
Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer


Abstract

1. Introduction
2. Model and Simulation Methodology
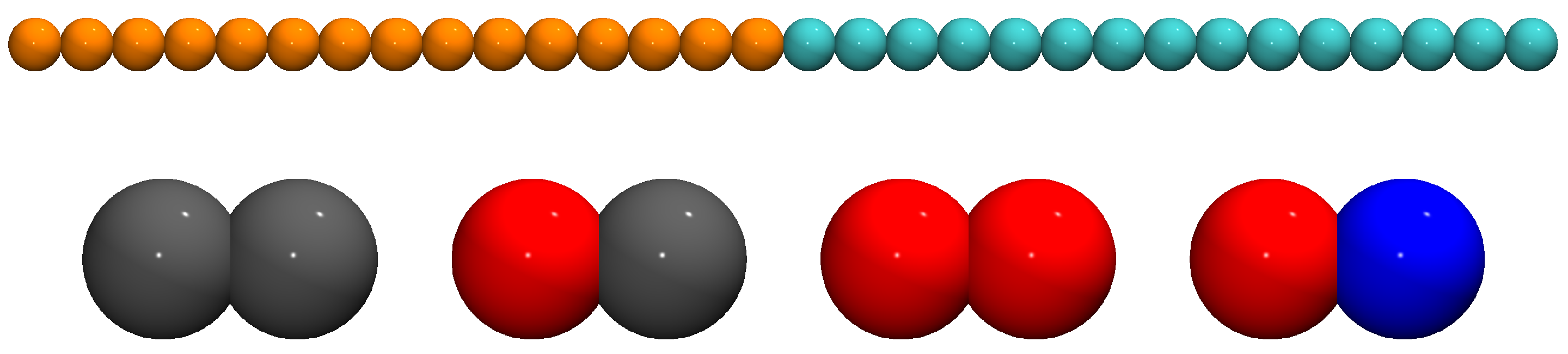
2.1. Molecular Model
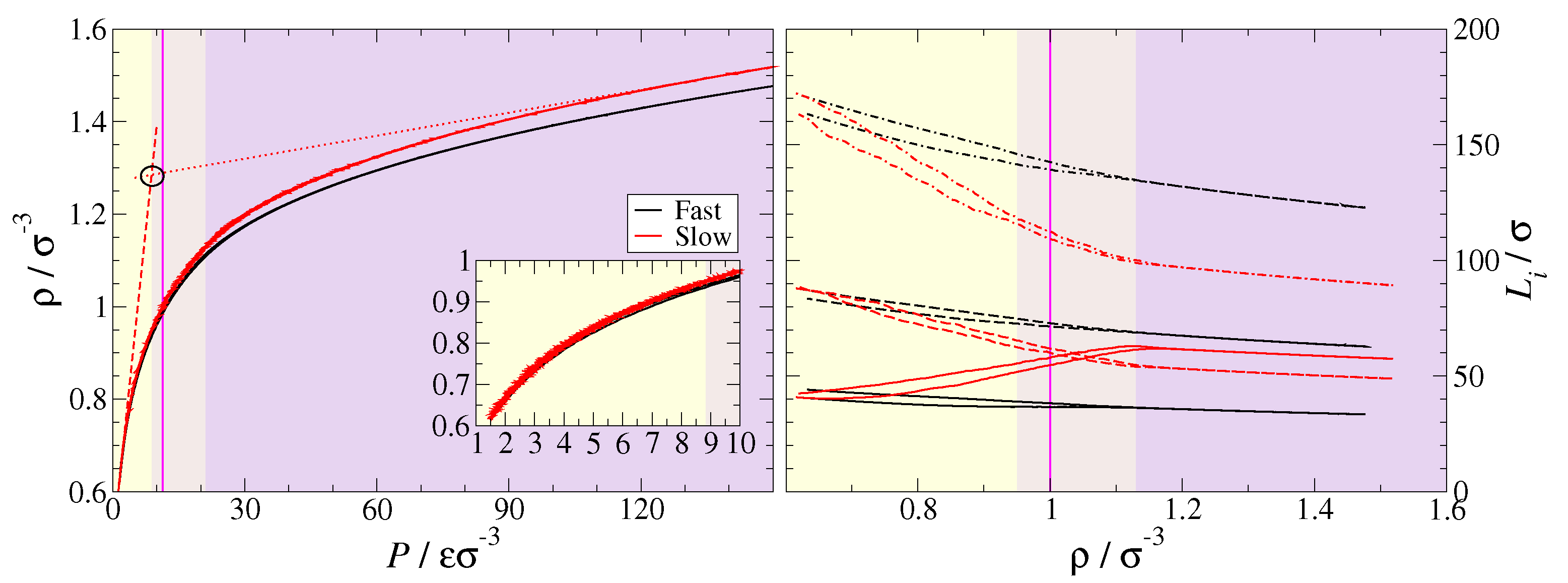
2.2. Simulation Methodology
2.3. Data Analysis
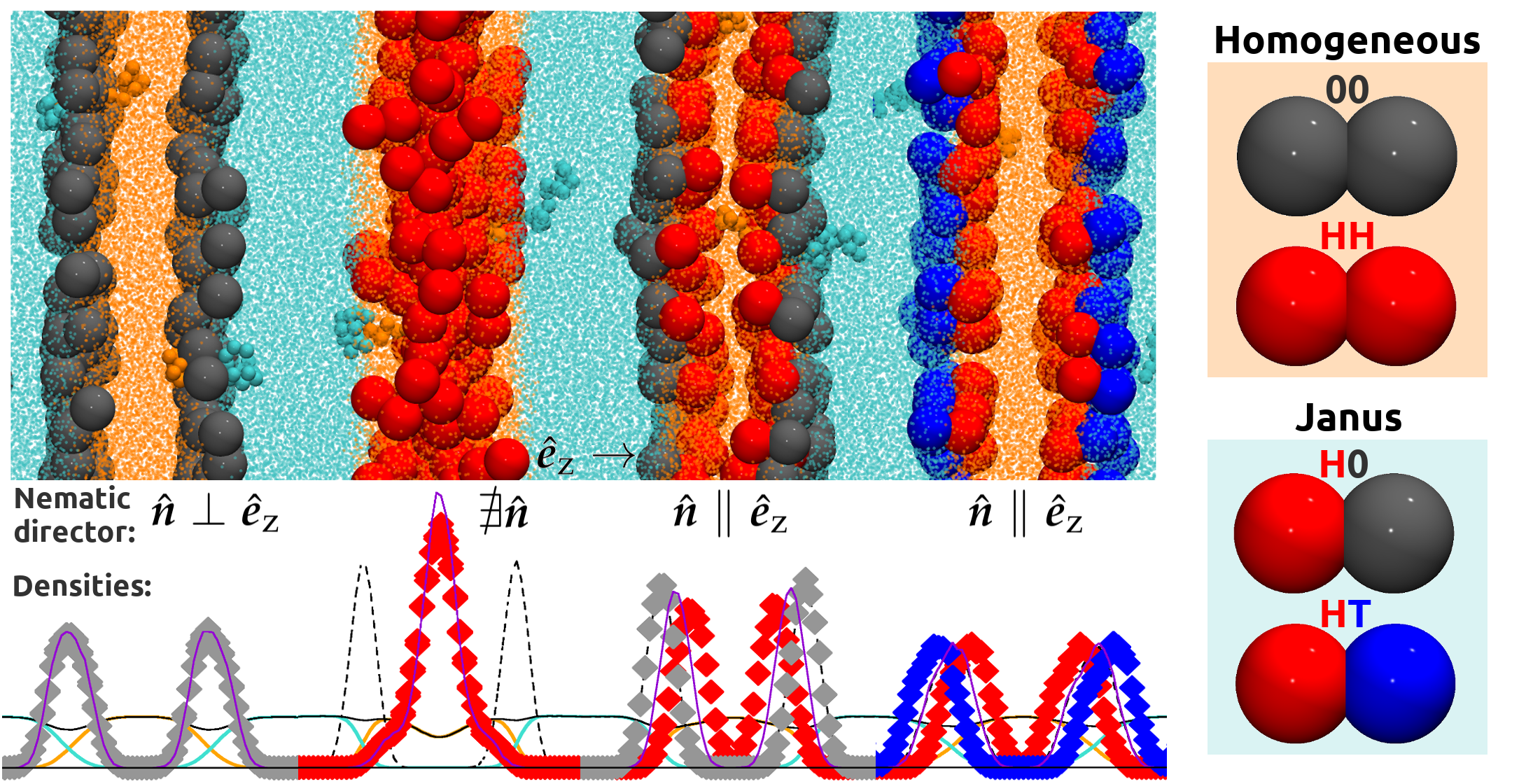
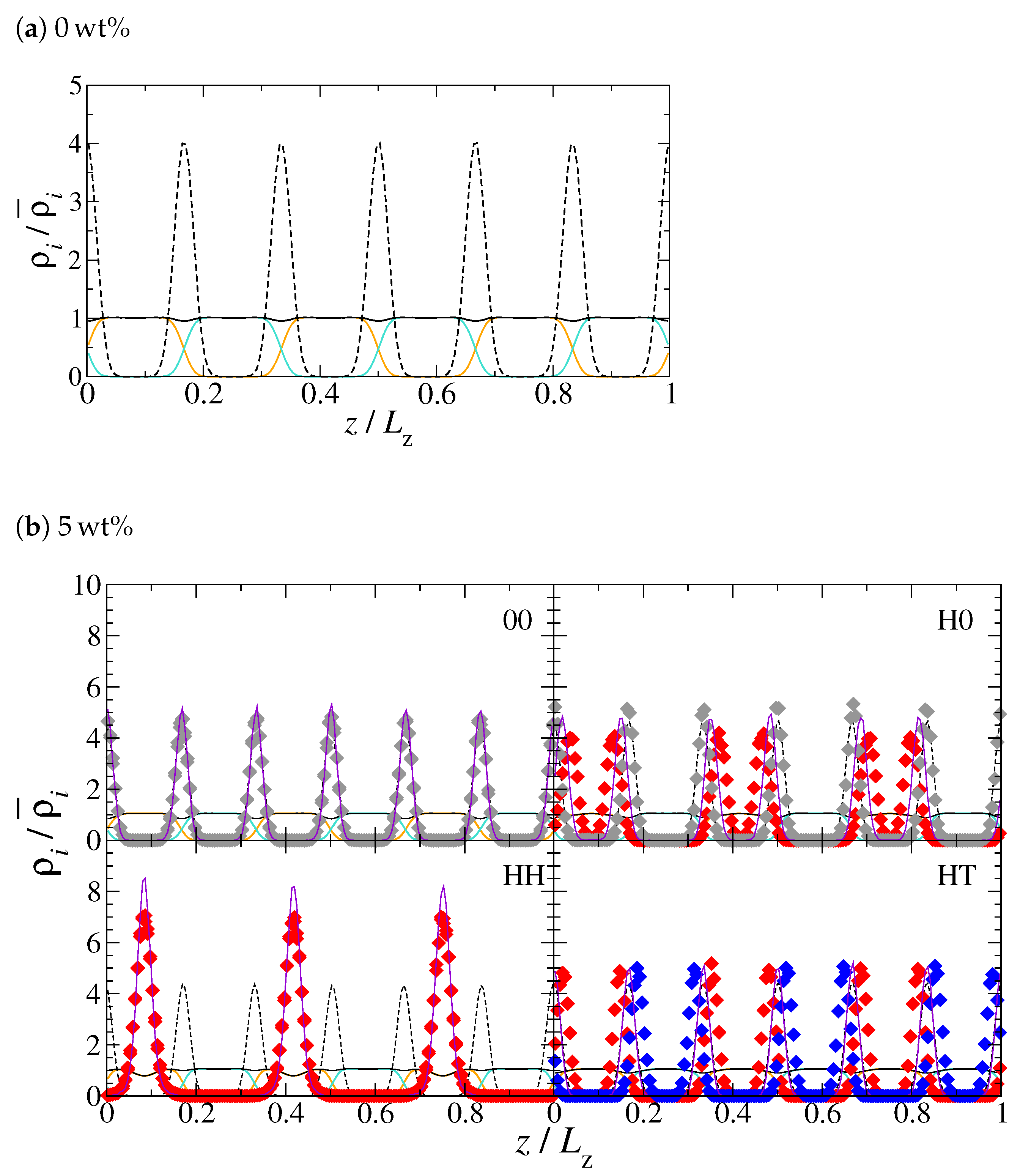
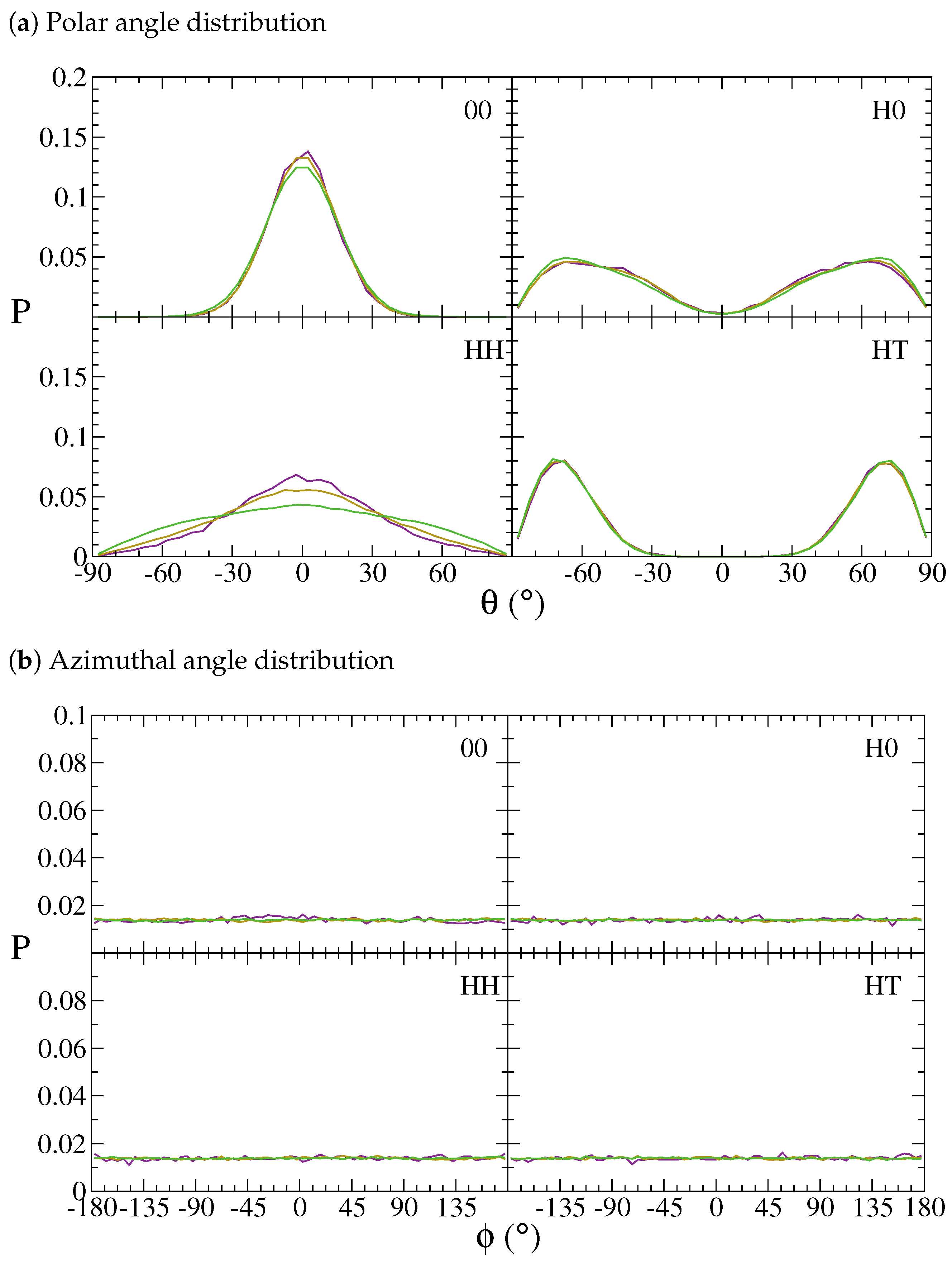
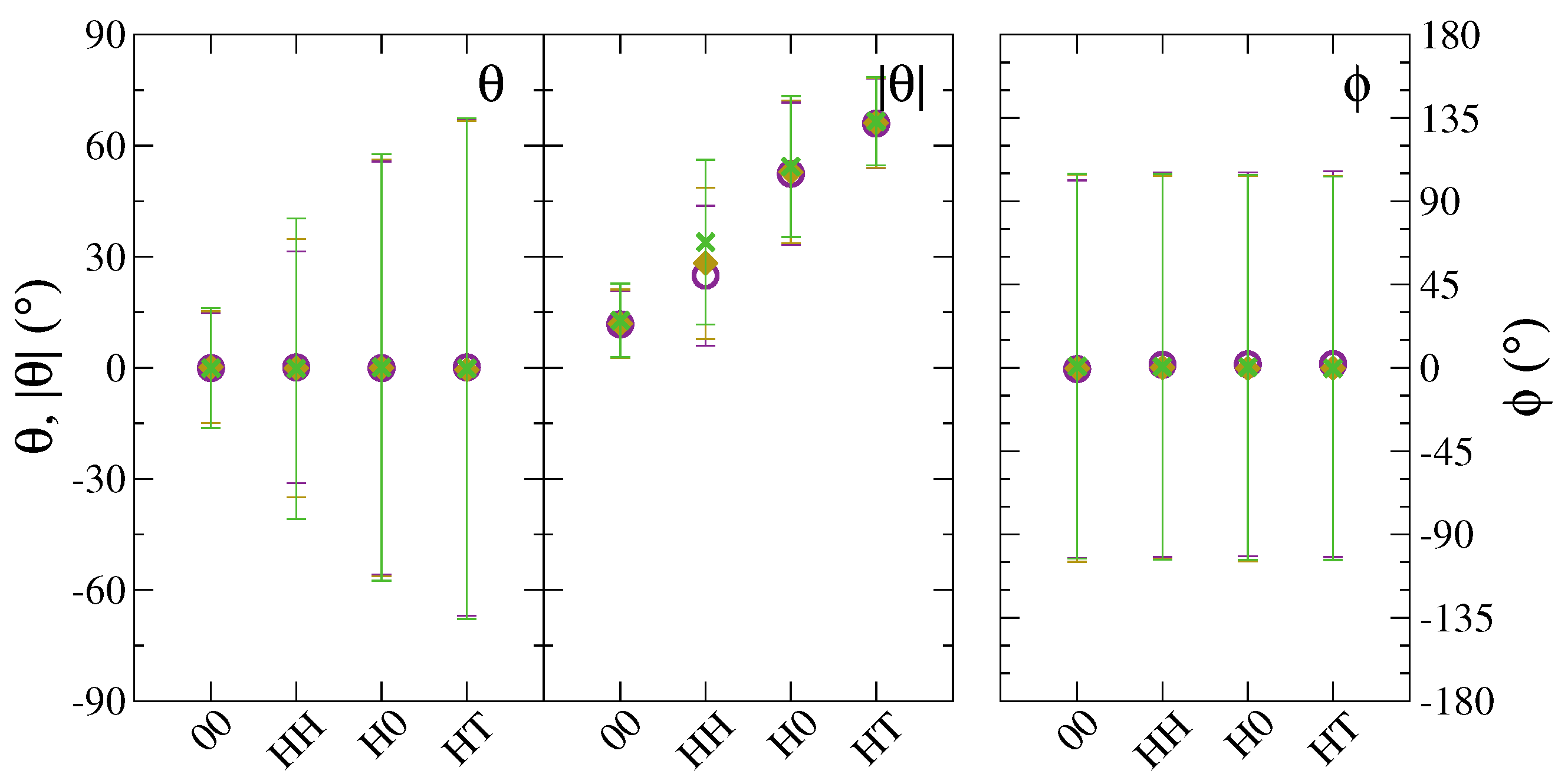
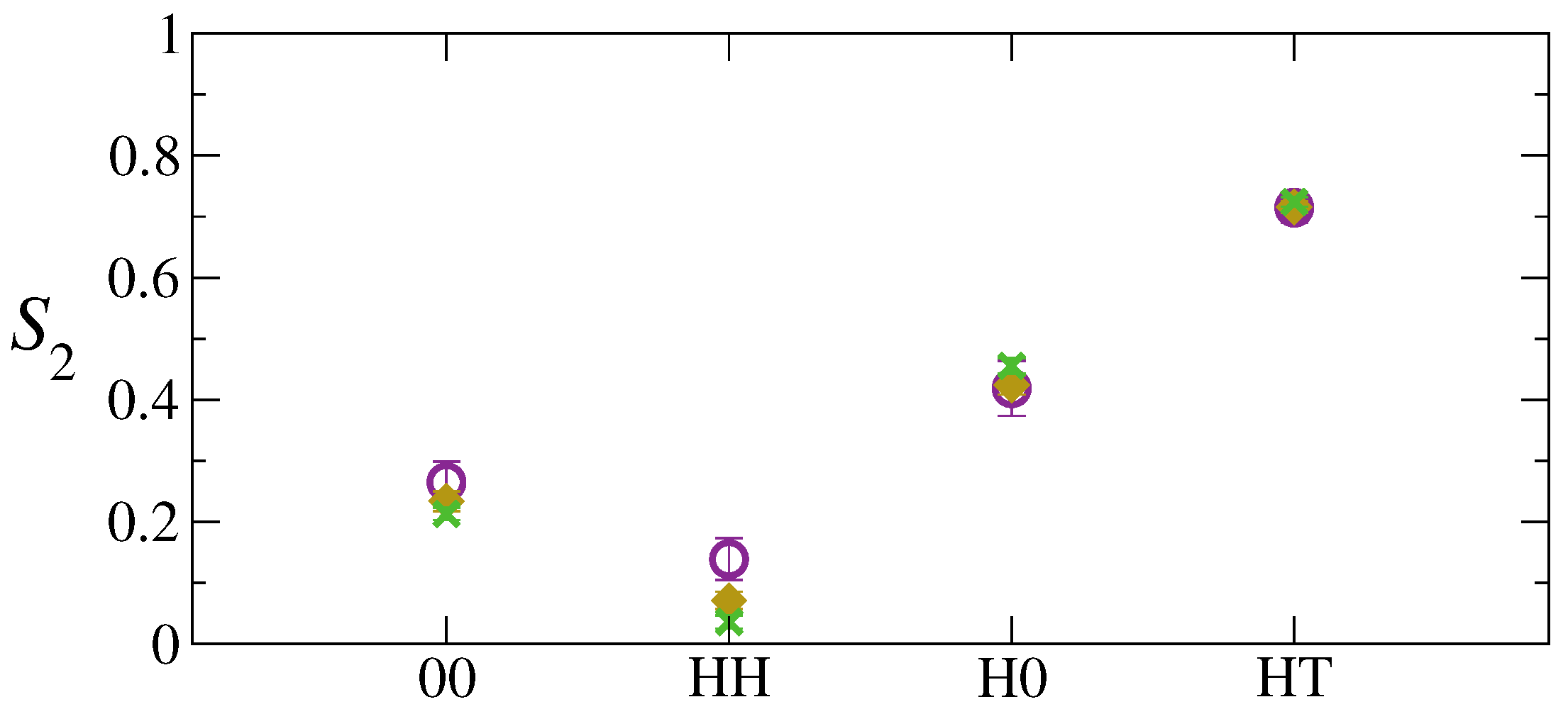
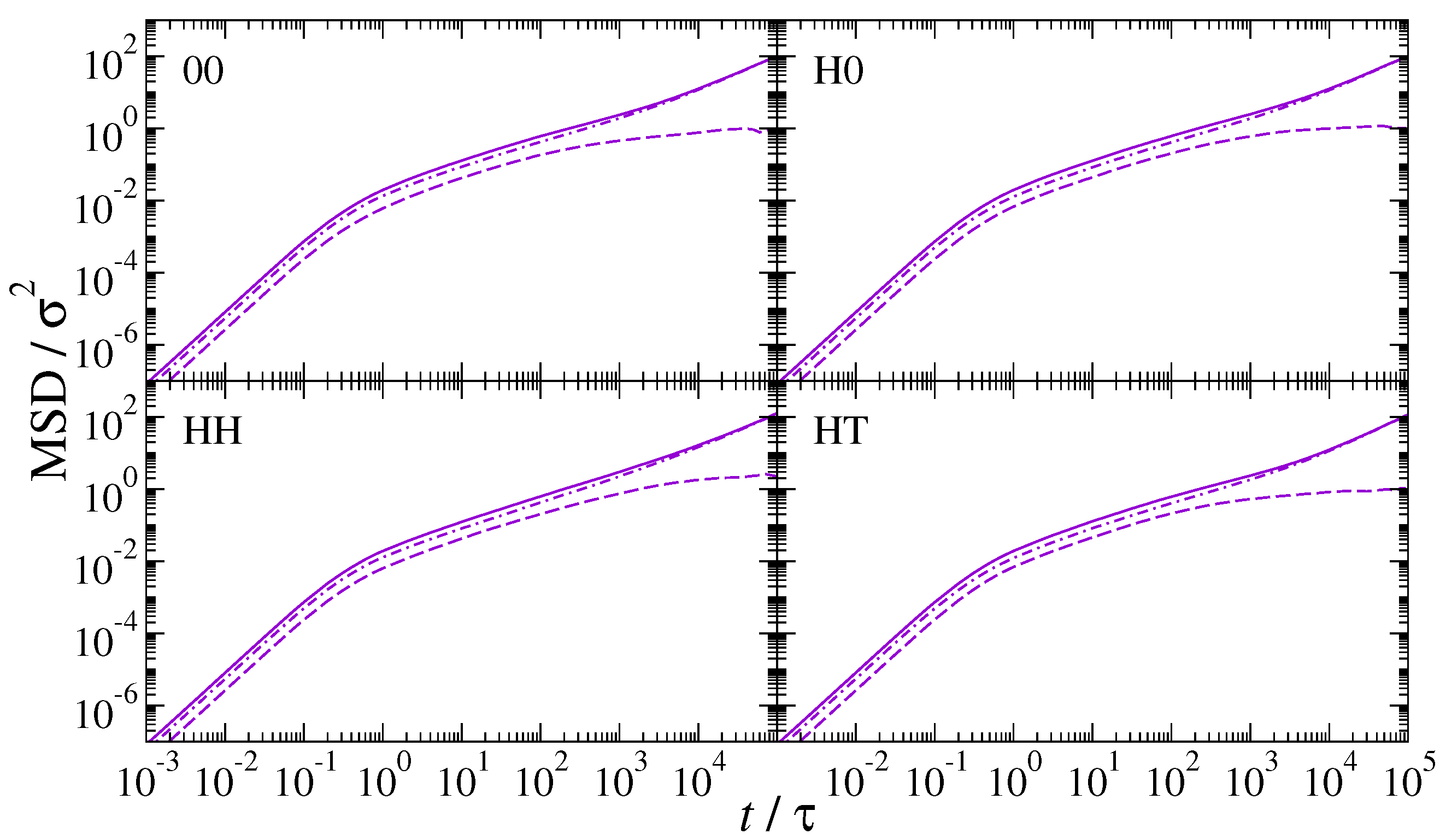
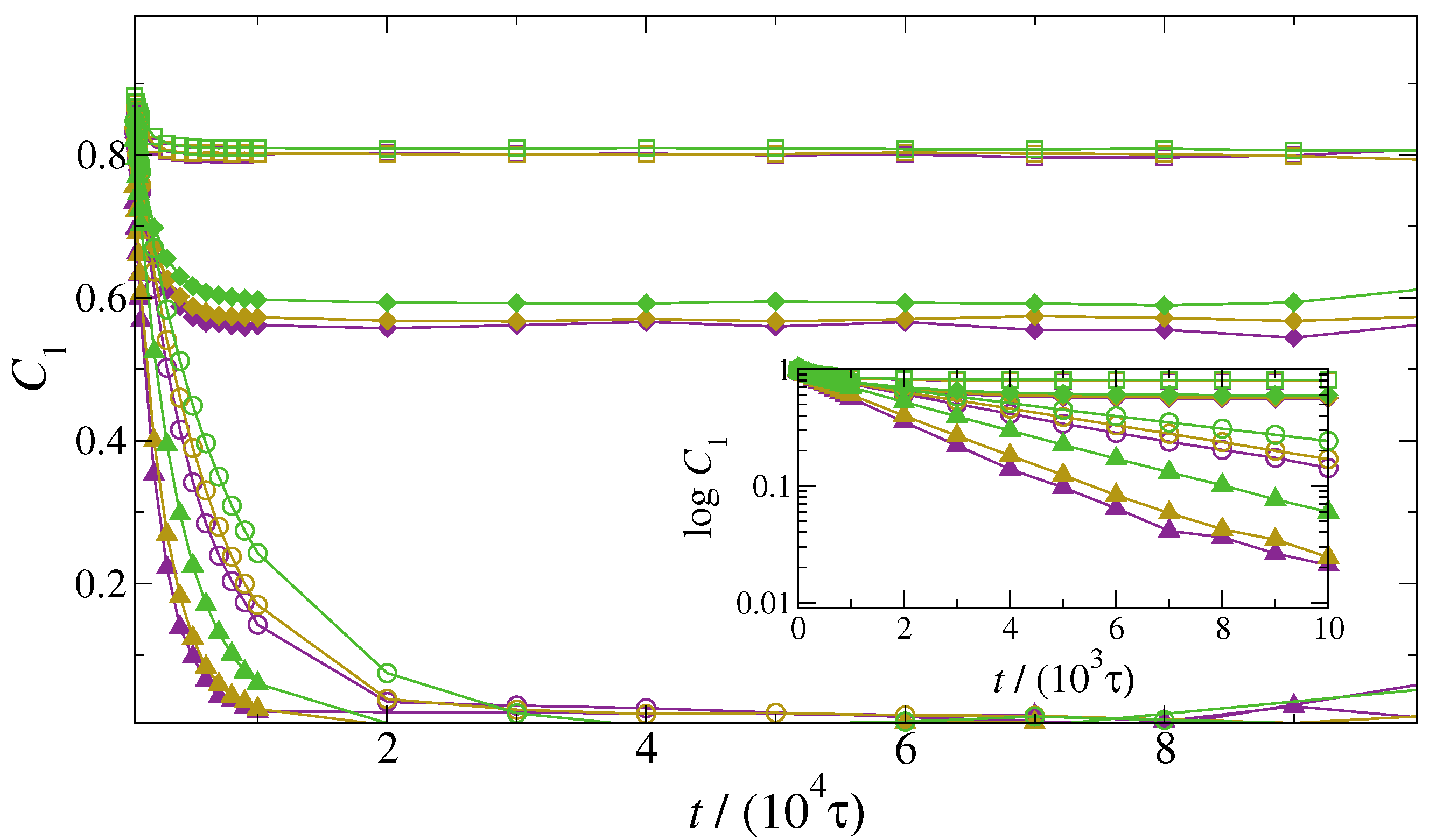
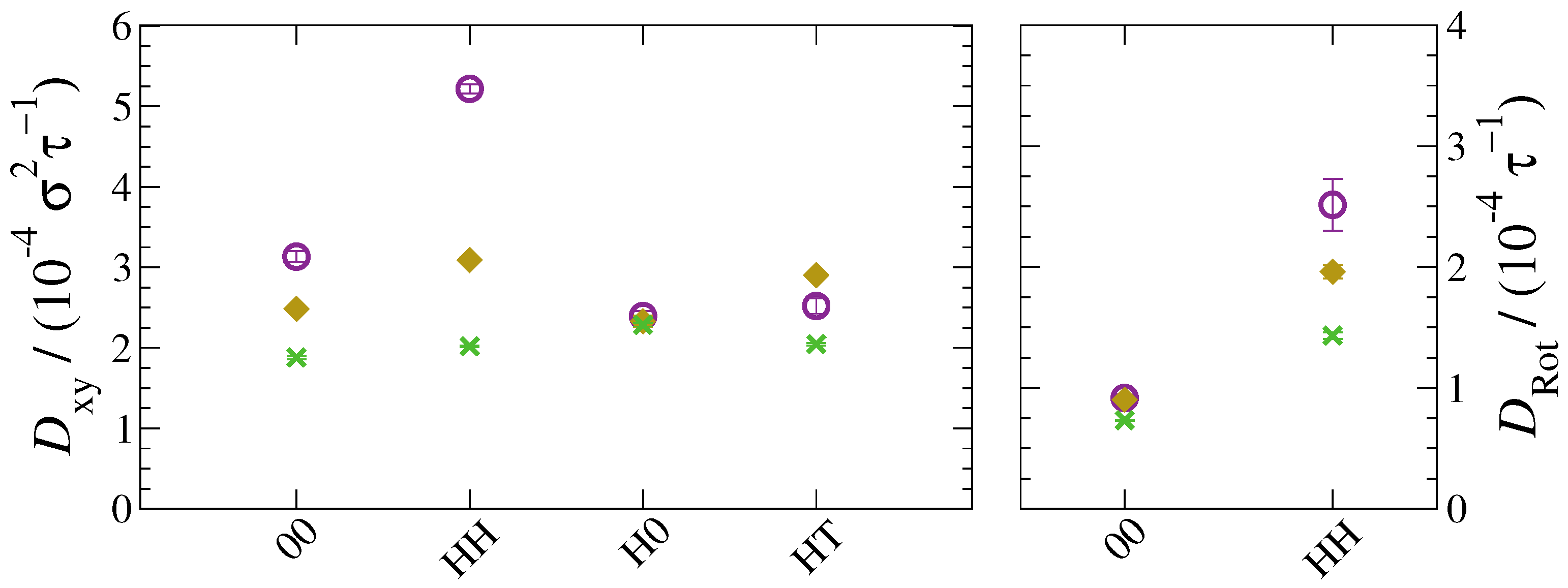
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PNC | Polymer nanocomposite |
| NP | Nanoparticle |
| BCP | Block copolymer |
| ND | Nano-dimer |
| MD | Molecular Dynamics |
| CG | Coarse-grain(ed) |
| LJ | Lennard–Jones (potential) |
| WCA | Weeks–Chandler–Andersen (potential) |
| FENE | Finitely Extensible Non-linear Elastic (potential) |
| NPT | Isothermal-isobaric or constant N, P and T (ensemble) |
| MSD | Mean-squared displacement |
References
- Yang, Q.; Loos, K. Janus nanoparticles inside polymeric materials: Interfacial arrangement toward functional hybrid materials. Polym. Chem. 2017, 8, 641–654. [Google Scholar] [CrossRef]
- Puglia, D.; Kenny, J. Chapter 7—Structure-property relationships of thermoset nanocomposites. In Thermosets, 2nd ed.; Guo, Q., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 231–276. [Google Scholar] [CrossRef]
- Müller, K.; Bugnicourt, E.; Latorre, M.; Jorda, M.; Echegoyen Sanz, Y.; Lagaron, J.M.; Miesbauer, O.; Bianchin, A.; Hankin, S.; Bölz, U.; et al. Review on the Processing and Properties of Polymer Nanocomposites and Nanocoatings and Their Applications in the Packaging, Automotive and Solar Energy Fields. Nanomaterials 2017, 7, 74. [Google Scholar] [CrossRef]
