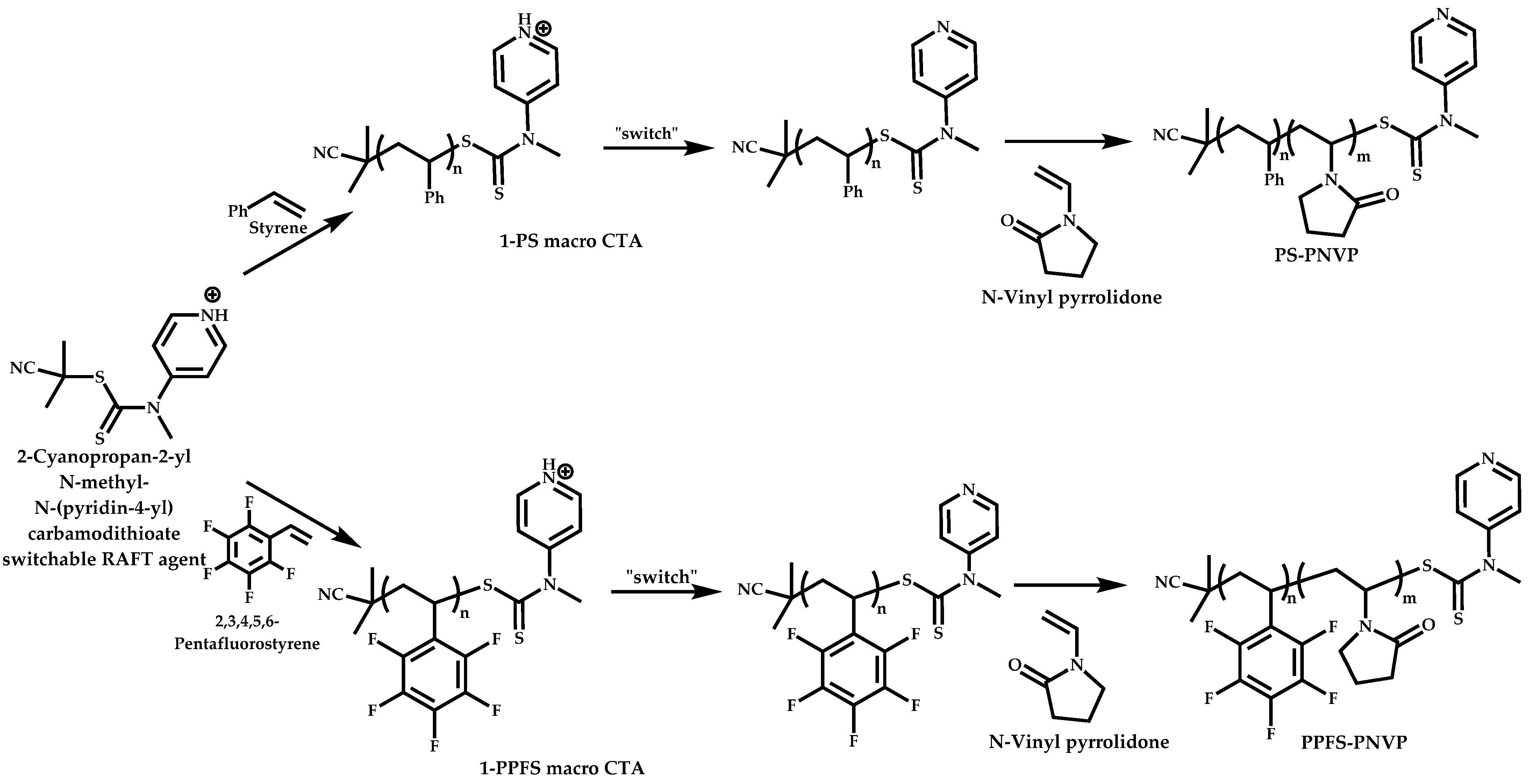
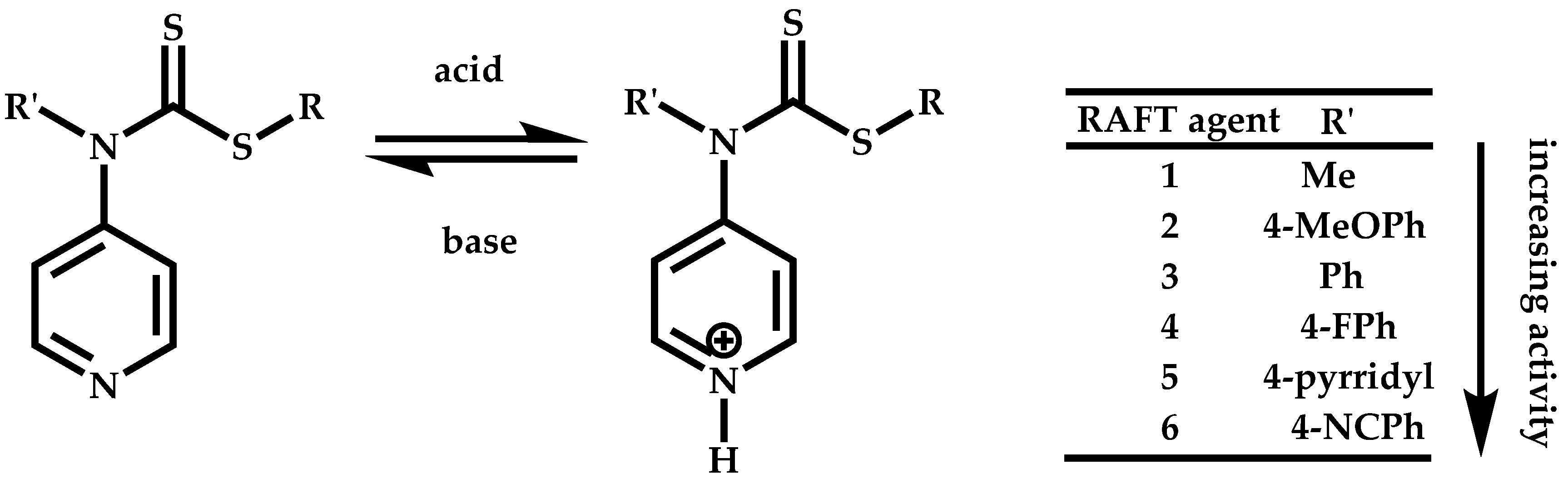
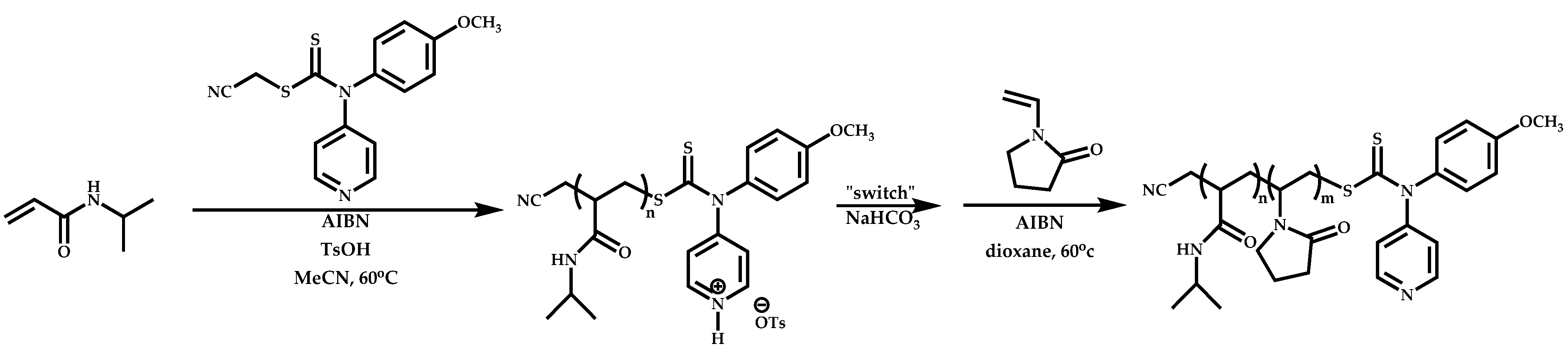
Employment of Switchable CTAs
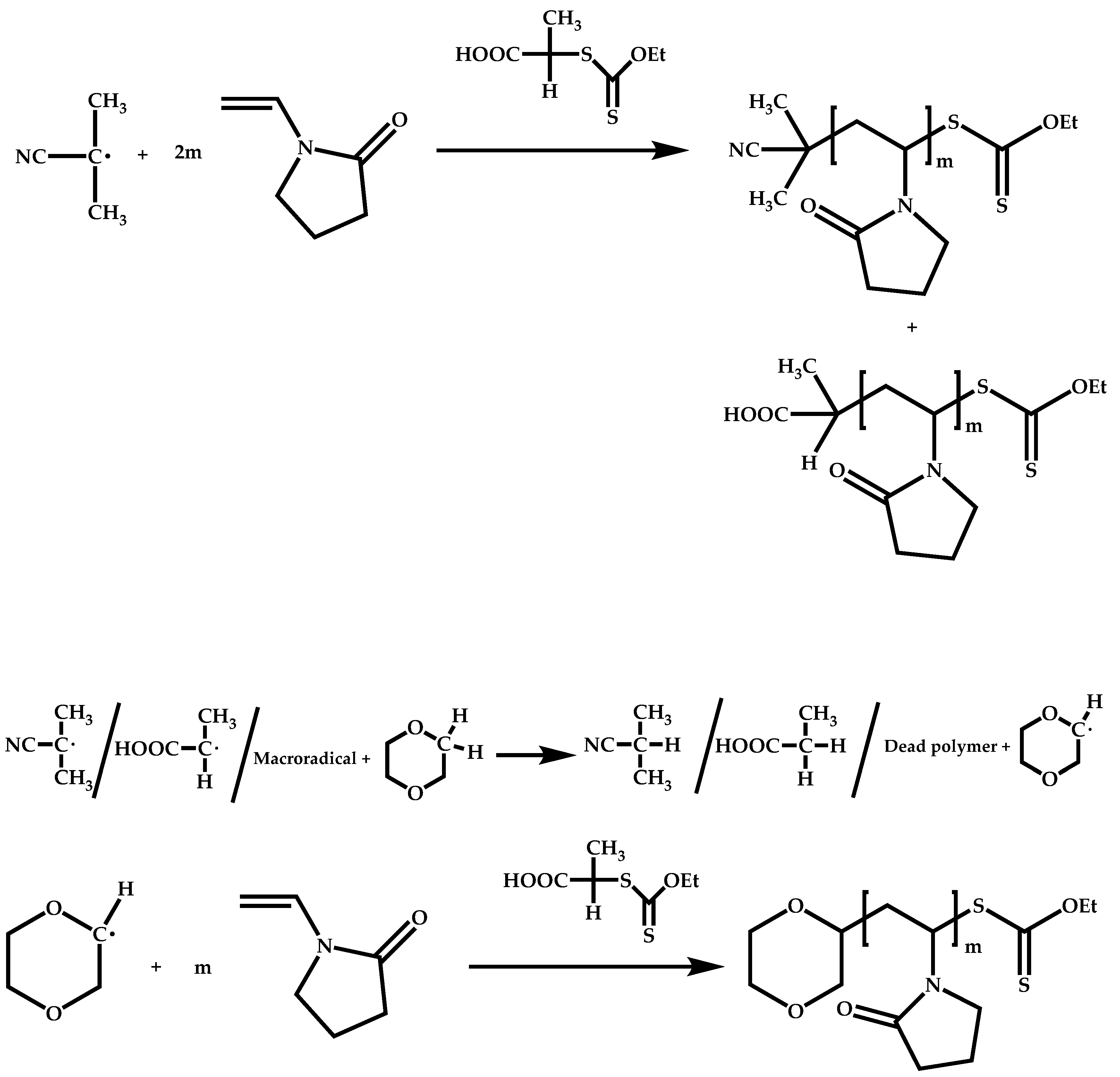
4.2.3. Combination of Different Polymerization Techniques
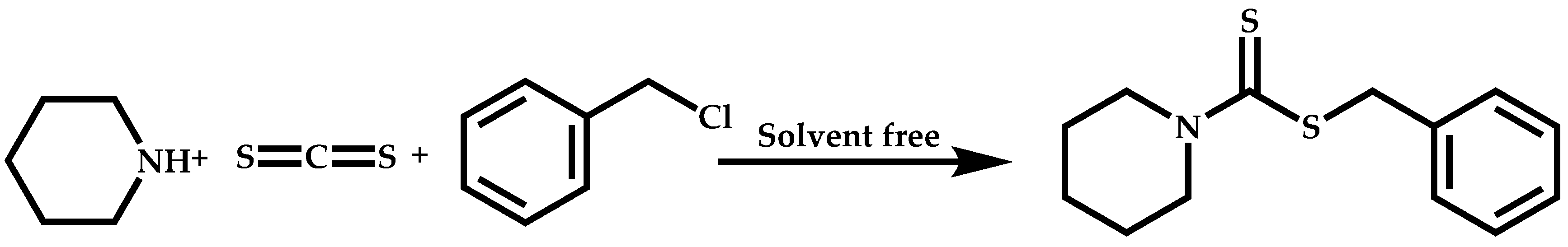
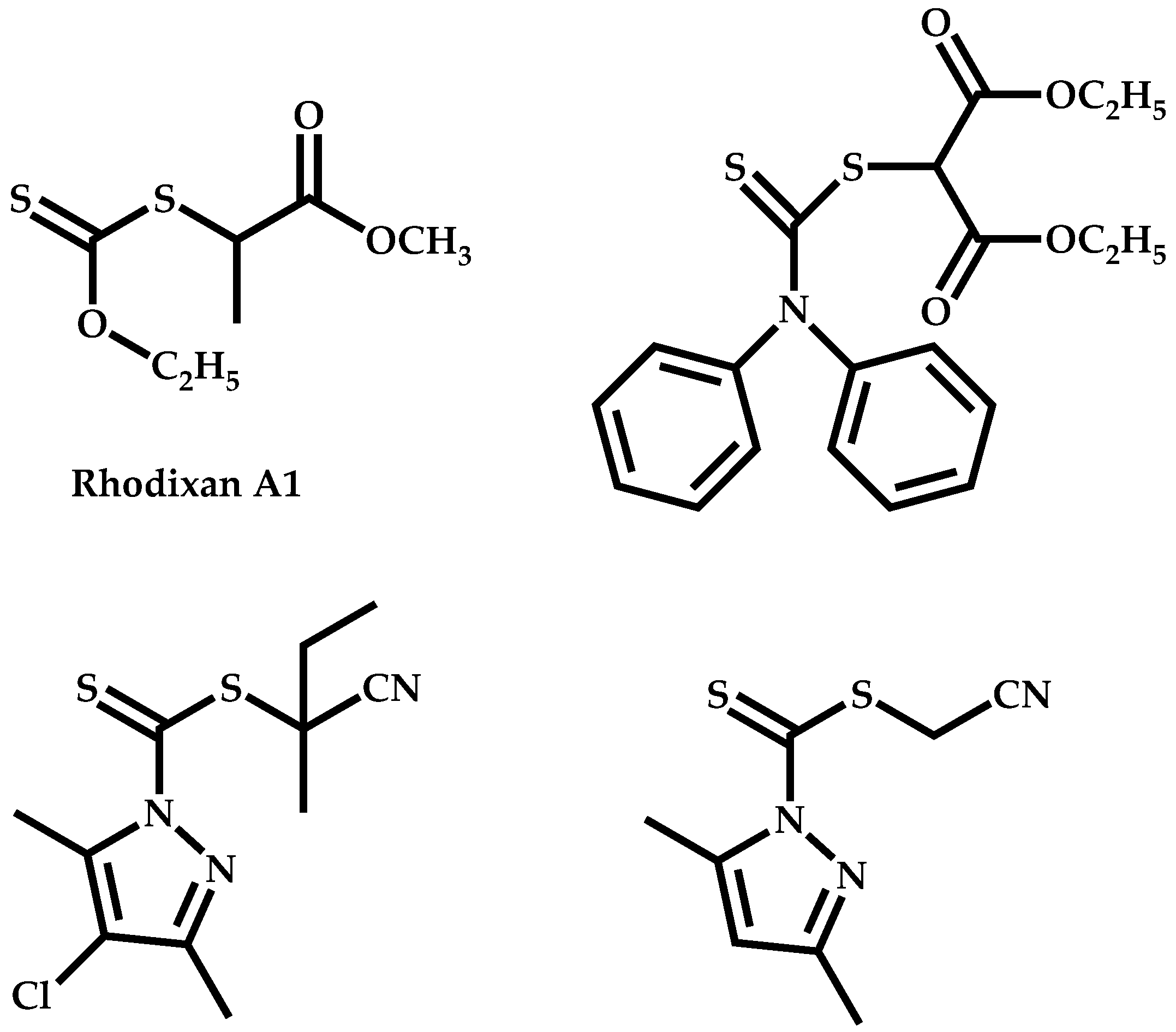
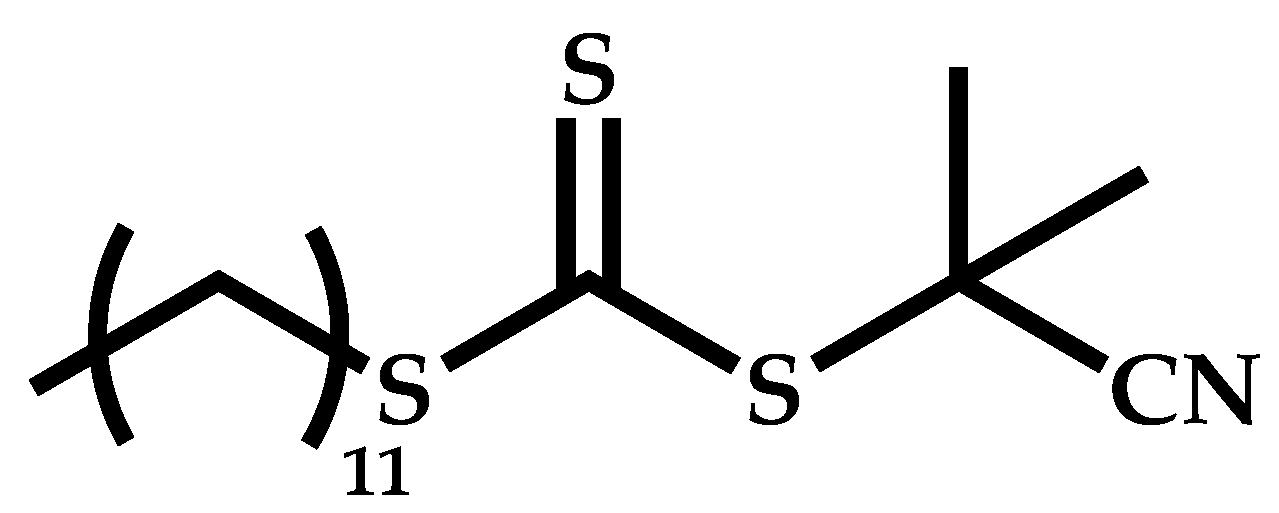
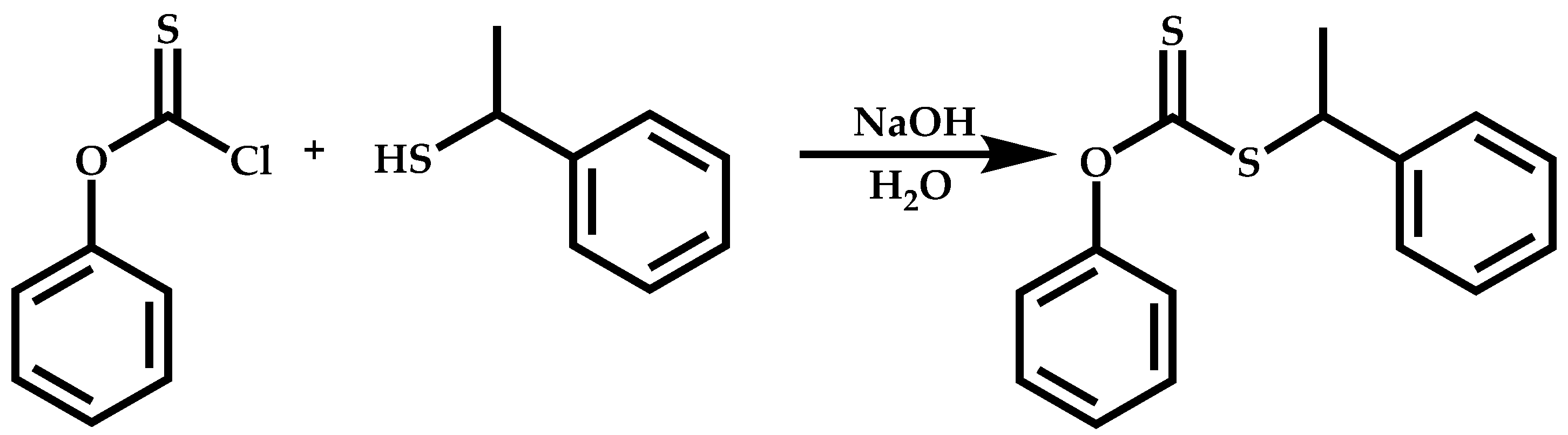
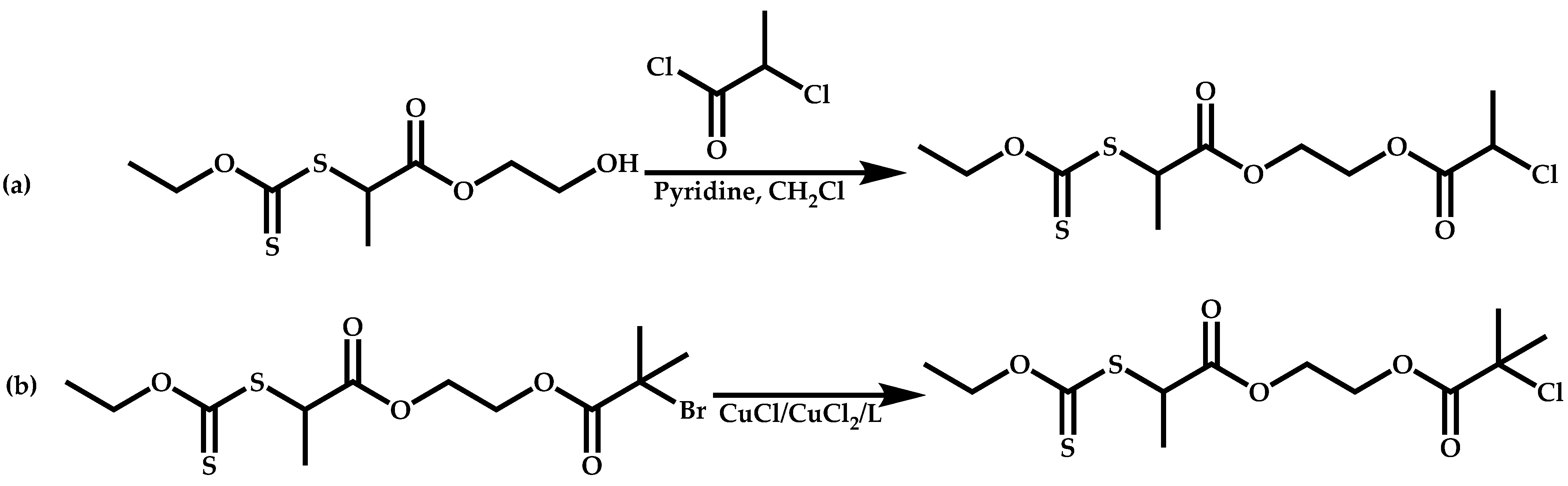
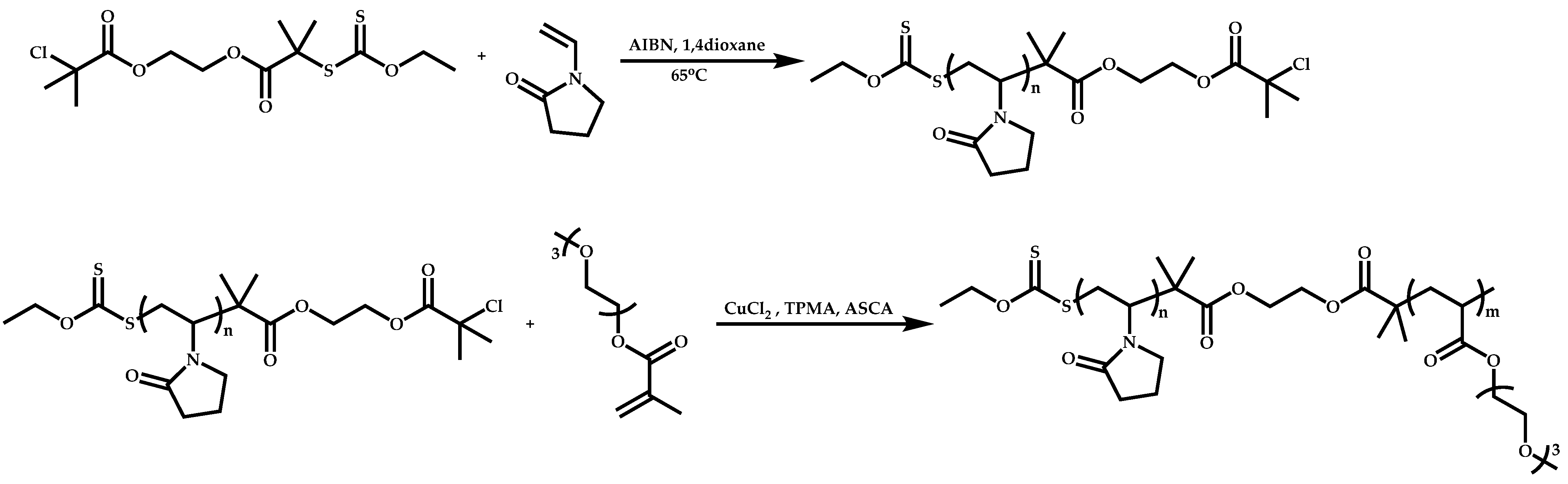
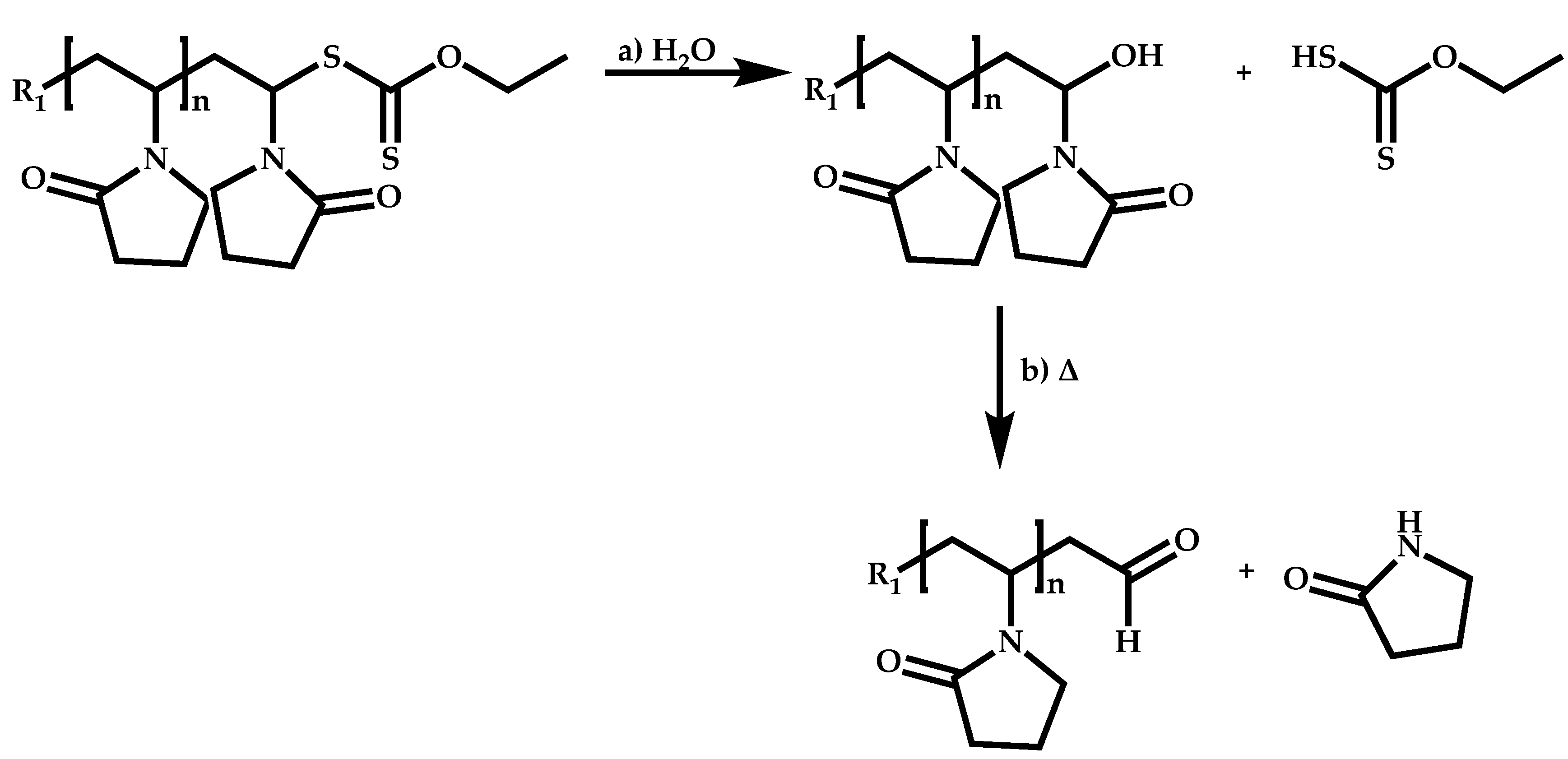
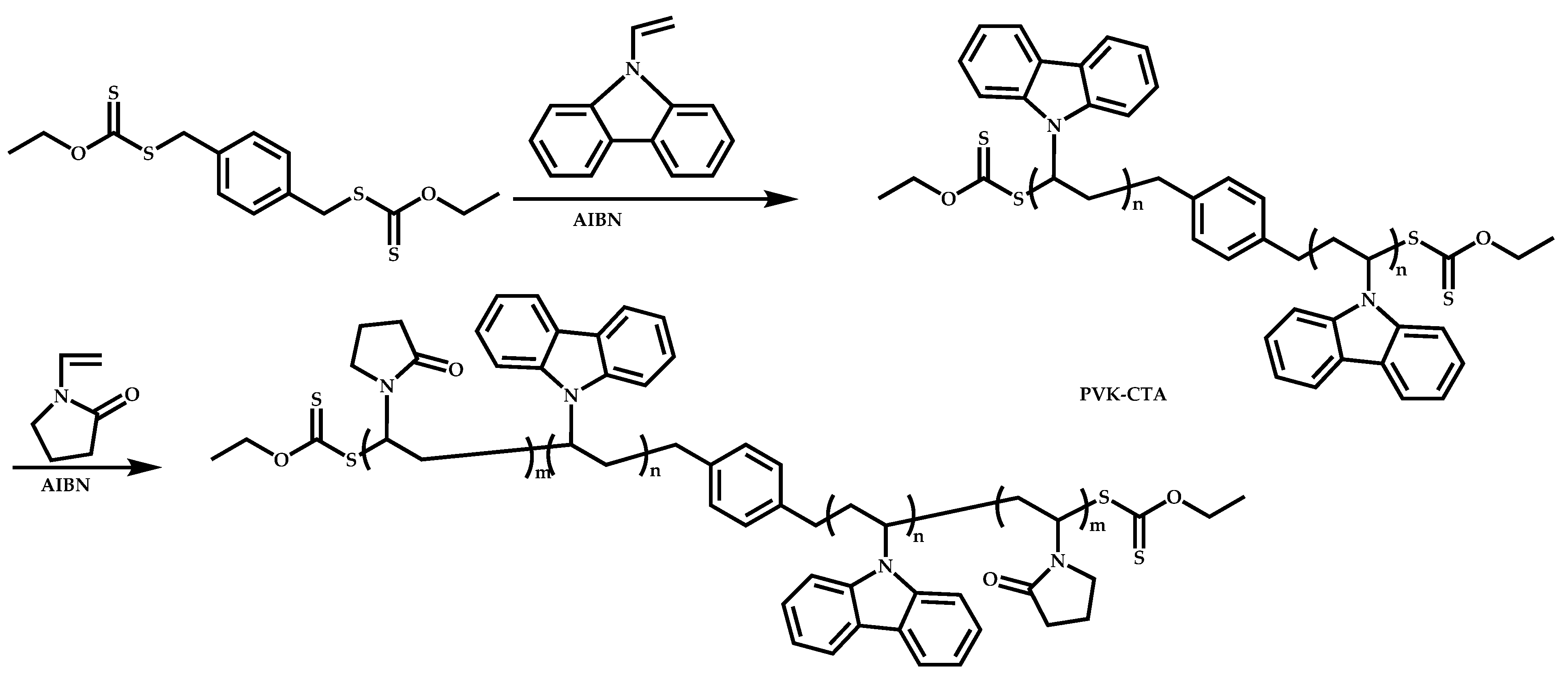
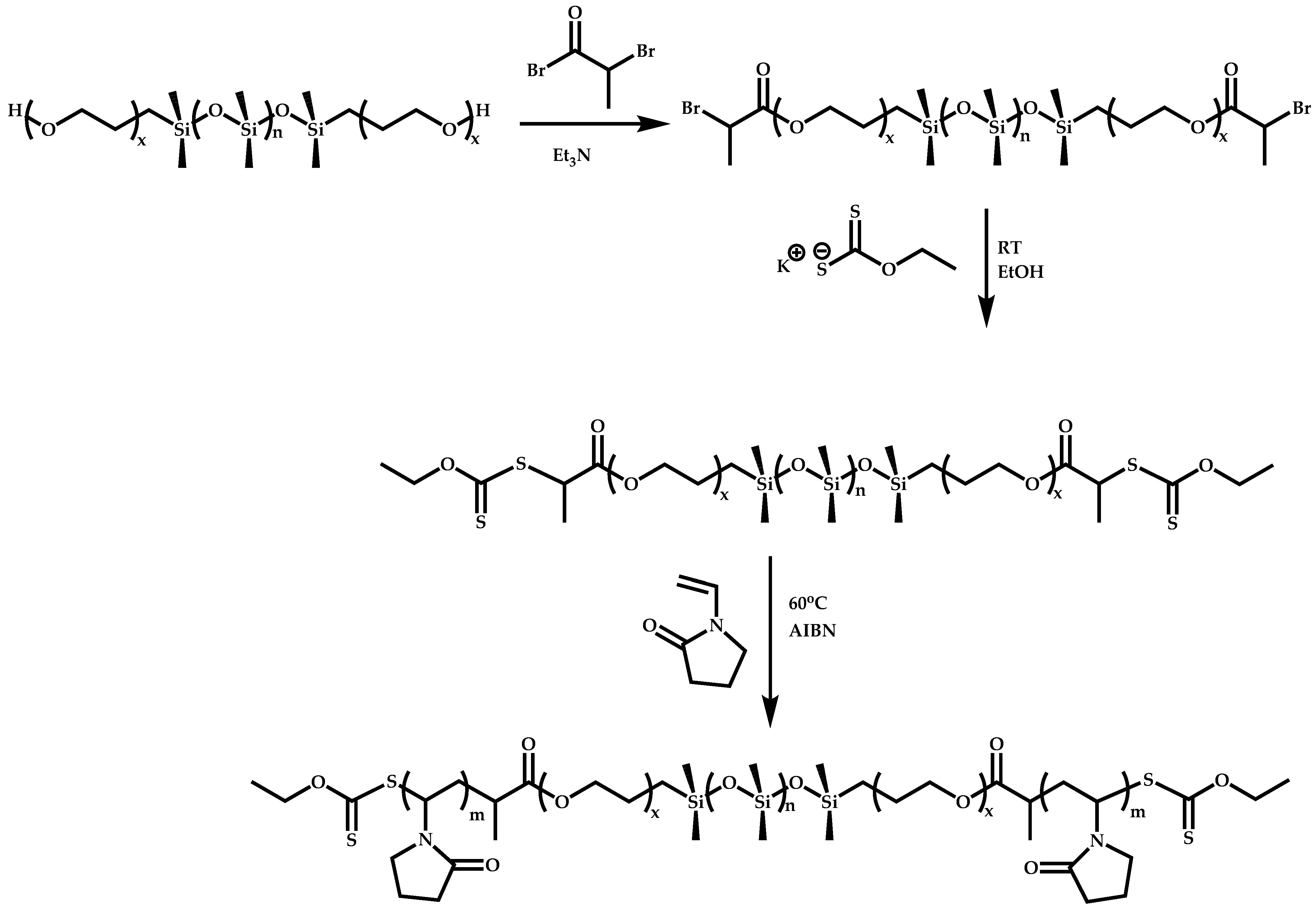
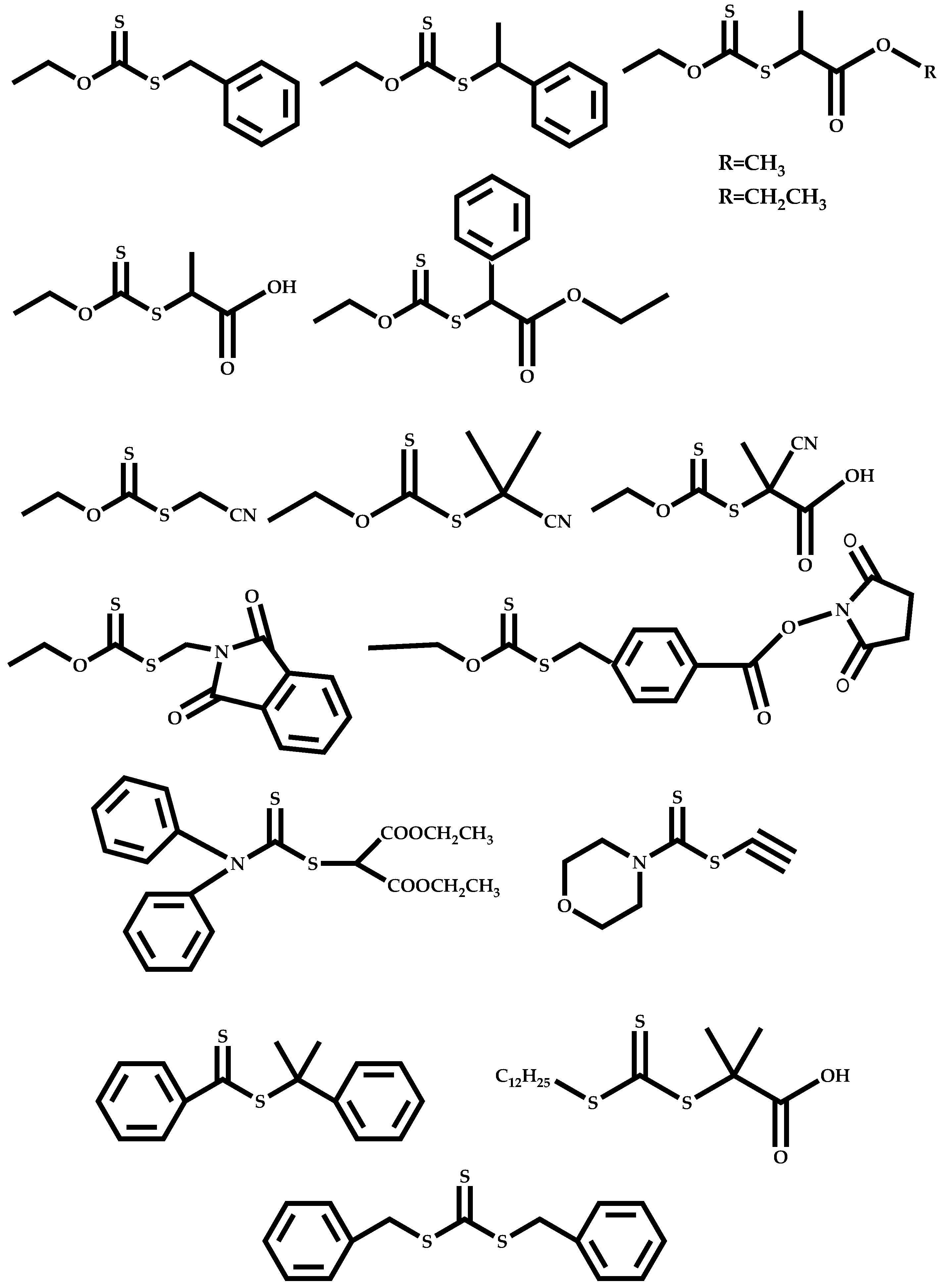
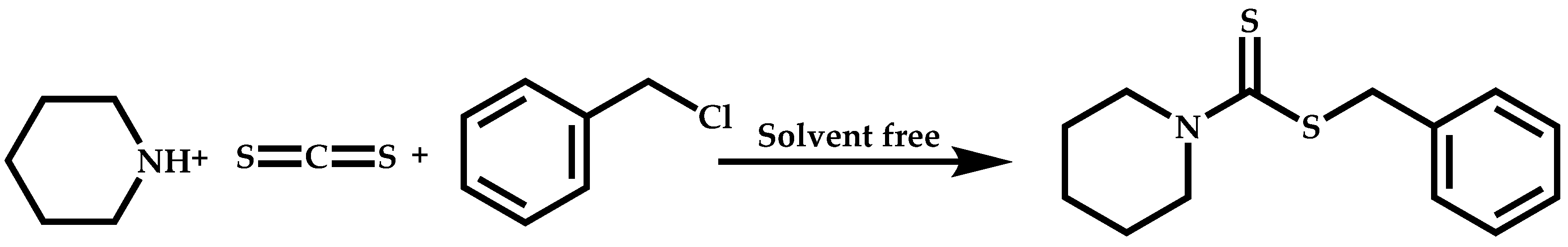
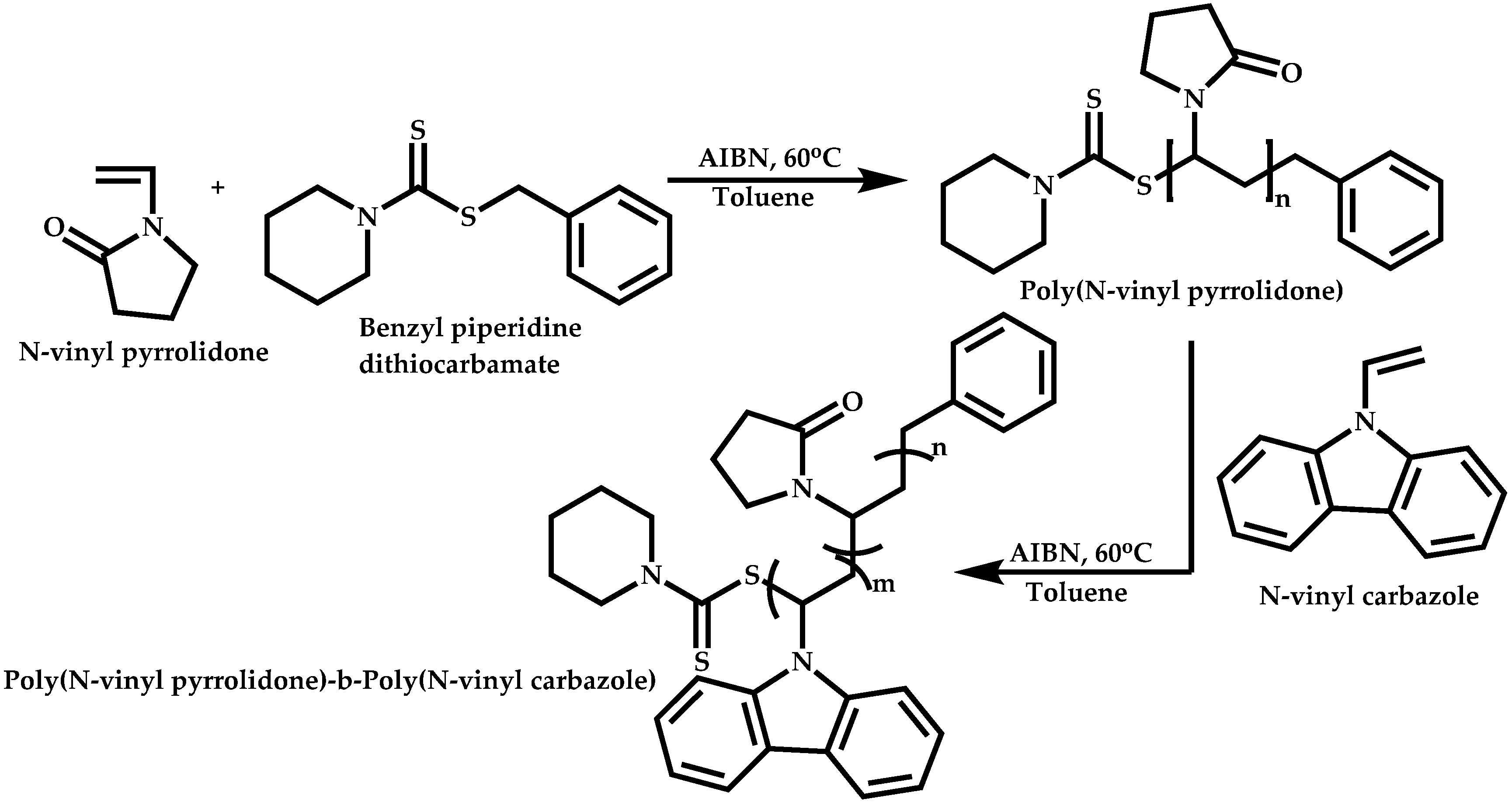
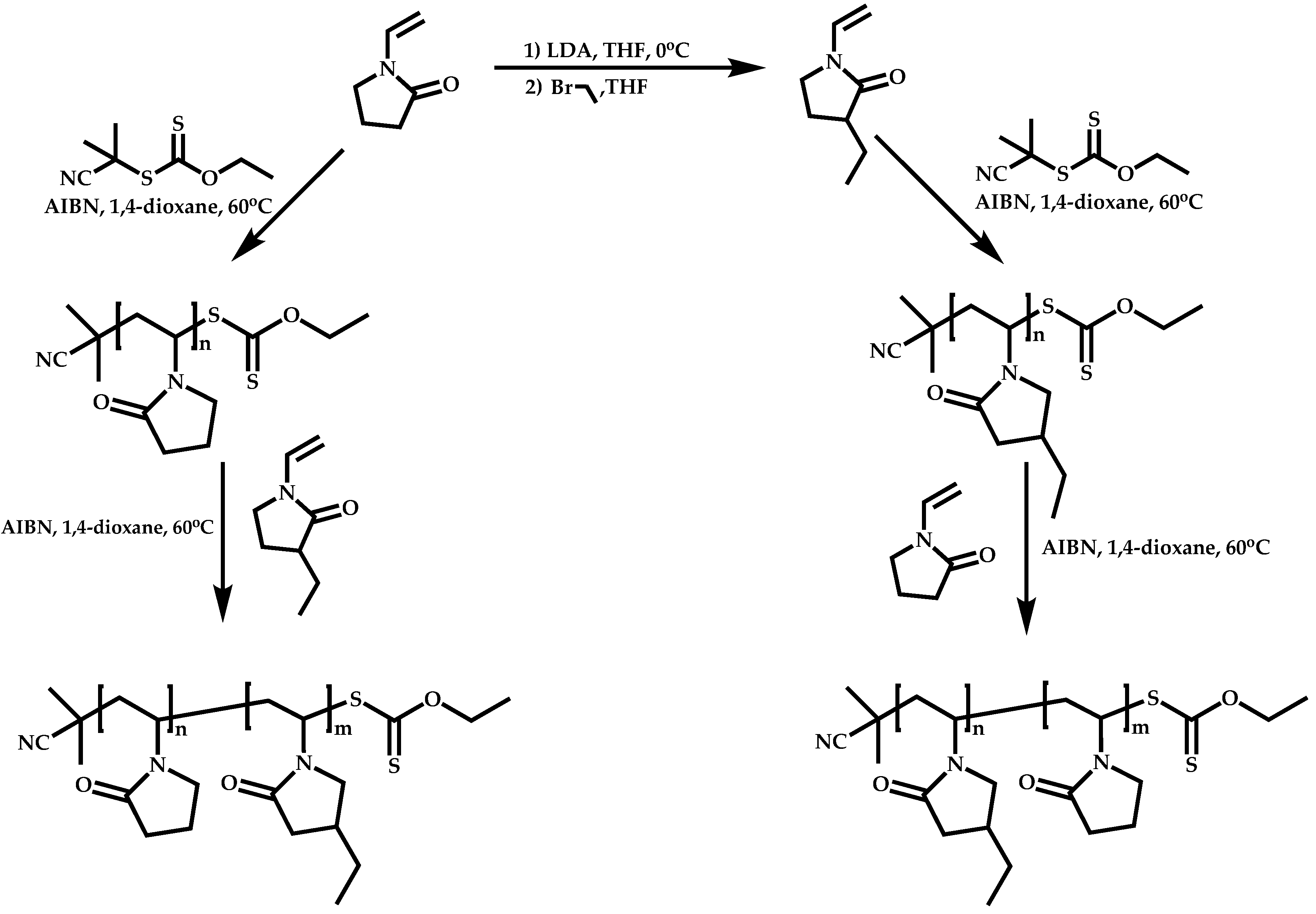
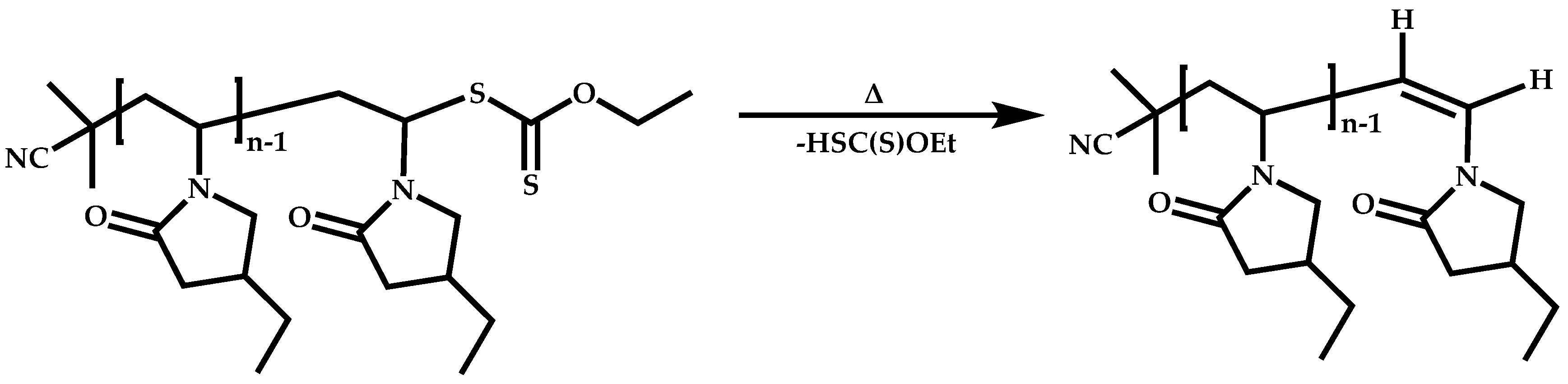
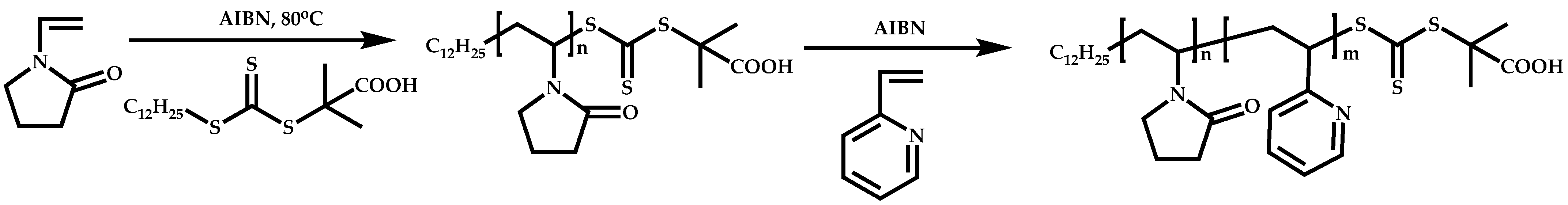
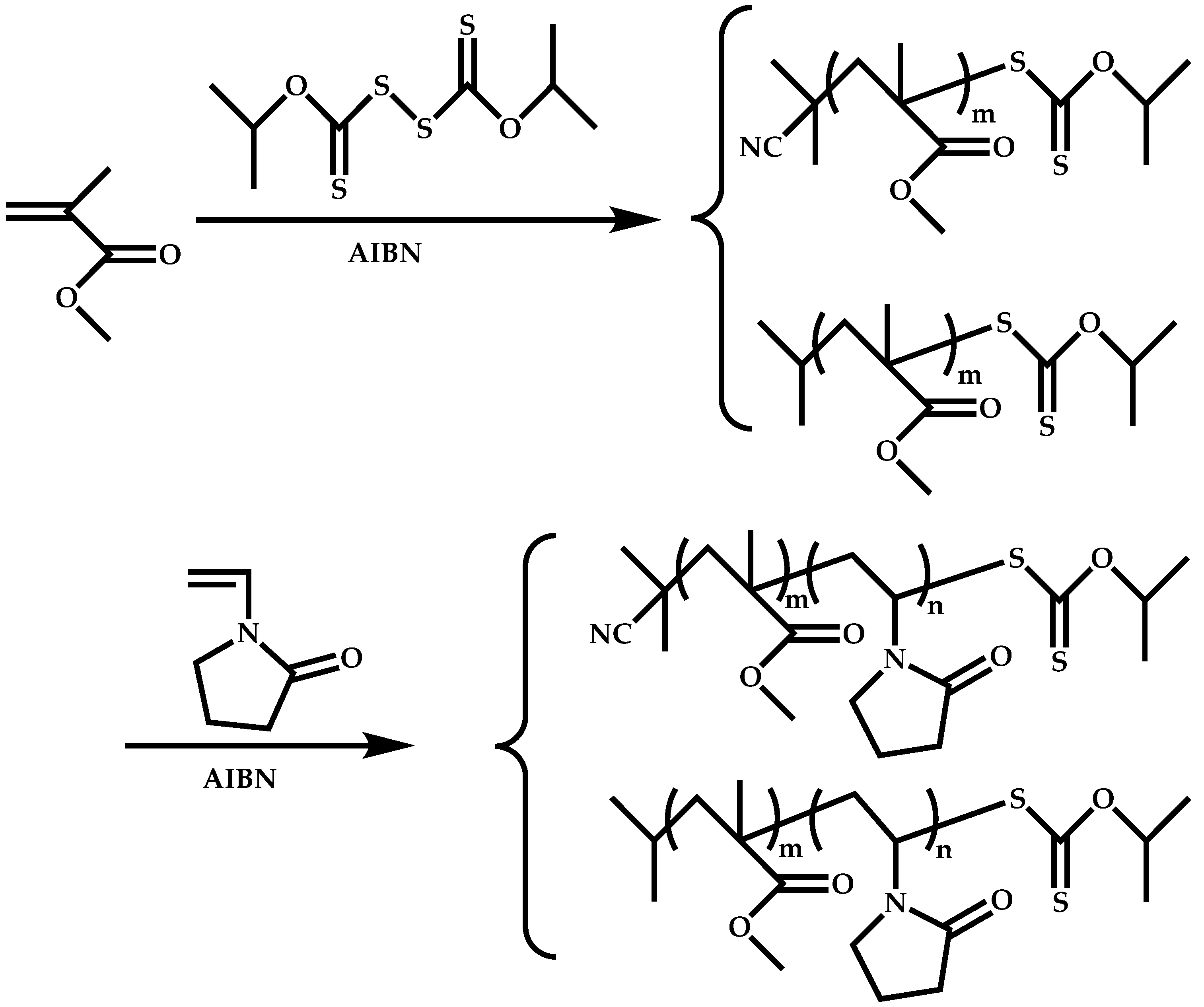
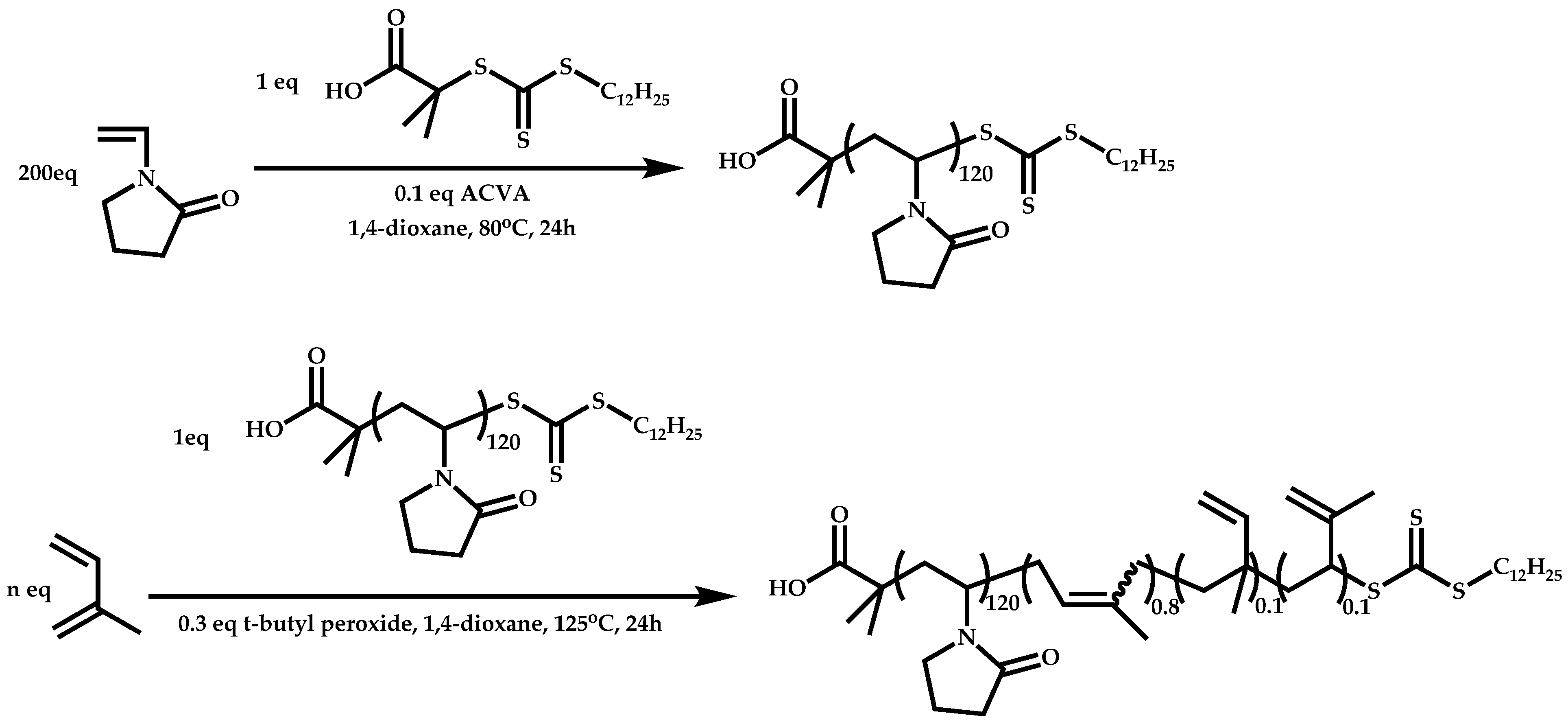
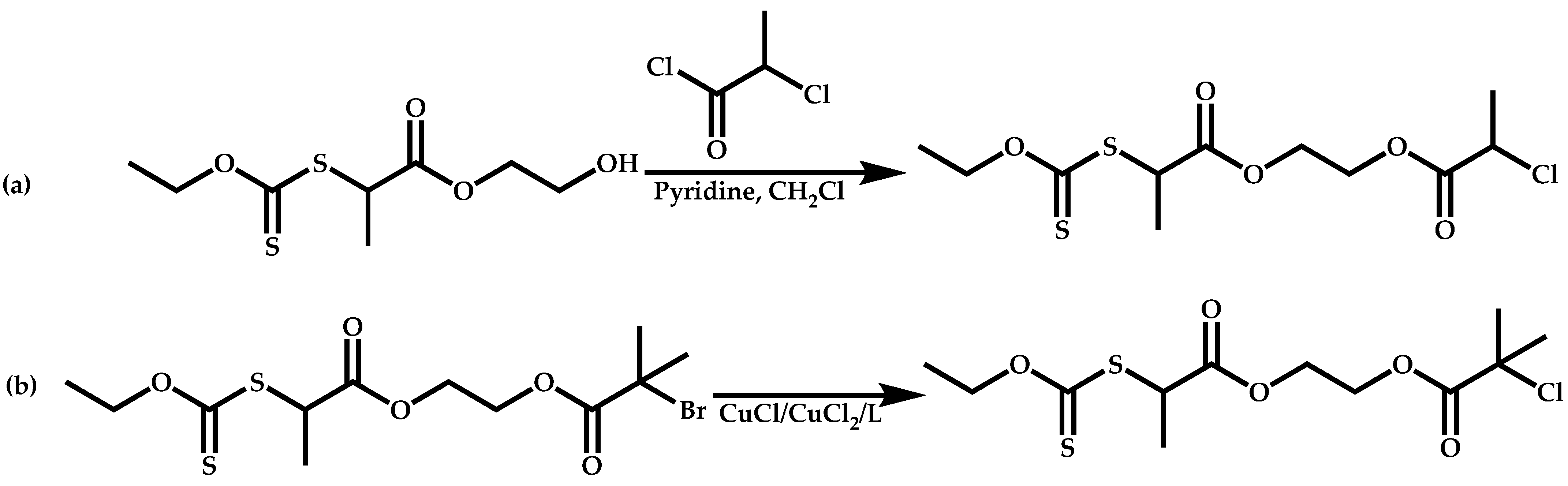
