
Phase Behavior of Ion-Containing Polymers in Polar Solvents: Predictions from a Liquid-State Theory with Local Short-Range Interactions



Abstract
1. Introduction
2. Model and Methods
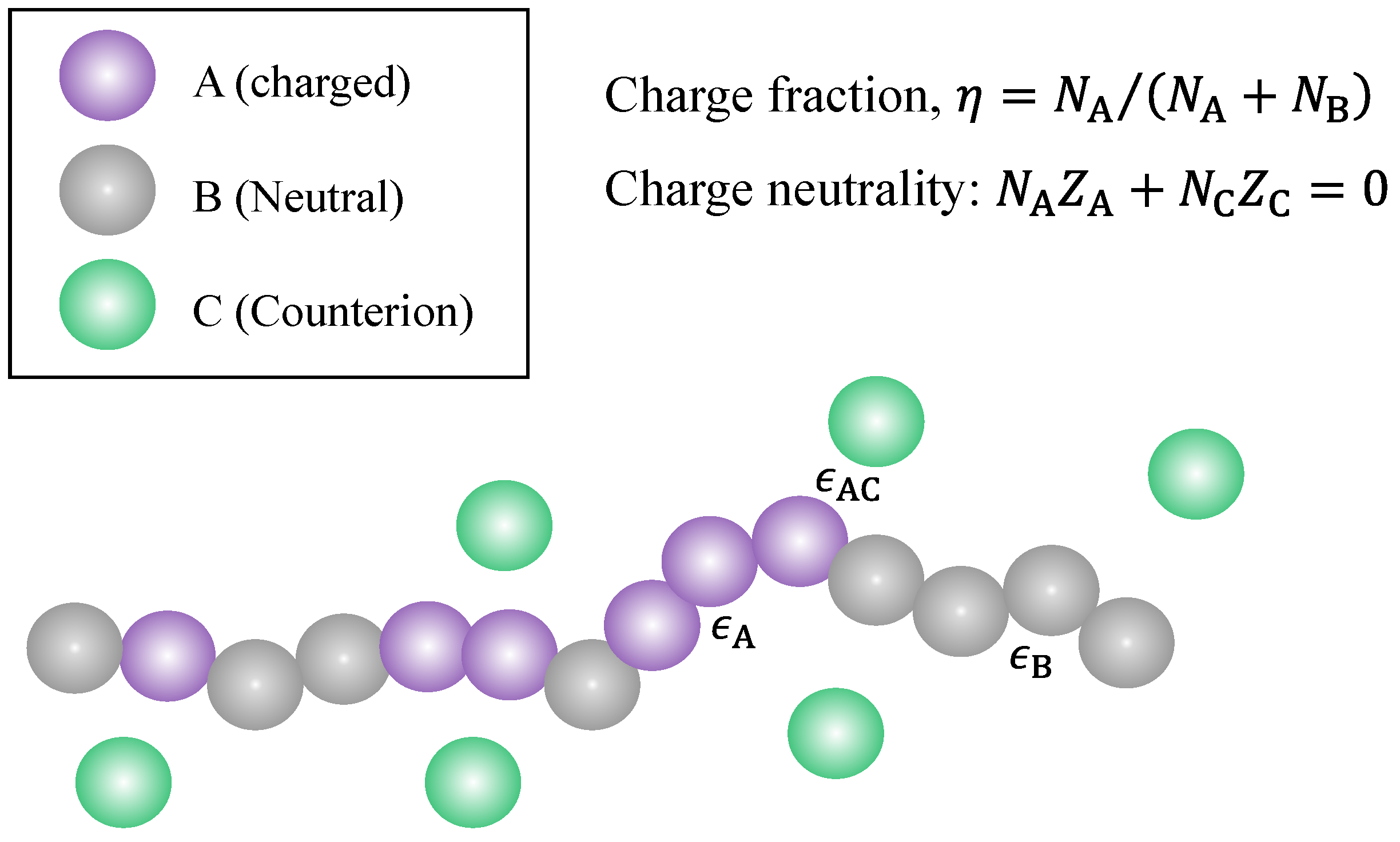
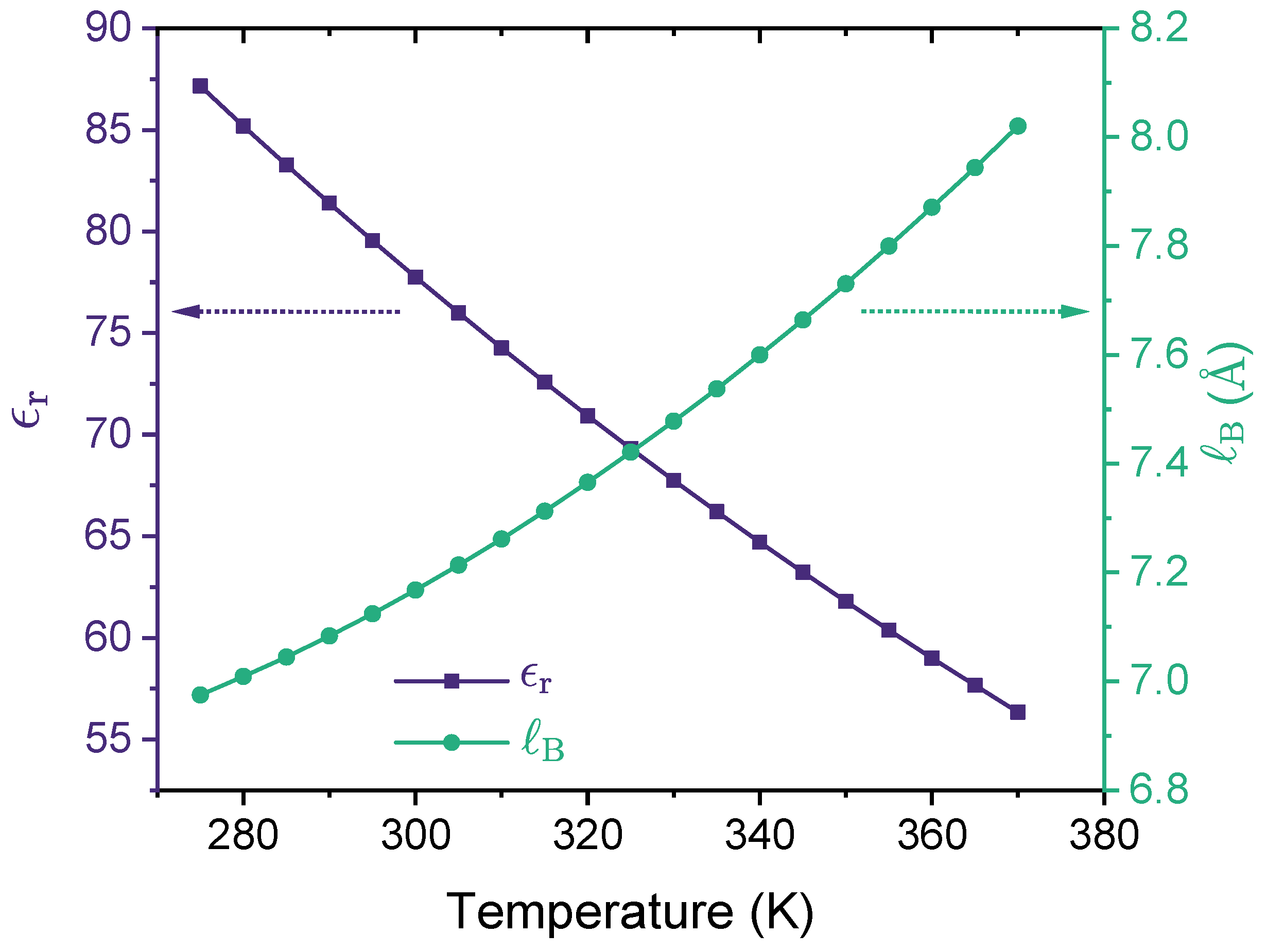
2.1. Polymer and Solution Models
2.2. Theoretical Formulation
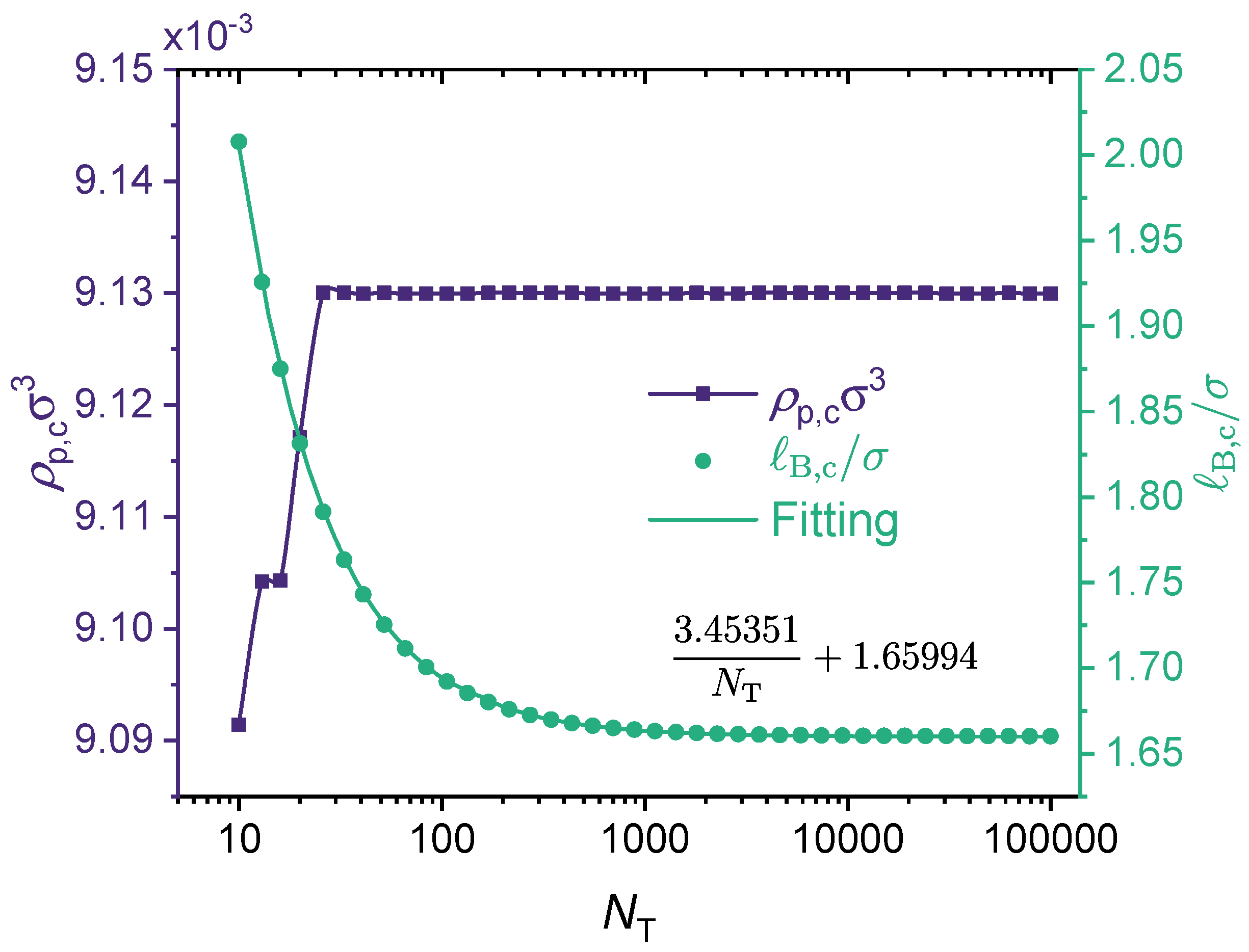
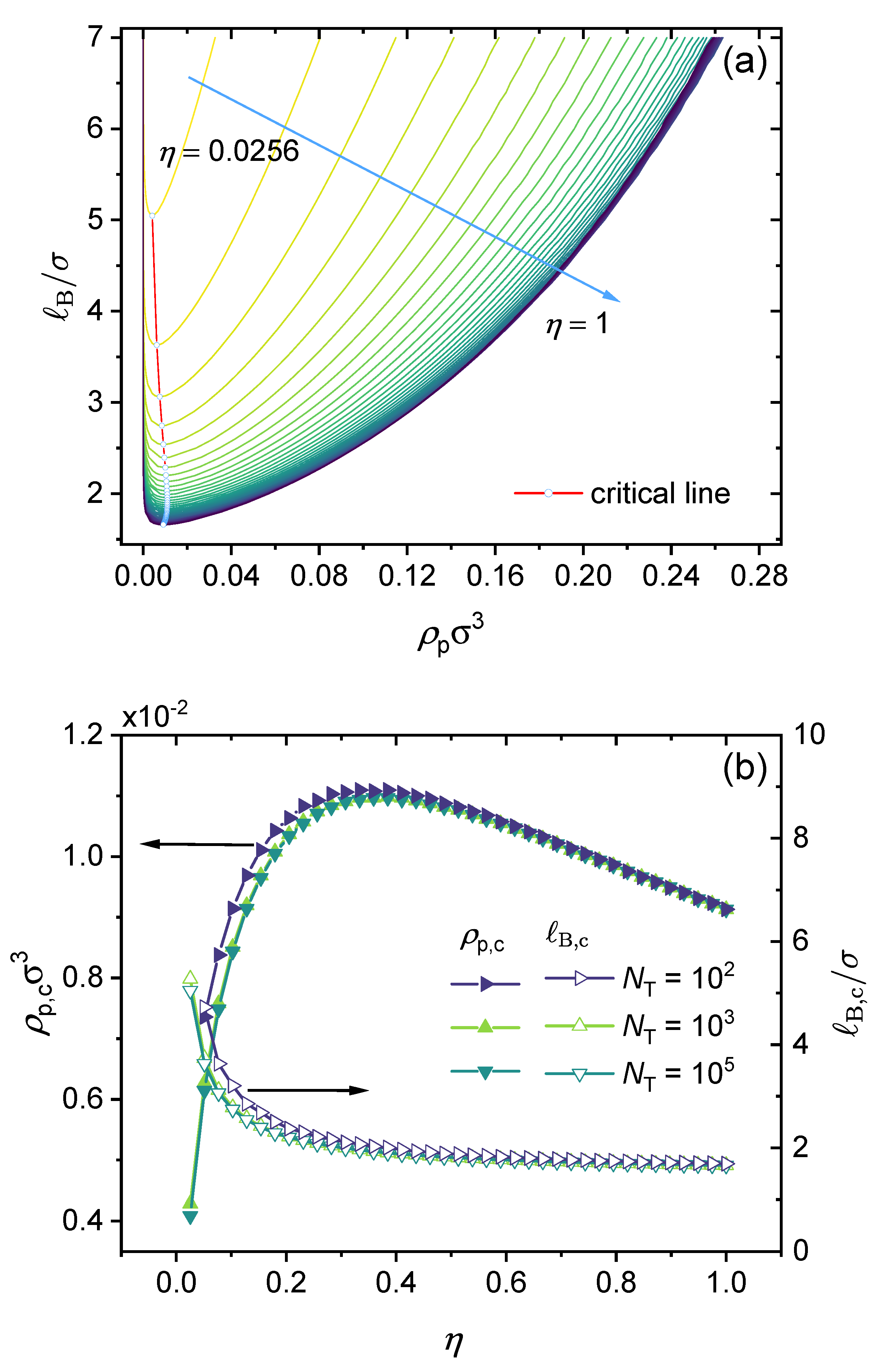
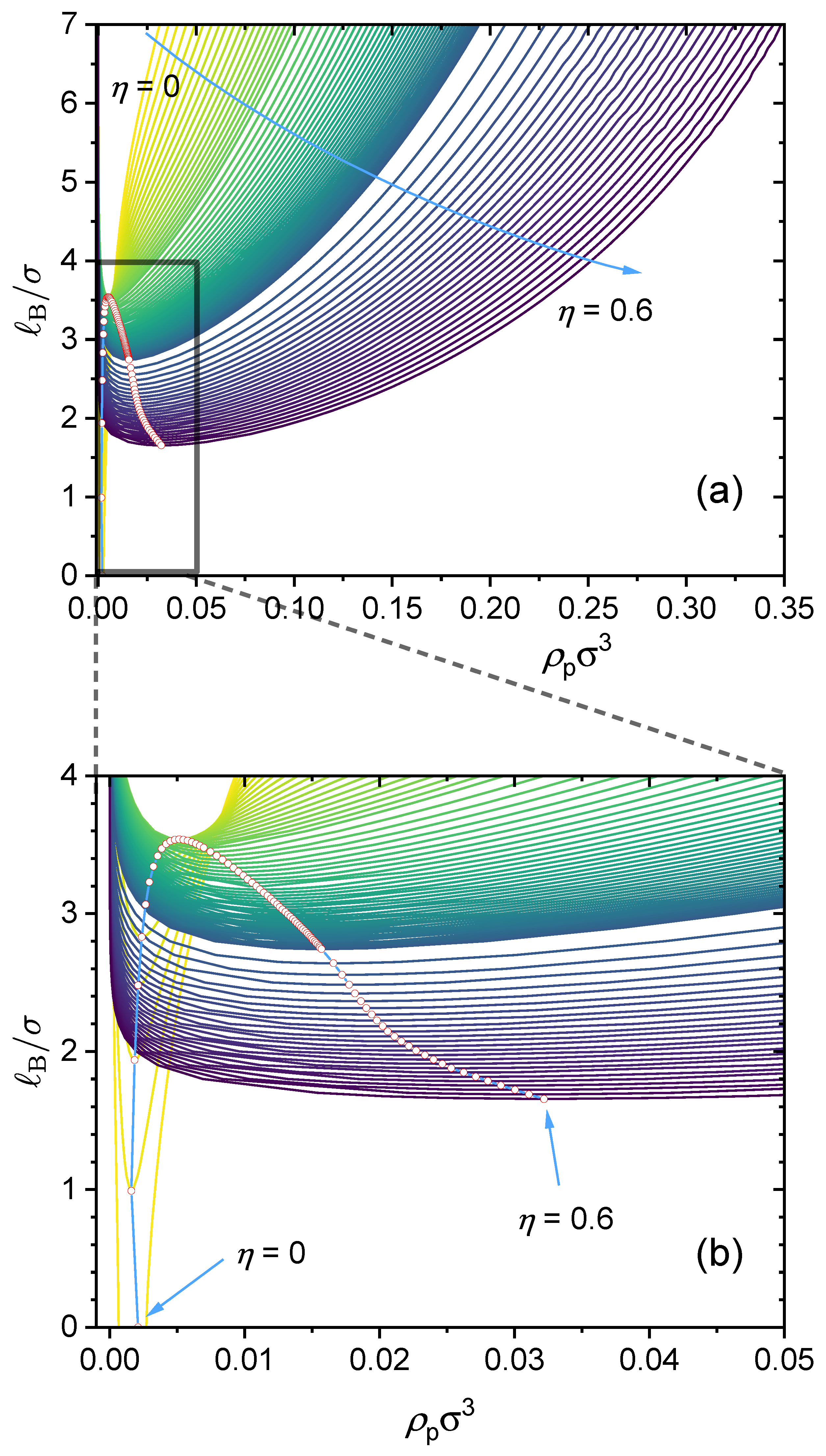
2.3. Construction of Phase Diagram
3. Results and Discussions
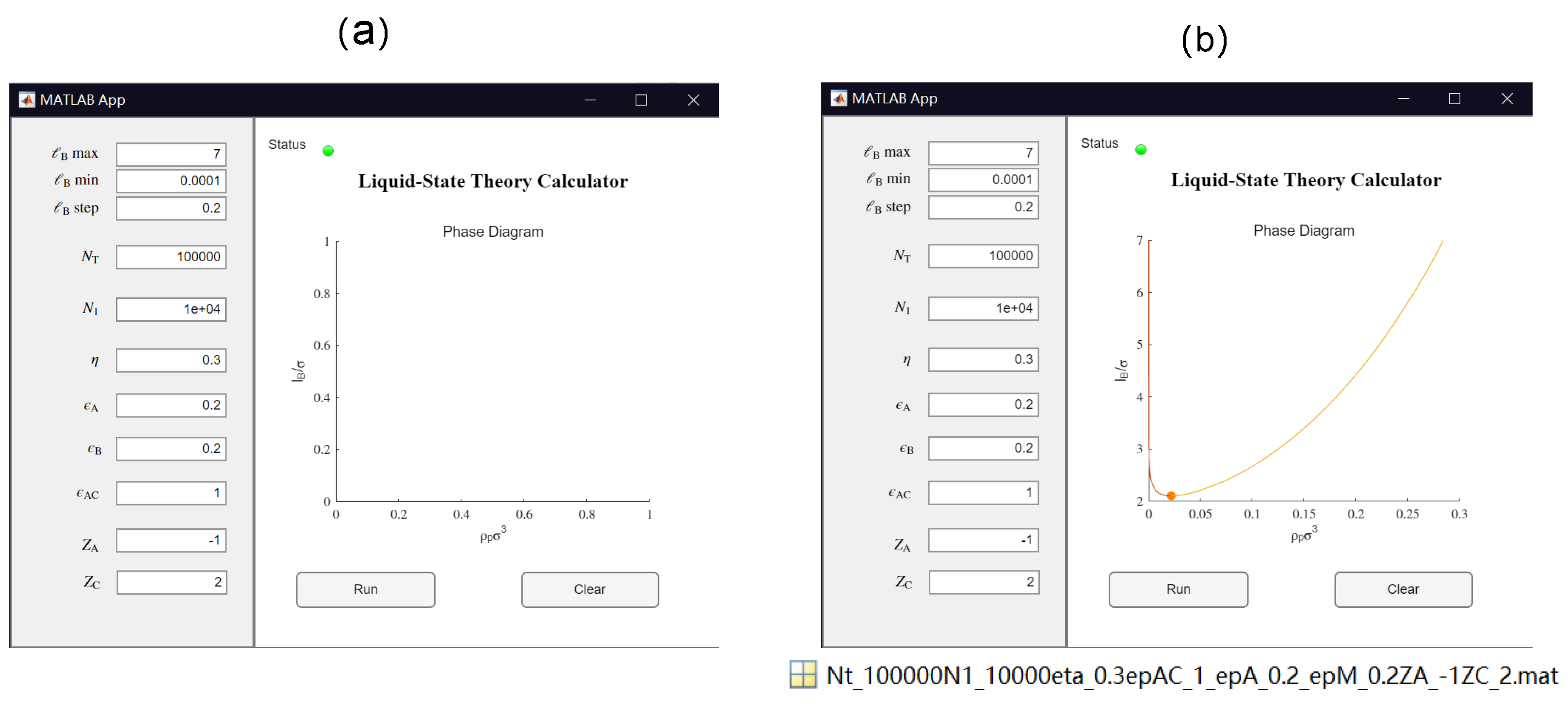
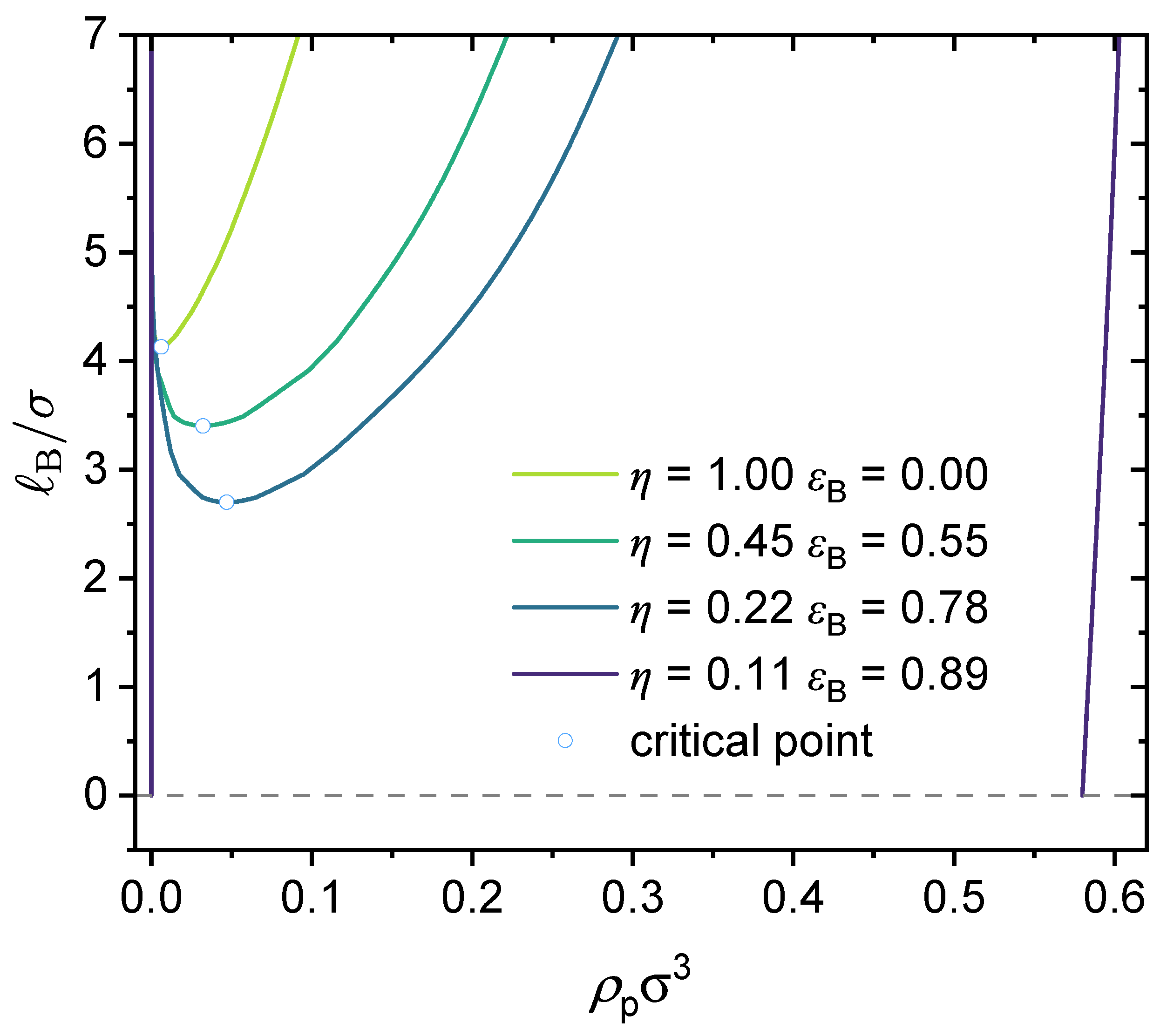
3.1. GUI App for the Salt-Free Case and Selected Sample Results
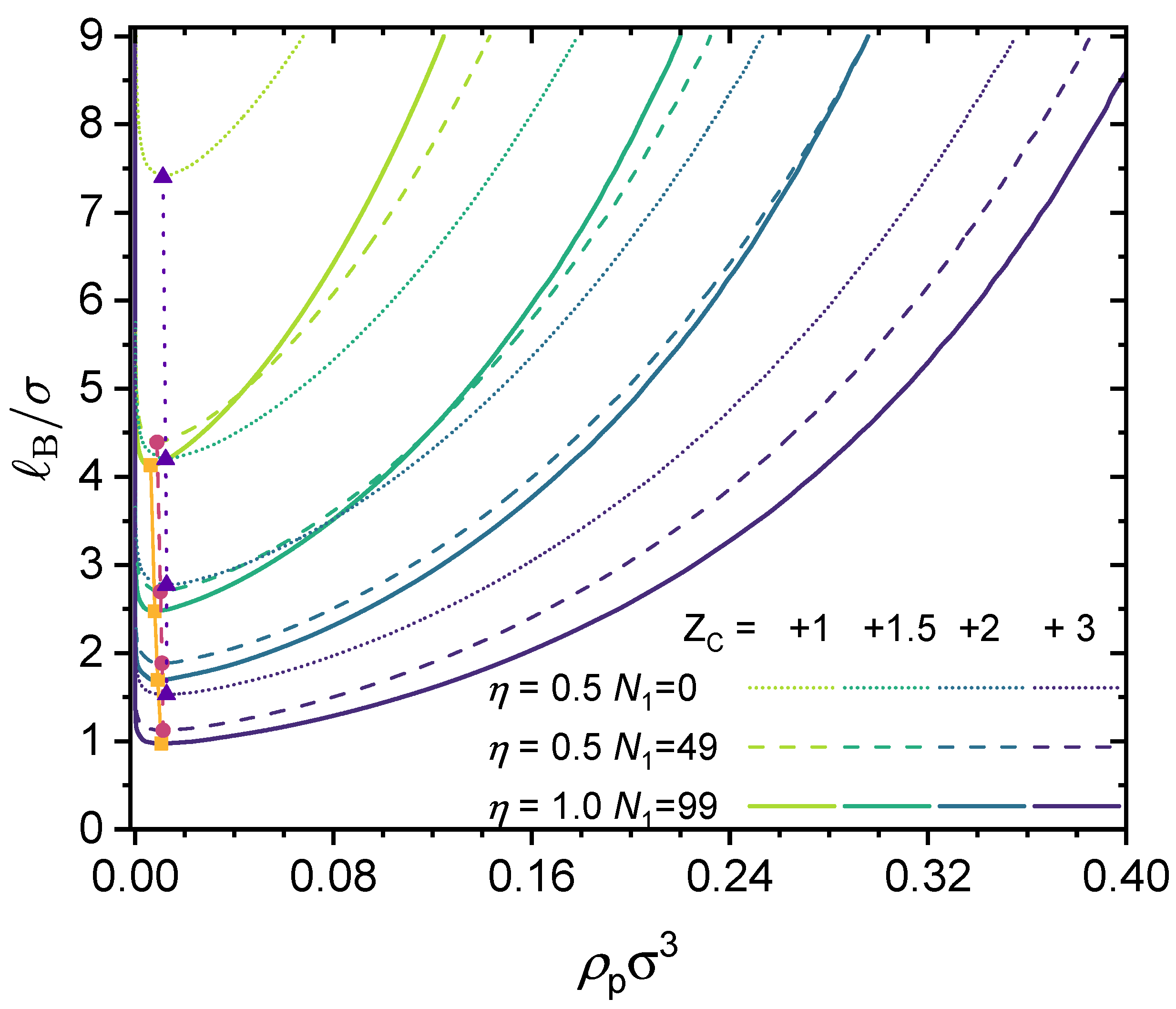
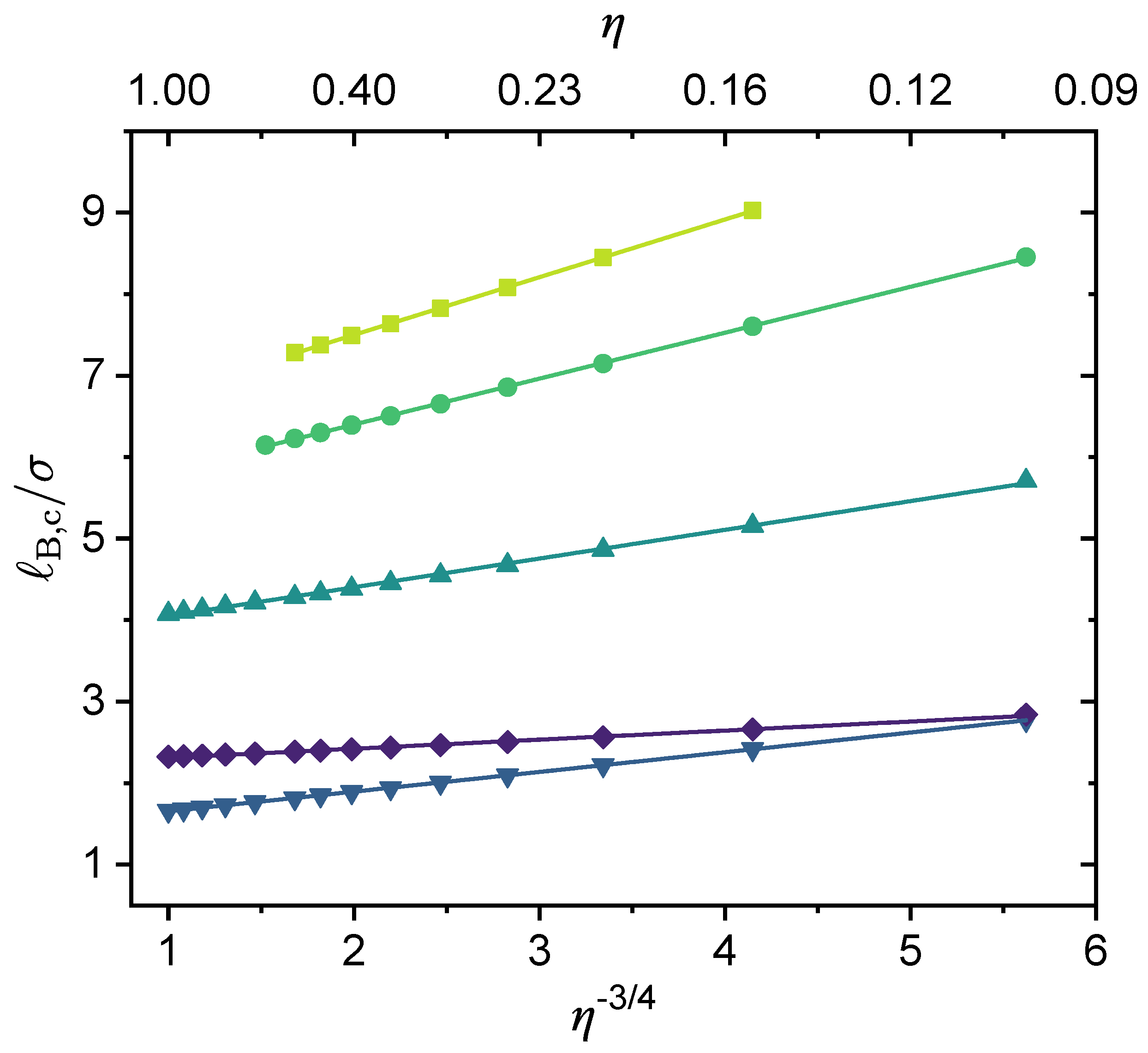
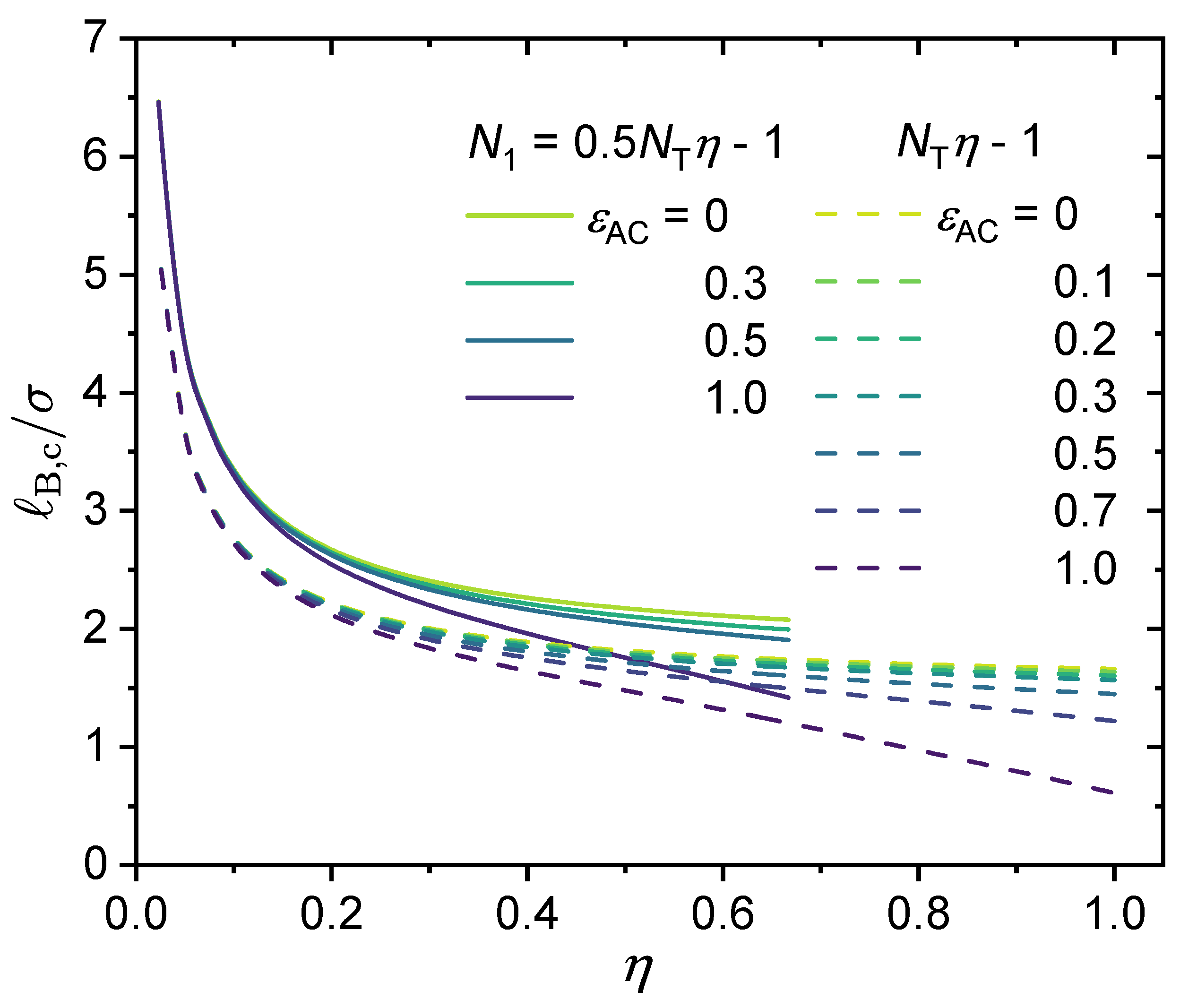
3.2. Effect of Chain Length and Charge Fraction
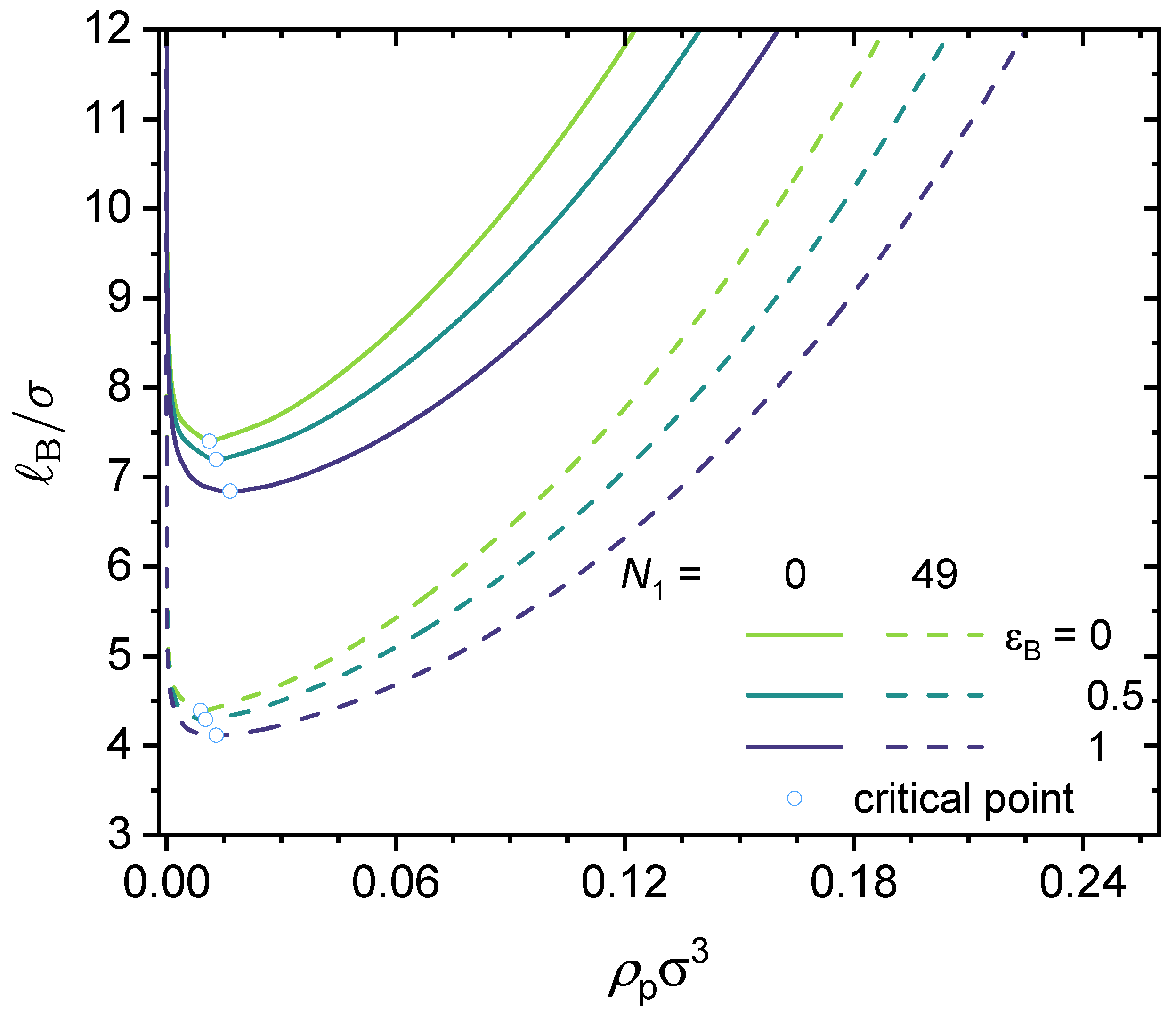
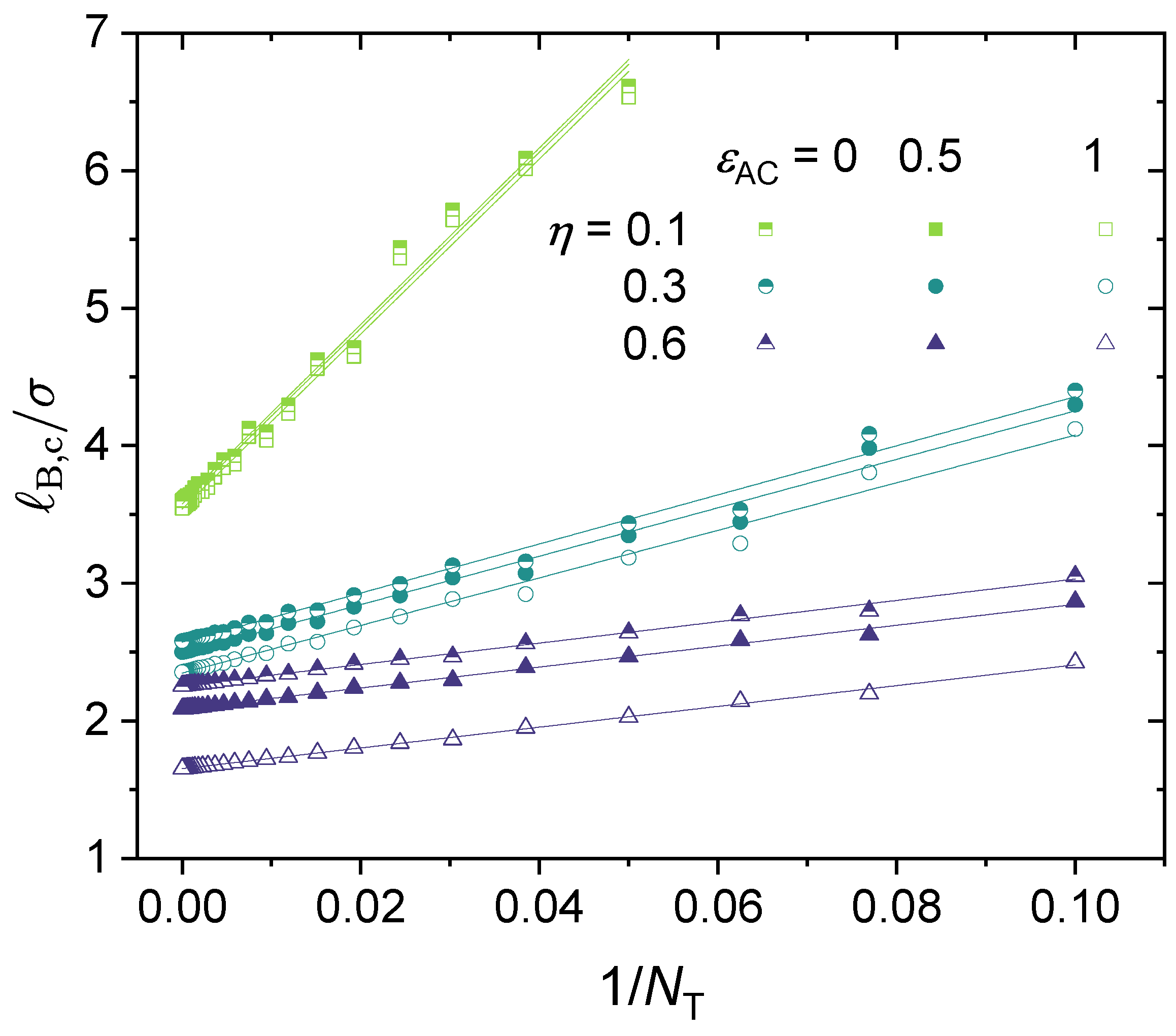
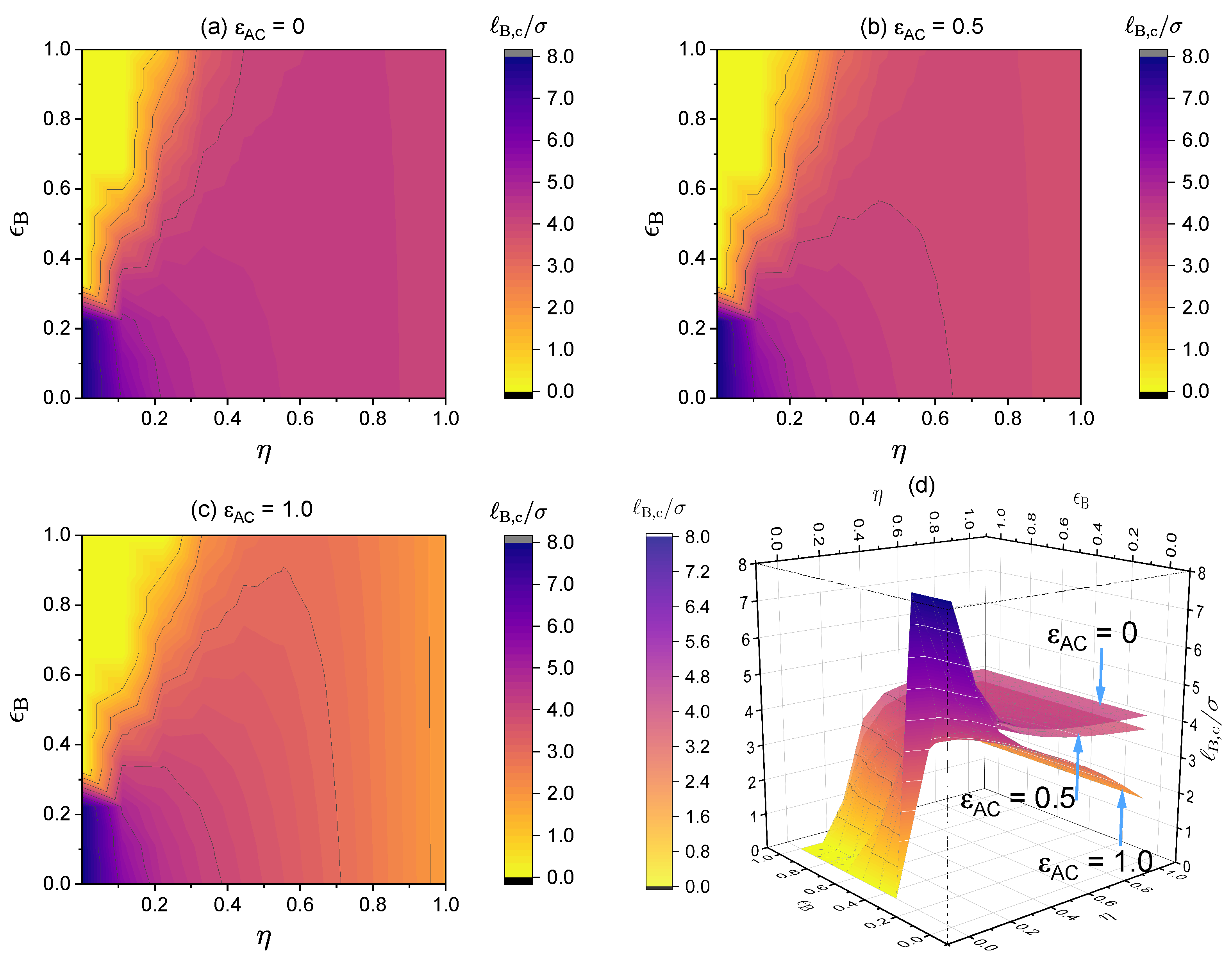
3.3. Effect of Local Short-Range Interactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BMCSL | Boublík–Mansoori–Carnahan–Starling–Leland |
| cDFT | classical Density Functional Theory |
| GUI App | Graphical User Interface Application |
| HPAM | partially hydrolyzed polyacrylamide |
| IUPAC | International Union of Pure and Applied Chemistry |
| LCST | Lower Critical Solution Temperature |
| LS | Liquid State |
| MSA | Mean-Spherical Approximation |
| PAA | Poly(acrylic acid) |
| PCEs | Polycarboxylate (ether/ester)-based Superplasticizers |
| PC-SAFT | Perturbed-Chain Statistical Associating Fluid Theory |
| PMAA | Poly(methacrylic acid) |
| TPT1 | first-order thermodynamic perturbation theory |
| TRUE | Transparent, Reproducible, Usable by others, and Extensible |
References
- Netz, R.R.; Andelman, D. Neutral and charged polymers at interfaces. Phys. Rep. 2003, 380, 1–95. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Theory and simulations of charged polymers: From solution properties to polymeric nanomaterials. Curr. Opin. Colloid Interface Sci. 2008, 13, 376–388. [Google Scholar] [CrossRef]
- Hickner, M.A. Ion-containing polymers: New energy & clean water. Mater. Today 2010, 13, 34–41. [Google Scholar] [CrossRef]
- Hess, M.; Jones, R.G.; Kahovec, J.; Kitayama, T.; Kratochvíl, P.; Kubisa, P.; Mormann, W.; Stepto, R.; Tabak, D.; Vohlídal, J.; et al. Terminology of polymers containing ionizable or ionic groups and of polymers containing ions (IUPAC Recommendations 2006). Pure Appl. Chem. 2006, 78, 2067–2074. [Google Scholar] [CrossRef]
- Kudaibergenov, S.E. Polyampholytes. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Polyelectrolytes: On the doorsteps of the second century. Polymer 2020, 202, 122714. [Google Scholar] [CrossRef]
- Hoagland, D. Polyelectrolytes. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003. [Google Scholar] [CrossRef]
- Mecerreyes, D. Polymeric ionic liquids: Broadening the properties and applications of polyelectrolytes. Prog. Polym. Sci. 2011, 36, 1629–1648. [Google Scholar] [CrossRef]
- Kocak, G.; Tuncer, C.; Bütün, V. pH-Responsive polymers. Polym. Chem. 2017, 8, 144–176. [Google Scholar] [CrossRef]
- Ofridam, F.; Tarhini, M.; Lebaz, N.; Gagniere, E.; Mangin, D.; Elaïssari, A. pH-sensitive polymers: Classification and some fine potential applications. Polym. Adv. Technol. 2021, 32, 1455–1484. [Google Scholar] [CrossRef]
