1. Introduction
The interest in the application of chitosan in industry, including in pharmaceuticals, is growing. The suitability and performance of chitosan as a component of formulations for drug delivery applications has been investigated in numerous studies. These include controlled drug delivery applications, use as a component of mucoadhesive dosage forms [
1,
2], rapid release dosage forms [
3,
4,
5], improved peptide delivery [
6,
7], colonic drug delivery systems [
8], and use for gene delivery [
9]. The use of biocomposites based on chitosan in the field of tissue engineering is being widely studied. In the processes of tissue healing, chitosan serves as an extracellular matrix for cellular organization, helping in the attachment, proliferation, and differentiation of living cells [
10]. Chitosan can also be used as a unique eco-friendly active bio-sorbent for the sorption of heavy metal ions from polluted water. It is commonly extracted from marine waste such as shrimp shells or crustacean waste [
11]. Chemically speaking, it is a polysaccharide comprising copolymers of glucosamine and N-acetylglucosamine [
12]. Whereas chitosan is soluble in acidic media, it shows no solubility in neutral and alkaline aqueous solutions. This is in contrast to a lot of other high-molecular-weight polymers that show a neutral or anionic character [
13]. The polycationic nature of chitosan makes it a bioadhesive that readily binds to negatively charged surfaces such as mucosal membranes. Thereby it increases the adhesion to the mucosa and, as a result, enhances the time of contact for penetration of drug molecules. Hence, chitosan has an absorption-promoting effect due to its mucoadhesion properties, but also due to transient opening of the tight junctions of the mucosal cell membrane [
14].
Alginates have been extensively studied for oral, parenteral, pulmonary, and transdermal drug delivery [
15]. They are mainly derived from brown algae, but can also be extracted from bacterial sources [
16]. Sodium alginate consists of the sodium salt of alginic acid, which is a mixture of polyuronic acids composed of residues of D-mannuronic acid and L-guluronic acid [
17,
18]. Sodium alginate can be used alone or in the presence of calcium ions, because one of the most important and useful properties of alginates is its ability to form gels in the presence of multivalent metal ions, such as calcium. The incorporation of calcium salts in pellet formulations altered the drug release, depending on the solubility of the calcium salts used. However, in most cases, it was only a slightly slower drug release [
15]. As the use of calcium ions did not have a significant influence on the drug release from pellets made by the extrusion–spheronization process, it was decided to prepare alginate pellets without calcium ions.
Methocel K100M (hydroxypropyl methylcellulose, HPMC, hypromellose) was used as a reference polymer, with the intention of comparing the physicochemical properties and dissolution behavior of the pellets containing natural polymers and pellets containing well-established cellulose ether. Hypromellose hydrophilic matrix systems have been widely studied and many successful products on the market utilize this versatile extended release technology [
19]. Hypromellose is an excellent excipient for formulation of classical dosage forms and advanced drug delivery systems. New methods of hypromellose processing include spray draying, hot-melt extrusion, 3D printing, and electrospinning [
20].
Pellets are described as geometrically-defined agglomerates, they are free-flowing, spherical or semi-spherical solid units with a size range of about 0.5–1.5 mm and are intended for oral administration [
21]. They differ from granules and conventional agglomerates in shape, surface area, and narrow particle size distribution [
22]. Pellets are used to control the drug release from the formulation or to prevent dose dumping. They offer reduced variation in gastric emptying rate and intestinal transit time, and also disperse freely in the gastrointestinal tract. Pelletization is a way to separate incompatible drugs or mask an unpleasant taste [
21]. Pellets can also reduce irritant effects on the gastric mucosa [
23]. Pharmaceutical pellets are typically manufactured via extrusion–spheronization. Afterwards, pellets are usually subjected to dissolution studies and screening, to achieve the desired physiochemical properties and size distribution; for which, we successfully applied dynamic image analysis (DIA) [
21]. DIA is a modern particle characterization method with exclusive 3D measurement software. Compared with the other traditional particle evaluation techniques, DIA has the major advantage that the instrument provides images of fast moving particles and is sensitive to differences in size and shape characteristics; therefore, it is being increasingly applied to particle evaluation in various processes, pharmaceuticals included [
24]. DIA has already been used in many studies for different types of materials: pharmaceutical excipients, minitablets, tailings, talc, concrete aggregates, sediments, volcanic ash, calcite, and coal [
25]. Pellets, often also coated, are administered in the form of hard gelatin capsules or multi-unit pellet system (MUPS) tablets that quickly disperse in the stomach [
26,
27].
