
Olefin-Metathesis-Derived Norbornene–Ethylene–Vinyl Acetate/Vinyl Alcohol Multiblock Copolymers: Impact of the Copolymer Structure on the Gas Permeation Properties

 ,
,  and
and
Abstract
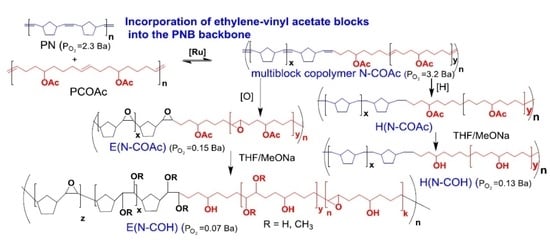
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Polymer Synthesis and Modification
3. Results and Discussion
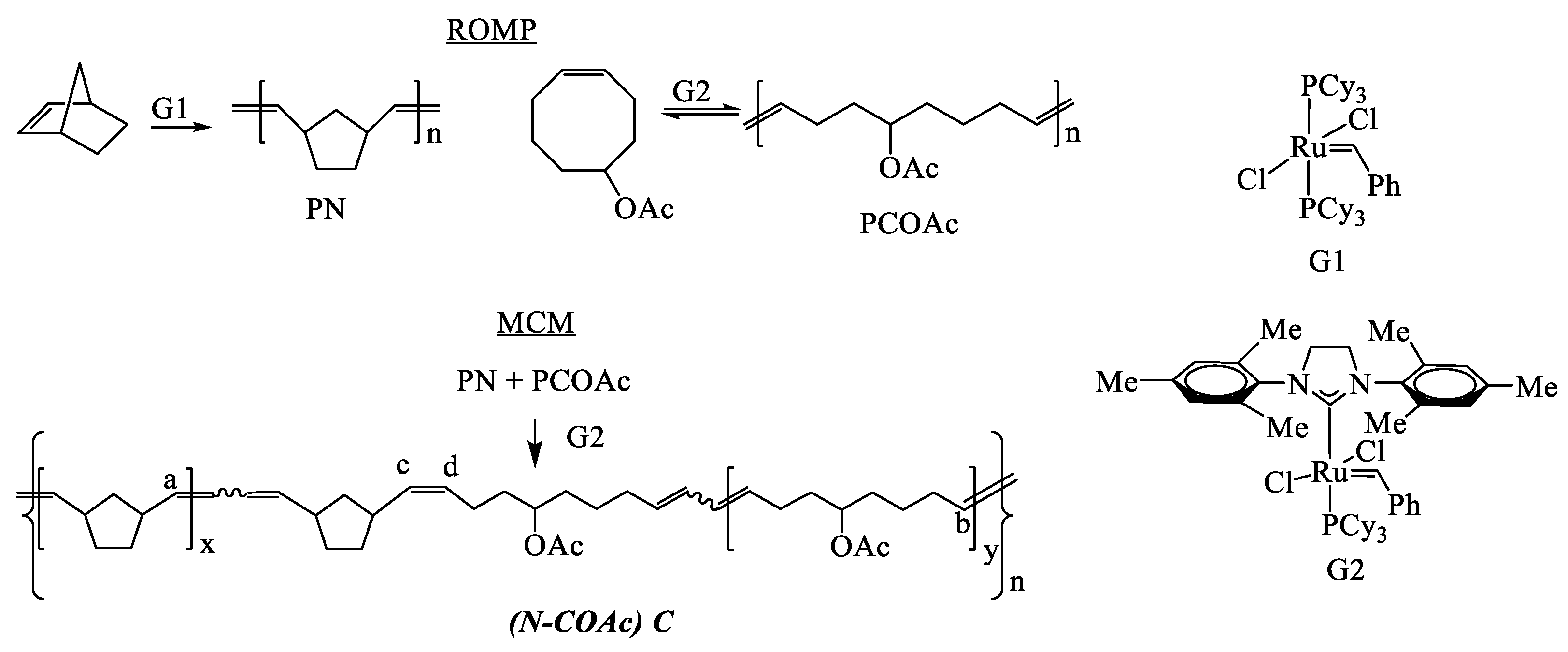
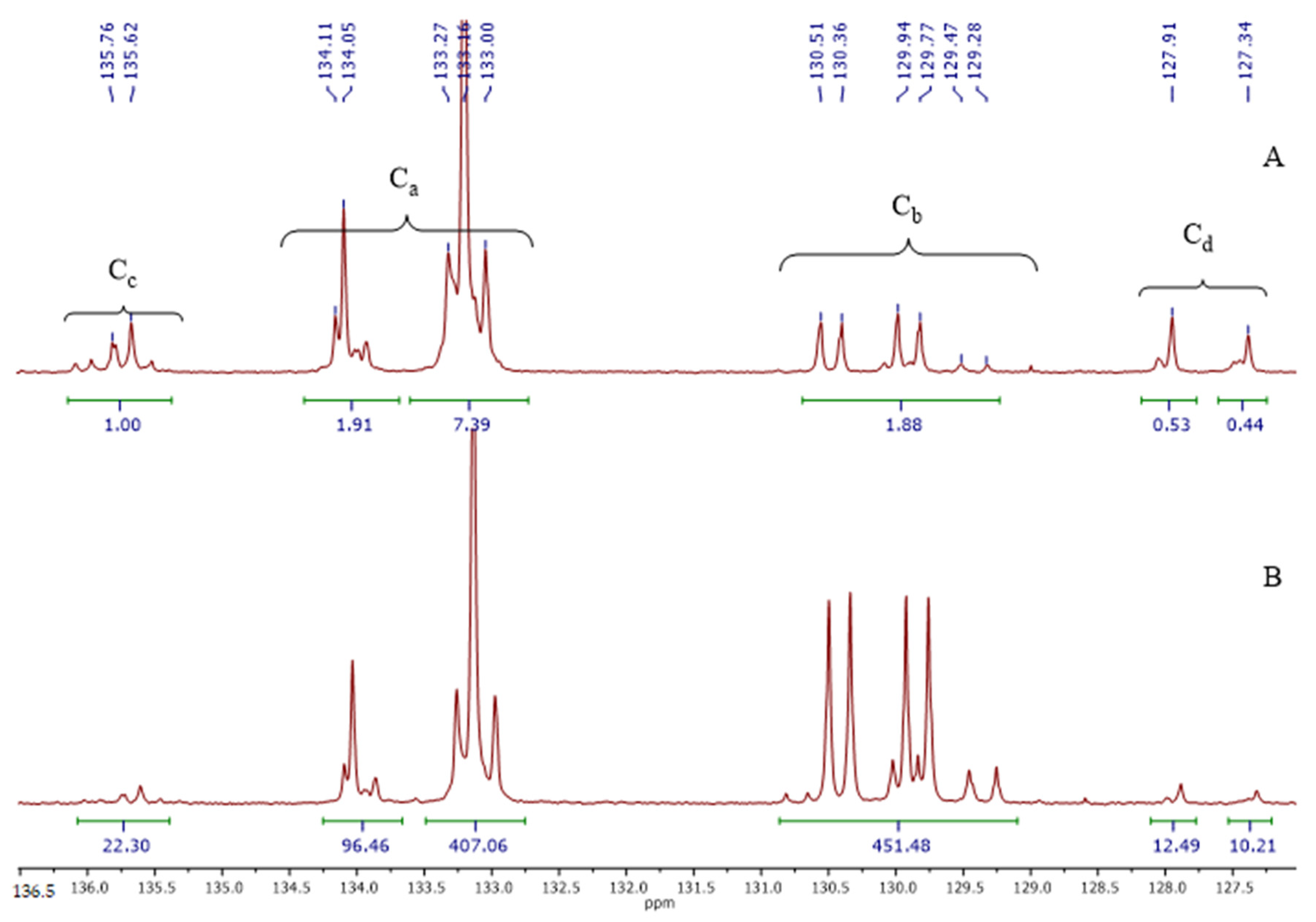
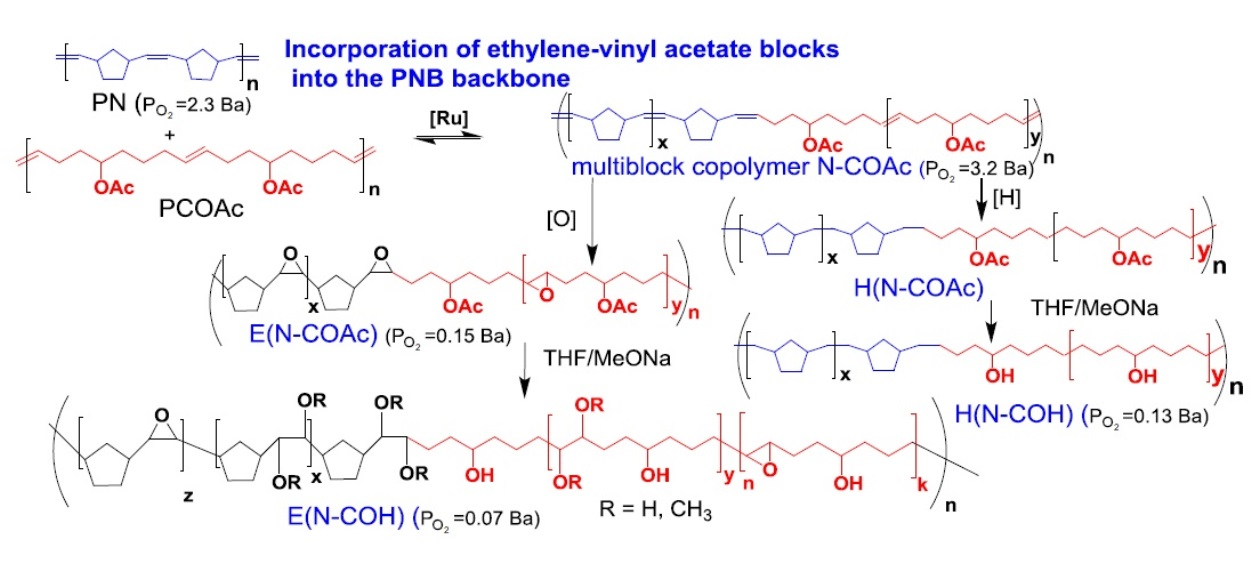
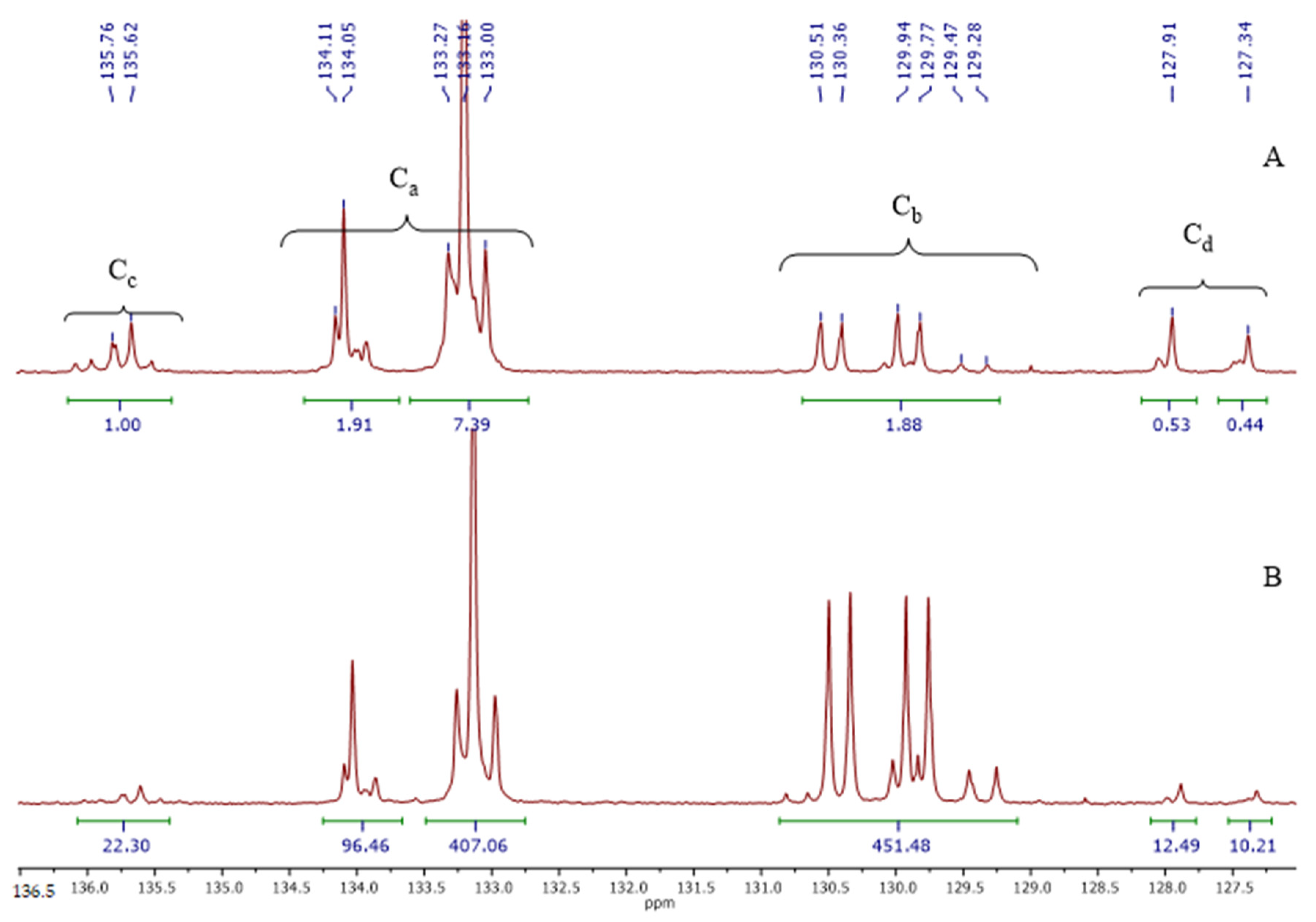
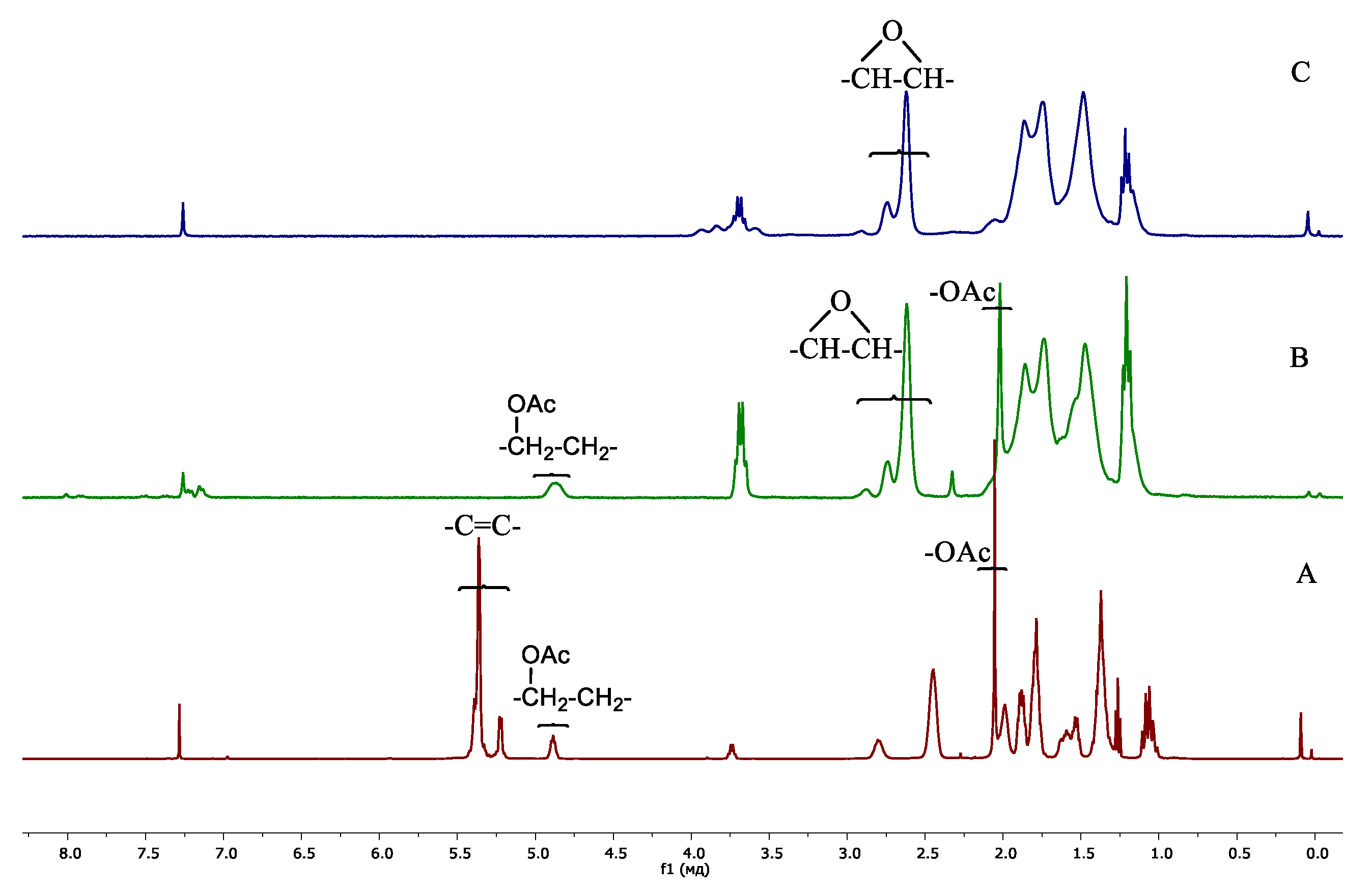
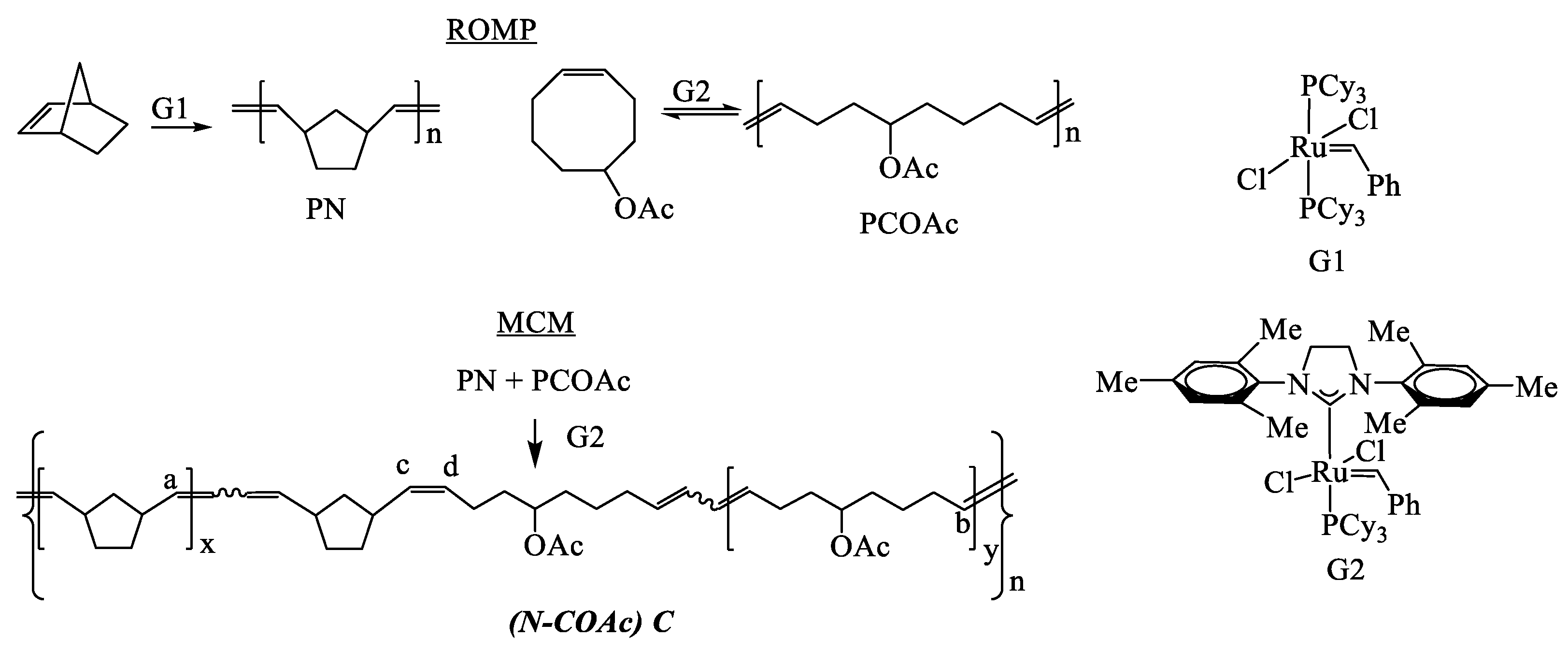
3.1. Polymer Synthesis
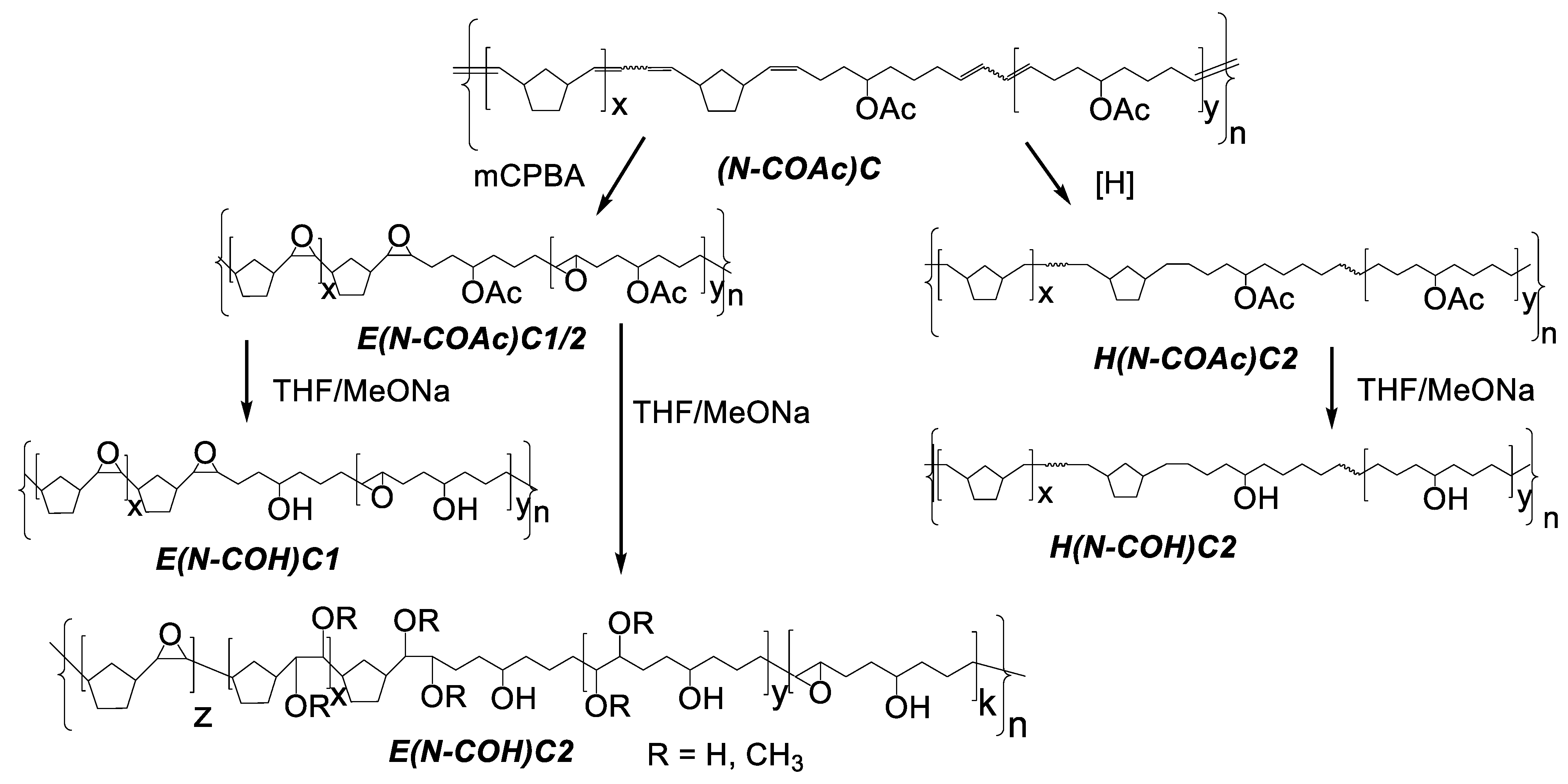
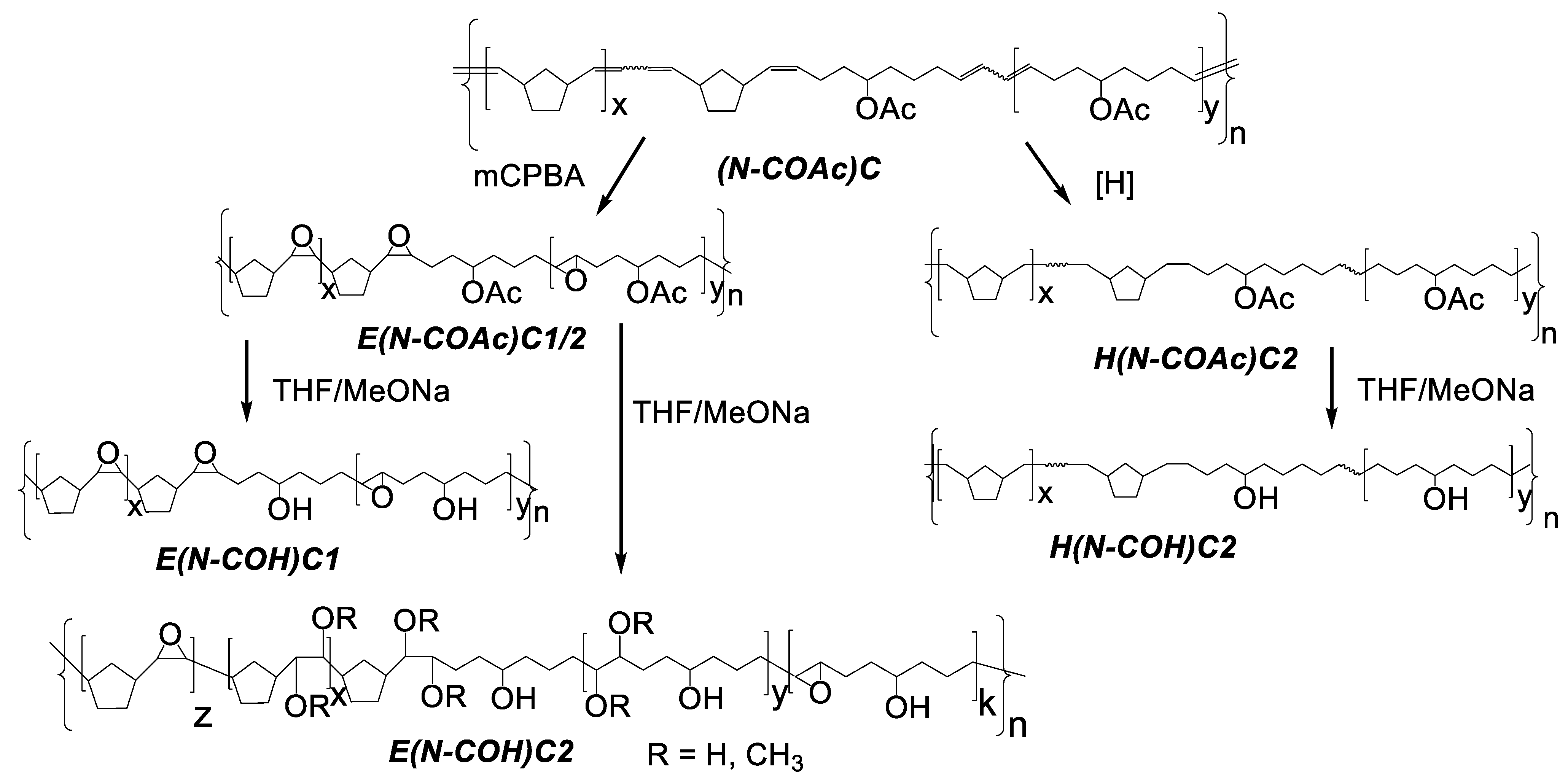
3.2. Copolymer Post-Modification
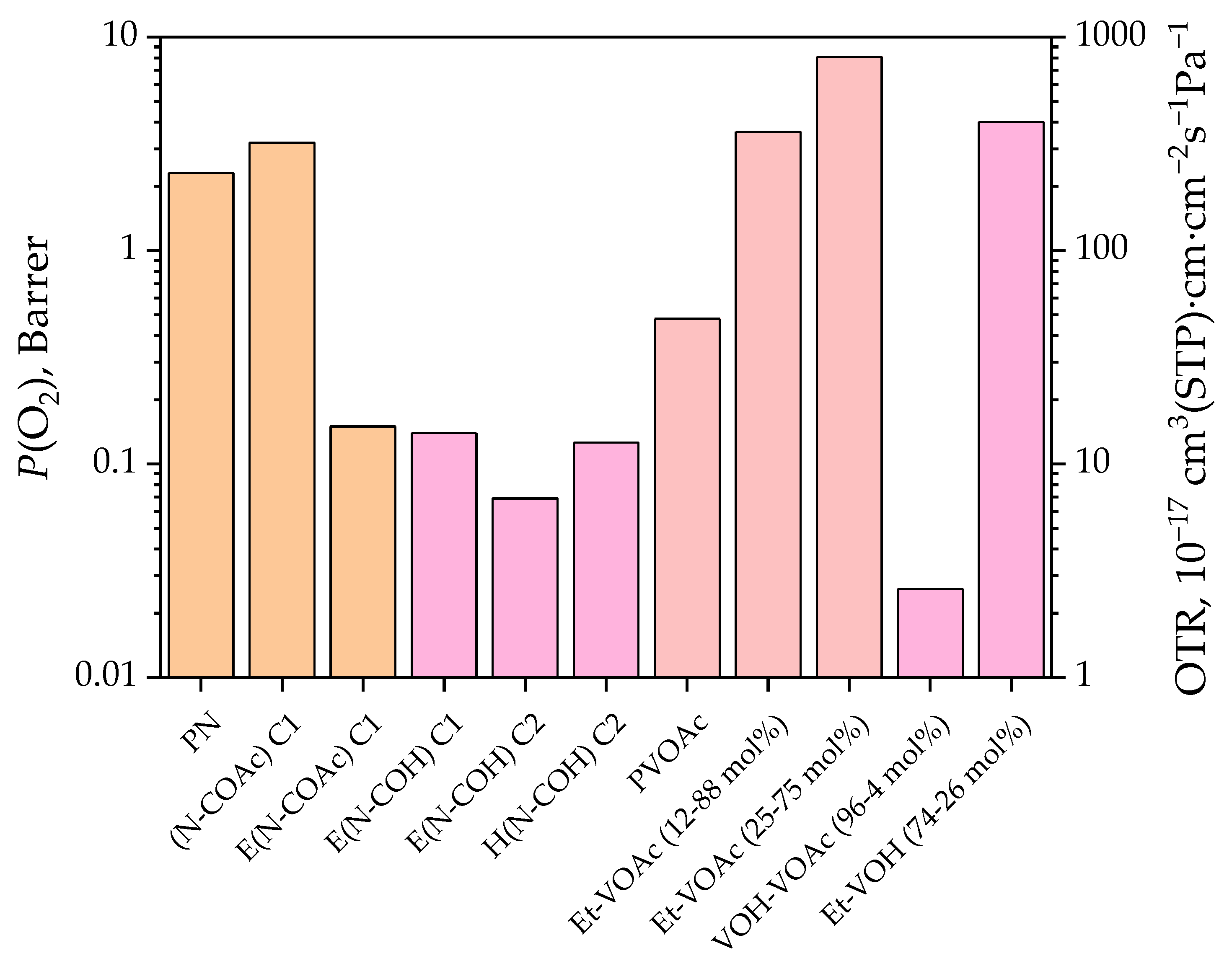
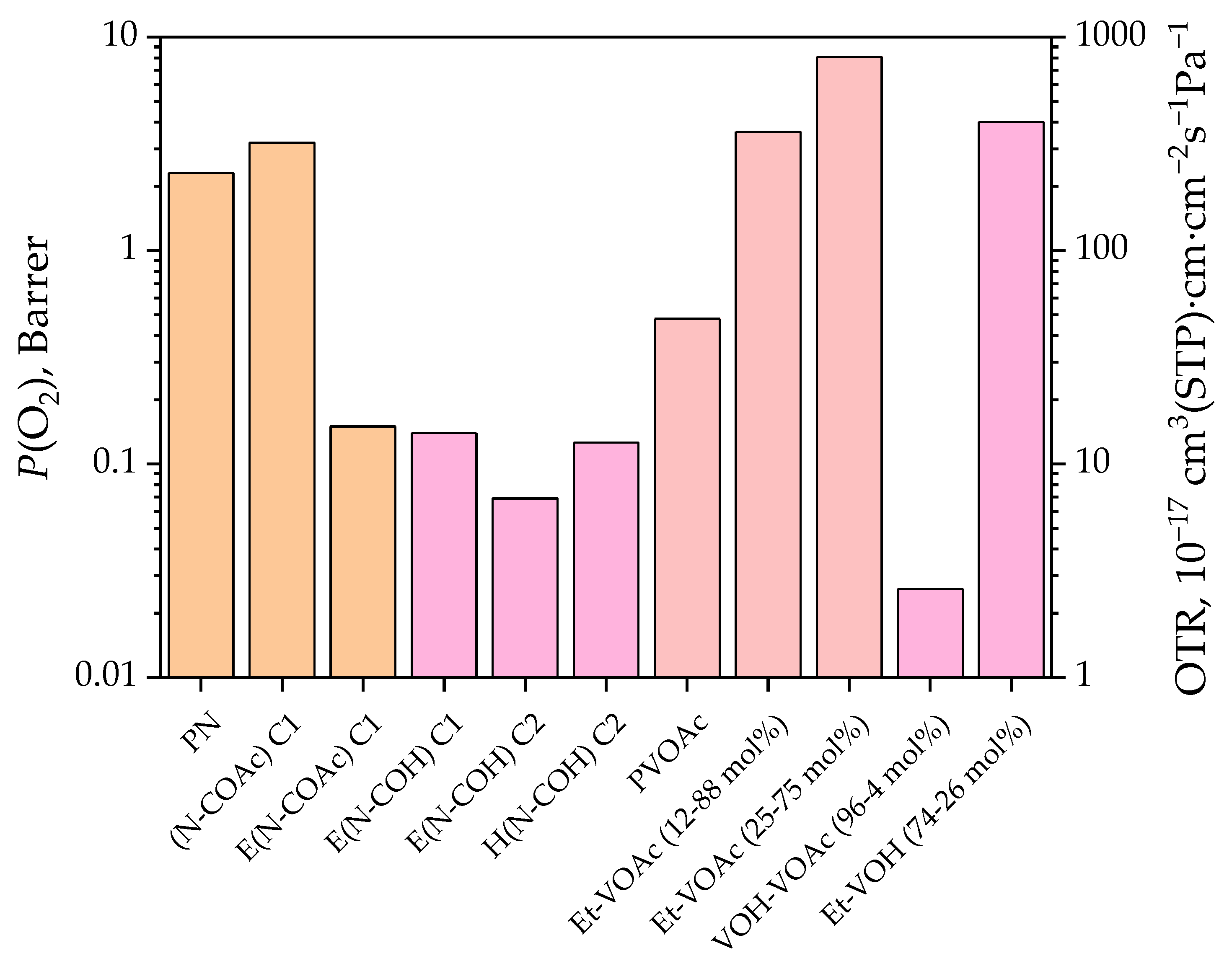
3.3. Gas Permeation and Other Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finkelshtein, E.; Gringolts, M.; Bermeshev, M.; Chapala, P. Polynorbornenes. In Membrane Materials for Gas and Vapor Separation: Synthesis and Application of Silicon-Containing Polymers; Yampolskii, Y., Finkelshtein, E., Eds.; Wiley: Chichester, UK, 2017; pp. 143–221. [Google Scholar] [CrossRef]
- Wang, X.; Wilson, T.J.; Alentiev, D.; Gringolts, M.; Finkelshtein, E.; Bermeshev, M.; Long, B.K. Substituted polynorbornene membranes: A modular template for targeted gas separations. Polym. Chem. 2021, 12, 2947–2977. [Google Scholar] [CrossRef]
- Belov, N.A.; Gringolts, M.L.; Morontsev, A.A.; Starannikova, L.E.; Yampolskii, Y.P.; Finkelstein, E.S. Gas-transport properties of epoxidated metathesis polynorbornenes. Polym. Sci. Ser. B 2017, 59, 560–569. [Google Scholar] [CrossRef]
- Katsumata, T.; Shiotsuki, M.; Sanda, F.; Masuda, T. Synthesis and properties of polynorbornenes bearing oligomeric siloxane pendant groups. Polymer 2009, 50, 1389–1394. [Google Scholar] [CrossRef]
- Morontsev, A.A.; Zhigarev, V.A.; Nikiforov, R.Y.; Belov, N.A.; Gringolts, M.L.; Finkelshtein, E.S.; Yampolskii, Y.P. A new approach to improvement of gas permeation properties of olefin metathesis derived poly(norbornenes): Gem-difluorocyclopropanation of backbone double bonds. Eur. Polym. J. 2018, 99, 340–349. [Google Scholar] [CrossRef]
- Hong, T.; Lai, S.; Mahurin, S.M.; Cao, P.; Voylov, D.N.; Meyer, H.M., III; Jacobs, C.B.; Carrillo, J.Y.; Kisliuk, A.; Ivanov, I.N.; et al. Highly Permeable Oligo(ethylene oxide)-co-poly(dimethylsiloxane) Membranes for Carbon Dioxide Separation. Adv. Sustain. Syst. 2018, 2, 1700113. [Google Scholar] [CrossRef]