- Duncan, T.V. Applications of nanotechnology in food packaging and food safety: Barrier materials, antimicrobials and sensors. J. Colloid Interface Sci. 2011, 363, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Muñoz, P.; Cerisuelo, J.P.; Domínguez, I.; López-Carballo, G.; Catalá, R.; Gavara, R. Chapter 8—Nanotechnology in Food Packaging. In Nanomaterials for Food Applications; López Rubio, A., Fabra Rovira, M.J., Martínez Sanz, M., Gómez-Mascaraque, L.G., Eds.; Micro and Nano Technologies; Elsevier: Amsterdam, The Netherlands, 2019; pp. 205–232. [Google Scholar] [CrossRef]
- Videira-Quintela, D.; Martin, O.; Montalvo, G. Recent advances in polymer-metallic composites for food packaging applications. Trends Food Sci. Technol. 2021, 109, 230–244. [Google Scholar] [CrossRef]
- Díez-Pascual, A.M.; Luceño Sánchez, J.A.; Peña Capilla, R.; García Díaz, P. Recent Developments in Graphene/Polymer Nanocomposites for Application in Polymer Solar Cells. Polymers 2018, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, I.E.; Akkuratov, A.V.; Troshin, P.A. Chapter 15—Polymer nanocomposites for solar cells: Research trends and perspectives. In Nanomaterials for Solar Cell Applications; Thomas, S., Sakho, E.H.M., Kalarikkal, N., Oluwafemi, S.O., Wu, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 557–600. [Google Scholar] [CrossRef]
- Kumar, S.K.; Ganesan, V.; Riggleman, R.A. Perspective: Outstanding theoretical questions in polymer-nanoparticle hybrids. J. Chem. Phys. 2017, 147, 020901. [Google Scholar] [CrossRef] [PubMed]
- Starr, F.W.; Douglas, J.F.; Glotzer, S.C. Origin of particle clustering in a simulated polymer nanocomposite and its impact on rheology. J. Chem. Phys. 2003, 119, 1777–1788. [Google Scholar] [CrossRef]
- Meng, D.; Kumar, S.K.; Cheng, S.; Grest, G.S. Simulating the miscibility of nanoparticles and polymer melts. Soft Matter 2013, 9, 5417–5427. [Google Scholar] [CrossRef]
- Zhao, D.; Di Nicola, M.; Khani, M.M.; Jestin, J.; Benicewicz, B.C.; Kumar, S.K. Role of block copolymer adsorption versus bimodal grafting on nanoparticle self-assembly in polymer nanocomposites. Soft Matter 2016, 12, 7241–7247. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.; Grest, G.S.; Kumar, S.K. Assembly of Polymer-Grafted Nanoparticles in Polymer Matrices. ACS Nano 2020, 14, 13491–13499. [Google Scholar] [CrossRef] [PubMed]