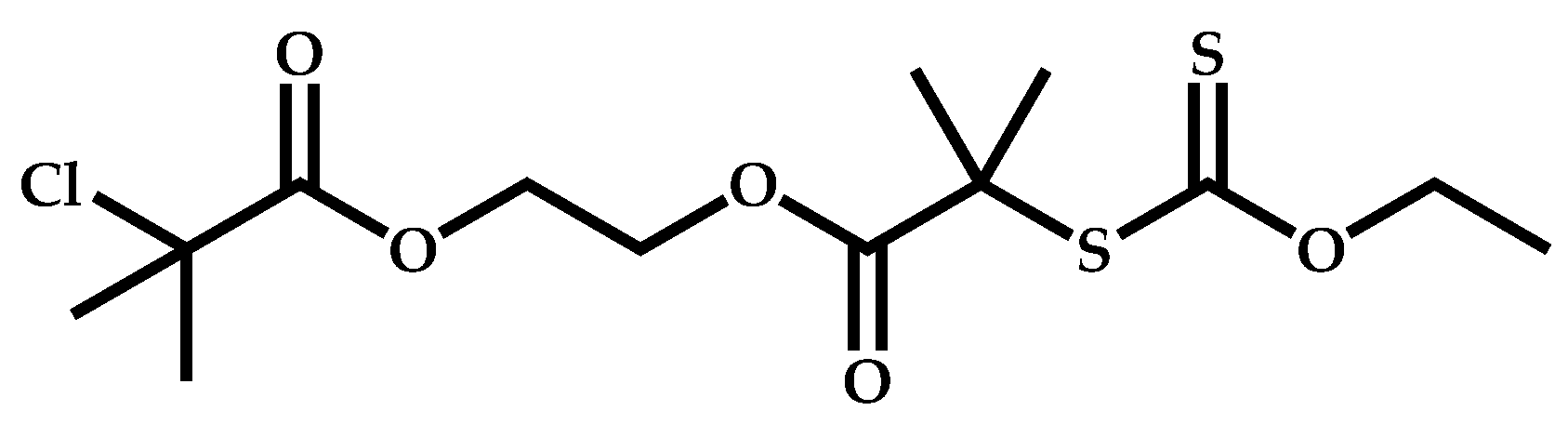
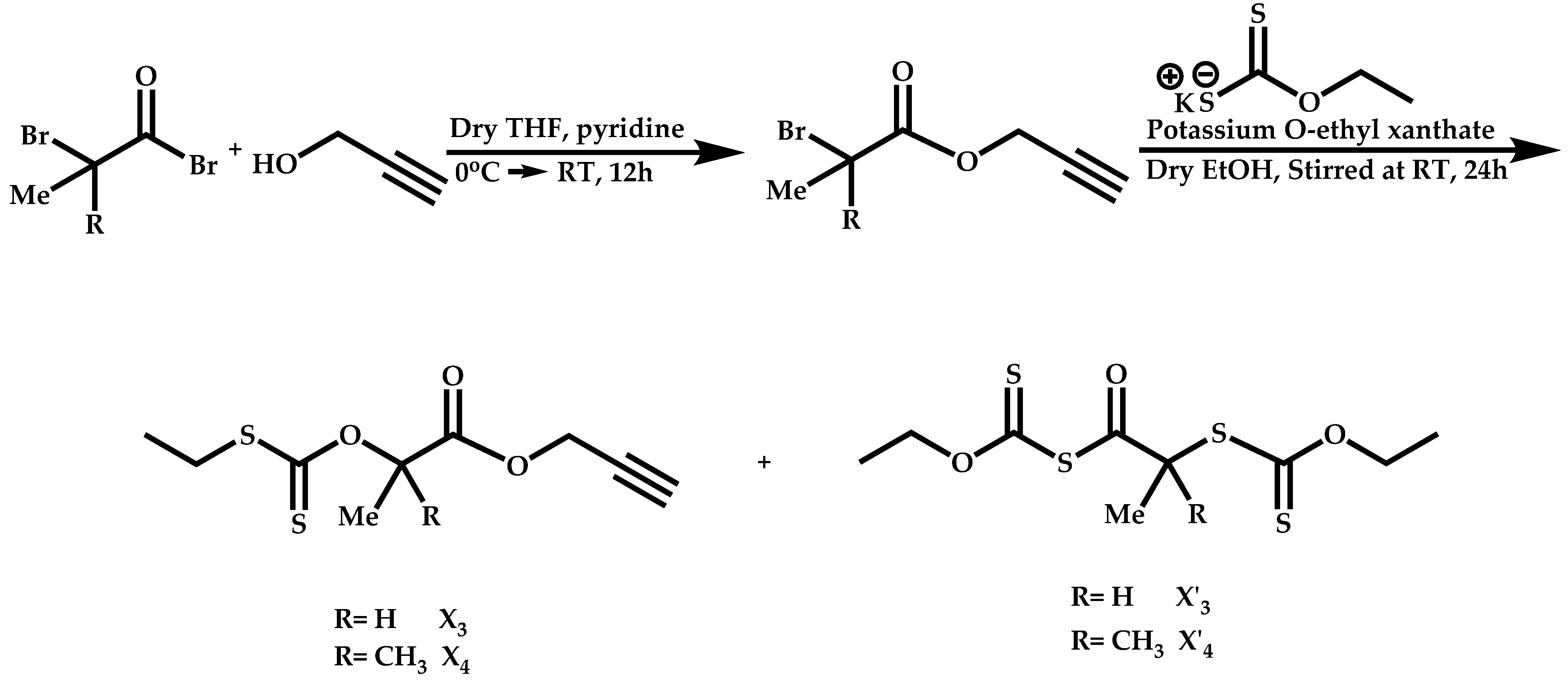
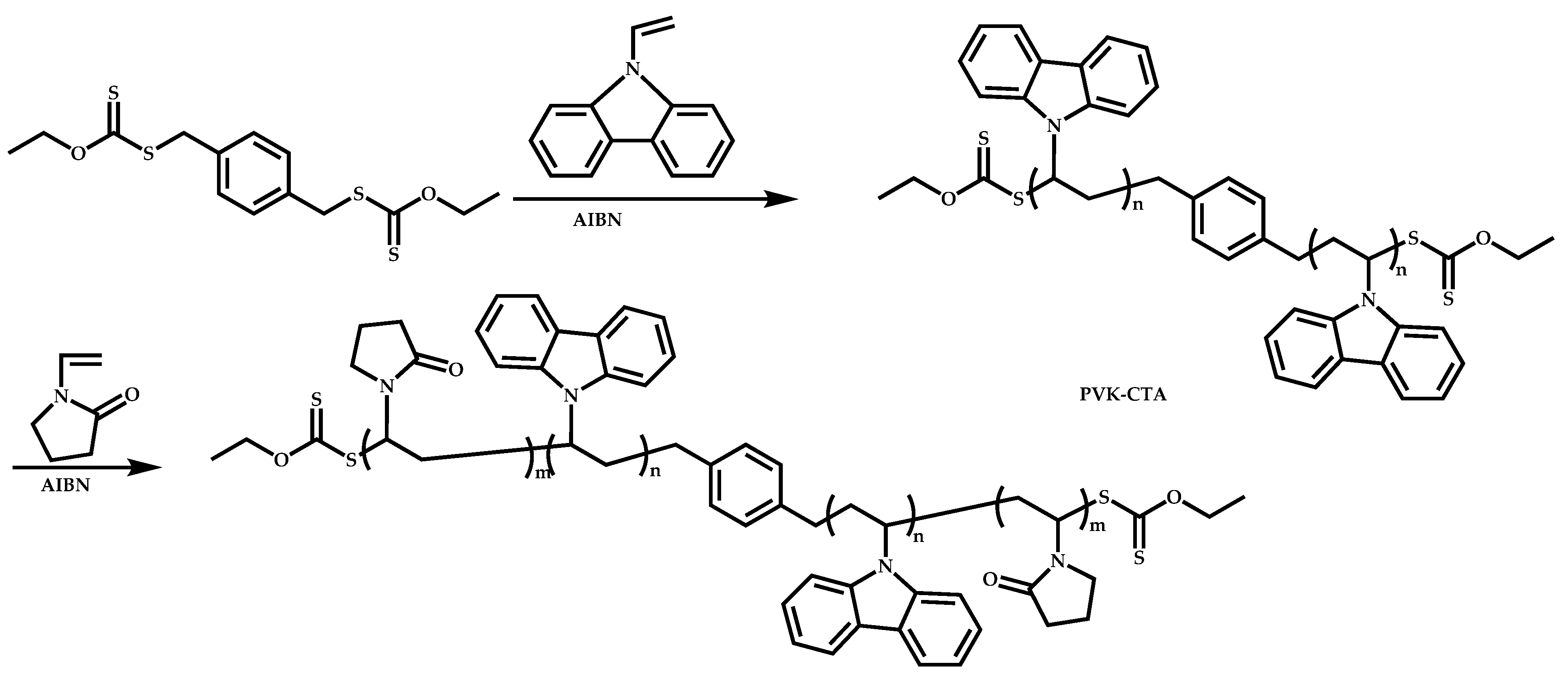
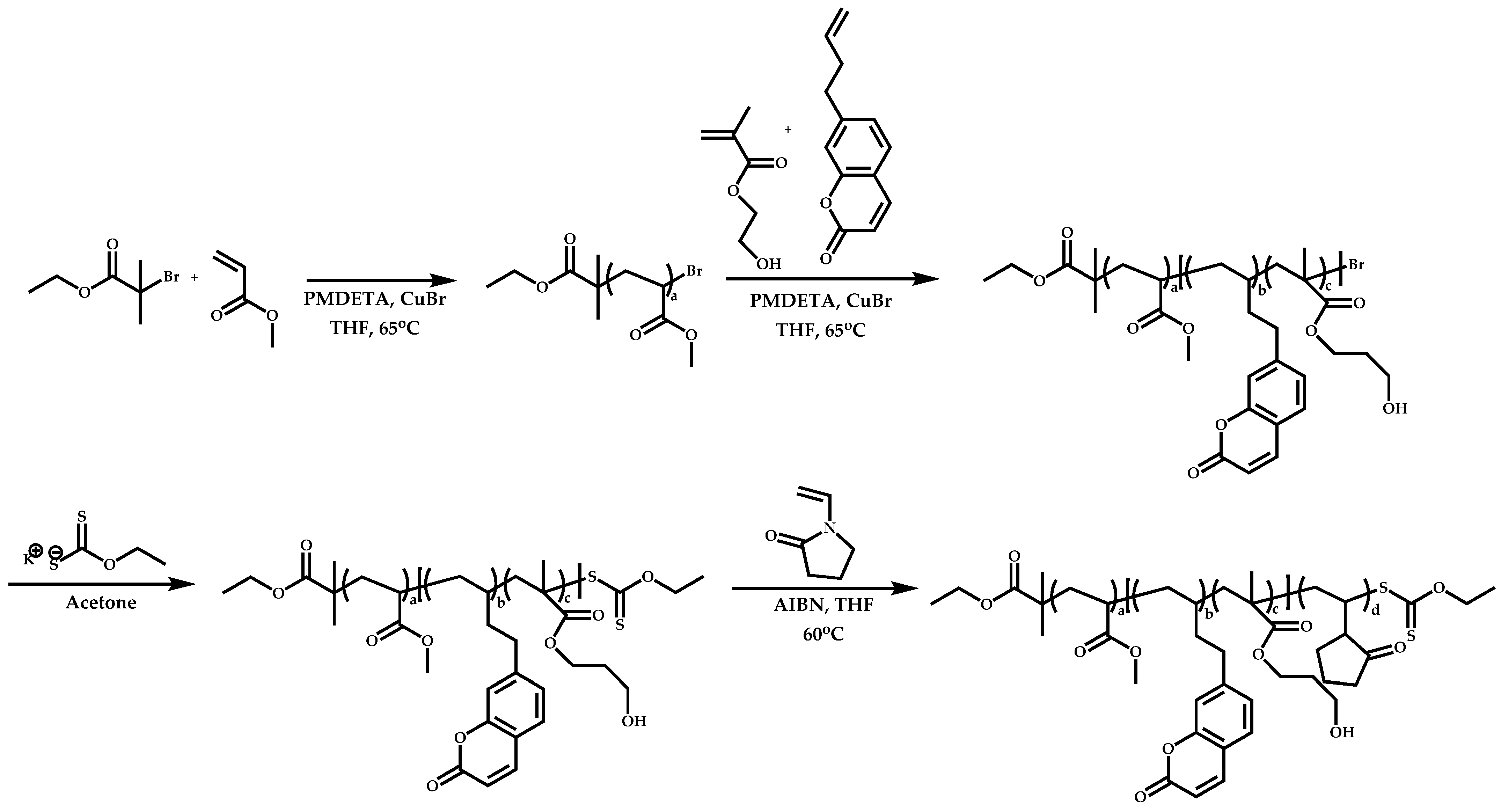
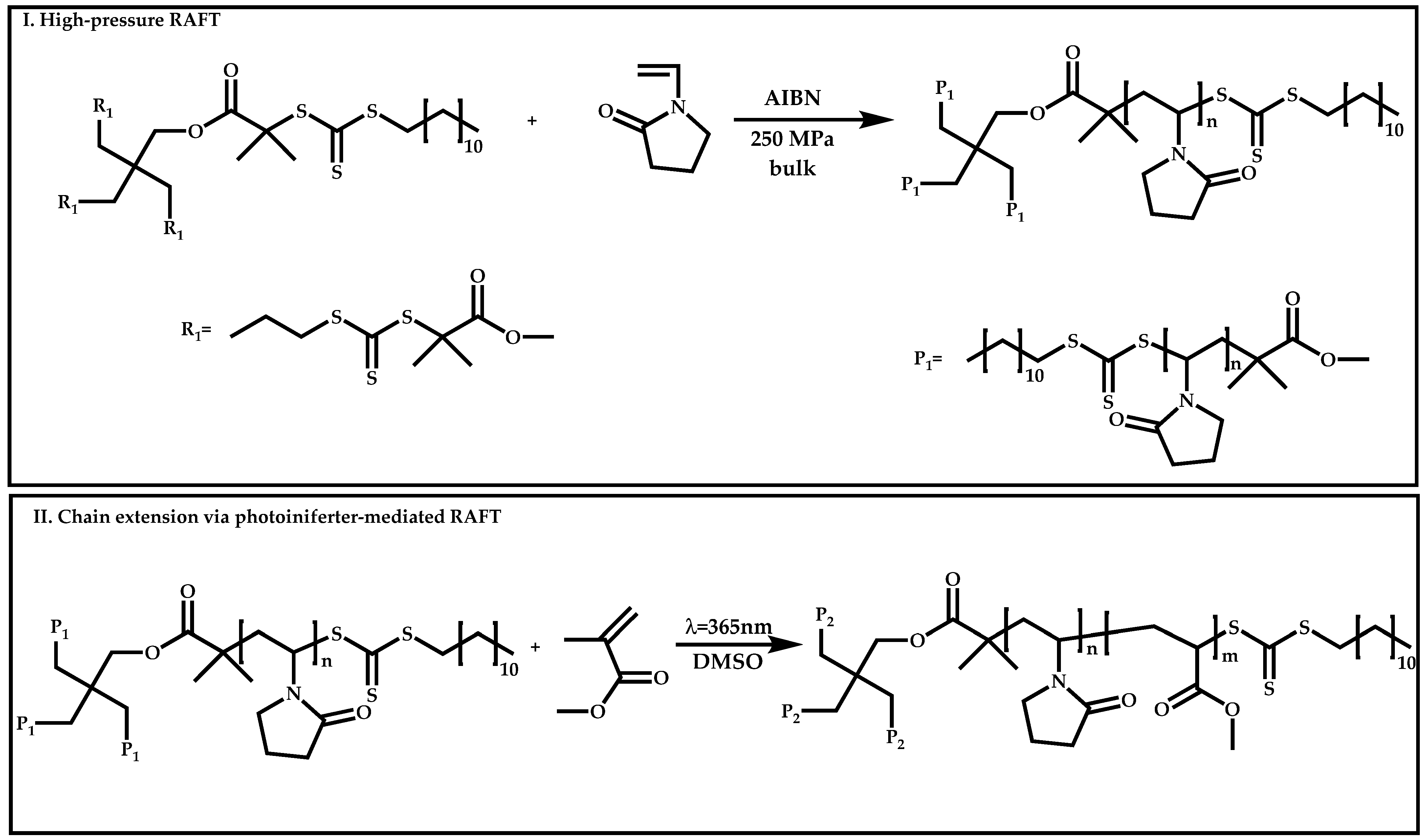
Dual Functions CTAs (Inifers)
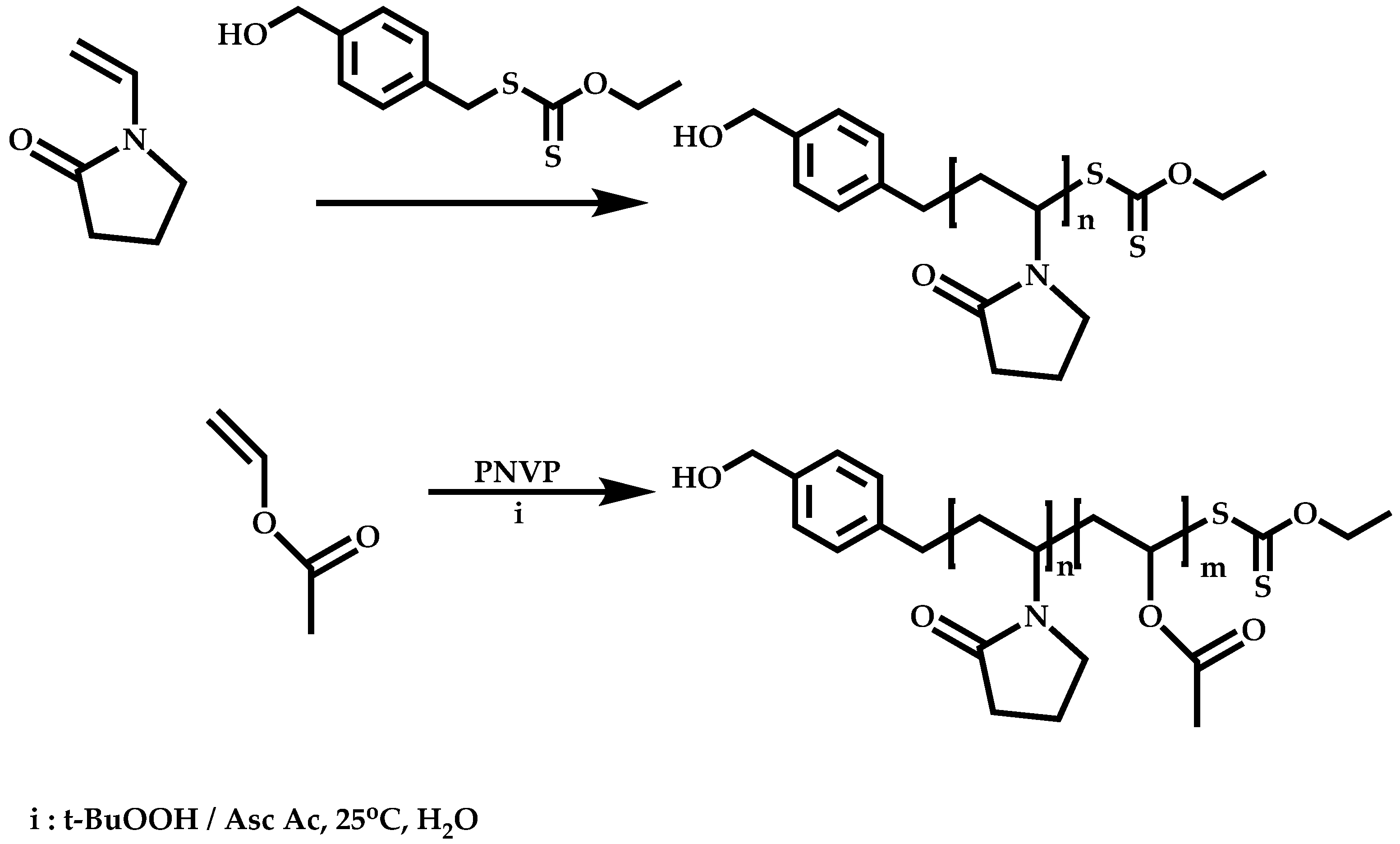
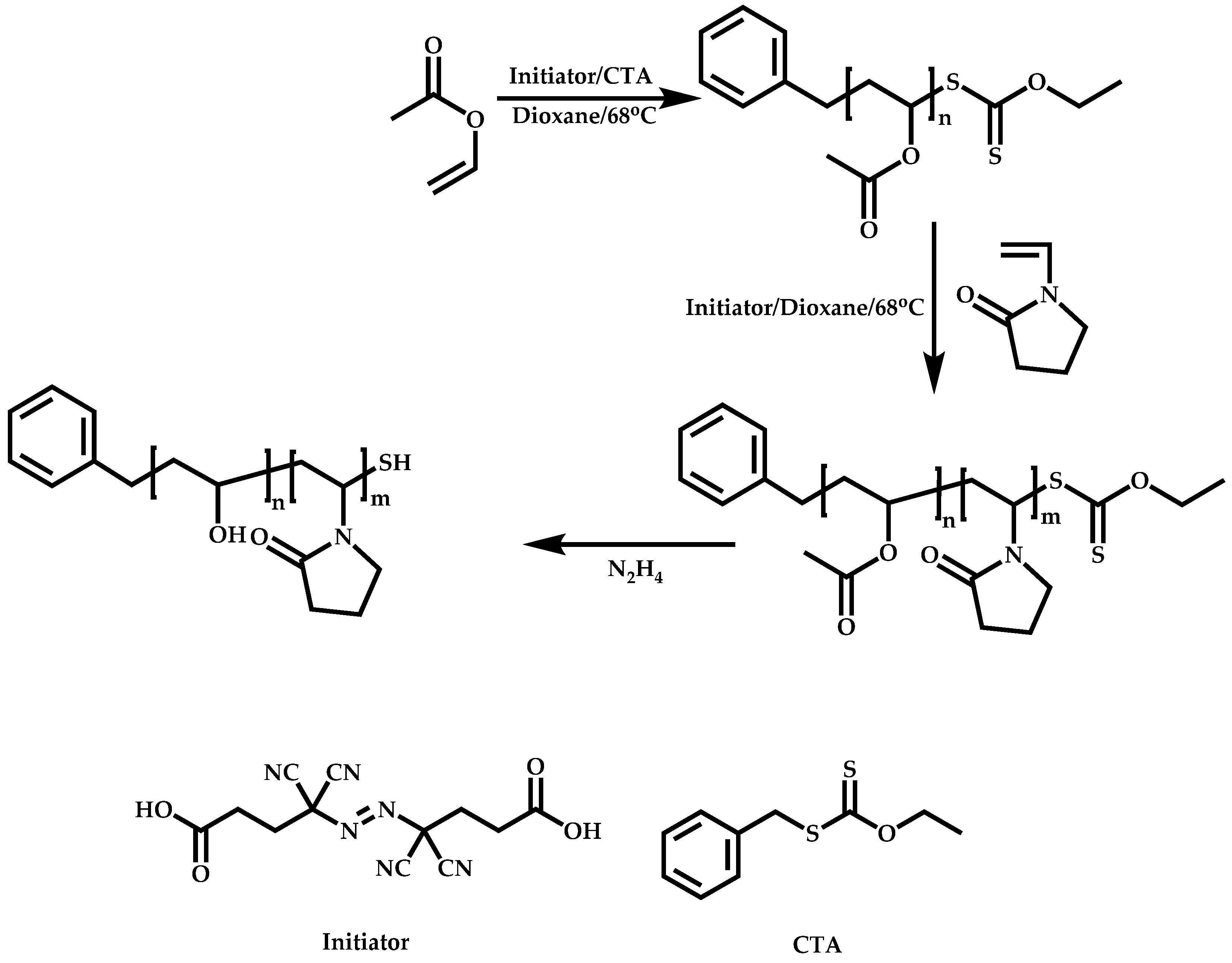
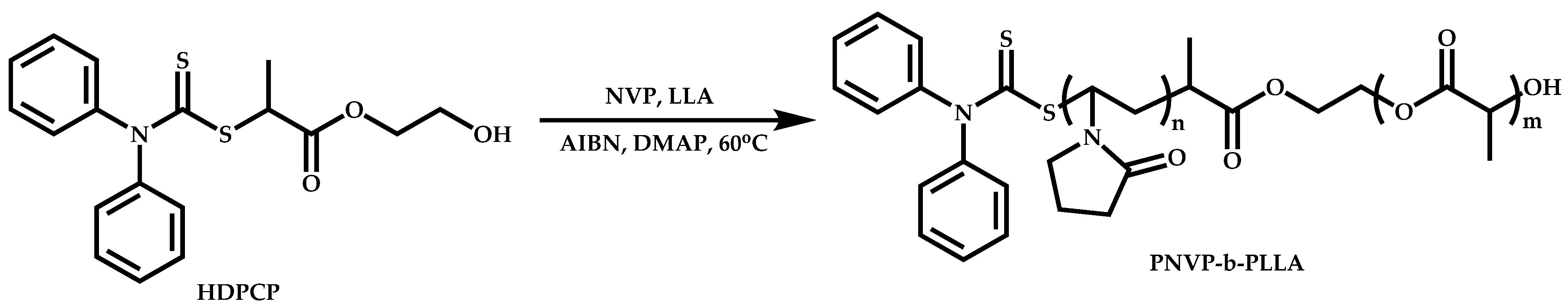
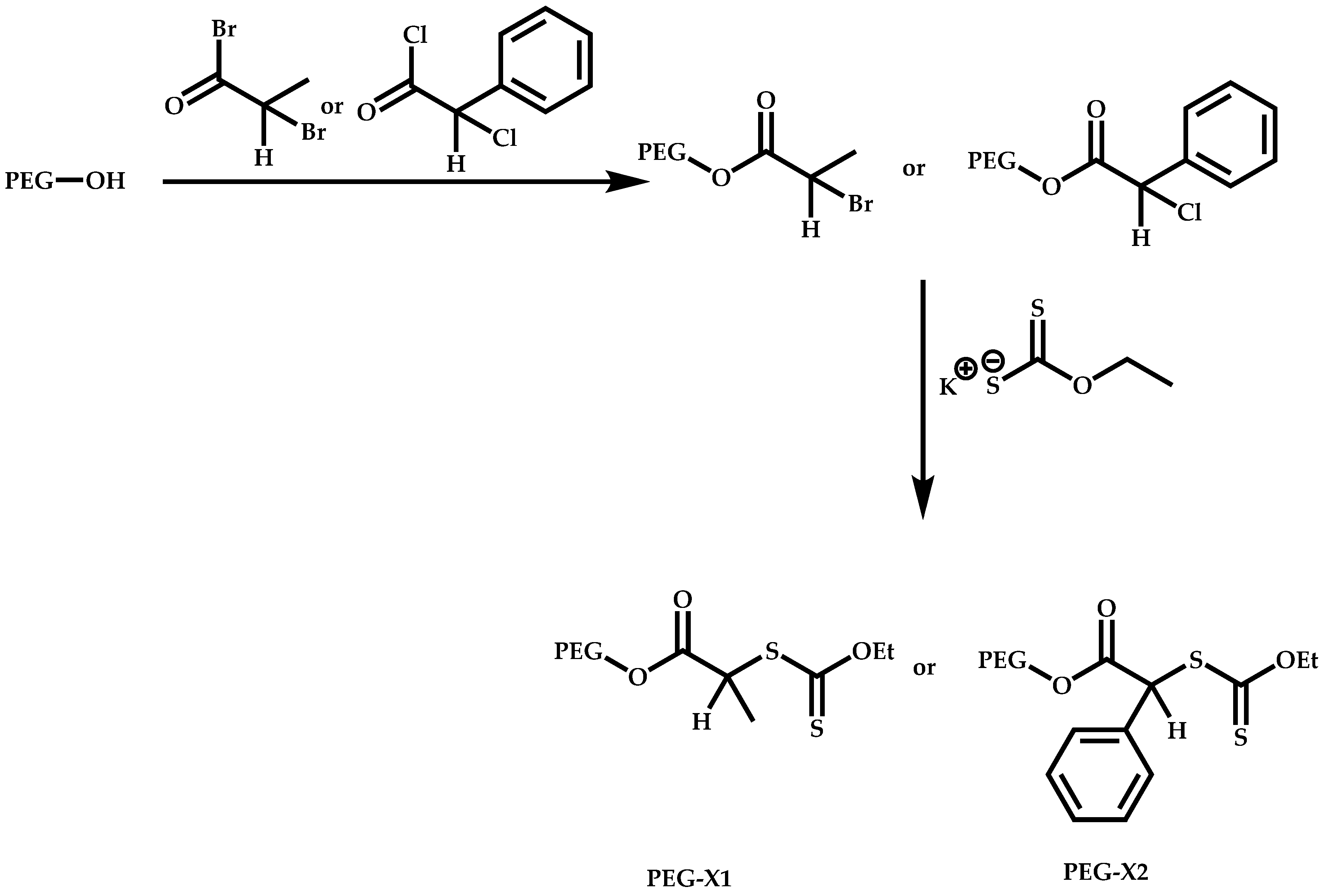
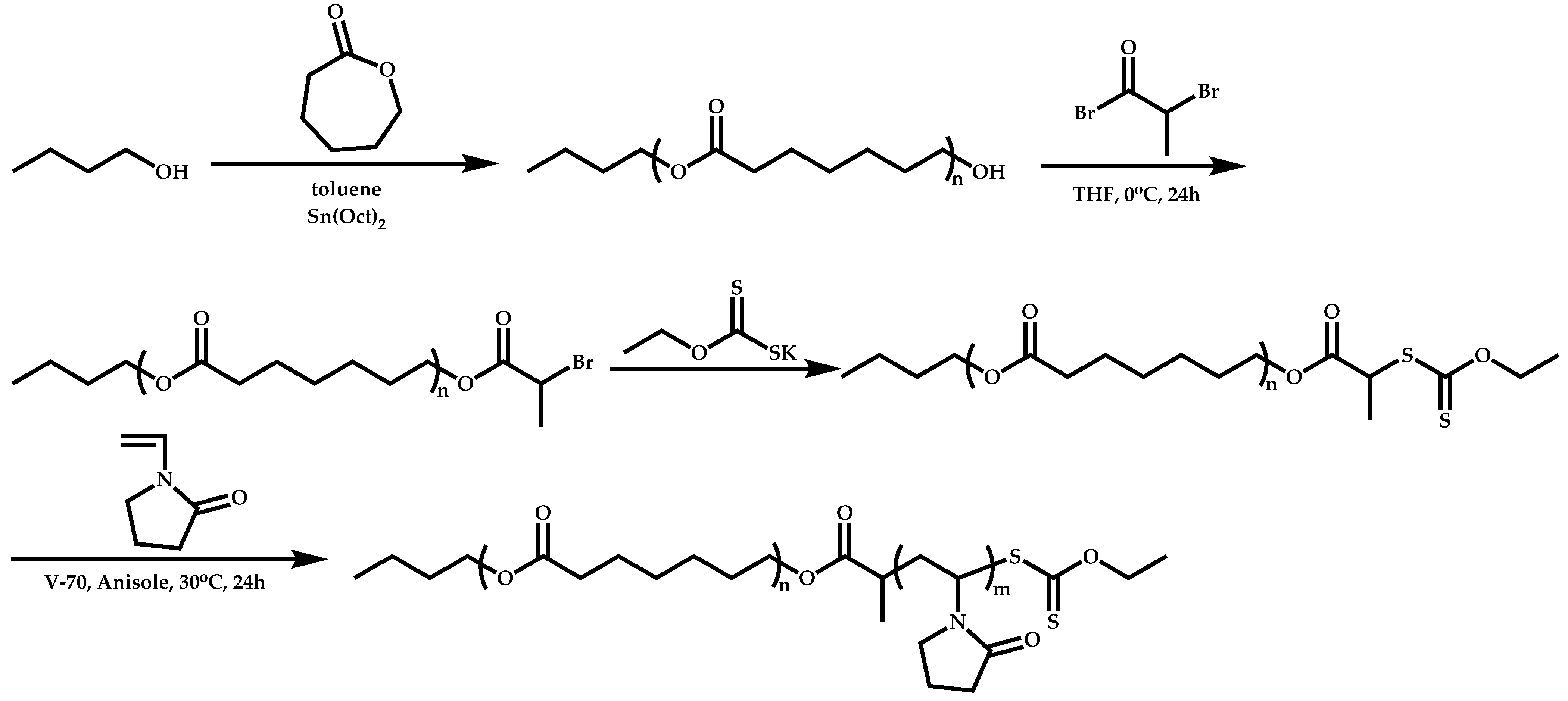
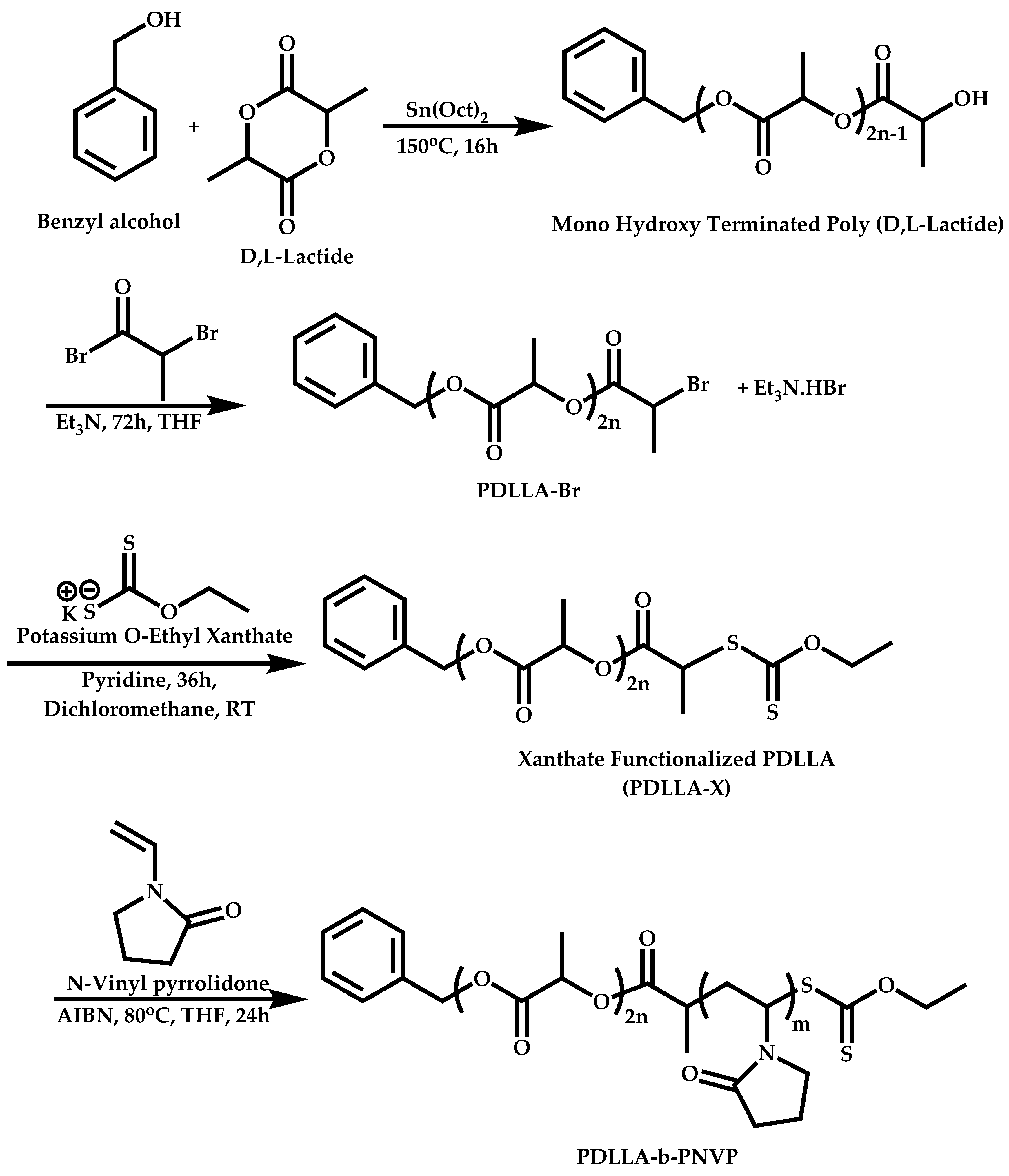
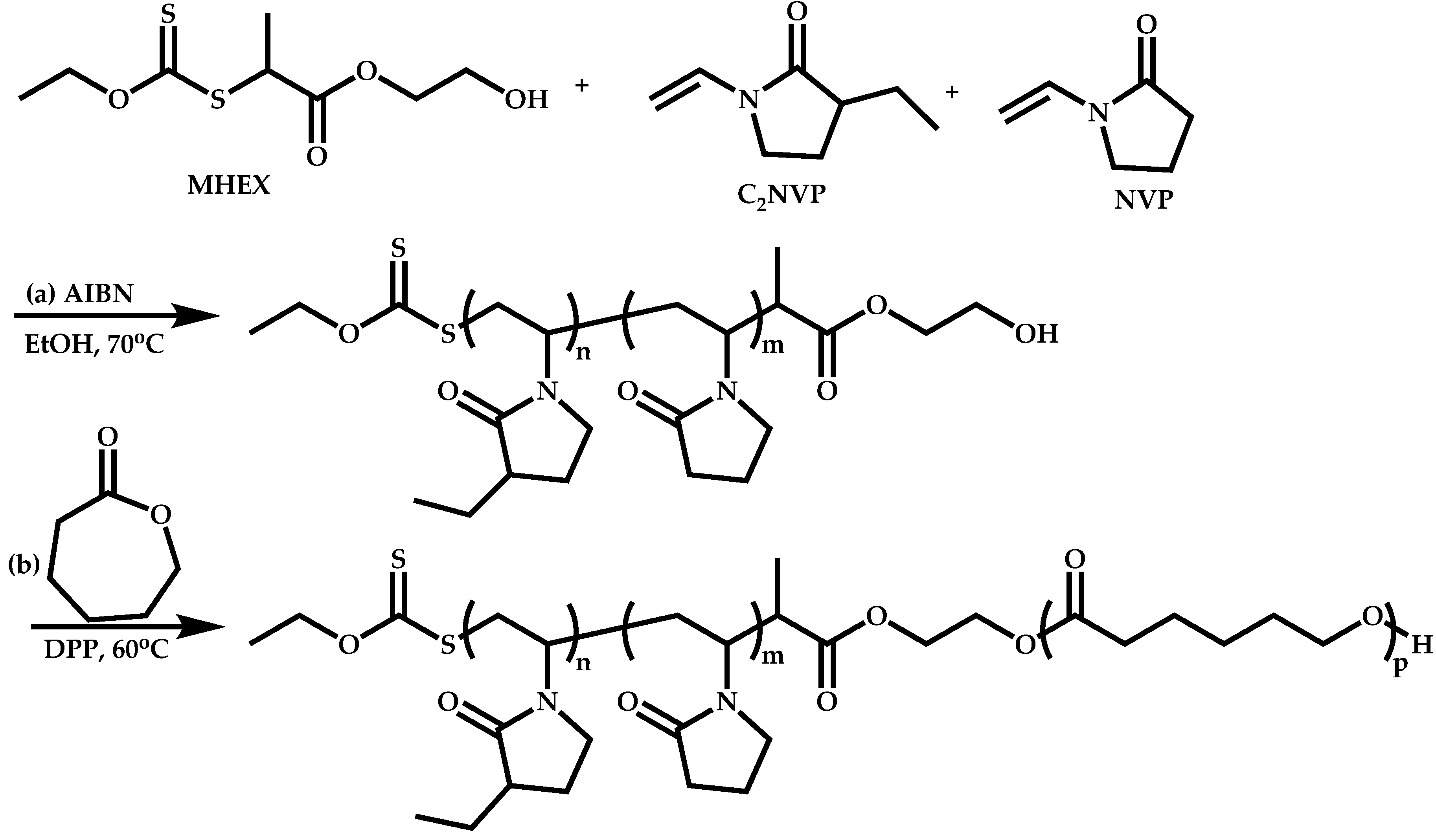
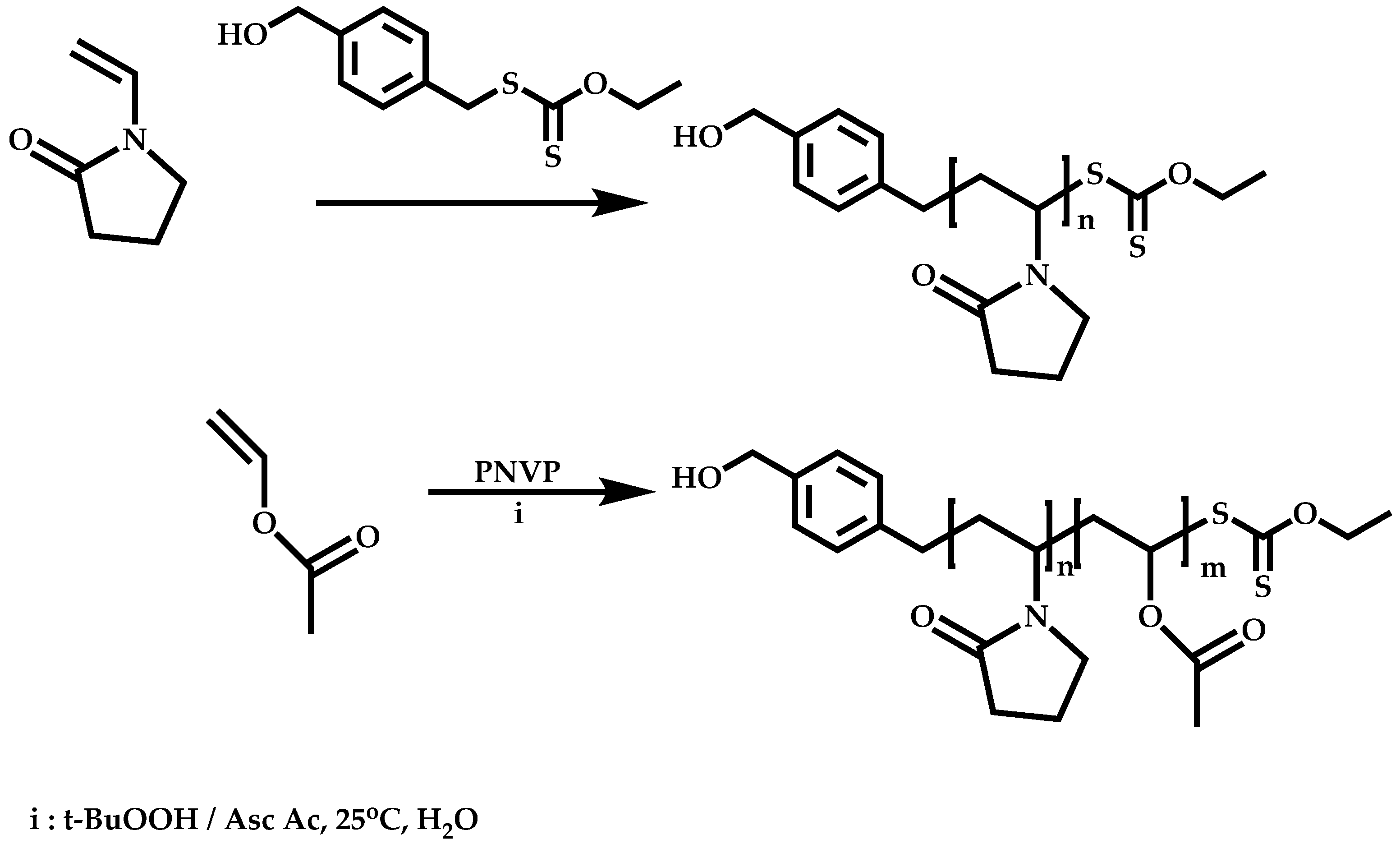
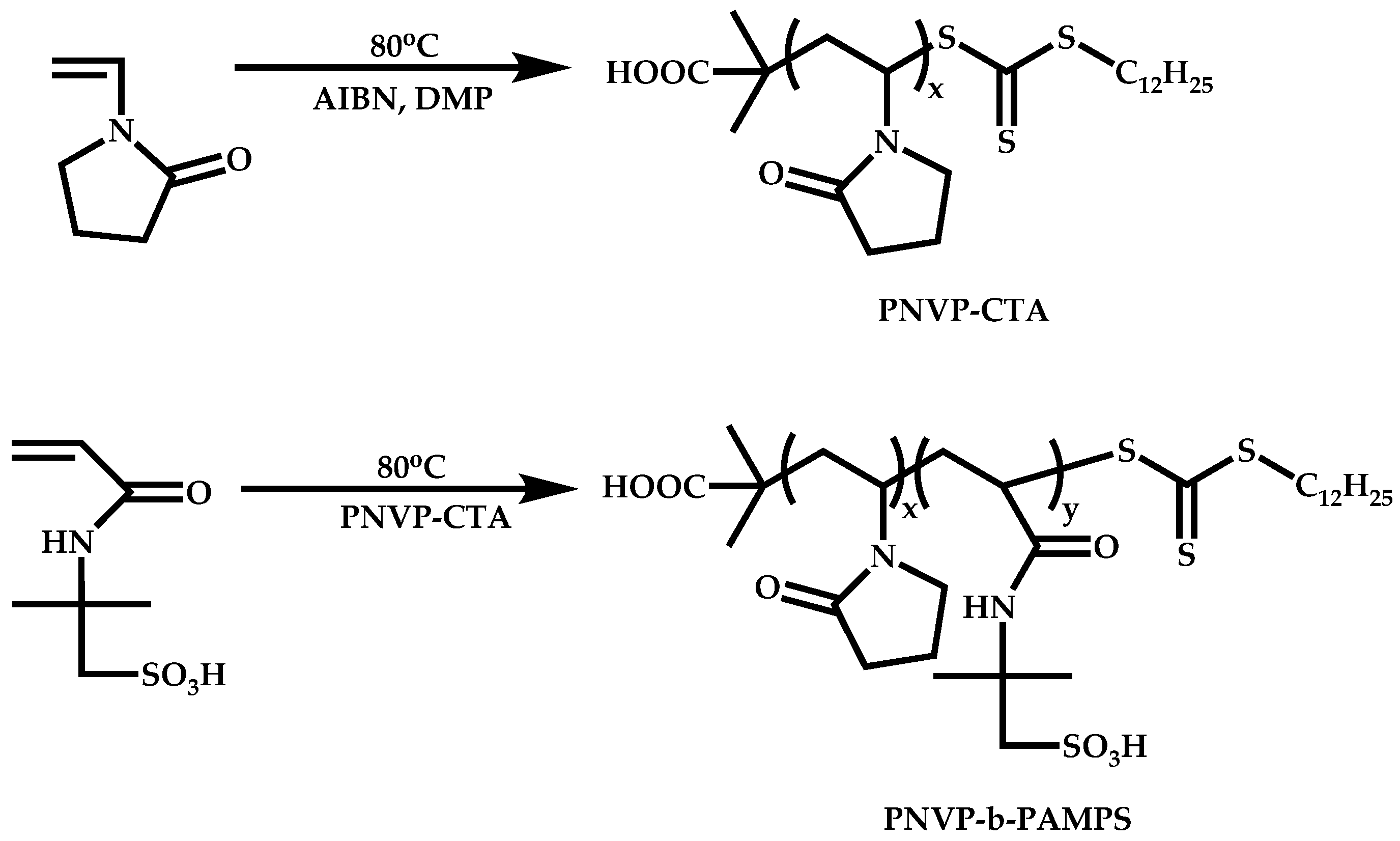
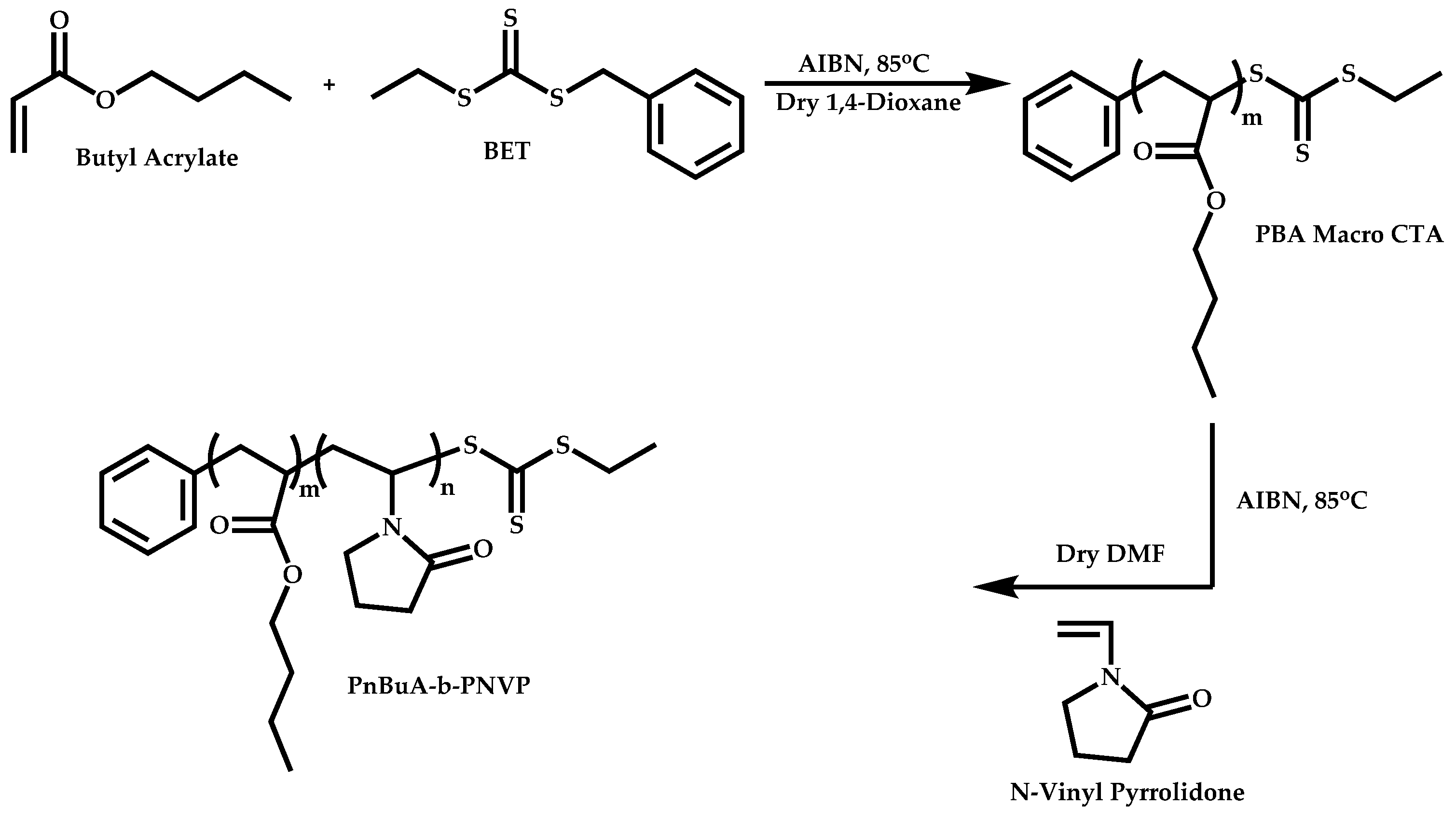
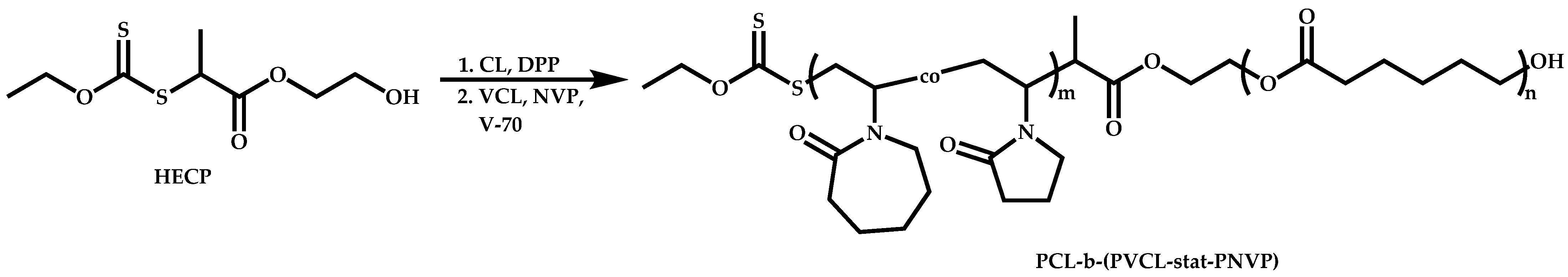
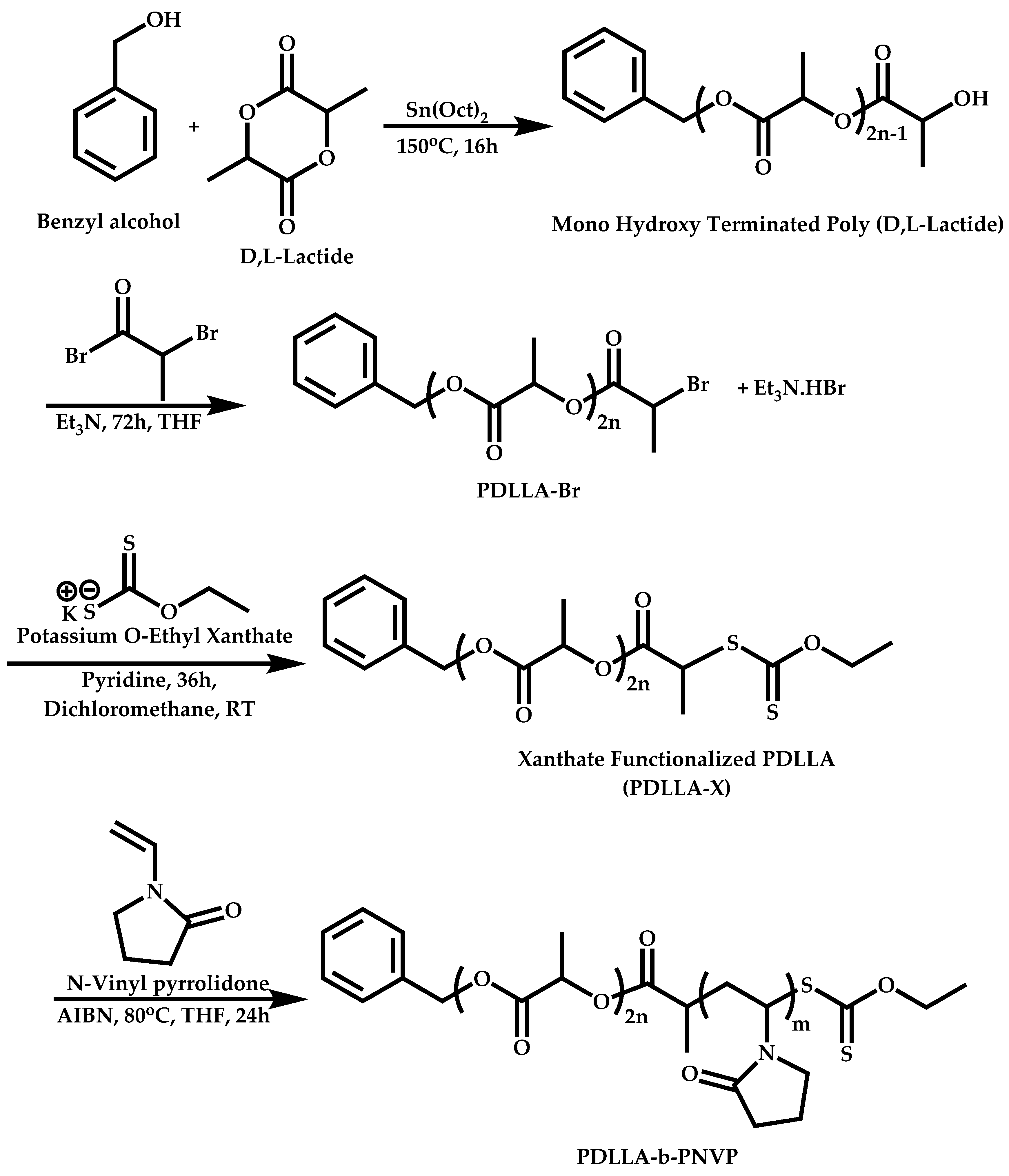
Transformation of the End-Group of the 1st Block to CTA, Suitable for the Polymerization of NVP
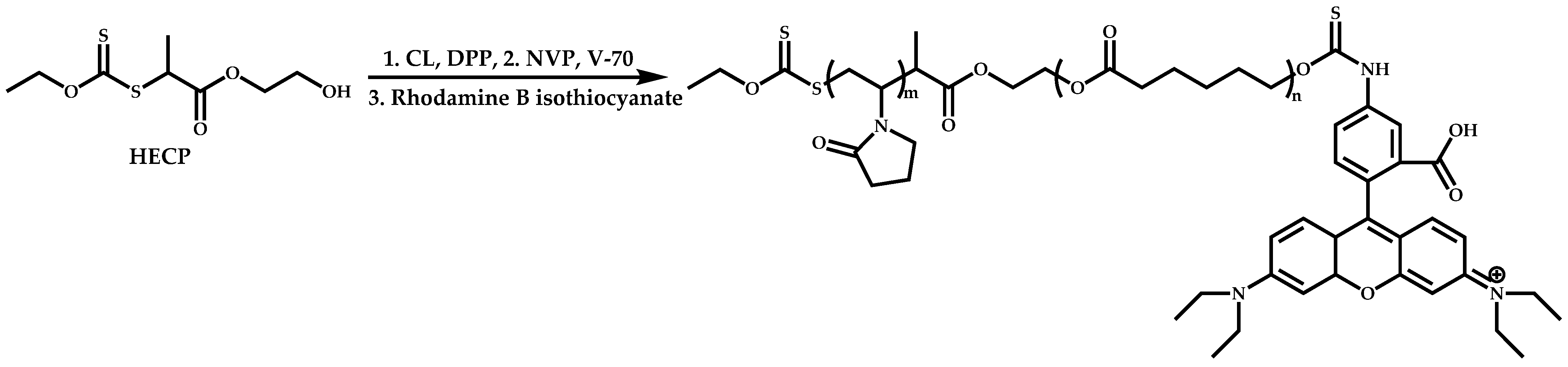
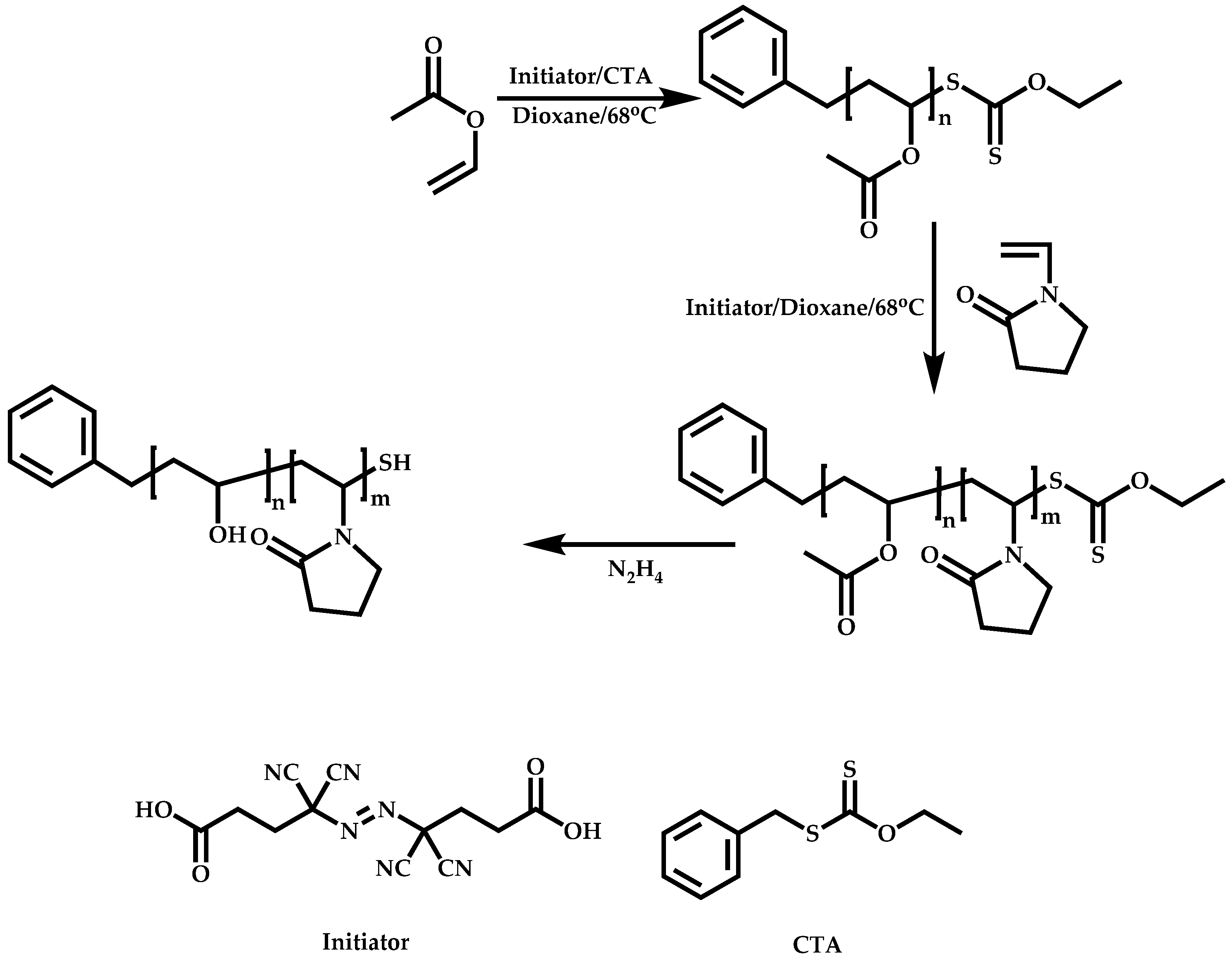
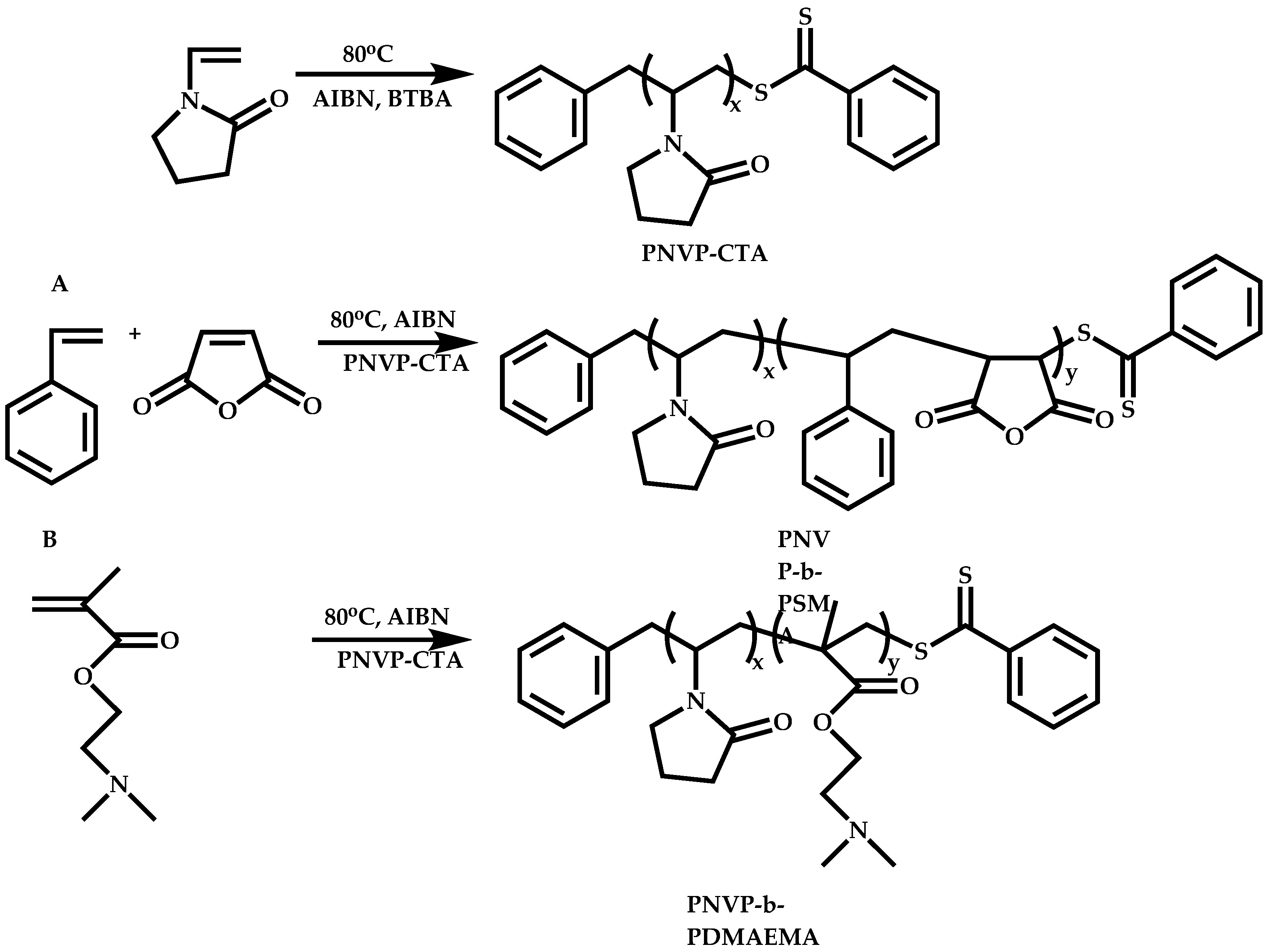
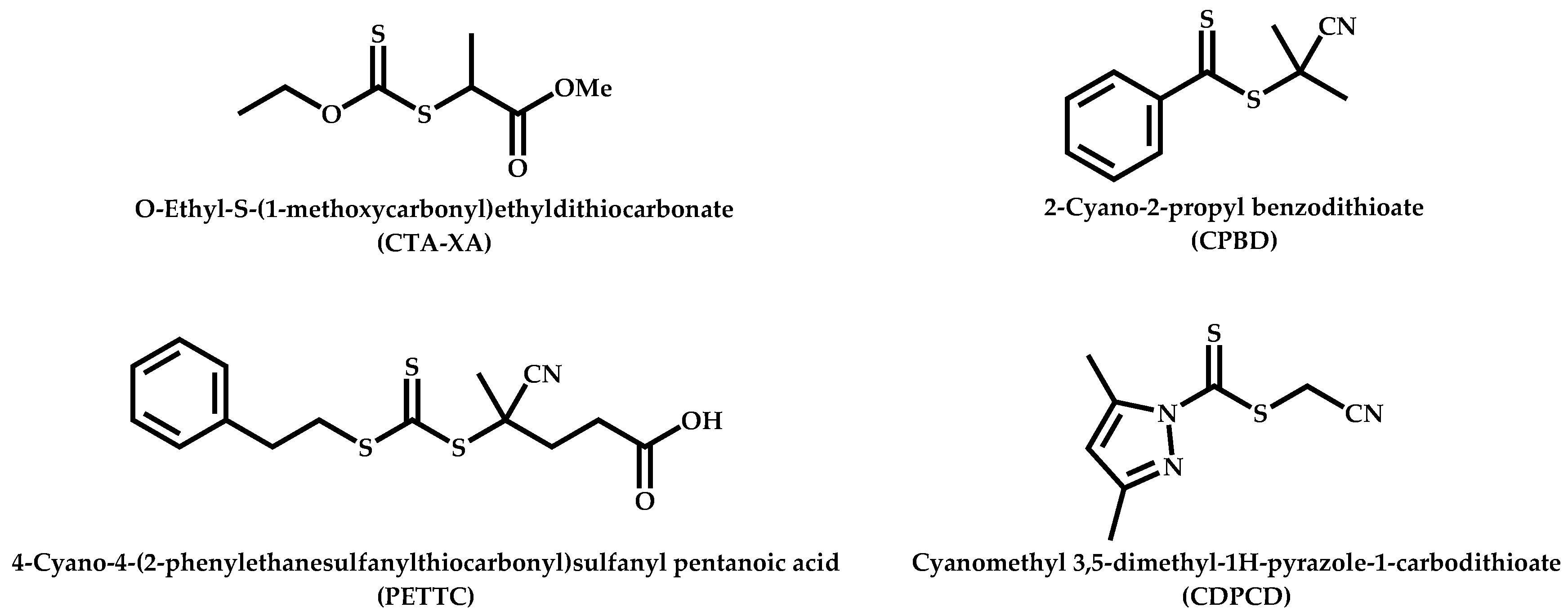
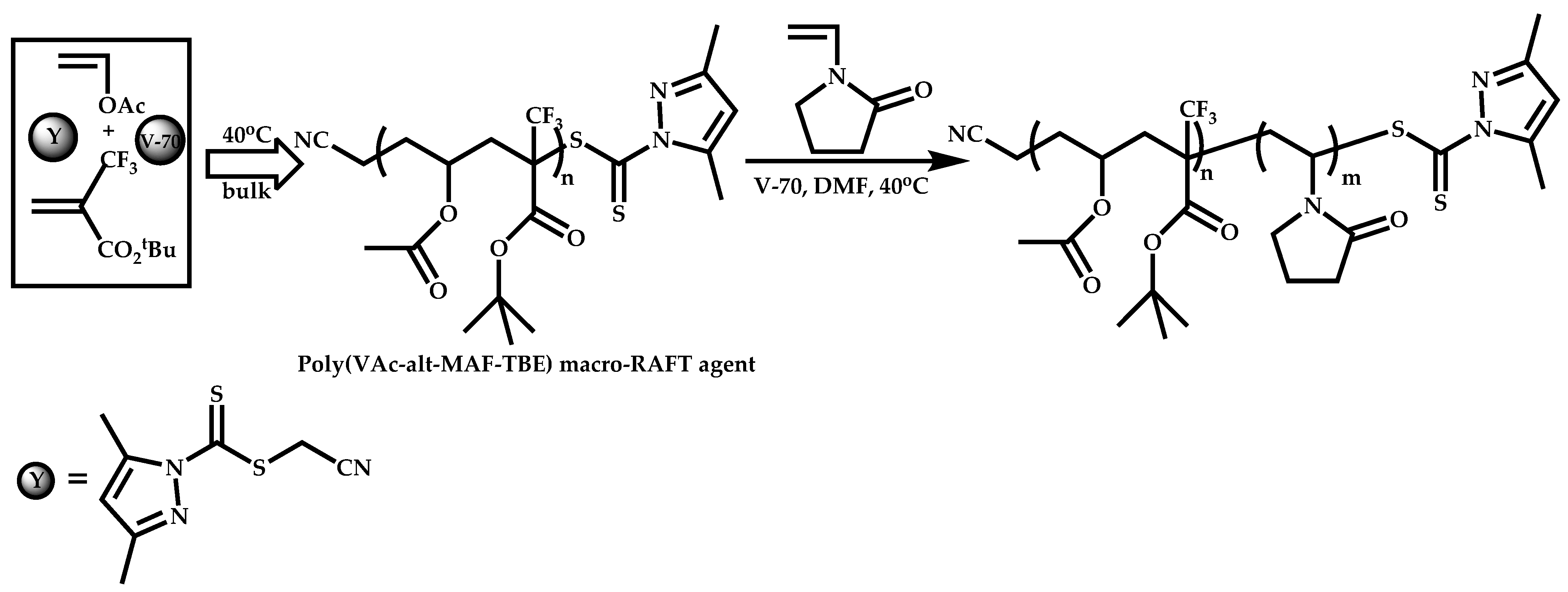