- Kim, D.; Hossain, I.; Kim, Y.; Choi, O.; Kim, T.-H. PEG/PPG-PDMS-Adamantane-Based Crosslinked Terpolymer Using the ROMP Technique to Prepare a Highly Permeable and CO2-Selective Polymer Membrane. Polymers 2020, 12, 1674. [Google Scholar] [CrossRef]
- Hong, T.; Niu, Z.; Hu, X.; Gmernicki, K.; Cheng, S.; Fan, F.; Casey Johnson, J.; Hong, E.; Mahurin, S.; Jiang, D.; et al. Effect of Cross-Link Density on Carbon Dioxide Separation in Polydimethylsiloxane-Norbornene Membranes. ChemSusChem 2015, 8, 3595–3604. [Google Scholar] [CrossRef]
- Hong, T.; Chatterjee, S.; Mahurin, S.M.; Fan, F.; Tian, Z.; Jiang, D.; Long, B.K.; Mays, J.W.; Sokolov, A.P.; Saito, T. Impact of tuning CO2-philicity in polydimethylsiloxane-based membranes for carbon dioxide separation. J. Membr. Sci. 2017, 530, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Hossain, I.; Kim, D.; Al Munsur, A.Z.; Roh, J.M.; Park, H.B.; Kim, T.-H. PEG/PPG–PDMS-Based Cross-Linked Copolymer Membranes Prepared by ROMP and In Situ Membrane Casting for CO2 Separation: An Approach to Endow Rubbery Materials with Properties of Rigid Polymers. ACS Appl. Mater. Interfaces 2020, 12, 27286–27299. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.; Santiago, A.A.; Gaviño, R.; Cerda, A.M.; Tlenkopatchev, M.A. Synthesis and ring-opening metathesis polymerization (ROMP) of new N-fluoro-phenylnorbornene dicarboximides by 2nd generation ruthenium alkylidene catalysts. Express Polym. Lett. 2007, 1, 274–282. [Google Scholar] [CrossRef]
- Gringolts, M.L.; Denisova, Y.I.; Shandryuk, G.A.; Krentsel, L.B.; Litmanovich, A.D.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Synthesis of norbornene–cyclooctene copolymers by the cross-metathesis of polynorbornene with polyoctenamer. RSC Adv. 2015, 5, 316–319. [Google Scholar] [CrossRef]
- Gringolts, M.L.; Denisova, Y.I.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Olefin metathesis in multiblock copolymer synthesis. Beilstein J. Org. Chem. 2019, 15, 218–235. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Roenko, A.V.; Gringolts, M.L.; Krentsel, L.B.; Peregudov, A.S.; Shandryuk, G.A.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Cross-Metathesis and Hydrogenation in Polynorbornene–Poly(5-hydroxyoctenamer) Mixture in the Presence of Grubbs’ Catalysts. Polym. Sci.-Ser. B 2018, 60, 735–745. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Gringolts, M.L.; Peregudov, A.S.; Krentsel, L.B.; Litmanovich, E.A.; Litmanovich, A.D.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Cross-metathesis of polynorbornene with polyoctenamer: A kinetic study. Beilstein J. Org. Chem. 2015, 11, 1796–1808. [Google Scholar] [CrossRef] [Green Version]
