
Molecular Targets for Antibody-Based Anti-Biofilm Therapy in Infective Endocarditis


Abstract
1. Introduction
1.1. The Background of IE and Its Modern Epidemiology
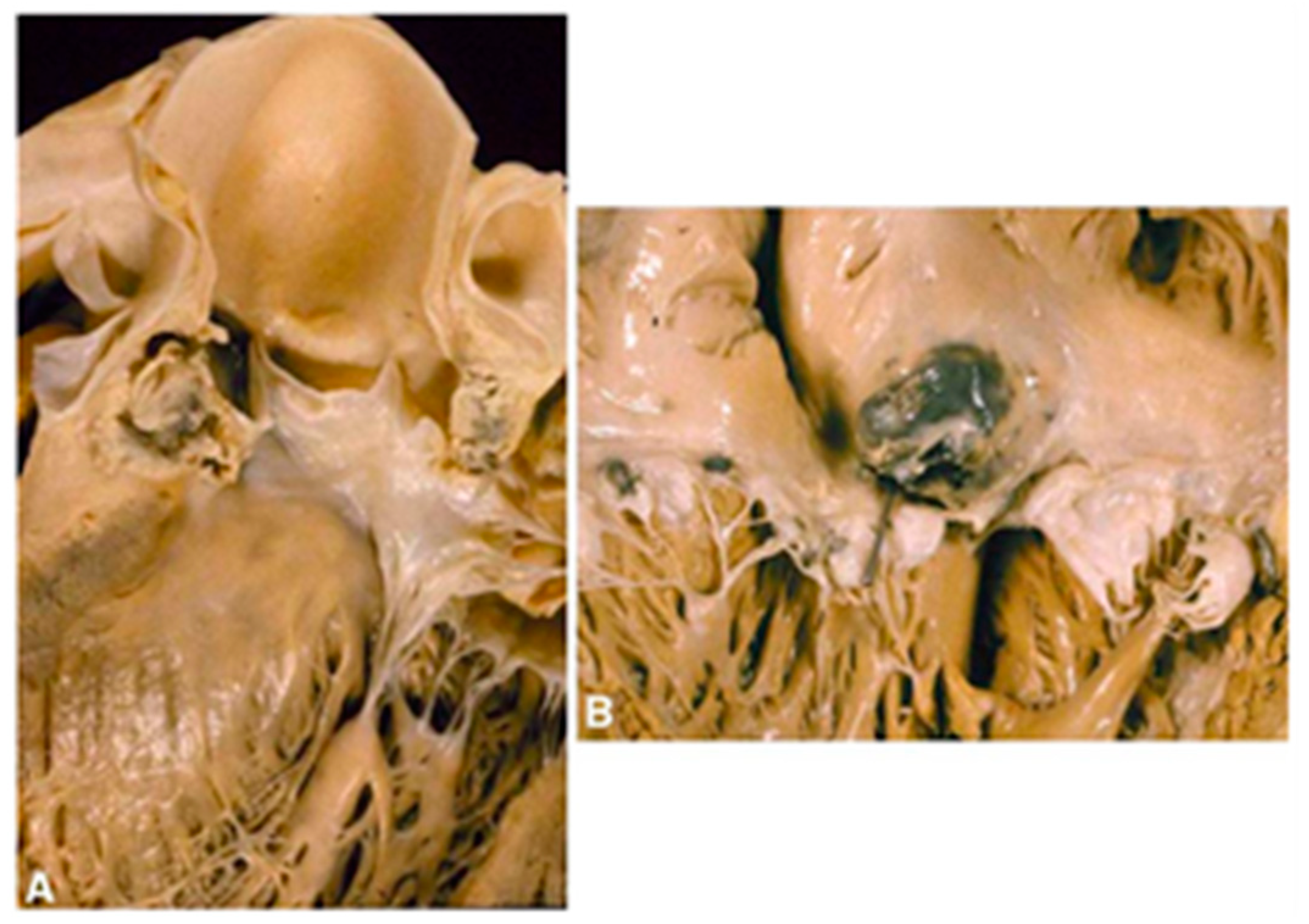
1.2. The Clinical Pathology of IE
1.3. The Challenges in Antibiotic Treatment and Prospective Anti-Biofilm Strategies
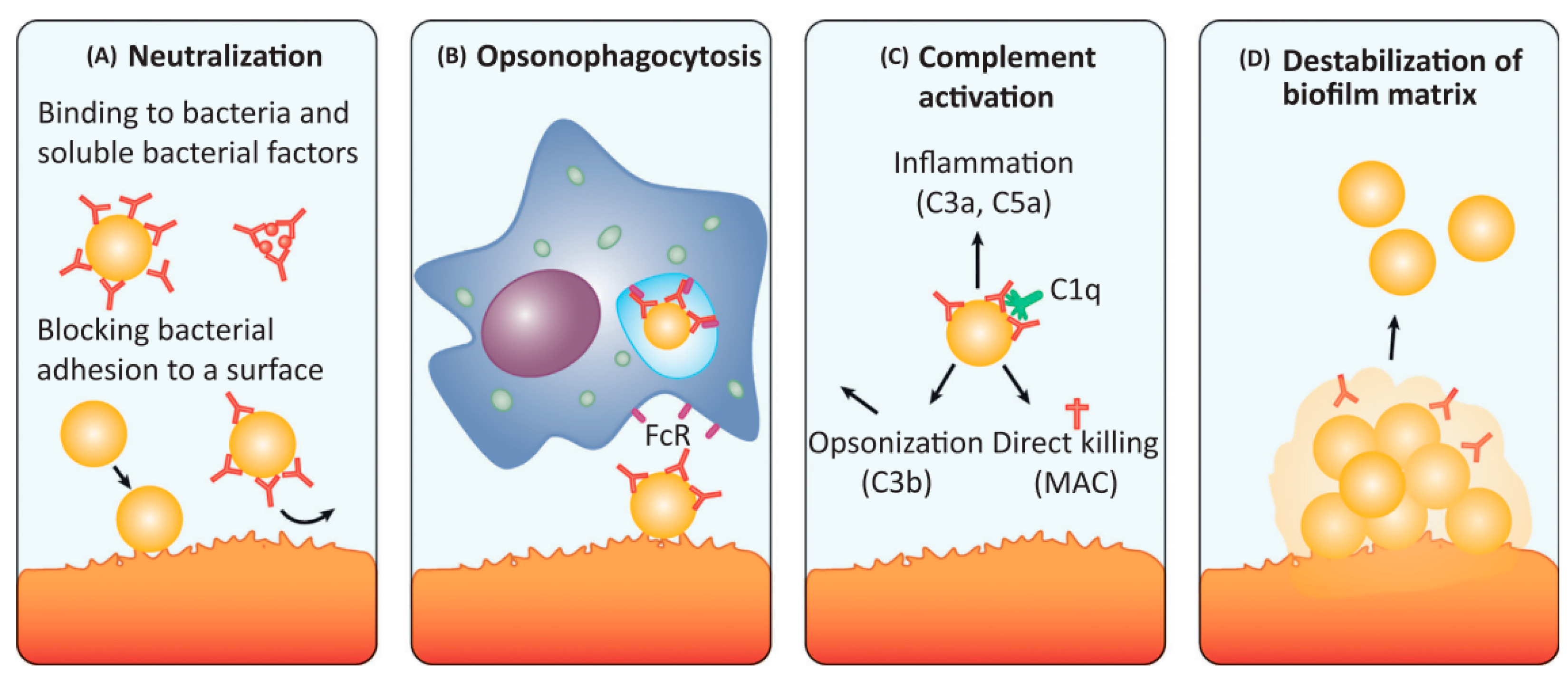
1.4. Antibodies as a Promising Approach for Anti-Biofilm IE Treatment
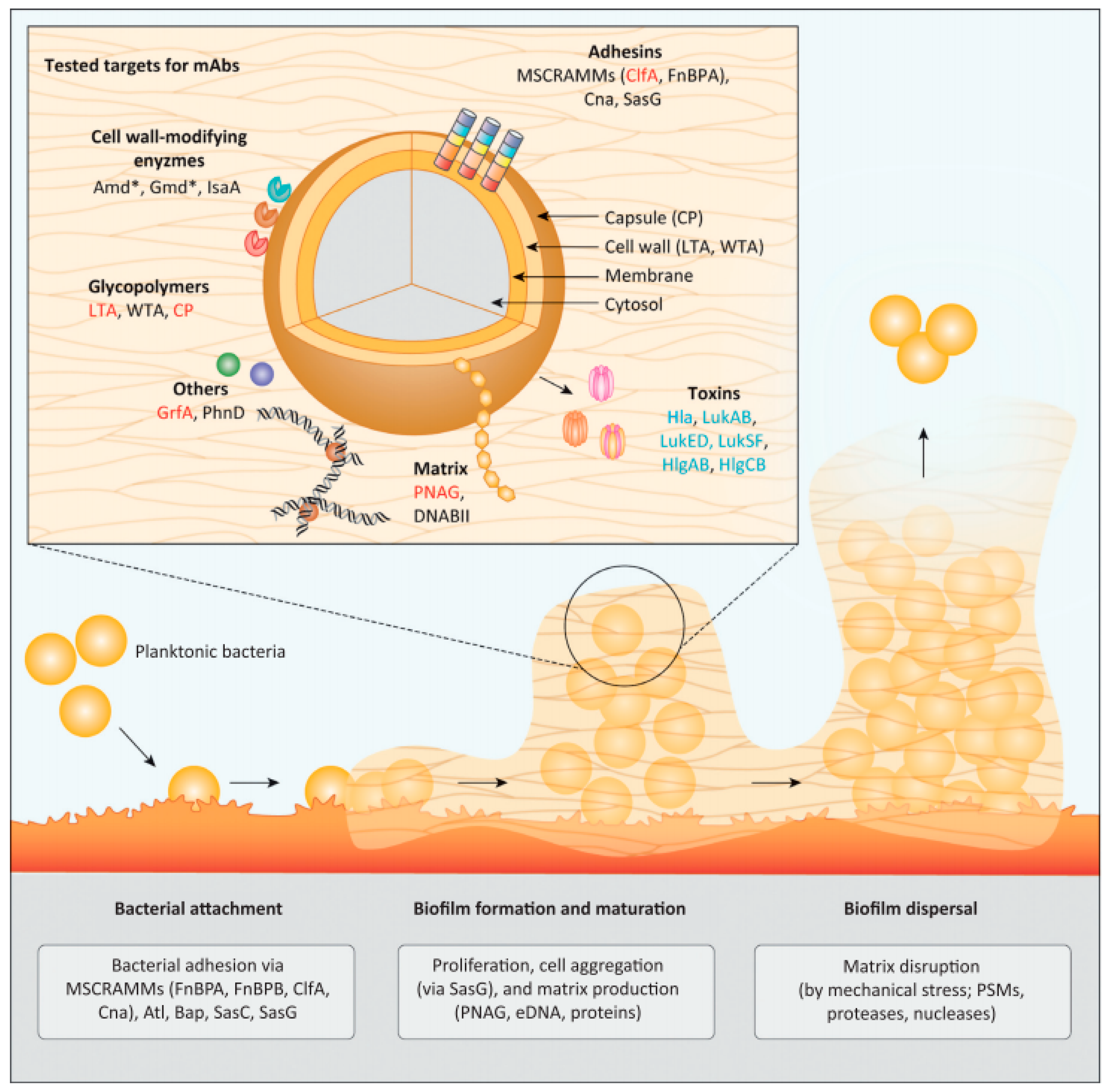
2. Molecular Targets for Monoclonal Antibodies Targeting Staphylococcus Biofilms
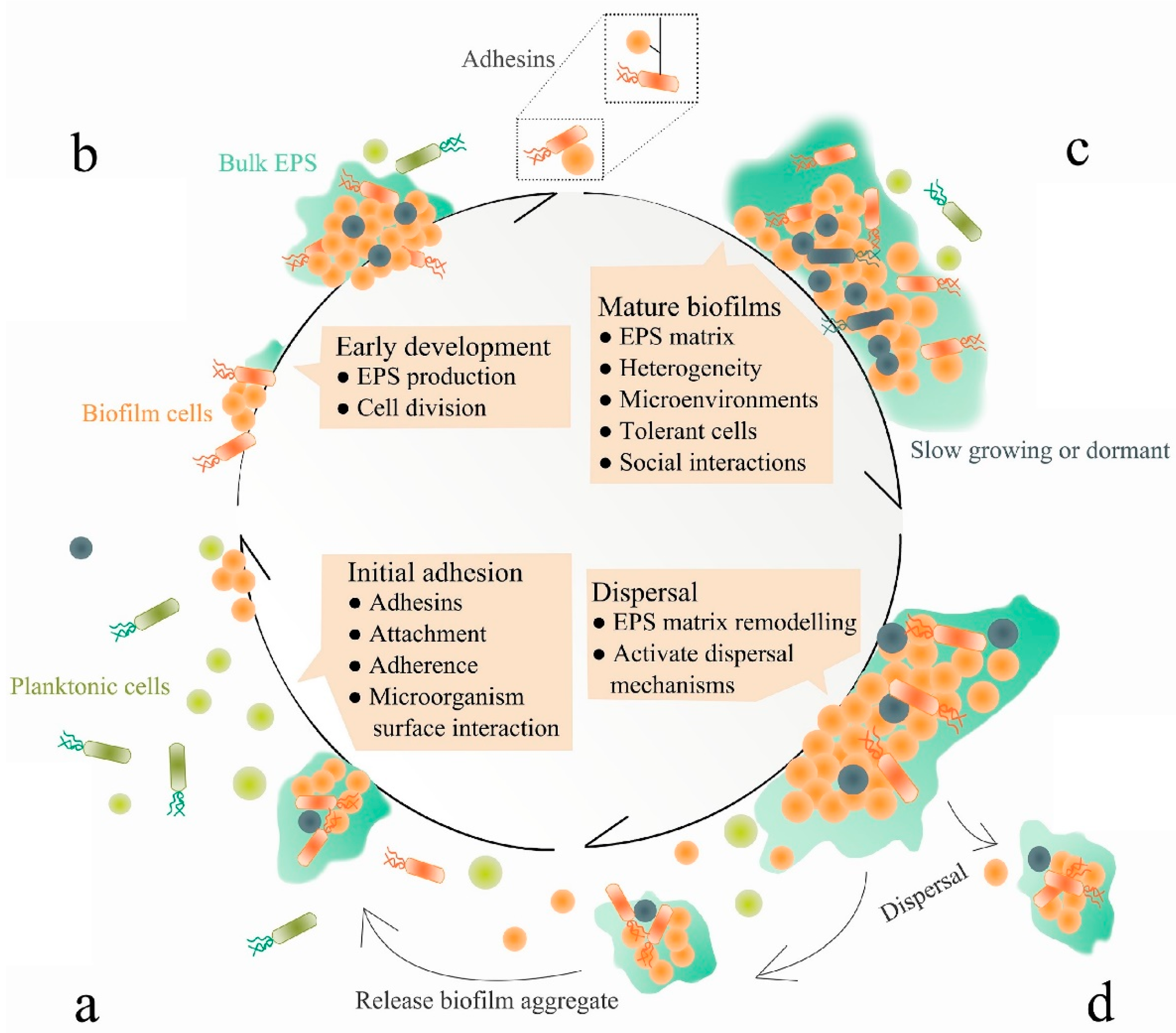
3. Anti-Biofilm Strategies
3.1. Inhibition of Bacterial Attachment
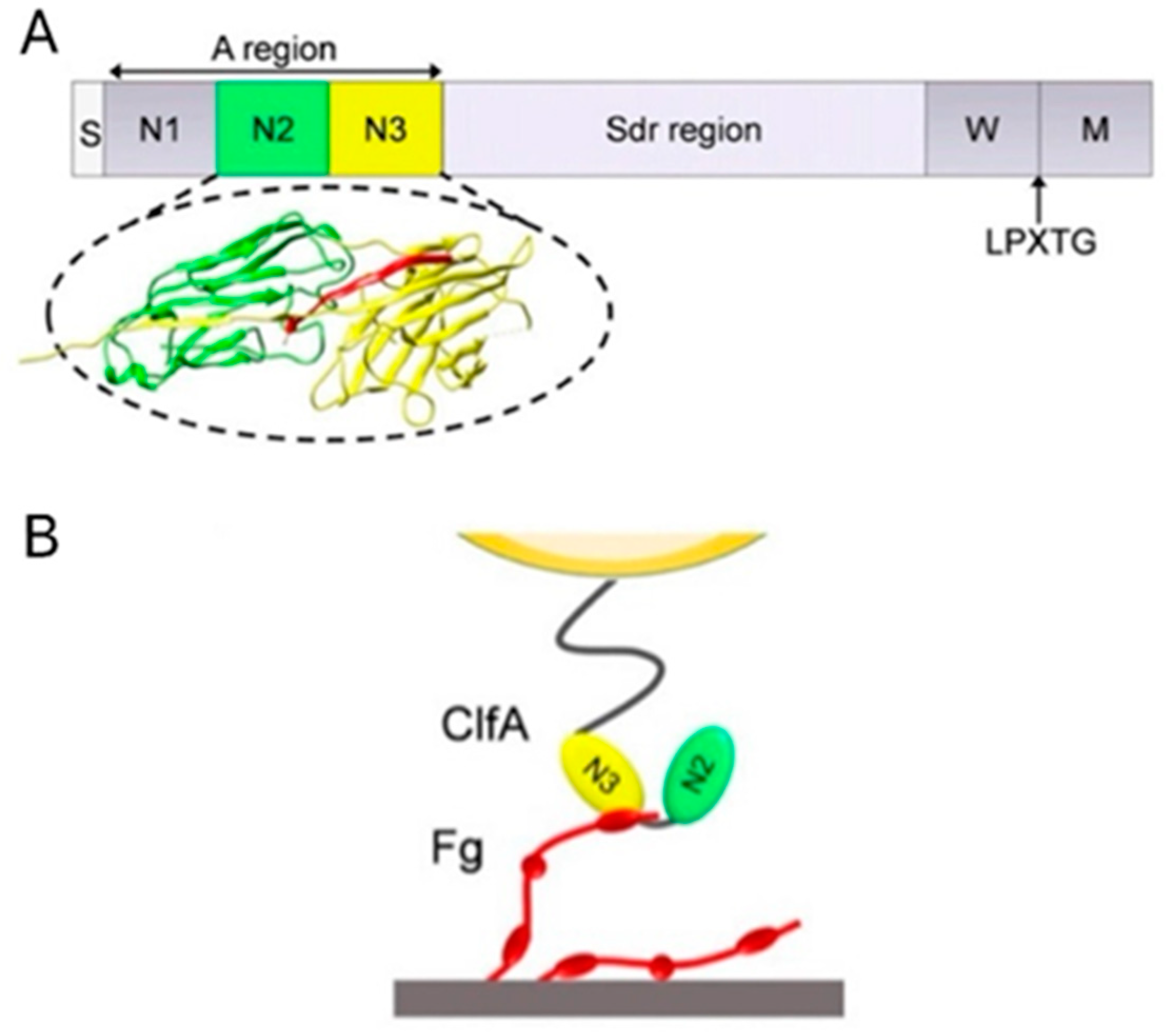
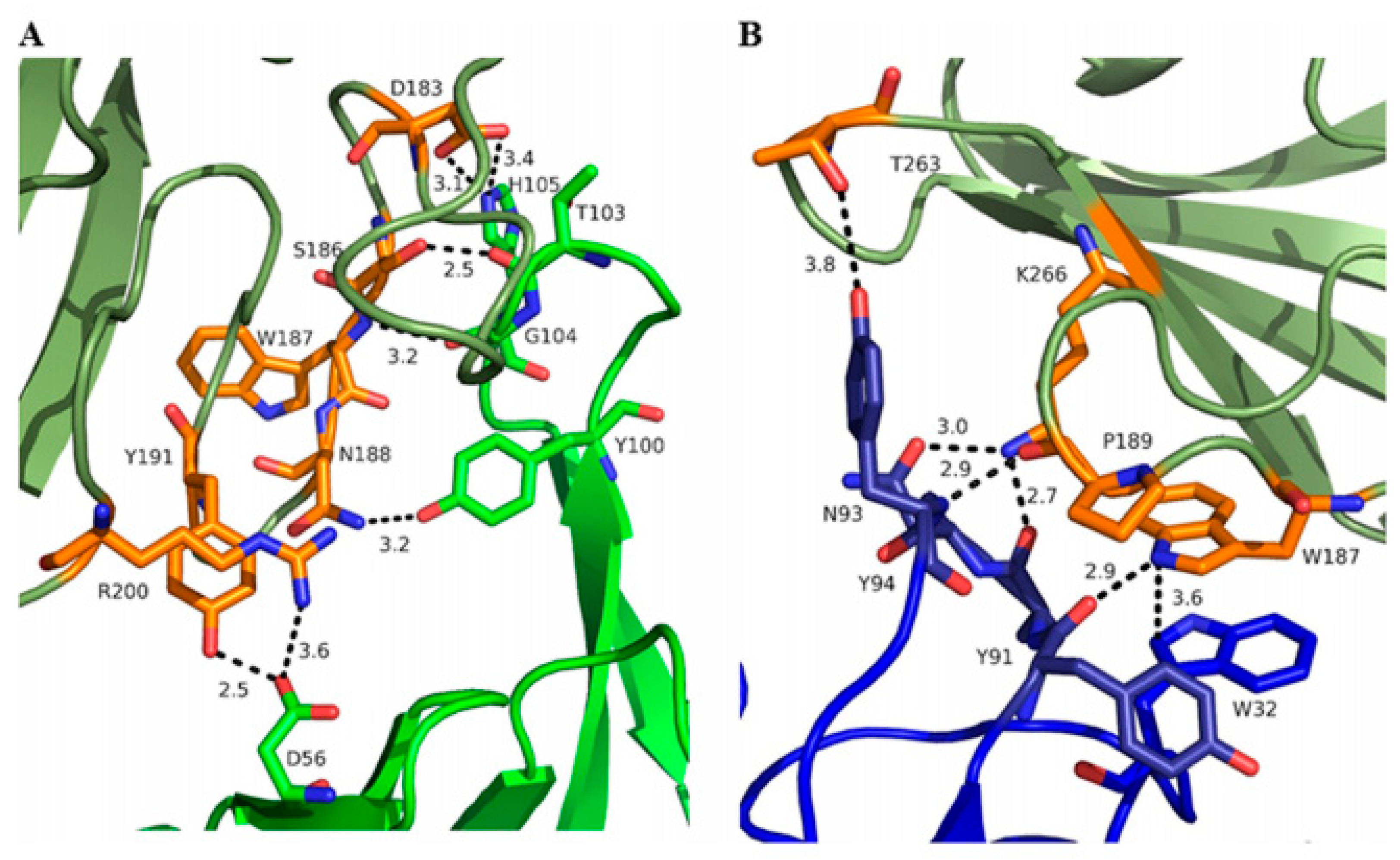
3.1.1. ClfA: Past Failure of Anti-ClfA Antibodies Enlightens Further Research
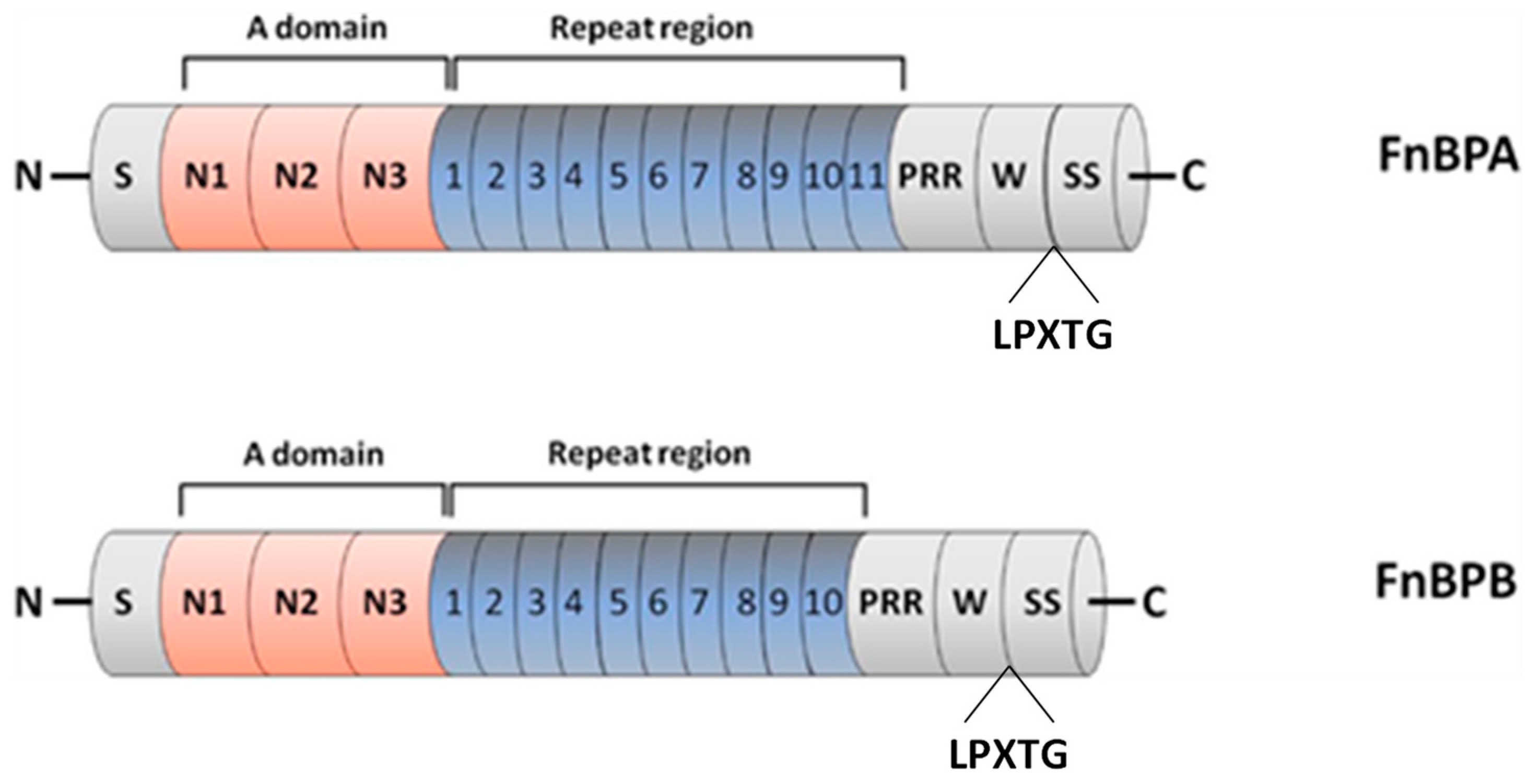
3.1.2. FnBP: The Possibility of Developing FnBP Antibody Is Waiting to Be Addressed
3.2. Decomposition of Biofilm Matrix
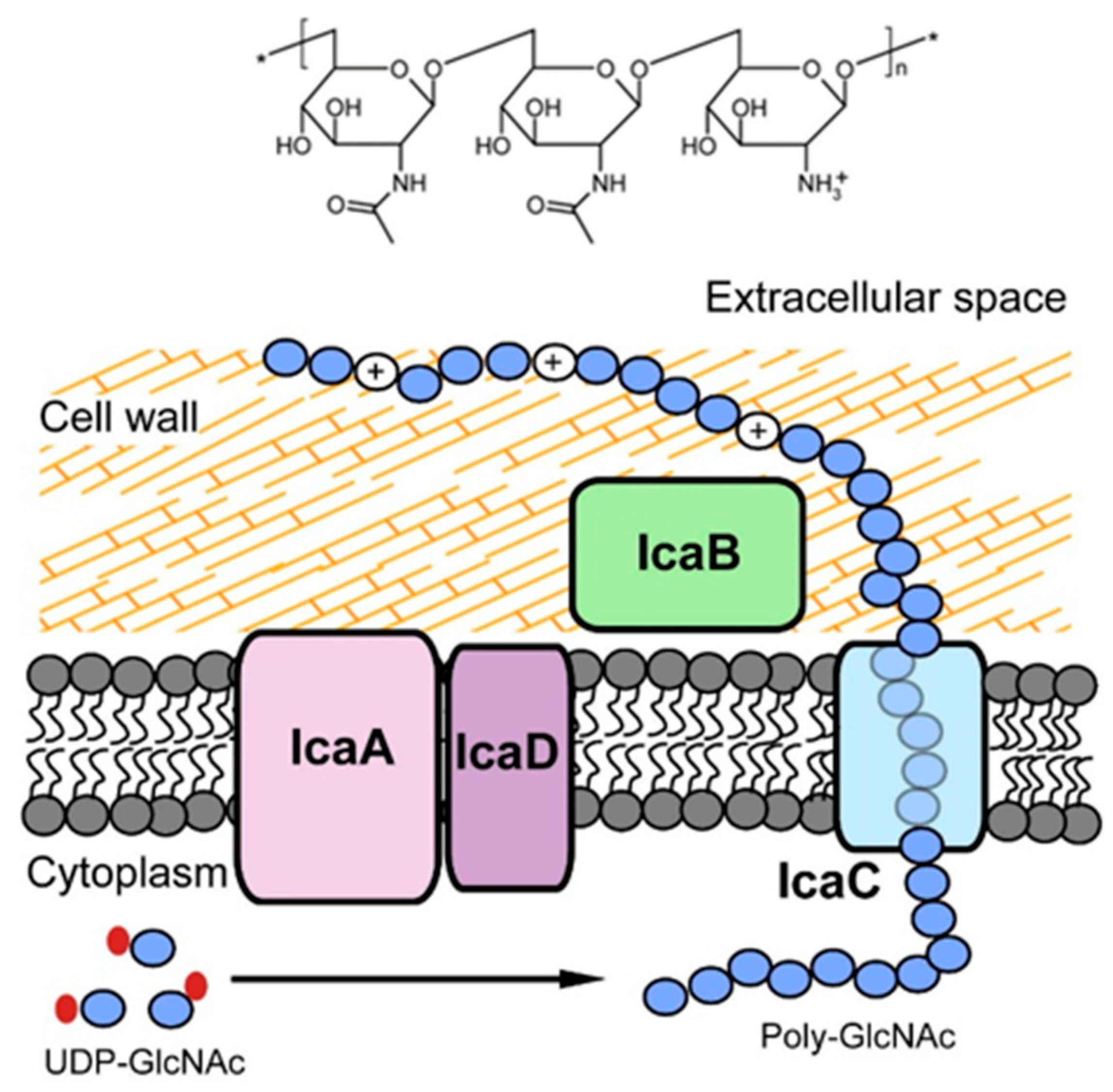
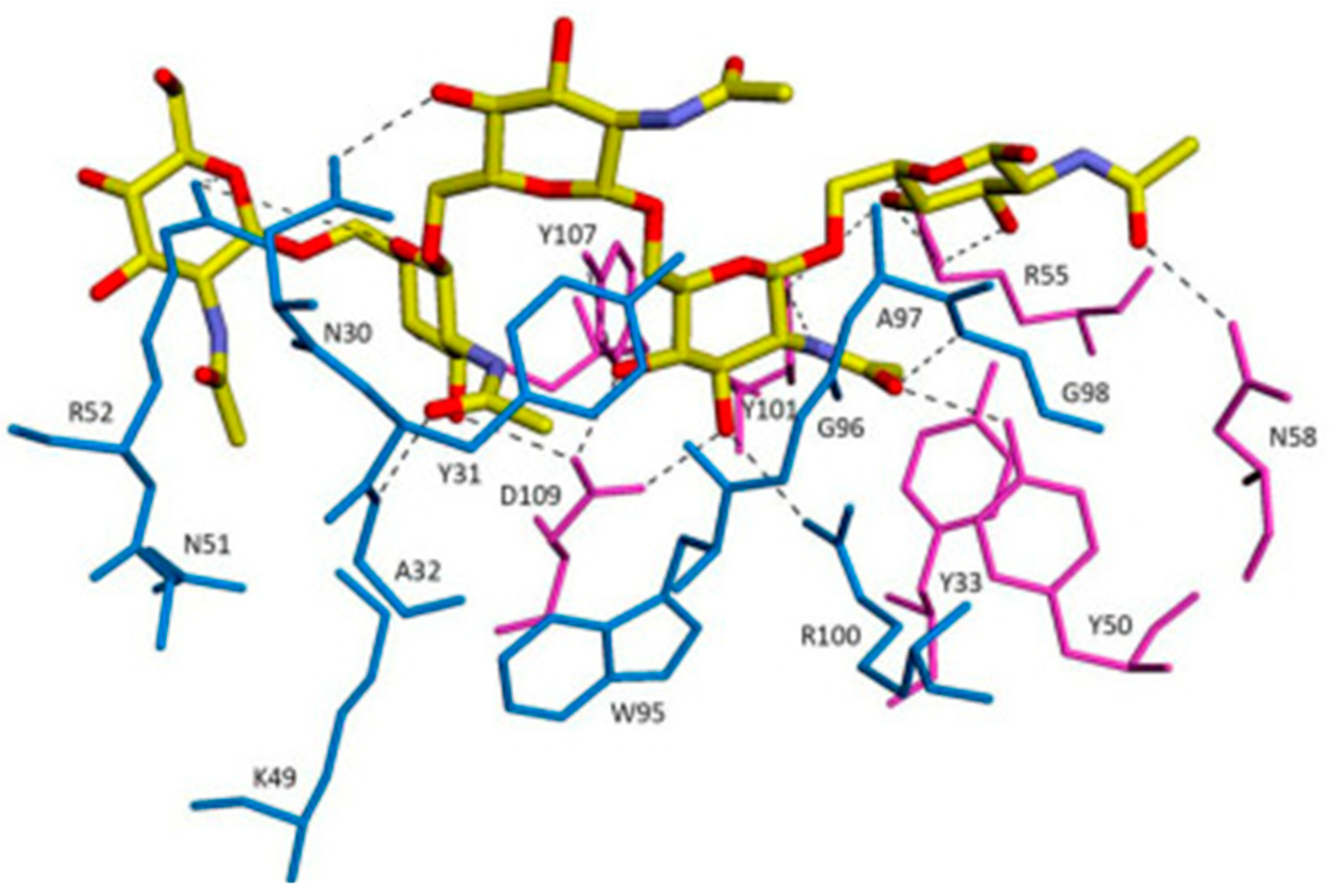
3.2.1. PNAG: The Antibody against PNAG/dPNAG Shows Optimal Anti-Biofilm Effect
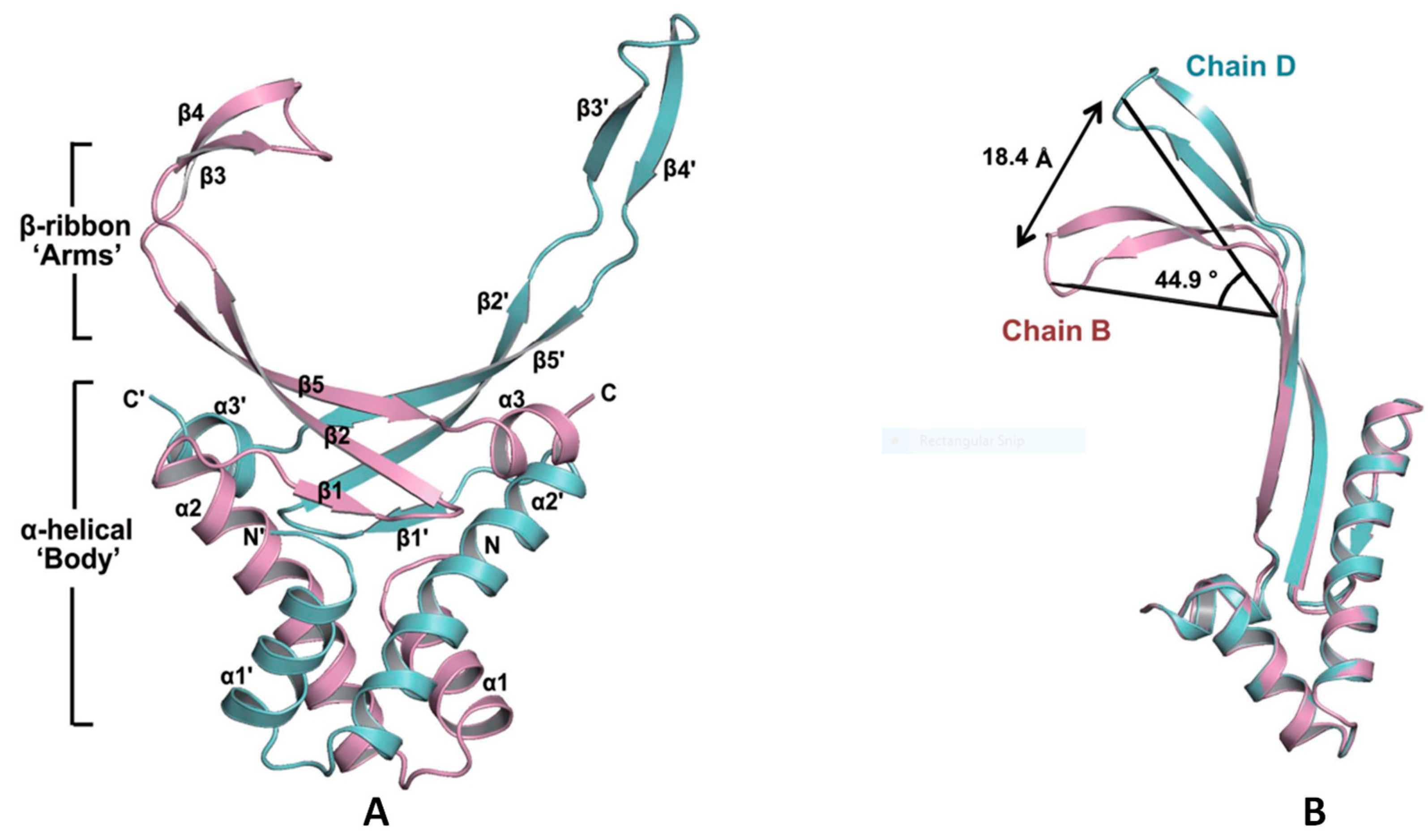
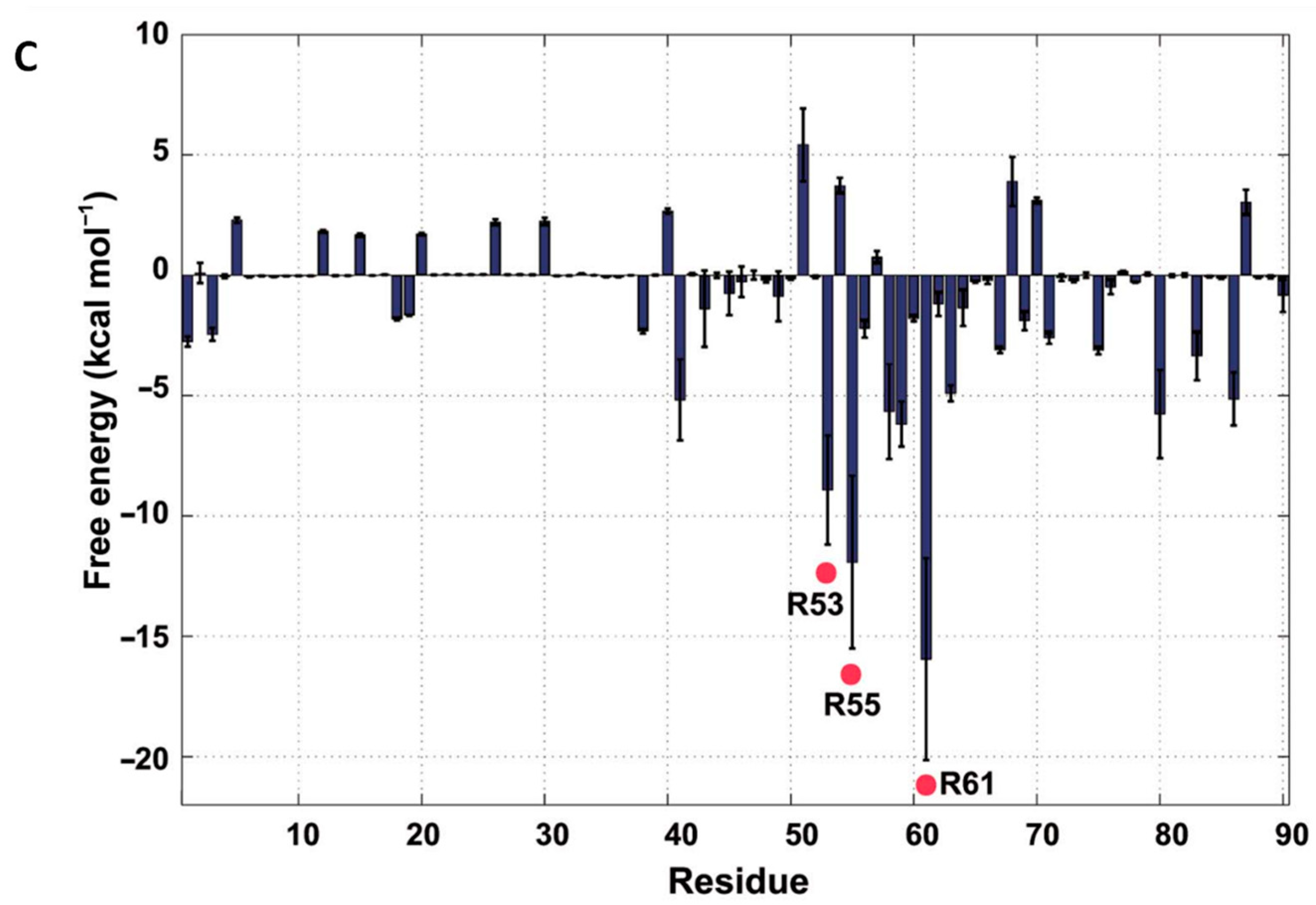
3.2.2. DNABII: A Promising Antibody Target for Anti-Biofilm Treatment
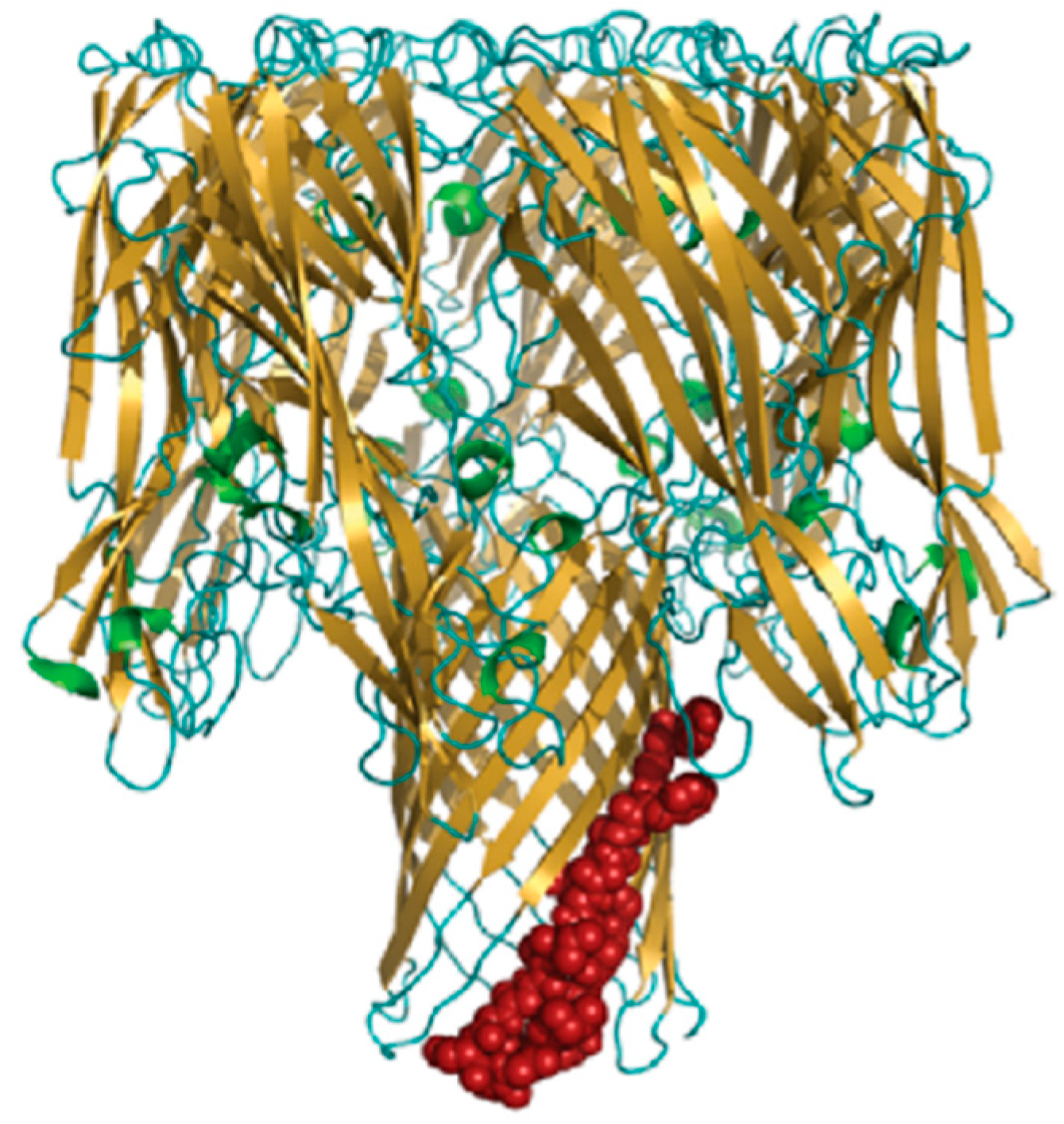
3.3. Targeting S. aureus Toxins as Supplemental Therapy
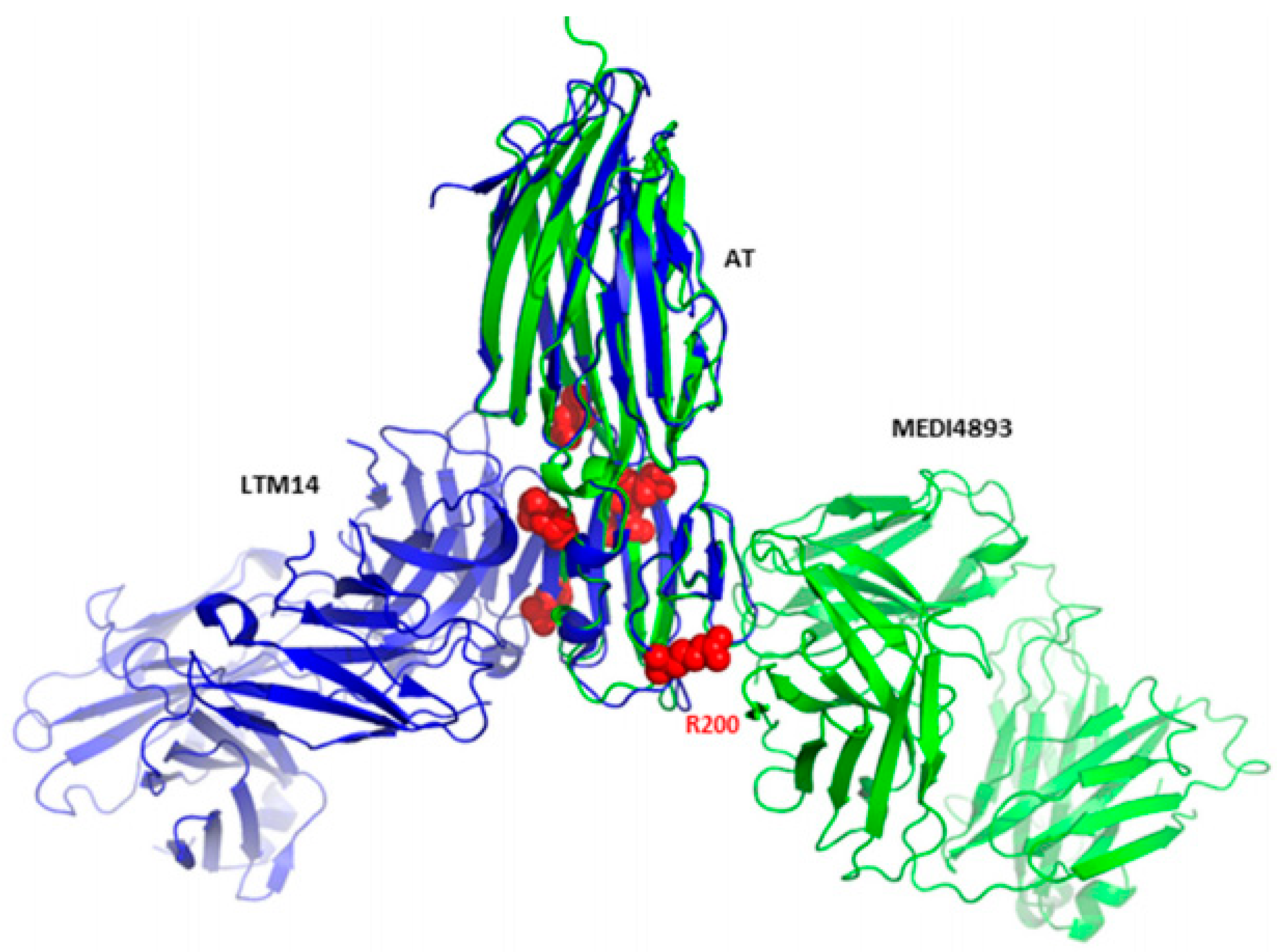
AT Antibody: The Only Type of Antibody Currently Successful in Clinical Trials
3.4. Other Targets for Anti-Biofilm Treatment
4. Conclusions and Future Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pant, S.; Patel, N.J.; Deshmukh, A.; Golwala, H.; Patel, N.; Badheka, A.; Hirsch, G.A.; Mehta, J.L. Trends in infective endocarditis incidence, microbiology, and valve replacement in the United States from 2000 to 2011. J. Am. Coll. Cardiol. 2015, 65, 2070–2076. [Google Scholar] [CrossRef]
- Bor, D.H.; Woolhandler, S.; Nardin, R.; Brusch, J.; Himmelstein, D.U. Infective endocarditis in the U.S., 1998–2009: A nationwide study. PLoS ONE 2013, 8, e60033. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, J.J.; Stearns, S.C.; Peppercorn, A.F.; Chu, V.H.; Fowler, V.G., Jr. Increasing US rates of endocarditis with Staphylococcus aureus: 1999–2008. Arch. Intern. Med. 2012, 172, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Loewe, L.; Rosenblatt, P.; Greene, H.J. Combined Penicillin and Heparin Therapy of Subacute Bacterial Endocarditis. Bull. N. Y. Acad. Med. 1946, 22, 270–272. [Google Scholar] [CrossRef] [PubMed]
- Cates, J.E.; Christie, R.V.; Garrod, L.P. Penicillin-resistant subacute bacterial endocarditis treated by a combination of penicillin and streptomycin. Br. Med. J. 1951, 1, 653–656. [Google Scholar] [CrossRef]
- Wallace, A.G.; Young, W.G., Jr.; Osterhout, S. Treatment of Acute Bacterial Endocarditis by Valve Excision and Replacement. Circulation 1965, 31, 450–453. [Google Scholar] [CrossRef]