- Rubinstein, M.; Papoian, G.A. Polyelectrolytes in biology and soft matter. Soft Matter 2012, 8, 9265–9267. [Google Scholar] [CrossRef]
- Kadajji, V.G.; Betageri, G.V. Water soluble polymers for pharmaceutical applications. Polymers 2011, 3, 1972–2009. [Google Scholar] [CrossRef]
- Plank, J.; Sakai, E.; Miao, C.; Yu, C.; Hong, J. Chemical admixtures—Chemistry, applications and their impact on concrete microstructure and durability. Cem. Concr. Res. 2015, 78, 81–99. [Google Scholar] [CrossRef]
- Kamcev, J.; Freeman, B.D. Charged polymer membranes for environmental/energy applications. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 111–133. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Kishimoto, S.; Nakamura, S.; Sato, Y.; Hattori, H. Polyelectrolyte complexes of natural polymers and their biomedical applications. Polymers 2019, 11, 672. [Google Scholar] [CrossRef]
- Sing, C.E.; Perry, S.L. Recent progress in the science of complex coacervation. Soft Matter 2020, 16, 2885–2914. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, N.S.; Haq, M.; Al-Shehri, D.; Rahaman, M.M.; Keshavarz, A.; Hossain, S. Comparative study of green and synthetic polymers for enhanced oil recovery. Polymers 2020, 12, 2429. [Google Scholar] [CrossRef]
- Mahajan, S.; Yadav, H.; Rellegadla, S.; Agrawal, A. Polymers for enhanced oil recovery: Fundamentals and selection criteria revisited. Appl. Microbiol. Biotechnol. 2021, 105, 8073–8090. [Google Scholar] [CrossRef]
- Gbadamosi, A.; Patil, S.; Kamal, M.S.; Adewunmi, A.A.; Yusuff, A.S.; Agi, A.; Oseh, J. Application of Polymers for Chemical Enhanced Oil Recovery: A Review. Polymers 2022, 14, 1433. [Google Scholar] [CrossRef]
- Kamal, M.S.; Hussein, I.A.; Sultan, A.S.; von Solms, N. Application of various water soluble polymers in gas hydrate inhibition. Renew. Sustain. Energy Rev. 2016, 60, 206–225. [Google Scholar] [CrossRef]
- Williams, P.A. Handbook of Industrial Water Soluble Polymers; Wiley-Blackwell: Oxford, UK, 2007. [Google Scholar]
- Jia, P.; Yang, Q.; Gong, Y.; Zhao, J. Dynamic exchange of counterions of polystyrene sulfonate. J. Chem. Phys. 2012, 136, 084904. [Google Scholar] [CrossRef]
- Shi, Y.; Peng, H.; Yang, J.; Zhao, J. Counterion binding dynamics of a polyelectrolyte. Macromolecules 2021, 54, 4926–4933. [Google Scholar] [CrossRef]
- Carrillo, J.M.Y.; Dobrynin, A.V. Polyelectrolytes in salt solutions: Molecular dynamics simulations. Macromolecules 2011, 44, 5798–5816. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. PolymerPhysics; Oxford University Press: New York, NY, USA, 2003; Volume 23. [Google Scholar]
- Teraoka, I. Polymer Solutions: An Introduction to Physical Properties; John Wiley & Sons, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Knychała, P.; Timachova, K.; Banaszak, M.; Balsara, N.P. 50th anniversary perspective: Phase behavior of polymer solutions and blends. Macromolecules 2017, 50, 3051–3065. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1941, 9, 660. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1942, 10, 51–61. [Google Scholar] [CrossRef]
- Huggins, M.L. Some properties of solutions of long-chain compounds. J. Phys. Chem. 1942, 46, 151–158. [Google Scholar] [CrossRef]
- Shultz, A.; Flory, P. Phase equilibria in polymer—Solvent systems1,2. J. Am. Chem. Soc. 1952, 74, 4760–4767. [Google Scholar] [CrossRef]
