


Optimization and Testing of Hybrid 3D Printing Vitrimer Resins

 ,
,
Abstract
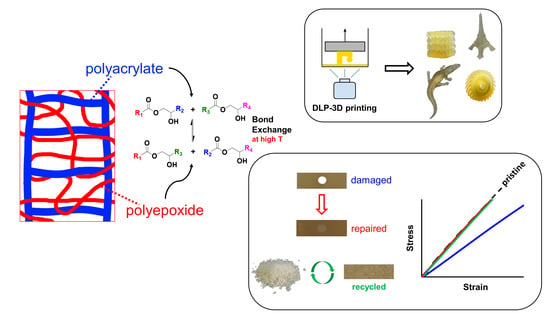
1. Introduction
2. Materials and Methods
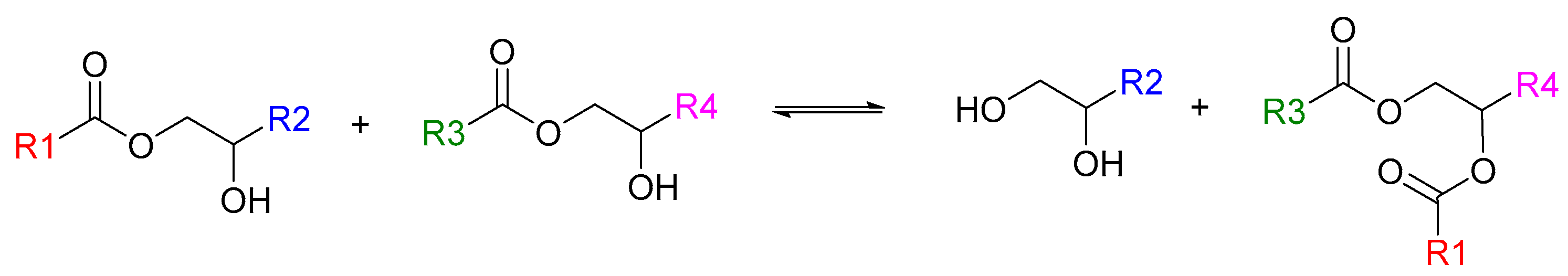
3. Results and Discussion
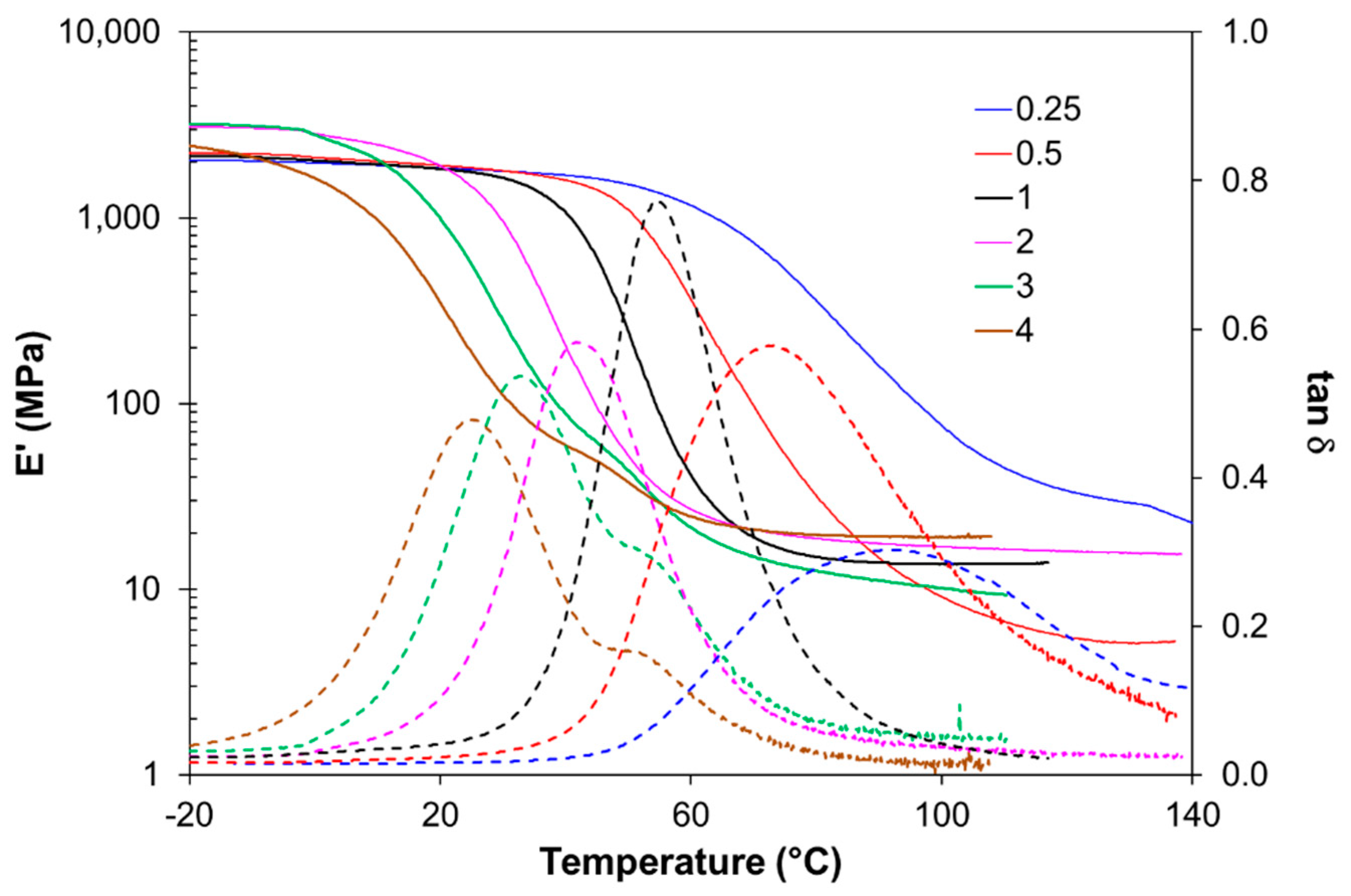
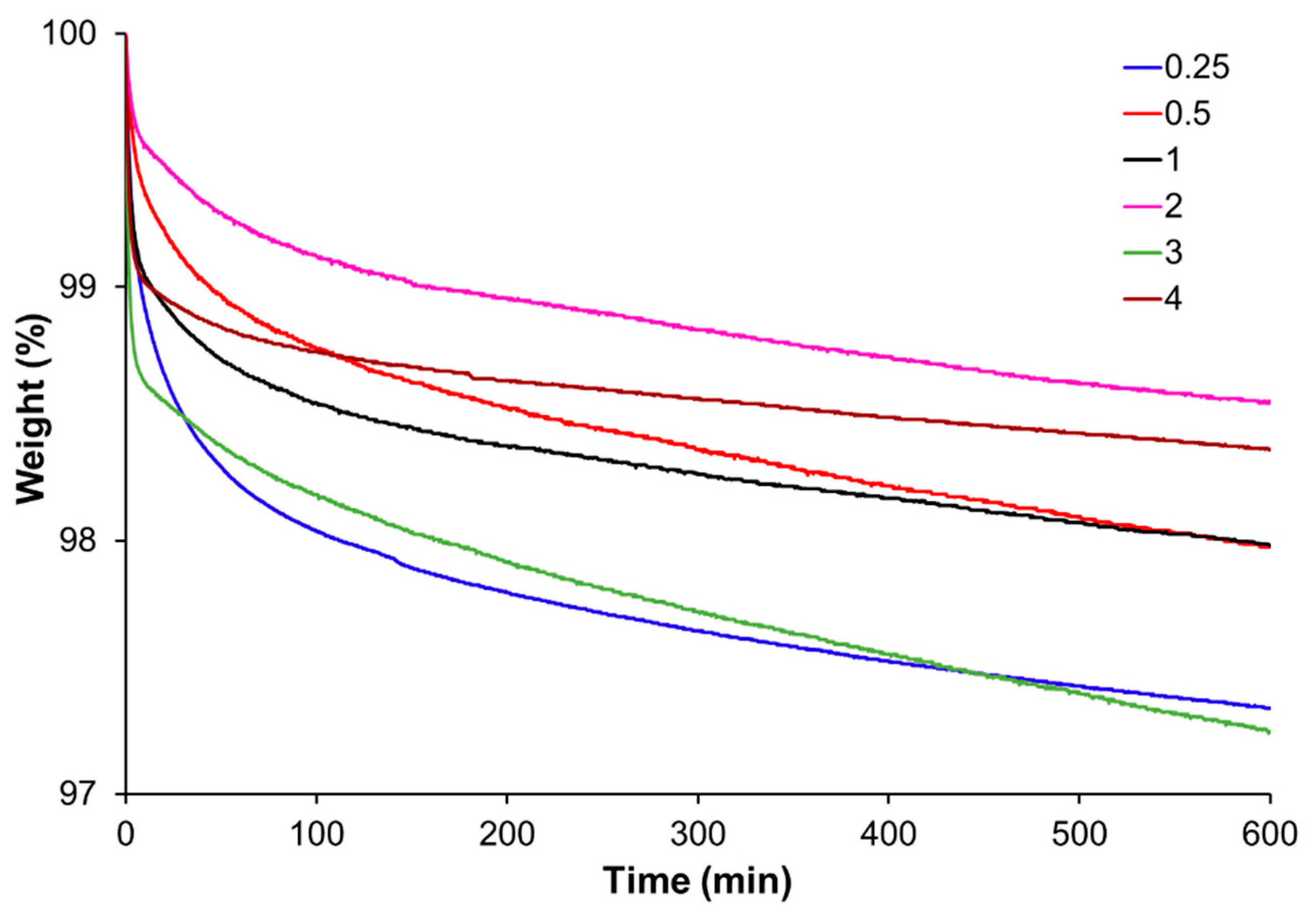
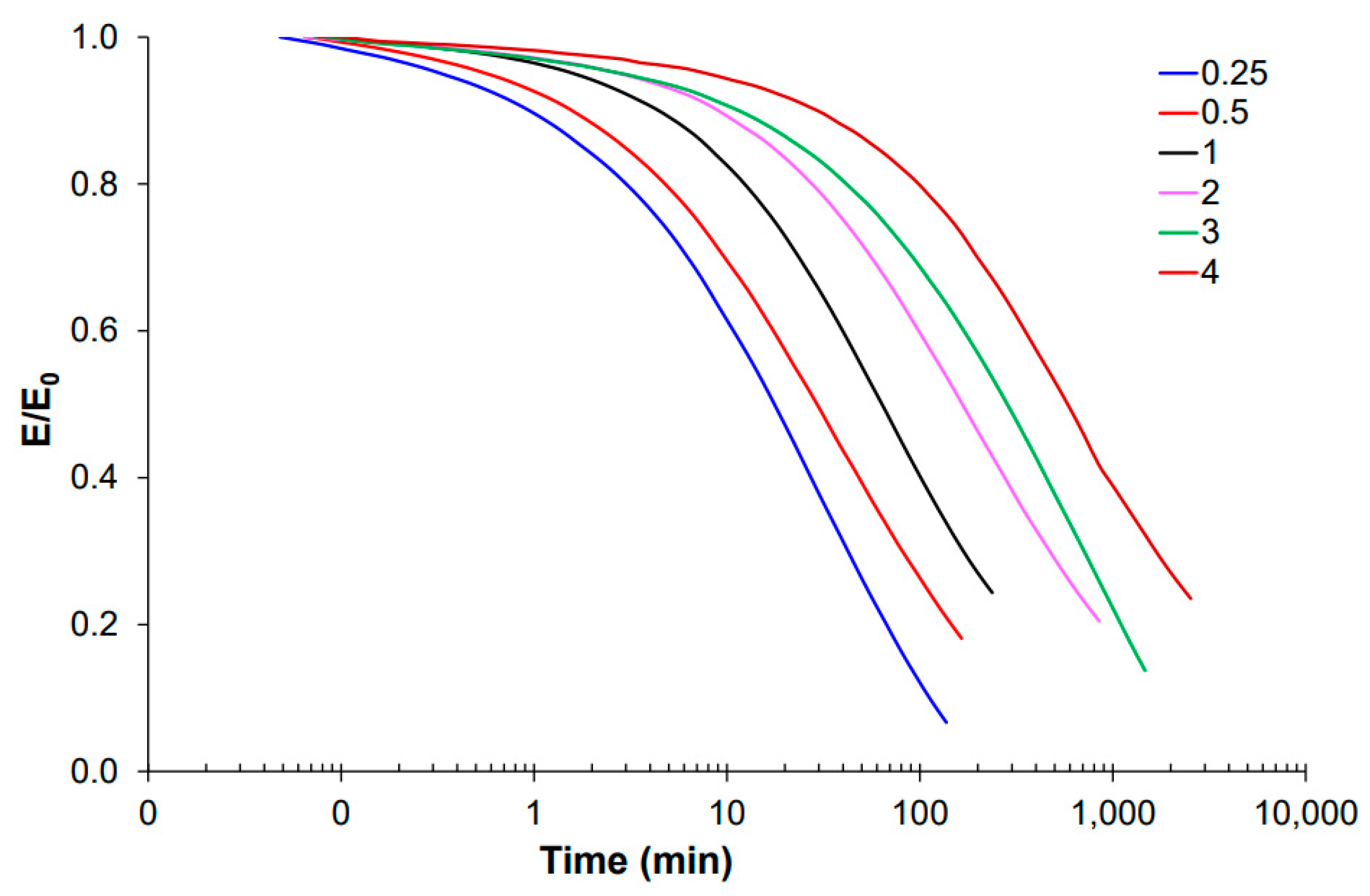
3.1. Effect of Acrylate to Epoxy Ratio
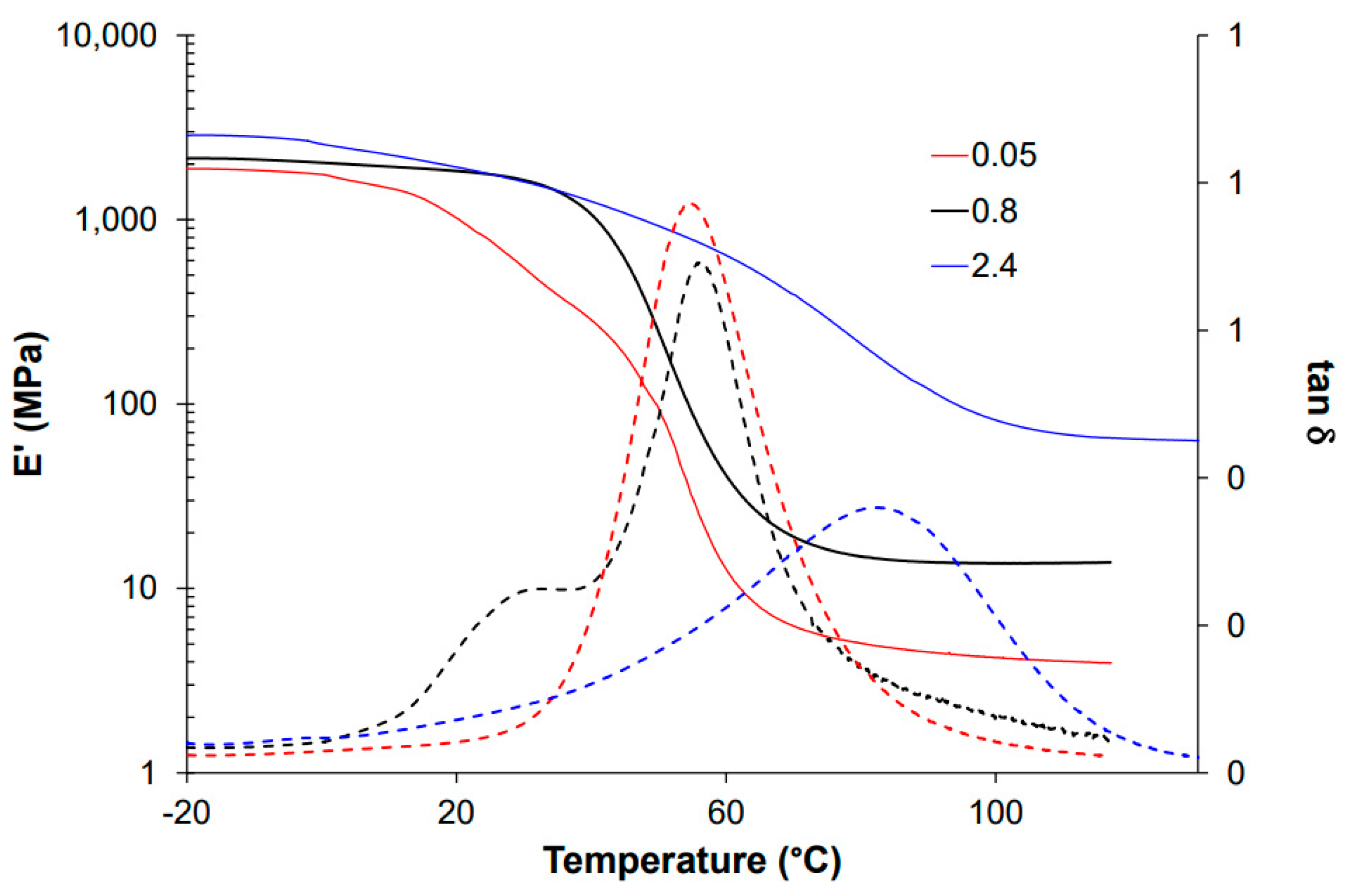
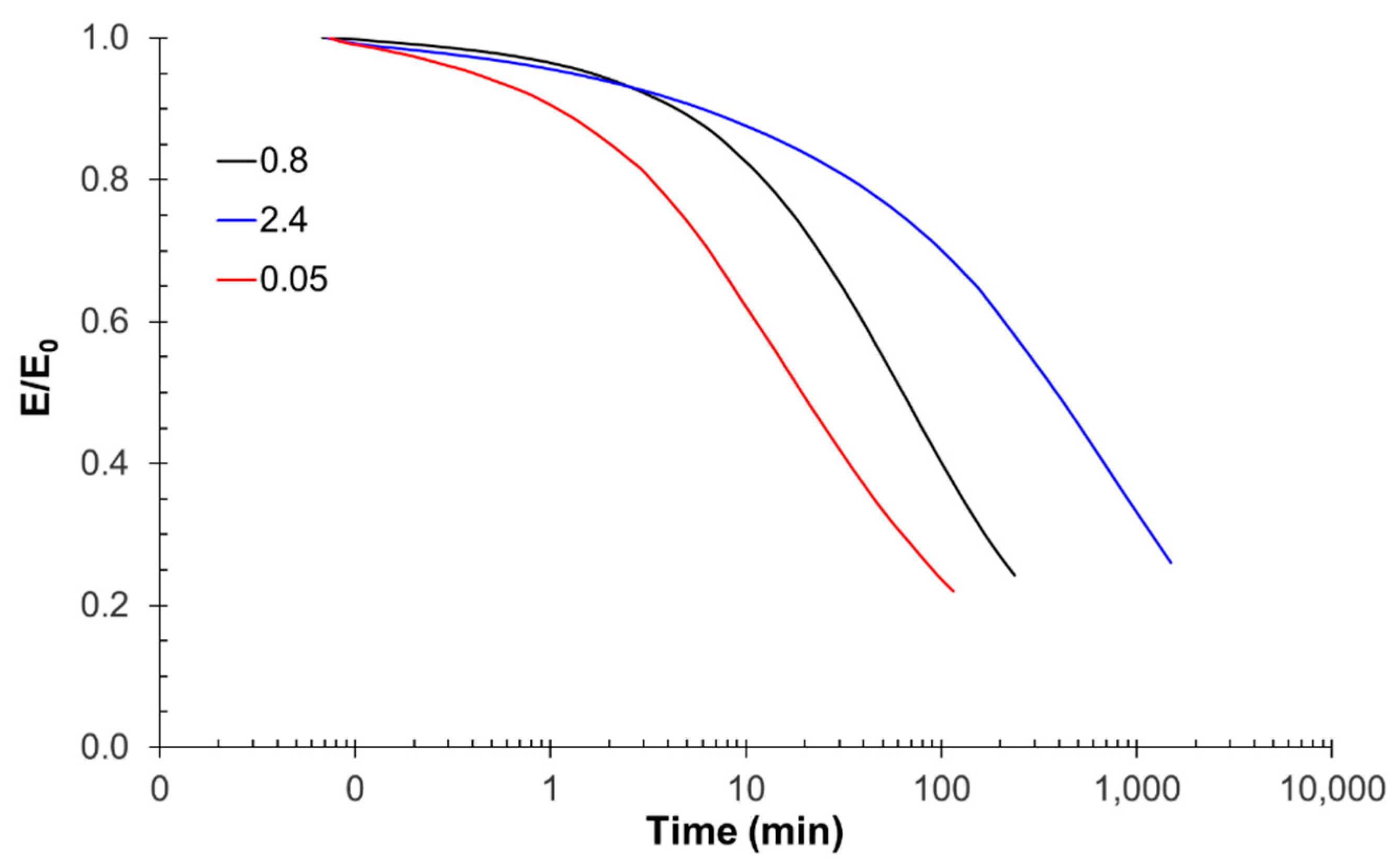
3.2. Effect of the Coupling Agent
3.3. Effect of Epoxide Type
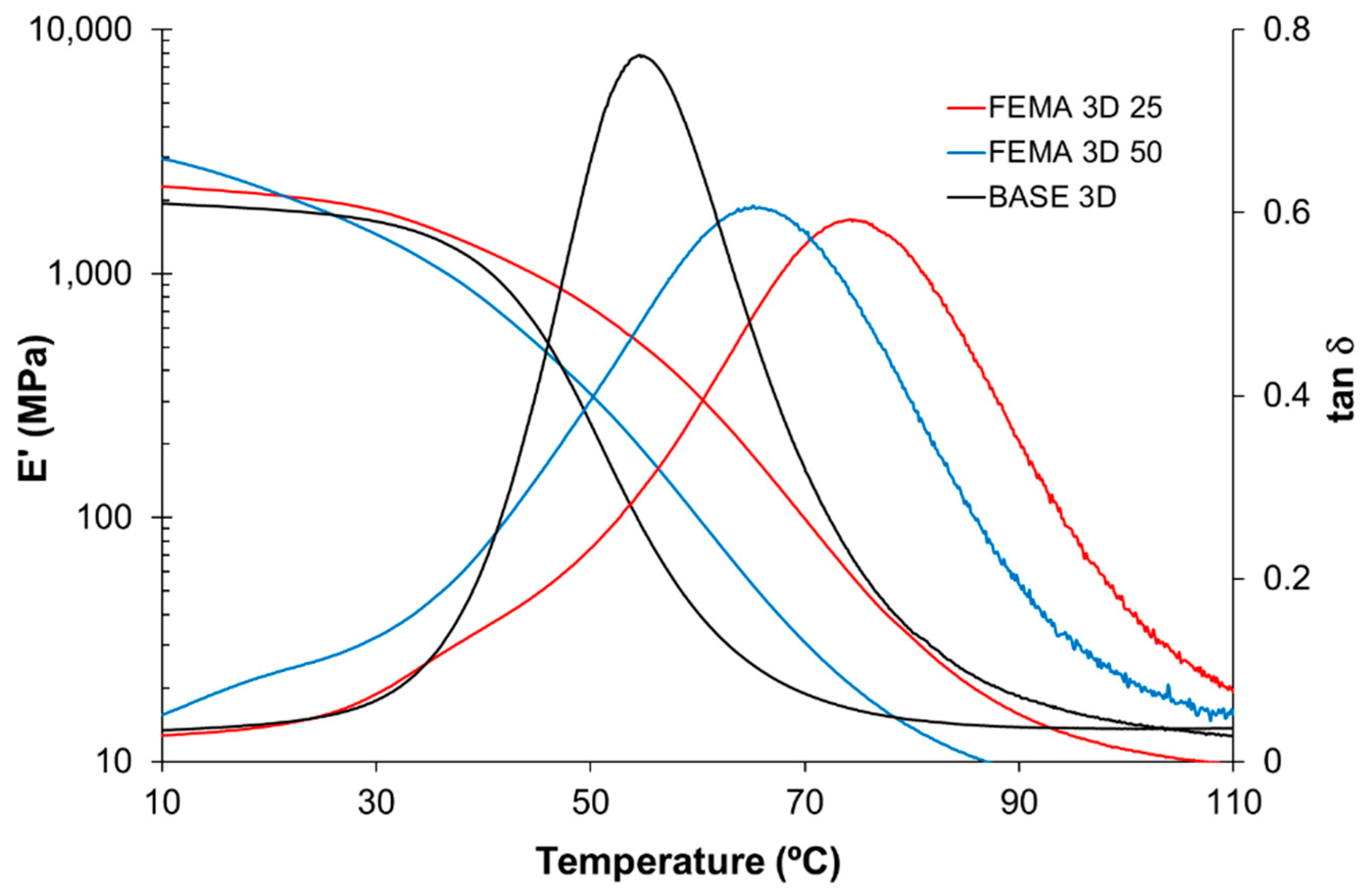
3.4. Optimization for 3D Printability
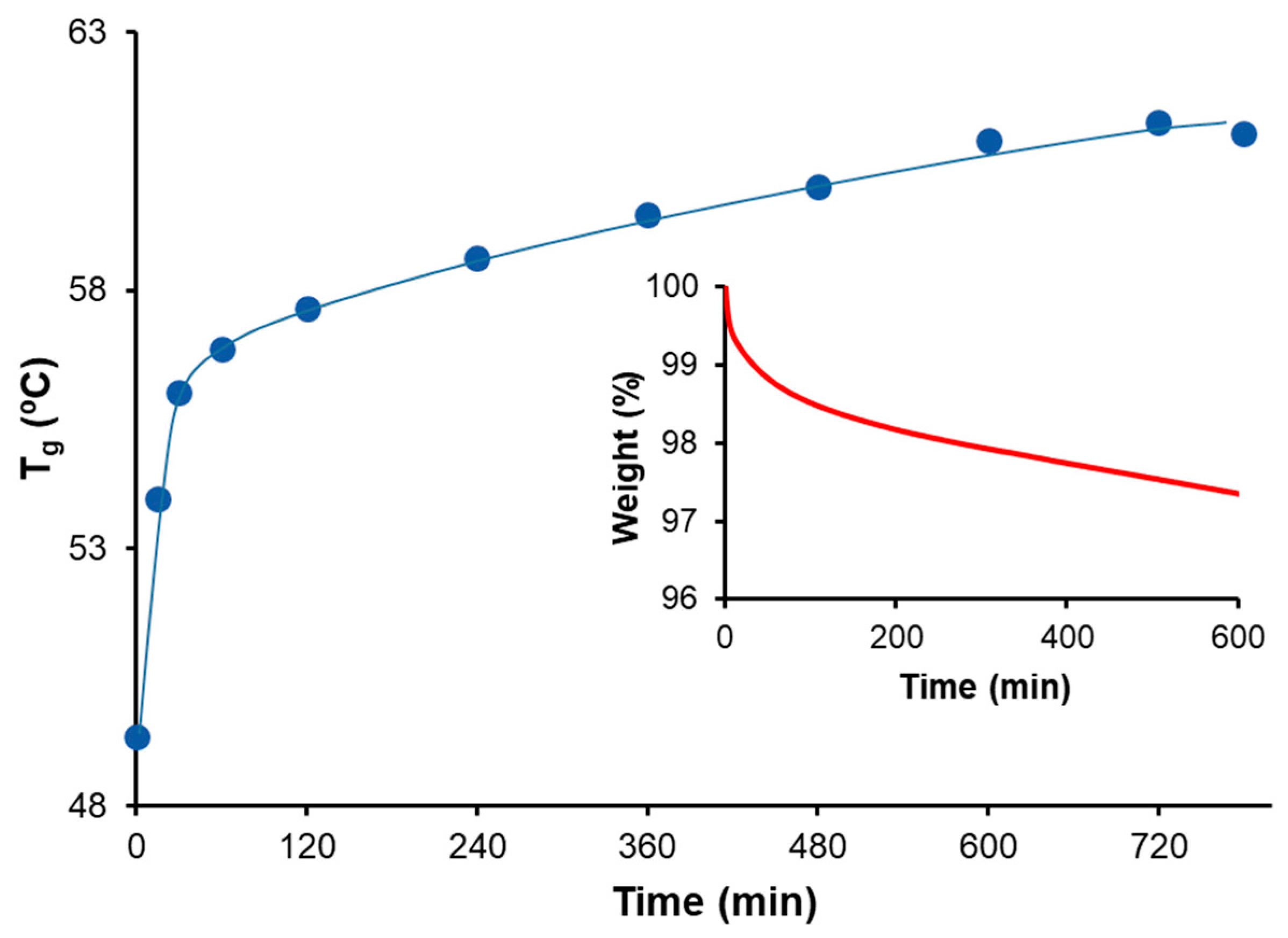
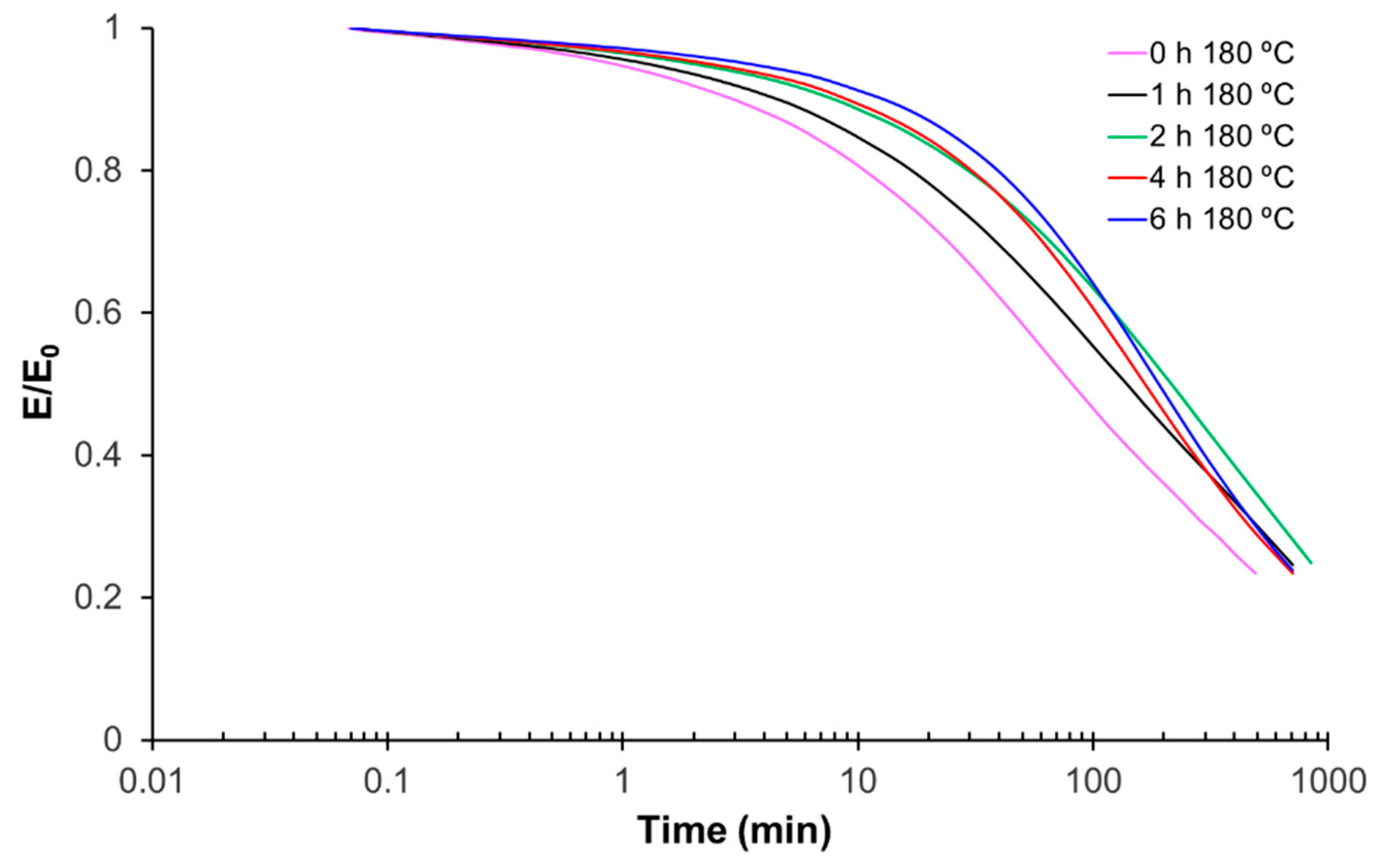
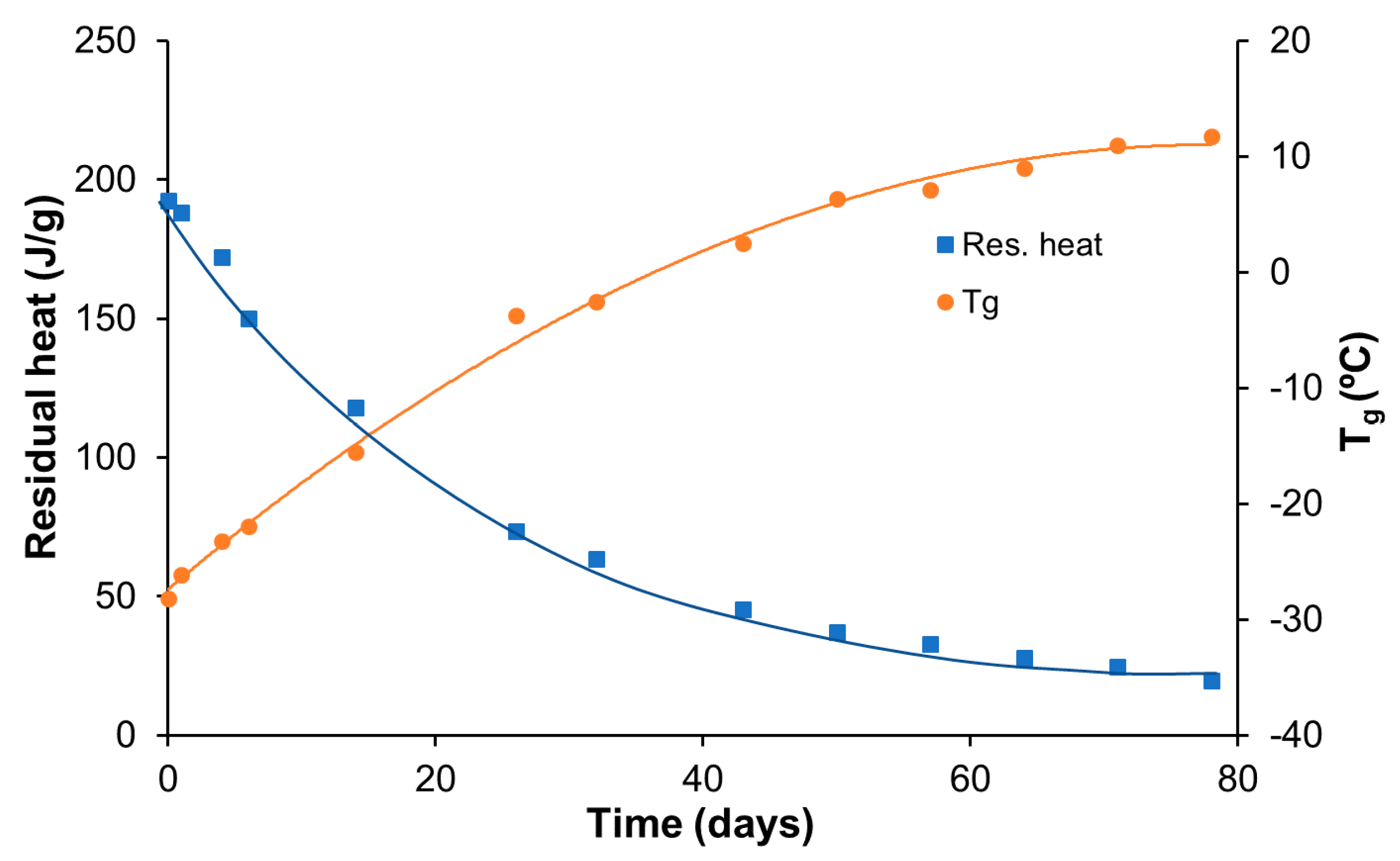
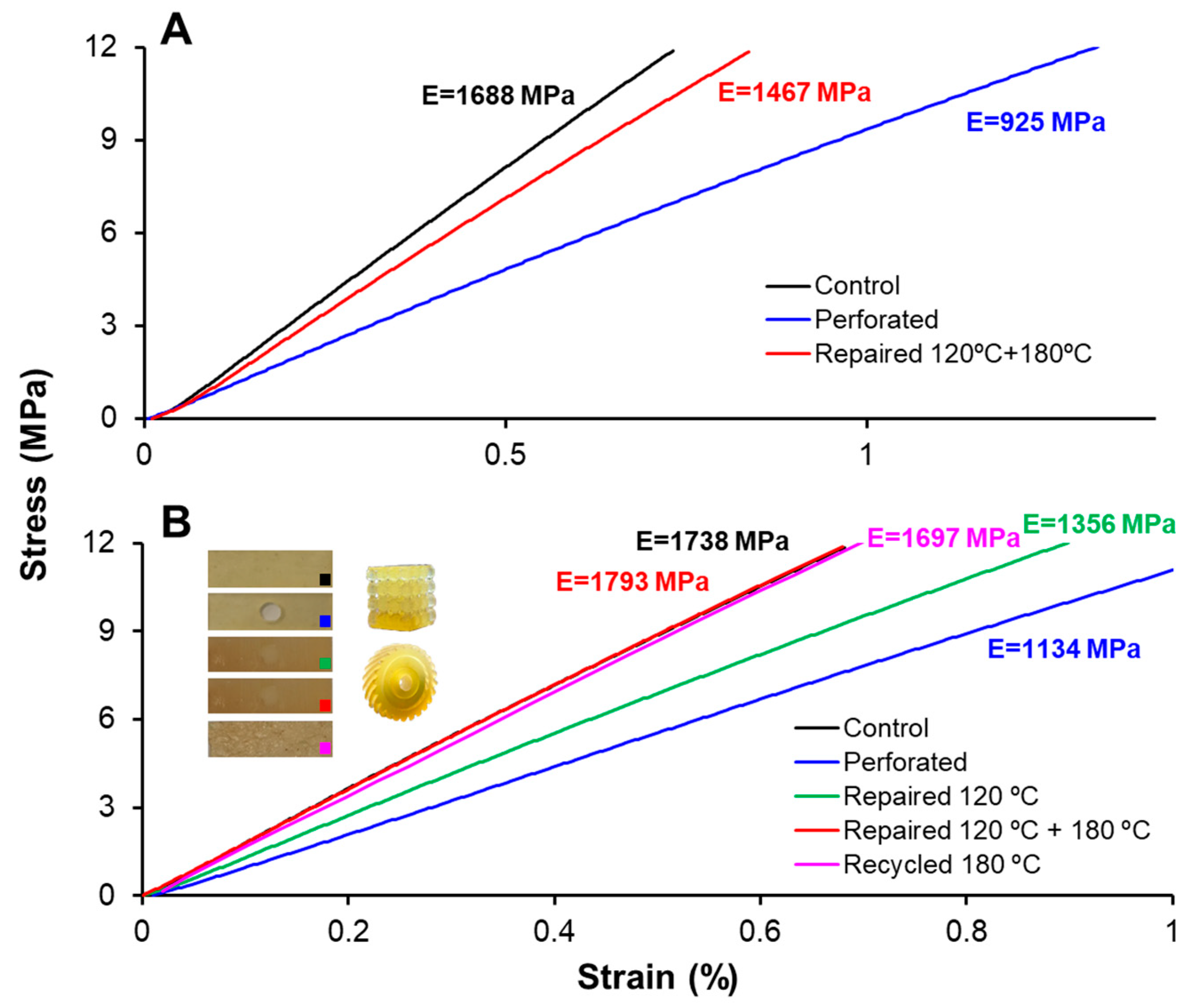
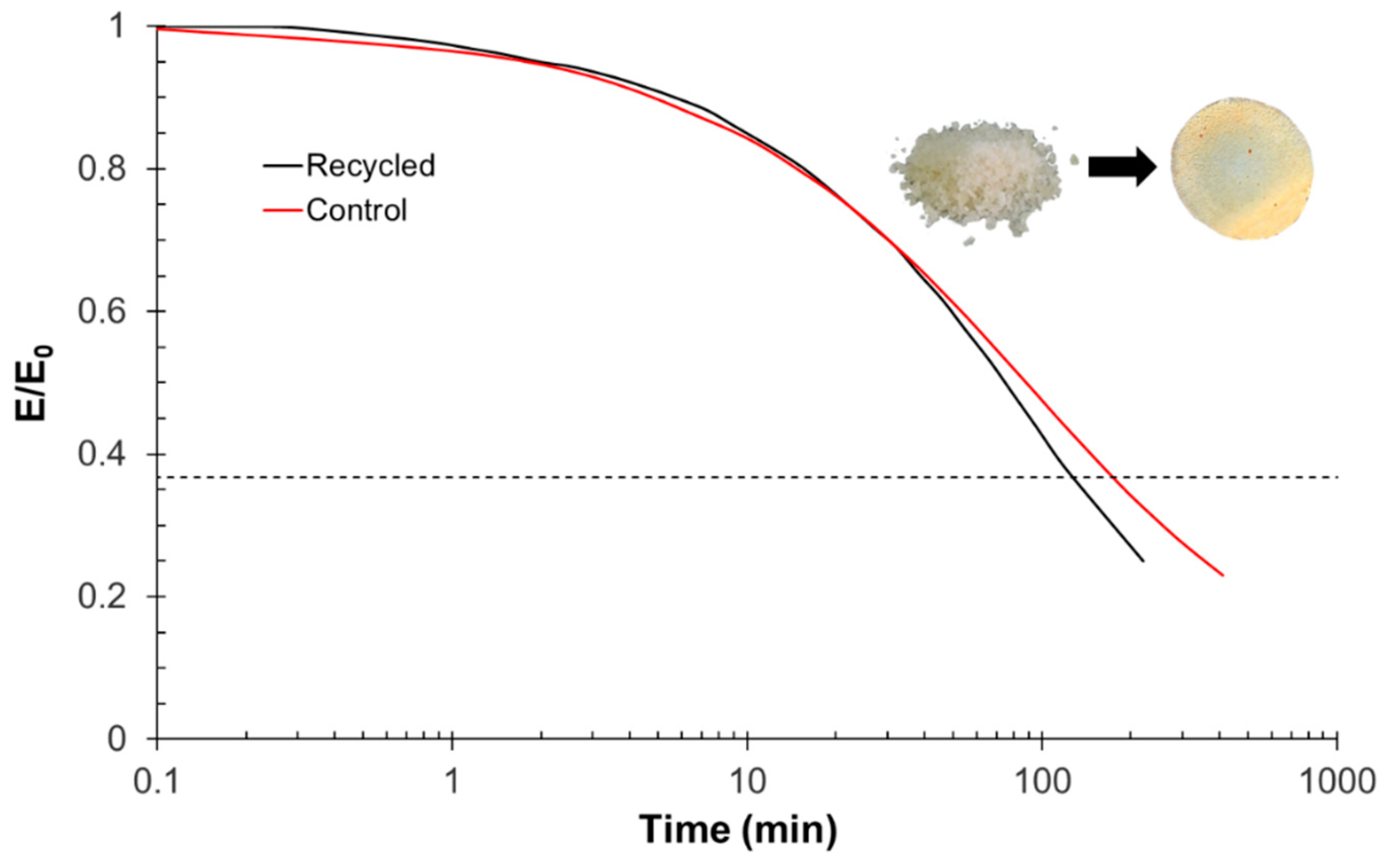
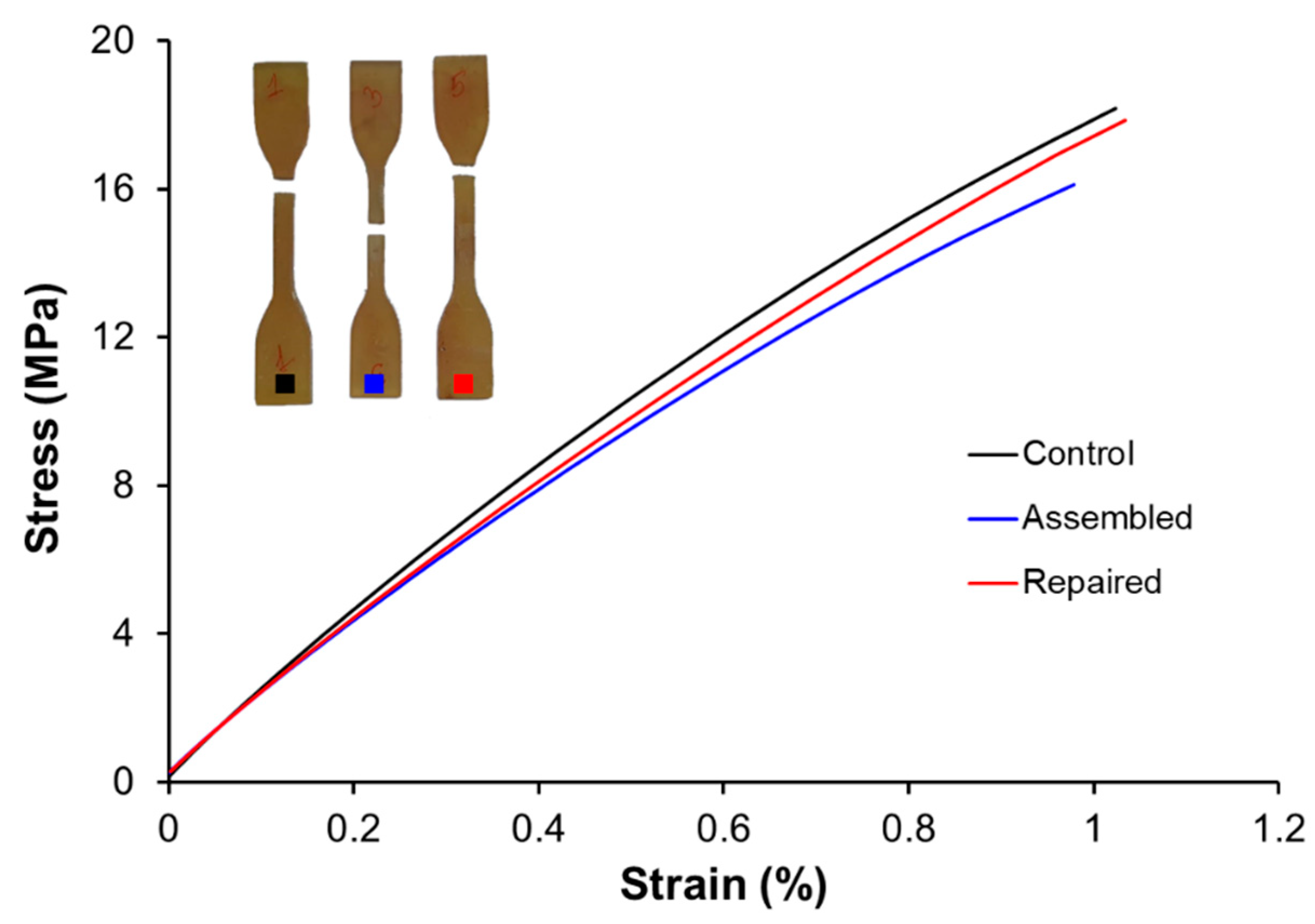
3.5. Printing, Repair, and Reprocessing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ligon, S.C.; Liska, R.; Stampfl, J.; Gurr, M.; Mülhaupt, R. Polymers for 3D printing and customized additive manufacturing. Chem. Rev. 2017, 117, 10212–10290. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M. 3D printing gets a boost and opportunities with polymer materials. ACS Macro Lett. 2014, 3, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Tumbleston, J.R.; Shirvanyants, D.; Ermoshkin, N.; Janusziewicz, R.; Johnson, A.R.; Kelly, D.; Chen, K.; Pinschmidt, R.; Rolland, J.P.; Ermoshkin, A.; et al. Continuous liquid interface production of 3D objects. Science 2015, 347, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.C.; Erkal, J.L.; Lockwood, S.Y.; Chen, C.; Spence, D.M. Evaluation of 3D printing and its potential impact on biotechnology and the chemical sciences. Anal. Chem. 2014, 86, 3240–3253. [Google Scholar] [CrossRef] [PubMed]
- Bekas, D.G.; Hou, Y.; Liu, Y.; Panesar, A. 3D printing to enable multifunctionality in polymer-based composites: A review. Compos. Part B Eng. 2019, 179, 107540. [Google Scholar] [CrossRef]
- Poelma, J.; Rolland, J. Rethinking digital manufacturing with polymers. Insights 2017, 358, 1384–1386. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.D.; Kashani, A.; Imbalzano, G.; Nguyen, K.T.Q.; Hui, D. Additive manufacturing (3D printing): A review of materials, methods, applications and challenges. Compos. Part B Eng. 2018, 143, 172–196. [Google Scholar] [CrossRef]