- Bieligmeyer, M.; Taheri, S.M.; German, I.; Boisson, C.; Probst, C.; Milius, W.; Altstädt, V.; Breu, J.; Schmidt, H.W.; D’Agosto, F.; et al. Completely Miscible Polyethylene Nanocomposites. J. Am. Chem. Soc. 2012, 134, 18157–18160. [Google Scholar] [CrossRef] [PubMed]
- Frischknecht, A.L.; Hore, M.J.A.; Ford, J.; Composto, R.J. Dispersion of Polymer-Grafted Nanorods in Homopolymer Films: Theory and Experiment. Macromolecules 2013, 46, 2856–2869. [Google Scholar] [CrossRef]
- Cobo Sánchez, C.; Wåhlander, M.; Taylor, N.; Fogelström, L.; Malmström, E. Novel Nanocomposites of Poly(lauryl methacrylate)-Grafted Al2O3 Nanoparticles in LDPE. ACS Appl. Mater. Interfaces 2015, 7, 25669–25678. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Su, Y.; Wen, X.; Wang, D. Manipulating crystallization behavior of poly(ethylene oxide) by functionalized nanoparticle inclusion. Polymer 2019, 165, 28–38. [Google Scholar] [CrossRef]
- Zhu, T.; Rahman, M.A.; Benicewicz, B.C. Synthesis of Well-Defined Polyolefin Grafted SiO2 Nanoparticles with Molecular Weight and Graft Density Control. ACS Macro Lett. 2020, 9, 1255–1260. [Google Scholar] [CrossRef]
- Matsen, M.W. The standard Gaussian model for block copolymer melts. J. Phys. Condens. Matter 2001, 14, R21–R47. [Google Scholar] [CrossRef]
- Fredrickson, G.H.; Ganesan, V.; Drolet, F. Field-Theoretic Computer Simulation Methods for Polymers and Complex Fluids. Macromolecules 2002, 35, 16–39. [Google Scholar] [CrossRef]
- García Daza, F.A.; Colville, A.J.; Mackie, A.D. Mean-Field Coarse-Grained Model for Poly(ethylene oxide)-Poly(propylene oxide)-Poly(ethylene oxide) Triblock Copolymer Systems. Langmuir 2015, 31, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Shou, Z.; Buxton, G.A.; Balazs, A.C. Predicting the self-assembled morphology and mechanical properties of mixtures of diblocks and rod-like nanoparticles. Compos. Interfaces 2003, 10, 343–368. [Google Scholar] [CrossRef]