- Work, W.J.; Horie, K.; Hess, M.; Stepto, R.F.T. Definition of terms related to polymer blends, composites, and multiphase polymeric materials (IUPAC Recommendations 2004). Pure Appl. Chem. 2004, 76, 1985–2007. [Google Scholar] [CrossRef]
- Michaeli, I.; Overbeek, J.T.G.; Voorn, M.J. Phase separation of polyelectrolyte solutions. J. Polym. Sci. 1957, 23, 443–450. [Google Scholar] [CrossRef]
- Overbeek, J.T.G.; Voorn, M. Phase separation in polyelectrolyte solutions. Theory of complex coacervation. J. Cell. Comp. Physiol. 1957, 49, 7–26. [Google Scholar] [CrossRef]
- Voorn, M.J. Phase separation in polymer solutions. In Fortschritte Der Hochpolymeren-Forschung; Springer: Berlin/Heidelberg, Germany, 1959; pp. 192–233. [Google Scholar]
- Overbeek, J.T.G. Polyelectrolytes, past, present and future. In Macromolecular Chemistry–11; Pergamon Press: Oxford, UK, 1977; pp. 91–101. [Google Scholar] [CrossRef]
- Veis, A.; Aranyi, C. Phase separation in polyelectrolyte systems. I. Complex coacervates of gelatin. J. Phys. Chem. 1960, 64, 1203–1210. [Google Scholar] [CrossRef]
- Veis, A. Phase separation in polyelectrolyte solutions. II. Interaction effects. J. Phys. Chem. 1961, 65, 1798–1803. [Google Scholar] [CrossRef]
- Veis, A. Phase separation in polyelectrolyte systems. III. Effect of aggregation and molecular weight heterogeneity. J. Phys. Chem. 1963, 67, 1960–1964. [Google Scholar] [CrossRef]
- Veis, A. A review of the early development of the thermodynamics of the complex coacervation phase separation. Adv. Colloid Interface Sci. 2011, 167, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, H.; Hu, Y.; Prausnitz, J.M. A molecular-thermodynamic model for polyelectrolyte solutions. J. Chem. Phys. 1998, 108, 780–784. [Google Scholar] [CrossRef]
- Jiang, J.; Blum, L.; Bernard, O.; Prausnitz, J.M. Thermodynamic properties and phase equilibria of charged hard sphere chain model for polyelectrolyte solutions. Mol. Phys. 2001, 99, 1121–1128. [Google Scholar] [CrossRef]
- Jiang, J.; Feng, J.; Liu, H.; Hu, Y. Phase behavior of polyampholytes from charged hard-sphere chain model. J. Chem. Phys. 2006, 124, 144908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Alsaifi, N.M.; Wu, J.; Wang, Z.G. Salting-out and salting-in of polyelectrolyte solutions: A liquid-state theory study. Macromolecules 2016, 49, 9720–9730. [Google Scholar] [CrossRef]
- Boublík, T. Hard-sphere equation of state. J. Chem. Phys. 1970, 53, 471–472. [Google Scholar] [CrossRef]
- Mansoori, G.A.; Carnahan, N.F.; Starling, K.E.; Leland, T.W., Jr. Equilibrium thermodynamic properties of the mixture of hard spheres. J. Chem. Phys. 1971, 54, 1523–1525. [Google Scholar] [CrossRef]
- Waisman, E.; Lebowitz, J.L. Exact solution of an integral equation for the structure of a primitive model of electrolytes. J. Chem. Phys. 1970, 52, 4307–4309. [Google Scholar] [CrossRef]
- Waisman, E.; Lebowitz, J.L. Mean spherical model integral equation for charged hard spheres I. Method of solution. J. Chem. Phys. 1972, 56, 3086–3093. [Google Scholar] [CrossRef]
- Waisman, E.; Lebowitz, J.L. Mean spherical model integral equation for charged hard spheres. II. Results. J. Chem. Phys. 1972, 56, 3093–3099. [Google Scholar] [CrossRef]
- Wertheim, M. Fluids with highly directional attractive forces. I. Statistical thermodynamics. J. Stat. Phys. 1984, 35, 19–34. [Google Scholar] [CrossRef]