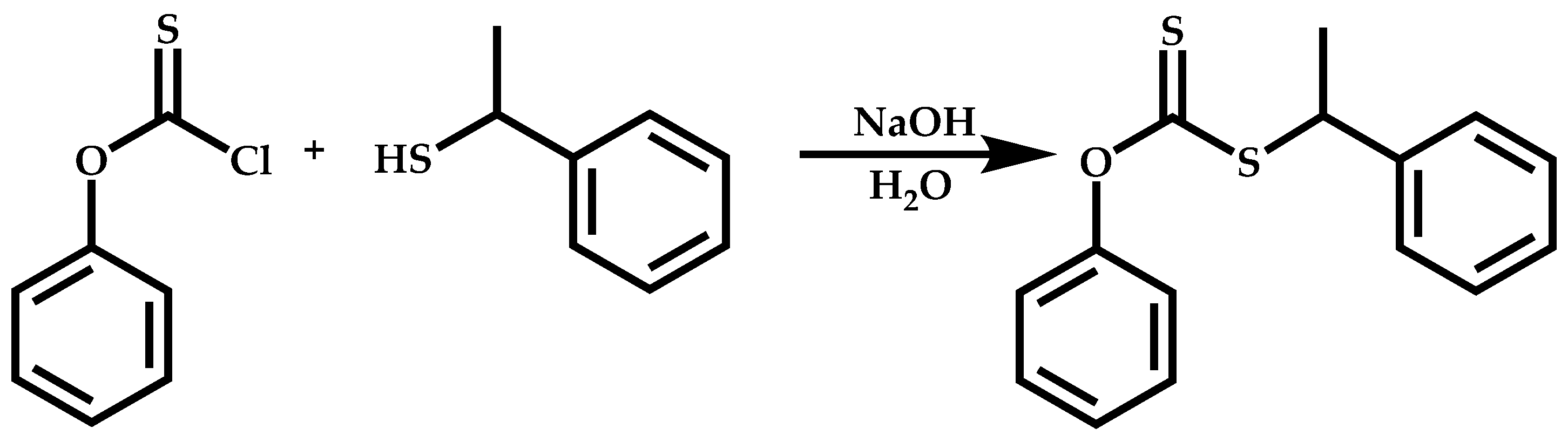
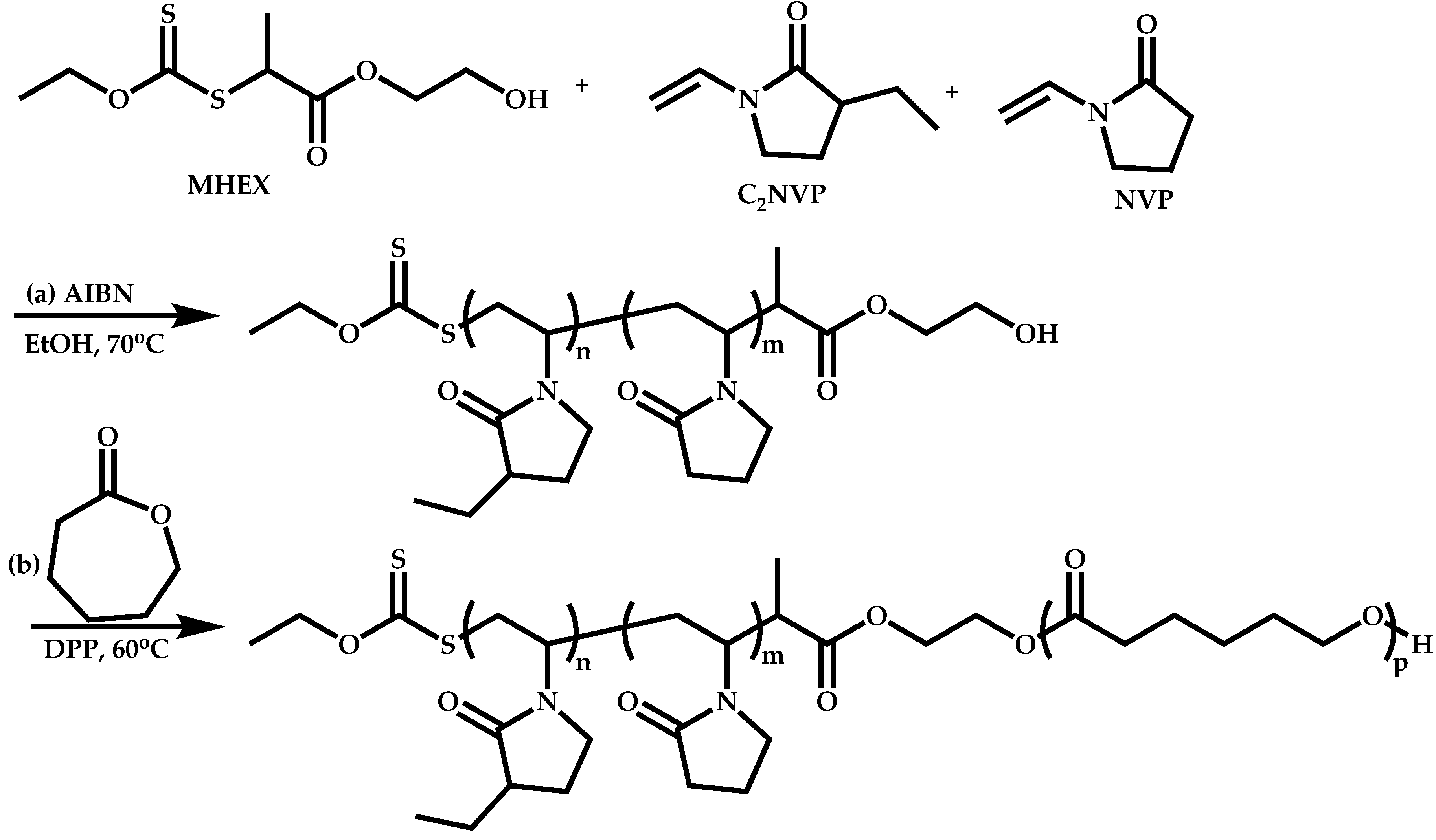
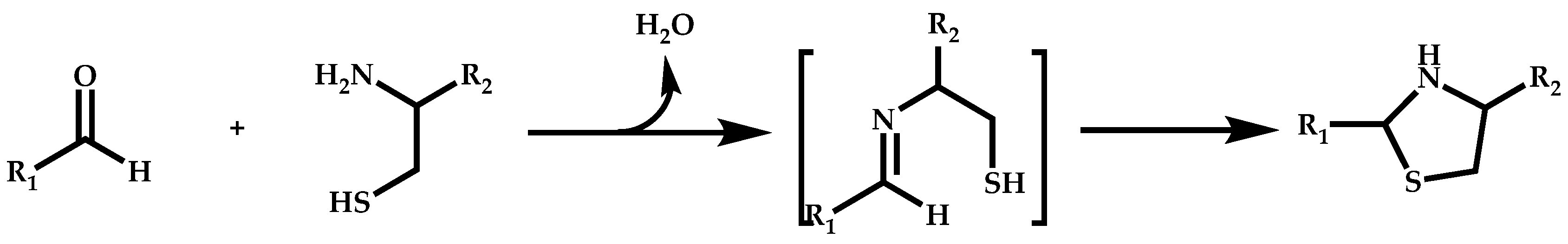
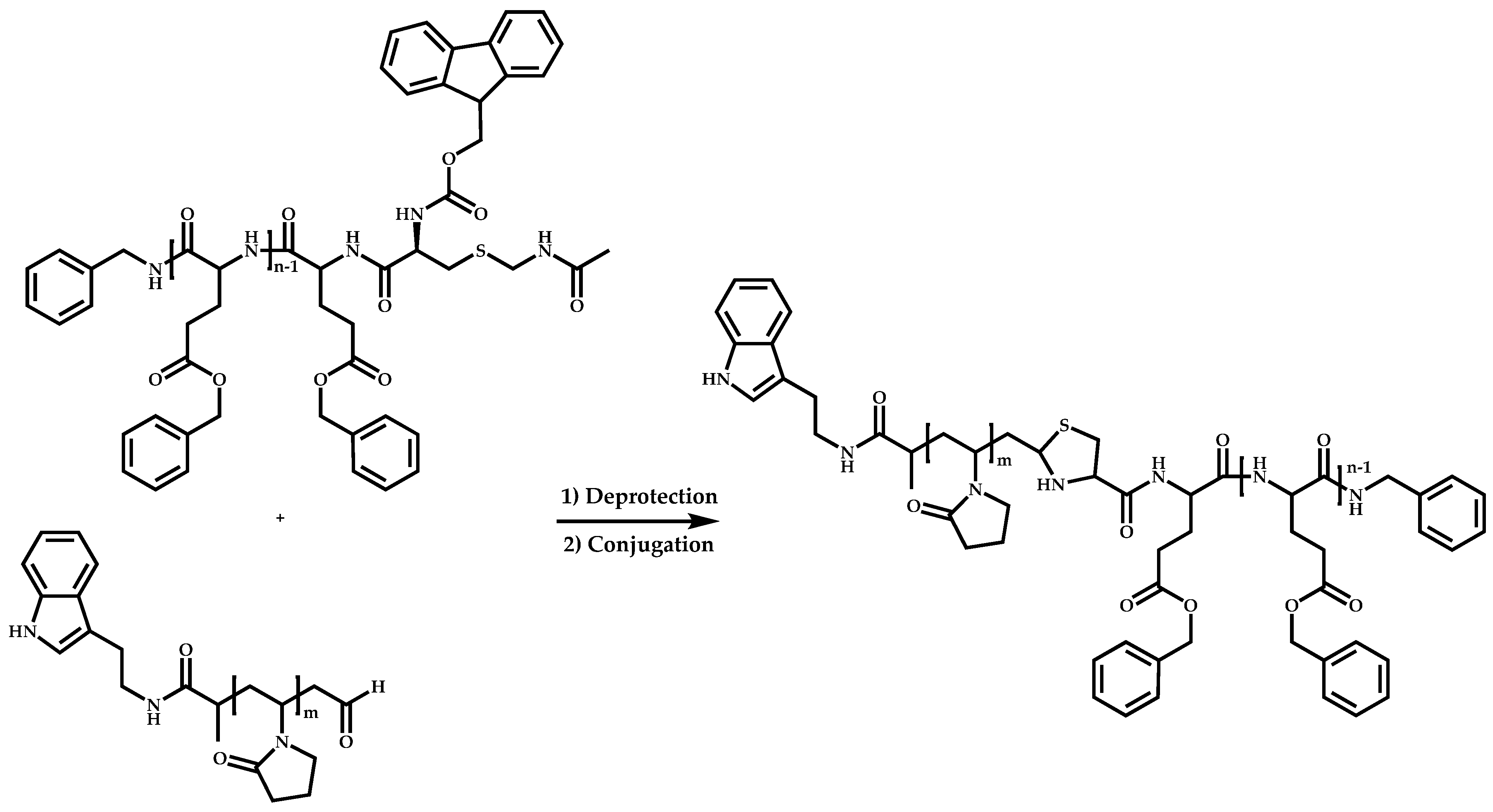
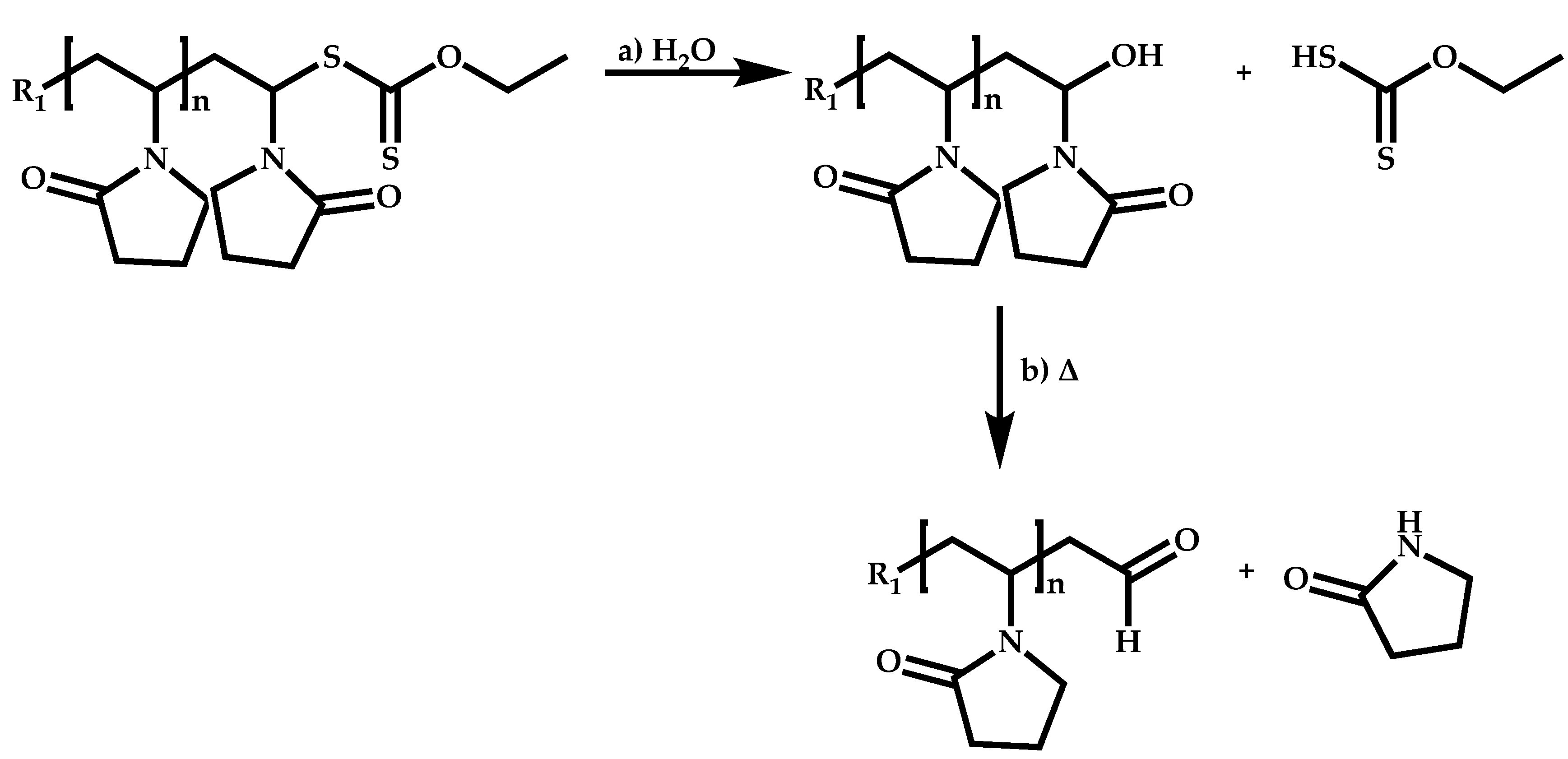
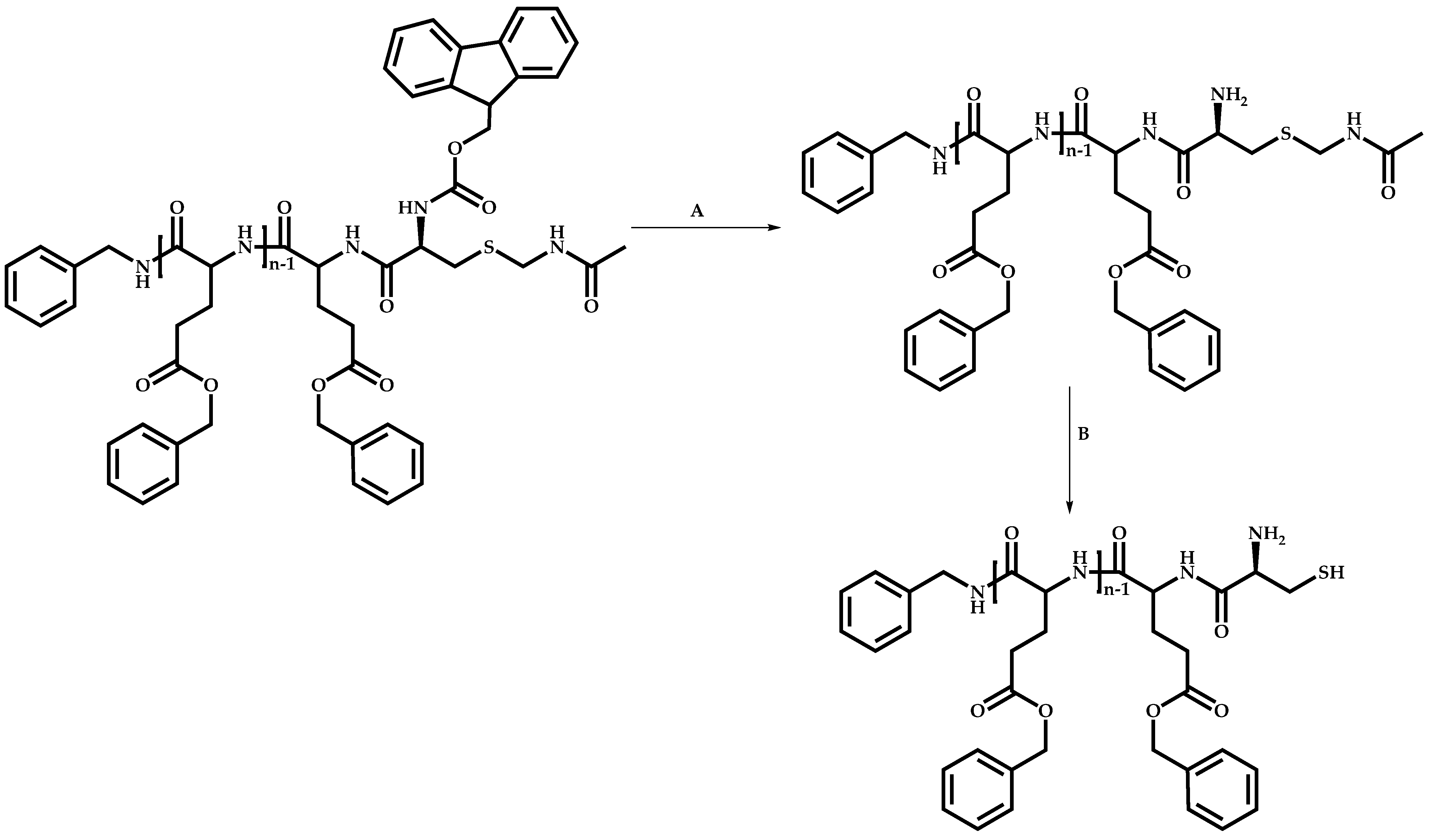
Functional CTAs
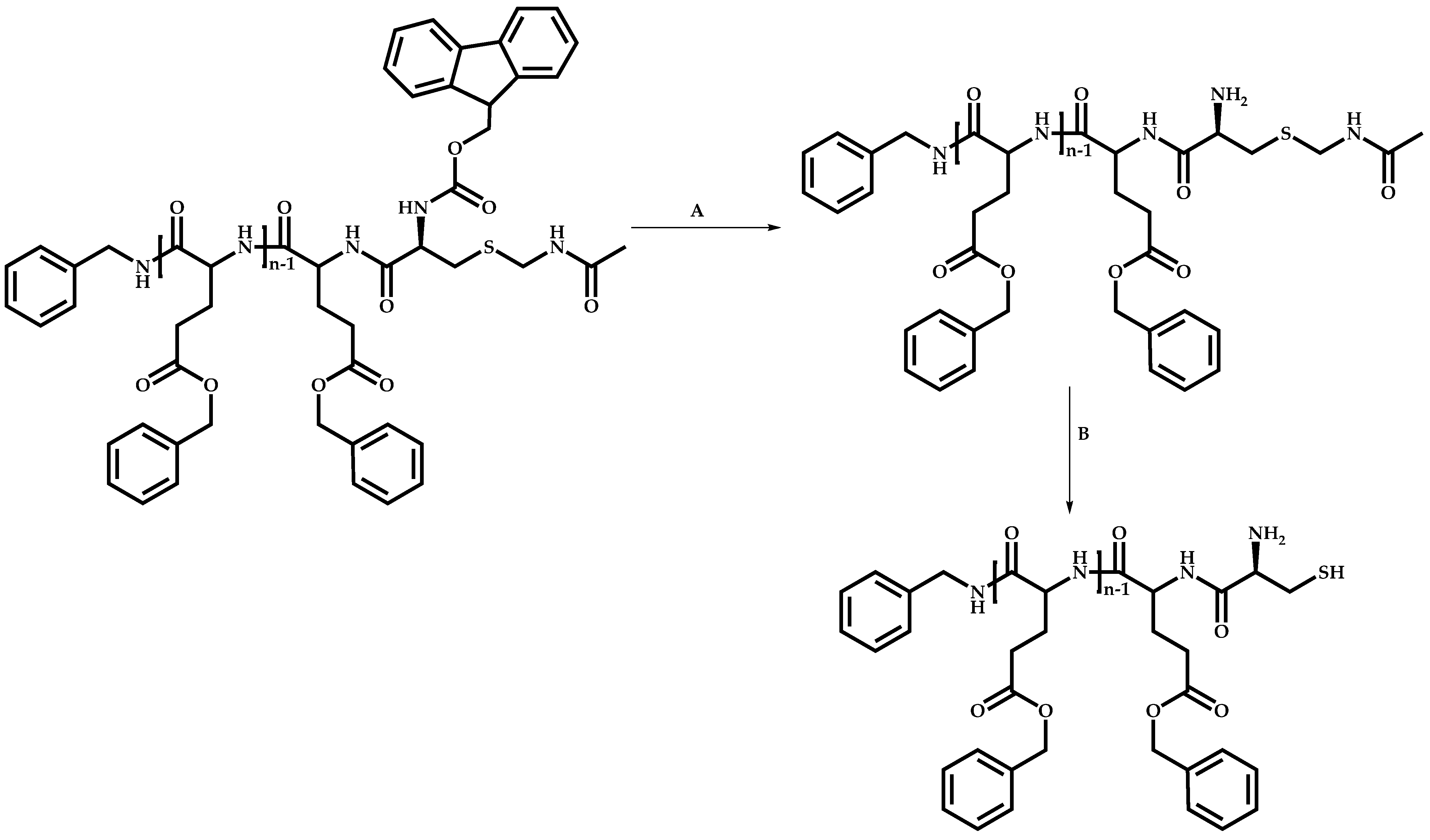
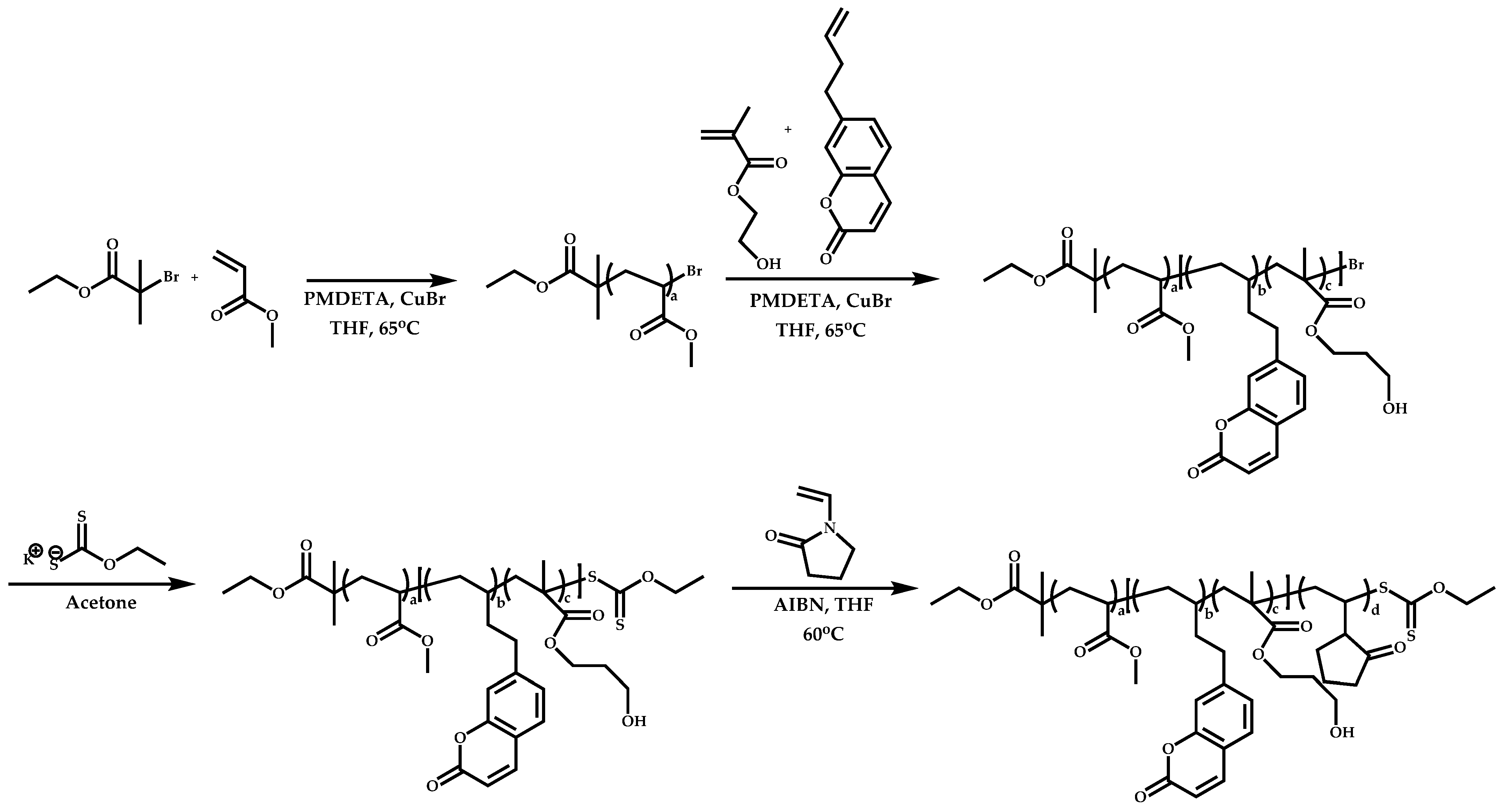
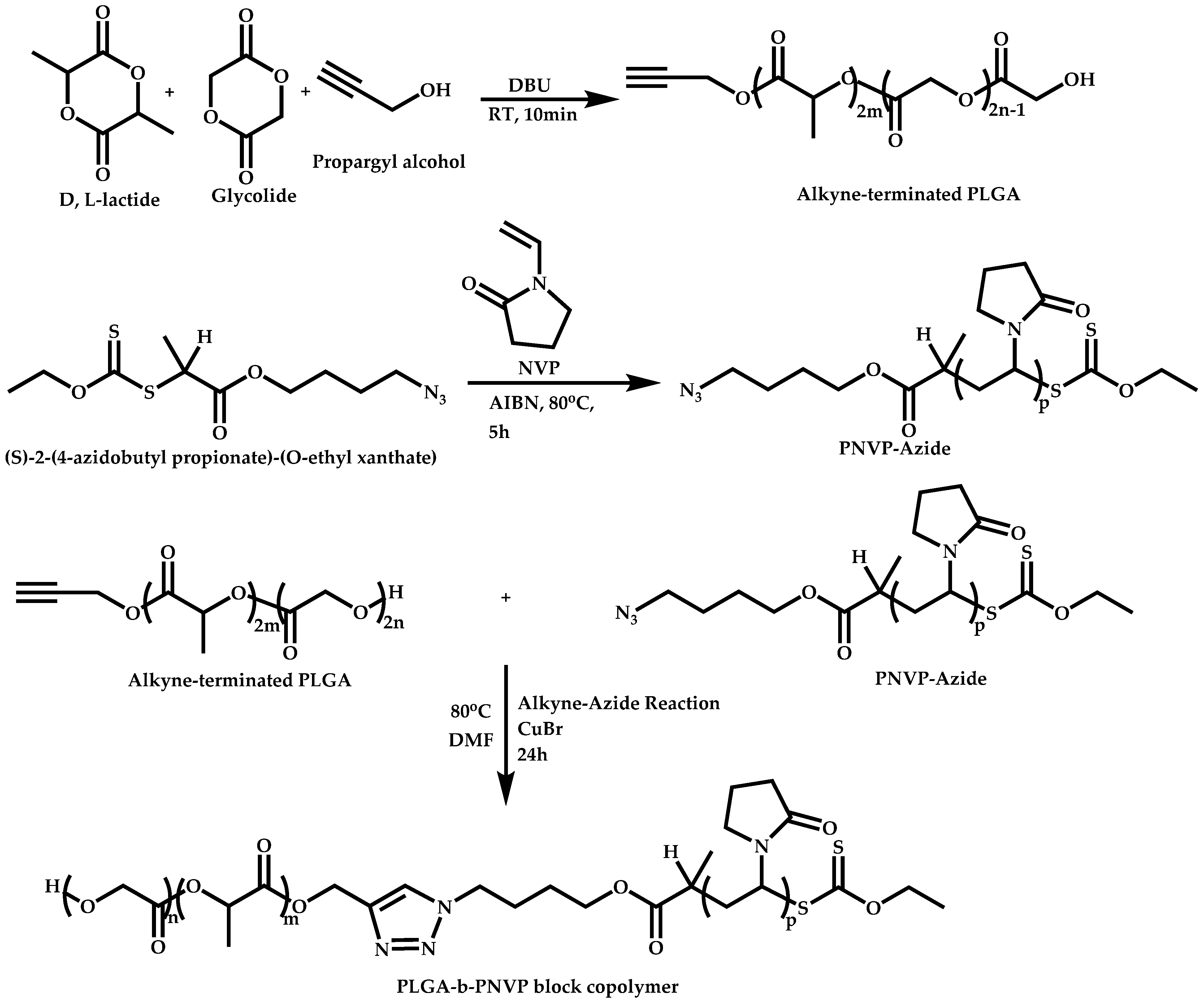
Linking Reactions of Individual Blocks
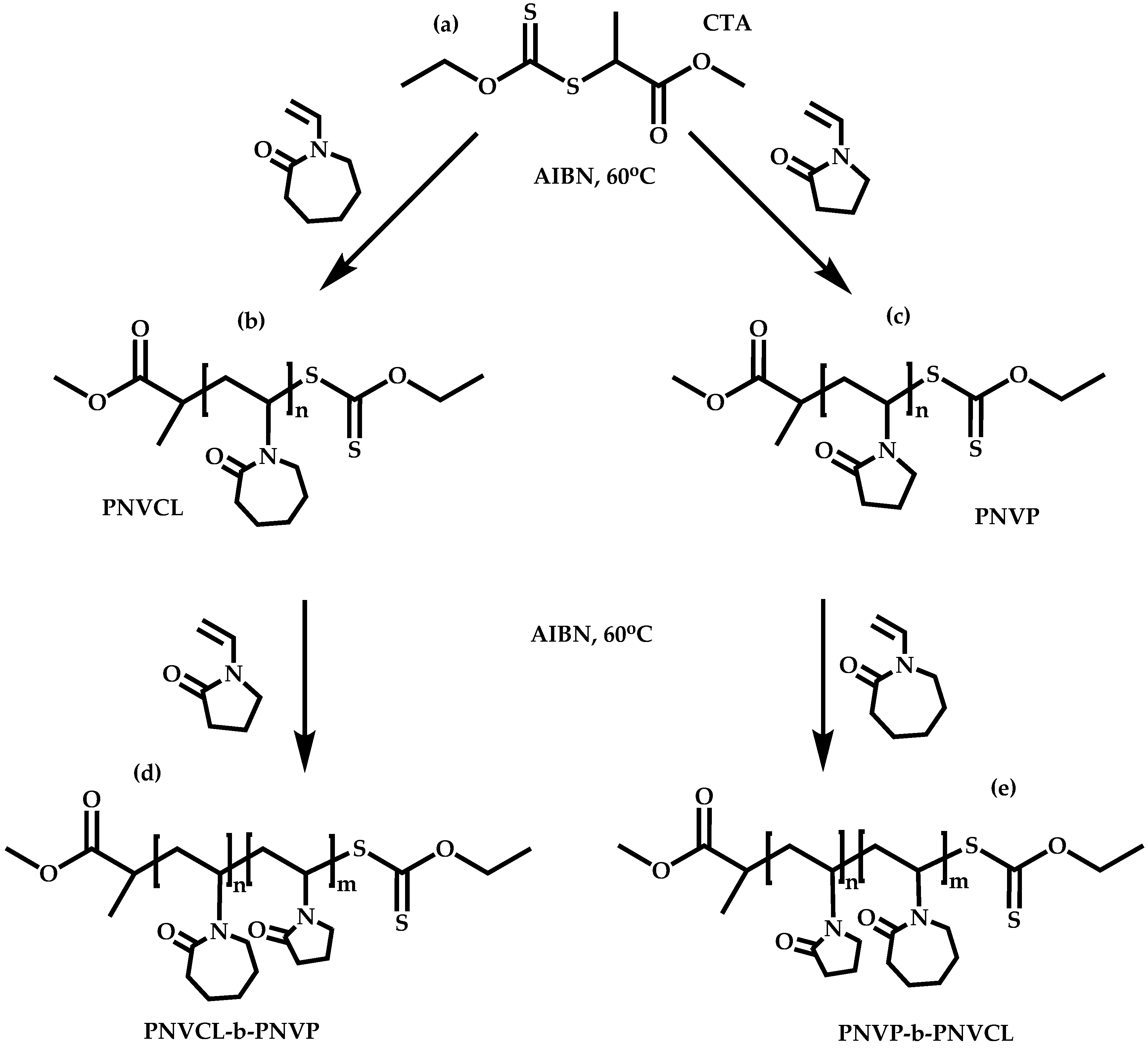
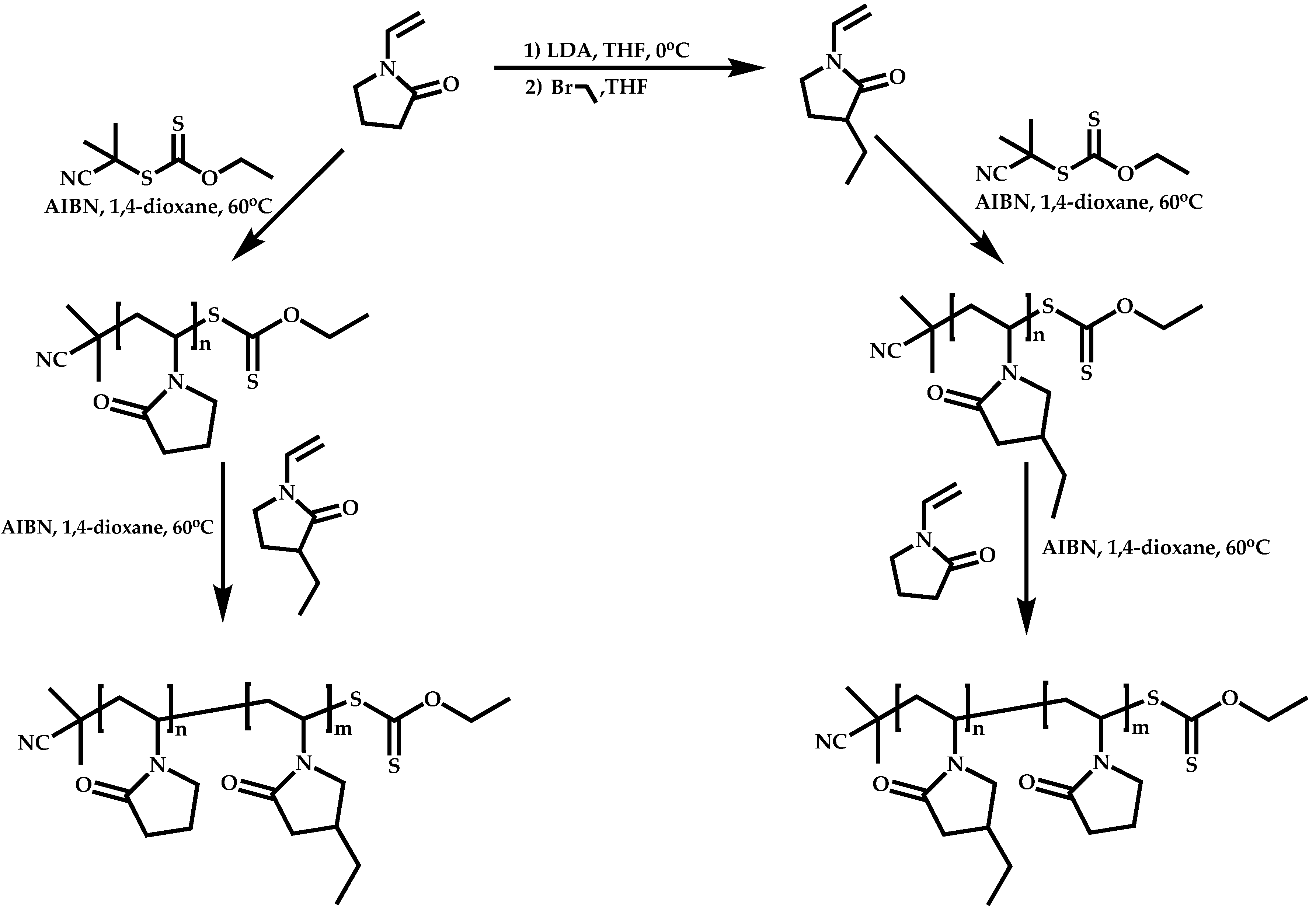
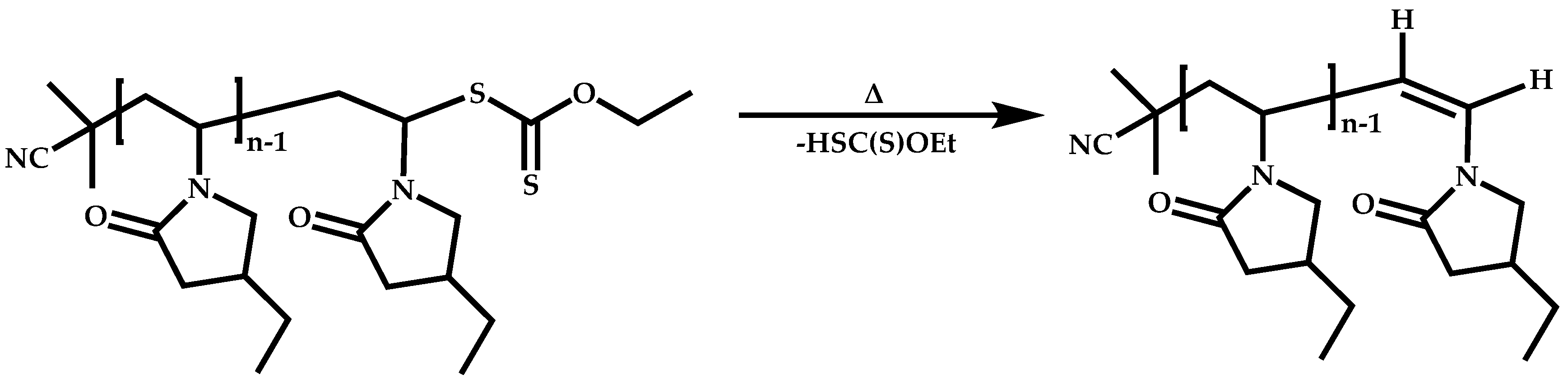
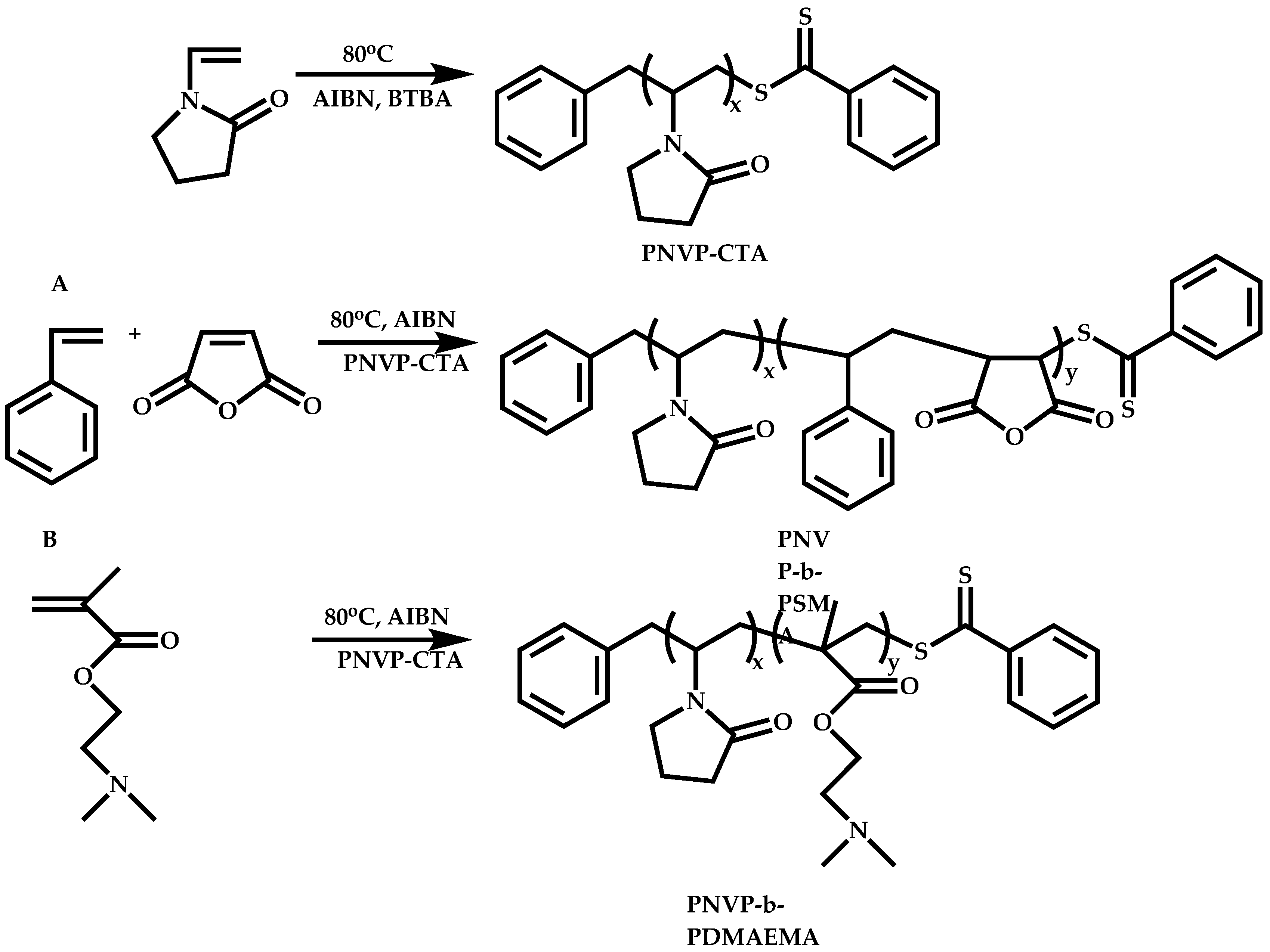
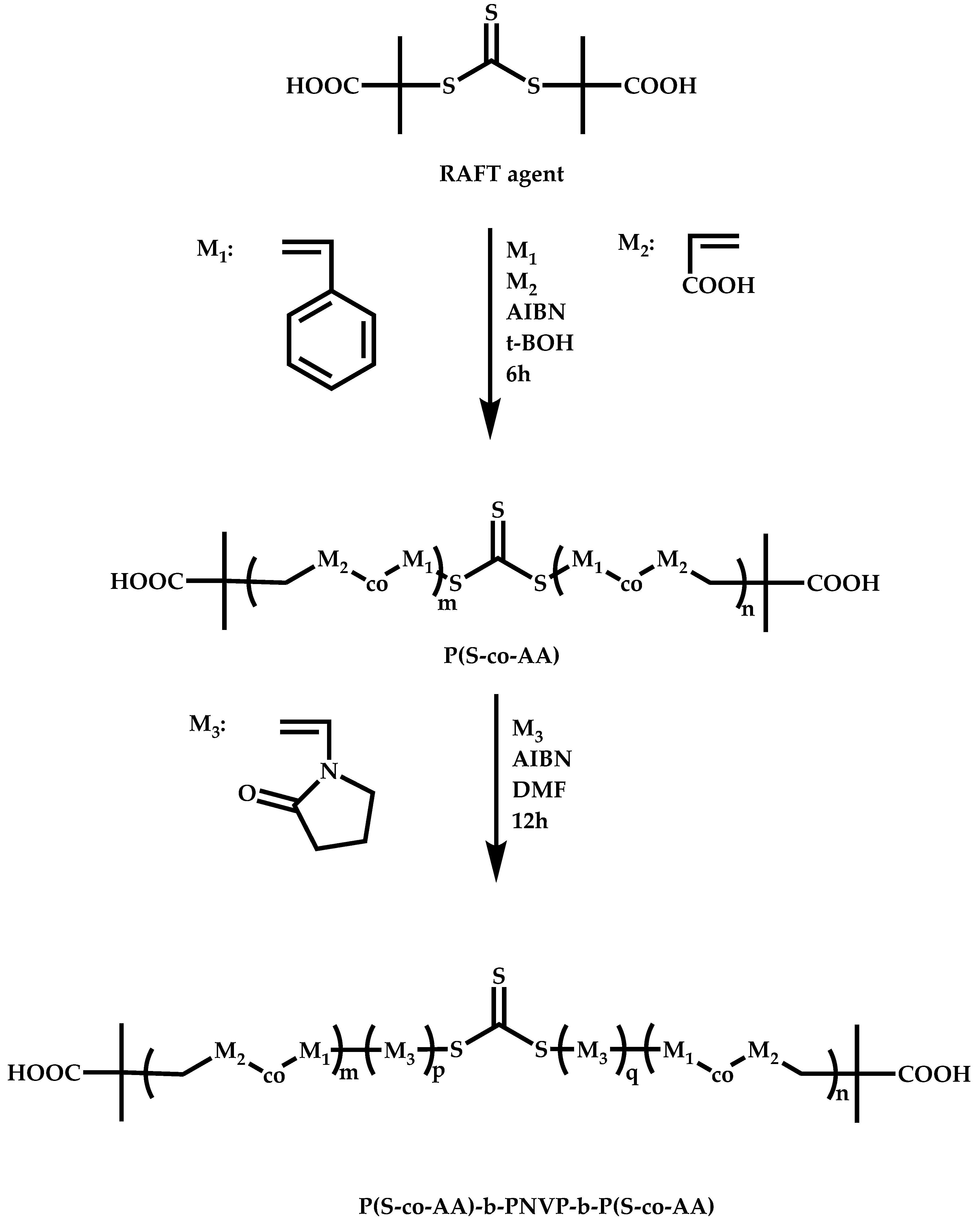
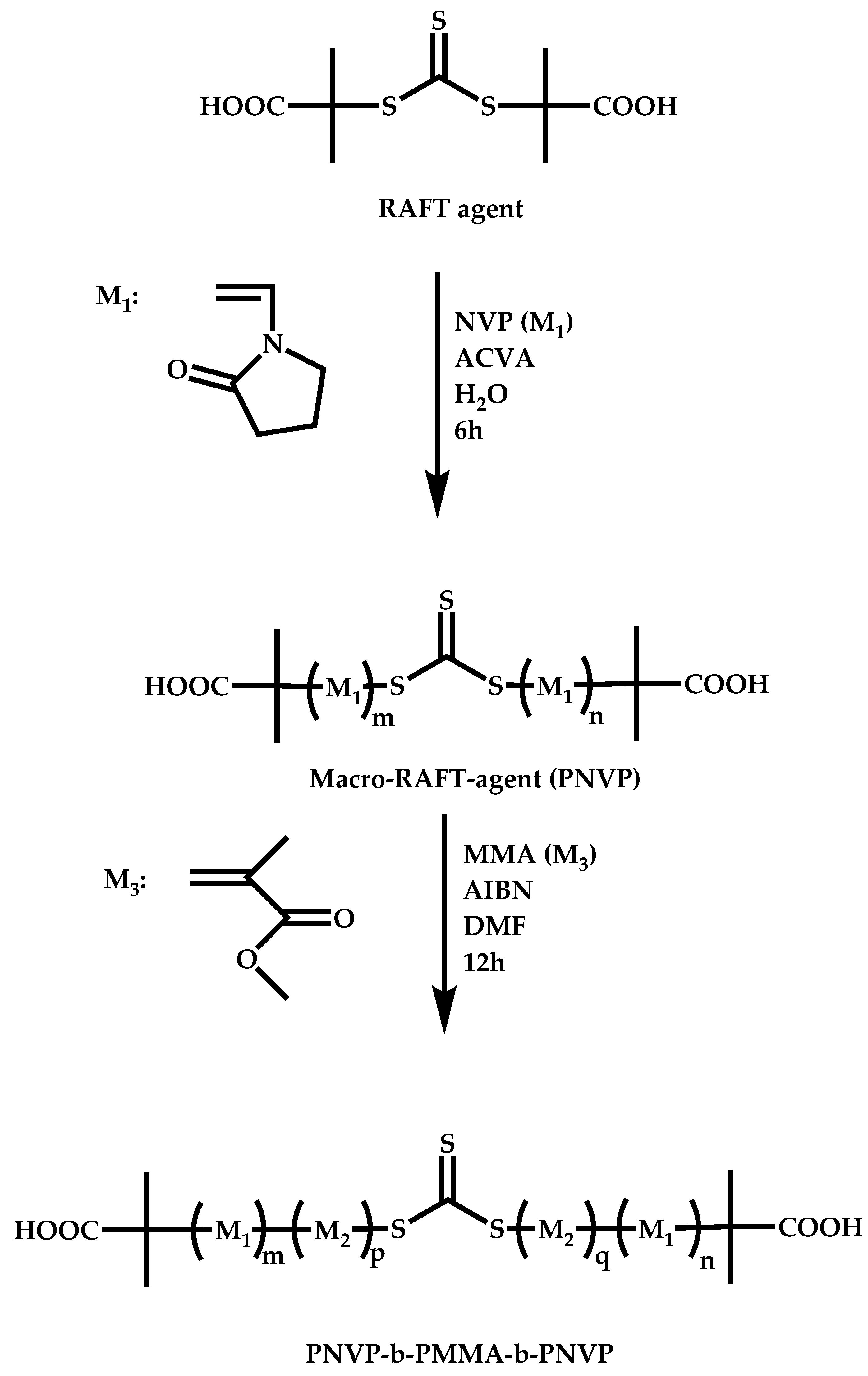
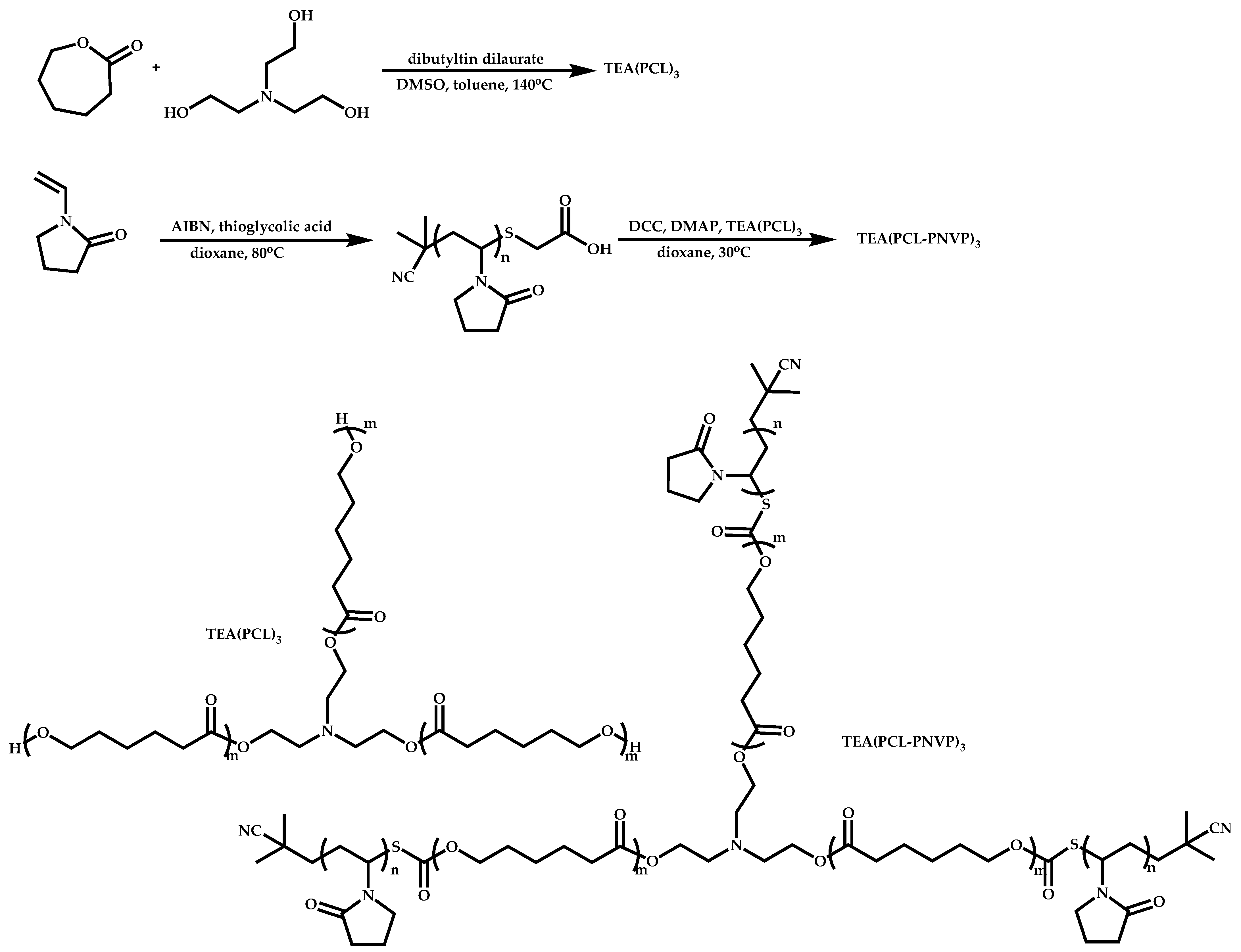
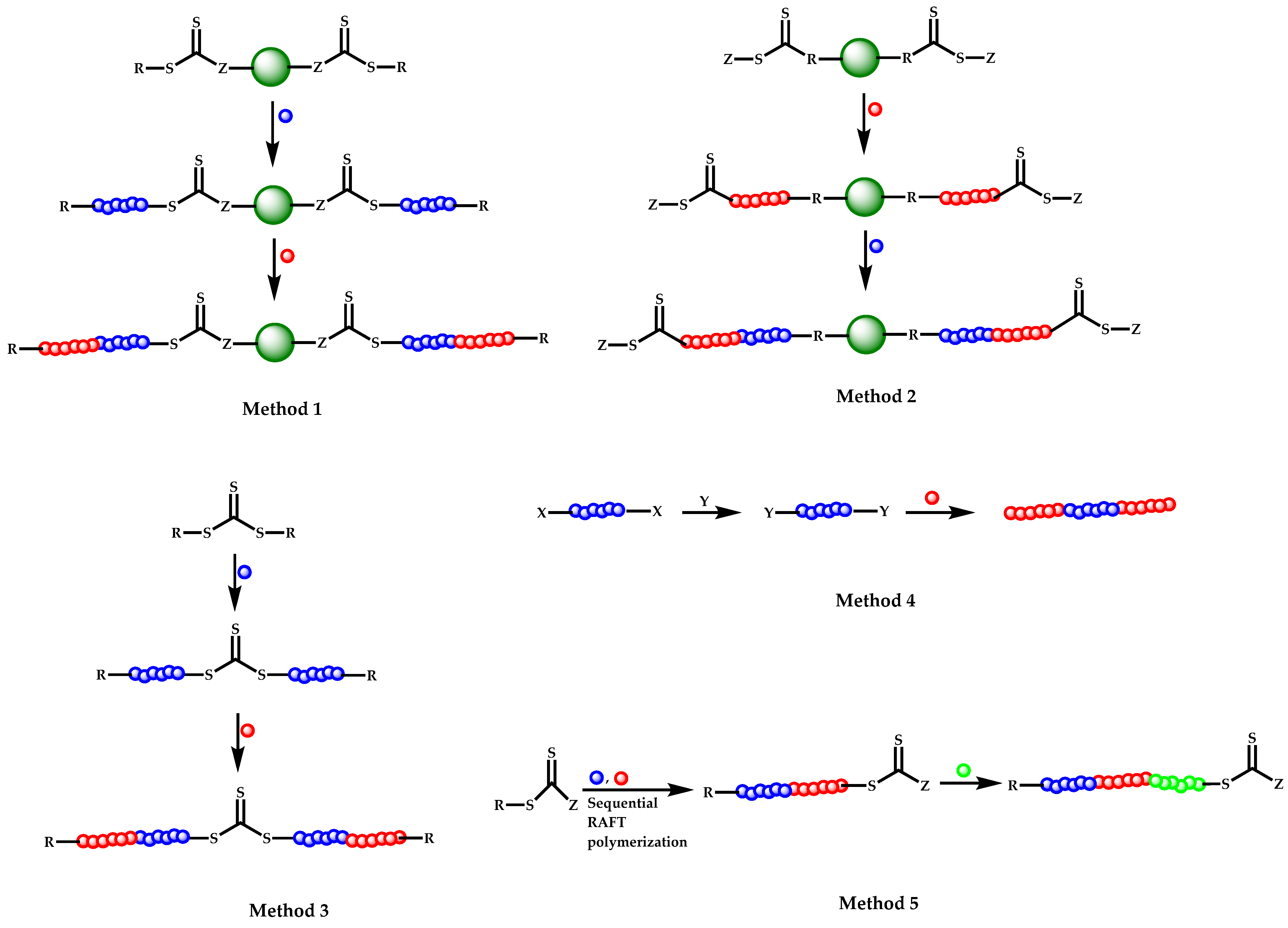
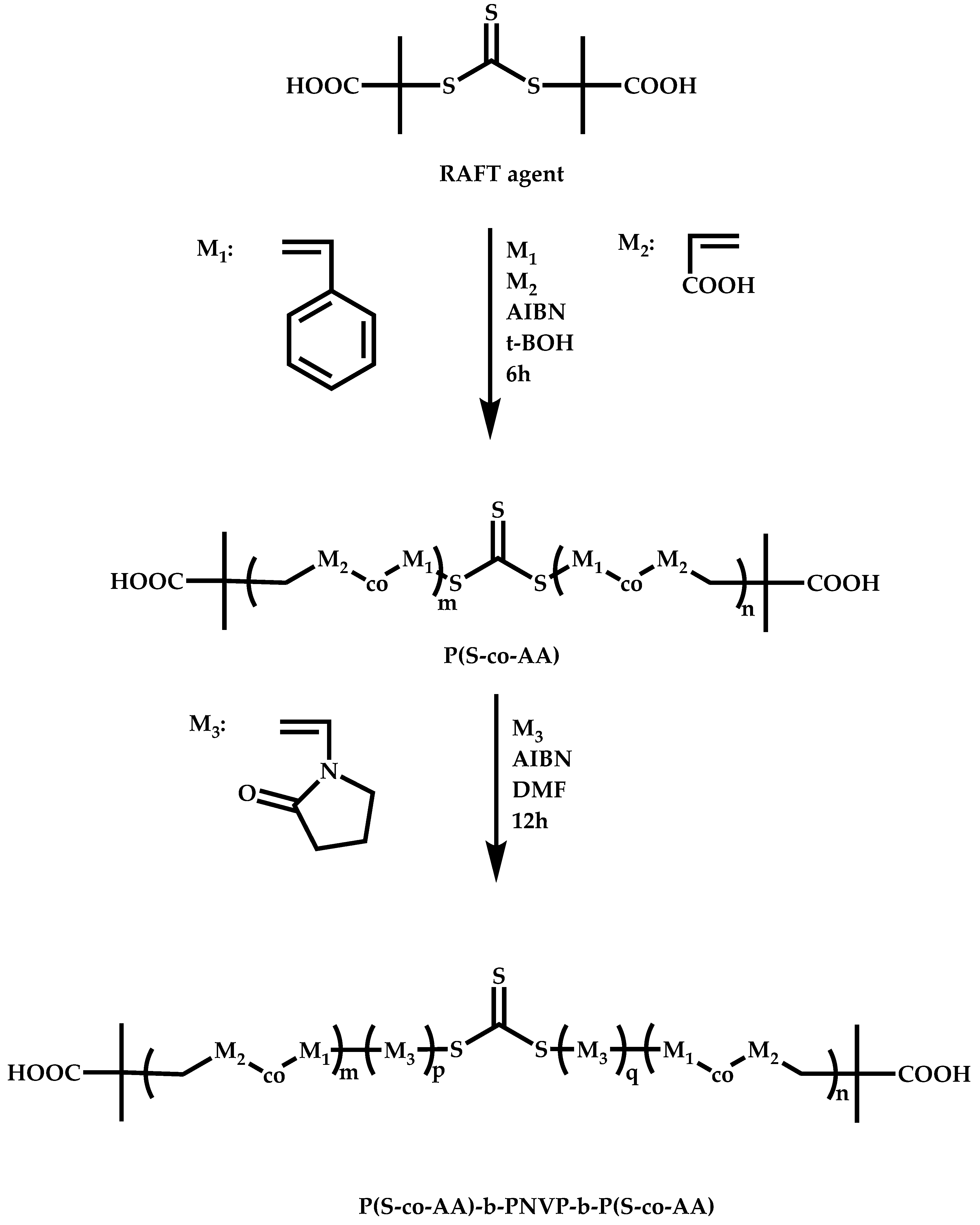
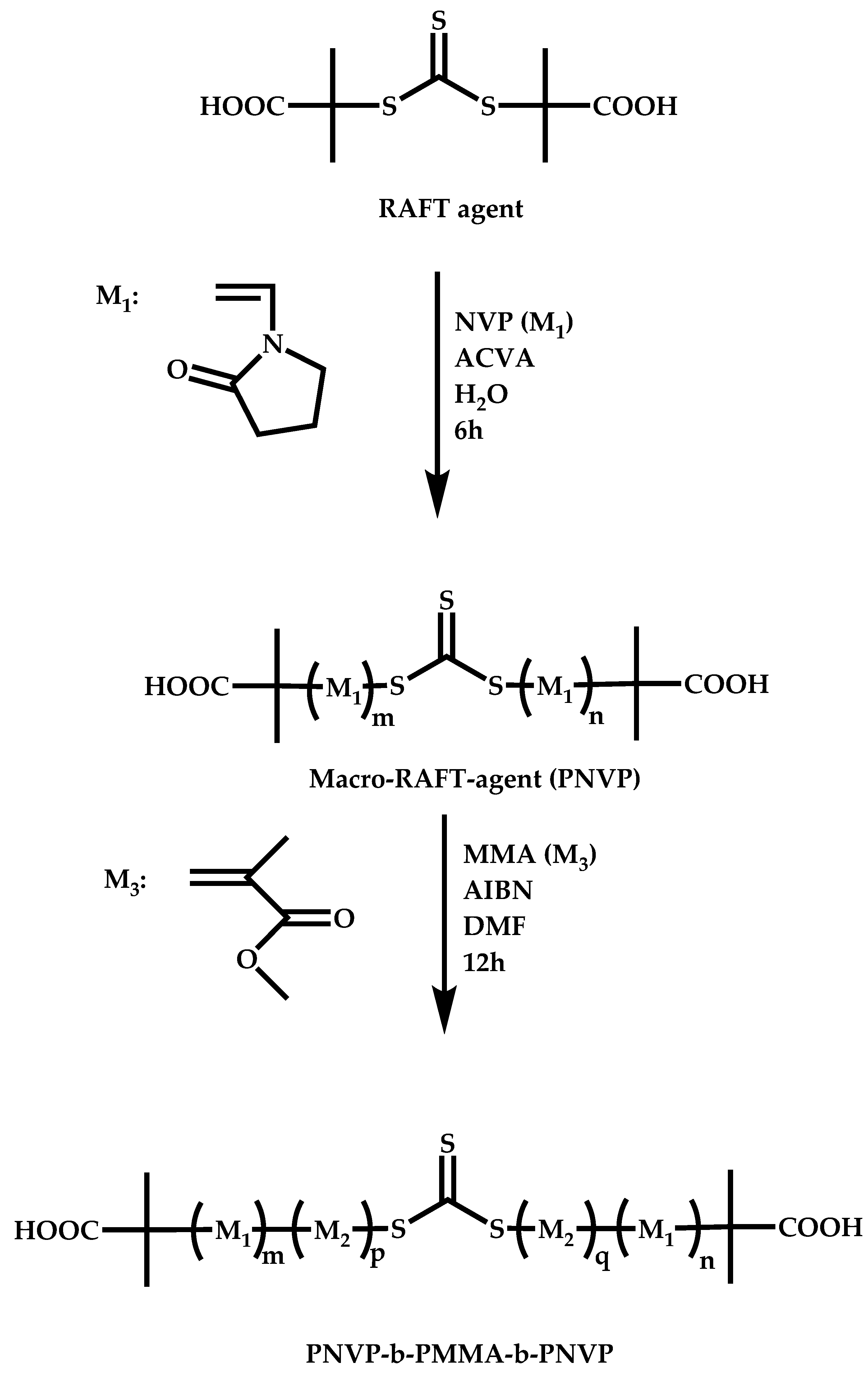
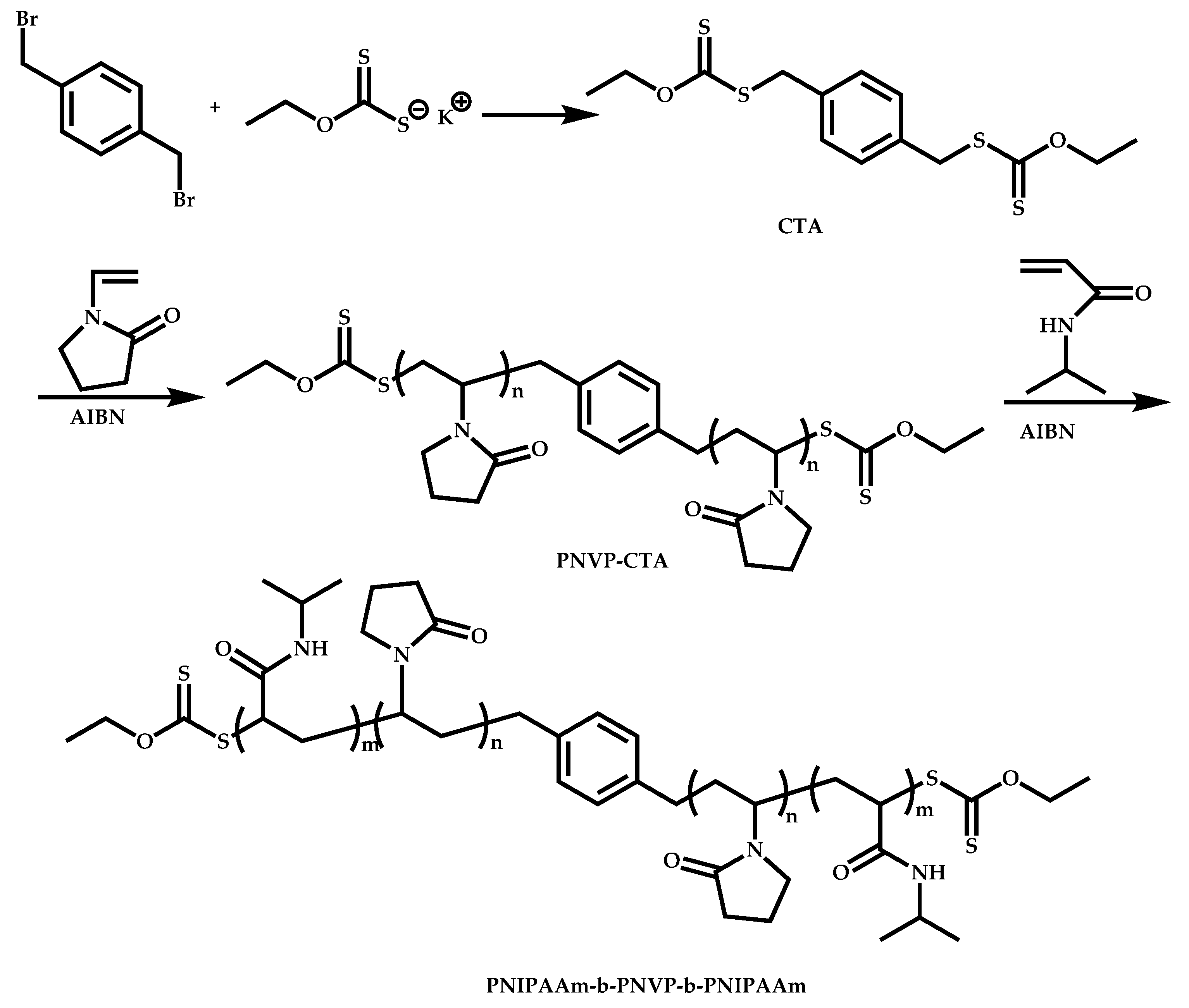
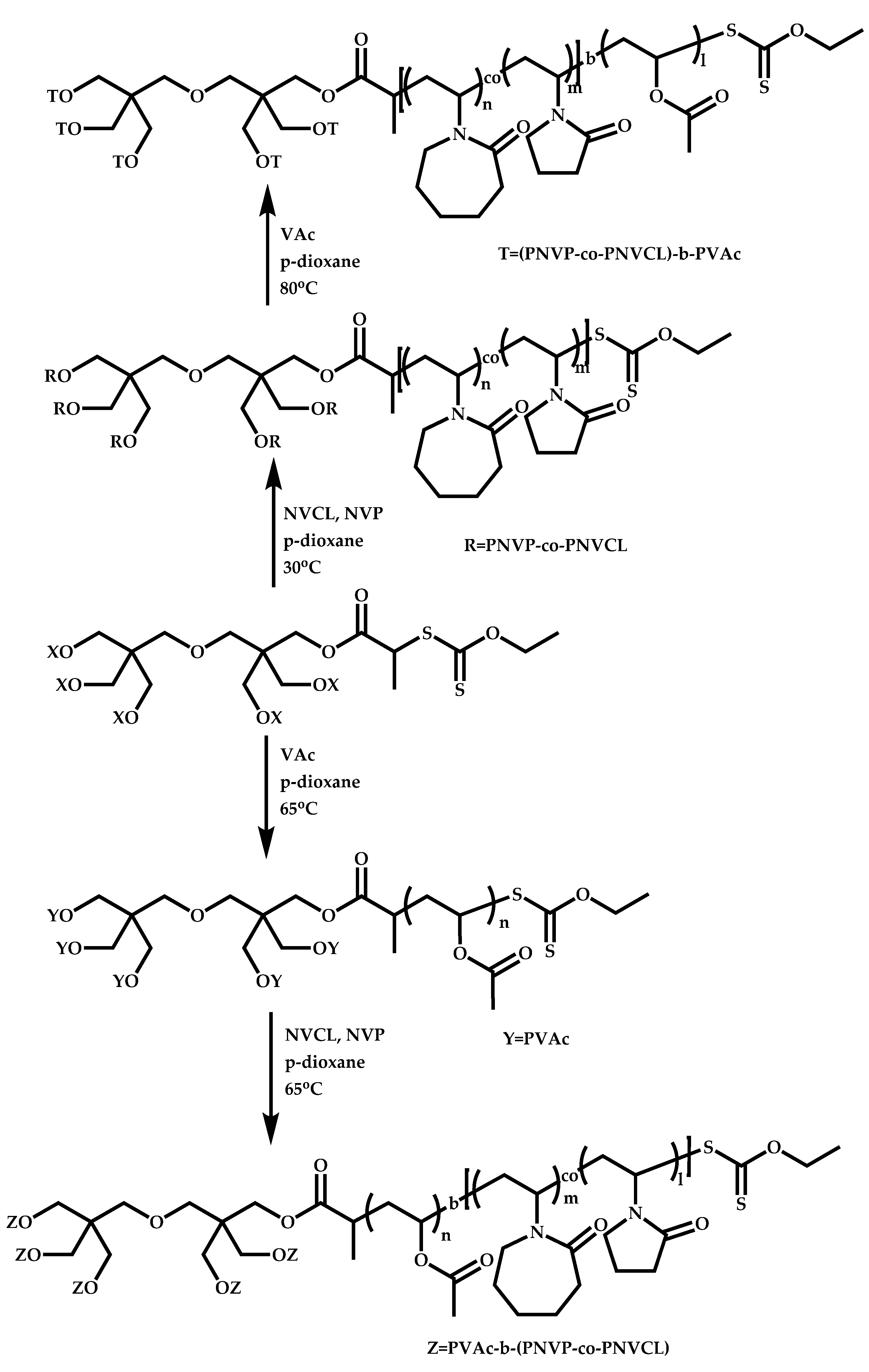
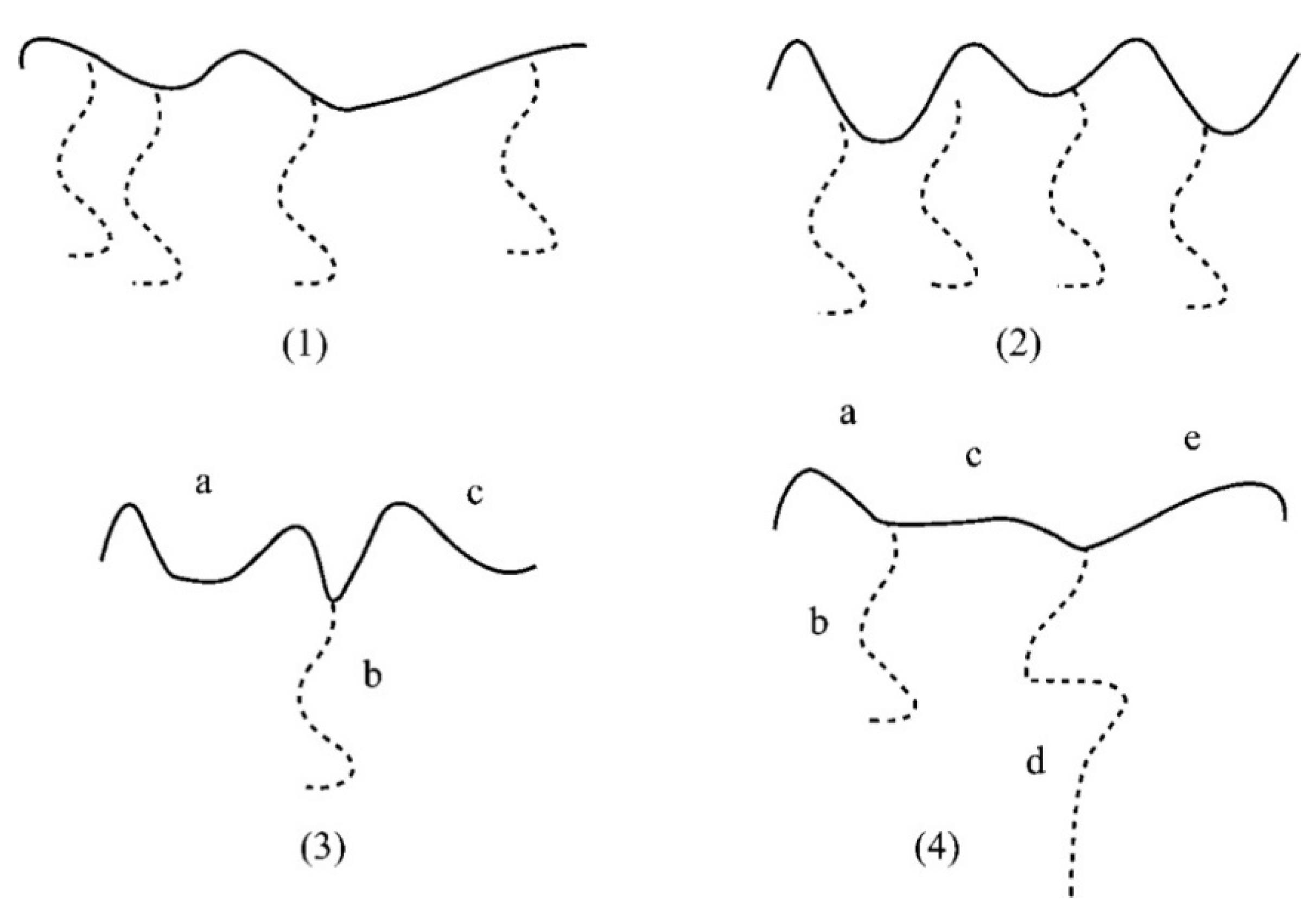