- Slipczuk, L.; Codolosa, J.N.; Davila, C.D.; Romero-Corral, A.; Yun, J.; Pressman, G.S.; Figueredo, V.M. Infective endocarditis epidemiology over five decades: A systematic review. PLoS ONE 2013, 8, e82665. [Google Scholar] [CrossRef]
- Martin, G.S.; Mannino, D.M.; Eaton, S.; Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2003, 348, 1546–1554. [Google Scholar] [CrossRef]
- Wisplinghoff, H.; Bischoff, T.; Tallent, S.M.; Seifert, H.; Wenzel, R.P.; Edmond, M.B. Nosocomial bloodstream infections in US hospitals: Analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis. 2004, 39, 309–317. [Google Scholar] [CrossRef]
- Fluit, A.C.; Jones, M.E.; Schmitz, F.J.; Acar, J.; Gupta, R.; Verhoef, J. Antimicrobial susceptibility and frequency of occurrence of clinical blood isolates in Europe from the SENTRY antimicrobial surveillance program, 1997 and 1998. Clin. Infect. Dis. 2000, 30, 454–460. [Google Scholar] [CrossRef]
- Cabell, C.H.; Heidenreich, P.A.; Chu, V.H.; Moore, C.M.; Stryjewski, M.E.; Corey, G.R.; Fowler, V.G., Jr. Increasing rates of cardiac device infections among Medicare beneficiaries: 1990–1999. Am. Heart J. 2004, 147, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Darouiche, R.O. Treatment of infections associated with surgical implants. N. Engl. J. Med. 2004, 350, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.T.; Steckelberg, J.M. Infective endocarditis in patients receiving long-term hemodialysis. Mayo Clin. Proc. 2000, 75, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Cahill, T.J.; Baddour, L.M.; Habib, G.; Hoen, B.; Salaun, E.; Pettersson, G.B.; Schafers, H.J.; Prendergast, B.D. Challenges in Infective Endocarditis. J. Am. Coll. Cardiol. 2017, 69, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, D.R.; Corey, G.R.; Hoen, B.; Miro, J.M.; Fowler, V.G., Jr.; Bayer, A.S.; Karchmer, A.W.; Olaison, L.; Pappas, P.A.; Moreillon, P.; et al. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: The International Collaboration on Endocarditis-Prospective Cohort Study. Arch. Intern. Med. 2009, 169, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Basso, C. Pathology and pathogenesis of infective endocarditis in native heart valves. Cardiovasc Pathol. 2006, 15, 256–263. [Google Scholar] [CrossRef]
- Freedman, L.R.; Valone, J., Jr. Experimental infective endocarditis. Prog. Cardiovasc Dis. 1979, 22, 169–180. [Google Scholar] [CrossRef]
- Gencbay, M.; Turan, F.; Degertekin, M.; Eksi, N.; Mutlu, B.; Unalp, A. High prevalence of hypercoagulable states in patients with recurrent thrombosis of mechanical heart valves. J. Heart Valve Dis. 1998, 7, 601–609. [Google Scholar]
- Selton-Suty, C.; Celard, M.; Le Moing, V.; Doco-Lecompte, T.; Chirouze, C.; Iung, B.; Strady, C.; Revest, M.; Vandenesch, F.; Bouvet, A.; et al. Preeminence of Staphylococcus aureus in infective endocarditis: A 1-year population-based survey. Clin. Infect. Dis. 2012, 54, 1230–1239. [Google Scholar] [CrossRef]