- Wertheim, M.S. Fluids with highly directional attractive forces. II. Thermodynamic perturbation theory and integral equations. J. Stat. Phys. 1984, 35, 35–47. [Google Scholar] [CrossRef]
- Wertheim, M.S. Fluids with highly directional attractive forces. III. Multiple attraction sites. J. Stat. Phys. 1986, 42, 459–476. [Google Scholar] [CrossRef]
- Wertheim, M.S. Fluids with highly directional attractive forces. IV. Equilibrium polymerization. J. Stat. Phys. 1986, 42, 477–492. [Google Scholar] [CrossRef]
- Wertheim, M. Thermodynamic perturbation theory of polymerization. J. Chem. Phys. 1987, 87, 7323–7331. [Google Scholar] [CrossRef]
- Zhang, P.; Shen, K.; Alsaifi, N.M.; Wang, Z.G. Salt partitioning in complex coacervation of symmetric polyelectrolytes. Macromolecules 2018, 51, 5586–5593. [Google Scholar] [CrossRef]
- Ylitalo, A.S.; Balzer, C.; Zhang, P.; Wang, Z.G. Electrostatic Correlations and Temperature-Dependent Dielectric Constant Can Model LCST in Polyelectrolyte Complex Coacervation. Macromolecules 2021, 54, 11326–11337. [Google Scholar] [CrossRef]
- Zhang, P.; Alsaifi, N.M.; Wu, J.; Wang, Z.G. Polyelectrolyte complex coacervation: Effects of concentration asymmetry. J. Chem. Phys. 2018, 149, 163303. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J. Density functional theory for polyelectrolytes near oppositely charged surfaces. Phys. Rev. Lett. 2006, 96, 048302. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Li, Z.; Wu, J. Structure and swelling of grafted polyelectrolytes: Predictions from a nonlocal density functional theory. Macromolecules 2007, 40, 334–343. [Google Scholar] [CrossRef]
- Jiang, J.; Ginzburg, V.V.; Wang, Z.G. Density functional theory for charged fluids. Soft Matter 2018, 14, 5878–5887. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, X.; Zhao, S. Complex coacervation in asymmetric solutions of polycation and polyanion. Langmuir 2019, 35, 15267–15274. [Google Scholar] [CrossRef]
- Xu, X.; Shi, H.; Wang, F. Near-critical phase behavior in polyelectrolyte solutions: Effect of charge fluctuations. J. Phys. Chem. B 2020, 124, 4203–4210. [Google Scholar] [CrossRef]
- Xu, X.; Qiu, Q.; Lu, C.; Zhao, S. Responsive Properties of Grafted Polyanion Chains: Effects of Dispersion Interaction and Salt. J. Chem. Eng. Data 2020, 65, 5708–5717. [Google Scholar] [CrossRef]
- Qiu, G.; Qiu, Q.; Qing, L.; Zhou, J.; Xu, X.; Zhao, S. Effects of Polyelectrolyte Surface Coating on the Energy Storage Performance in Supercapacitors. J. Phys. Chem. C 2022, 126, 19. [Google Scholar] [CrossRef]
- Jiang, J. Software Package: An Advanced Theoretical Tool for Inhomogeneous Fluids (Atif). Chin. J. Polym. Sci. 2022, 40, 220–230. [Google Scholar] [CrossRef]
- Muthukumar, M. 50th anniversary perspective: A perspective on polyelectrolyte solutions. Macromolecules 2017, 50, 9528–9560. [Google Scholar] [CrossRef]
- Vlachy, V.; Hribar-Lee, B.; Kalyuzhnyi, Y.V.; Dill, K.A. Short-range interactions: From simple ions to polyelectrolyte solutions. Curr. Opin. Colloid Interface Sci. 2004, 9, 128–132. [Google Scholar] [CrossRef]
- Bishop, K.J.; Wilmer, C.E.; Soh, S.; Grzybowski, B.A. Nanoscale forces and their uses in self-assembly. Small 2009, 5, 1600–1630. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, E.; Abe, K. Interactions Between Macromolecules in Solution and Intermacromolecular Complexes. In Interactions Between Macromolecules in Solution and Intermacromolecular Complexes, Advances in Polymer Science; Tsuchida, E., Abe, K., Eds.; Springer: Berlin/Heidelberg, Germany, 1982; Volume 45, pp. 1–119. [Google Scholar] [CrossRef]