- Jung, K.; Corrigan, N.; Ciftci, M.; Xu, J.; Seo, S.E.; Hawker, C.J.; Boyer, C. Designing with Light: Advanced 2D, 3D, and 4D Materials. Adv. Mater. 2020, 32, 1–21. [Google Scholar] [CrossRef]
- Layani, M.; Wang, X.; Magdassi, S. Novel Materials for 3D Printing by Photopolymerization. Adv. Mater. 2018, 30, 1–7. [Google Scholar] [CrossRef]
- Conjaerts, A.; Willaert, R.G. Gravity-driven adaptive evolution of an industrial brewer’s yeast strain towards a snowflake phenotype in a 3d-printed mini tower fermentor. Fermentation 2017, 3, 4. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kloxin, C.J.; Bowman, C.N. Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev. 2013, 42, 7161–7173. [Google Scholar] [CrossRef]
- Kloxin, C.J.; Scott, T.F.; Adzima, B.J.; Bowman, C.N. Covalent adaptable networks (CANs): A unique paradigm in cross-linked polymers. Macromolecules 2010, 43, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, B.; Sanka, R.V.S.P.; Binder, W.H.; Parthasarthy, V.; Rana, S.; Karak, N. Vitrimers: Associative dynamic covalent adaptive networks in thermoset polymers. Chem. Eng. J. 2020, 385, 123820. [Google Scholar] [CrossRef]