- Friedrich, A.W. Control of hospital acquired infections and antimicrobial resistance in Europe: The way to go. Wien. Med. Wochenschr. 2019, 169, 25–30. [Google Scholar] [CrossRef]
- Cassini, A.; Hogberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef]
- Jia, K.; Fang, T.; Wang, X.; Liu, Y.; Sun, W.; Wang, Y.; Ding, T.; Wang, J.; Li, C.; Xu, D.; et al. Antibiotic Resistance Patterns of Staphylococcus aureus Isolates from Retail Foods in Mainland China: A Meta-Analysis. Foodborne Pathog. Dis. 2020, 17, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.L.; Apisarnthanarak, A.; Madriaga, G. The Burden of Healthcare-Associated Infections in Southeast Asia: A Systematic Literature Review and Meta-analysis. Clin. Infect. Dis. 2015, 60, 1690–1699. [Google Scholar] [CrossRef]
- Stapleton, P.D.; Taylor, P.W. Methicillin resistance in Staphylococcus aureus: Mechanisms and modulation. Sci. Prog. 2002, 85, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Deleo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Kishii, K.; Ito, T.; Watanabe, S.; Okuzumi, K.; Hiramatsu, K. Recurrence of heterogeneous methicillin-resistant Staphylococcus aureus (MRSA) among the MRSA clinical isolates in a Japanese university hospital. J. Antimicrob. Chemother. 2008, 62, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, K.; Hanaki, H.; Ino, T.; Yabuta, K.; Oguri, T.; Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997, 40, 135–136. [Google Scholar] [CrossRef]
- Jensen, S.O.; Lyon, B.R. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009, 4, 565–582. [Google Scholar] [CrossRef]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Pinto, R.M.; Lopes-de-Campos, D.; Martins, M.C.L.; Van Dijck, P.; Nunes, C.; Reis, S. Impact of nanosystems in Staphylococcus aureus biofilms treatment. FEMS Microbiol. Rev. 2019, 43, 622–641. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.; Foulston, L.; DeFrancesco, A.S.; Losick, R. An Electrostatic Net Model for the Role of Extracellular DNA in Biofilm Formation by Staphylococcus aureus. J. Bacteriol. 2015, 197, 3779–3787. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.N.C.; Yildiz, F.H. Biofilm Matrix Proteins. Microbiol Spectr 2015, 3, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Geng, M.; Bai, L. Targeting Biofilms Therapy: Current Research Strategies and Development Hurdles. Microorganisms 2020, 8, 1222. [Google Scholar] [CrossRef]
- Boles, B.R.; Horswill, A.R. Staphylococcal biofilm disassembly. Trends Microbiol. 2011, 19, 449–455. [Google Scholar] [CrossRef]
- Colque-Navarro, P.; Jacobsson, G.; Andersson, R.; Flock, J.I.; Mollby, R. Levels of antibody against 11 Staphylococcus aureus antigens in a healthy population. Clin. Vaccine Immunol. 2010, 17, 1117–1123. [Google Scholar] [CrossRef]
- Feuillie, C.; Formosa-Dague, C.; Hays, L.M.C.; Vervaeck, O.; Derclaye, S.; Brennan, M.P.; Foster, T.J.; Geoghegan, J.A.; Dufrêne, Y.F. Molecular interactions and inhibition of the staphylococcal biofilm-forming protein SdrC. Proc. Natl. Acad. Sci. USA 2017, 114, 3738–3743. [Google Scholar] [CrossRef]