- Batys, P.; Kivistö, S.; Lalwani, S.M.; Lutkenhaus, J.L.; Sammalkorpi, M. Comparing water-mediated hydrogen-bonding in different polyelectrolyte complexes. Soft Matter 2019, 15, 7823–7831. [Google Scholar] [CrossRef] [PubMed]
- Boas, M.; Vasilyev, G.; Vilensky, R.; Cohen, Y.; Zussman, E. Structure and rheology of polyelectrolyte complexes in the presence of a hydrogen-bonded co-solvent. Polymers 2019, 11, 1053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Batys, P.; O’Neal, J.T.; Li, F.; Sammalkorpi, M.; Lutkenhaus, J.L. Molecular origin of the glass transition in polyelectrolyte assemblies. ACS Cent. Sci. 2018, 4, 638–644. [Google Scholar] [CrossRef]
- Jha, P.K.; Desai, P.S.; Li, J.; Larson, R.G. pH and salt effects on the associative phase separation of oppositely charged polyelectrolytes. Polymers 2014, 6, 1414–1436. [Google Scholar] [CrossRef]
- Yamazoe, K.; Higaki, Y.; Inutsuka, Y.; Miyawaki, J.; Cui, Y.T.; Takahara, A.; Harada, Y. Enhancement of the hydrogen-bonding network of water confined in a polyelectrolyte brush. Langmuir 2017, 33, 3954–3959. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Rubinstein, M. Hydrophobic polyelectrolytes. Macromolecules 1999, 32, 915–922. [Google Scholar] [CrossRef]
- Popa-Nita, S.; Rochas, C.; David, L.; Domard, A. Structure of natural polyelectrolyte solutions: Role of the hydrophilic/hydrophobic interaction balance. Langmuir 2009, 25, 6460–6468. [Google Scholar] [CrossRef]
- Khan, N.; Brettmann, B. Intermolecular interactions in polyelectrolyte and surfactant complexes in solution. Polymers 2018, 11, 51. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, W.; Yu, J.; Li, Y.; Zhao, C. Recent development of pH-responsive polymers for cancer nanomedicine. Molecules 2018, 24, 4. [Google Scholar] [CrossRef]
- Ristroph, K.D.; Prud’homme, R.K. Hydrophobic ion pairing: Encapsulating small molecules, peptides, and proteins into nanocarriers. Nanoscale Adv. 2019, 1, 4207–4237. [Google Scholar] [CrossRef]
- Afolabi, R.O.; Oluyemi, G.F.; Officer, S.; Ugwu, J.O. Hydrophobically associating polymers for enhanced oil recovery–Part A: A review on the effects of some key reservoir conditions. J. Pet. Sci. Eng. 2019, 180, 681–698. [Google Scholar] [CrossRef]
- Gregory, K.P.; Elliott, G.R.; Robertson, H.; Kumar, A.; Wanless, E.J.; Webber, G.B.; Craig, V.S.; Andersson, G.G.; Page, A.J. Understanding specific ion effects and the Hofmeister series. Phys. Chem. Chem. Phys. 2022, 24, 12682–12718. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, G. Ionic effects on synthetic polymers: From solutions to brushes and gels. Soft Matter 2020, 16, 4087–4104. [Google Scholar] [CrossRef]
- Smiatek, J. Theoretical and computational insight into solvent and specific ion effects for polyelectrolytes: The importance of local molecular interactions. Molecules 2020, 25, 1661. [Google Scholar] [CrossRef]
- Moghaddam, S.Z.; Thormann, E. The Hofmeister series: Specific ion effects in aqueous polymer solutions. J. Colloid Interface Sci. 2019, 555, 615–635. [Google Scholar] [CrossRef]