- Denisova, Y.I.; Gringolts, M.L.; Krentsel’, L.B.; Shandryuk, G.A.; Litmanovich, A.D.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Regulation of the degree of blockiness of the norbornene–cyclooctene copolymer synthesized via the cross-metathesis reaction. Polym. Sci. Ser. B 2016, 58, 292–297. [Google Scholar] [CrossRef]
- Roenko, A.V.; Denisova, Y.I.; Gringolts, M.L.; Peregudov, A.S.; Shandryuk, G.A.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Cross-Metathesis between Polynorbornene and Poly(5,6-epoxy-1-octenamer). Polym. Sci. Ser. C 2019, 61, 134–144. [Google Scholar] [CrossRef]
- Kim, D.-G.; Takigawa, T.; Kashino, T.; Burtovyy, O.; Bell, A.; Register, R.A. Hydroxyhexafluoroisopropylnorbornene Block and Random Copolymers via Vinyl Addition Polymerization and Their Application as Biobutanol Pervaporation Membranes. Chem. Mater. 2015, 27, 6791–6801. [Google Scholar] [CrossRef]
- Wang, C.; Feng, Z.; Zhao, Y.; Li, X.; Li, W. Preparation and properties of ion exchange membranes for PEMFC with sulfonic and carboxylic acid groups based on polynorbornenes. Int. J. Hydrogen Energy 2017, 42, 29988–29994. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Roenko, A.V.; Adzhieva, O.A.; Gringolts, M.L.; Shandryuk, G.A.; Peregudov, A.S.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Facile synthesis of norbornene-ethylene-vinyl acetate/vinyl alcohol multiblock copolymers by the olefin cross-metathesis of polynorbornene with poly(5-acetoxy-1-octenylene). Polym. Chem. 2020, 11, 7063–7077. [Google Scholar] [CrossRef]
- Mokwena, K.K.; Tang, J. Ethylene Vinyl Alcohol: A Review of Barrier Properties for Packaging Shelf Stable Foods. Crit. Rev. Food Sci. Nutr. 2012, 52, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Gerhard Karall Engineering. Norsorex. 2021. Available online: https://www.norsorex.com (accessed on 24 December 2021).
- Slugovc, C. Industrial Applications of Olefin Metathesis Polymerization. In Olefin Metathesis: Theory and Practice; Grela, K., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 329–333. [Google Scholar] [CrossRef]
- Lee, D.; Choi, M.; Ha, C. Polynorbornene Dicarboximide/Amine Functionalized Graphene Hybrids for Potential Oxygen Barrier Films. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1611–1621. [Google Scholar] [CrossRef]
- Wang, Y.; Hasegawa, Y.; Serikawa, T.; Oyaizu, K.; Nishide, H. Ultrahigh oxygen-scavenging norbornene copolymers bearing imidazolyl iron complexes for fabricating active and sustainable packaging films. Chem. Commun. 2020, 56, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Hillmyer, M.A.; Laredo, W.R.; Grubbs, R.H. Ring-Opening Metathesis Polymerization of Functionalized Cyclooctenes by a Ruthenium-Based Metathesis Catalyst. Macromolecules 1995, 28, 6311–6316. [Google Scholar] [CrossRef]