- Hur, K.; Hennig, R.G.; Escobedo, F.A.; Wiesner, U. Mesoscopic structure prediction of nanoparticle assembly and coassembly: Theoretical foundation. J. Chem. Phys. 2010, 133, 194108. [Google Scholar] [CrossRef] [PubMed]
- Berezkin, A.V.; Kudryavtsev, Y.V.; Gorkunov, M.V.; Osipov, M.A. Ordering of anisotropic nanoparticles in diblock copolymer lamellae: Simulations with dissipative particle dynamics and a molecular theory. J. Chem. Phys. 2017, 146, 144902. [Google Scholar] [CrossRef] [PubMed]
- Sides, S.W.; Kim, B.J.; Kramer, E.J.; Fredrickson, G.H. Hybrid Particle-Field Simulations of Polymer Nanocomposites. Phys. Rev. Lett. 2006, 96, 250601. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.U.; Matsen, M.W. Positioning Janus Nanoparticles in Block Copolymer Scaffolds. Phys. Rev. Lett. 2009, 102, 078303. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Hagberg, B.A.; Riggleman, R.A. The distribution of homogeneously grafted nanoparticles in polymer thin films and blends. Soft Matter 2014, 10, 8083–8094. [Google Scholar] [CrossRef]
- Beaudoin, E.; Abecassis, B.; Constantin, D.; Degrouard, J.; Davidson, P. Strain-controlled fluorescence polarization in a CdSe nanoplatelet–block copolymer composite. Chem. Commun. 2015, 51, 4051–4054. [Google Scholar] [CrossRef]
- Beaudoin, E.; Davidson, P.; Abecassis, B.; Bizien, T.; Constantin, D. Reversible strain alignment and reshuffling of nanoplatelet stacks confined in a lamellar block copolymer matrix. Nanoscale 2017, 9, 17371–17377. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, M.; Dubois, P. Polymer-layered silicate nanocomposites: Preparation, properties and uses of a new class of materials. Mater. Sci. Eng. R Rep. 2000, 28, 1–63. [Google Scholar] [CrossRef]
- Chang, K.C.; Chen, S.T.; Lin, H.F.; Lin, C.Y.; Huang, H.H.; Yeh, J.M.; Yu, Y.H. Effect of clay on the corrosion protection efficiency of PMMA/Na+-MMT clay nanocomposite coatings evaluated by electrochemical measurements. Eur. Polym. J. 2008, 44, 13–23. [Google Scholar] [CrossRef]