- Volodkin, D.; von Klitzing, R. Competing mechanisms in polyelectrolyte multilayer formation and swelling: Polycation–polyanion pairing vs. polyelectrolyte–ion pairing. Curr. Opin. Colloid Interface Sci. 2014, 19, 25–31. [Google Scholar] [CrossRef]
- Van Der Vegt, N.F.; Haldrup, K.; Roke, S.; Zheng, J.; Lund, M.; Bakker, H.J. Water-mediated ion pairing: Occurrence and relevance. Chem. Rev. 2016, 116, 7626–7641. [Google Scholar] [CrossRef]
- Collins, K.D. Why continuum electrostatics theories cannot explain biological structure, polyelectrolytes or ionic strength effects in ion–protein interactions. Biophys. Chem. 2012, 167, 43–59. [Google Scholar] [CrossRef]
- Salis, A.; Ninham, B.W. Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited. Chem. Soc. Rev. 2014, 43, 7358–7377. [Google Scholar] [CrossRef]
- Parsons, D.F.; Boström, M.; Nostro, P.L.; Ninham, B.W. Hofmeister effects: Interplay of hydration, nonelectrostatic potentials, and ion size. Phys. Chem. Chem. Phys. 2011, 13, 12352–12367. [Google Scholar] [CrossRef] [PubMed]
- Travesset, A.; Vangaveti, S. Electrostatic correlations at the Stern layer: Physics or chemistry? J. Chem. Phys. 2009, 131, 11B608. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Xu, X.; Wang, Y. Phase Behavior of Partially Charged Polyelectrolyte Solutions with Salt: A Theoretical Study. Macromol. Theory Simulations 2021, 30, 2000098. [Google Scholar] [CrossRef]
- Peng, S.; Wu, C. Light scattering study of the formation and structure of partially hydrolyzed poly (acrylamide)/calcium (II) complexes. Macromolecules 1999, 32, 585–589. [Google Scholar] [CrossRef]
- Molnar, F.; Rieger, J. “Like-charge attraction” between anionic polyelectrolytes: Molecular dynamics simulations. Langmuir 2005, 21, 786–789. [Google Scholar] [CrossRef]
- Turesson, M.; Labbez, C.; Nonat, A. Calcium mediated polyelectrolyte adsorption on like-charged surfaces. Langmuir 2011, 27, 13572–13581. [Google Scholar] [CrossRef]
- Nap, R.J.; Park, S.H.; Szleifer, I. Competitive calcium ion binding to end-tethered weak polyelectrolytes. Soft Matter 2018, 14, 2365–2378. [Google Scholar] [CrossRef]
- Nap, R.J.; Szleifer, I. Effect of calcium ions on the interactions between surfaces end-grafted with weak polyelectrolytes. J. Chem. Phys. 2018, 149, 163309. [Google Scholar] [CrossRef]
- Mehandzhiyski, A.Y.; Riccardi, E.; van Erp, T.S.; Trinh, T.T.; Grimes, B.A. Ab initio molecular dynamics study on the interactions between carboxylate ions and metal ions in water. J. Phys. Chem. B 2015, 119, 10710–10719. [Google Scholar] [CrossRef]
- Chen, S.S.; Kreglewski, A. Applications of the augmented van der Waals theory of fluids.: I. Pure fluids. Ber. Bunsenges. Phys. Chem. 1977, 81, 1048–1052. [Google Scholar] [CrossRef]
- Gross, J.; Sadowski, G. Perturbed-chain SAFT: An equation of state based on a perturbation theory for chain molecules. Ind. Eng. Chem. Res. 2001, 40, 1244–1260. [Google Scholar] [CrossRef]
- Müller-Plathe, F. Coarse-graining in polymer simulation: From the atomistic to the mesoscopic scale and back. ChemPhysChem 2002, 3, 754–769. [Google Scholar] [CrossRef]
- Kotelyanskii, M.; Theodorou, D. Simulation Methods for Polymers; Marcel Dekker, Inc.: New York, NY, USA, 2004. [Google Scholar]
- Gartner, T.E., III; Jayaraman, A. Modeling and simulations of polymers: A roadmap. Macromolecules 2019, 52, 755–786. [Google Scholar] [CrossRef]