- Barrer, R.M.; Rideal, E.K. Permeation, diffusion and solution of gases in organic polymers. Trans. Faraday Soc. 1939, 35, 628–643. [Google Scholar] [CrossRef]
- Denisova, Y.I.; Zhigarev, V.A.; Gringolts, M.L.; Shandryuk, G.A.; Peregudov, A.S.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Cyclododecene in Olefin Metathesis: Polymerization and Macromolecular Cross-Metathesis with Polynorbornene. Polym. Sci. Ser. C 2019, 61, 120–133. [Google Scholar] [CrossRef]
- Pitet, L.M.; Zhang, J.; Hillmyer, M.A. Sequential ROMP of cyclooctenes as a route to linear polyethylene block copolymers. Dalton Trans. 2013, 42, 9079–9088. [Google Scholar] [CrossRef] [PubMed]
- Morontsev, A.A.; Gringolts, M.L.; Filatova, M.P.; Finkelshtein, E.S. Modification of silicon-substituted polynorbornenes by epoxidation of main chain double bonds. Polym. Sci. Ser. B 2016, 58, 695–702. [Google Scholar] [CrossRef]
- Morontsev, A.A.; Denisova, Y.I.; Gringolts, M.L.; Filatova, M.P.; Shandryuk, G.A.; Finkelshtein, E.S.; Kudryavtsev, Y.V. Epoxidation of Multiblock Copolymers of Norbornene and Cyclooctene. Polym. Sci. Ser. B 2018, 60, 688–698. [Google Scholar] [CrossRef]
- Hayano, S.; Takeyama, Y.; Tsunogae, Y.; Igarashi, I. Hydrogenated Ring-Opened Poly(endo-dicyclopentadiene)s Made via Stereoselective ROMP Catalyzed by Tungsten Complexes: Crystalline Tactic Polymers and Amorphous Atactic Polymer. Macromolecules 2006, 39, 4663–4670. [Google Scholar] [CrossRef]
- Kwon, O.J.; Vo, H.T.; Lee, S.B.; Kim, T.K.; Kim, H.S.; Lee, H. Ring-Opening Metathesis Polymerization and Hydrogenation of Ethyl-substituted Tetracyclododecene. Bull. Korean Chem. Soc. 2011, 32, 2737–2742. [Google Scholar] [CrossRef] [Green Version]
- Autenrieth, B.; Jeong, H.; Forrest, W.P.; Axtell, J.C.; Ota, A.; Lehr, T.; Buchmeiser, M.R.; Schrock, R.R. Stereospecific Ring-Opening Metathesis Polymerization (ROMP) of endo -Dicyclopentadiene by Molybdenum and Tungsten Catalysts. Macromolecules 2015, 48, 2480–2492. [Google Scholar] [CrossRef]
- Xue, X.; Tian, L.; Hong, S.; Zhang, S.; Wu, Y. Effects of Composition and Sequence of Ethylene-Vinyl Acetate Copolymers on Their Alcoholysis and Oxygen Barrier Property of Alcoholyzed Copolymers. Ind. Eng. Chem. Res. 2019, 58, 4125–4136. [Google Scholar] [CrossRef]
- Lee, L.-B.W.; Register, R.A. Hydrogenated Ring-Opened Polynorbornene: A Highly Crystalline Atactic Polymer. Macromolecules 2005, 38, 1216–1222. [Google Scholar] [CrossRef]