- Alfarhan, S.; Brown, J.; Liu, B.; Long, T.; Jin, K. Chemically recyclable crosslinked thiol-ene photopolymers via thiol-disulfide exchange reactions. J. Polym. Sci. 2022, 1–12. [Google Scholar] [CrossRef]
- Bongiardina, N.J.; Soars, S.M.; Podgorski, M.; Bowman, C.N. Radical-disulfide exchange in thiol–ene–disulfidation polymerizations. Polym. Chem. 2022, 13, 3991–4003. [Google Scholar] [CrossRef]
- Alabiso, W.; Hron, T.M.; Reisinger, D.; Bautista-Anguís, D.; Schlögl, S. Shape memory-assisted self-healing of dynamic thiol-acrylate networks. Polym. Chem. 2021, 12, 5704–5714. [Google Scholar] [CrossRef]
- Zhang, B.; Kowsari, K.; Serjouei, A.; Dunn, M.L.; Ge, Q. Reprocessable thermosets for sustainable three-dimensional printing. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Casado, J.; Konuray, O.; Roig, A.; Fernández-Francos, X.; Ramis, X. 3D printable hybrid acrylate-epoxy dynamic networks. Eur. Polym. J. 2022, 173, 111256. [Google Scholar] [CrossRef]
- Salaeh, S.; Das, A.; Wießner, S.; Stapor, M. Vitrimer-like material based on a biorenewable elastomer crosslinked with a dimeric fatty acid. Eur. Polym. J. 2021, 151, 110452. [Google Scholar] [CrossRef]
- Liu, T.; Hao, C.; Zhang, S.; Yang, X.; Wang, L.; Han, J.; Li, Y.; Xin, J.; Zhang, J. A Self-Healable High Glass Transition Temperature Bioepoxy Material Based on Vitrimer Chemistry. Macromolecules 2018, 51, 5577–5585. [Google Scholar] [CrossRef]
- Niu, X.; Wang, F.; Li, X.; Zhang, R.; Wu, Q.; Sun, P. Using Zn2+ Ionomer to Catalyze Transesterification Reaction in Epoxy Vitrimer. Ind. Eng. Chem. Res. 2019, 58, 5698–5706. [Google Scholar] [CrossRef]
- Demongeot, A.; Mougnier, S.J.; Okada, S.; Soulié-Ziakovic, C.; Tournilhac, F. Coordination and catalysis of Zn2+ in epoxy-based vitrimers. Polym. Chem. 2016, 7, 4486–4493. [Google Scholar] [CrossRef]
- Fernández-francos, X.; Konuray, O.; Ramis, X.; Serra, À.; De, S. Enhancement of 3D-printable materials by dual-curing procedures. Materials 2021, 14, 107. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