4.2.4. Triblock Copolymers and Terpolymers Based on NVP Synthesized via RAFT Polymerization
- (a)
- Symmetric triblock copolymers of the type A-b-B-b-A, where the A blocks have the same molecular weight.
- (b)
- Asymmetric triblock copolymers of the type A-b-B-b-A’, where the A and A’ blocks are chemically identical but have different molecular weight.
- (c)
- Triblock terpolymers of the type A-b-B-b-C.
- (a)
- Use of two CTAs chemically connected through the Z group.
- (b)
- Use of two CTAs chemically connected through the R group.
- (c)
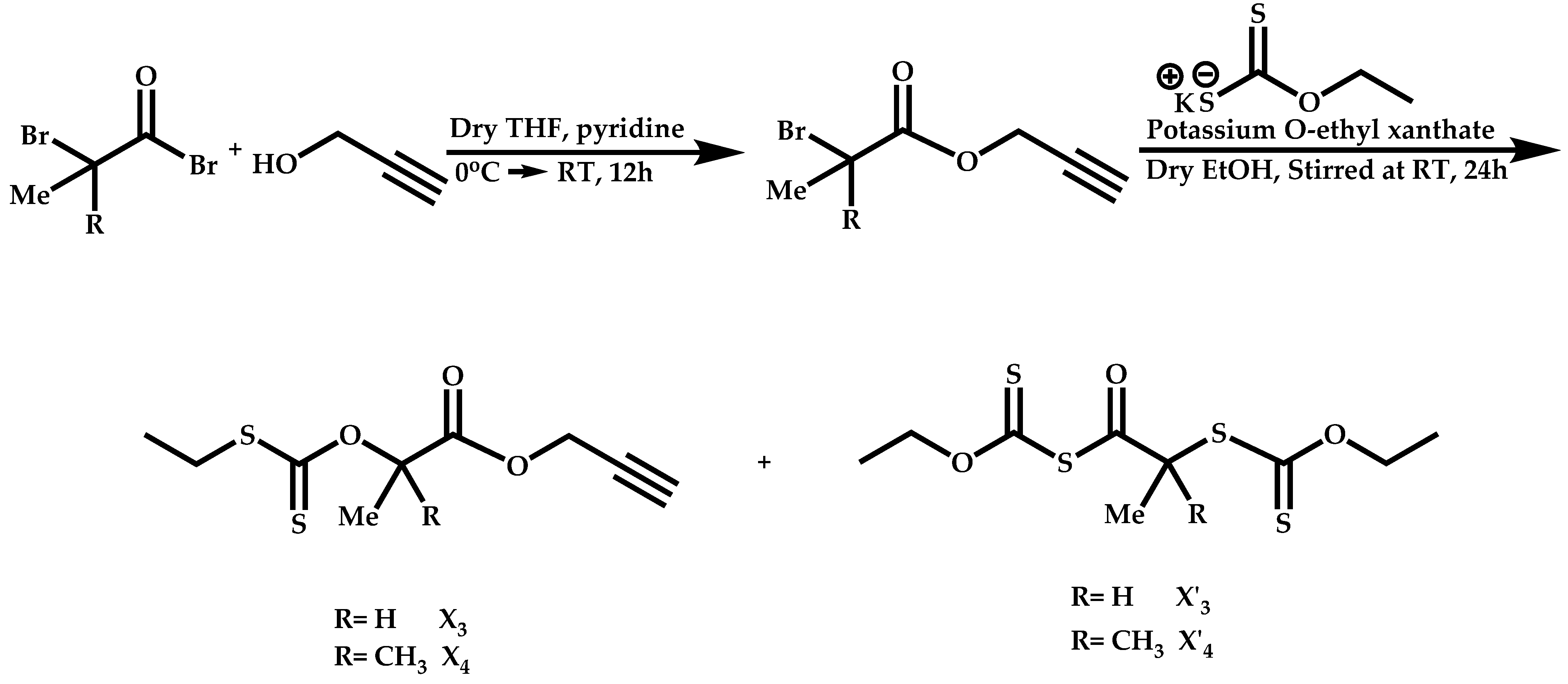
- Use of double CTAs, e.g., CTAs with two leaving groups Z on the same molecule.
- (d)
- Sequential addition of monomers using the same monofunctional CTA.
- (e)
- End group functionalization of a polymer prepared by a non-RAFT methodology in order to incorporate a suitable CTA moiety, followed by RAFT polymerization of one or two suitable monomers.
- (f)
- Use of a functional CTA able to promote RAFT and another type of polymerization.