- Leonard, A.C.; Petrie, L.E.; Cox, G. Bacterial Anti-adhesives: Inhibition of Staphylococcus aureus Nasal Colonization. ACS Infect. Dis. 2019, 5, 1668–1681. [Google Scholar] [CrossRef]
- Dryla, A.; Prustomersky, S.; Gelbmann, D.; Hanner, M.; Bettinger, E.; Kocsis, B.; Kustos, T.; Henics, T.; Meinke, A.; Nagy, E. Comparison of antibody repertoires against Staphylococcus aureus in healthy individuals and in acutely infected patients. Clin. Diagn. Lab. Immunol. 2005, 12, 387–398. [Google Scholar] [CrossRef]
- Thomer, L.; Emolo, C.; Thammavongsa, V.; Kim, H.K.; McAdow, M.E.; Yu, W.; Kieffer, M.; Schneewind, O.; Missiakas, D. Antibodies against a secreted product of Staphylococcus aureus trigger phagocytic killing. J. Exp. Med. 2016, 213, 293–301. [Google Scholar] [CrossRef]
- Kumar, A.; Ray, P.; Kanwar, M.; Sharma, M.; Varma, S. A comparative analysis of antibody repertoire against Staphylococcus aureus antigens in patients with deep-seated versus superficial staphylococcal infections. Int. J. Med. Sci. 2005, 2, 129–136. [Google Scholar] [CrossRef]
- Raafat, D.; Otto, M.; Reppschlager, K.; Iqbal, J.; Holtfreter, S. Fighting Staphylococcus aureus Biofilms with Monoclonal Antibodies. Trends Microbiol. 2019, 27, 303–322. [Google Scholar] [CrossRef] [PubMed]
- Belyi, Y.; Rybolovlev, I.; Polyakov, N.; Chernikova, A.; Tabakova, I.; Gintsburg, A. Staphylococcus aureus Surface Protein G is An Immunodominant Protein and a Possible Target in An Anti-Biofilm Drug Development. Open Microbiol. J. 2018, 12, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Domanski, P.J.; Patel, P.R.; Bayer, A.S.; Zhang, L.; Hall, A.E.; Syribeys, P.J.; Gorovits, E.L.; Bryant, D.; Vernachio, J.H.; Hutchins, J.T.; et al. Characterization of a humanized monoclonal antibody recognizing clumping factor A expressed by Staphylococcus aureus. Infect. Immun. 2005, 73, 5229–5232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tkaczyk, C.; Kasturirangan, S.; Minola, A.; Jones-Nelson, O.; Gunter, V.; Shi, Y.Y.; Rosenthal, K.; Aleti, V.; Semenova, E.; Warrener, P.; et al. Multimechanistic Monoclonal Antibodies (MAbs) Targeting Staphylococcus aureus Alpha-Toxin and Clumping Factor A: Activity and Efficacy Comparisons of a MAb Combination and an Engineered Bispecific Antibody Approach. Antimicrob. Agents Chemother. 2017, 61, e00629-17. [Google Scholar] [CrossRef]
- Varshney, A.K.; Kuzmicheva, G.A.; Lin, J.; Sunley, K.M.; Bowling, R.A., Jr.; Kwan, T.Y.; Mays, H.R.; Rambhadran, A.; Zhang, Y.; Martin, R.L.; et al. A natural human monoclonal antibody targeting Staphylococcus Protein A protects against Staphylococcus aureus bacteremia. PLoS ONE 2018, 13, e0190537. [Google Scholar] [CrossRef]
- Franca, A.; Vilanova, M.; Cerca, N.; Pier, G.B. Monoclonal antibody raised against PNAG has variable effects on static S. epidermidis biofilm accumulation in vitro. Int. J. Biol. Sci. 2013, 9, 518–520. [Google Scholar] [CrossRef]