- Krook, N.M.; Ford, J.; Maréchal, M.; Rannou, P.; Meth, J.S.; Murray, C.B.; Composto, R.J. Alignment of Nanoplates in Lamellar Diblock Copolymer Domains and the Effect of Particle Volume Fraction on Phase Behavior. ACS Macro Lett. 2018, 7, 1400–1407. [Google Scholar] [CrossRef]
- Merritt, S.M.J.; Wemyss, A.M.; Farris, S.; Patole, S.; Patias, G.; Haddleton, D.M.; Shollock, B.; Wan, C. Gas Barrier Polymer Nanocomposite Films Prepared by Graphene Oxide Encapsulated Polystyrene Microparticles. ACS Appl. Polym. Mater. 2020, 2, 725–731. [Google Scholar] [CrossRef]
- Hore, M.J.A.; Composto, R.J. Functional Polymer Nanocomposites Enhanced by Nanorods. Macromolecules 2014, 47, 875–887. [Google Scholar] [CrossRef]
- White, S.I.; DiDonna, B.A.; Mu, M.; Lubensky, T.C.; Winey, K.I. Simulations and electrical conductivity of percolated networks of finite rods with various degrees of axial alignment. Phys. Rev. B 2009, 79, 024301. [Google Scholar] [CrossRef]
- Ma, X.; Zhu, X.; You, F.; Feng, J.; Wang, M.C.; Zhao, X. Preparation and optical polarization of Ag/epoxy composite films with aligned Ag nanowires. J. Alloys Compd. 2014, 592, 57–62. [Google Scholar] [CrossRef]
- Duan, S.k.; Niu, Q.l.; Wei, J.f.; He, J.b.; Yin, Y.a.; Zhang, Y. Water-bath assisted convective assembly of aligned silver nanowire films for transparent electrodes. Phys. Chem. Chem. Phys. 2015, 17, 8106–8112. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Bian, R.; Guo, C.; Xu, B.; Liu, H.; Jiang, L. Aligning Ag Nanowires by a Facile Bioinspired Directional Liquid Transfer: Toward Anisotropic Flexible Conductive Electrodes. Adv. Mater. 2018, 30, 1706938. [Google Scholar] [CrossRef]
- Xu, Y.; Ge, D.; Calderon-Ortiz, G.A.; Exarhos, A.L.; Bretz, C.; Alsayed, A.; Kurz, D.; Kikkawa, J.M.; Dreyfus, R.; Yang, S.; et al. Highly conductive and transparent coatings from flow-aligned silver nanowires with large electrical and optical anisotropy. Nanoscale 2020, 12, 6438–6448. [Google Scholar] [CrossRef]