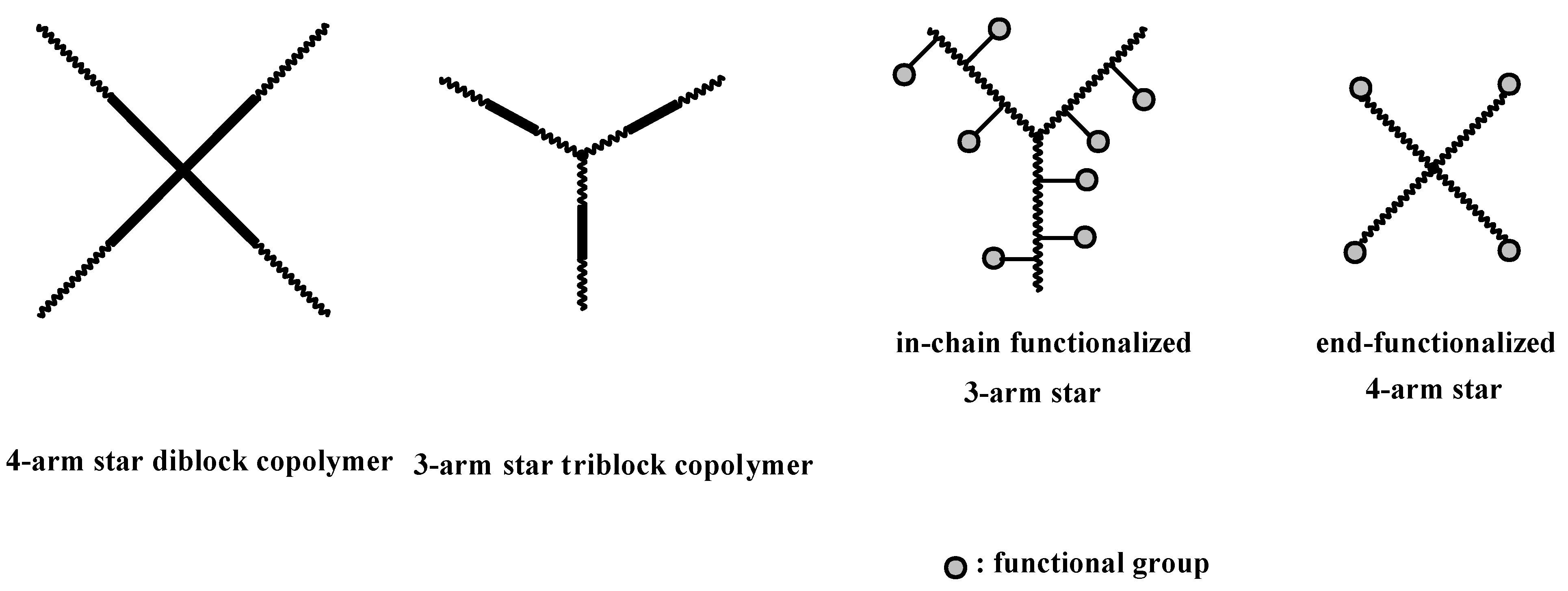
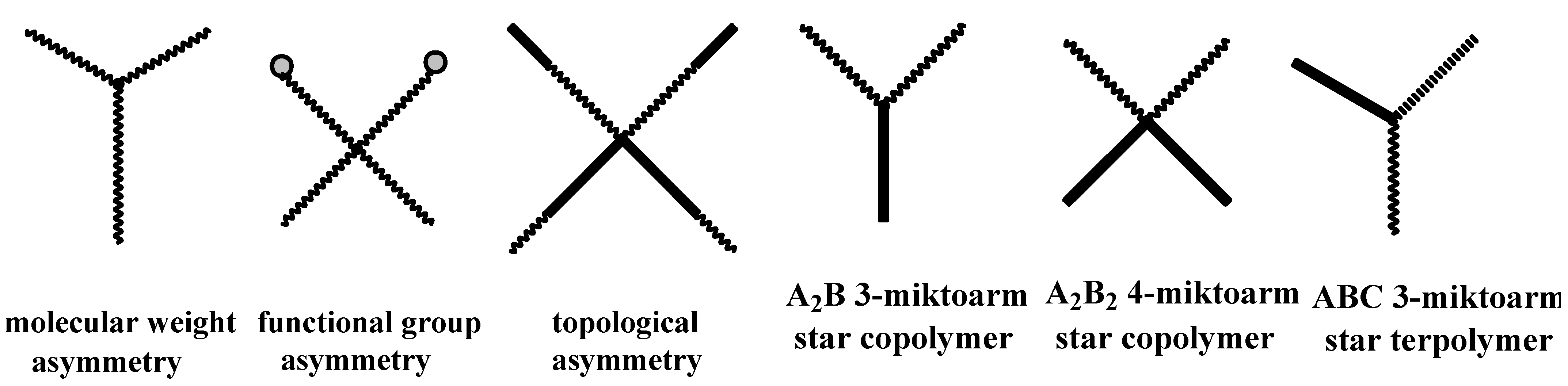
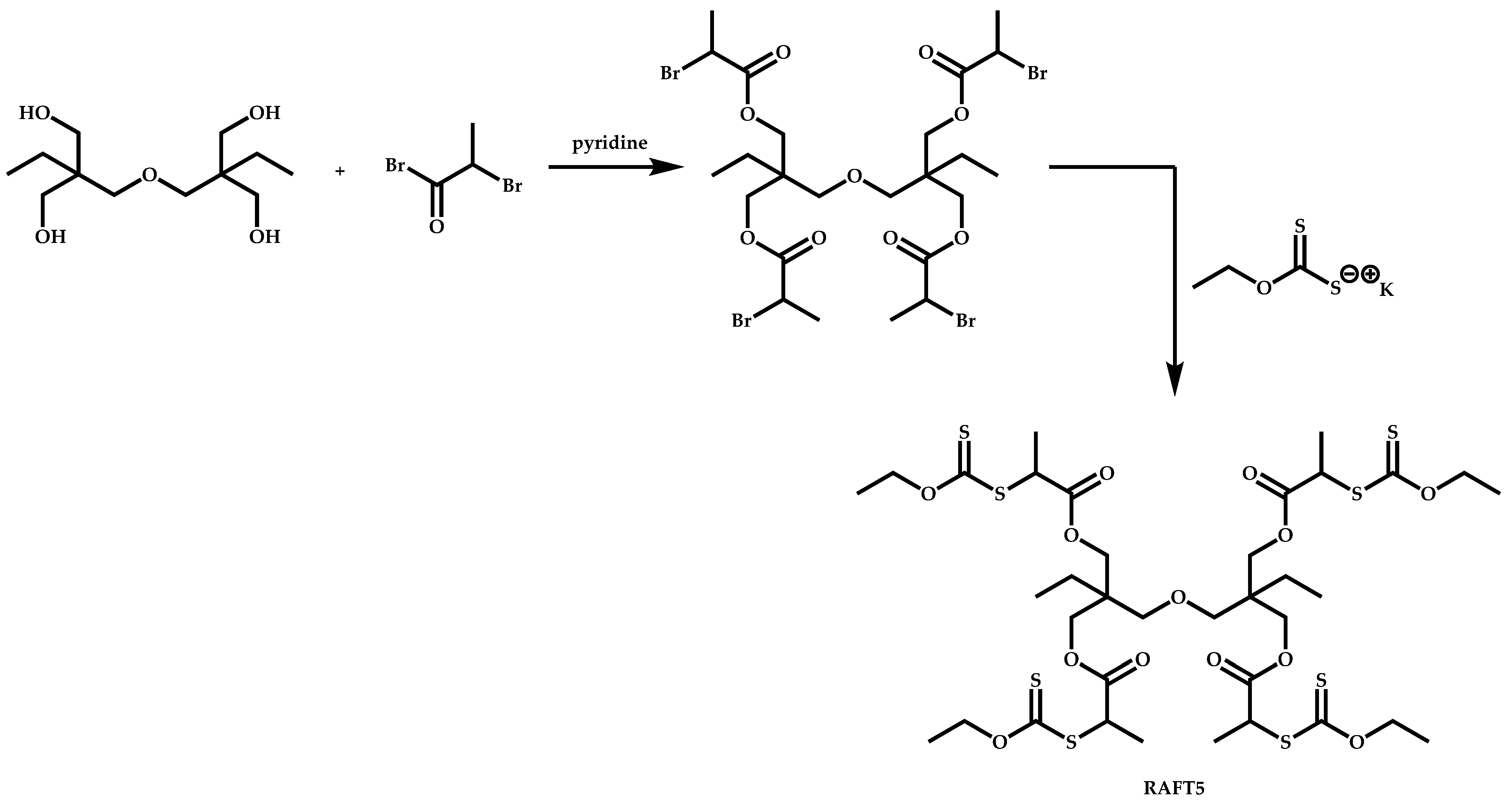
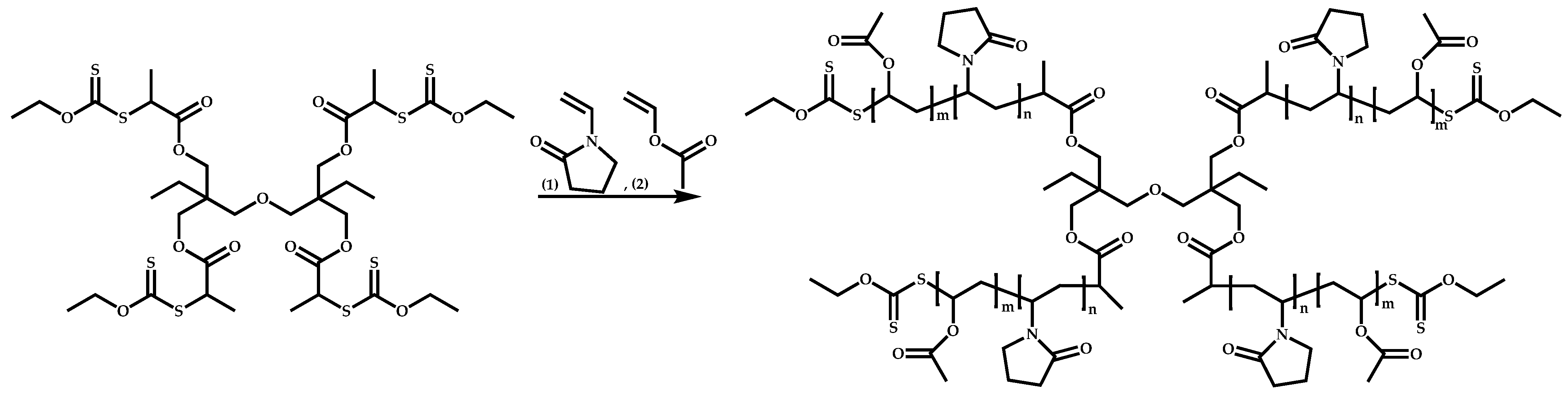
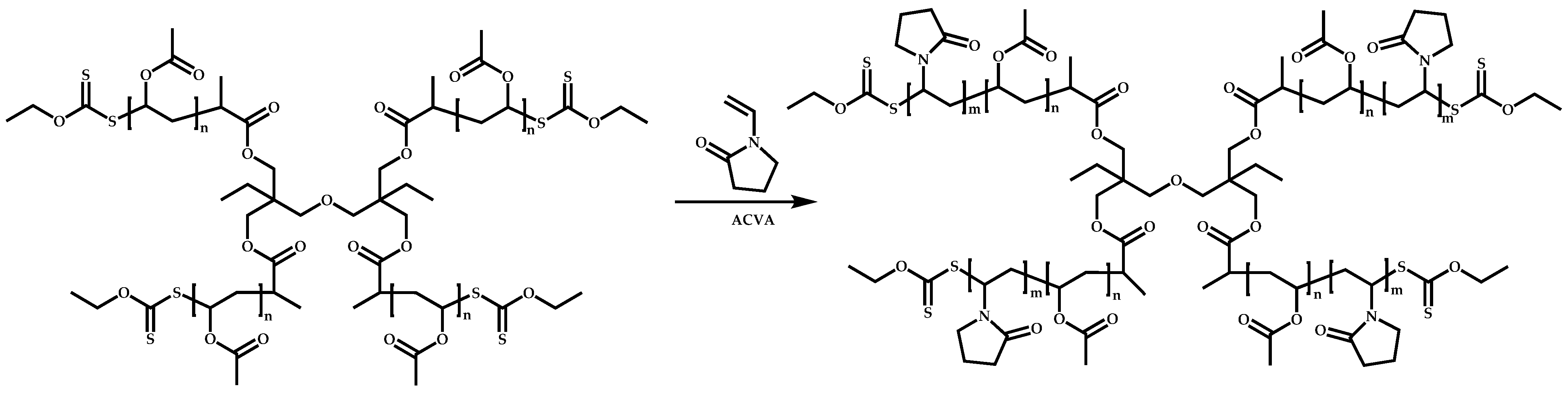
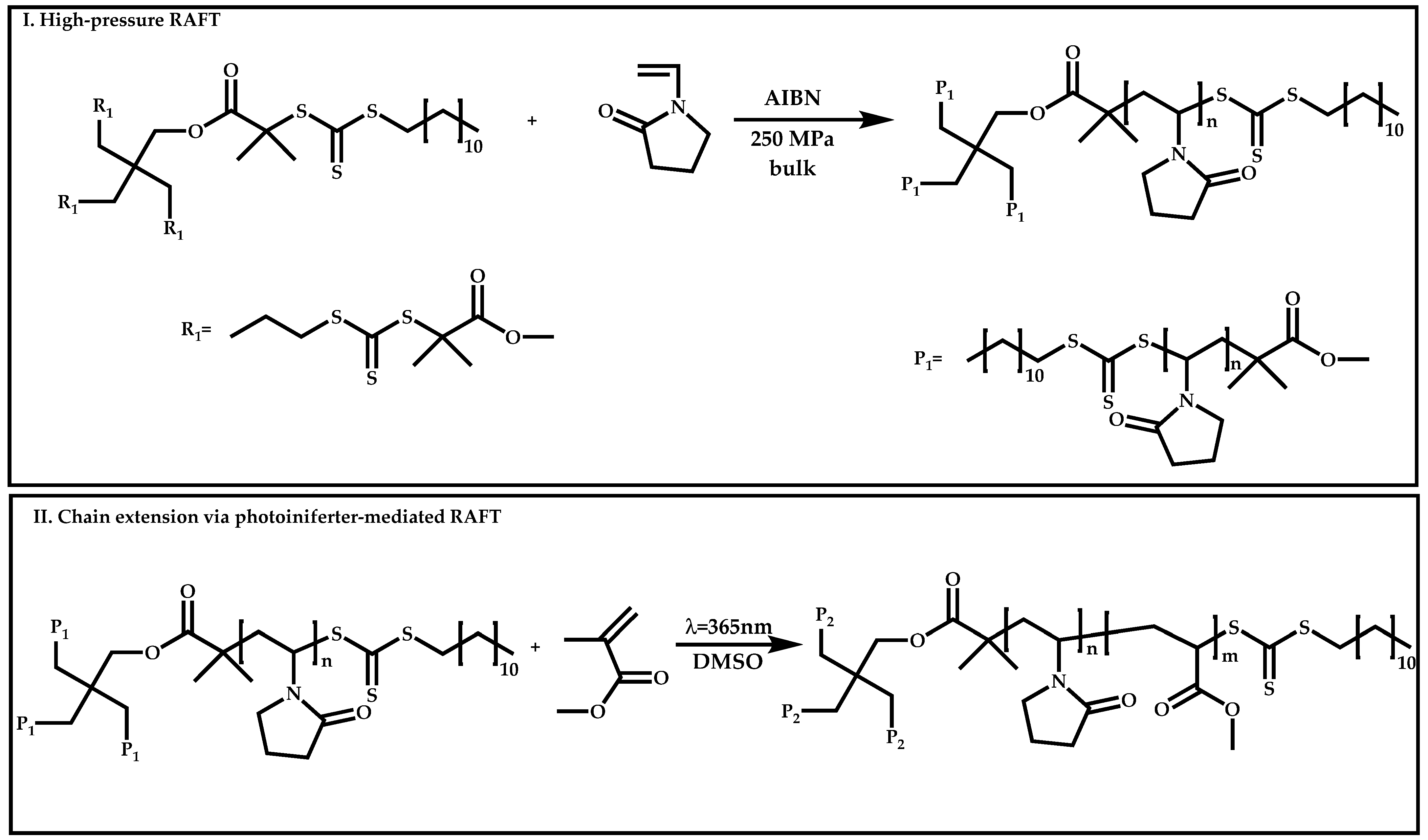
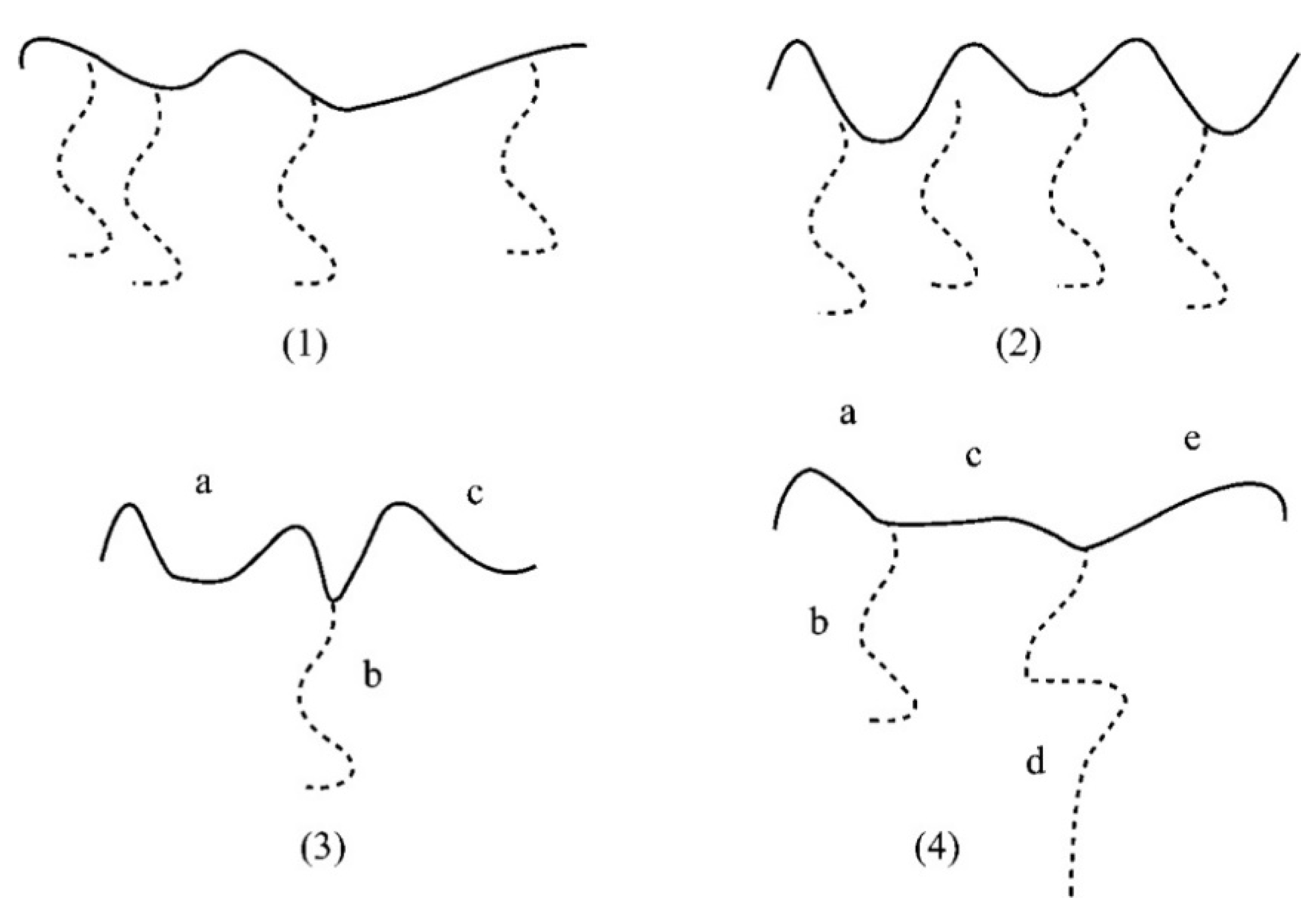
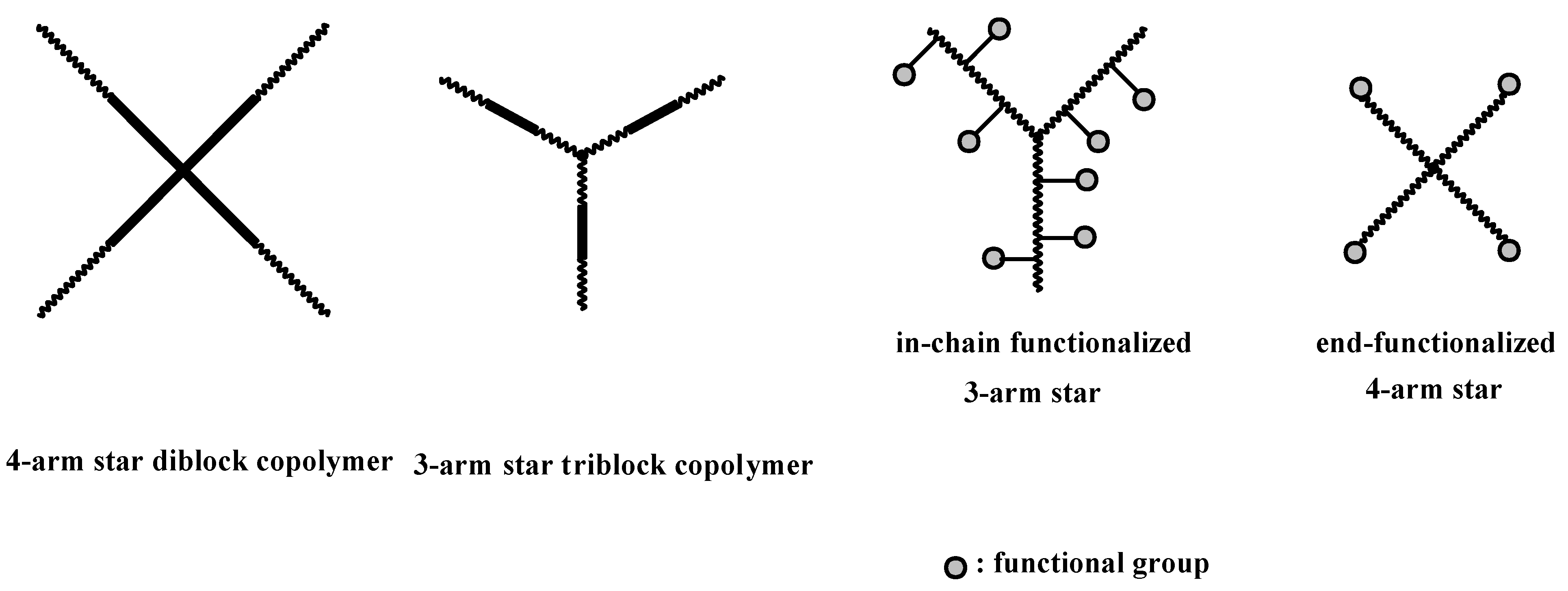
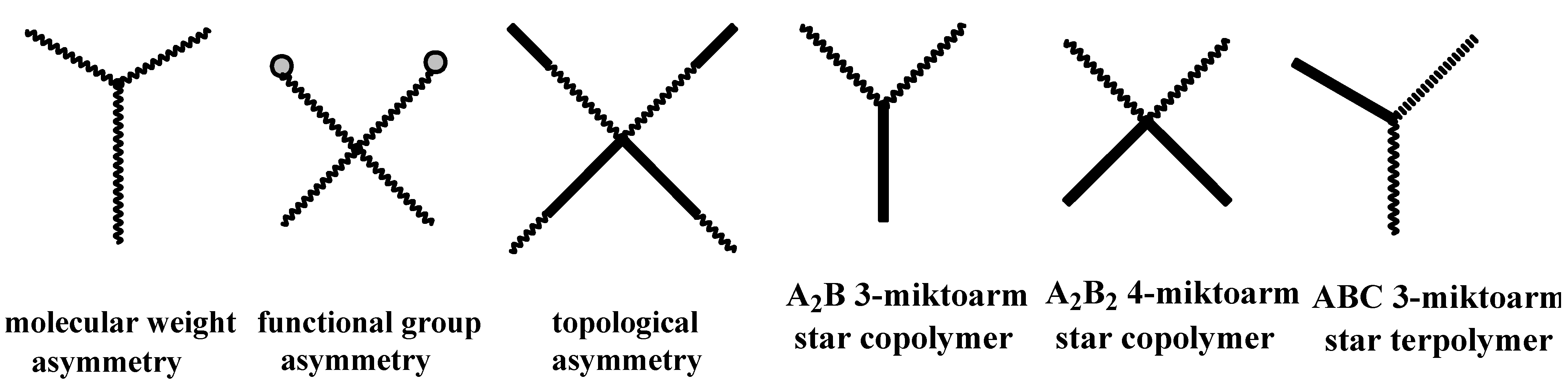
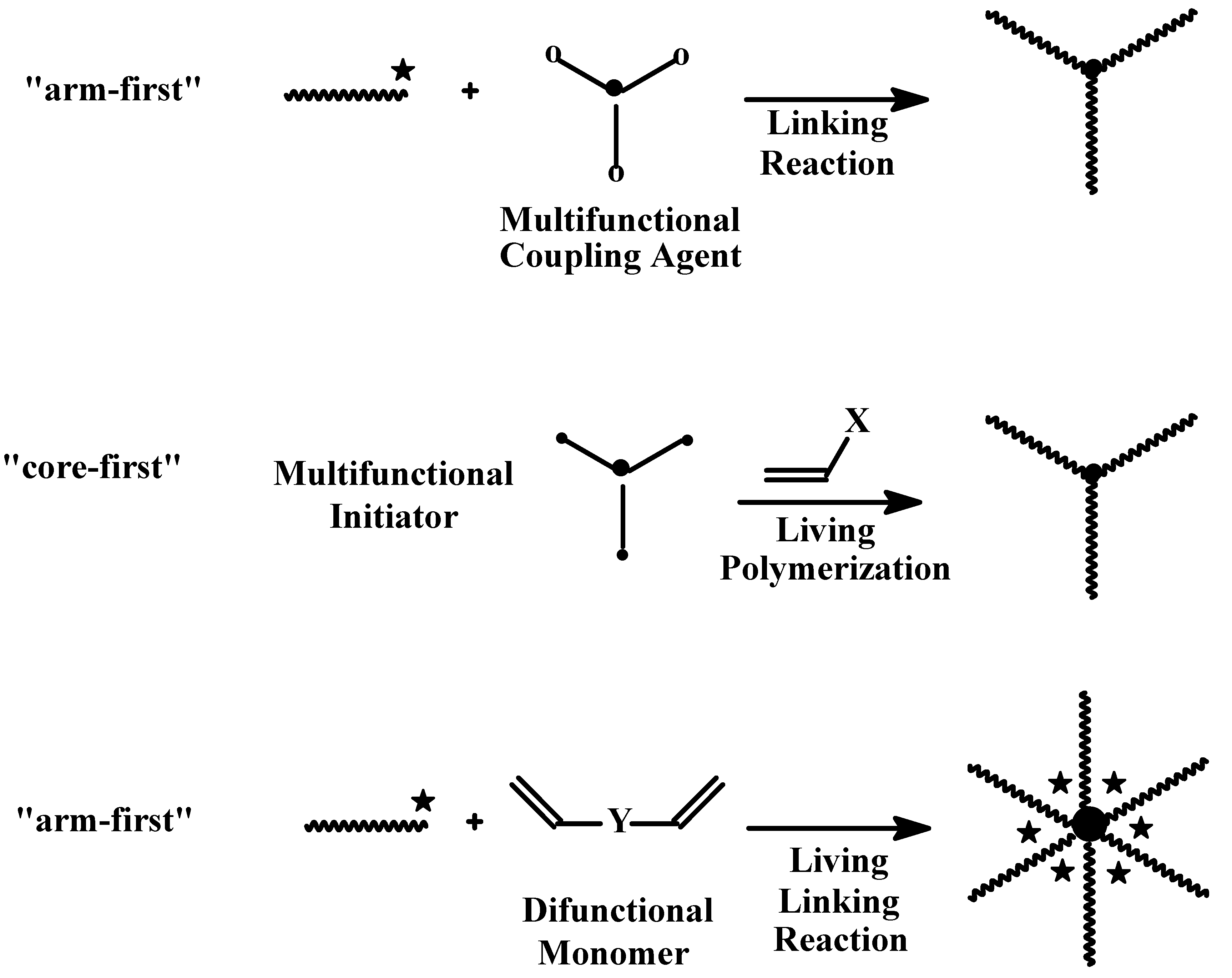
4.3. Star Polymers
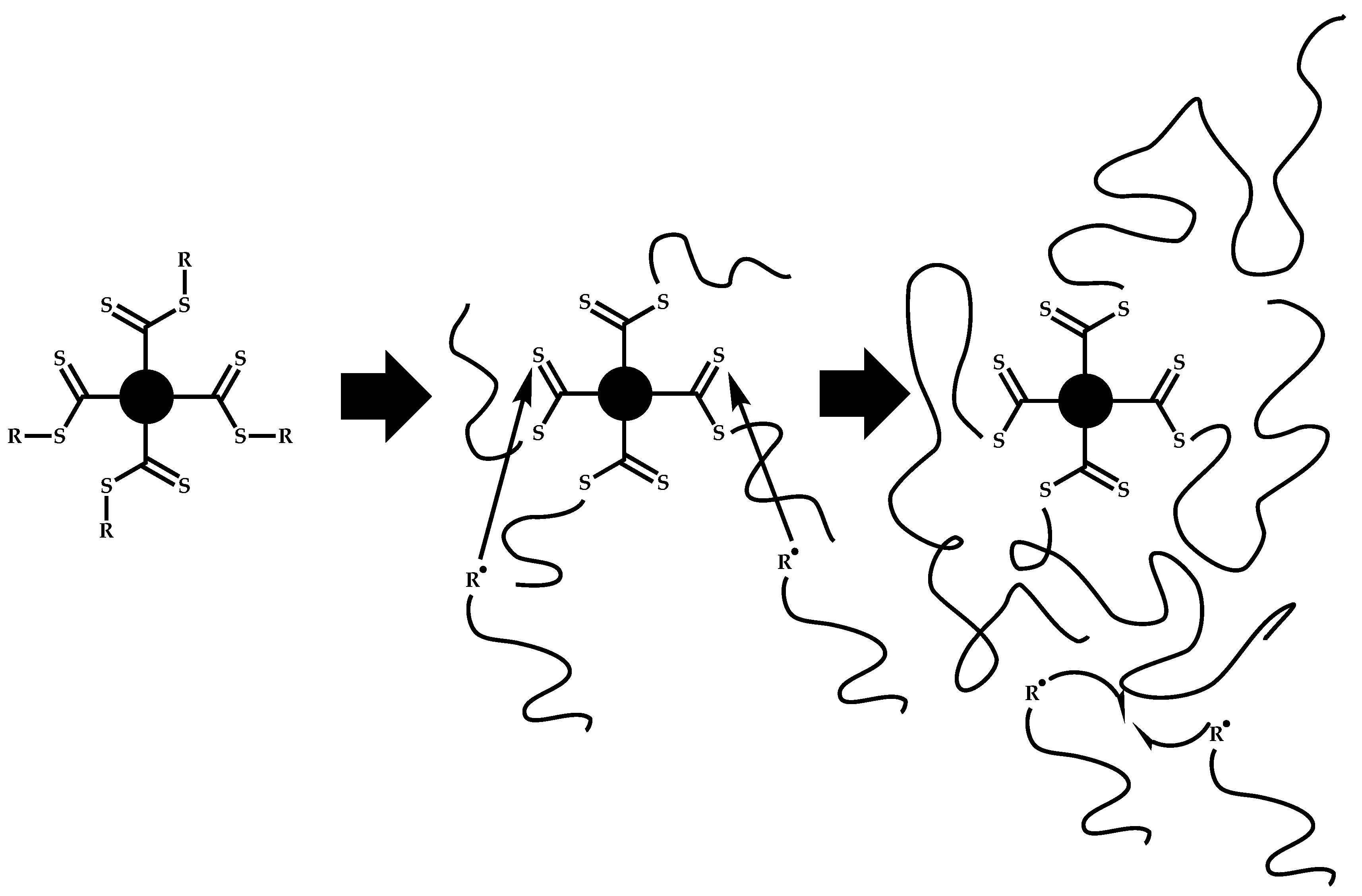
4.3.1. Introduction
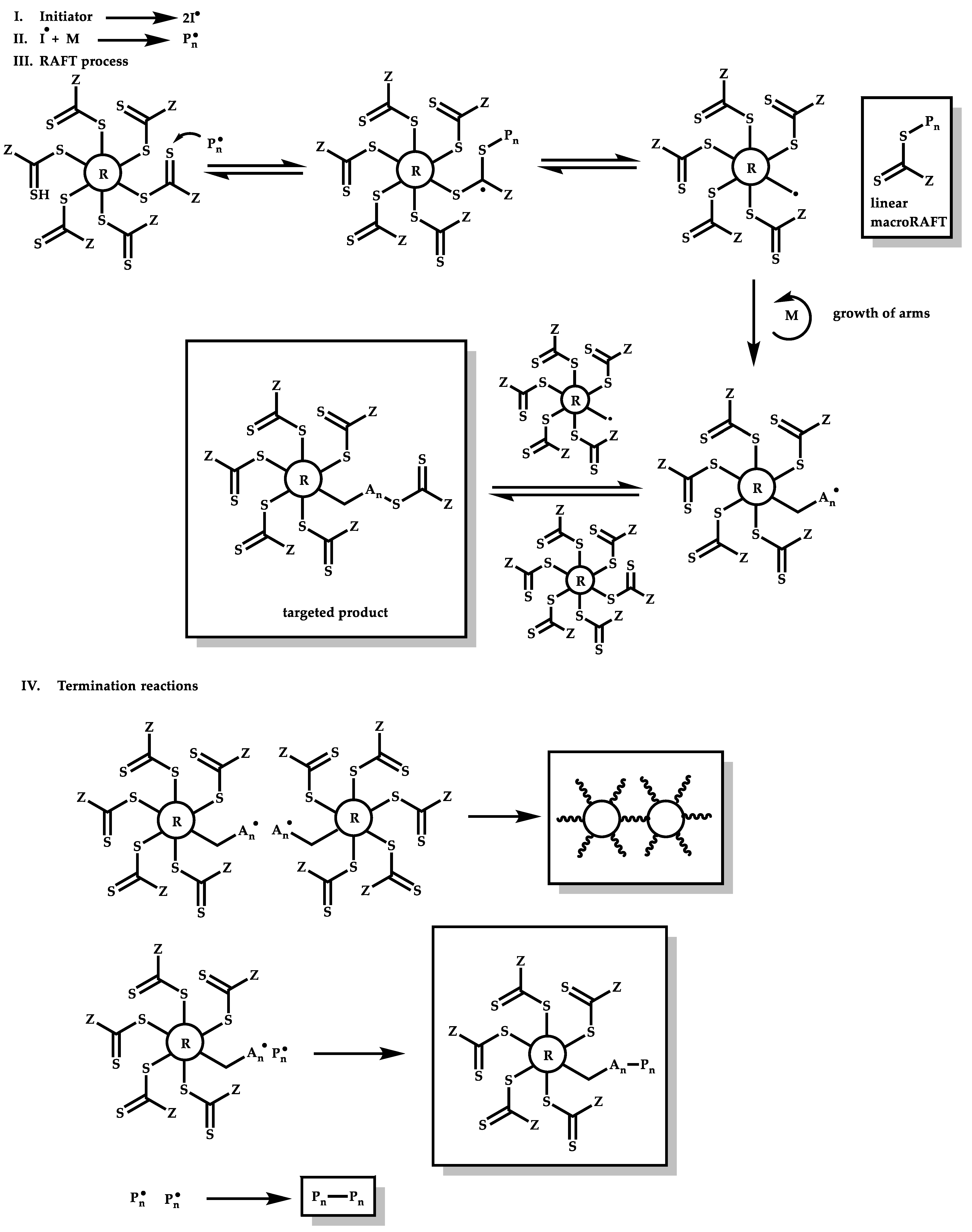
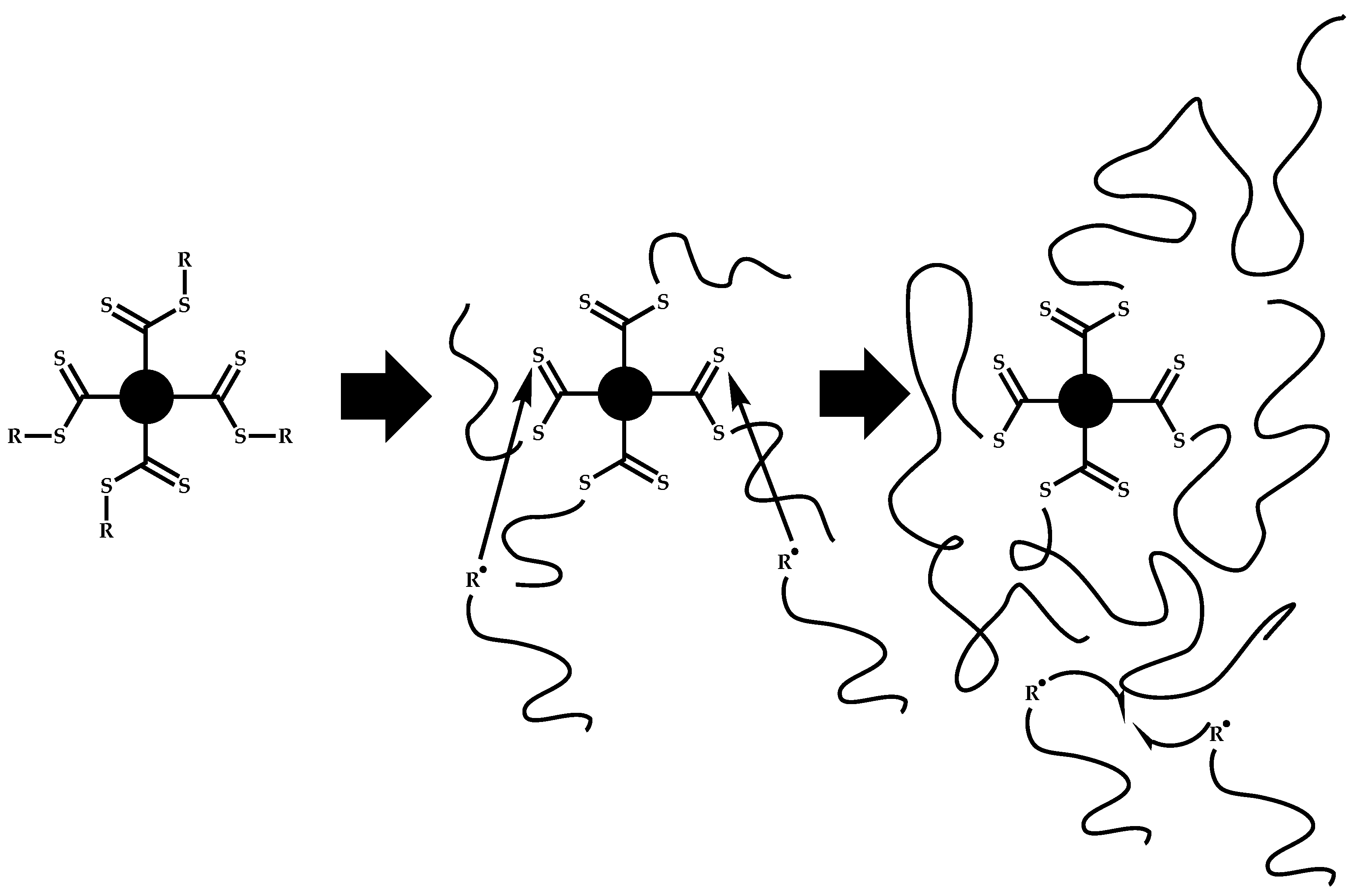
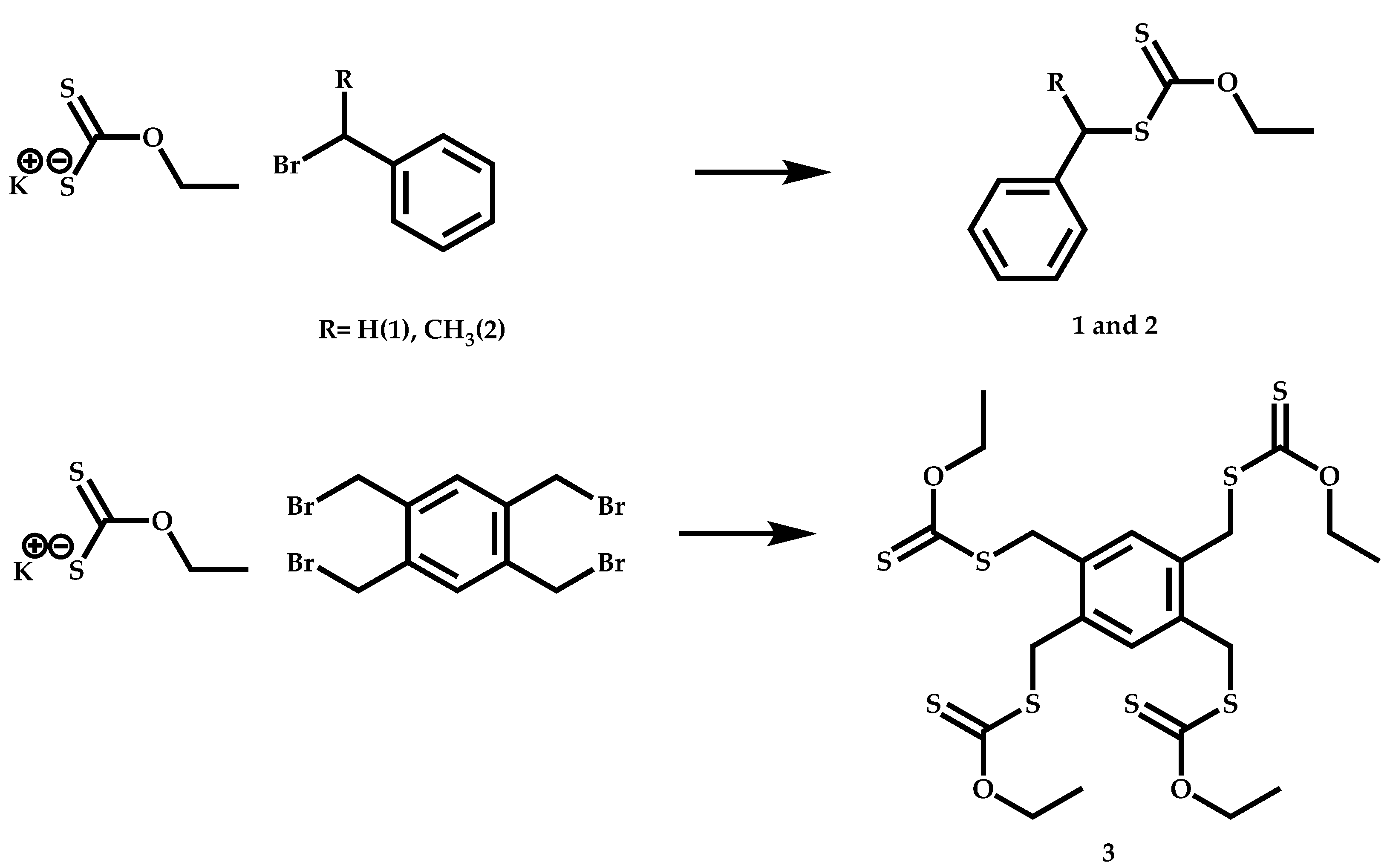
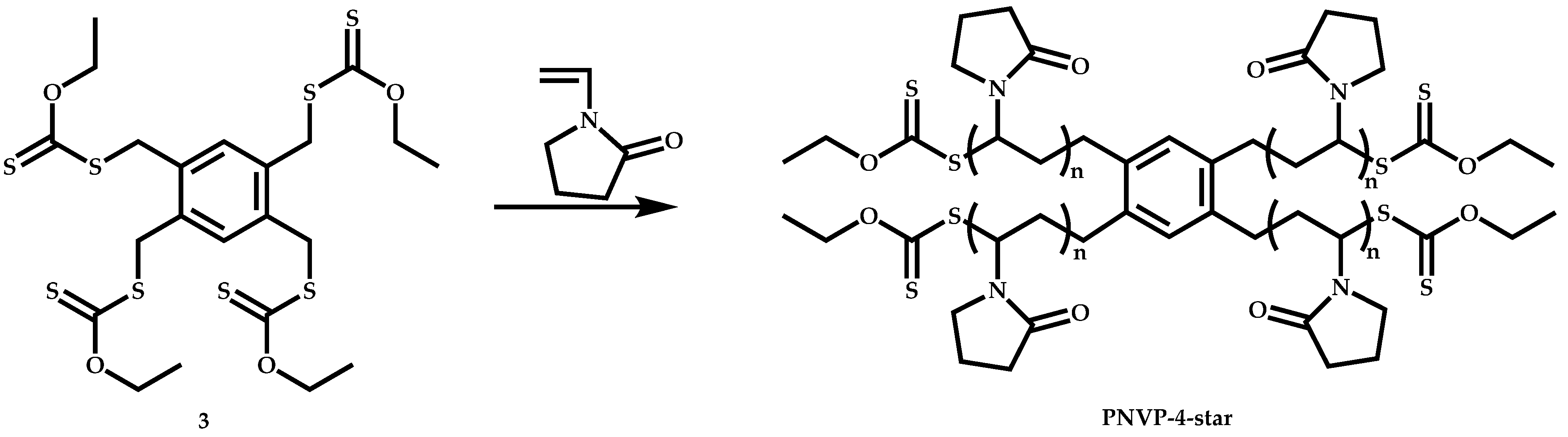
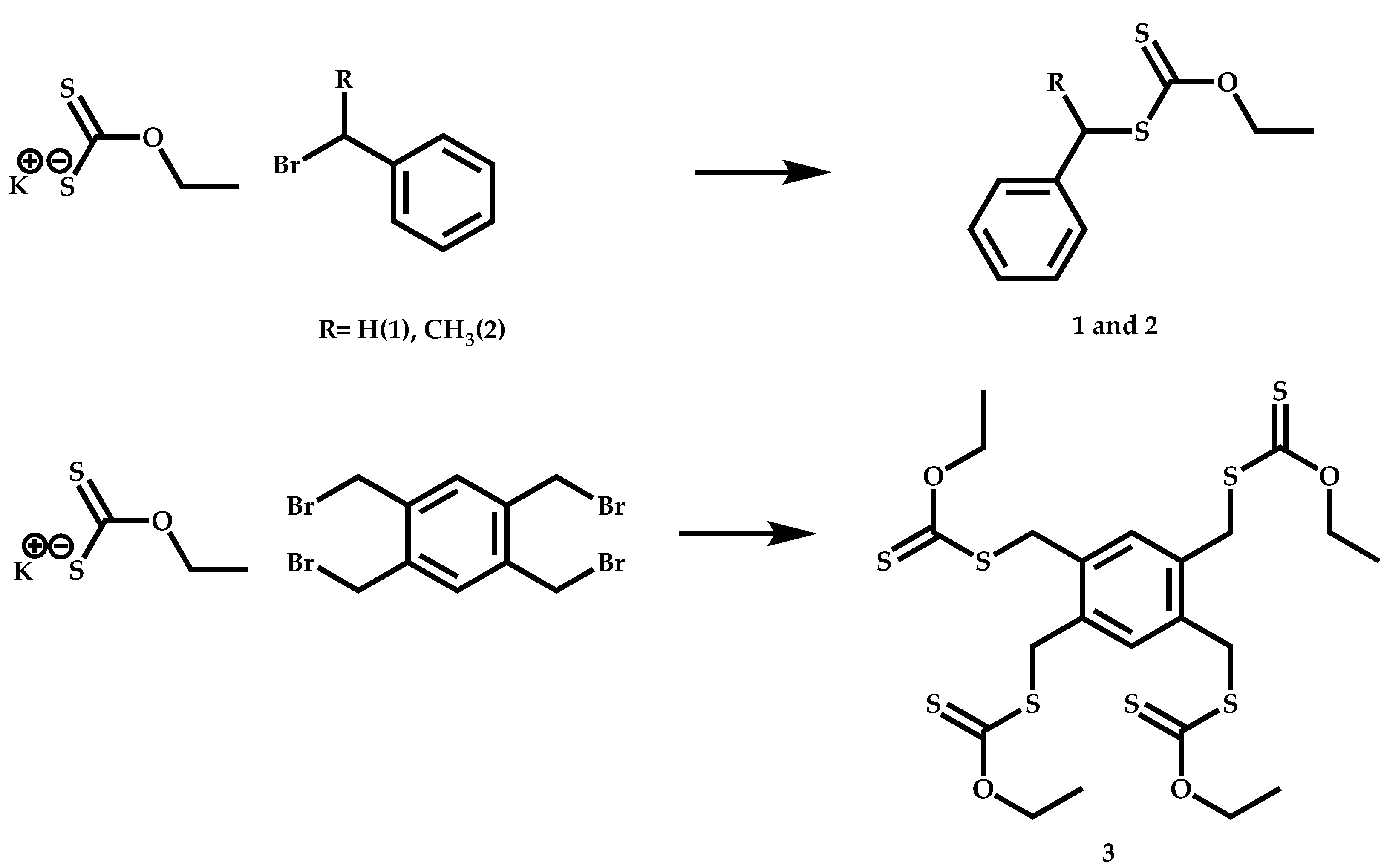
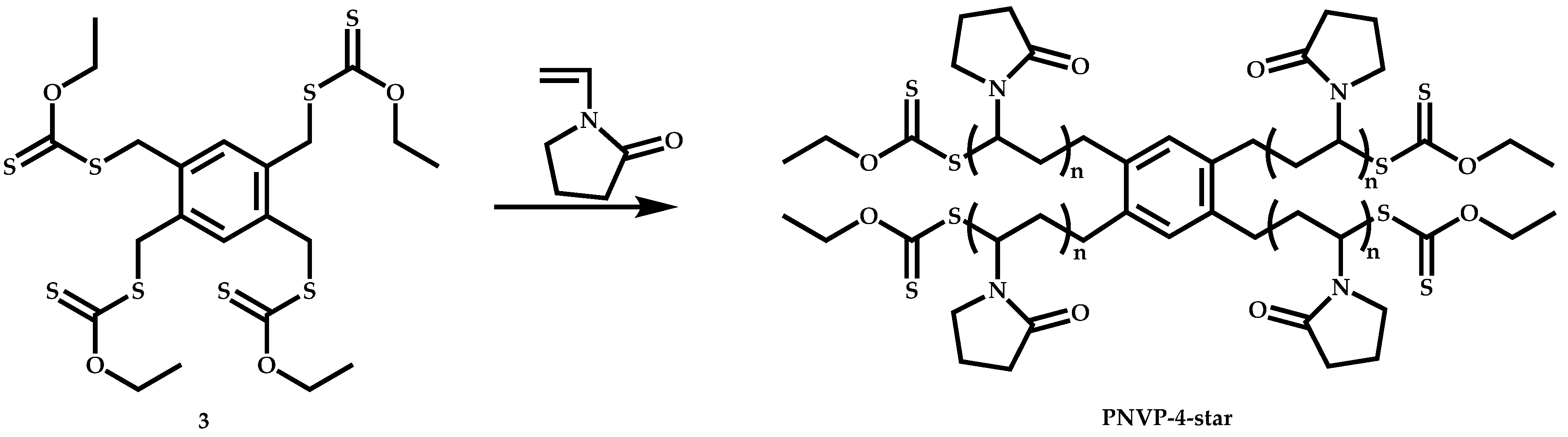
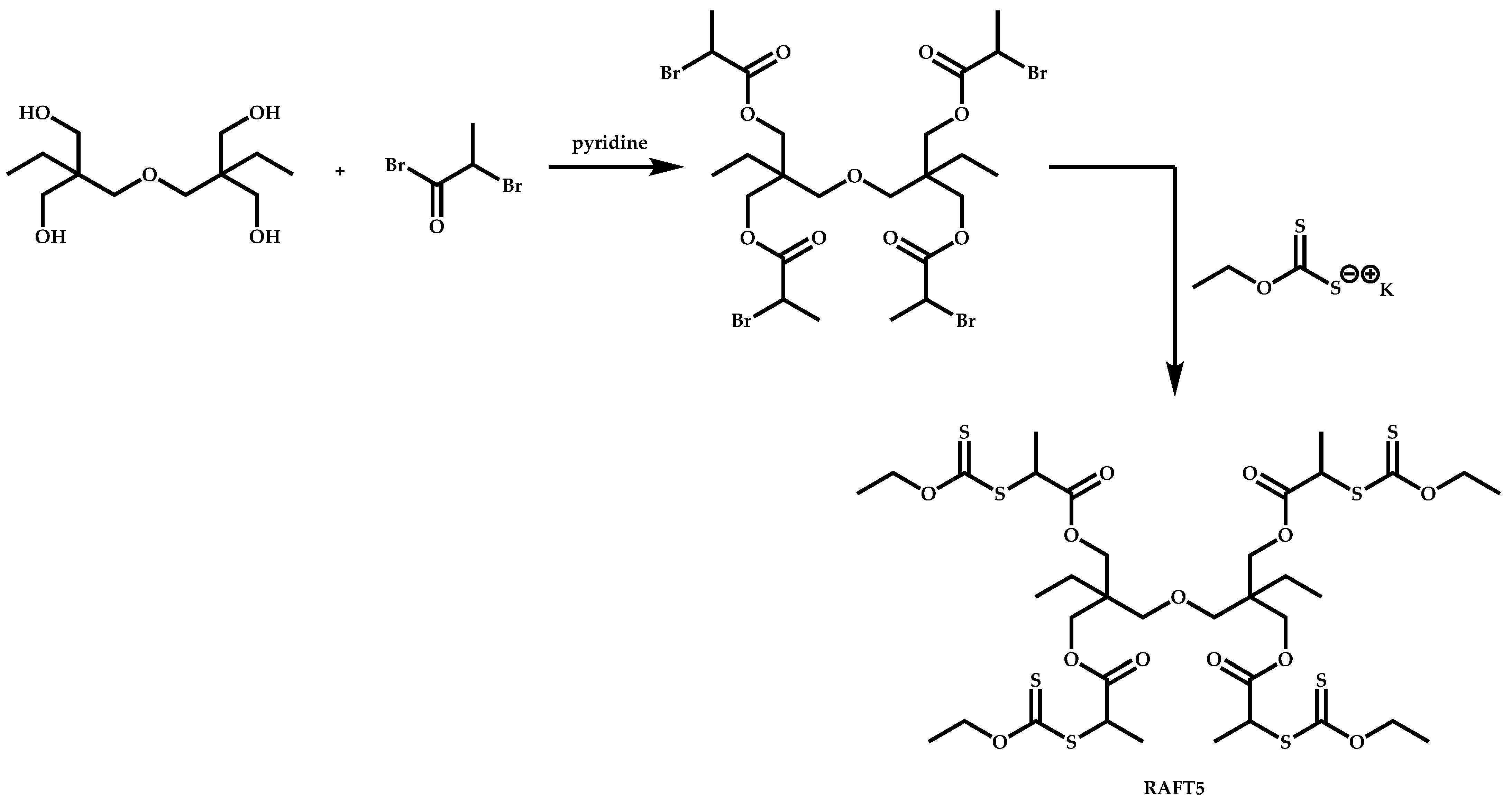
4.3.2. Star Polymers following the Core-First Technique
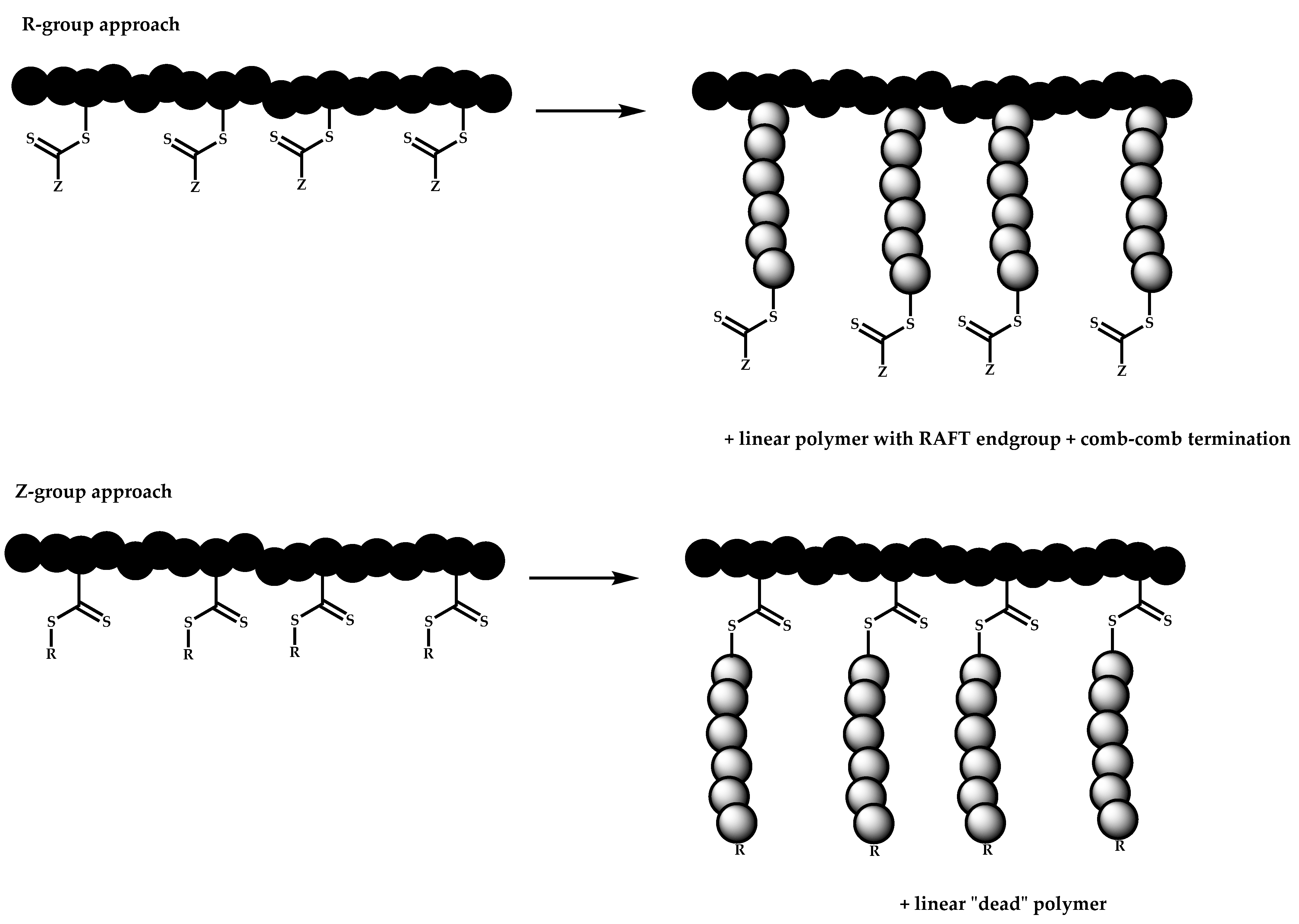
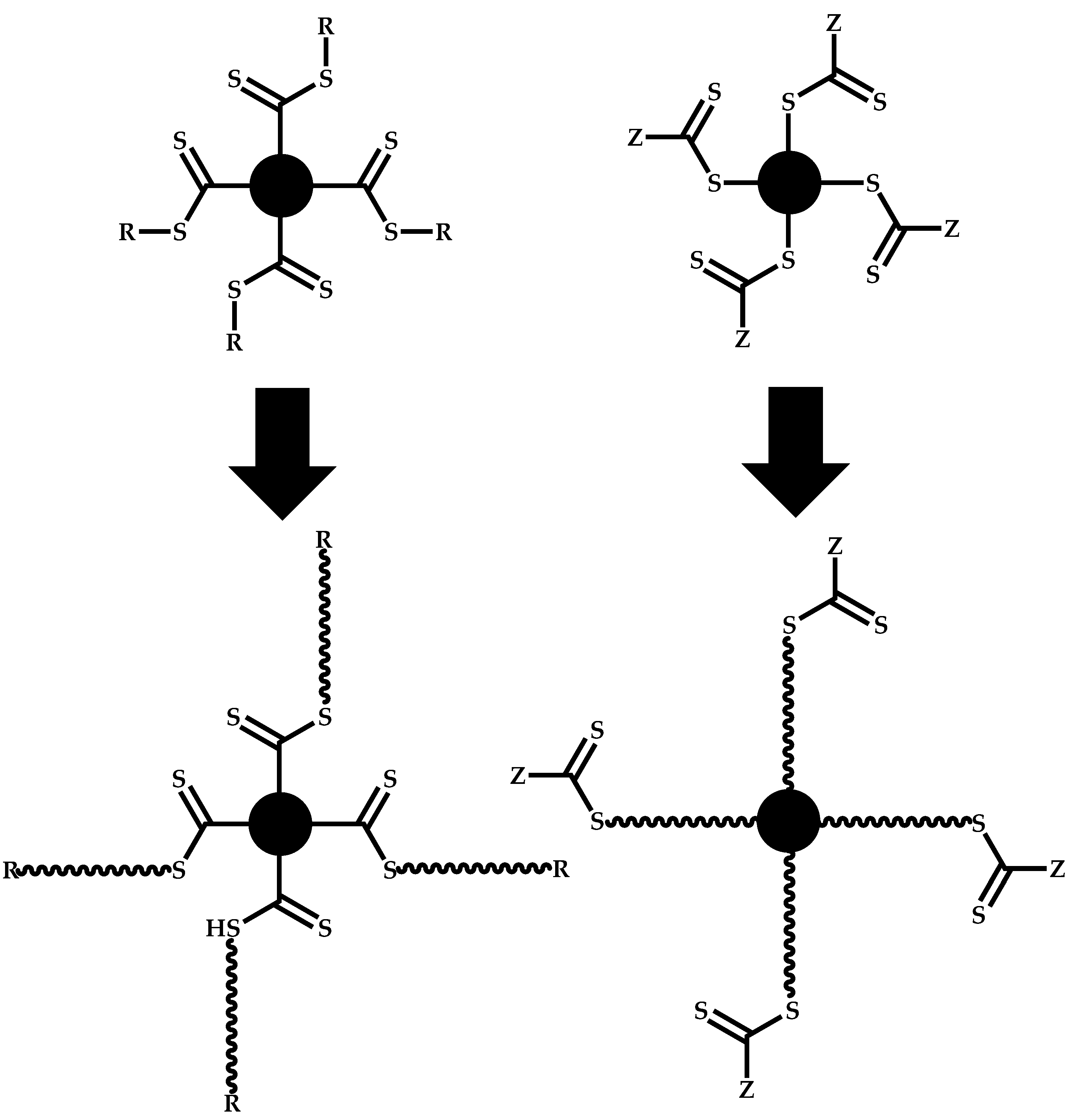
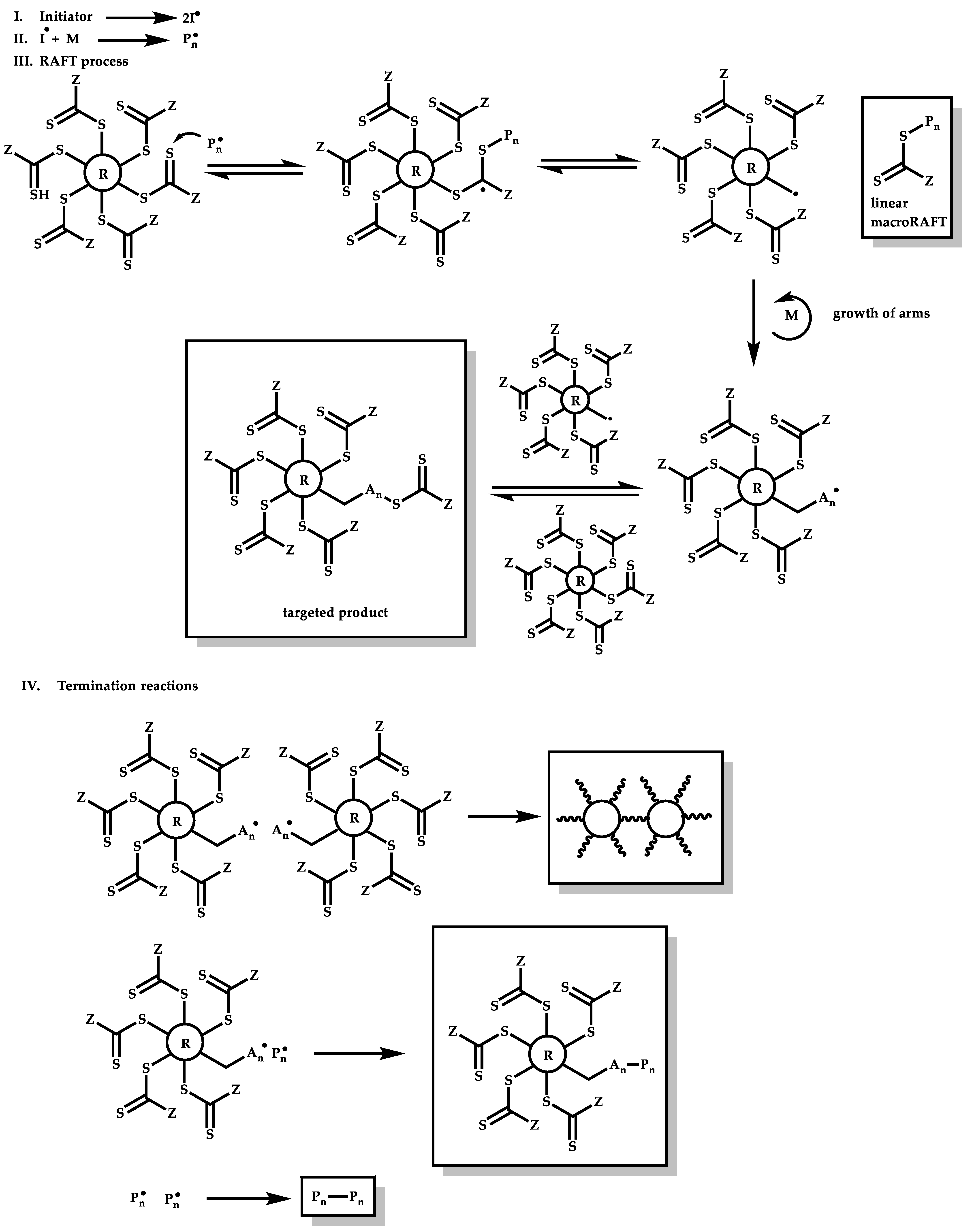
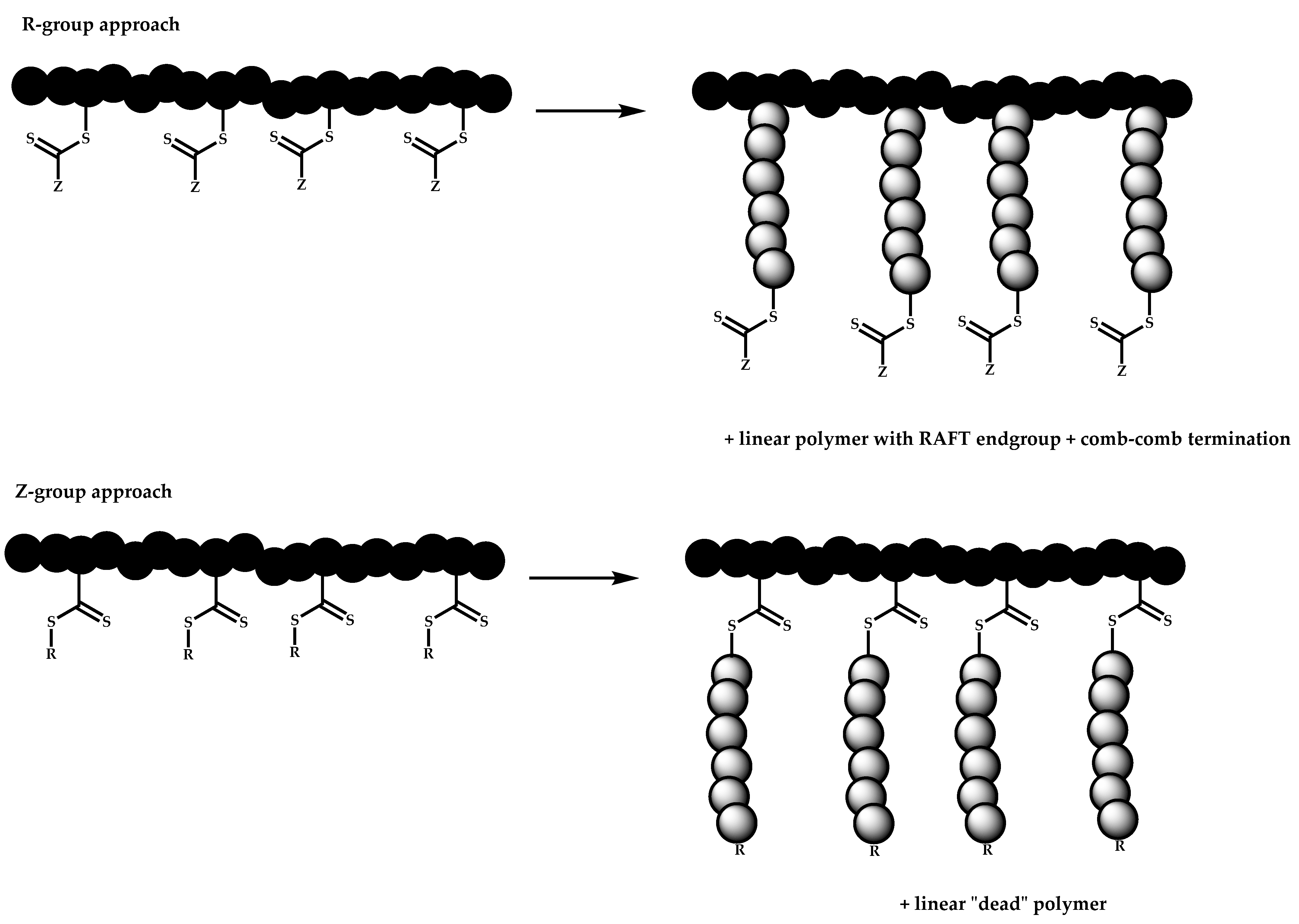
R-Group Approach
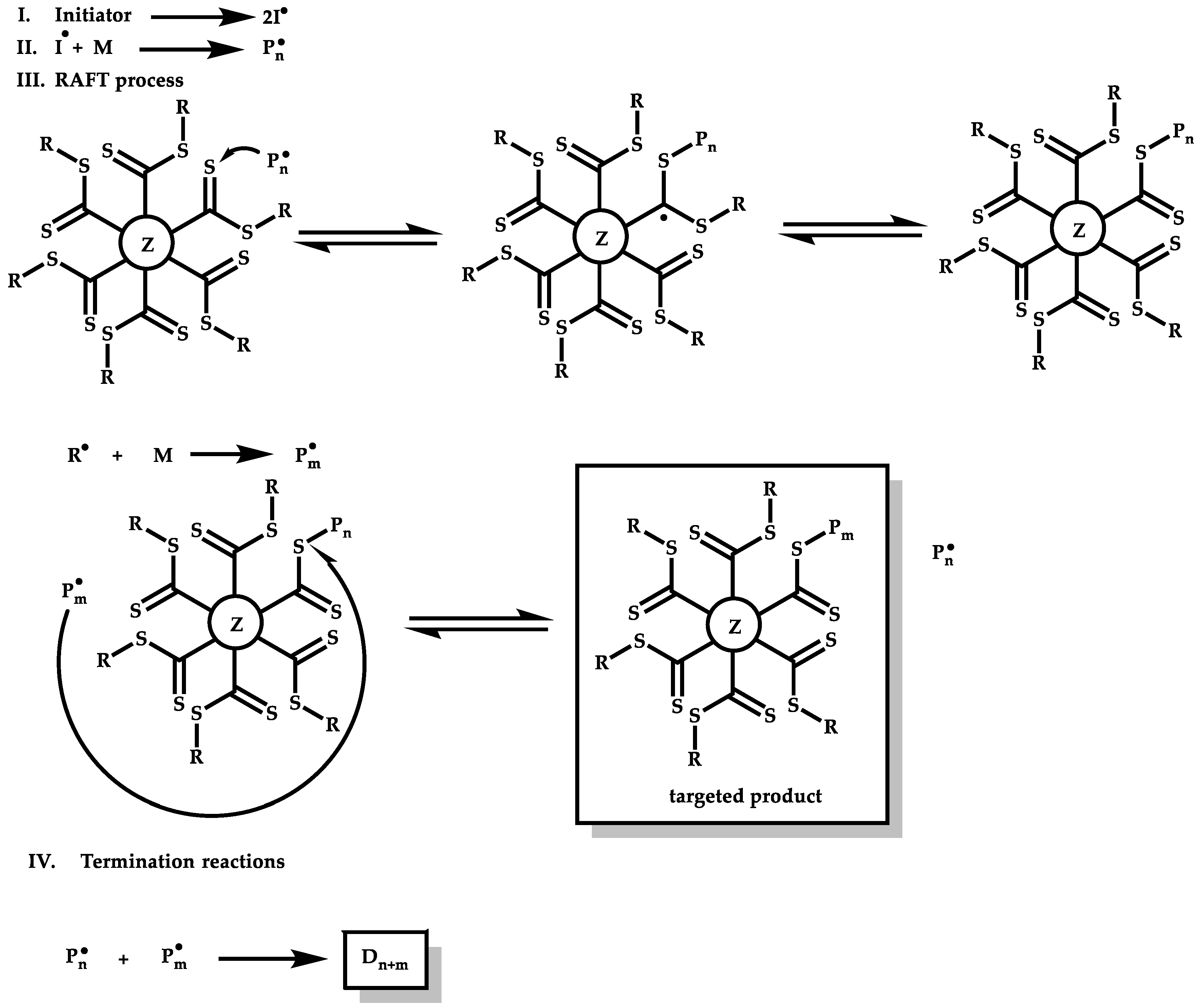
Z-Group Approach
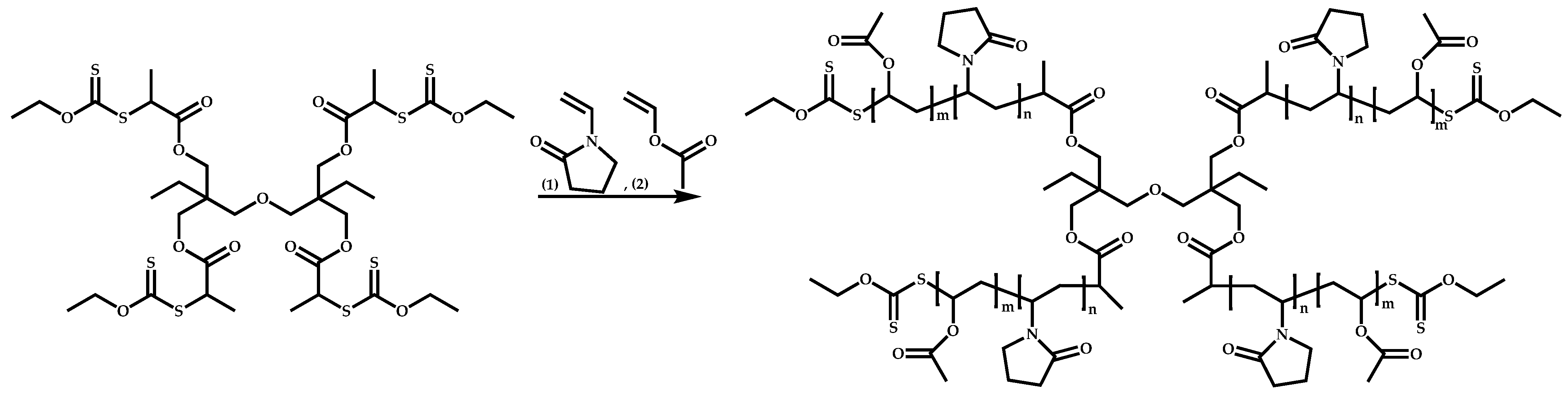
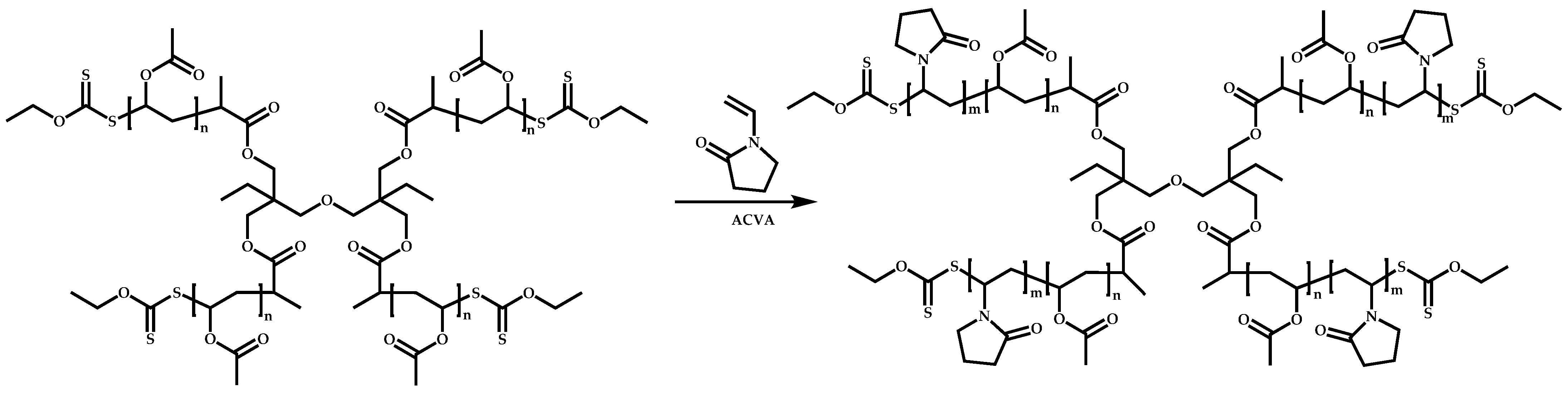
4.3.3. Star Polymers Following the Arm-First Technique
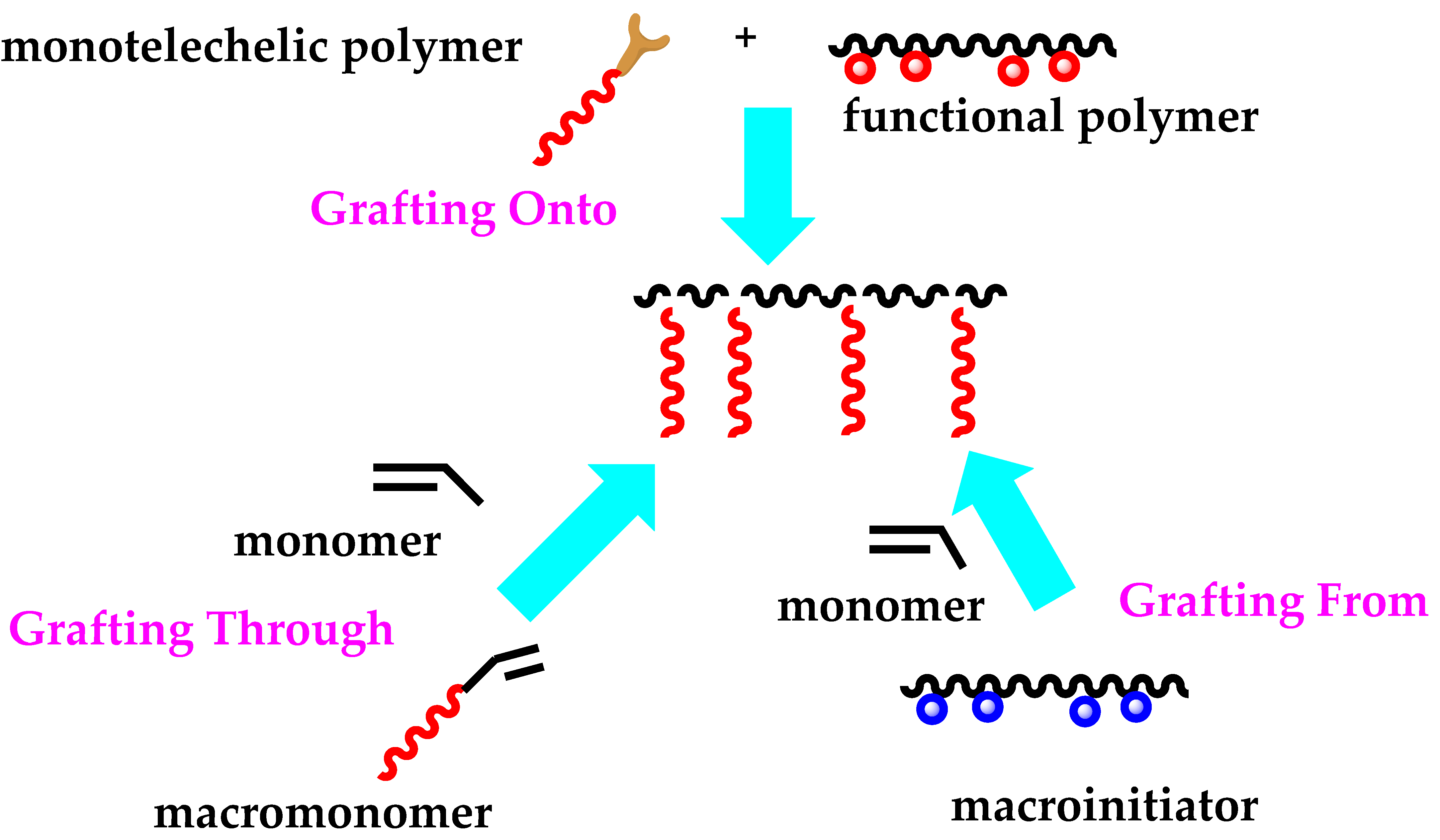
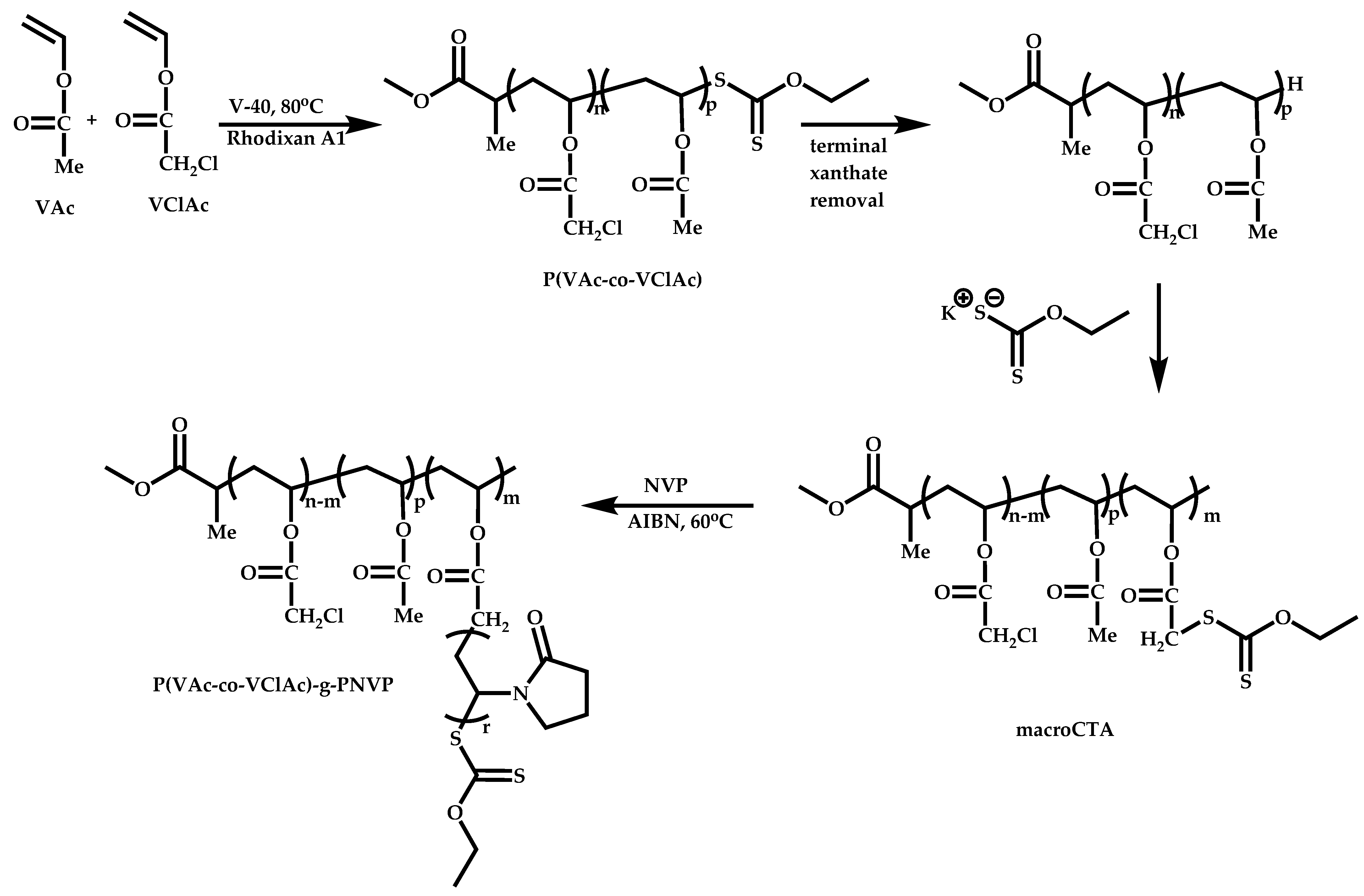
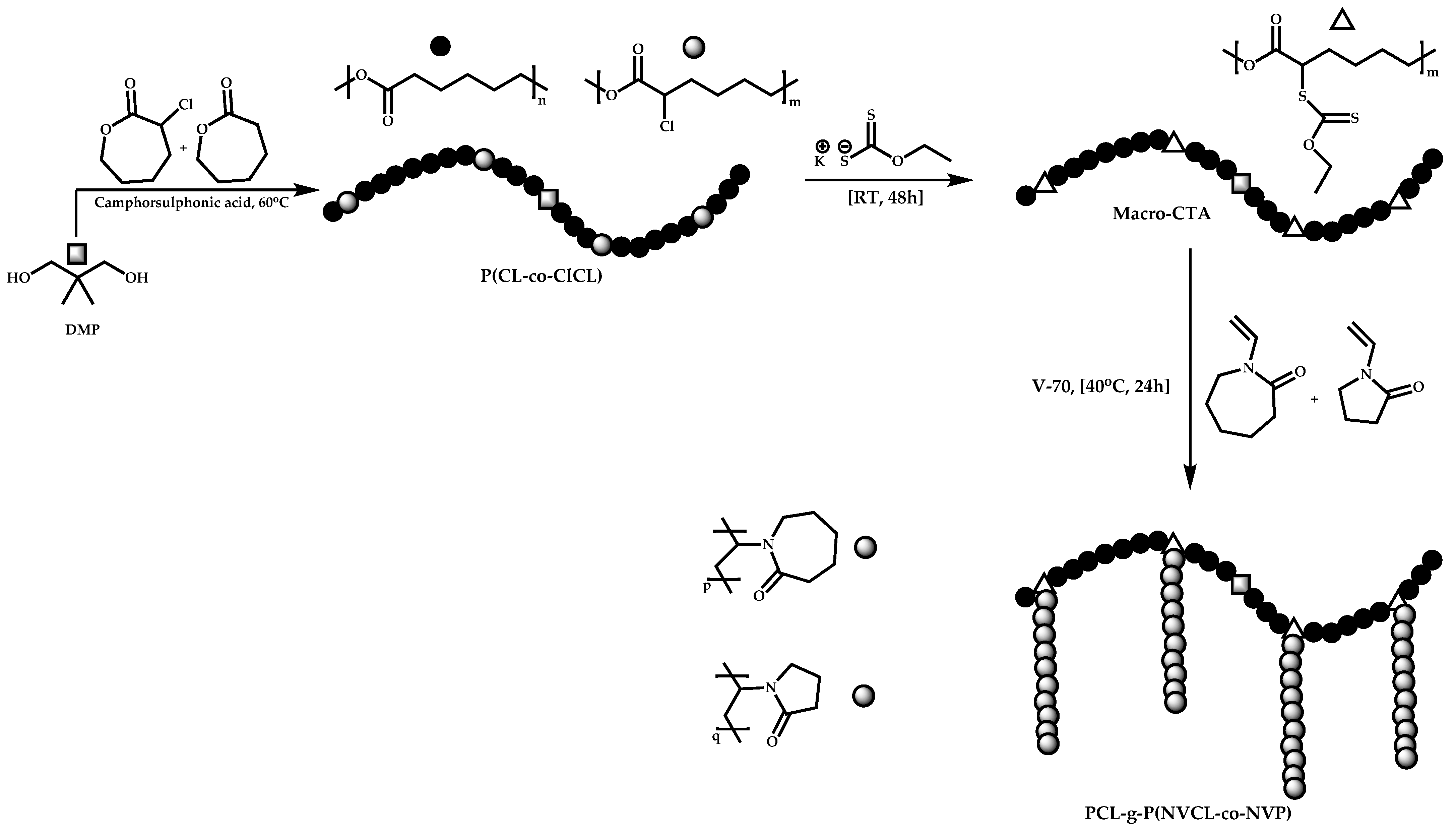
4.4. Graft Copolymers
Introduction
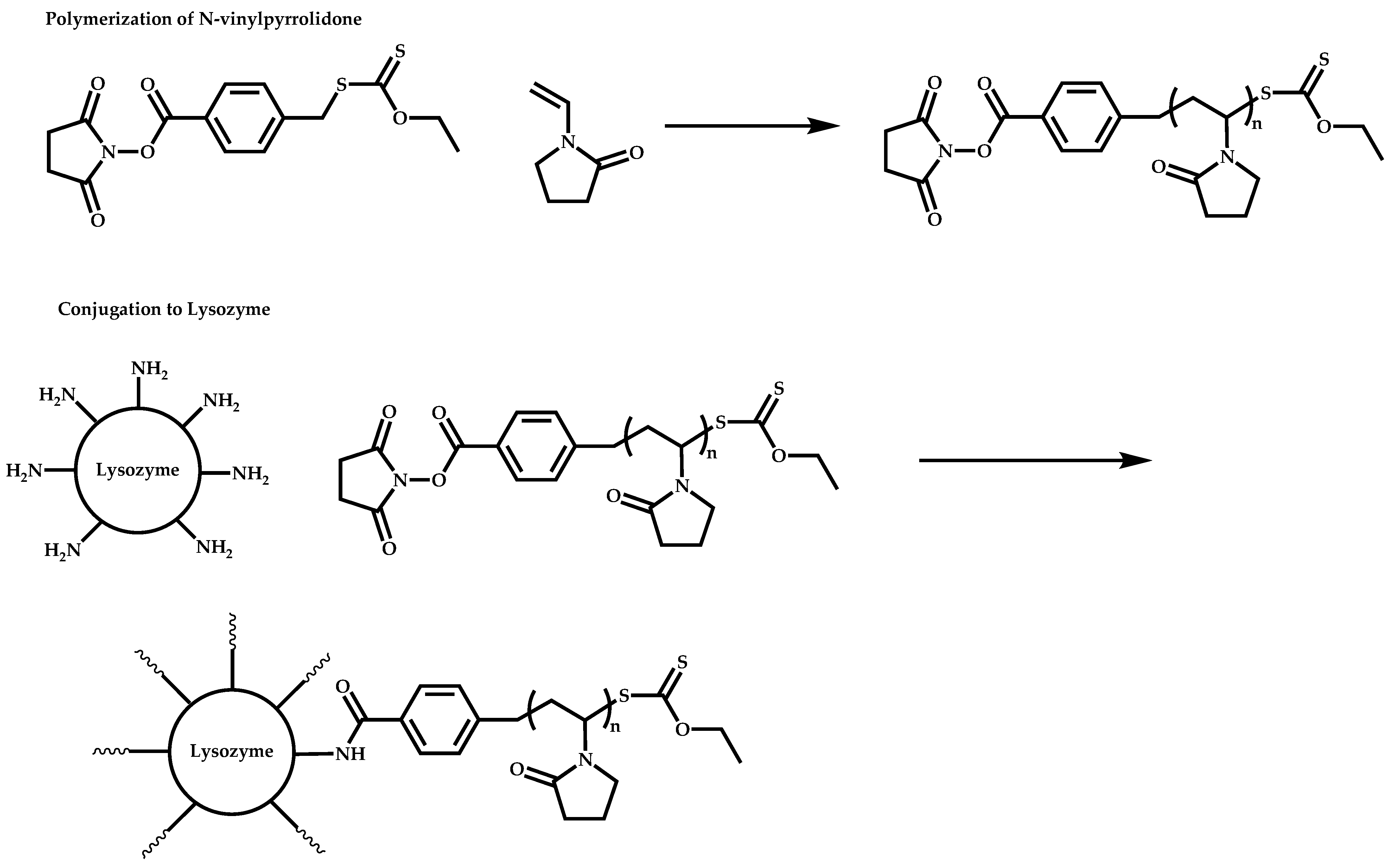
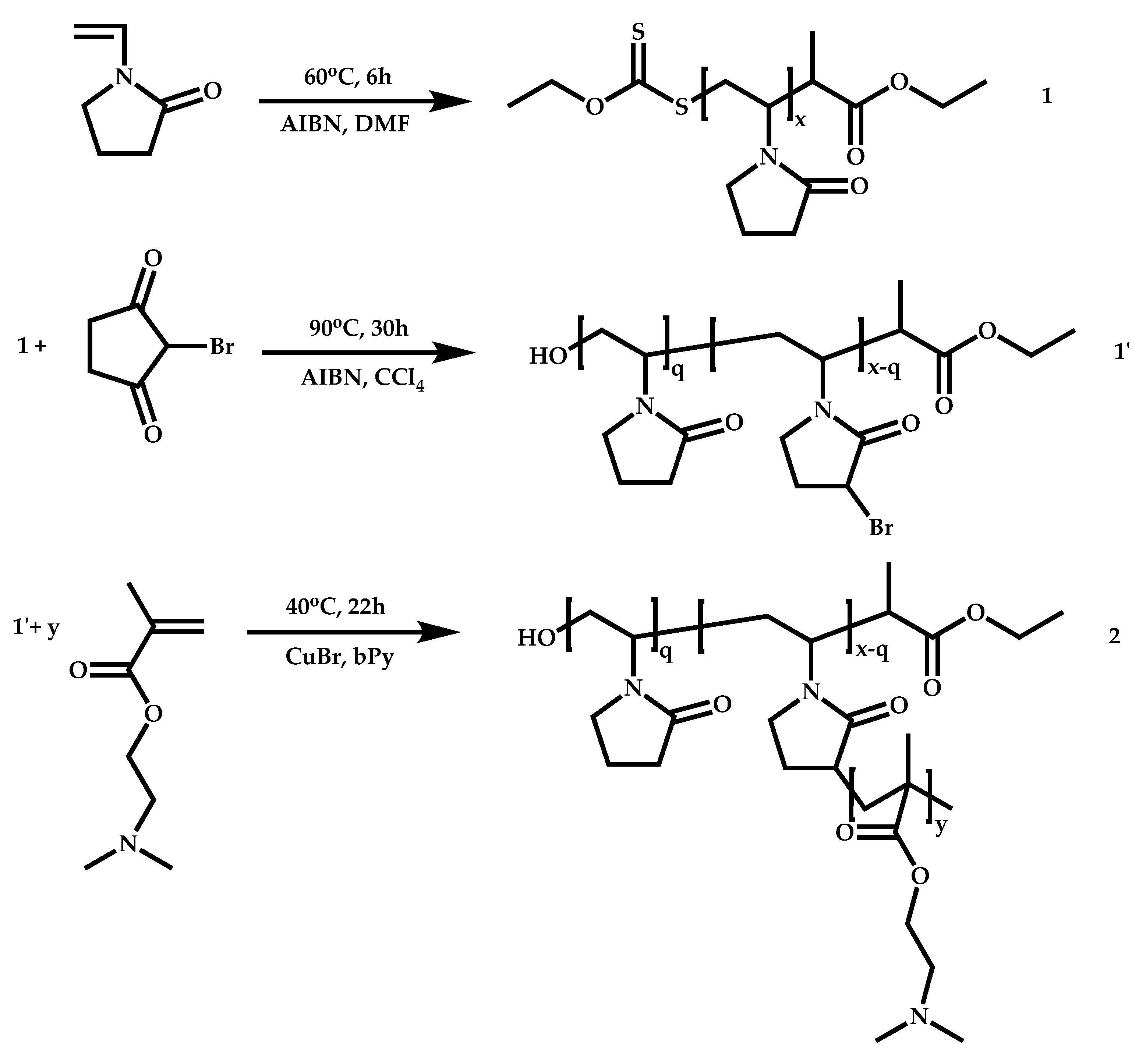
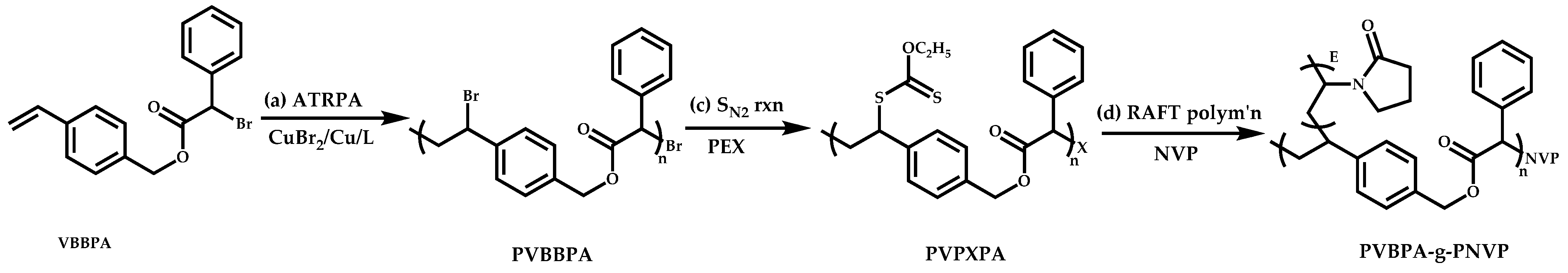
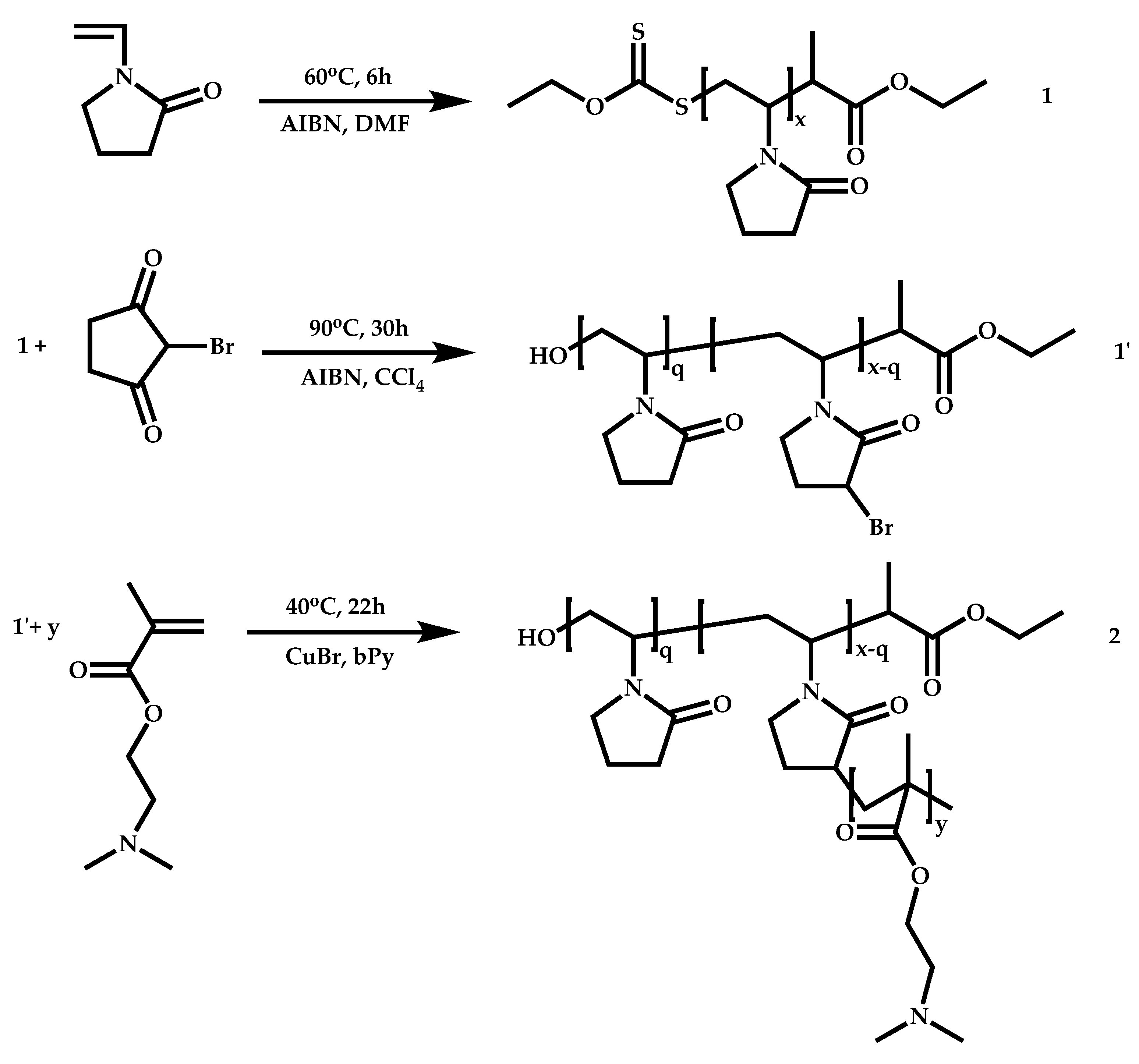
- The attachment of the RAFT agent to the backbone. This approach adopts the main synthetic paths that are followed in its star equivalent synthesis. The CTA can be attached to the backbone either from the Z (Z-group approach) or the R group (R-group approach), following in each case the same mechanistic course with the advantages and disadvantages of each method (Scheme 87). In the R approach for the synthesis of graft copolymers, the main difference in the graft synthesis is the concentration of the CTA on the backbone, which is considerably higher in the case of graft monomers than in stars. This leads to a greater occurrence of side reactions, especially graft–graft coupling. In graft polymers, there is a vastly greater number of branches than in star polymers, resulting in increased termination reactions. Therefore, the amount of graft–graft coupling increases upon increasing the number of side chains. Thus, the amount of side products has an immediate effect on the molecular weight distribution of the final product, which can be controlled through various pathways, including (a) keeping the molecular weight of the side chains as low as possible, (b) reducing the concentration of the radicals in the polymerization reaction and (c) lowering the reaction temperature. Similar drawbacks are faced by the Z-group approach for the synthesis of graft copolymers. The most important is the steric shielding effect leading to pronounced termination reactions of two linear macro-radicals.