- Hillmyer, M.A.; Nguyen, S.T.; Grubbs, R.H. Utility of a Ruthenium Metathesis Catalyst for the Preparation of End-Functionalized Polybutadiene. Macromolecules 1997, 30, 718–721. [Google Scholar] [CrossRef]
- Yoo, J.; Cho, I.; Kim, S.Y. Sequence-controlled ethylene/vinyl cinnamate copolymers: Synthesis and application to the photoalignment of liquid crystals. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5401–5406. [Google Scholar] [CrossRef]
- Merekalova, N.D.; Bondarenko, G.N.; Denisova, Y.I.; Krentsel, L.B.; Litmanovich, A.D.; Kudryavtsev, Y.V. Effect of chain structure on hydrogen bonding in vinyl acetate-vinyl alcohol copolymers. J. Mol. Struct. 2017, 1134, 475–481. [Google Scholar] [CrossRef]
- Kobayashi, S.; Fukuda, K.; Kataoka, M.; Tanaka, M. Regioselective Ring-Opening Metathesis Polymerization of 3-Substituted Cyclooctenes with Ether Side Chains. Macromolecules 2016, 49, 2493–2501. [Google Scholar] [CrossRef]
- Tretinnikov, O.N.; Zagorskaya, S.A. Determination of the degree of crystallinity of poly(vinyl alcohol) by FTIR spectroscopy. J. Appl. Spectrosc. 2012, 79, 521–526. [Google Scholar] [CrossRef]
- Cataldo, F. FTIR spectroscopic characterization of hydrogenated polyoctenamer and polynorbornene and DSC study of their thermal properties. Polym. Int. 1994, 34, 49–57. [Google Scholar] [CrossRef]
- Takahashi, M.; Tashiro, K.; Amiya, S. Crystal Structure of Ethylene-Vinyl Alcohol Copolymers. Macromolecules 1999, 32, 5860–5871. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Pérez, E.; Pereña, J.M.; Benavente, R. Wide-Angle X-ray Diffraction Study of the Phase Behavior of Vinyl Alcohol–Ethylene Copolymers. Macromolecules 1998, 31, 2559–2564. [Google Scholar] [CrossRef]
- Bott, R.H.; Kuphal, J.A.; Robeson, L.M.; Sagl, D. Miscibility of poly(vinyl acetate) and vinyl acetate-ethylene copolymers with styrene-acrylic acid and acrylate-acrylic acid copolymers. J. Appl. Polym. Sci. 1995, 58, 1593–1605. [Google Scholar] [CrossRef]
- Dos Santos, W.N.; de Sousa, J.A.; Gregorio, R. Thermal conductivity behaviour of polymers around glass transition and crystalline melting temperatures. Polym. Test. 2013, 32, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Plate, N.A.; Durgarjan, S.G.; Khotimskii, V.S.; Teplyakov, V.V.; Yampol’skii, Y.P. Novel poly(silicon olefins) for gas separations. J. Membr. Sci. 1990, 52, 289–304. [Google Scholar] [CrossRef]
- Marais, S.; Saiter, J.M.; Devallencourt, C.; Nguyen, Q.T.; Métayer, M. Study of transport of small molecules through ethylene-co-vinyl acetate copolymers films. Part B: CO2 and O2 gases. Polym. Test. 2002, 21, 425–431. [Google Scholar] [CrossRef]