- Yan, L.T.; Popp, N.; Ghosh, S.K.; Böker, A. Self-Assembly of Janus Nanoparticles in Diblock Copolymers. ACS Nano 2010, 4, 913–920. [Google Scholar] [CrossRef]
- Thorkelsson, K.; Nelson, J.H.; Alivisatos, A.P.; Xu, T. End-to-End Alignment of Nanorods in Thin Films. Nano Lett. 2013, 13, 4908–4913. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Z.; Guo, R.; Xu, G.; Huang, Z.; Yan, L.T. Entropy-Mediated Mechanical Response of the Interfacial Nanoparticle Patterning. Nano Lett. 2014, 14, 6910–6916. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Huang, Z.; Xu, Z.; Yan, L.T. Tailoring Interfacial Nanoparticle Organization through Entropy. Acc. Chem. Res. 2018, 51, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.; Pinna, M.; Zvelindovsky, A.; Pagonabarraga, I. Co-assembly of Janus nanoparticles in block copolymer systems. Soft Matter 2019, 15, 6400–6410. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.; Pinna, M.; Zvelindovsky, A.V.; Pagonabarraga, I. Nonspherical Nanoparticles in Block Copolymer Composites: Nanosquares, Nanorods, and Diamonds. Macromolecules 2019, 52, 8285–8294. [Google Scholar] [CrossRef]
- Rasin, B.; Lindsay, B.J.; Ye, X.; Meth, J.S.; Murray, C.B.; Riggleman, R.A.; Composto, R.J. Nanorod position and orientation in vertical cylinder block copolymer films. Soft Matter 2020, 16, 3005–3014. [Google Scholar] [CrossRef]
- Sarkar, B.; Alexandridis, P. Block copolymer–nanoparticle composites: Structure, functional properties, and processing. Prog. Polym. Sci. 2015, 40, 33–62. [Google Scholar] [CrossRef]
- Thorkelsson, K.; Mastroianni, A.J.; Ercius, P.; Xu, T. Direct Nanorod Assembly Using Block Copolymer-Based Supramolecules. Nano Lett. 2012, 12, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Krook, N.M.; Tabedzki, C.; Elbert, K.C.; Yager, K.G.; Murray, C.B.; Riggleman, R.A.; Composto, R.J. Experiments and Simulations Probing Local Domain Bulge and String Assembly of Aligned Nanoplates in a Lamellar Diblock Copolymer. Macromolecules 2019, 52, 8989–8999. [Google Scholar] [CrossRef]