Acyclovir, the model drug used in this study, is the most widely used drug for infections of cutaneous herpes, genital herpes, chicken pox, varicella zoster infections, and herpes keratitis. Oral acyclovir is generally used five times a day (200 or 400 mg tablets) [
28]. Frequent dosing of acyclovir is based on the physicochemical properties of acyclovir. Acyclovir is described as ‘‘slightly soluble in water’’ in different pharmacopoeias. The partition coefficient (log P) in n-octanol at 22 °C is 1.57. A log P value greater than that of metoprolol (1.72) indicates high permeability. As the log P value reported for acyclovir lies far below that value, it is expected to have low permeability. Acyclovir’s absolute bioavailability following oral administration has been reported to be in a range of 10–30% [
29].
This study aimed to determine whether the natural polymers, sodium alginate and chitosan, are full-fledged substitutes for semi-synthetic HPMC in the role of a wetting agent in the formulation of pellets and to what extent they affect the quality properties of pellets, compressed MUPS tablets, as well as the in vitro dissolution of a model drug, acyclovir.
Some research papers have focused on chitosan pellet production by the extrusion–spheronization process, but there is a lack of the papers which have also focused on the transformation process into the final MUPS tablet dosage form [
13,
30,
31]. MUPS tablets are composed of tablets containing uncoated or coated pellets. The compaction process of pellets to form the MUPS tablet can cause variations of the mechanical properties and dissolution behavior of the drug. According to some studies, MUPS tablet formulations could enhance the membrane permeation of drugs, specifically by inclusion of functional excipients in MUPS tablet formulations. Interest has been shown in the use of mucoadhesive polymers as functional excipients for this purpose [
32]. This might be a way to enhance the bioavailability of poorly bioavailable APIs such as acyclovir; therefore, we formulated pellets with a content of chitosan or sodium alginate.
2. Materials and Methods
Materials used for the pellet formulation were acyclovir of European Pharmacopea quality, which was obtained from Union Quimico Farmaceutica S.A., Barcelona, Spain. Chitosan (medium molecular weight 90–310 kDa, degree of deacetylation 82%) and sodium alginate (molecular weight 120,000–190,000 g/mol) were purchased from Sigma-Aldrich Chemie GmbH, Steinheim, Germany, Methocel K100M; co-processed microcrystalline cellulose with lactose monohydrate and natrium carboxymethyl cellulose (Specicell®140) were supplied by The Dow Chemical company, Michigan, USA. Acetic acid, sodium chloride, and hydrochloric acid were purchased from Centralchem s.r.o, Bratislava, Slovakia. Pellets were filled into hard gelatin capsules, which were provided by Interpharm a.s., Bratislava, Slovakia. Purified water was freshly prepared by distillation.
2.1. Preparation of Pellets
The binder solutions were prepared by dispersing polymers in purified water (chitosan 2%, w/w, sodium alginate 2%, w/w, hydroxypropyl methylcellulose 2%, w/w). Chitosan solution was acidified using acetic acid (3%, w/w). The powdered components, specifically acyclovir and co-processed microcrystalline cellulose with lactose monohydrate, were homogenized and subsequently wetted with the solutions of binders in a mixer–granulator Diosna planetary mixer P1-6 (Diosna GmbH, Osnabrück, Germany). The wet homogenized blend was extruded with a Gabler Laboratory extruder (Gabler Engineering GmbH, Malsch, Germany) through a shaping die of 0.8 mm diameter, while the speed of the rotating screw was 40 rpm. The extruded product was broken and spheronized with a Gabler Spheronizer (Gabler Engineering GmbH, Malsch, Germany), with the speed of the rotating plate 1200 rpm for 3 min. Pellets were dried in a fluid bed dryer Glatt GPCG2 Lab System (Glatt GmbH, Binzen, Germany). The temperature of the inlet air was 50 °C, and the drying process was maintained till the pellet moisture was less than 3%, measured using a Mettler-Toledo Halogen Moisture Analyzer HG63 (Mettler-Toledo GmbH, Greifensee, Switzerland). Three different batches of pellets were prepared, varying in the kind of binder used for preparation (Sodium alginate, chitosan, Methocel K100M hereinafter referred to as HPMC).
2.2. Characterisation of Pellets
2.2.1. Surface Morphology
The morphology of the pellets was characterized in greater depth using a scanning electron microscope, Tescan Vega3 (Tescan Orsay Holding, a.s., Brno, Czech Republic). It was used for examination at magnifications of 45×, 150×, and 300×.
2.2.2. Measurement of Pellet Size, Shape, and Sphericity
There are several methods for determining the shape and size of particles based on the principle of image analysis. One such method is dynamic image analysis (DIA), in which particles are captured in motion by digitalizing photos of each particle from a camera and storing them in an image file. The particles tumble and rotate, and are illuminated by stroboscopic light. The images are used to calculate morphological parameters based on the known size and location of the pixels in each image. DIA, unlike other techniques, can report different size parameters as it processes the image of particles. The size distribution and shape of the pellets were analyzed using a PartAn 3D device (Microtrac MRB, Haan, Germany).