- Anwand, D.; Müller, F.W.; Strehmel, B.; Schiller, K. Determination of the molecular mobility and the free volume of thin polymeric films with fluorescence probes. Die Makromol. Chem. 1991, 192, 1981–1991. [Google Scholar] [CrossRef]
- Ito, K.; Hong-ling, L.; Saito, Y.; Yamamoto, T.; Nishihara, Y.; Ujihira, Y.; Nomura, K. Free-volume study of ethylene-vinyl alcohol copolymer evaluated through positronium lifetime measurement. J. Radioanal. Nucl. Chem. 2003, 255, 437–441. [Google Scholar] [CrossRef]
- Pye, D.G.; Hoehn, H.H.; Panar, M. Measurement of gas permeability of polymers. I. Permeabilities in constant volume/variable pressure apparatus. J. Appl. Polym. Sci. 1976, 20, 1921–1931. [Google Scholar] [CrossRef]
- Meares, P. The Diffusion of Gases Through Polyvinyl Acetate1. J. Am. Chem. Soc. 1954, 76, 3415–3422. [Google Scholar] [CrossRef]
- Stone, M.; Orme, C.; Peterson, E.; Benson, M.; Kaszuba, J.; Mroz, E.; Haga, M. Gas Permeation Testing Results from the Mixed Waste Focus Area Improved Hydrogen Getter Program. Sep. Sci. Technol. 2005, 40, 419–431. [Google Scholar] [CrossRef]
- Haraya, K.; Hwang, S.-T. Permeation of oxygen, argon and nitrogen through polymer membranes. J. Membr. Sci. 1992, 71, 13–27. [Google Scholar] [CrossRef]
- Toi, K.; Maeda, Y.; Tokuda, T. Mechanism of diffusion and sorption of carbon dioxide in poly(vinyl acetate) above and below the glass transition temperature. J. Membr. Sci. 1983, 13, 15–27. [Google Scholar] [CrossRef]
- Yuan, Y.; Teja, A.S. Quantification of specific interactions between CO2 and the carbonyl group in polymers via ATR-FTIR measurements. J. Supercrit. Fluids 2011, 56, 208–212. [Google Scholar] [CrossRef]
- Pérez, E.; Luján, M.; Salazar, J. Preparation and properties of terpolymers of ethylene, vinyl acetate and vinyl alcohol. Macromol. Chem. Phys. 2000, 201, 1323–1328. [Google Scholar] [CrossRef]
- Puleo, A.C.; Paul, D.R.; Kelley, S.S. The effect of degree of acetylation on gas sorption and transport behavior in cellulose acetate. J. Membr. Sci. 1989, 47, 301–332. [Google Scholar] [CrossRef]
- Galperin, I.; Carter, J.H.; Hein, P.R. Poly(norbornene): Tensile and dynamic-mechanical properties. J. Appl. Polym. Sci. 1968, 12, 1751–1754. [Google Scholar] [CrossRef]
- EVAL TM, a Unique Kuraray Technology. Available online: https://eval.kuraray.com/wp-content/uploads/2021/11/tds_eval_F171B.pdf (accessed on 24 December 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymers, P | Cat | [PN]/[PCOAc], wt/wt | Time, h | Yield, % | Mw·10−3 | Ð | Tg, °C | LN | LCOAc |
|---|---|---|---|---|---|---|---|---|---|
| PN 1 | G1 | - | 1 | 98 | 424 | 2.0 | 39 | - | - |
| PCOAc 2 | G2 | - | 24 | 88 | 417 | 1.9 | −37 | - | - |