- Rasin, B.; Chao, H.; Jiang, G.; Wang, D.; Riggleman, R.A.; Composto, R.J. Dispersion and alignment of nanorods in cylindrical block copolymer thin films. Soft Matter 2016, 12, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.W.; Xu, T. Tailoring Co-assembly of Nanodiscs and Block Copolymer-Based Supramolecules by Manipulating Interparticle Interactions. Macromolecules 2019, 52, 2833–2842. [Google Scholar] [CrossRef]
- Dong, B.; Huang, Z.; Chen, H.; Yan, L.T. Chain-Stiffness-Induced Entropy Effects Mediate Interfacial Assembly of Janus Nanoparticles in Block Copolymers: From Interfacial Nanostructures to Optical Responses. Macromolecules 2015, 48, 5385–5393. [Google Scholar] [CrossRef]
- Chen, P.; Yang, Y.; Dong, B.; Huang, Z.; Zhu, G.; Cao, Y.; Yan, L.T. Polymerization-Induced Interfacial Self-Assembly of Janus Nanoparticles in Block Copolymers: Reaction-Mediated Entropy Effects, Diffusion Dynamics, and Tailorable Micromechanical Behaviors. Macromolecules 2017, 50, 2078–2091. [Google Scholar] [CrossRef]
- Osipov, M.; Ushakova, A. Orientational ordering and spatial distribution of Janus nanoparticles in lamellae diblock copolymers. J. Mol. Liq. 2018, 267, 330–336, Special Issue Dedicated to the Memory of Professor Y. Reznikov. [Google Scholar] [CrossRef]
- Patti, A.; Siperstein, F.R.; Mackie, A.D. Phase Behavior of Model Surfactants in the Presence of Hybrid Particles. J. Phys. Chem. C 2007, 111, 16035–16044. [Google Scholar] [CrossRef]
- Patti, A.; Mackie, A.D.; Siperstein, F.R. Monte Carlo Simulation of Self-Assembled Ordered Hybrid Materials. Langmuir 2007, 23, 6771–6780. [Google Scholar] [CrossRef] [PubMed]
- Patti, A.; Mackie, A.D.; Zelenak, V.; Siperstein, F.R. One-pot synthesis of amino functionalized mesoporous silica materials: Using simulations to understand transitions between different structures. J. Mater. Chem. 2009, 19, 724–732. [Google Scholar] [CrossRef]