The characteristic dimension for determining the size of a pellet was the area equivalent diameter (
Da) Equation (1), obtained from multiple photos of individual pellets.
where
A is the area of the projected particle.
Mean pellet size (dmean) was calculated according to the relationship in Equation (2).
Various shape parameters such as sphericity (φ) were used to determine the shape of the pellets, Equation (3).
A sphericity value φ = 1 corresponds to a perfect sphere [
25].
We also observed the mean pellet size change and sphericity change during the dissolution testing of the pellets, which were withdrawn from the dissolution apparatus after 2, 4, and 6 h.
2.2.3. Particle-Size Distribution Estimation by Analytical Sieving
A sieve analysis was performed on a Test Sieve Shaker Haver EML 200 digital T (Haver & Boecker, Oelde, Germany). Sieve sizes used for the analysis were 250 µm, 355 µm, 500 µm, 710 µm, 900 µm, and 1250 µm, and the time of agitation was 5 min. The weight of material retained on each sieve was accurately determined. This test gives the weight percentage of pellets in each sieve size range.
2.2.4. Mechanical Resistance of Pellets
The pellets (15 g) were poured into a glass vial and put into a laboratory shaker to agitate for 10 min. Then the pellets were sieved through the sieve with a nominal aperture of 250 µm and weighed again. The pellet loss after agitation, expressed as a percentage mean of three parallel measurements, corresponds to the mechanical resistance.
2.2.5. Determination of Matrix Erosion
Determination of matrix erosion was performed in the same apparatus, under the same conditions, as the dissolution testing. The baskets containing 0.4 g of pellets were removed from the dissolution medium after 2, 4, and 6 h and dried to a constant weight at 50 °C. The difference between the initial and the final weight was expressed as the percentage of matrix erosion [
30].
2.3. Resistance to Crushing of MUPS Tablets
The resistance to crushing of MUPS tablets was tested using a tablet hardness tester Schleuniger 2E (Dr. K. Schleuniger & Co., Solothurn, Switzerland). Ten tablets were placed into the apparatus horizontally, the force (N) needed for their crushing/destruction was recorded and expressed as a mean ± SD.
2.4. Dissolution Profiles
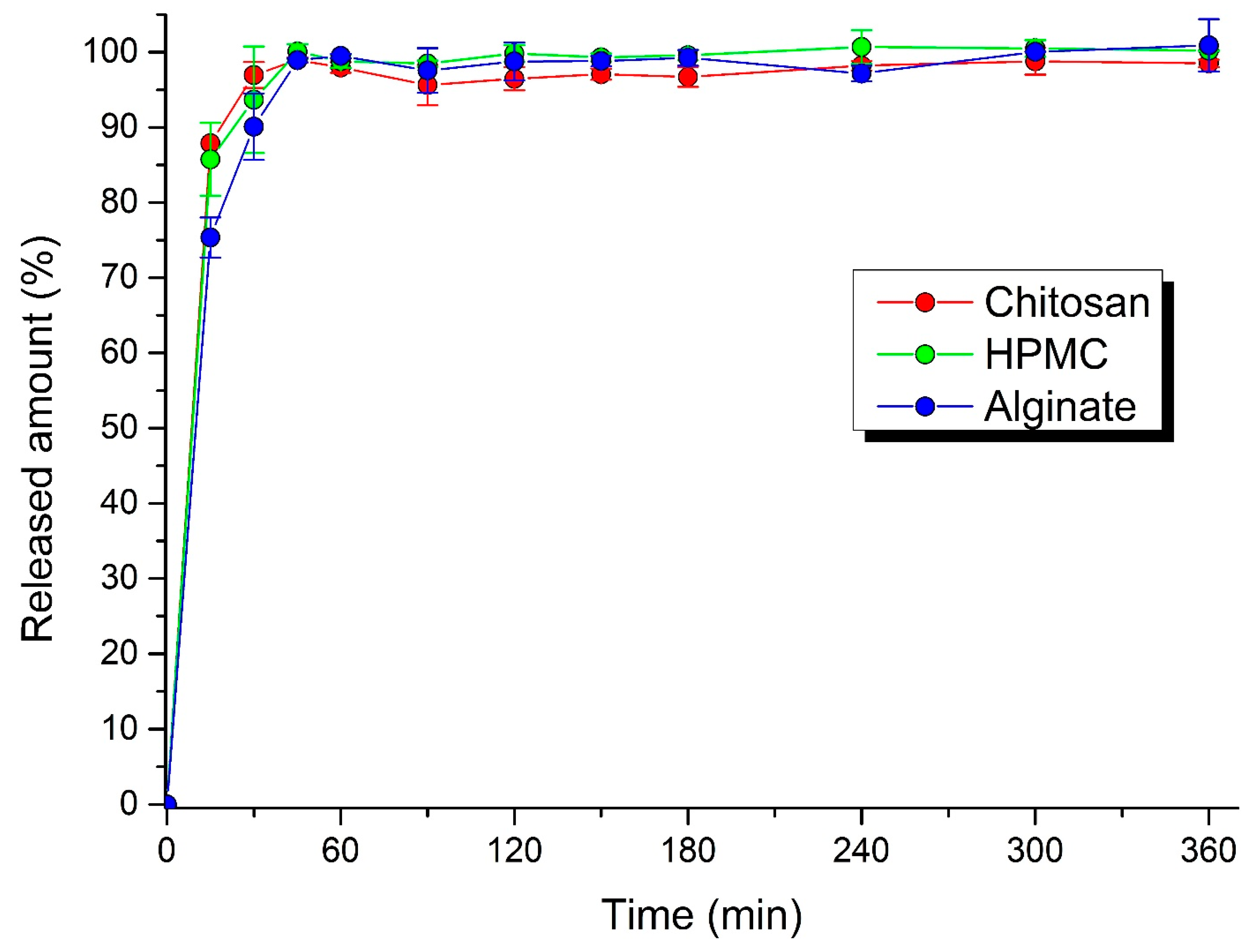
The pellets were filled into hard gelatin capsules or compressed into MUPS tablets. A rotary tablet press machine (Romaco Kilian, Cologne, Germany) was set up to produce tablets of average weight 0.300 g and a hardness between 50 to 80 N. Gelatin capsules were filled manually with 0.400 g of pellets. In vitro dissolution testing was performed using a basket-type dissolution tester Erweka DT 6 (Erweka GmbH, Langen, Germany). A simulated gastric fluid without pepsin, prepared by dissolving 2.0 g of sodium chloride in 7.0 mL of hydrochloric acid, replenished with purified water up to 1000 mL, was used as the dissolution medium (pH value approximately 1.2). The rotation of baskets was set to 50 rpm. The dissolution testing lasted 6 h, while the dissolution medium was permanently heated to 37 ± 0.5 °C. The samples of dissolution medium were withdrawn at fixed intervals (5, 10, 15, 30, 45, 60, 90, 120, 180, 240, 300, 360 min). The released amount of acyclovir in solutions was detected spectrophotometrically at 255 nm on a Genesys™ 10S UV-Visible Spectrophotometer (Thermo Scientific, Waltham, MA, USA) against a blank (the dissolution medium).