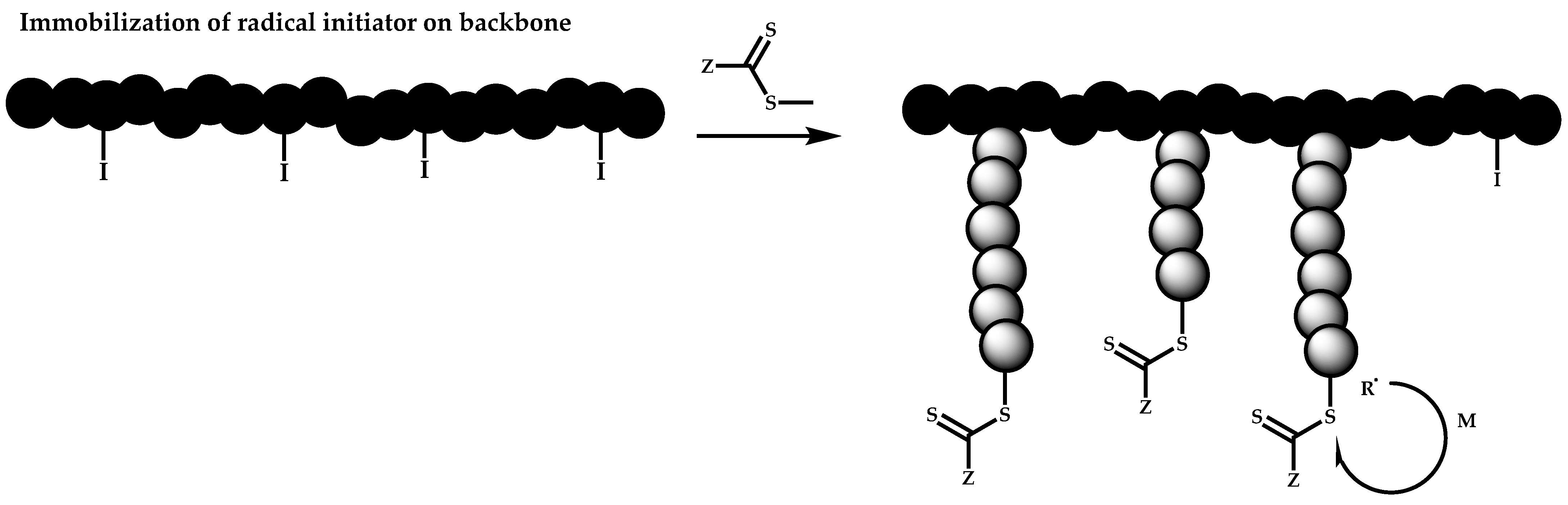
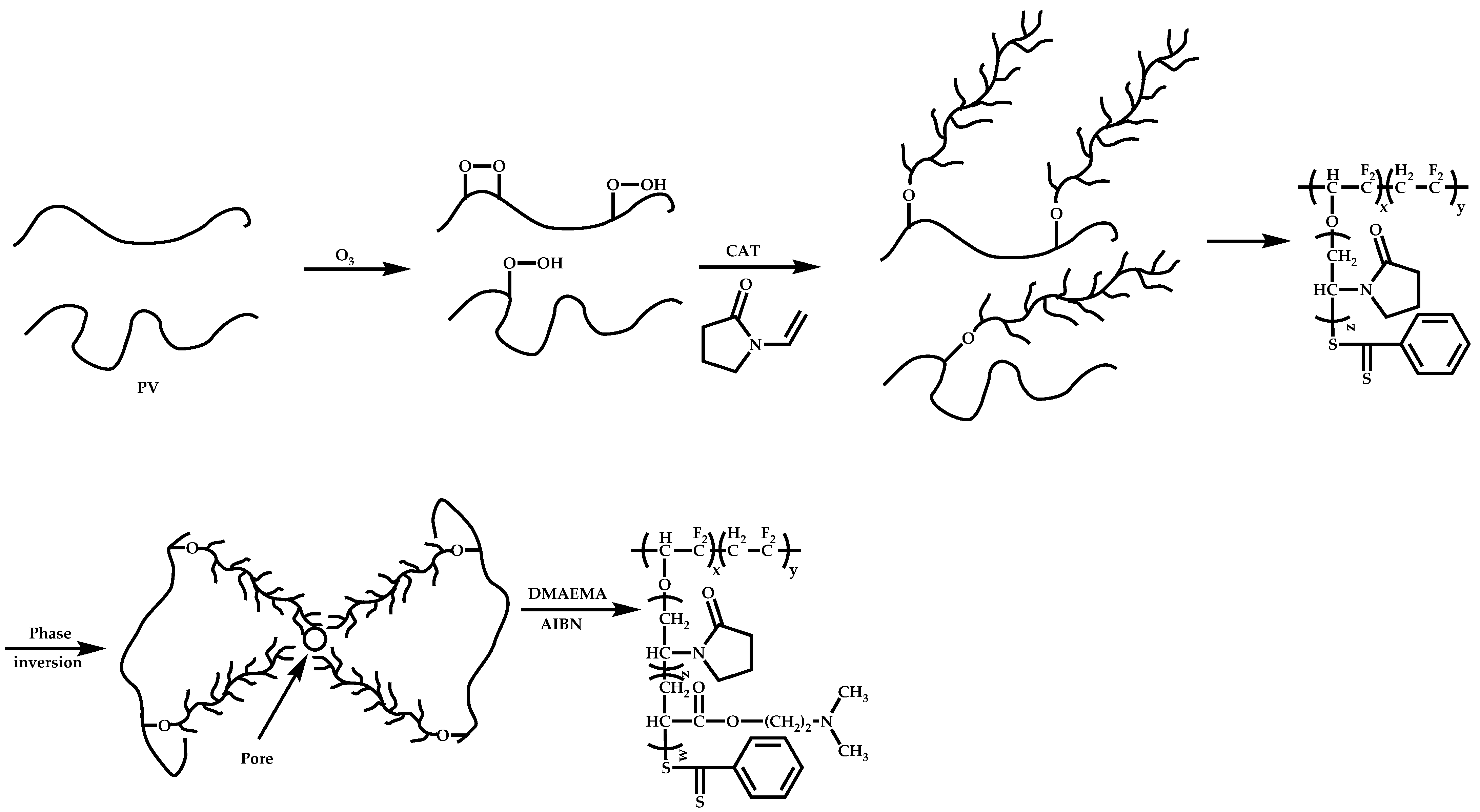
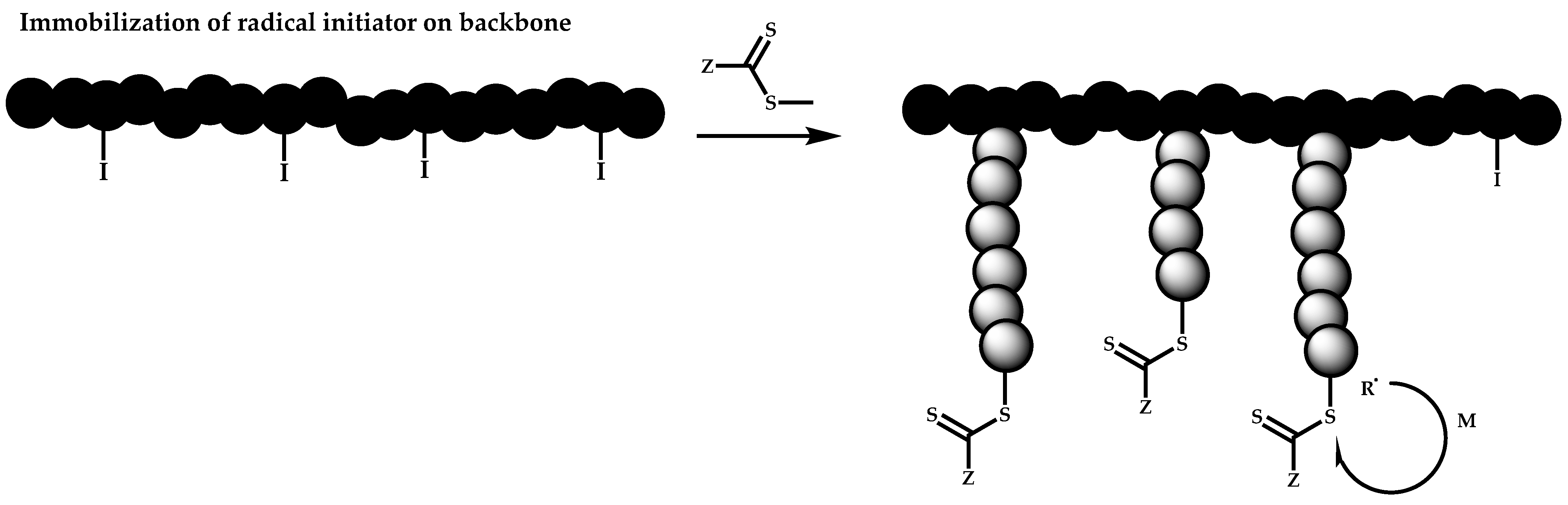
- The attachment of radical initiator fragments to the backbone (Scheme 88). Important parameters in this case are the amount of the CTA over the initiation sites, which controls the molecular weight of the side chains and the rate of activation of the initiator. The most commonly observed byproducts in this case are linear macro-RAFT agents generated from the leaving R group of the CTA.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teodorescu, M.; Bercea, M. Poly(vinylpyrrolidone)—A Versatile Polymer for Biomedical and Beyond Medical Applications. Polym. Plast. Technol. Eng. 2015, 549, 923–943. [Google Scholar] [CrossRef]
- Franco, F.; De Marco, I. The Use of Poly(N-vinyl pyrrolidone) in the Delivery of Drugs: A Review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef] [PubMed]
- Moulay, S. Molecular iodine/polymer complexes. J. Polym. Eng. 2013, 33, 389–443. [Google Scholar] [CrossRef]
- Husain, M.S.B.; Gupta, A.; Alashwal, B.Y.; Sharma, S. Synthesis of PVA/PVP based hydrogel for biomedical applications: A review. Energy Sour. Part A Recovery Util. Environ. Effects 2018, 40, 2388–2393. [Google Scholar] [CrossRef]
- Reis, C.P.; Silva, C.; Martinho, N.; Rosado, C. Drug carriers for oral delivery of peptides and proteins: Accomplishments and future Perspectives. Therap. Deliv. 2013, 4, 251–265. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef]
- Dolman, M.E.M.; Harmsen, S.; Storm, G.; Hennink, W.E.; Kok, R.J. Drug targeting to the kidney: Advances in the active targeting of therapeutics to proximal tubular cells. Adv. Drug Deliv. Rev. 2010, 62, 1344–1357. [Google Scholar] [CrossRef]
- Kurakula, M.; Koteswara Rao, G.S.N. Pharmaceutical assessment of polyvinylpyrrolidone (PVP): As excipient from conventional to controlled delivery systems with a spotlight on COVID-19 inhibition. J. Drug Deliv. Sci. Technol. 2020, 60, 102046. [Google Scholar] [CrossRef]
- Liang, D.; Hsiao, B.S.; Chu, B. Functional electrospun nanofibrous scaffolds for biomedical applications. Adv. Drug Deliv. Rev. 2007, 59, 1392–1412. [Google Scholar] [CrossRef] [Green Version]
- Culbertson, B.M. Glass-ionomer dental restoratives. Prog. Polym. Sci. 2001, 26, 577–604. [Google Scholar] [CrossRef]
- Godlewska, K.; Stepnowski, P.; Paszkiewicz, M. Pollutant analysis using passive samplers: Principles, sorbents, calibration and applications: A review. Environ. Chem. Lett. 2020, 19, 465–520. [Google Scholar] [CrossRef]
- Lowe, S.; O’Brien-Simpson, N.M.; Connal, L.A. Antibiofouling polymer interfaces: Poly(ethylene glycol) and other promising candidates. Polym. Chem. 2015, 6, 198. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.-M.; Chiu, T.-C.; Tseng, W.-L.; Chang, H.-T. Analysis of Nucleic Acids and Proteins in Capillary Electrophoresis and Microchip Capillary Electrophoresis Using Polymers as Additives of the Background Electrolytes. Curr. Anal. Chem. 2006, 2, 17–33. [Google Scholar] [CrossRef]
- Kumar, A.; Galaev, I.Y.; Mattiasson, B. Polymer displacement/shielding in protein chromatography. J. Chromatogr. B 2000, 741, 103–113. [Google Scholar] [CrossRef]
- Halake, K.; Birajdar, M.; Soo Kim, B.; Bae, H.; Lee, C.-C.; Jin Kim, Y.; Kim, S.; Jin Kim, H.; Ahn, S.; Yeoung An, S.; et al. Recent application developments of water-soluble synthetic polymers. J. Industr. Engin. Chem. 2014, 20, 3913–3918. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Wu, Z.; Chen, H. Poly(N-vinylpyrrolidone)-Modified Surfaces for Biomedical Applications. Macromol. Biosci. 2013, 13, 147–154. [Google Scholar] [CrossRef]
- Haaf, F.; Sanner, A.; Straub, F. Polymers of N-Vinylpyrrolidone: Synthesis, Characterization and Uses. Polym. J. 1985, 17, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Afzal, A.; Kausar, A.; Siddiq, M. A Review on Polymer/Cement Composite with Carbon Nanofiller and Inorganic Filler. Polym. Plast. Technol. Eng. 2016, 55, 1299–1323. [Google Scholar] [CrossRef]
- Ye, Y.-S.; Rick, J.; Hwang, B.-J. Water Soluble Polymers as Proton Exchange Membranes for Fuel Cells. Polymers 2012, 4, 913–963. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, H.; Zhou, Q.; Qu, H.; Dong, T.; Zhang, M.; Tang, B.; Zhang, J.; Cui, G. Safety-Enhanced Polymer Electrolytes for Sodium Batteries: Recent Progress and Perspectives. ACS Appl. Mater. Interfaces 2019, 11, 17109–17127. [Google Scholar] [CrossRef]
- Koczkur, K.M.; Mourdikoudis, S.; Polavarapu, L.; Skrabalak, S.E. Polyvinylpyrrolidone (PVP) in nanoparticle synthesis. Dalton Trans. 2015, 44, 17883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, B.; Sun, Y.; Mayers, B.; Xia, Y. Shape-Controlled Synthesis of Metal Nanostructures: The Case of Silver. Chem. Eur. J. 2005, 11, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living Radical Polymerization by the RAFT Process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT Agent Design and Synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Corrigan, N.; Jung, K.; Moad, G.; Hawker, C.J.; Matyjaszewski, K.; Boyer, C. Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications. Progr. Polym. Sci. 2020, 111, 101311. [Google Scholar] [CrossRef]
- Moad, G. RAFT polymerization to form stimuli-responsive polymers. Polym. Chem. 2017, 8, 177. [Google Scholar] [CrossRef]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Mori, H. Recent progress in controlled radical polymerization of N-vinyl monomers. Eur. Polym. J. 2013, 49, 2808–2838. [Google Scholar] [CrossRef] [Green Version]
- Radić, D.; Gargallo, L. Synthesis, Reactivity Ratios, and Solution Behavior of Vinylpyrrolidone-co-Monoalkyl Itaconate and Vinylpyrrolidone-co-Dialkyl Itaconate Copolymers. Macromolecules 1997, 30, 817–825. [Google Scholar] [CrossRef]
- Gatica, N.; Gargallo, L.; Radić, D. 2-Vinylpyridine-co-N-vinyl-2-pyrrolidone and 4-vinylpyridine-co-N-vinyl-2-pyrrolidone copolymers: Synthesis and reactivity ratios. Polym. Int. 1998, 45, 285–290. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process a third update. Austr. J. Chem. 2012, 65, 985–1076. [Google Scholar] [CrossRef]
- Vana, P. Kinetic aspects of RAFT polymerization. Macromol. Symp. 2007, 248, 71–81. [Google Scholar] [CrossRef]
- Pound, G.; Eksteen, Z.; Pfukwa, R.; McKenzie, J.M.; Lange, R.F.M.; Klumperman, B. Unexpected reactions associated with the xanthate-mediated polymerization of N-vinylpyrrolidone. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6575–6593. [Google Scholar] [CrossRef]
- Barner-Kowolik, C. Handbook of RAFT Polymerization; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Bergius, W.N.A.; Hutchings, L.R.; Sarih, N.M.; Thompson, R.L.; Jeschke, M.; Fisher, R. Synthesis and characterisation of end-functionalised poly(N-vinylpyrrolidone) additives by reversible addition-fragmentation transfer polymerization. Polym. Chem. 2013, 4, 2815–2827. [Google Scholar] [CrossRef] [Green Version]
- Reader, P.W.; Pfukwa, R.; Jokonya, S.; Arnott, G.E.; Klumperman, B. Synthesis of α,ω-heterotelechelic PVP for bioconjugation,: Via a one-pot orthogonal end-group modification procedure. Polym. Chem. 2016, 7, 6450–6456. [Google Scholar] [CrossRef] [Green Version]
- Elias, H.-G. Macromolecules: Volume 1, 2: Synthesis, Materials, and Technology; Springer Science & Business Media: New York, NY, USA, 1977. [Google Scholar]
- Hagiopol, C. CoPolym.ization: Toward a Systematic Approach; Springer Science & Business Media: New York, NY, USA, 1999. [Google Scholar]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Macromol. Sci. Part A Chem. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdos, F. Analysis of the Linear Methods for Determining Copolymerization Reactivity Ratios. I. A New Improved Linear Graphic Method. J. Macromol. Sci. Chem. 1975, 9, 1–27. [Google Scholar] [CrossRef]
- Beckingham, B.S.; Sanoja, G.E.; Lynd, N.A. Simple and Accurate Determination of Reactivity Ratios Using a Nonterminal Model of Chain Copolymerization. Macromolecules 2015, 48, 6922–6930. [Google Scholar] [CrossRef] [Green Version]
- Devasia, R.; Reghunadhan Nair, C.P.; Ninan, K.N. Polyacrylonitrile precursors for carbon fiber with imidocarboxylic acid units: Copolymerization of acrylonitrile with maleimidobenzoic acid. J. Macromol. Sci. Part A 2002, 39, 693–708. [Google Scholar] [CrossRef]
- Barson, C.A.; Fenn, D.R. A method for determining reactivity ratios when copolymerizations are influenced by penultimate group effects. Eur. Polym. J. 1987, 23, 833–834. [Google Scholar] [CrossRef]
- Lowry, G.G. The effect of depropagation on copolymer composition. I. General theory for one depropagating monomer. J. Polym. Sci. 1960, 42, 463–477. [Google Scholar] [CrossRef]
- Seiner, J.A.; Litt, M. The role of monomer charge-transfer complexes in free radical copolymerization. I. Derivation of terminal complex model equations. Macromolecules 1971, 4, 308. [Google Scholar] [CrossRef]
- Beginn, U. COPOINT—A simple computer program to determine copolymerization parameters by numerical integration. e-Polymers 2005, 5, 1–12. [Google Scholar] [CrossRef]
- Tidwell, P.W.; Mortimer, G.A. An improved method of calculating copolymerization reactivity ratios. J. Polym. Sci. Part A 1965, 3, 369. [Google Scholar] [CrossRef]
- Van Den Brink, M.; Smulders, W.; Van Herk, A.M.; German, A.L. Nonlinear regression by visualization of the sum of residual space applied to the integrated copolymerization equation with errors in all variables. I. Introduction of the model, simulations and design of experiments. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 3804–3816. [Google Scholar] [CrossRef]
- Igarashi, S. Representation of composition and blockiness of the copolymer by a triangular coordinate system. J. Polym. Sci. Part B Polym. Lett. 1963, 1, 359–363. [Google Scholar] [CrossRef]
- Bovey, F.A.; Mirau, P.A. NMR of Polymers; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Kokkorogianni, O.; Kontoes-Georgoudakis, P.; Athanasopoulou, M.; Polizos, N.; Pitsikalis, M. Statistical Copolymers of N-Vinylpyrrolidone and Isobornyl Methacrylate via Free Radical and RAFT Polymerization: Monomer Reactivity Ratios, Thermal Properties, and Kinetics of Thermal Decomposition. Polymers 2021, 13, 778. [Google Scholar] [CrossRef] [PubMed]
- Mitsoni, E.; Roka, N.; Pitsikalis, M. Statistical copolymerization of N-vinyl-pyrrolidone and alkyl methacrylates via RAFT: Reactivity ratios and thermal analysis. J. Polym. Res. 2019, 26, 118. [Google Scholar] [CrossRef]
- Gikarakis, T.; Pappas, I.; Arvanitaki, P.; Pantazi, E.; Mitsoni, E.; Roka, N.; Pitsikalis, M. Thermal Stability and Kinetics of Thermal Decomposition of Statistical Copolymers of N-Vinylpyrrolidone and Alkyl Methacrylates Synthesized via RAFT Polymerization. J. Chem. 2021, 2021, 6633052. [Google Scholar] [CrossRef]
- Roka, N.; Pitsikalis, M. Statistical copolymers of N-vinylpyrrolidone and benzyl methacrylate via RAFT: Monomer reactivity ratios, thermal properties and kinetics of thermal decomposition. J. Macromol. Sci. Part A Pure Appl. Chem. 2018, 55, 222–230. [Google Scholar] [CrossRef]
- Roka, N.; Kokkorogianni, O.; Pitsikalis, M. Statistical copolymers of N-vinylpyrrolidone and 2-(dimethylamino)ethyl methacrylate via RAFT: Monomer reactivity ratios, thermal properties, and kinetics of thermal decomposition. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3776–3787. [Google Scholar] [CrossRef]
- Knapp, K.A.; Nuñez, I.M.; Shipp, D.A. Effect of polymer chain architecture on the aqueous solution properties of amphiphilic copolymers: A study of poly(N-vinylpyrrolidone-co-vinyl laurate). Polymer 2018, 141, 54–61. [Google Scholar] [CrossRef]
- Ang, M.T.C.; Gumbau-Brisa, R.; Allan, D.S.; McDonald, R.; Ferguson, M.J.; Holbein, B.E.; Bierenstiel, M. DIBI, a 3-hydroxypyridin-4-one chelator iron-binding polymer with enhanced antimicrobial activity. Medchemcomm 2018, 9, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Chen, J.K.; Chen, T.; Huang, C.F. Synthesis of PNVP-Based Copolymers with tunable thermosensitivity by sequential reversible addition-fragmentation chain transfer copolymerization and ring-opening polymerization. Polymers 2017, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Xu, W.; Pich, A. Temperature and pH dual-responsive poly(vinyl lactam) copolymers functionalized with amine side groups: Via RAFT polymerization. Polym. Chem. 2016, 7, 5011–5022. [Google Scholar] [CrossRef]
- Peng, H.; Huang, X.; Melle, A.; Karperien, M.; Pich, A. Redox-responsive degradable prodrug nanogels for intracellular drug delivery by crosslinking of amine-functionalized poly(N-vinylpyrrolidone) copolymers. J. Coll. Interfaces Sci. 2019, 540, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Kather, M.; Rübsam, K.; Jakob, F.; Schwaneberg, U.; Pich, A. Water-Soluble Reactive Copolymers Based on Cyclic N-Vinylamides with Succinimide Side Groups for Bioconjugation with Proteins. Macromolecules 2015, 48, 4256–4268. [Google Scholar] [CrossRef]
- Peng, H.; Rübsam, K.; Jakob, F.; Schwaneberg, U.; Pich, A. Tunable Enzymatic Activity and Enhanced Stability of Cellulase Immobilized in Biohybrid Nanogels. Biomacromolecules 2016, 17, 3619–3631. [Google Scholar] [CrossRef]
- Peng, H.; Rübsam, K.; Huang, X.; Jakob, F.; Karperien, M.; Schwaneberg, U.; Pich, A. Reactive copolymers based on N-vinyl lactams with pyridyl disulfide side groups via RAFT polymerization and postmodification via thiol-disulfide exchange reaction. Macromolecules 2016, 49, 7141–7154. [Google Scholar] [CrossRef]
- Zhao, X.; Coutelier, O.; Nguyen, H.H.; Delmas, C.; Destarac, M.; Marty, J.D. Effect of copolymer composition of RAFT/MADIX-derived N-vinylcaprolactam/N-vinylpyrrolidone statistical copolymers on their thermoresponsive behavior and hydrogel properties. Polym. Chem. 2015, 6, 5233–5243. [Google Scholar] [CrossRef]
- Maji, S.; Zhang, Z.; Voorhaar, L.; Pieters, S.; Stubbe, B.; Van Vlierberghe, S.; Dubruel, P.; Geest, B.; Hoogenboom, R. Thermoresponsive polymer coated gold nanoparticles: From MADIX/RAFT copolymerization of N-vinylpyrrolidone and N-vinylcaprolactam to salt and temperature induced nanoparticle aggregation. RSC Adv. 2015, 5, 42388–42398. [Google Scholar] [CrossRef]
- Ponce-Vargas, S.M.; Cortez-Lemus, N.A.; Licea-Claveríe, A. Preparation of poly(N-Vinylcaprolactam) (NVCL) and statistical copolymers of NVCL with variable cloud point temperature by using a trithiocarbonate RAFT agent. Macromol. Symp. 2013, 325, 56–70. [Google Scholar] [CrossRef]
- Wohl, B.M.; Smith, A.A.A.; Kryger, M.B.L.; Zelikin, A.N. Narrow therapeutic window of ribavirin as an inhibitor of nitric oxide synthesis is broadened by macromolecular prodrugs. Biomacromolecules 2013, 14, 3916–3926. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, Z.; Young, D.J.; Liu, M.; Wu, C.; Wu, Y.L.; Loh, X.J. Towards the Prevention of Coronavirus Infection: What Role Can Polymers Play? Mater. Today Adv. 2021, 10, 100140. [Google Scholar] [CrossRef] [PubMed]
- Zaitsev, S.D.; Semchikov, Y.D.; Chernikova, E.V. Controlled radical copolymerization of N-vinylpyrrolidone with 1,1,1,3,3,3-hexafluoroisopropyl-α-fluoroacrylate. Polym. Sci. Ser. B 2009, 51, 84–88. [Google Scholar] [CrossRef]
- Gasilova, E.R.; Solomin, I.V.; Kulikov, E.E.; Zotova, O.S.; Zaitsev, S.D.; Semchikov, Y.D. Conformations of Alternating Partially Fluorinated Copolymers in Dilute Ethanol Solutions. Inter. J. Polym. Anal. Charact. 2013, 18, 510–519. [Google Scholar] [CrossRef]
- Riess, G.; Hurtrez, G.; Bahadur, P. Block Copolymers. In Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H.F., Bikales, N., Overberger, C.G., Menges, G., Kroschwitz, J.I., Eds.; John Wiley &Sons: Hoboken, NJ, USA, 1985; Volume 2, pp. 324–434. [Google Scholar]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H. Synthesis of block copolymers. Adv. Polym. Sci. 2005, 189, 1–124. [Google Scholar]
- Theodosopoulos, G.; Pitsikalis, M. Block Copolymers: Recent Synthetic Routes and Developments. In Anionic Polymerization: Principles, Practice, Strength, Consequences and Applications; Hadjichristidis, N., Hirao, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 541–623. [Google Scholar]
- Hamley, I.W. The Physics of Block Copolymers; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Abetz, V.; Simon, P.F.W. Phase behavior and morphologies of block copolymers. Adv. Polym. Sci. 2005, 189, 125–212. [Google Scholar]
- Hamley, I.W. (Ed.) Developments in Block Copolymer Science and Technology; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Gohy, J.F. Block Copolymer Micelles. Adv. Polym. Sci. 2005, 190, 65–136. [Google Scholar]
- Riess, G. Micellization of block copolymers. Progr. Polym. Sci. 2003, 28, 1107–1170. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P. Structure and design of polymeric surfactant-based drug delivery systems. J. Control. Release 2001, 73, 137–172. [Google Scholar] [CrossRef]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Kabanov, A.V.; Batrakova, E.V.; Alakhov, V.Y. Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J. Control. Release 2002, 82, 189–212. [Google Scholar] [CrossRef]
- Kakizawa, Y.; Kataoka, K. Block copolymer micelles for delivery of gene and related compounds. Adv. Drug Deliv. Rev. 2002, 54, 203–222. [Google Scholar] [CrossRef]
- Munch, M.R.; Gast, A.P. Kinetics of block copolymer adsorption on dielectric surfaces from a selective solvent. Macromolecules 1990, 23, 2313–2320. [Google Scholar] [CrossRef]
- Xu, R.; D’Oliveira, J.M.R.; Winnik, M.A.; Riess, G.; Croucher, M.D. Characterization of Block Copolymer Micelles and Their Adsorption Onto Latex Particles. J. Appl. Polym. Sci. Appl. Polym. Symp. 1992, 51, 135–149. [Google Scholar] [CrossRef]
- Breulman, M.; Förster, S.; Antonietti, M. Mesoscopic surface patterns formed by block copolymer micelles. Macromol. Chem. Phys. 2000, 201, 204–211. [Google Scholar] [CrossRef]
- Cao, T.; Yin, W.; Armstrong, J.L.; Webber, S.E. Adsorption of Photoactive Amphiphilic Polymers onto Hydrophobic Polymer Films: Polystyrene-block-poly(2-vinylnaphthalene)-block-poly(methacrylic acid). Langmuir 1994, 10, 1841–1847. [Google Scholar] [CrossRef]
- Spatz, J.P.; Sheiko, S.; Möller, M. Ion-Stabilized Block Copolymer Micelles: Film Formation and Intermicellar Interaction. Macromolecules 1996, 29, 3220–3226. [Google Scholar] [CrossRef]
- Andrew, G.; Martina, H.S. Complex polymer architectures via RAFT polymerization: From fundamental process to extending the scope using click chemistry and nature’s building blocks. Progr. Polym. Sci. 2012, 37, 38–105. [Google Scholar]
- Moad, G.; Rizzardo, E.; Thang, S.H. Radical addition–fragmentation chemistry in polymer synthesis. Polymer 2008, 49, 1079–1131. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.K. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 495–505. [Google Scholar]
- Hawker, C.J.; Bosman, A.W.; Harth, E. New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerizations. Chem. Rev. 2001, 101, 3661–3688. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Xia, J. Atom Transfer Radical Polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Davis, T.P. (Eds.) Handbook of Radical Polymerization; Wiley-Interscience: Hoboken, NJ, USA, 2002. [Google Scholar]
- Renjith, D.; Raveendra, L.B.; Redouane, B.; Nathalie, M.; Yves, G. Controlled Radical Polymerization of N-Vinylpyrrolidone by Reversible Addition-Fragmentation Chain Tranfer Process. Macromol. Symp. 2005, 229, 8–17. [Google Scholar]
- Zard, S.Z. Discovery of the RAFT/MADIX Process: Mechanistic Insights and Polymer Chemistry Implications. Macromolecules 2020, 53, 8144–8159. [Google Scholar] [CrossRef]
- Fandrich, N.; Falkenhagen, J.; Weidner, S.M.; Pfeifer, D.; Staal, B.; Thunemann, A.F.; Laschewsky, A. Characterization of New Amphiphilic Block Copolymers of N-Vinyl Pyrrolidone and Vinyl Acetate, 1—Analysis of Copolymer Composition, End Groups, Molar Masses and Molar Mass Distributions. Macromol. Chem. Phys. 2010, 211, 869–878. [Google Scholar] [CrossRef]
- Fandrich, N.; Falkenhagen, J.; Weidner, S.M.; Staal, B.; Thunemann, A.F.; Laschewsky, A. Characterization of New Amphiphilic Block Copolymers of N-Vinylpyrrolidone and Vinyl Acetate, 2—Chromatographic Seperation and Analysis by MALDI-TOF and FT-IR Coupling. Macromol. Chem. Phys. 2010, 211, 1678–1688. [Google Scholar] [CrossRef]
- Bailly, N.; Thomas, M.; Klumperman, B. Poly(N-ninylpyrrolidone)-block-poly(vinyl acetate) as a Drug Delivery Vehicle for Hydrophobic Drugs. Biomacromolecules 2012, 13, 4109–4117. [Google Scholar] [CrossRef]
- Binauld, S.; Delafresnaye, L.; Charleux, B.; D’Agosto, F.; Lansalot, M. Emulsion Polymerization of Vinyl Acetate in Presence of Different Hydrophilic Polymers Obtained by RAFT/MADIX. Macromolecules 2014, 47, 3461–3472. [Google Scholar] [CrossRef]
- Pound, G.; Eksteen, Z.; Pfukwa, R.; McKenzie, J.M.; Lange, R.F.M.; Klumperman, B. Enhancing control of macromolecular architecture by synergistic combination of atom transfer radical polymerization, nitroxide mediated polymerization, and reversible addition-fragmentation chain transfer polymerization. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6575–6593. [Google Scholar] [CrossRef]
- Corgdon, T.R.; Notman, R.; Gibson, M.I. Influence of Block Copolymerization on the Antifreeze Protein Mimetic Ice Recrystallization Inhibition Activity of Poly(vinyl alcohol). Biomacromolecules 2016, 17, 3033–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deepak; Sharma, S.; Kumar, R.; Nandy, K.; Srivastava, A.; Tomar, S.M.; Acharya, A. Developing a nontoxic and biocompatible polymeric self-assembly by using RAFT methodology for biomedical application. Mater. Today Commun. 2019, 18, 14–24. [Google Scholar] [CrossRef]
- Liang, X.; Kozlovskaya, V.; Cox, C.P.; Wang, Y.; Saeed, M.; Kharlampieva, E. Synthesis and Self-Assembly of Thermosensitive Double-Hydrophilic Poly(N-vinylcaprolactam)-b-poly(N-vinyl-2-pyrrolidone) Diblock Copolymers. J. Polym. Chem. 2014, 52, 2725–2737. [Google Scholar] [CrossRef]
- Huang, Z.; Pan, P.; Bao, Y. Solution and Aqueous Miniemulsion Polymerization of Vinyl Chloride Mediated by a Fluorinated Xanthate. J. Polym. Chem. 2016, 54, 2092–2101. [Google Scholar] [CrossRef]
- Giliomee, J.; Pfukwa, R.; Gule, N.P.; Klumperman, B. Smart block copolymers of PVP and an alkylated PVP derivative: Synthesis, characterization, thermoresponsive behavior and self-assembly. Polym. Chem. 2016, 7, 1138–1146. [Google Scholar] [CrossRef]
- Sugihara, S.; Kawamoto, Y.; Maeda, Y. Direct Radical Polymerization of Vinyl Ethers: Reversible Addition-Fragmentation Chain Transfer Polymerization of Hdroxy-Functional Vinyl Ethers. Macromolecules 2016, 49, 1563–1574. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Umeda, A.; Sato, Y.; Mori, H. Synthesis of 1,2,4-triazolium salt-based polymers and block copolymers by RAFT polymerization: Ion conductivity and assembled structures. Polymer 2016, 96, 81–93. [Google Scholar] [CrossRef]
- Li, L.; Li, J.; Zheng, S. Poly(vinylidene fluoride)-block-poly(N-vinylpyrrolidone) deblock copolymers: Synthesis via sequential RAFT/MADIX polymerization and self-assembly behavior. Polymer 2018, 142, 61–71. [Google Scholar] [CrossRef]
- Aoshima, H.; Uchiyama, M.; Satoh, K.; Kamigaito, M. Interconvertible Livinh Radical and Cationic Polymerization through Reversible Activation of Dormant Species with Dual Activity. Angew. Chem. Int. Ed. 2014, 53, 10932–10936. [Google Scholar] [CrossRef]
- Uchiyama, M.; Satoh, K.; Kamigaito, M. Thioether-Mediated Degenerative Chain-Transfer Chain-Transfer Cationic Polymerization: A Simple Metal-Free System for Living Cationic Polymerization. Macromolecules 2015, 48, 5533–5542. [Google Scholar] [CrossRef]
- Uchiyama, M.; Satoh, K.; Kamigaito, M. Cationic RAFT Polymerization Using ppm Concentrations of Organic Acid. Angew. Chem., Int. Ed. 2015, 54, 1924–1928. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, M.; Satoh, K.; Kamigaito, M. Diversifying Cationic RAFT Polymerization with Various Counteranions: Generation of Cationic Species from Organic Halides and Various Metal Salts. ACS Macromol. Lett. 2016, 5, 1157–1161. [Google Scholar] [CrossRef]