- Romero Pastrana, F.; Neef, J.; Koedijk, D.; de Graaf, D.; Duipmans, J.; Jonkman, M.F.; Engelmann, S.; van Dijl, J.M.; Buist, G. Human antibody responses against non-covalently cell wall-bound Staphylococcus aureus proteins. Sci. Rep. 2018, 8, 3234. [Google Scholar] [CrossRef]
- Giersing, B.K.; Dastgheyb, S.S.; Modjarrad, K.; Moorthy, V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine 2016, 34, 2962–2966. [Google Scholar] [CrossRef]
- Tkaczyk, C.; Hamilton, M.M.; Sadowska, A.; Shi, Y.; Chang, C.S.; Chowdhury, P.; Buonapane, R.; Xiao, X.; Warrener, P.; Mediavilla, J.; et al. Targeting Alpha Toxin and ClfA with a Multimechanistic Monoclonal-Antibody-Based Approach for Prophylaxis of Serious Staphylococcus aureus Disease. mBio 2016, 7, e00528-16. [Google Scholar] [CrossRef]
- Hall, A.E.; Domanski, P.J.; Patel, P.R.; Vernachio, J.H.; Syribeys, P.J.; Gorovits, E.L.; Johnson, M.A.; Ross, J.M.; Hutchins, J.T.; Patti, J.M. Characterization of a protective monoclonal antibody recognizing Staphylococcus aureus MSCRAMM protein clumping factor A. Infect. Immun. 2003, 71, 6864–6870. [Google Scholar] [CrossRef] [PubMed]
- Weems, J.J., Jr.; Steinberg, J.P.; Filler, S.; Baddley, J.W.; Corey, G.R.; Sampathkumar, P.; Winston, L.; John, J.F.; Kubin, C.J.; Talwani, R.; et al. Phase II, randomized, double-blind, multicenter study comparing the safety and pharmacokinetics of tefibazumab to placebo for treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2006, 50, 2751–2755. [Google Scholar] [CrossRef] [PubMed]
- Squibb, B.-M. A Phase IIa Dose Escalation Study to Assess Safety and Pharmacokinetics of Aurexis® in Cystic Fibrosis Subjects Chronically Colonized with Staphylococcus aureus in Their Lungs. Available online: https://clinicaltrials.gov/ct2/show/NCT00198289 (accessed on 21 April 2022).
- O’Neill, E.; Pozzi, C.; Houston, P.; Humphreys, H.; Robinson, D.A.; Loughman, A.; Foster, T.J.; O’Gara, J.P. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J. Bacteriol. 2008, 190, 3835–3850. [Google Scholar] [CrossRef]
- Rennermalm, A.; Li, Y.H.; Bohaufs, L.; Jarstrand, C.; Brauner, A.; Brennan, F.R.; Flock, J.I. Antibodies against a truncated Staphylococcus aureus fibronectin-binding protein protect against dissemination of infection in the rat. Vaccine 2001, 19, 3376–3383. [Google Scholar] [CrossRef]
- Visai, L.; Xu, Y.; Casolini, F.; Rindi, S.; Hook, M.; Speziale, P. Monoclonal antibodies to CNA, a collagen-binding microbial surface component recognizing adhesive matrix molecules, detach Staphylococcus aureus from a collagen substrate. J. Biol. Chem. 2000, 275, 39837–39845. [Google Scholar] [CrossRef]
- Pharma, N. A Multi Centre, Double-Blind, Randomised, Placebo Controlled Prospective Study on the Safety and Efficacy of Aurograb in Patients with Severe, Deep-Seated Staphylococcal Infections Receiving Vancomycin. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00217841 (accessed on 21 April 2022).