2.5. The Similarity of Dissolution Profiles
The dissolution profiles were compared through the determination of the difference factor
f1 and similarity factor
f2. The difference factor
f1 expresses the percent difference between the two dissolution profiles at each time point. It is a measurement of the relative error between the two profiles [
33]. It is calculated as a sum of the absolute values for the differences between the tested product (
T) and the reference product (
R), relative to the sum of the mean percentage of the released drug from the reference product, as Equation (4) describes:
The similarity factor
f2 is a logarithmic reciprocal square root transformation of the sum of squared differences between the profiles of the tested and the reference product. It represents the measurement of similarity in the percentage (%) of dissolution between the “average” dissolution profiles, where n is the number of time points and
Rt and
Tt are the mean percentages of the released drug from the (
R) and (
T) products [
34]:
4. Conclusions
Pellets, as a microparticulate delivery system, provide significant benefits for oral administration. Compared to oral granules, pellets have a regular spherical shape and, due to more complex technological manufacturing procedures, they are also more mechanically resistant. These facts were confirmed by the excellent results of the sphericity test and the mechanical resistance, while the highest sphericity was achieved under the influence of the chitosan binder. The prepared pellets, with drug release influenced by matrix erosion, were mechanically resistant (near 100%), no matter which binder was used. In contrast, the mechanical resistance of the MUPS tablets, pressed under the same conditions, altered significantly, depending on the binder used in the pellets, while the lowest was recorded with chitosan. Another important finding is that the dissolution profiles, depicting the release of acyclovir under in vitro conditions from pellets filled into capsules and MUPS tablets, were remarkably similar, as evidenced by the similarity factor f2 moving from 61.19 to 65.41. Due to the detection process, it can be concluded that pressing pellets will not cause their destruction. To slow down the drug release, either the technique of coating the pellets themselves, or directly coating MUPS tablets in polymers that slowly dissolve in gastric and intestinal fluid, have to be used for this purpose. Finally, it can be stated that chitosan and alginate sodium are suitable natural binder alternatives to HPMC, ensuring a similar quality, physical properties, and drug release from pellets.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}