| (N–COAc)C1 3 | G2 | 2:1 | 3.5 | 93 | 289 | 1.7 | −1 | 10 | 3 |
| (N–COAc)C2 4 | G1 | 1:2 | 24 | 80 | 144 | 2.0 | −29 | 24 | 21 |
| # | Modified Copolymer | Yield, % | Mw·10−3 | Mn·10−3 | Ð | Tg, °C | Tm, °C | −ΔH, J⋅g−1 | Solubility |
|---|---|---|---|---|---|---|---|---|---|
| Epoxidation 1 | |||||||||
| 1 | E(N–COAc)C1 | 66 | 119 | 72 | 1.6 | 31 | abs | abs | CHCl3 |
| 2 | E(N–COAc)C2 | 80 | 86 | 51 | 1.6 | 6 | abs | abs | CHCl3 |
| Hydrogenation 2 | |||||||||
| 3 | H(N–COAc)C2 | 67 | 144 | 70 | 2 | −36 | abs | abs | CHCl3 |
| Deacetylation 3 | |||||||||
| 4 | E(N–COH)C1 | 85 | 105 | 67 | 1.6 | 45 | abs | abs | CHCl3: MeOH 5:1 vol, RT |
| 5 | E(N–COH)C2 | 71 | 152 | 57 | 2.6 | 90 | abs | abs | THF, RT |
| 6 | H(N–COH)C2 | 87 | n/d | n/d | n/d | 28 | 120 | 33 | THF or CHCl3:MeOH= 5:1 vol, T ≥ 55 °C |
| Characteristics | Copolymer | ||||
|---|---|---|---|---|---|
| (N–COAc)C1 | E(N–COAc)C1 | E(N–COH)C1 | E(N–COH)C2 | H(N–COH)C2 | |
| Mw·10−3 | 289 | 119 | 105 | 152 | n/d |
| Mn·10−3 | 169 | 72 | 67 | 57 | n/d |
| DM | 1.7 | 1.6 | 1.6 | 2.6 | n/d |
| Tg, °C | −1 | 31 | 45 | 90 | 28 |
| Tm, °C | abs | abs | abs | abs | 120 |
| Gas | Permeability Coefficient P, Barrer | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PVOAc | Et–VOAc 12/88 [48] | Et–VOAc 25/75 [48] * | Et–VOAc 40/60 [48] * | Et–VOAc 58/42 [48] * | LDPE [48] | VOH–VOAc 96/4 [49] | Et–VOH 74/26 [50] | PVOH [51] | |
| He | 12.9 | - | - | - | - | - | - | - | 7.1 × 10−3 |
| H2 | 8.91 [52] 14.6 [53] | - | - | - | - | - | - | - | 0.65 × 10−3 |
| O2 | 0.479 [52] 0.359 (25 °C) [54] | 3.6 | 8.1 | 10.5 | 5.3 | 2.3 | 0.026 | 4.0 | 0.03 × 10−3 |
| N2 | 0.052 (25 °C) [54] | - | - | - | - | - | - | - | - |
| CO2 | 11.9 [53] | 30 | 70 | 30 | 57 | 16 | |||
| CH4 | 0.80 [53] | - | - | - | - | - | - | - | 0.03 × 10−3 |
| Diffusivity D·108, cm2·s−1 | |||||||||
| He | 977 | - | - | - | - | - | - | - | - |
| H2 | 263 [52] | - | - | - | - | - | - | - | - |
| O2 | 5.62 [52] 2.7 (25 °C) [54] | 25 | 63 | 45 | 39 | 35 | - | - | - |
| N2 | 0.5 (25 °C) [54] | - | - | - | - | - | - | - | - |
| CO2 | 0.81 (36 °C) [55] | 7.8 | 23 | 18 | 46 | 40 | - | - | - |
| Gas | Permeability Coefficient P, Barrer | |||||
|---|---|---|---|---|---|---|
| PN | (N–COAc) C1 | E(N–COAc) C1 | E(N–COH)C1 | E(N–COH)C2 | H(N–COH)C2 | |
| He | 14.9 | 14 | 3.8 | 3.1 | 2.1 | 1.4 |
| H2 | 18 | 18 | 3.1 | 2.6 | 1.7 | 1.2 |
| O2 | 2.3 | 3.2 | 0.15 | 0.14 | 0.069 | 0.126 |
| N2 | 0.43 | 0.73 | 0.024 | 0.015 | 0.0070 | 0.010 |
| CO2 | 9.3 | 20 | 0.96 | 0.73 | 0.31 | 0.54 |
| CH4 | 0.78 | 1.8 | 0.037 | 0.033 | 0.010 | - |