- Gong, P.; Genzer, J.; Szleifer, I. Phase behavior and charge regulation of weak polyelectrolyte grafted layers. Phys. Rev. Lett. 2007, 98, 018302. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Avni, Y.; Andelman, D.; Podgornik, R. Phase separation of polyelectrolytes: The effect of charge regulation. J. Phys. Chem. B 2021, 125, 7863–7870. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Bera, A.; Ojha, K.; Mandal, A. Effects of alkali, salts, and surfactant on rheological behavior of partially hydrolyzed polyacrylamide solutions. J. Chem. Eng. Data 2010, 55, 4315–4322. [Google Scholar] [CrossRef]
- Kamal, M.S.; Sultan, A.S.; Al-Mubaiyedh, U.A.; Hussein, I.A. Review on polymer flooding: Rheology, adsorption, stability, and field applications of various polymer systems. Polym. Rev. 2015, 55, 491–530. [Google Scholar] [CrossRef]
- Quezada, G.R.; Saavedra, J.H.; Rozas, R.E.; Toledo, P.G. Molecular dynamics simulations of the conformation and diffusion of partially hydrolyzed polyacrylamide in highly saline solutions. Chem. Eng. Sci. 2020, 214, 115366. [Google Scholar] [CrossRef]
- Guenoun, P.; Davis, H.T.; Tirrell, M.; Mays, J.W. Aqueous micellar solutions of hydrophobically modified polyelectrolytes. Macromolecules 1996, 29, 3965–3969. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Rubinstein, M. Hydrophobically modified polyelectrolytes in dilute salt-free solutions. Macromolecules 2000, 33, 8097–8105. [Google Scholar] [CrossRef]
- Feng, Y.; Grassl, B.; Billon, L.; Khoukh, A.; François, J. Effects of NaCl on steady rheological behaviour in aqueous solutions of hydrophobically modified polyacrylamide and its partially hydrolyzed analogues prepared by post-modification. Polym. Int. 2002, 51, 939–947. [Google Scholar] [CrossRef]
- Shu, X.; Ran, Q.; Liu, J.; Zhao, H.; Zhang, Q.; Wang, X.; Yang, Y.; Liu, J. Tailoring the solution conformation of polycarboxylate superplasticizer toward the improvement of dispersing performance in cement paste. Constr. Build. Mater. 2016, 116, 289–298. [Google Scholar] [CrossRef]
- Shu, X.; Zhao, H.; Wang, X.; Zhang, Q.; Yang, Y.; Ran, Q.; Liu, J. Effect of hydrophobic units of polycarboxylate superplasticizer on the flow behavior of cement paste. J. Dispers. Sci. Technol. 2017, 38, 256–264. [Google Scholar] [CrossRef]
- Fernandez, D.P.; Goodwin, A.R.H.; Lemmon, E.W.; Levelt Sengers, J.M.H.; Williams, R.C. A formulation for the static permittivity of water and steam at temperatures from 238 K to 873 K at pressures up to 1200 MPa, including derivatives and Debye–Hückel coefficients. J. Phys. Chem. Ref. Data 1997, 26, 1125–1166. [Google Scholar] [CrossRef]
- Malmberg, C.; Maryott, A. Dielectric constant of water from 0 to 100 C. J. Res. Natl. Bur. Stand. 1956, 56, 1–8. [Google Scholar] [CrossRef]
- Xu, X.; Cristancho, D.E.; Costeux, S.; Wang, Z.G. Density-functional theory for polymer-carbon dioxide mixtures: A perturbed-chain SAFT approach. J. Chem. Phys. 2012, 137, 054902. [Google Scholar] [CrossRef]
- Blum, L. Mean spherical model for asymmetric electrolytes: I. Method of solution. Mol. Phys. 1975, 30, 1529–1535. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Z.G. Interfacial Structure and Tension of Polyelectrolyte Complex Coacervates. Macromolecules 2021, 54, 10994–11007. [Google Scholar] [CrossRef]
- Jahan, M.; Uline, M.J. Quantifying Mg2+ Binding to ssDNA Oligomers: A Self-Consistent Field Theory Study at Varying Ionic Strengths and Grafting Densities. Polymers 2018, 10, 1403. [Google Scholar] [CrossRef]