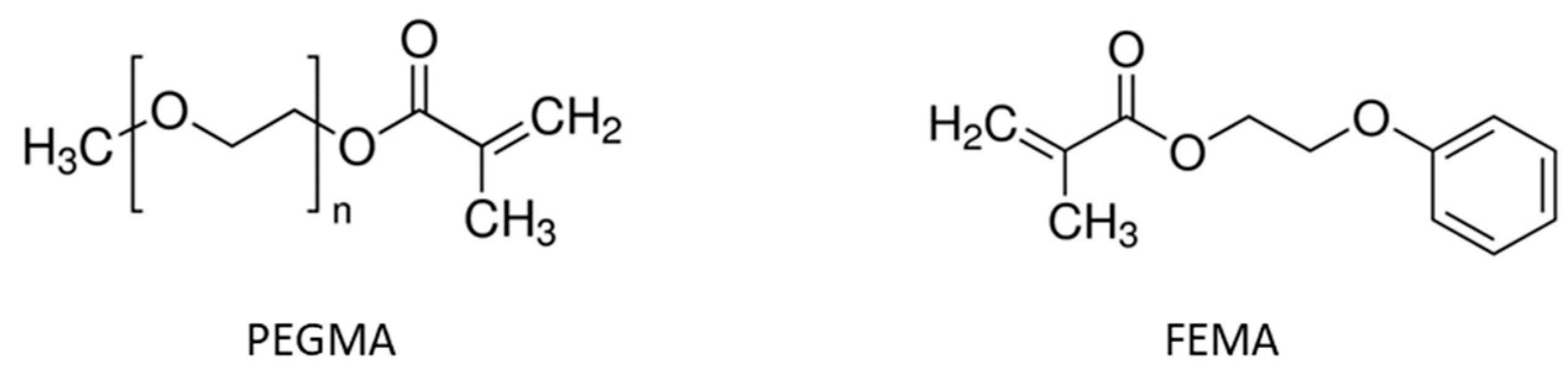{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formn. | GLYDA | PEGMA | GMA | DG | GLU | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| g | eq·103 | g | eq·103 | g | eq·103 | g | eq·103 | g | eq·103 | |
| 0.25 | 0.25 | 1.44 | 0.25 | 0.83 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| 0.5 | 0.5 | 2.87 | 0.5 | 1.66 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| 1 * | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| 2 | 2 | 11.5 | 2 | 6.66 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| 3 | 3 | 17.2 | 3 | 10 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| 4 | 4 | 23 | 4 | 13.3 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| Formn. | GLYDA | PEGMA | GMA | DG | GLU | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| g | eq·103 | g | eq·103 | g | eq·103 | g | eq·103 | g | eq·103 | |
| 0.05 | 1 | 5.74 | 1 | 3.33 | 0.056 | 0.394 | 3 | 16 | 1.086 | 16.4 |
| 0.8 * | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| 2.4 | 1 | 5.74 | 1 | 3.33 | 2.34 | 16.4 | 0 | 0 | 1.086 | 16.4 |
| Formn. | GLYDA | PEGMA | GMA | Epoxy | GLU | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| g | eq·103 | g | eq·103 | g | eq·103 | g | eq·103 | g | eq·103 | |