- Nilsson, I.M.; Patti, J.M.; Bremell, T.; Hook, M.; Tarkowski, A. Vaccination with a recombinant fragment of collagen adhesin provides protection against Staphylococcus aureus-mediated septic death. J. Clin. Investig. 1998, 101, 2640–2649. [Google Scholar] [CrossRef]
- McCarthy, H.; Waters, E.M.; Bose, J.L.; Foster, S.; Bayles, K.W.; O’Neill, E.; Fey, P.D.; O’Gara, J.P. The major autolysin is redundant for Staphylococcus aureus USA300 LAC JE2 virulence in a murine device-related infection model. FEMS Microbiol. Lett. 2016, 363, fnw087. [Google Scholar] [CrossRef]
- Nair, N.; Vinod, V.; Suresh, M.K.; Vijayrajratnam, S.; Biswas, L.; Peethambaran, R.; Vasudevan, A.K.; Biswas, R. Amidase, a cell wall hydrolase, elicits protective immunity against Staphylococcus aureus and S. epidermidis. Int. J. Biol. Macromol. 2015, 77, 314–321. [Google Scholar] [CrossRef]
- Haghighat, S.; Siadat, S.D.; Sorkhabadi, S.M.; Sepahi, A.A.; Mahdavi, M. Cloning, expression and purification of autolysin from methicillin-resistant Staphylococcus aureus: Potency and challenge study in Balb/c mice. Mol. Immunol. 2017, 82, 10–18. [Google Scholar] [CrossRef]
- Varrone, J.J.; de Mesy Bentley, K.L.; Bello-Irizarry, S.N.; Nishitani, K.; Mack, S.; Hunter, J.G.; Kates, S.L.; Daiss, J.L.; Schwarz, E.M. Passive immunization with anti-glucosaminidase monoclonal antibodies protects mice from implant-associated osteomyelitis by mediating opsonophagocytosis of Staphylococcus aureus megaclusters. J. Orthop. Res. 2014, 32, 1389–1396. [Google Scholar] [CrossRef]
- van den Berg, S.; Bonarius, H.P.; van Kessel, K.P.; Elsinga, G.S.; Kooi, N.; Westra, H.; Bosma, T.; van der Kooi-Pol, M.M.; Koedijk, D.G.; Groen, H.; et al. A human monoclonal antibody targeting the conserved staphylococcal antigen IsaA protects mice against Staphylococcus aureus bacteremia. Int. J. Med. Microbiol. 2015, 305, 55–64. [Google Scholar] [CrossRef]
- Oesterreich, B.; Lorenz, B.; Schmitter, T.; Kontermann, R.; Zenn, M.; Zimmermann, B.; Haake, M.; Lorenz, U.; Ohlsen, K. Characterization of the biological anti-staphylococcal functionality of hUK-66 IgG1, a humanized monoclonal antibody as substantial component for an immunotherapeutic approach. Hum. Vaccin. Immunother. 2014, 10, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Ohlsen, K.; Lorenz, U. Immunotherapeutic strategies to combat staphylococcal infections. Int. J. Med. Microbiol. 2010, 300, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Jung, D.-J.; An, J.-H.; Kurokawa, K.; Jung, Y.-C.; Kim, M.-J.; Aoyagi, Y.; Matsushita, M.; Takahashi, S.; Lee, H.-S.; Takahashi, K.; et al. Specific Serum Ig Recognizing Staphylococcal Wall Teichoic Acid Induces Complement-Mediated Opsonophagocytosis against Staphylococcus aureus. J. Immunol. 2012, 189, 4951. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, J.; Lee, J.C. Antibodies to capsular polysaccharides are not protective against experimental Staphylococcus aureus endocarditis. Infect. Immun. 1995, 63, 375–380. [Google Scholar] [CrossRef]
- Zhou, C.; Lehar, S.; Gutierrez, J.; Rosenberger, C.M.; Ljumanovic, N.; Dinoso, J.; Koppada, N.; Hong, K.; Baruch, A.; Carrasco-Triguero, M.; et al. Pharmacokinetics and pharmacodynamics of DSTA4637A: A novel THIOMAB antibody antibiotic conjugate against Staphylococcus aureus in mice. MAbs 2016, 8, 1612–1619. [Google Scholar] [CrossRef]
- Wang-Lin, S.X.; Zhou, C.; Kamath, A.V.; Hong, K.; Koppada, N.; Saad, O.M.; Carrasco-Triguero, M.; Khojasteh, C.; Deng, R. Minimal physiologically-based pharmacokinetic modeling of DSTA4637A, A novel THIOMAB antibody antibiotic conjugate against Staphylococcus aureus, in a mouse model. MAbs 2018, 10, 1131–1143. [Google Scholar] [CrossRef]
- Lee, J.C.; Park, J.S.; Shepherd, S.E.; Carey, V.; Fattom, A. Protective efficacy of antibodies to the Staphylococcus aureus type 5 capsular polysaccharide in a modified model of endocarditis in rats. Infect. Immun. 1997, 65, 4146–4151. [Google Scholar] [CrossRef]
- Liu, B.; Park, S.; Thompson, C.D.; Li, X.; Lee, J.C. Antibodies to Staphylococcus aureus capsular polysaccharides 5 and 8 perform similarly in vitro but are functionally distinct in vivo. Virulence 2017, 8, 859–874. [Google Scholar] [CrossRef]
- Rupp, M.E.; Holley, H.P., Jr.; Lutz, J.; Dicpinigaitis, P.V.; Woods, C.W.; Levine, D.P.; Veney, N.; Fowler, V.G., Jr. Phase II, randomized, multicenter, double-blind, placebo-controlled trial of a polyclonal anti-Staphylococcus aureus capsular polysaccharide immune globulin in treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 2007, 51, 4249–4254. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.K.; Schelonka, R.; White, R.; Holley, H.P.; Bifano, E.; Cummings, J.; Adcock, K.; Kaufman, D.; Puppala, B.; Riedel, P.; et al. A blinded, randomized, multicenter study of an intravenous Staphylococcus aureus immune globulin. J. Perinatol. 2006, 26, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Weisman, L.E. Antibody for the prevention of neonatal noscocomial staphylococcal infection: A review of the literature. Arch. De Pédiatrie 2007, 14, S31–S34. [Google Scholar] [CrossRef]
- Weisman, L.E.; Thackray, H.M.; Steinhorn, R.H.; Walsh, W.F.; Lassiter, H.A.; Dhanireddy, R.; Brozanski, B.S.; Palmer, K.G.; Trautman, M.S.; Escobedo, M.; et al. A randomized study of a monoclonal antibody (pagibaximab) to prevent staphylococcal sepsis. Pediatrics 2011, 128, 271–279. [Google Scholar] [CrossRef]
- Patel, M.; Kaufman, D.A. Anti-lipoteichoic acid monoclonal antibody (pagibaximab) studies for the prevention of staphylococcal bloodstream infections in preterm infants. Expert Opin. Biol. Ther. 2015, 15, 595–600. [Google Scholar] [CrossRef]
- Cerca, N.; Jefferson, K.K.; Maira-Litran, T.; Pier, D.B.; Kelly-Quintos, C.; Goldmann, D.A.; Azeredo, J.; Pier, G.B. Molecular basis for preferential protective efficacy of antibodies directed to the poorly acetylated form of staphylococcal poly-N-acetyl-beta-(1-6)-glucosamine. Infect. Immun. 2007, 75, 3406–3413. [Google Scholar] [CrossRef]
- Kelly-Quintos, C.; Cavacini, L.A.; Posner, M.R.; Goldmann, D.; Pier, G.B. Characterization of the opsonic and protective activity against Staphylococcus aureus of fully human monoclonal antibodies specific for the bacterial surface polysaccharide poly-N-acetylglucosamine. Infect. Immun. 2006, 74, 2742–2750. [Google Scholar] [CrossRef]
- Sanofi-Aventis. A Randomized, Double-blind, Placebo-controlled Trial to Assess the Pharmacokinetics, Pharmacodynamics and Safety of a Single Dose of SAR279356 in Patients Hospitalized in Intensive Care Unit and on Mechanical Ventilation. Available online: https://clinicaltrials.gov/ct2/show/NCT.T01389700 (accessed on 21 April 2022).
- Estelles, A.; Woischnig, A.K.; Liu, K.; Stephenson, R.; Lomongsod, E.; Nguyen, D.; Zhang, J.; Heidecker, M.; Yang, Y.; Simon, R.J.; et al. A High-Affinity Native Human Antibody Disrupts Biofilm from Staphylococcus aureus Bacteria and Potentiates Antibiotic Efficacy in a Mouse Implant Infection Model. Antimicrob. Agents Chemother. 2016, 60, 2292–2301. [Google Scholar] [CrossRef]
- Xiong, Y.Q.; Estelles, A.; Li, L.; Abdelhady, W.; Gonzales, R.; Bayer, A.S.; Tenorio, E.; Leighton, A.; Ryser, S.; Kauvar, L.M. A Human Biofilm-Disrupting Monoclonal Antibody Potentiates Antibiotic Efficacy in Rodent Models of both Staphylococcus aureus and Acinetobacter baumannii Infections. Antimicrob. Agents Chemother. 2017, 61, e00904-17. [Google Scholar] [CrossRef]
- LLC, T.B. Study to Evaluate Safety and Activity of TRL1068 in Prosthetic Joint Infections. Available online: https://clinicaltrials.gov/ct2/show/NCT04763759 (accessed on 21 April 2022).
- Wang, Y.; Cheng, L.I.; Helfer, D.R.; Ashbaugh, A.G.; Miller, R.J.; Tzomides, A.J.; Thompson, J.M.; Ortines, R.V.; Tsai, A.S.; Liu, H.; et al. Mouse model of hematogenous implant-related Staphylococcus aureus biofilm infection reveals therapeutic targets. Proc. Natl. Acad. Sci. USA 2017, 114, E5094–E5102. [Google Scholar] [CrossRef]
- Anderson, M.J.; Schaaf, E.; Breshears, L.M.; Wallis, H.W.; Johnson, J.R.; Tkaczyk, C.; Sellman, B.R.; Sun, J.; Peterson, M.L. Alpha-Toxin Contributes to Biofilm Formation among Staphylococcus aureus Wound Isolates. Toxins 2018, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, V.; Peng, L.; Damschroder, M.M.; Cheng, L.; Sadowska, A.; Tkaczyk, C.; Sellman, B.R.; Wu, H.; Dall’Acqua, W.F. Mechanisms of neutralization of a human anti-alpha-toxin antibody. J. Biol. Chem. 2014, 289, 29874–29880. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Robbie, G.J.; Wu, Y.; Esser, M.T.; Jensen, K.; Schwartz, H.I.; Bellamy, T.; Hernandez-Illas, M.; Jafri, H.S. Safety, Tolerability, and Pharmacokinetics of MEDI4893, an Investigational, Extended-Half-Life, Anti-Staphylococcus aureus Alpha-Toxin Human Monoclonal Antibody, in Healthy Adults. Antimicrob. Agents Chemother. 2017, 61, e01020-16. [Google Scholar] [CrossRef] [PubMed]
- Pharmaceuticals, A. AR-301 (Tosatoxumab). Available online: https://aridispharma.com/ar-301/ (accessed on 21 April 2022).