| Gas | D·108, cm2·s−1 | S, cm3(STP)·cm−3·atm−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PN [3] | N–COAc | E(N–COAc) | E(N–COH) C1 | E(N–COH) C2 | PN [3] | N–COAc C1 | E(N–COAc) C1 | E(N–COH) C1 | E(N–COH) C2 | |
| He | - | - | 350 | 377 | - | - | - | 0.008 | 0.006 | - |
| H2 | - | - | 97 | 52.3 | 78 | - | - | 0.024 | 0.04 | 0.016 |
| O2 | 12.8 | 19 | 1.4 | 2.1 | 0.82 | 0.14 | 0.143 | 0.079 | 0.05 | 0.064 |
| N2 | 3.6 | 8.5 | 0.54 | 0.2 | 0.30 | 0.09 | 0.07 | 0.034 | 0.06 | 0.018 |
| CO2 | 5.5 | 11 | 0.34 | 0.3 | 0.22 | 1.3 | 1.4 | 2.1 | 1.96 | 1.1 |
| CH4 | 1.5 | 4.6 | 0.16 | 0.1 | 0.057 | 0.4 | 0.3 | 0.18 | 0.23 | 0.135 |
| Tensile Properties | PN 1 [59] | EVAL 2 [60] | E(N–COAc) C1 | E(N–COH) C1 | E(N–COH) C2 |
|---|---|---|---|---|---|
| Tg °C | 39 | 53–63 | 31 | 45 | 90 |
| Tensile strength, б, MPa | 21–45 | 29–41 | 15.1 ± 2.4 | 11.8 ± 2.9 | 26.3 ± 1.5 |
| Elongation at break, ε,% | 16–300 | 12–18 | 505 ± 16 | 522 ± 18 | 13.5 ± 1.4 |
| Young’s modulus, E, MPa | 20–90 | 3500–4900 | 108 ± 12 | 254 ± 19 | 2755 ± 260 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roenko, A.V.; Nikiforov, R.Y.; Gringolts, M.L.; Belov, N.A.; Denisova, Y.I.; Shandryuk, G.A.; Bondarenko, G.N.; Kudryavtsev, Y.V.; Finkelshtein, E.S. Olefin-Metathesis-Derived Norbornene–Ethylene–Vinyl Acetate/Vinyl Alcohol Multiblock Copolymers: Impact of the Copolymer Structure on the Gas Permeation Properties. Polymers 2022, 14, 444. https://doi.org/10.3390/polym14030444
Roenko AV, Nikiforov RY, Gringolts ML, Belov NA, Denisova YI, Shandryuk GA, Bondarenko GN, Kudryavtsev YV, Finkelshtein ES. Olefin-Metathesis-Derived Norbornene–Ethylene–Vinyl Acetate/Vinyl Alcohol Multiblock Copolymers: Impact of the Copolymer Structure on the Gas Permeation Properties. Polymers. 2022; 14(3):444. https://doi.org/10.3390/polym14030444
Chicago/Turabian StyleRoenko, Alexey V., Roman Y. Nikiforov, Maria L. Gringolts, Nikolay A. Belov, Yulia I. Denisova, Georgiy A. Shandryuk, Galina N. Bondarenko, Yaroslav V. Kudryavtsev, and Eugene S. Finkelshtein. 2022. "Olefin-Metathesis-Derived Norbornene–Ethylene–Vinyl Acetate/Vinyl Alcohol Multiblock Copolymers: Impact of the Copolymer Structure on the Gas Permeation Properties" Polymers 14, no. 3: 444. https://doi.org/10.3390/polym14030444
APA StyleRoenko, A. V., Nikiforov, R. Y., Gringolts, M. L., Belov, N. A., Denisova, Y. I., Shandryuk, G. A., Bondarenko, G. N., Kudryavtsev, Y. V., & Finkelshtein, E. S. (2022). Olefin-Metathesis-Derived Norbornene–Ethylene–Vinyl Acetate/Vinyl Alcohol Multiblock Copolymers: Impact of the Copolymer Structure on the Gas Permeation Properties. Polymers, 14(3), 444. https://doi.org/10.3390/polym14030444