- Tagliazucchi, M.; De La Cruz, M.O.; Szleifer, I. Self-organization of grafted polyelectrolyte layers via the coupling of chemical equilibrium and physical interactions. Proc. Natl. Acad. Sci. USA 2010, 107, 5300–5305. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes 3rd Edition: The Art of Scientific Computing, 3rd ed.; Cambridge University Press: Cambridge, UK, 2007; pp. 477–483. [Google Scholar]
- Thompson, M.W.; Gilmer, J.B.; Matsumoto, R.A.; Quach, C.D.; Shamaprasad, P.; Yang, A.H.; Iacovella, C.R.; McCabe, C.; Cummings, P.T. Towards molecular simulations that are transparent, reproducible, usable by others, and extensible (TRUE). Mol. Phys. 2020, 118, e1742938. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, M. Phase diagram of polyelectrolyte solutions: Weak polymer effect. Macromolecules 2002, 35, 9142–9145. [Google Scholar] [CrossRef]
- Ermoshkin, A.; Olvera de La Cruz, M. A modified random phase approximation of polyelectrolyte solutions. Macromolecules 2003, 36, 7824–7832. [Google Scholar] [CrossRef]
- Orkoulas, G.; Kumar, S.K.; Panagiotopoulos, A.Z. Monte Carlo study of coulombic criticality in polyelectrolytes. Phys. Rev. Lett. 2003, 90, 048303. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Solveyra, E.; Nap, R.J.; Huang, K.; Szleifer, I. Theoretical modeling of chemical equilibrium in weak polyelectrolyte layers on curved nanosystems. Polymers 2020, 12, 2282. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | A | B | C | D |
|---|---|---|---|---|
| Number density | ||||
| Valence |
| Notions | Definition |
|---|---|
| max | Upper bound of the range of the Bjerrum length |
| min | Lower bound of the range of the Bjerrum length |
| step | Step length (bin size) of the Bjerrum length |
| Total number of (A+B) segments of the polymer chain | |
| Number of bond connections between charged segments (A) | |
| Charge fraction of the polymer chain | |
| Strength of dispersion interaction between A and C | |
| Strength of dispersion interaction between A and A | |
| Strength of dispersion interaction between B and B | |
| Valence of individual ionized groups of the polymer | |
| Valence of counterions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Qiu, Q.; Yedilbayeva, A.; Kairula, D.; Dai, L. Phase Behavior of Ion-Containing Polymers in Polar Solvents: Predictions from a Liquid-State Theory with Local Short-Range Interactions. Polymers 2022, 14, 4421. https://doi.org/10.3390/polym14204421
Wang Y, Qiu Q, Yedilbayeva A, Kairula D, Dai L. Phase Behavior of Ion-Containing Polymers in Polar Solvents: Predictions from a Liquid-State Theory with Local Short-Range Interactions. Polymers. 2022; 14(20):4421. https://doi.org/10.3390/polym14204421
Chicago/Turabian StyleWang, Yanwei, Qiyuan Qiu, Arailym Yedilbayeva, Diana Kairula, and Liang Dai. 2022. "Phase Behavior of Ion-Containing Polymers in Polar Solvents: Predictions from a Liquid-State Theory with Local Short-Range Interactions" Polymers 14, no. 20: 4421. https://doi.org/10.3390/polym14204421
APA StyleWang, Y., Qiu, Q., Yedilbayeva, A., Kairula, D., & Dai, L. (2022). Phase Behavior of Ion-Containing Polymers in Polar Solvents: Predictions from a Liquid-State Theory with Local Short-Range Interactions. Polymers, 14(20), 4421. https://doi.org/10.3390/polym14204421