- Thomsen, I.P.; Sapparapu, G.; James, D.B.A.; Cassat, J.E.; Nagarsheth, M.; Kose, N.; Putnam, N.; Boguslawski, K.M.; Jones, L.S.; Wood, J.B.; et al. Monoclonal Antibodies Against the Staphylococcus aureus Bicomponent Leukotoxin AB Isolated Following Invasive Human Infection Reveal Diverse Binding and Modes of Action. J. Infect. Dis. 2017, 215, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Burnie, J.P.; Matthews, R.C.; Carter, T.; Beaulieu, E.; Donohoe, M.; Chapman, C.; Williamson, P.; Hodgetts, S.J. Identification of an immunodominant ABC transporter in methicillin-resistant Staphylococcus aureus infections. Infect. Immun. 2000, 68, 3200–3209. [Google Scholar] [CrossRef]
- Lam, H.; Kesselly, A.; Stegalkina, S.; Kleanthous, H.; Yethon, J.A. Antibodies to PhnD inhibit staphylococcal biofilms. Infect. Immun. 2014, 82, 3764–3774. [Google Scholar] [CrossRef]
- Cucarella, C.; Solano, C.; Valle, J.; Amorena, B.; Lasa, I.; Penades, J.R. Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J. Bacteriol. 2001, 183, 2888–2896. [Google Scholar] [CrossRef]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Hook, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef]
- Broker, B.M.; Holtfreter, S.; Bekeredjian-Ding, I. Immune control of Staphylococcus aureus-regulation and counter-regulation of the adaptive immune response. Int. J. Med. Microbiol. 2014, 304, 204–214. [Google Scholar] [CrossRef]
- Lindsay, J.A.; Moore, C.E.; Day, N.P.; Peacock, S.J.; Witney, A.A.; Stabler, R.A.; Husain, S.E.; Butcher, P.D.; Hinds, J. Microarrays reveal that each of the ten dominant lineages of Staphylococcus aureus has a unique combination of surface-associated and regulatory genes. J. Bacteriol. 2006, 188, 669–676. [Google Scholar] [CrossRef]
- Murphy, E.; Lin, S.L.; Nunez, L.; Andrew, L.; Fink, P.S.; Dilts, D.A.; Hoiseth, S.K.; Jansen, K.U.; Anderson, A.S. Challenges for the evaluation of Staphylococcus aureus protein based vaccines: Monitoring antigenic diversity. Hum. Vaccin. 2011, 7 (Suppl. S1), 51–59. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herman-Bausier, P.; Labate, C.; Towell, A.M.; Derclaye, S.; Geoghegan, J.A.; Dufrene, Y.F. Staphylococcus aureus clumping factor A is a force-sensitive molecular switch that activates bacterial adhesion. Proc. Natl. Acad. Sci. USA 2018, 115, 5564–5569. [Google Scholar] [CrossRef] [PubMed]
- McDevitt, D.; Francois, P.; Vaudaux, P.; Foster, T.J. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol. Microbiol. 1994, 11, 237–248. [Google Scholar] [CrossRef]
- Ni Eidhin, D.; Perkins, S.; Francois, P.; Vaudaux, P.; Hook, M.; Foster, T.J. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol. Microbiol. 1998, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Wann, E.R.; Gurusiddappa, S.; Hook, M. The fibronectin-binding MSCRAMM FnbpA of Staphylococcus aureus is a bifunctional protein that also binds to fibrinogen. J. Biol. Chem. 2000, 275, 13863–13871. [Google Scholar] [CrossRef] [PubMed]
- Ponnuraj, K.; Bowden, M.G.; Davis, S.; Gurusiddappa, S.; Moore, D.; Choe, D.; Xu, Y.; Hook, M.; Narayana, S.V. A "dock, lock, and latch" structural model for a staphylococcal adhesin binding to fibrinogen. Cell 2003, 115, 217–228. [Google Scholar] [CrossRef]
- Brady, R.A.; Mocca, C.P.; Burns, D.L. Immunogenicity analysis of Staphylococcus aureus clumping factor A genetic variants. Clin. Vaccine Immunol. 2013, 20, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Becsek, B.; Pietrasanta, L.; Obrist, D. Turbulent Systolic Flow Downstream of a Bioprosthetic Aortic Valve: Velocity Spectra, Wall Shear Stresses, and Turbulent Dissipation Rates. Front. Physiol. 2020, 11, 577188. [Google Scholar] [CrossRef]
- Zhang, P.; Yeo, J.H.; Qian, P.; Hwang, N.H. Shear stress investigation across mechanical heart valve. ASAIO J. 2007, 53, 530–536. [Google Scholar] [CrossRef]
- Hawkins, J.; Kodali, S.; Matsuka, Y.V.; McNeil, L.K.; Mininni, T.; Scully, I.L.; Vernachio, J.H.; Severina, E.; Girgenti, D.; Jansen, K.U.; et al. A recombinant clumping factor A-containing vaccine induces functional antibodies to Staphylococcus aureus that are not observed after natural exposure. Clin. Vaccine Immunol. 2012, 19, 1641–1650. [Google Scholar] [CrossRef]
- Ganesh, V.K.; Liang, X.; Geoghegan, J.A.; Cohen, A.L.V.; Venugopalan, N.; Foster, T.J.; Hook, M. Lessons from the Crystal Structure of the S. aureus Surface Protein Clumping Factor A in Complex With Tefibazumab, an Inhibiting Monoclonal Antibody. EBioMedicine 2016, 13, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Patti, J.M. A humanized monoclonal antibody targeting Staphylococcus aureus. Vaccine 2004, 22 (Suppl. S1), S39–S43. [Google Scholar] [CrossRef] [PubMed]
- Rozalska, B.; Wadstrom, T. Protective opsonic activity of antibodies against fibronectin-binding proteins (FnBPs) of Staphylococcus aureus. Scand. J. Immunol. 1993, 37, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Tkaczyk, C.; Semenova, E.; Shi, Y.Y.; Rosenthal, K.; Oganesyan, V.; Warrener, P.; Stover, C.K.; Sellman, B.R. Alanine Scanning Mutagenesis of the MEDI4893 (Suvratoxumab) Epitope Reduces Alpha Toxin Lytic Activity In Vitro and Staphylococcus aureus Fitness in Infection Models. Antimicrob. Agents Chemother. 2018, 62, e01033-18. [Google Scholar] [CrossRef]
- Loughman, A.; Sweeney, T.; Keane, F.M.; Pietrocola, G.; Speziale, P.; Foster, T.J. Sequence diversity in the A domain of Staphylococcus aureus fibronectin-binding protein A. BMC Microbiol. 2008, 8, 74. [Google Scholar] [CrossRef]
- Burke, F.M.; McCormack, N.; Rindi, S.; Speziale, P.; Foster, T.J. Fibronectin-binding protein B variation in Staphylococcus aureus. BMC Microbiol. 2010, 10, 160. [Google Scholar] [CrossRef]
- Herman-Bausier, P.; El-Kirat-Chatel, S.; Foster Timothy, J.; Geoghegan Joan, A.; Dufrêne Yves, F.; Torres, V.; Pier Gerald, B. Staphylococcus aureus Fibronectin-Binding Protein A Mediates Cell-Cell Adhesion through Low-Affinity Homophilic Bonds. mBio 2015, 6, e00413-15. [Google Scholar] [CrossRef]
- Devaraj, A.; Buzzo, J.; Rocco, C.J.; Bakaletz, L.O.; Goodman, S.D. The DNABII family of proteins is comprised of the only nucleoid associated proteins required for nontypeable Haemophilus influenzae biofilm structure. Microbiologyopen 2018, 7, e00563. [Google Scholar] [CrossRef]
- Provenza, G.; Provenzano, M.; Visai, L.; Burke, F.M.; Geoghegan, J.A.; Stravalaci, M.; Gobbi, M.; Mazzini, G.; Arciola, C.R.; Foster, T.J.; et al. Functional analysis of a murine monoclonal antibody against the repetitive region of the fibronectin-binding adhesins fibronectin-binding protein A and fibronectin-binding protein B from Staphylococcus aureus. FEBS J. 2010, 277, 4490–4505. [Google Scholar] [CrossRef]
- Zheng, S.; Bawazir, M.; Dhall, A.; Kim, H.E.; He, L.; Heo, J.; Hwang, G. Implication of Surface Properties, Bacterial Motility, and Hydrodynamic Conditions on Bacterial Surface Sensing and Their Initial Adhesion. Front. Bioeng. Biotechnol. 2021, 9, 643722. [Google Scholar] [CrossRef]
- O’Gara, J.P. ica and beyond: Biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol. Lett. 2007, 270, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Cywes-Bentley, C.; Skurnik, D.; Zaidi, T.; Roux, D.; Deoliveira, R.B.; Garrett, W.S.; Lu, X.; O’Malley, J.; Kinzel, K.; Zaidi, T.; et al. Antibody to a conserved antigenic target is protective against diverse prokaryotic and eukaryotic pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, E2209–E2218. [Google Scholar] [CrossRef] [PubMed]
- Burgui, S.; Gil, C.; Solano, C.; Lasa, I.; Valle, J. A Systematic Evaluation of the Two-Component Systems Network Reveals That ArlRS Is a Key Regulator of Catheter Colonization by Staphylococcus aureus. Front. Microbiol. 2018, 9, 342. [Google Scholar] [CrossRef]
- Atkin, K.E.; MacDonald, S.J.; Brentnall, A.S.; Potts, J.R.; Thomas, G.H. A different path: Revealing the function of staphylococcal proteins in biofilm formation. FEBS Lett. 2014, 588, 1869–1872. [Google Scholar] [CrossRef] [PubMed]
- Roux, D.; Cywes-Bentley, C.; Zhang, Y.F.; Pons, S.; Konkol, M.; Kearns, D.B.; Little, D.J.; Howell, P.L.; Skurnik, D.; Pier, G.B. Identification of Poly-N-acetylglucosamine as a Major Polysaccharide Component of the Bacillus subtilis Biofilm Matrix. J. Biol. Chem. 2015, 290, 19261–19272. [Google Scholar] [CrossRef]
- Haji-Ghassemi, O.; Blackler, R.J.; Martin Young, N.; Evans, S.V. Antibody recognition of carbohydrate epitopesdagger. Glycobiology 2015, 25, 920–952. [Google Scholar] [CrossRef]
- Cerca, N.; Maira-Litran, T.; Jefferson, K.K.; Grout, M.; Goldmann, D.A.; Pier, G.B. Protection against Escherichia coli infection by antibody to the Staphylococcus aureus poly-N-acetylglucosamine surface polysaccharide. Proc. Natl. Acad. Sci. USA 2007, 104, 7528–7533. [Google Scholar] [CrossRef]
- D’Andrea, M.M.; Lau, G.W. DNABII targeting antibodies as vaccines against biofilm diseases. EBioMedicine 2020, 58, 102921. [Google Scholar] [CrossRef]
- Turnbull, L.; Toyofuku, M.; Hynen, A.L.; Kurosawa, M.; Pessi, G.; Petty, N.K.; Osvath, S.R.; Carcamo-Oyarce, G.; Gloag, E.S.; Shimoni, R.; et al. Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat. Commun. 2016, 7, 11220. [Google Scholar] [CrossRef]
- Maira-Litran, T.; Kropec, A.; Goldmann, D.A.; Pier, G.B. Comparative opsonic and protective activities of Staphylococcus aureus conjugate vaccines containing native or deacetylated Staphylococcal Poly-N-acetyl-beta-(1-6)-glucosamine. Infect. Immun. 2005, 73, 6752–6762. [Google Scholar] [CrossRef]
- Skurnik, D.; Cywes-Bentley, C.; Pier, G.B. The exceptionally broad-based potential of active and passive vaccination targeting the conserved microbial surface polysaccharide PNAG. Expert Rev. Vaccines 2016, 15, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Gening, M.L.; Maira-Litran, T.; Kropec, A.; Skurnik, D.; Grout, M.; Tsvetkov, Y.E.; Nifantiev, N.E.; Pier, G.B. Synthetic {beta}-(1->6)-linked N-acetylated and nonacetylated oligoglucosamines used to produce conjugate vaccines for bacterial pathogens. Infect. Immun. 2010, 78, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Soliman, C.; Walduck, A.K.; Yuriev, E.; Richards, J.S.; Cywes-Bentley, C.; Pier, G.B.; Ramsland, P.A. Structural basis for antibody targeting of the broadly expressed microbial polysaccharide poly-N-acetylglucosamine. J. Biol. Chem. 2018, 293, 5079–5089. [Google Scholar] [CrossRef] [PubMed]
- Mann, E.E.; Rice, K.C.; Boles, B.R.; Endres, J.L.; Ranjit, D.; Chandramohan, L.; Tsang, L.H.; Smeltzer, M.S.; Horswill, A.R.; Bayles, K.W. Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS ONE 2009, 4, e5822. [Google Scholar] [CrossRef]
- Rocco, C.J.; Davey, M.E.; Bakaletz, L.O.; Goodman, S.D. Natural antigenic differences in the functionally equivalent extracellular DNABII proteins of bacterial biofilms provide a means for targeted biofilm therapeutics. Mol. Oral Microbiol. 2017, 32, 118–130. [Google Scholar] [CrossRef]
- Swinger, K.K.; Rice, P.A. IHF and HU: Flexible architects of bent DNA. Curr. Opin. Struct. Biol. 2004, 14, 28–35. [Google Scholar] [CrossRef]
- Devaraj, A.; Justice, S.S.; Bakaletz, L.O.; Goodman, S.D. DNABII proteins play a central role in UPEC biofilm structure. Mol. Microbiol. 2015, 96, 1119–1135. [Google Scholar] [CrossRef]