- Bilalis, P.; Pitsikalis, M.; Hadjichristidis, N. Controlled Nitroxide-Mediated and Reversible Addition-Fragmentation Chain Transfer Polymerization of N-Vinylpyrrolidone: Synthesis of Block Copolymers with Styrene and 2-Vinylpyridine. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 659–665. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, X.; Yuan, J.; Ding, T.; Gao, Q. Preparation and drug controlled-release of polyion complex micelles as drug delivery systems. Colloids Surf. B Biointerf. 2009, 68, 218–224. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Xie, D.; Liu, D.; Chen, Z.; Li, K.; Li, Z.; Tichnell, B.; Liu, Z. Design and synthesis study of the thermosensitive poly(N-vinylpyrrolidone-b-N, N-diethylacrylamide). Des. Monom. Polym. 2018, 21, 43–54. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, T.; Song, X.; Zhang, L.; Zhang, C.; Jin, S.; Xing, J.; Liang, X.J. Structural impact of graft and block copolymers based on poly(N-vinylpyrrolidone) and poly(2-dimethylaminoethyl methacrylate) in gene delivery. J. Mater. Chem. B. 2015, 3, 4027–4035. [Google Scholar] [CrossRef]
- Zhang, C.; Li, L.; Cong, H.; Zheng, S. Poly(methyl methacrylate)-block-poly(N-vinyl pyrrolidone) Diblock Copolymer: A Facile Synthesis via Sequential Radical Polymerization Mediated by Isopropylxanthic Disulfide and Its Nanostructuring Polybenzoxazine Thermosets. J. Polym. Chem. 2014, 52, 952–962. [Google Scholar] [CrossRef]
- Luo, Y.L.; Yuan, J.F.; Liu, X.J.; Xie, H.; Gao, Q.Y. Self-assembled Polyion Complex Micelles based on PVP-b-PAMPS and PVP-b-PDMAEMA for Drug Delivery. J. Bioact. Compat. Polym. 2010, 25, 292–304. [Google Scholar] [CrossRef]
- Zhang, P.; Zhong, X.; Chai, Y.; Liu, Y. The effect of PVP-b-PMAA block copolymer on morphologies control of calcium carbonate. Colloid Polym. Sci. 2008, 286, 1135–1141. [Google Scholar] [CrossRef]
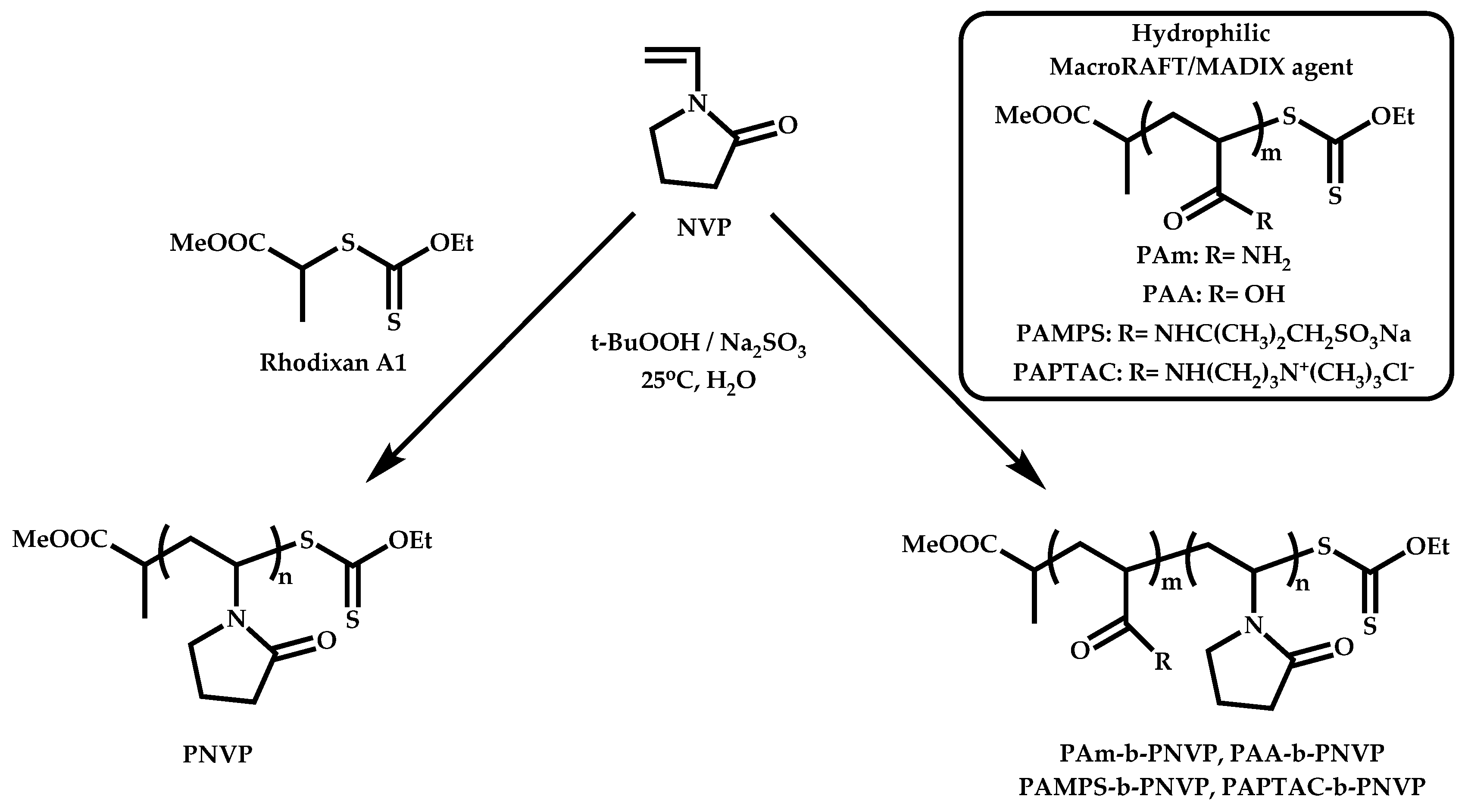
- Guinaudeau, A.; Coutelier, O.; Sandeau, A.; Mazieres, S.; Nguyen Thi, H.D.; Le Drogo, V.; Wilson, D.J.; Destarac, M. Facile Access to Poly(N-vinylpyrrolidone)-Based Double Hydrophilic Block Copolymers by Aqueous Ambient RAFT/MADIX Polymerization. Macromolecules 2014, 47, 41–50. [Google Scholar] [CrossRef]
- Bartels, J.W.; Billings, B.L.; Ghosh, B.; Urban, M.W.; Greenlief, M.; Wooley, K.L. Amphiphilic Cross-Linked Networks Produced from the Vulcanization of Nanodomains within Thin Films of Poly(N-vinylpyrrolidone)-b-Poly(isoprene). Langmuir 2009, 25, 9535–9544. [Google Scholar] [CrossRef] [PubMed]
- Nandy, K.; Srivastava, A.; Afgan, S.; Deepak; Kumar, R.; Rawar, A.K.; Singh, R.; Singh, R.K. The benzyl ethyl trithiocarbonate mediated control synthesis of a block copolymer containing N-vinyl pyrrolidone by RAFT methodology: Influence of polymer composition on cell cytotoxicity and cell viability. Eur. Polym. J. 2020, 122, 109387. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, H.; Zhang, L.; Zhou, Y.; Zhang, W.; Pan, X.; Zhang, Z.; Zhu, X. RAFT polymerization of N-vinylpyrrolidone mediated by cyanoprop-2-yl-1-dithionaphthalate in the presence of a fluoroalcohol: The possibility of altering monomer properties by hydrogen bonding? Polym. Chem. 2016, 7, 2015–2021. [Google Scholar] [CrossRef]
- Banerjee, S.; Guerre, M.; Ameduri, B.; Ladmiral, V. Synthesis of 2-(trifluoromethyl)acrylate-containg block copolymers via RAFT polymerization using a universal chain transfer agent. Polym. Chem. 2018, 9, 3511–3521. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Bai, L.; Zhang, L.; Cheng, Z.; Zhu, X. Facile Synthesis of poly(N-vinyl pyrrolidone) block copolymers with “more activated” monomers by using photoinduced successive RAFT polymerization. Polym. Chem. 2020, 11, 2080–2088. [Google Scholar] [CrossRef]
- Benaglia, M.; Chiefari, J.; Chong, Y.K.; Moad, G.; Rizzardo, E.; Thang, S.H. Universal (Switchable) RAFT Agent. J. Am. Chem. Soc. 2009, 131, 6914–6915. [Google Scholar] [CrossRef]
- Moad, G.; Keddie, D.; Guerrero-Sanchez, C.; Rizzardo, E.; Thang, S.H. Advances in Switchable RAFT Polymerization. Macromol. Symp. 2015, 350, 34–42. [Google Scholar] [CrossRef]
- Keddie, D.J.; Guerrero-Sanchez, C.; Moad, G.; Rizzardo, E.; Thnag, S.H. Switchable Reversible Addition—Fragmentation Chain Transfer (RAFT) Polymerization in Aqueous Solution, N,N-Dimethylacrylamide. Macromolecules 2011, 44, 6738–6745. [Google Scholar] [CrossRef]
- Pan, X.; Guo, X.; Choi, B.; Feng, A.; Wei, X.; Thang, S.H. A facile synthesis of pH stimuli biocompatible block copolymer poly(methacrylic acid)-block-poly(N-vinylpyrrolidone) utilizing switchable RAFT agents. Polym. Chem. 2019, 10, 2083–2090. [Google Scholar] [CrossRef]
- Rajput, F.; Colantuoni, A.; Bayahya, S.; Dhane, R.; Servio, P.; Maric, M. Poly(styrene/pentaflyorostyrene)-block-Poly(vinyl alcohol/vinylpyrrolidone) Amphiphilic Block Copolymers for Kinetic Gas Hydrate Inhibitors: Synthesis, Micellization Behavior and Methane Hydrate Kinetic Inhibition. Polym. Chem. 2018, 56, 2445–2457. [Google Scholar] [CrossRef]
- Stace, S.J.; Moad, G.; Fellows, C.M.; Keddie, D.J. The effect of Z-group modification on the RAFT polymerization of N-vinylpyrrolidone controlled by “switchable” N-pyridyl-functional dithiocarbamates. Polym. Chem. 2015, 6, 7119–7126. [Google Scholar] [CrossRef]
- Bernaerts, K.V.; Du Prez, F.E. Dual/heterofunctional initiators for the combination of mechanistically distinct polymerization techniques. Prog. Polym. Sci. 2006, 31, 671–722. [Google Scholar] [CrossRef]
- Yagci, Y.; Tasdelen, M.A. Mechanistic transformations involving living and controlled/living polymerization methods. Prog. Polym. Sci. 2006, 31, 1133–1170. [Google Scholar] [CrossRef]
- Huang, C.F.; Nicolay, R.; Kwak, Y.; Chang, F.C.; Matyjaszewski, K. Homopolymerization and Block Copolymerization of N-Vinylpyrrolidone by ATRP and RAFT with Haloxanthate Inifers. Macromolecules 2009, 42, 8198–8210. [Google Scholar] [CrossRef]
- Jumeaux, C.; Chapman, R.; Chandrawati, R.; Stevens, M.M. Synthesis and self-assembly of temperature-responsive copolymers based on N-vinylpyrrolidone and triethylene glycol methacrylate. Polym. Chem. 2015, 6, 4116–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang Yu, Y.; Li, G.; Kim, J.; Youk, J.H. One-pot synthesis of poly(N-vinylcaprolactam) -based biocompatible block copolymers using a dual initiator for ROP and RAFT polymerization. Polymer 2013, 54, 6119–6124. [Google Scholar]
- Kang, H.U.; Chang Yu, Y.; Shin, S.J.; Kim, J.; Youk, J.H. One-Pot Synthesis of Poly(N-vinylpyrrolidone)-b-poly(ε-caprolactone) Block Copolymers Using a Dual Initiator for RAFT Polymerization and ROP. Macromolecules 2013, 46, 1291–1295. [Google Scholar] [CrossRef]
- Shin, S.J.; Yu, Y.C.; Seo, J.D.; Cho, S.J.; Youk, J.H. One-Step Synthesis of Poly(N-vinylpyrrolidone)-b-poly(L-lactide) Block Copolymers Using a Dual Initiator for RAFT Polymerization and ROP. J. Polym. Sci. 2014, 52, 1607–1613. [Google Scholar] [CrossRef]
- Pound, G.; Aguesse, F.; McLeary, J.B.; Lange, R.F.M.; Klumperman, B. Xanthate-Mediated Copolymerization of Vinyl Monomers for Amphiphilic and Double-Hydrophilic Block Copolymers with Poly(ethylene glycol). Macromolecules 2007, 40, 8861–8871. [Google Scholar] [CrossRef]
- Yu, Y.C.; Kang, H.W.; Lee, D.W.; Ki, B.R.; Youk, J.H. Synthesis of amphiphilic poly(N-vinylpyrrolidone)-b-poly(ε-caprolactone) block copolymers by a combination of ROP and RAFT. In Proceedings of the 18th International Conference on Composite Materials, Jeju, Korea, 21–26 August 2011. [Google Scholar]
- Mishra, A.K.; Patel, V.K.; Vishwakarma, N.K.; Biswas, C.S.; Raula, M.; Misra, A.; Mandal, T.K.; Ray, B. Synthesis of Well-Defined Amphiphilic Poly(ε-caprolactone)-b-poly(N-vinylpyrrolidone) Block Copolymers via the Combination of ROP and Xanthate-Mediated RAFT Polymerization. Macromolecules 2011, 44, 2465–2473. [Google Scholar] [CrossRef]
- Ramesh, K.; Mishra, A.K.; Patel, V.K.; Vishwakarma, N.K.; Biswas, C.S.; Paira, T.K.; Mandal, T.K.; Maiti, P.; Misra, N.; Ray, B. Synthesis of well-defined amphiphilic poly(D,L-Lactide)-b-poly(N-vinylpyrrolidone) block copolymers using ROP and xanthate-mediated RAFT polymerization. Polymer 2012, 53, 5743–5753. [Google Scholar] [CrossRef]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ Chemistry in Polymer and Materials Science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Xi, W.; Scott, T.F.; Kloxin, C.J.; Bowman, C.N. Click Chemistry in Materials Science. Adv. Funct. Mater. 2014, 24, 2572–2590. [Google Scholar] [CrossRef] [Green Version]
- Geng, Z.; Shin, J.J.; Xi, Y.; Hawker, C.J. Click chemistry strategies for the accelerated synthesis of functional macromolecules. J. Polym. Sci. 2021, 59, 963–1042. [Google Scholar] [CrossRef]
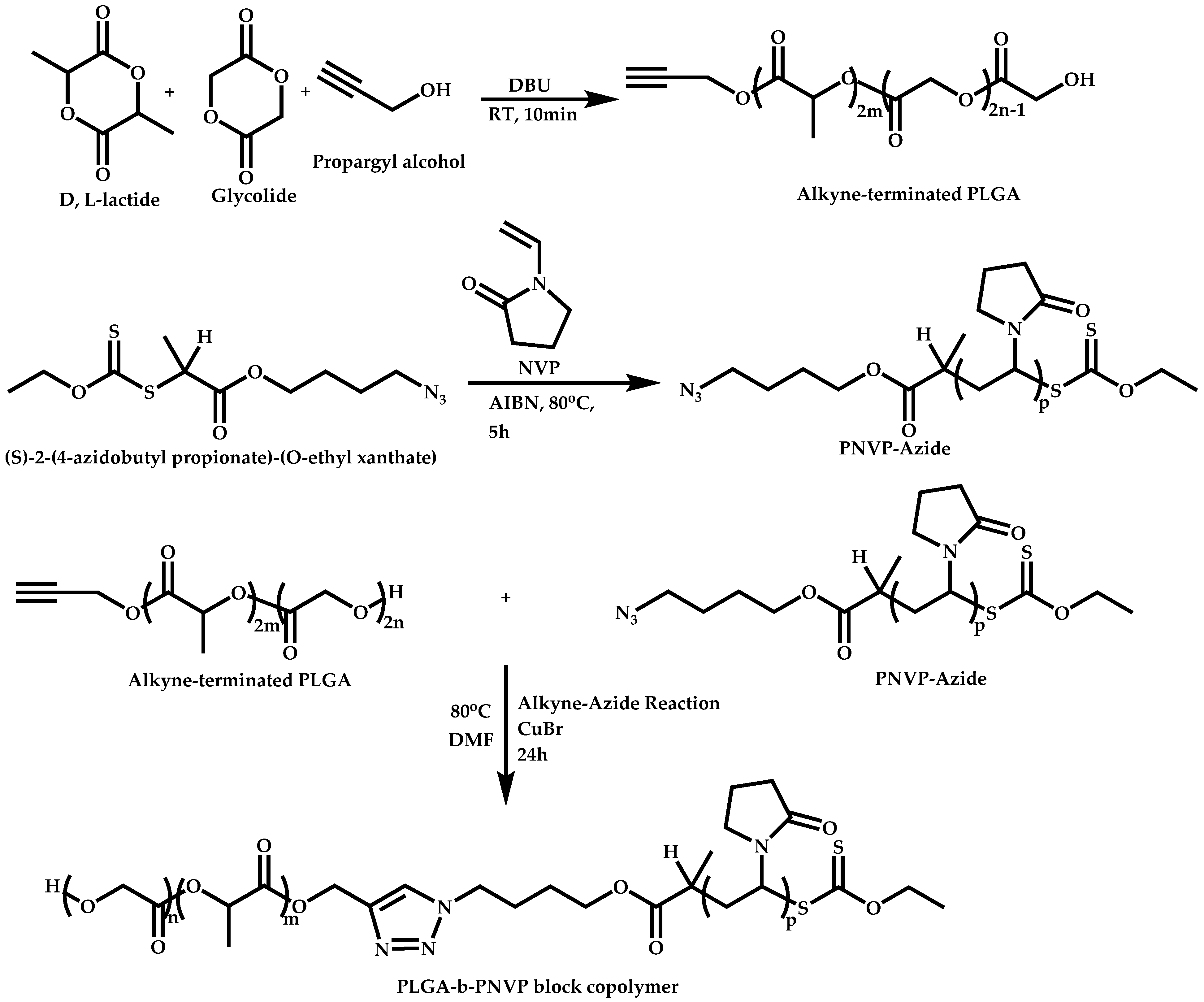
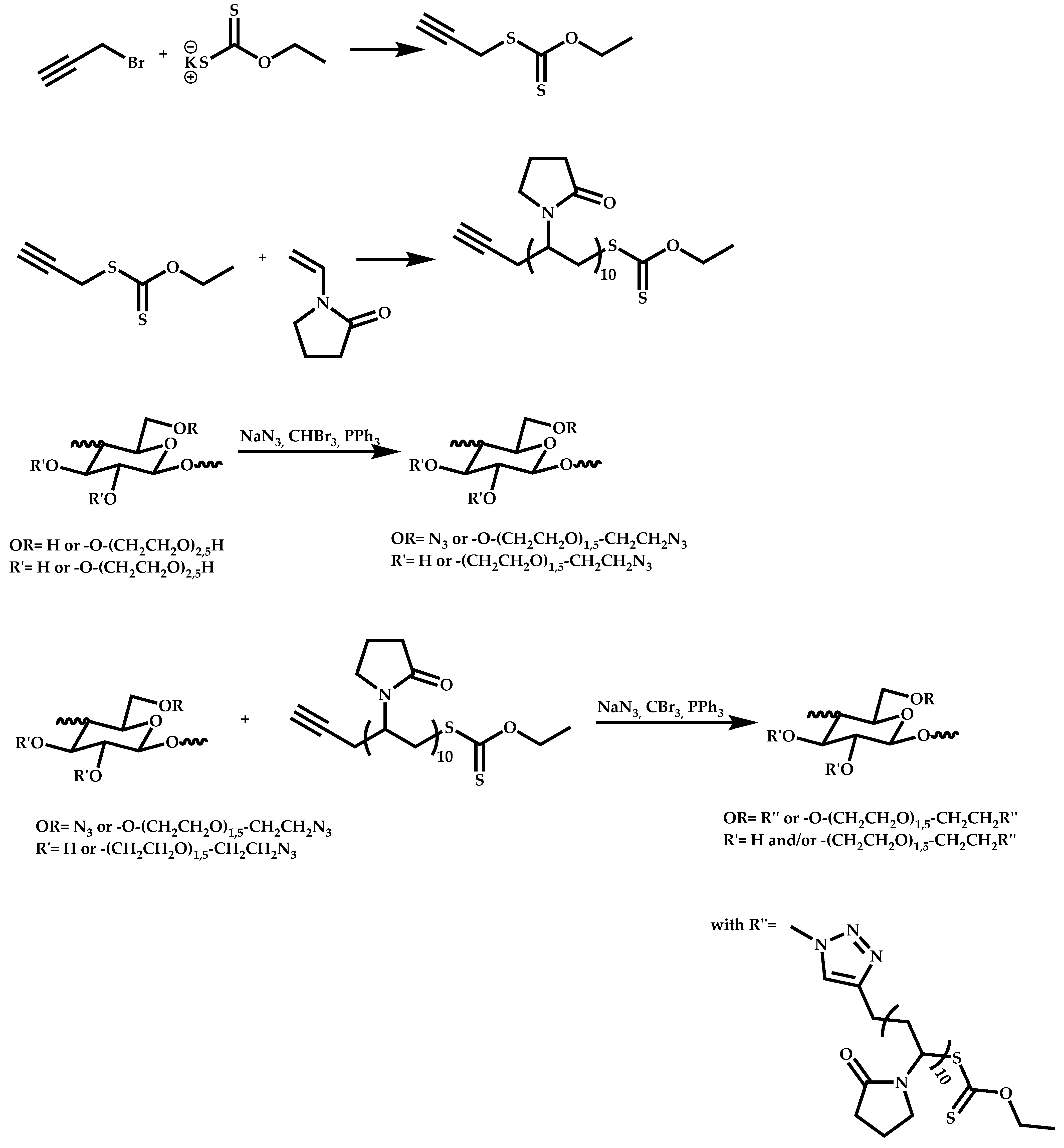
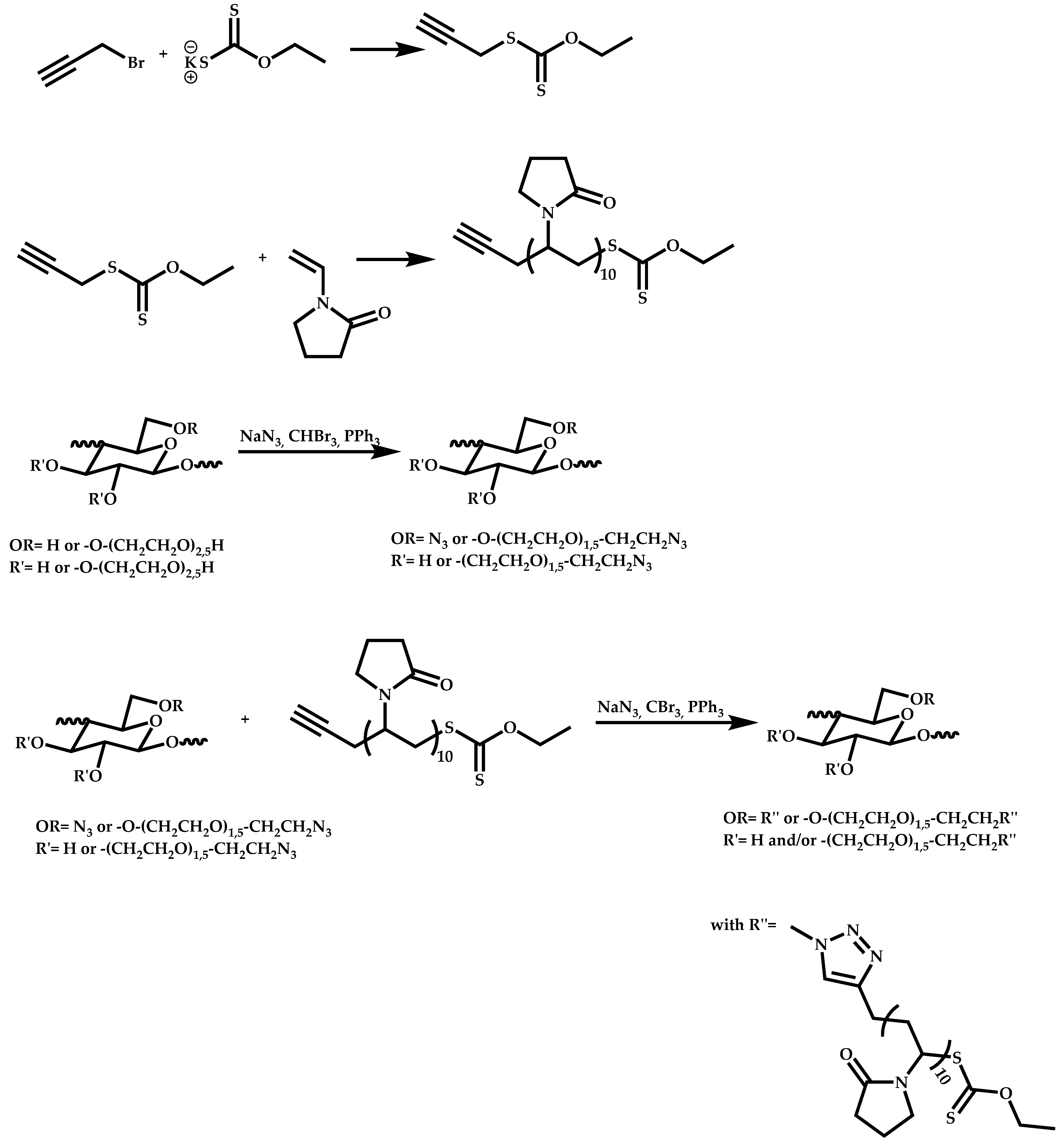
- Patel, V.K.; Vishwakarma, N.K.; Mishra, A.K.; Biswas, C.S.; Maiti, P.; Ray, B. Synthesis of Alkyne-Terminated Xanthated RAFT Agents and Their Uses for the Controlled Radical Polymerization of N-Vinylpyrrolidone and the Synthesis of Its Block Copolymer Using Click Chemistry. J. Appl. Polym. Sci. 2013, 127, 4305–4317. [Google Scholar] [CrossRef]
- Ramesh, K.; Singh, S.; Mitra, K.; Chattopadhyay, D.; Misra, N.; Ray, B. Self-assembly of Novel Poly(D,L-Lactide-co-Glycolide)-b-Poly(N-vinylpyrrolidone) (PLGA-b-PNVP) Amphiphilic Diblock Copolymers. Colloid Polym. Sci. 2016, 294, 399–407. [Google Scholar] [CrossRef]
- Jacobs, J.; Pound-Lana, G.; Klumperman, B. Poly(N-vinylpyrrolidone-b-(γ-benzyl-L-glutamate))—synthesis and self-assembly into pH-sensitive micelles. Polym. Chem. 2012, 3, 2551–2560. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Pispas, S.; Avgeropoulos, A. Linear and non-linear triblock terpolymers. Synthesis, self-assembly in selective solvents and in bulk. Prog. Polym. Sci. 2005, 30, 725–782. [Google Scholar] [CrossRef]
- Nie, S.; Xue, J.; Lu, Y.; Liu, Y.; Wang, D.; Sun, S.; Ran, F.; Zhao, C. Improved blood compatibility of polyethersulfone membrane with a hydrophilic and anionic surface. Coloids Surf. B Biointerf. 2012, 100, 116–125. [Google Scholar] [CrossRef]
- Ran, F.; Nie, S.; Zhao, W.; Li, J.; Su, B.; Sun, S.; Zhao, C. Biocompatibility of modified polyethersulfone membranes by blending an amphiphilic triblock co-polymer of poly(vinyl pyrrolidone)-b-poly(methyl methacrylate)-b-poly(vinyl pyrrolidone). Acta Biomater. 2011, 7, 3370–3381. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.Q.; Ran, F.; He, C.; Zhao, P.F.; Wei, X.H.; Li, J.; Zhao, C.S. Synthesis of amphiphilic tri-block copolymer poly(vinylpyrrolidone)-b-poly(methyl methacrylate)-b-poly(vinylpyrrolidone) for the modification of polyethersulfone membrane. Chin. Chem. Lett. 2011, 22, 370–373. [Google Scholar] [CrossRef]
- Cong, H.; Li, L.; Zheng, S. Poly(N-isopropylacrylamide)-block-poly(vinyl pyrrolidone) block copolymer networks: Synthesis and rapid thermoresponse of hudrogels. Polymer 2013, 54, 1370–1380. [Google Scholar] [CrossRef]
- Xiang, Y.; Li, L.; Zheng, S. Photophysical and dielectric properties of nanostructured epoxy thermosets containing poly(N-vinylcarbazole) nanophases. Polymer 2016, 98, 344–352. [Google Scholar] [CrossRef]
- Xiang, Y.; Cong, H.; Li, L.; Zheng, S. Poly(N-vinyl pyrrolidone)-block-Poly(N-vinyl carbazole)-block-Poly(N-vinyl pyrrolidone) Triblock Copolymers: Synthesis via RAFT/MADIX Process, Self-assembly Behavior, and Photphysical Properties. J. Polym. Chem. 2016, 54, 1852–1863. [Google Scholar] [CrossRef]
- Pavlovic, D.; Lou, Q.; Linhardt, J.G.; Kunzler, J.F.; Shipp, D.A. Poly(N-vinylpyrrolidone)-Polydimethylsiloxane Amphiphilic ABA Triblock Copolymers. J. Polym. Chem. 2017, 55, 3387–3394. [Google Scholar] [CrossRef]
- Ehsan, R.T.E.; Marzieh, G.; Mehdi, S.K.; Hossein, R.M.; Amir, K.K. Temperature-induced self-assembly of amphiphilic triblock terpolymers to low cytotoxic spherical and cubic structures as curcumin carriers. J. Mol. Liq. 2020, 313, 113504. [Google Scholar]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H. Polymers with Star-related Structures. In Macromolecular Engineering. Precise Synthesis, Materials Properties, Applications; Matyjaszewski, C., Gnanou, Y., Leibler, L., Eds.; Chapter 6; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007. [Google Scholar]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Sakellariou, G. Macromolecular Architectures by Living and Controlled/Living Polymerization. In Controlled and Living Polymerizations: From Mechanisms to Applications; Matyjaszewski, C., Muller, A.H.E., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 343–443. [Google Scholar]
- Hadjichristidis, N.; Pitsikalis, M.; Pispas, S.; Iatrou, H. Polymers with Complex Architecture by Living Anionic Polymerization. Chem. Rev. 2001, 101, 3747–3792. [Google Scholar] [CrossRef]
- Pitsikalis, M.; Pispas, S.; Mays, J.W.; Hadjichristidis, N. Nonlinear Block Copolymer Architectures. Adv. Polym. Sci. 1998, 135, 1–37. [Google Scholar]
- Hadjichristidis, N.; Pispas, S.; Iatrou, H.; Pitsikalis, M. Linking Chemistry and Anionic Polymerization. Curr. Org. Chem. 2002, 6, 155–176. [Google Scholar] [CrossRef]
- Iatrou, H.; Avgeropoulos, A.; Sakellariou, G.; Pitsikalis, M.; Hadjichristidis, N. Miktoarm Star (μ-Star) Polymers: A Successful Story. In Miktoarm Star Polymers: From Basics of Branched Architecture to Synthesis, Self-Assembly and Applications; Kakkar, A., Ed.; Polymer Chemistry Series No 25; The Royal Society of Chemistry: Croydon, UK, 2017; Chapter 1; pp. 1–30. [Google Scholar]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Mays, J. Macromolecular architectures by living and controlled/living polymerization. Prog. Polym. Sci. 2006, 31, 1068–1132. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Driva, P.; Sakellariou, G.; Chatzichristidi, M. Polymer with Star-Related Structures: Synthesis, Properties and Application. Polym. Sci. A Comp. Ref. 2012, 6, 29–109. [Google Scholar]
- Chaffey-Millar, H.; Bush, M.; Davis, T.P.; Stenzel, M.H.; Barner-Kowollik, C. Advanced computational strategies for modelling the evolution of full molecular weight distributions formed during multiarmed (star) polymerizations. Macromol. Theory Simul. 2005, 14, 143–157. [Google Scholar] [CrossRef]
- Chaffey-Millar, H.; Stenzel, M.H.; Davis, T.P.; Coote, M.L.; Barner-Kowollik, C. Design criteria for star polymer formation processes via living free radical polymerization. Macromolecules 2006, 39, 6406–6419. [Google Scholar] [CrossRef]
- Uyen Nguyen, T.L.; Eagles, K.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Investigation of the influence of the Architectures of Poly(vinyl pyrrolidone) Polymers Made via the Reversible Addition-Fragmentation Chain Transfer/Macromolecular Design via the Interchange of Xanthates Mechanism on the Stabilization of Suspension Polymerizations. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 4372–4383. [Google Scholar]
- Johnson, I.J.; Khosravi, E.; Musa, O.M.; Simnett, R.E.; Eissa, A.M. Xanthates Designed for the Preparation of N-Vinyl Pyrrolidone-Based Linear and Star Architectures via RAFT Polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 775–786. [Google Scholar] [CrossRef]
- Maksym, P.; Tarnacka, M.; Bernat, R.; Bielas, R.; Mielanczyk, A.; Hachula, B.; Kaminski, K.; Paluch, M. Pressure-assisted strategy for the synthesis of vinyl pyrrolidone-based macro-star photoiniferters. A route to star block copolymers. J. Polym. Sci. 2020, 58, 1393–1399. [Google Scholar] [CrossRef]
- Cortez-Lemus, M.A.; Licea-Claverie, A. Preparation of a Mini-Library of Thermo-Responsive Star (NVCL/NVP-Vac) Polymers with Tailored Properties Using a Hexafunctional Xanthate RAFT Agent. Polymers 2018, 10, 20. [Google Scholar] [CrossRef] [Green Version]
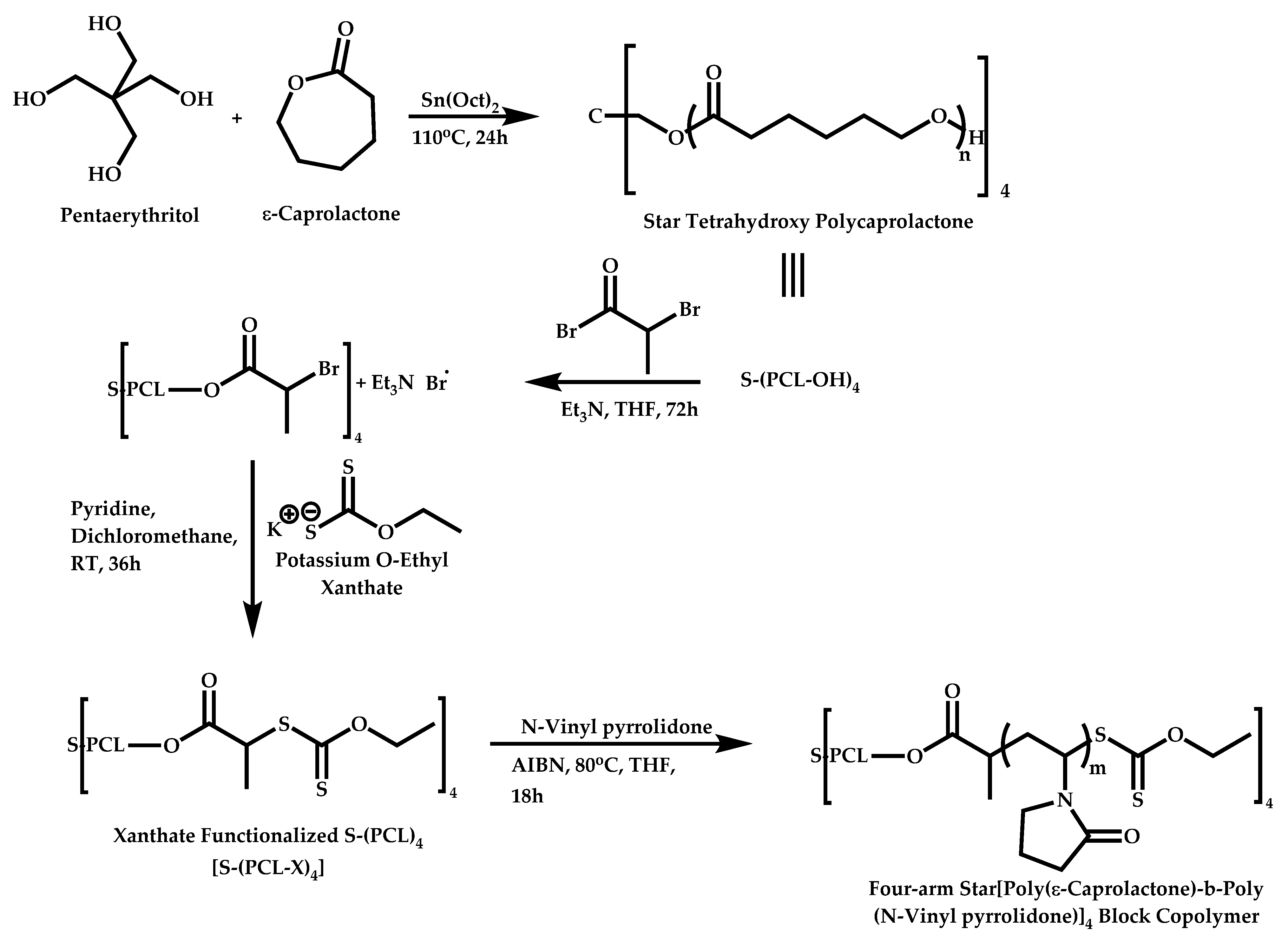
- Mishra, A.K.; Ramesh, K.; Paira, T.K.; Srivastava, D.N.; Mandal, T.K.; Misra, N.; Ray, B. Synthesis and self-assembly properties of well-defined four-arm star poly(ε-caprolactone)-b-poly(N-vinylpyrrolidone) amphiphilic block copolymers. Polym. Bull. 2013, 70, 3201–3220. [Google Scholar] [CrossRef]
- Hira, S.K.; Ramesh, K.; Gupta, U.K.; Mitra, K.; Misra, N.; Ray, B.; Manna, P.P.P. Methotrexate-loaded four-arm star amphiphilic block copolymer elicits CD8+ T cell response against a highly aggressive and metastatic experimental lymphoma. ACS Appl. Matter. Interfaces 2015, 7, 20021–20033. [Google Scholar] [CrossRef]
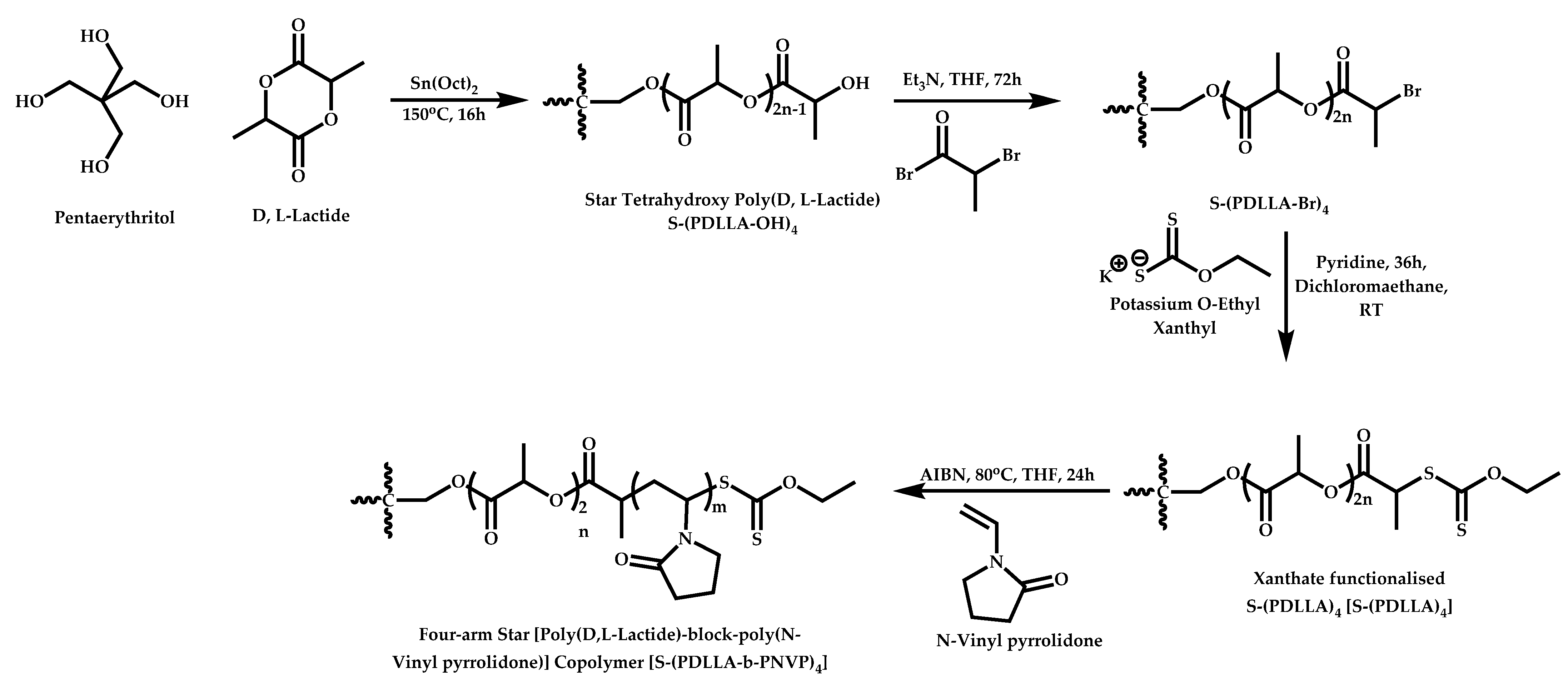
- Zhai, C.; Liu, X.; Yuan, J.; Gao, Q. Synthesis, characterization and drug delivery research of an amphiphilic biodegradable star-shaped block copolymer. Polym. Bull. 2013, 70, 419–429. [Google Scholar] [CrossRef]
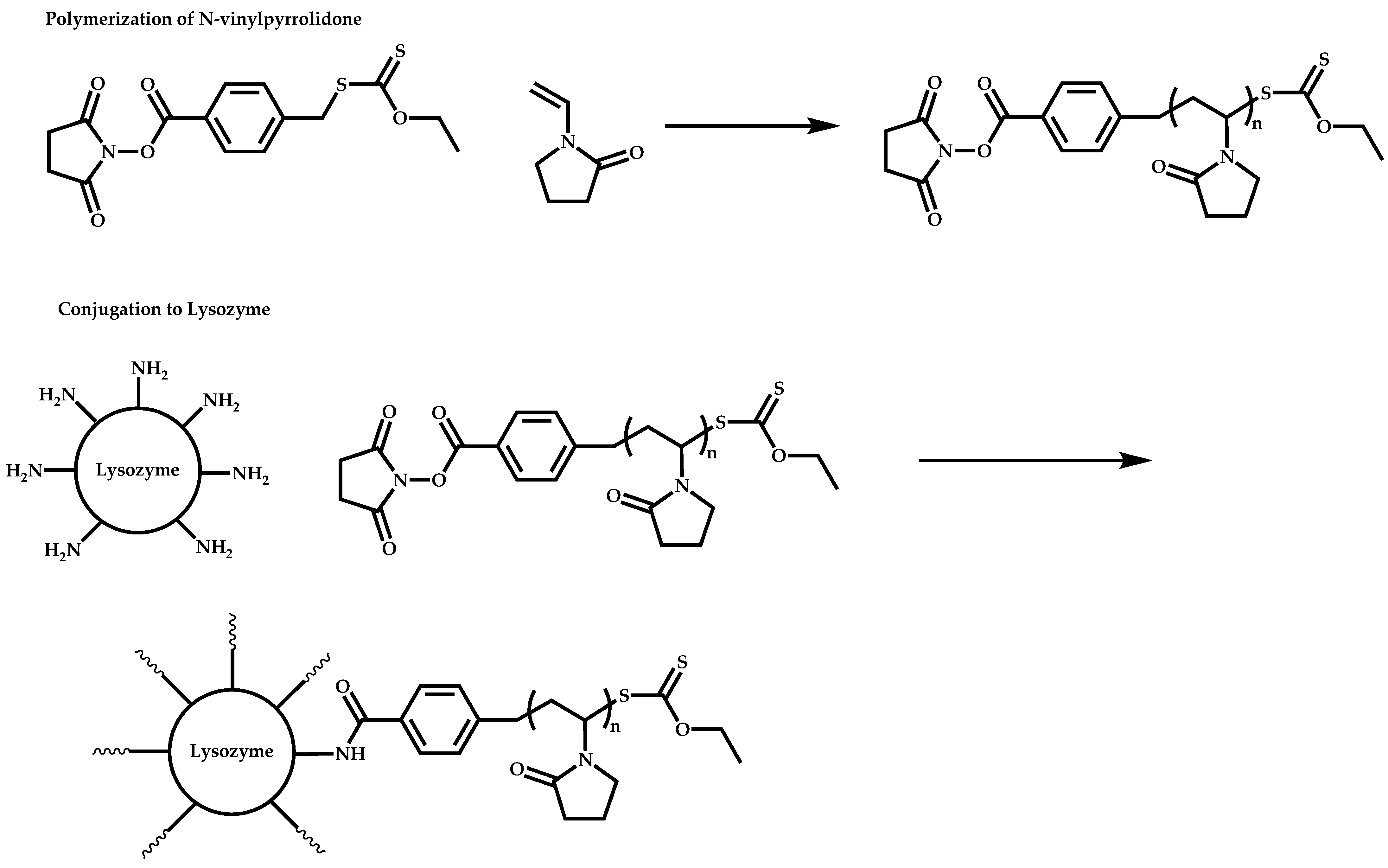
- McDowall, L.; Chen, G.; Stenzel, M.H. Synthesis of Seven-Arm Poly(vinyl pyrrolidone) Star Polymers with Lysozyme Core Prepared by MADIX/RAFT Polymerization. Macromol. Rapid Commun. 2008, 29, 1666–1671. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Driva, P.; Chatzichristidi, M.; Sakellariou, G.; Lohse, D. Graft Copolymers. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Hadjichristidis, N.; Pitsikalis, M.; Iatrou, H.; Pispas, S. The Strength of the Macromonomer Strategy for Complex Macromolecular Architecture: Molecular Characterization, Properties and Application of Polymacromonomers. Macromol. Rapid Commun. 2003, 24, 979–1013. [Google Scholar] [CrossRef]
- Joubert, F.; Musa, O.; Hodgson, D.R.W.; Cameron, N.R. Graft copolymers of hydroxyethyl cellulose by a ‘grafting to’ method: 15N labelling as a powerful characterization tool in ‘click’ polymer chemistry. Polym. Chem. 2015, 6, 1567–1575. [Google Scholar] [CrossRef] [Green Version]
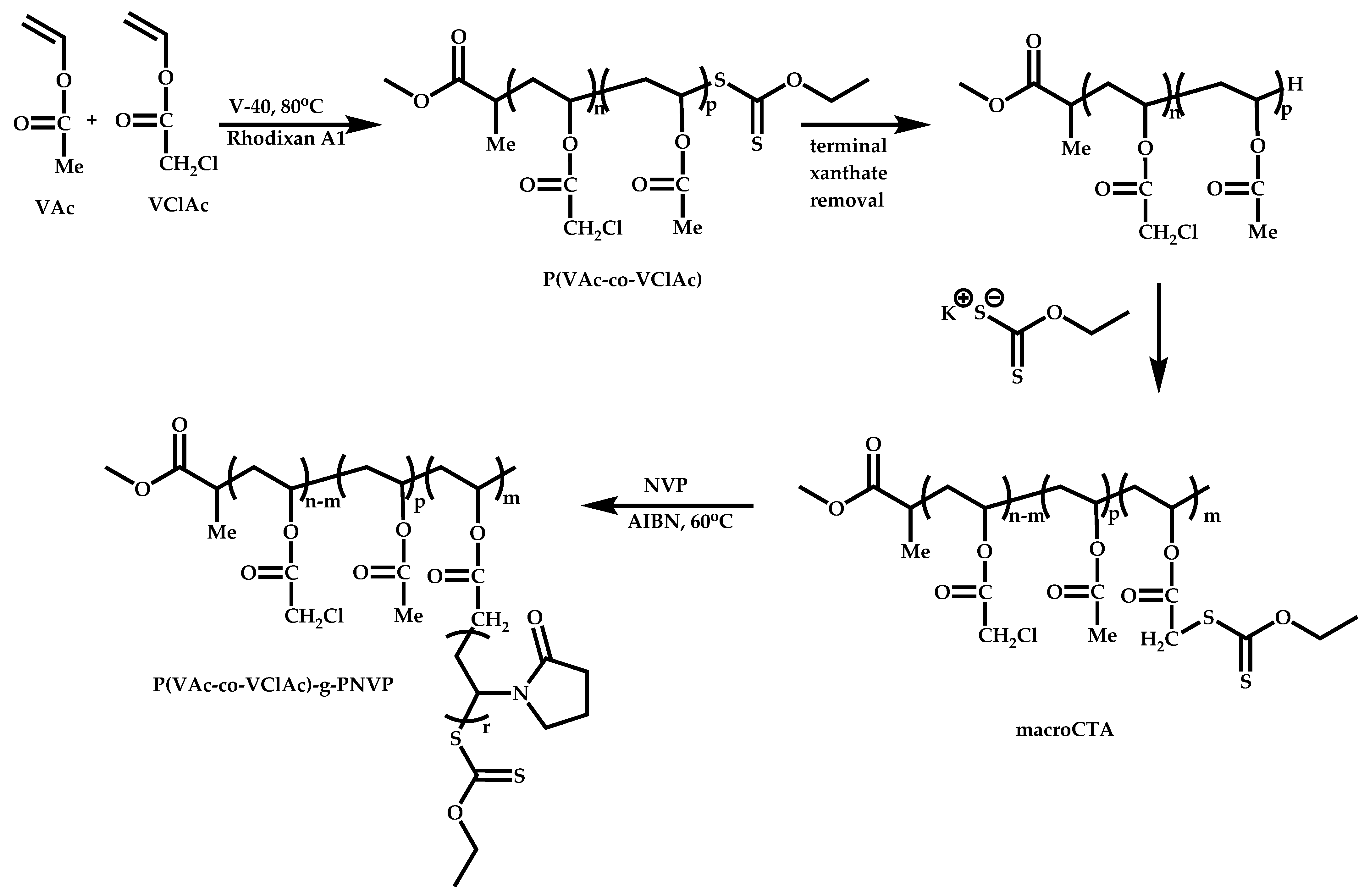
- Atanase, L.I.; Winninger, J.; Delaite, C.; Riess, G. Reversible addition-fragmentation chain transfer synthesis and micellar characteristics of biocompatible amphiphilic poly(vinyl acetate)-graft-poly(N-vinyl-2-pyrrolidone) copolymers. Eur. Polym. J. 2014, 53, 109–117. [Google Scholar] [CrossRef]
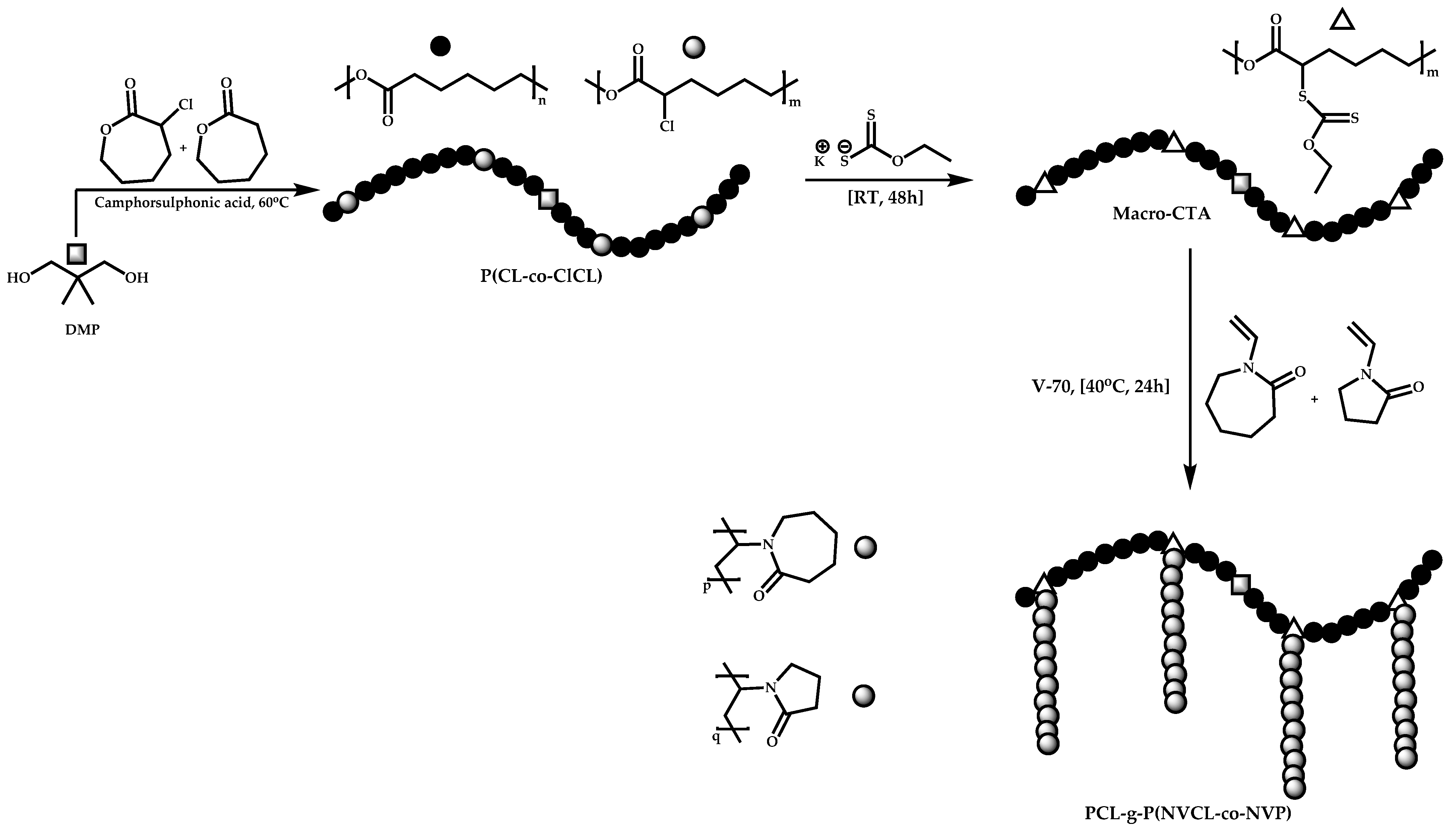
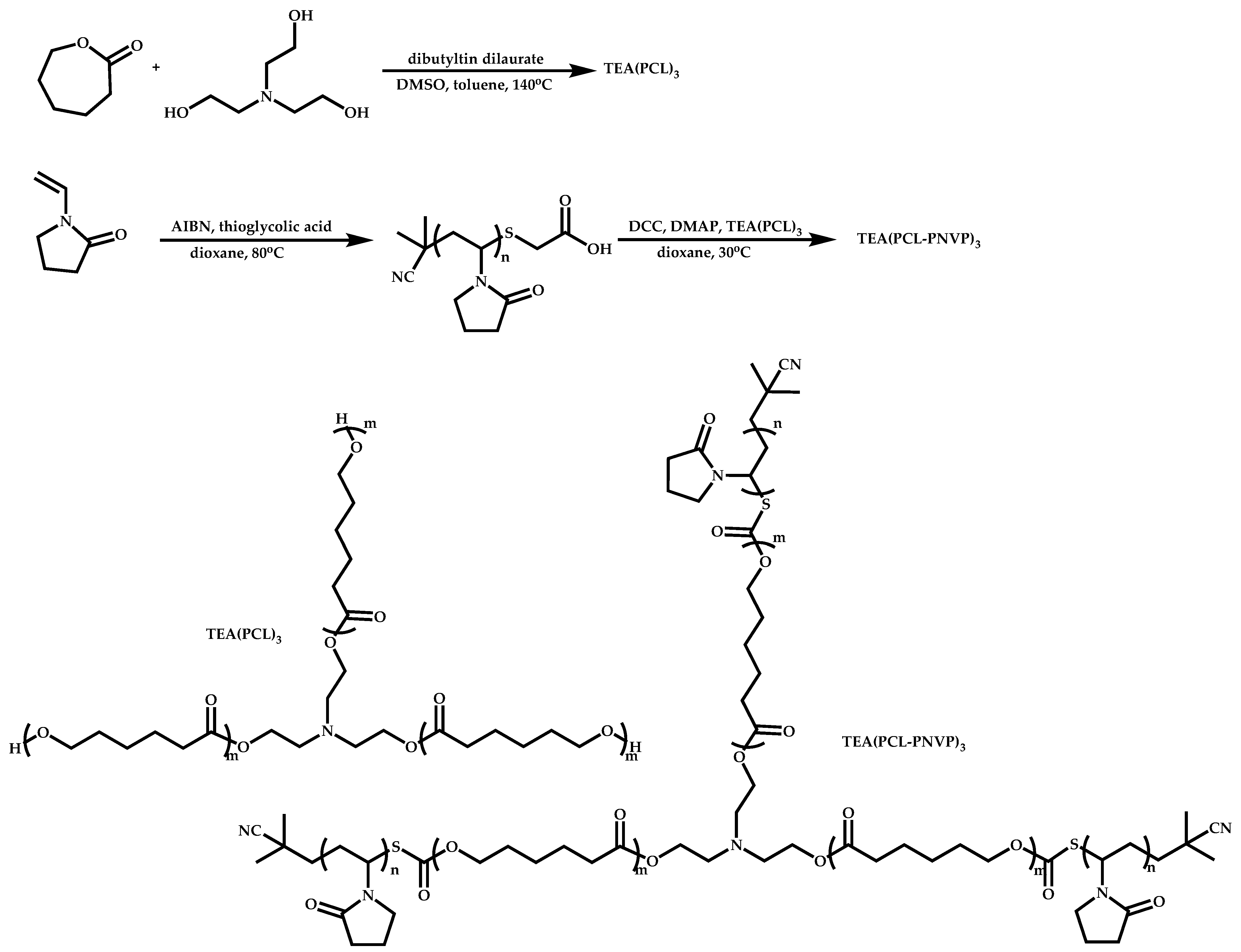
- Winninger, J.; Iurea, D.M.; Atanase, L.I.; Salhi, S.; Delaite, C.; Reiss, G. Micellization of novel biocompatible thermo-sensitive graft copolymers based on poly(ε-caprolactone), poly(N-vinylcaprolactam) and poly(N-vinylpyrrolidone). Eur. Polym. J. 2019, 119, 74–82. [Google Scholar] [CrossRef]
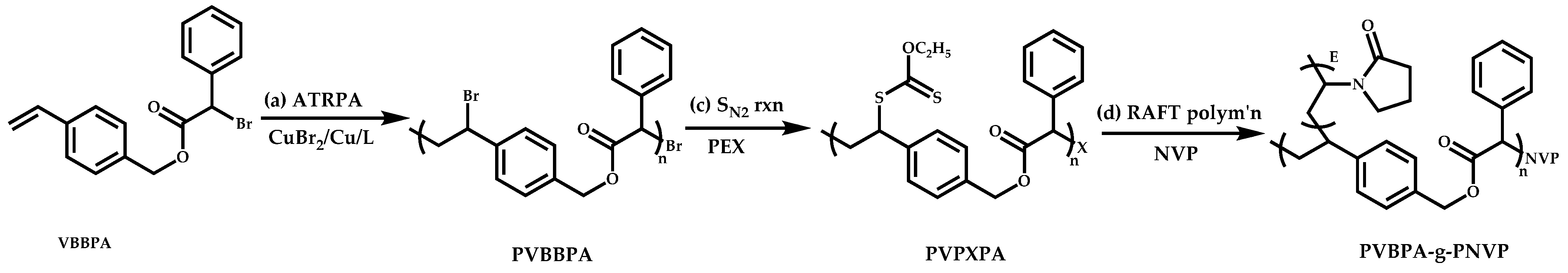
- Huang, Y.S.; Chen, J.K.; Kuo, S.W.; Hsieh, Y.A.; Yamamoto, S.; Nakanishi, J.; Huang, C.F. Synthesis of Poly(N-vinylpyrrolidone)-Based Polymer Bottlebrushes by ATRPA and RAFT Polymerization: Toward Drug Delivery Application. Polymers 2019, 11, 1079. [Google Scholar] [CrossRef] [Green Version]
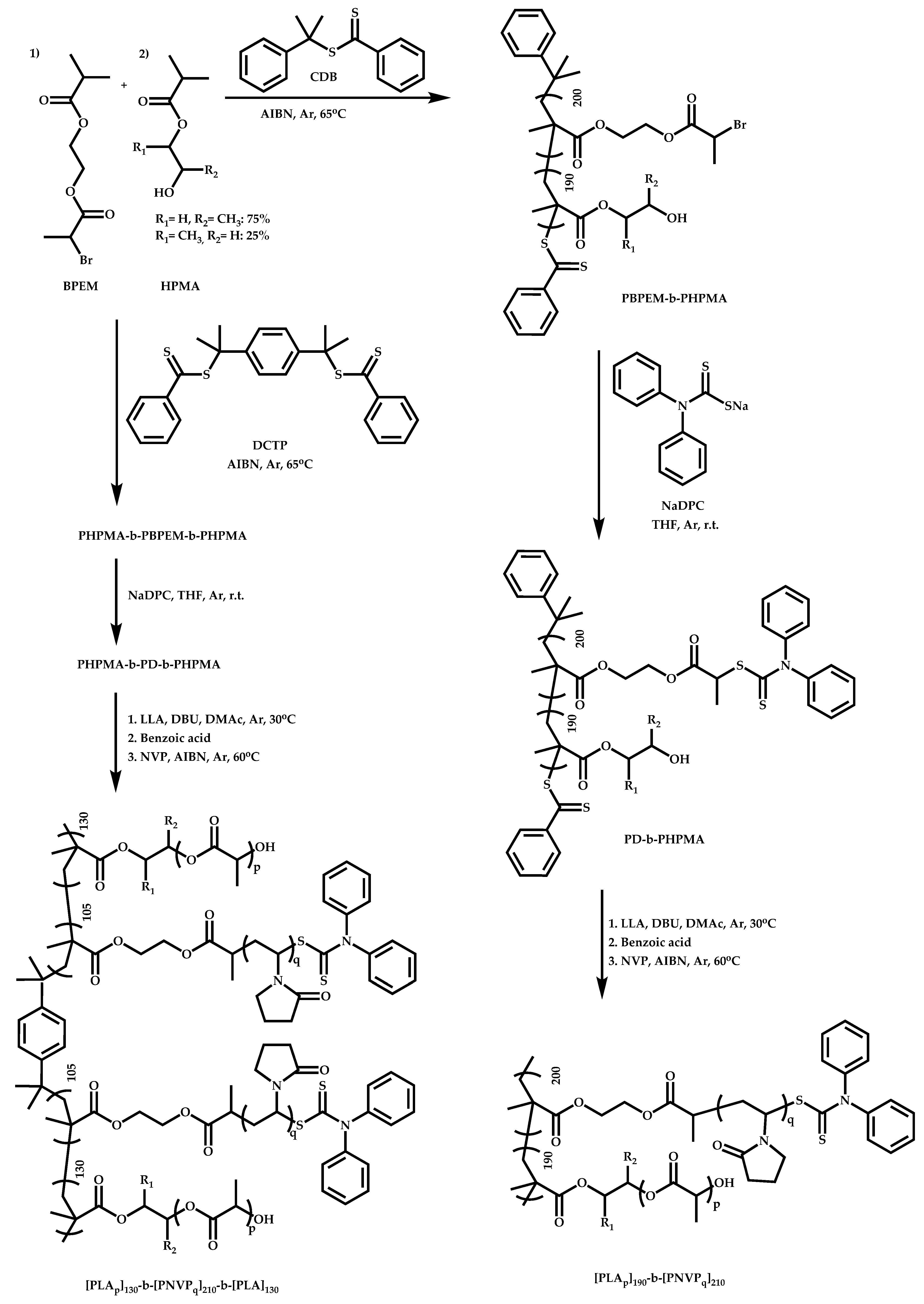
- Wang, Y.; Ren, R.; Ling, J.; Sun, W.; Shen, Z. One-pot “grafting-from” synthesis of amphiphilic bottlebrush block copolymers containing PLA and PVP side chains via tandem ROP and RAFT polymerization. Polymer 2018, 138, 378–386. [Google Scholar] [CrossRef]
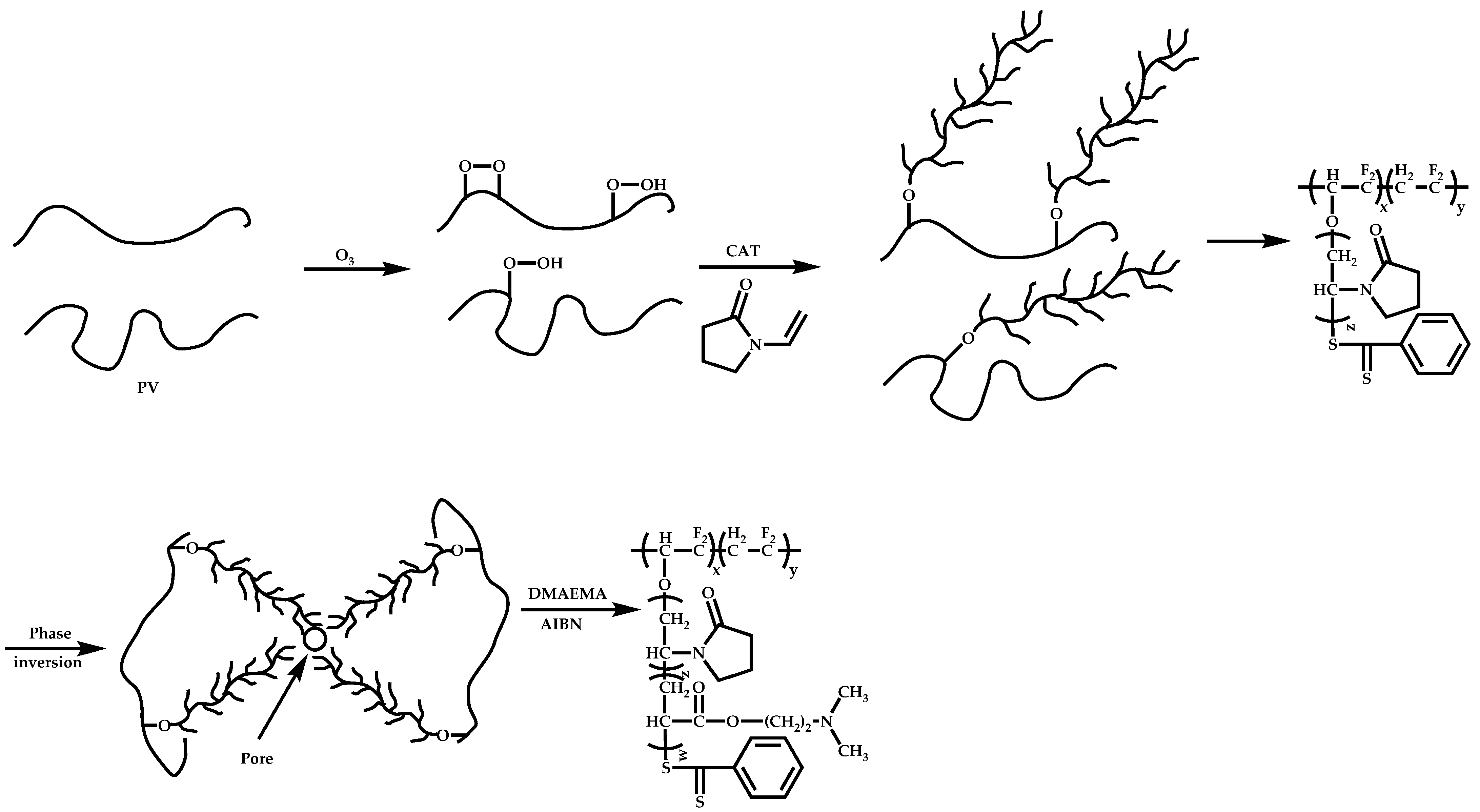
- Peng, Q.; Lu, S.; Chen, D.; Wu, X.; Fan, P.; Zhong, R.; Xu, Y. Poly(vinylidene fluoride)-graft-Poly(N-vinyl-2-pyrrolidone) Copolymers Prepared via a RAFT-Mediated Process and their Use in Antifouling and Antibacterial Membranes. Macromol. Biosci. 2007, 7, 1149–1159. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
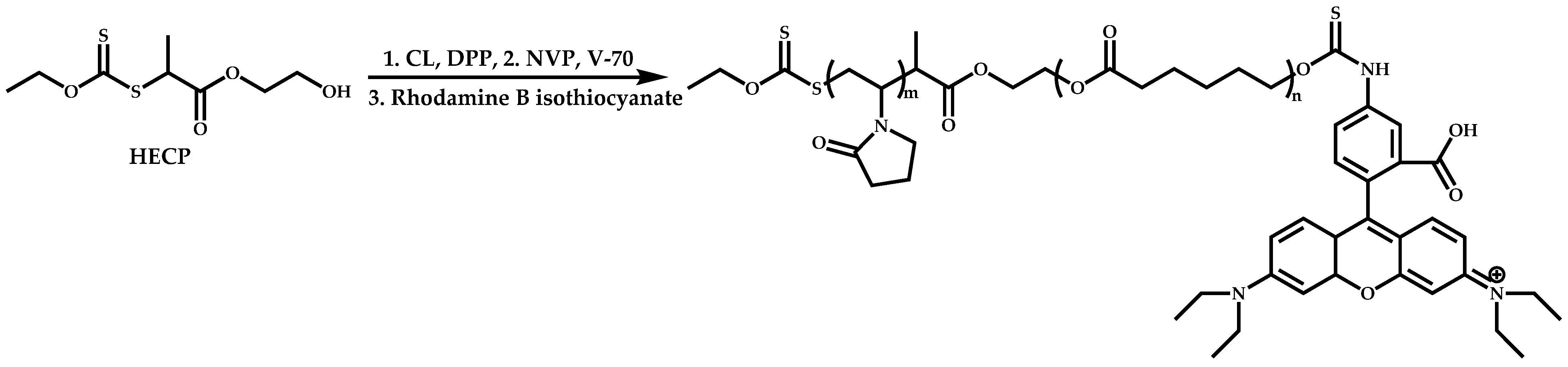{kind=link}
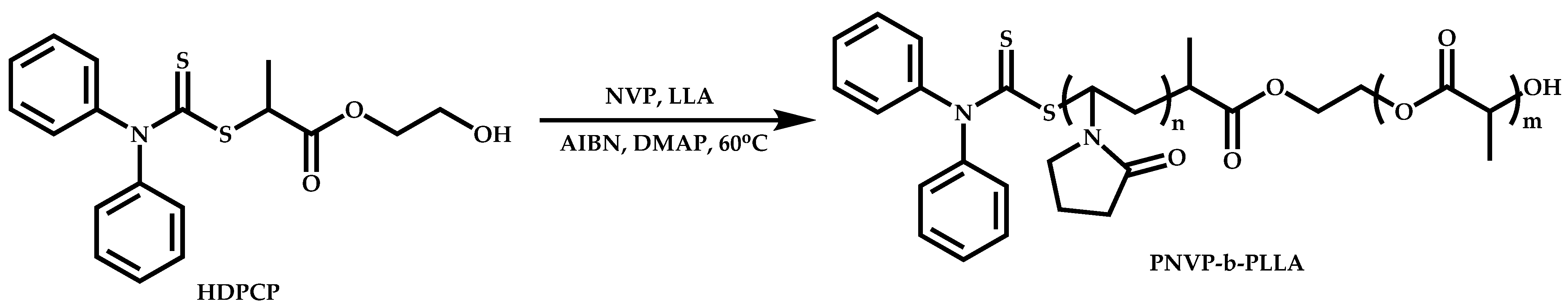{kind=link}
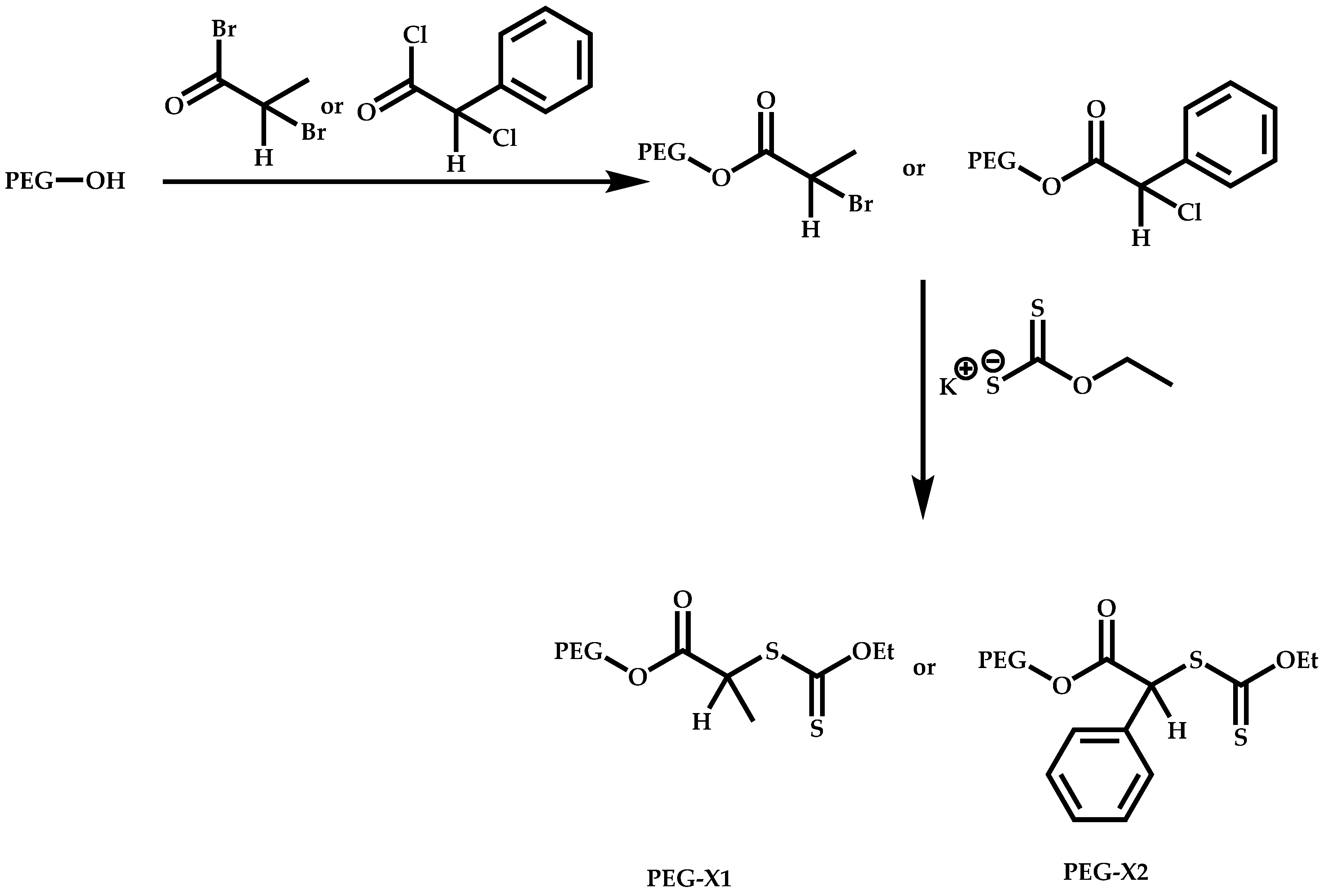{kind=link}
{kind=link}
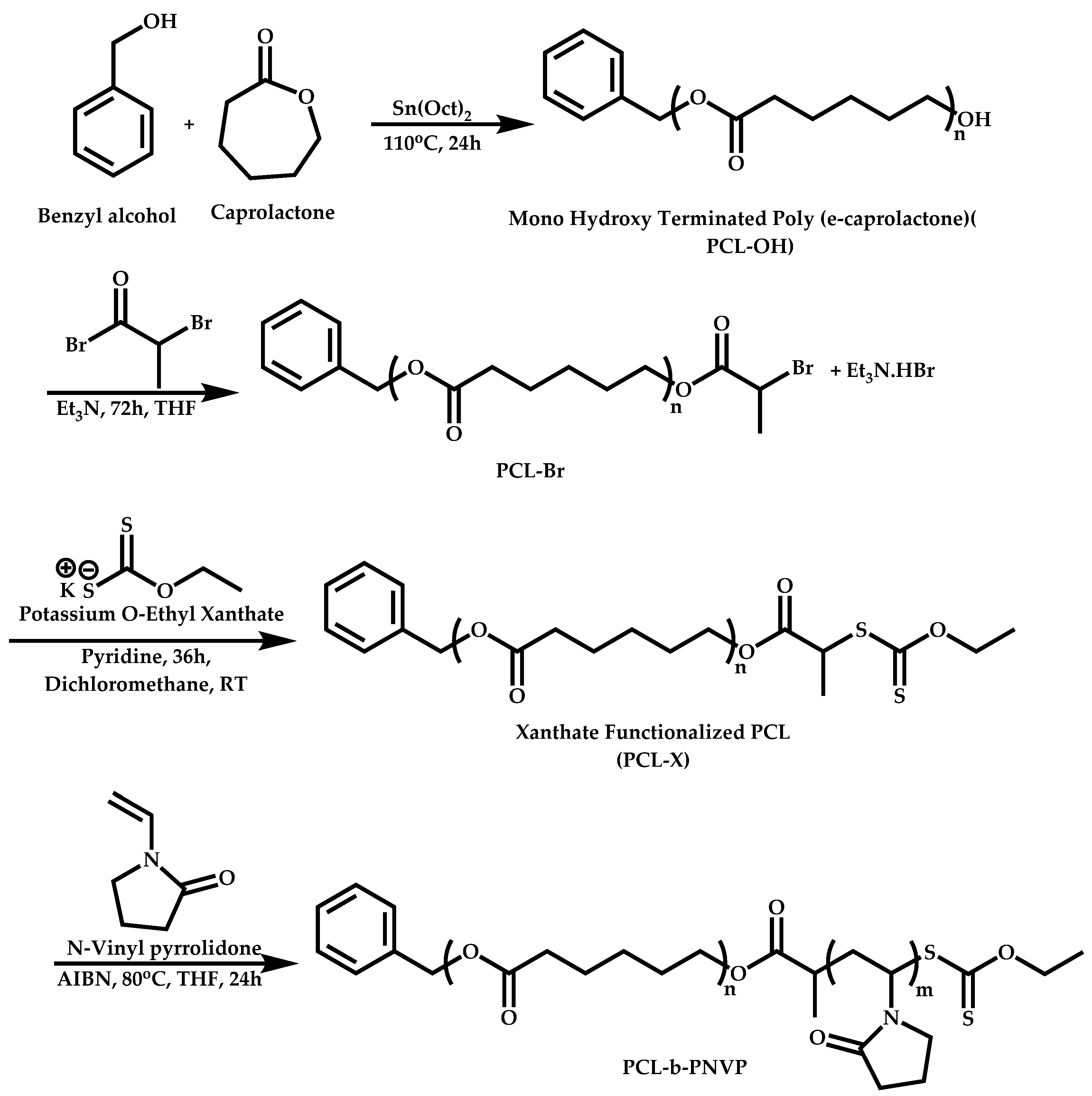{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Comonomer | CTA | Ref. |
|---|---|---|---|
| 1 | 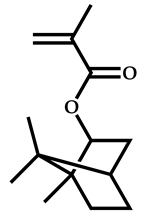 Isobornyl methacrylate | 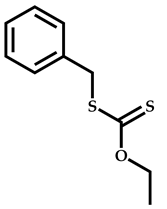 [(O-ethylxanthyl)methyl]benzene | [51] |
| 2 | 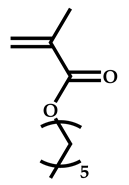 Hexyl methacrylate | 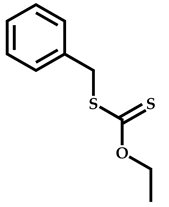 [(O-ethylxanthyl)methyl]benzene | [52,53] |
| 3 |  Hexyl methacrylate | 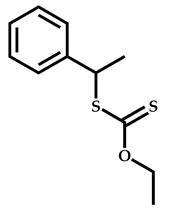 [(O-ethylxanthyl)ethyl]benzene | [52,53] |
| 4 | 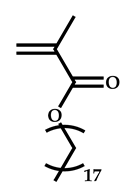 Stearyl methacrylate | 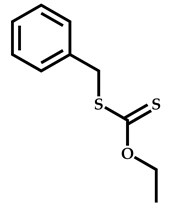 [(O-ethylxanthyl)methyl]benzene | [52,53] |
| 5 | 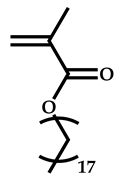 Stearyl methacrylate | 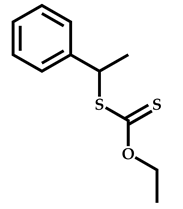 [(O-ethylxanthyl)ethyl]benzene | [52,53] |
| 6 |  Benzyl methacrylate | 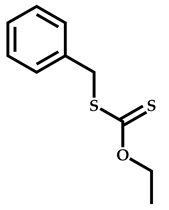 [(O-ethylxanthyl)methyl]benzene | [54] |
| 7 | 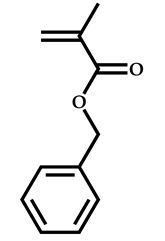 Benzyl methacrylate | 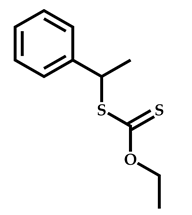 [(O-ethylxanthyl)ethyl]benzene | [54] |
| 8 | 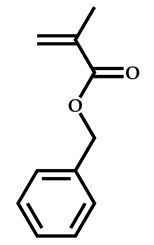 Benzyl methacrylate | 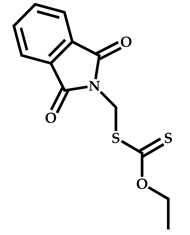 O-ethyl-S-(phthalimidylmethyl) xanthate | [54] |
| 9 | 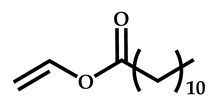 Vinyl laurate |  S-(2-ethyl propionate)-O-ehtyl xanthate | [56] |
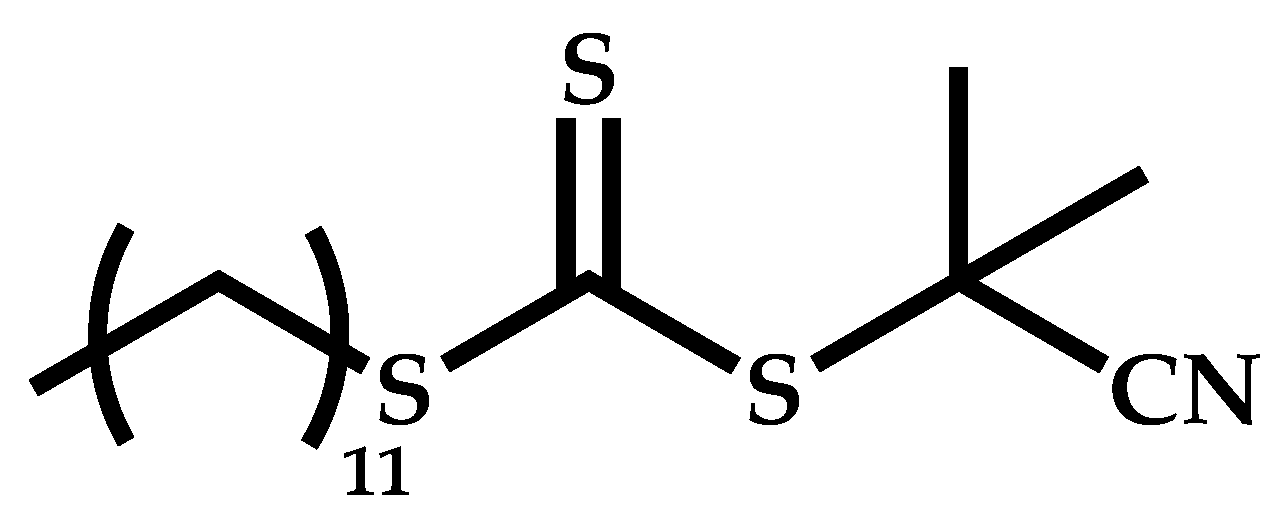
| 10 | 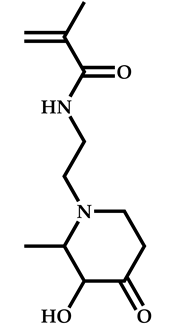 N-[2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)yl)ethyl]-2-methylprop-2-enamide | 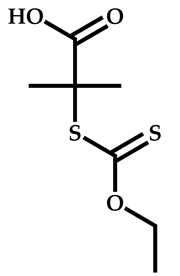 2-[(ethoxymethanethioyl)sulfanyl]-2-methylpropanoic acid | [57] |
| 11 | 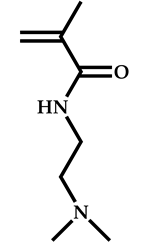 2-(Dimethylamino)ethyl methacrylate | 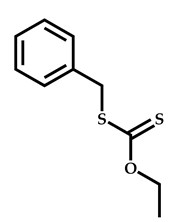 [(O-ethylxanthyl)methyl]benzene | [55] |
| 12 | 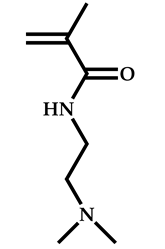 2-(Dimethylamino)ethyl methacrylate | 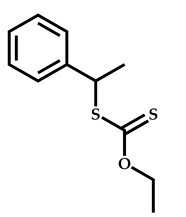 [(O-ethylxanthyl)ethyl]benzene | [55] |
| 13 | 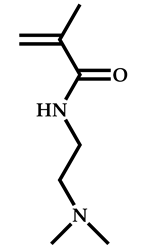 2-(Dimethylamino)ethyl methacrylate | 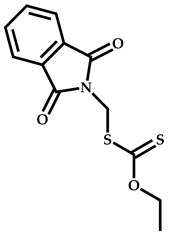 O-ethyl-S-(phthalimidylmethyl) xanthate | [55] |
| 14 | 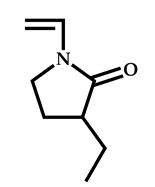 3-ethyl-1-vinyl-2-pyrrolidone | 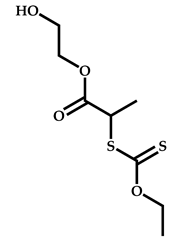 S-(1-methyl-4-hydroxyentyl acetate)-O-ethyl xanthate | [58] |
| 15 | 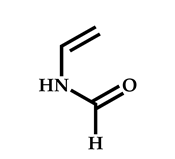 N-Vinylformamide | 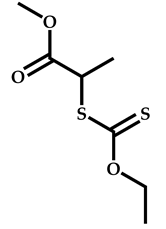 Methyl-2-(ethoxycarbonothiolthio)propanoate | [59,60] |
| 16 | 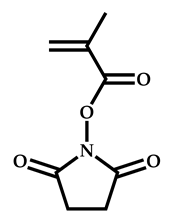 N-(methacryloxy)succinimide |  Methyl-2-(ethoxycarbonothiolthio)propanoate | [61,62] |
| 17 | 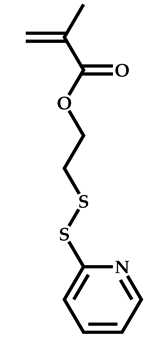 Pyridyl disulfide ethyl methacrylate | 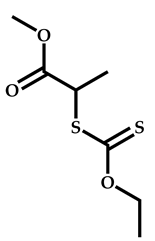 Methyl-2-(ethoxycarbonothiolthio)propanoate | [63] |
| 18 | 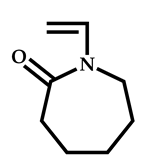 N-vinylcaprolactam |  Methyl-2-(ethoxycarbonothiolthio)propanoate | [64] |
| 19 |  N-vinylcaprolactam |  S-2-ethylpropionate-O-ethyl xanthate | [65] |
| 20 | 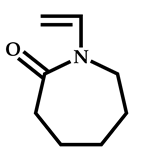 N-vinylcaprolactam |  2-cyano-5-hydroxypentan-2-yl dodecyl carbonotrithioate | [66] |
| 21 | 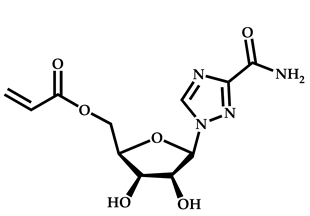 Ribavirin acrylate | 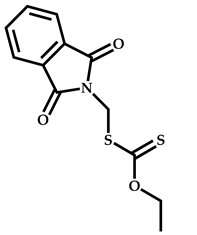 O-ethyl-S-(phthalimidylmethyl) xanthate | [67,68] |
| 22 | 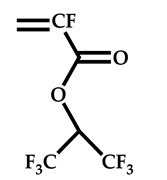 1,1,1-3,3,3-hexafluoroisopropyl-x-fluoroacrylate |  Benzyl dithiobenzoate | [60,70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roka, N.; Kokkorogianni, O.; Kontoes-Georgoudakis, P.; Choinopoulos, I.; Pitsikalis, M. Recent Advances in the Synthesis of Complex Macromolecular Architectures Based on Poly(N-vinyl pyrrolidone) and the RAFT Polymerization Technique. Polymers 2022, 14, 701. https://doi.org/10.3390/polym14040701
Roka N, Kokkorogianni O, Kontoes-Georgoudakis P, Choinopoulos I, Pitsikalis M. Recent Advances in the Synthesis of Complex Macromolecular Architectures Based on Poly(N-vinyl pyrrolidone) and the RAFT Polymerization Technique. Polymers. 2022; 14(4):701. https://doi.org/10.3390/polym14040701
Chicago/Turabian StyleRoka, Nikoletta, Olga Kokkorogianni, Philippos Kontoes-Georgoudakis, Ioannis Choinopoulos, and Marinos Pitsikalis. 2022. "Recent Advances in the Synthesis of Complex Macromolecular Architectures Based on Poly(N-vinyl pyrrolidone) and the RAFT Polymerization Technique" Polymers 14, no. 4: 701. https://doi.org/10.3390/polym14040701
APA StyleRoka, N., Kokkorogianni, O., Kontoes-Georgoudakis, P., Choinopoulos, I., & Pitsikalis, M. (2022). Recent Advances in the Synthesis of Complex Macromolecular Architectures Based on Poly(N-vinyl pyrrolidone) and the RAFT Polymerization Technique. Polymers, 14(4), 701. https://doi.org/10.3390/polym14040701