| NG | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 1.28 | 10.7 | 1.086 | 16.4 |
| CE | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 2.14 | 10.7 | 1.086 | 16.4 |
| TGE | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 1.49 | 10.7 | 1.086 | 16.4 |
| PPGGE | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 3.42 | 10.7 | 1.086 | 16.4 |
| PPGGEs | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 2.03 | 10.7 | 1.086 | 16.4 |
| TFG | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 1.29 | 10.7 | 1.086 | 16.4 |
| DG * | 1 | 5.74 | 1 | 3.33 | 0.816 | 5.74 | 2 | 10.7 | 1.086 | 16.4 |
| Post-Treatment | NG | CE | TGE | PPGGE | PPGGEs | TFG | DG |
|---|---|---|---|---|---|---|---|
| Before | −18 | 8 | 8 | −45 | −5 | 38 | 42 |
| After | 4 | 32 | 24 | −42 | −3 | 43 | 47 |
| Formn. | GMA | PPGGEc | DG | GLU | ||||
|---|---|---|---|---|---|---|---|---|
| g | eq·103 | g | eq·103 | g | eq·103 | g | eq·103 | |
| FEMA 3D 25 * | 0.816 | 5.74 | 0.5 | 2.67 | 1.5 | 8.02 | 1.086 | 16.4 |
| FEMA 3D 50 | 0.816 | 5.74 | 1 | 5.35 | 1 | 5.35 | 1.086 | 16.4 |
| BASE 3D | 0.816 | 5.74 | 0 | 0 | 2 | 10.7 | 1.086 | 16.4 |