- Goodman, S.D.; Obergfell, K.P.; Jurcisek, J.A.; Novotny, L.A.; Downey, J.S.; Ayala, E.A.; Tjokro, N.; Li, B.; Justice, S.S.; Bakaletz, L.O. Biofilms can be dispersed by focusing the immune system on a common family of bacterial nucleoid-associated proteins. Mucosal Immunol. 2011, 4, 625–637. [Google Scholar] [CrossRef]
- Kamashev, D.; Balandina, A.; Rouviere-Yaniv, J. The binding motif recognized by HU on both nicked and cruciform DNA. EMBO J. 1999, 18, 5434–5444. [Google Scholar] [CrossRef]
- Azam, T.A.; Ishihama, A. Twelve species of the nucleoid-associated protein from Escherichia coli. Sequence recognition specificity and DNA binding affinity. J. Biol. Chem. 1999, 274, 33105–33113. [Google Scholar] [CrossRef]
- Thomas, V.C.; Thurlow, L.R.; Boyle, D.; Hancock, L.E. Regulation of autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular proteases influences biofilm development. J. Bacteriol. 2008, 190, 5690–5698. [Google Scholar] [CrossRef]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Nagaraja, V.; Ramakumar, S. Structural and evolutionary analyses reveal determinants of DNA binding specificities of nucleoid-associated proteins HU and IHF. Mol. Phylogenet Evol. 2017, 107, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Im, H.; Jee, J.G.; Jang, S.B.; Yoon, H.J.; Kwon, A.R.; Kang, S.M.; Lee, B.J. beta-Arm flexibility of HU from Staphylococcus aureus dictates the DNA-binding and recognition mechanism. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 3273–3289. [Google Scholar] [CrossRef] [PubMed]
- Novotny, L.A.; Jurcisek, J.A.; Goodman, S.D.; Bakaletz, L.O. Monoclonal antibodies against DNA-binding tips of DNABII proteins disrupt biofilms in vitro and induce bacterial clearance in vivo. EBioMedicine 2016, 10, 33–44. [Google Scholar] [CrossRef]
- Oscherwitz, J.; Cease, K.B. Identification and validation of a linear protective neutralizing epitope in the beta-pore domain of alpha toxin. PLoS ONE 2015, 10, e0116882. [Google Scholar] [CrossRef]
- Walker, B.; Bayley, H. Key residues for membrane binding, oligomerization, and pore forming activity of staphylococcal alpha-hemolysin identified by cysteine scanning mutagenesis and targeted chemical modification. J. Biol. Chem. 1995, 270, 23065–23071. [Google Scholar] [CrossRef]
- Biswas, R.; Voggu, L.; Simon, U.K.; Hentschel, P.; Thumm, G.; Gotz, F. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett. 2006, 259, 260–268. [Google Scholar] [CrossRef]
- Koedijk, D.; Pastrana, F.R.; Hoekstra, H.; Berg, S.V.D.; Back, J.W.; Kerstholt, C.; Prins, R.C.; Bakker-Woudenberg, I.; van Dijl, J.M.; Buist, G. Differential epitope recognition in the immunodominant staphylococcal antigen A of Staphylococcus aureus by mouse versus human IgG antibodies. Sci. Rep. 2017, 7, 8141. [Google Scholar] [CrossRef]
- Brown, S.; Santa Maria, J.P., Jr.; Walker, S. Wall teichoic acids of gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef]
- Percy, M.G.; Grundling, A. Lipoteichoic acid synthesis and function in gram-positive bacteria. Annu. Rev. Microbiol. 2014, 68, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Tabor, D.E.; Yu, L.; Mok, H.; Tkaczyk, C.; Sellman, B.R.; Wu, Y.; Oganesyan, V.; Slidel, T.; Jafri, H.; McCarthy, M.; et al. Staphylococcus aureus Alpha-Toxin Is Conserved among Diverse Hospital Respiratory Isolates Collected from a Global Surveillance Study and Is Neutralized by Monoclonal Antibody MEDI4893. Antimicrob. Agents Chemother. 2016, 60, 5312–5321. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Valour, F.; Nguyen, N.T.Q.; Doan, T.M.N.; Koelkebeck, H.; Richardson, C.; Cheng, L.I.; Sellman, B.R.; Tkaczyk, C.; Diep, B.A. Multimechanistic Monoclonal Antibody Combination Targeting Key Staphylococcus aureus Virulence Determinants in a Rabbit Model of Prosthetic Joint Infection. Antimicrob. Agents Chemother. 2021, 65, e0183220. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.; Turner, A.P.; Piletsky, S.A. Advances in the manufacture of MIP nanoparticles. Trends Biotechnol. 2010, 28, 629–637. [Google Scholar] [CrossRef]
- Canfarotta, F.; Poma, A.; Guerreiro, A.; Piletsky, S. Solid-phase synthesis of molecularly imprinted nanoparticles. Nat. Protoc. 2016, 11, 443–455. [Google Scholar] [CrossRef]
- Piletsky, S.; Canfarotta, F.; Poma, A.; Bossi, A.M.; Piletsky, S. Molecularly Imprinted Polymers for Cell Recognition. Trends Biotechnol. 2020, 38, 368–387. [Google Scholar] [CrossRef]
- Liu, R.; Poma, A. Advances in Molecularly Imprinted Polymers as Drug Delivery Systems. Molecules 2021, 26, 3589. [Google Scholar] [CrossRef]
- Poma, A.; Whitcombe, M.J.; Piletsky, S. Plastic Antibodies. In Designing Receptors for the Next Generation of Biosensors; Piletsky, S.A., Whitcombe, M.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 105–129. [Google Scholar]
- Poma, A.; Guerreiro, A.; Whitcombe, M.J.; Piletska, E.V.; Turner, A.P.; Piletsky, S.A. Solid-Phase Synthesis of Molecularly Imprinted Polymer Nanoparticles with a Reusable Template-“Plastic Antibodies”. Adv. Funct. Mater. 2013, 23, 2821–2827. [Google Scholar] [CrossRef]
- Moczko, E.; Poma, A.; Guerreiro, A.; Perez de Vargas Sansalvador, I.; Caygill, S.; Canfarotta, F.; Whitcombe, M.J.; Piletsky, S. Surface-modified multifunctional MIP nanoparticles. N. Nanoscale 2013, 5, 3733–3741. [Google Scholar] [CrossRef]
- Subrahmanyam, S.; Guerreiro, A.; Poma, A.; Moczko, E.; Piletska, E.; Piletsky, S. Optimisation of experimental conditions for synthesis of high affinity MIP nanoparticles. Eur. Polym. J. 2013, 49, 100–105. [Google Scholar] [CrossRef]
- Poma, A.; Guerreiro, A.; Caygill, S.; Moczko, E.; Piletsky, S. Automatic reactor for solid-phase synthesis of molecularly imprinted polymeric nanoparticles (MIP NPs) in water. RSC Adv. 2014, 4, 4203–4206. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, A.; Poma, A.; Karim, K.; Moczko, E.; Takarada, J.; de Vargas-Sansalvador, I.P.; Turner, N.; Piletska, E.; de Magalhaes, C.S.; Glazova, N.; et al. Influence of surface-imprinted nanoparticles on trypsin activity. Adv. Healthc. Mater. 2014, 3, 1426–1429. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.; Brahmbhatt, H.; Watts, J.K.; Turner, N.W. Nucleoside-Tailored Molecularly Imprinted Polymeric Nanoparticles (MIP NPs). Macromolecules 2014, 47, 6322–6330. [Google Scholar] [CrossRef]
- Poma, A.; Brahmbhatt, H.; Pendergraff, H.M.; Watts, J.K.; Turner, N.W. Generation of novel hybrid aptamer-molecularly imprinted polymeric nanoparticles. Adv. Mater. 2015, 27, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Brahmbhatt, H.; Poma, A.; Pendergraff, H.M.; Watts, J.K.; Turner, N.W. Improvement of DNA recognition through molecular imprinting: Hybrid oligomer imprinted polymeric nanoparticles (oligoMIP NPs). Biomater. Sci. 2016, 4, 281–287. [Google Scholar] [CrossRef]
- Canfarotta, F.; Lezina, L.; Guerreiro, A.; Czulak, J.; Petukhov, A.; Daks, A.; Smolinska-Kempisty, K.; Poma, A.; Piletsky, S.; Barlev, N.A. Specific Drug Delivery to Cancer Cells with Double-Imprinted Nanoparticles against Epidermal Growth Factor Receptor. Nano Lett. 2018, 18, 4641–4646. [Google Scholar] [CrossRef]
- Bedwell, T.S.; Anjum, N.; Ma, Y.; Czulak, J.; Poma, A.; Piletska, E.; Whitcombe, M.J.; Piletsky, S.A. New protocol for optimisation of polymer composition for imprinting of peptides and proteins. RSC Adv. 2019, 9, 27849–27855. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody and Target | Clinical Trial | ||||||
|---|---|---|---|---|---|---|---|
| Targets | Antibody Functions | Antibody | Refs | Name [Company; NCT Number] | Status | Intervention | Refs |
| ClfA | Block Fg binding/agglutination of human plasma; displace FBG-bound bacteria; promote OPK | Mu/mAb (mAb 12-9, 11H10); Huz/mAb (Tefibazumab) | [44,51] | Tefibazumab (Aurexis) [Inhibitex] | Phase II (failed) | Huz/mAb (IgG1) | [52] |
| Tefibazumab (Aurexis1) [Inhibitex; NCT00198289] | Phase IIa (failed) | Huz/mAb (IgG1) | [53] | ||||
| FnBPB | Block Fn binding; promote OPK and nGr activation; reduce biofilm formation | Mu/mAb | [54,55] | ||||
| Cna | Block CN binding; displace CN from bacterial surface; promote OPK; block laminin and C1q binding | Mu/pAb; Mu/mAb | [56,57,58] | ||||
| SasG | Reduce biofilm formation | Ra/pAb | [43] | ||||
| Atl | Inhibit biofilm formation; promote OPK | Mu/pAb | [59,60,61] | ||||
| Atl-Amd | Promote OPK | Mu/pAb | [60] | ||||
| Atl-Gmd | Promote OPK; block bacterial division (binary fission); induce agglutination | IgG1 Mu/mAb (1C11) | [62] | ||||
| IsaA | Promote nGr activation (oxidative burst) and OPK by nGr (UK-66); promote OPK in whole blood (hUK-66); promote nGr activation, but not phagocytosis (1D9) | Mu/mAb (UK-66); Huz/mAb (hUK-66); Hu/mAb (1D9) | [63,64,65,66] | ||||
| WTA | Promote C3 deposition and OPK by nGr (Hu/pAb) | Hu/mAb; IgG Hu/mAb (THIOMAB) | [67,68] | DSTA4637S [Roche/Genentech; NCT03162250] | Phase Ib (ongoing) | THIOMABTM antibody (Hu/mAb; IgG1)-antibiotic conjugate | [69,70] |
| CP | Promote OPK (Mu/mAb) | Mu/mAb; Ra/pAb; Mu/pAb | [71,72] | AltaStaphTM [Nabi Biopharmaceuticals; NCT00063089] | Phase II (halted) | Polyclonal human IgG | [73] |
| AltaStaphTM [Nabi Biopharmaceuticals; NCT00066989] | Phase II (failed) | Polyclonal human IgG | [74] | ||||
| LTA | Promote OPK | Murine/human chimeric mAb (Pagibaximab) | [75] | Pagibaximab1 [Biosynexus; NCT00631800] | Phase II (finished) | Murine/human chimeric mAb | [76] |
| Pagibaximab1 [Biosynexus; NCT00646399] | Phase III (failed) | Murine/human chimeric mAb | [77] | ||||
| PNAG/ dPNAG | Promote OPK | IgG1 Hu/mAb (F598) | [78,79] | SAR279356 [Sanofi-Aventis; NCT01389700] | Phase IIa (terminated due to difficulty in patient recruitment) | Hu/mAb | [80] |
| DNABII | Disrupt established biofilms | Native Hu/mAb (TRL1068) | [81,82] | TRL1068 [Trellis BioscienceLLC; NCT04763759] | Phase I (recruiting) | Hu/mAb | [83] |
| AT | Neutralise toxin activity; modestly inhibit biofilm formation | Hu/mAb (MEDI4893) | [84,85,86] | MEDI4893 (Suvratoxumab) [MedImmune LLC; NCT02296320] | Phase II (successful) | Hu/mAb (IgG1) | [87] |
| AR-301 (Salvecin) [Aridis Pharmaceuticals; NCT01589185] | Phase IIa (successful) | Hu/mAb (IgG1) | [88] | ||||
| LukAB | Neutralise LukAB-mediated cytotoxicity; inhibit LukAB binding to I domain of CD11b | Hu/mAb (SA-13, -15 and -17) | [89] | ||||
| GrfA | Reduce colonies in organ | Recombinant human scFv | [90] | Aurograb [NeuTec Pharma Ltd/Novartis Pharma AG; NCT00217841] | Phase II (failed) | Single-chain antibody fragment (Fab) | [57,90] |
| PhnD | Inhibit biofilm formation under shear flow (S. aureus and S. epidermidis), promote OPK by nGr | Ra/pAb | [91] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Poma, A. Molecular Targets for Antibody-Based Anti-Biofilm Therapy in Infective Endocarditis. Polymers 2022, 14, 3198. https://doi.org/10.3390/polym14153198
Han J, Poma A. Molecular Targets for Antibody-Based Anti-Biofilm Therapy in Infective Endocarditis. Polymers. 2022; 14(15):3198. https://doi.org/10.3390/polym14153198
Chicago/Turabian StyleHan, Jiahe, and Alessandro Poma. 2022. "Molecular Targets for Antibody-Based Anti-Biofilm Therapy in Infective Endocarditis" Polymers 14, no. 15: 3198. https://doi.org/10.3390/polym14153198
APA StyleHan, J., & Poma, A. (2022). Molecular Targets for Antibody-Based Anti-Biofilm Therapy in Infective Endocarditis. Polymers, 14(15), 3198. https://doi.org/10.3390/polym14153198