| Specimens | Control | Assembled | Repaired |
|---|---|---|---|
| Young’s modulus (MPa) | 1741 | 1628 | 1710 |
| Fracture stress (MPa) | 17.9 | 15.9 | 17.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casado, J.; Konuray, O.; Benet, G.; Fernández-Francos, X.; Morancho, J.M.; Ramis, X. Optimization and Testing of Hybrid 3D Printing Vitrimer Resins. Polymers 2022, 14, 5102. https://doi.org/10.3390/polym14235102
Casado J, Konuray O, Benet G, Fernández-Francos X, Morancho JM, Ramis X. Optimization and Testing of Hybrid 3D Printing Vitrimer Resins. Polymers. 2022; 14(23):5102. https://doi.org/10.3390/polym14235102
Chicago/Turabian StyleCasado, Jaime, Osman Konuray, Gerard Benet, Xavier Fernández-Francos, José Maria Morancho, and Xavier Ramis. 2022. "Optimization and Testing of Hybrid 3D Printing Vitrimer Resins" Polymers 14, no. 23: 5102. https://doi.org/10.3390/polym14235102
APA StyleCasado, J., Konuray, O., Benet, G., Fernández-Francos, X., Morancho, J. M., & Ramis, X. (2022). Optimization and Testing of Hybrid 3D Printing Vitrimer Resins. Polymers, 14(23), 5102. https://doi.org/10.3390/polym14235102