- Patti, A.; Mackie, A.D.; Siperstein, F.R. Monte Carlo simulations of self-assembling hexagonal and cage-like bifunctional periodic mesoporous materials. J. Mater. Chem. 2009, 19, 7848–7855. [Google Scholar] [CrossRef]
- Patti, A. Monte Carlo simulations of self-assembling star-block copolymers in dilute solutions. Colloids Surf. A Physicochem. Eng. Asp. 2010, 361, 81–89. [Google Scholar] [CrossRef]
- Zeleňák, V.; Skřínska, M.; Siperstein, F.R.; Patti, A. Phase evolution during one-pot synthesis of amine modified mesoporous silica materials: Preparation, properties, carbon dioxide adsorption. Appl. Surf. Sci. 2019, 476, 886–896. [Google Scholar] [CrossRef]
- Campos-Villalobos, G.; Siperstein, F.R.; Charles, A.; Patti, A. Solvent-induced morphological transitions in methacrylate-based block-copolymer aggregates. J. Colloid Interface Sci. 2020, 572, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Patti, A. Molecular Dynamics of Spherical Nanoparticles in Dense Polymer Melts. J. Phys. Chem. B 2014, 118, 3731–3742. [Google Scholar] [CrossRef]
- Burgos-Mármol, J.J.; Álvarez-Machancoses, Ó.; Patti, A. Modeling the Effect of Polymer Chain Stiffness on the Behavior of Polymer Nanocomposites. J. Phys. Chem. B 2017, 121, 6245–6256. [Google Scholar] [CrossRef]
- Burgos-Mármol, J.J.; Patti, A. Unveiling the impact of nanoparticle size dispersity on the behavior of polymer nanocomposites. Polymer 2017, 113, 92–104. [Google Scholar] [CrossRef]
- Burgos-Mármol, J.J.; Solans, C.; Patti, A. Effective short-range Coulomb correction to model the aggregation behavior of ionic surfactants. J. Chem. Phys. 2016, 144, 234904. [Google Scholar] [CrossRef]
- Larson, R.G.; Scriven, L.E.; Davis, H.T. Monte Carlo simulation of model amphiphile-oil–water systems. J. Chem. Phys. 1985, 83, 2411–2420. [Google Scholar] [CrossRef]
- Wang, Y.; Gkeka, P.; Fuchs, J.E.; Liedl, K.R.; Cournia, Z. DPPC-cholesterol phase diagram using coarse-grained Molecular Dynamics simulations. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Salerno, K.M.; Agrawal, A.; Perahia, D.; Grest, G.S. Resolving Dynamic Properties of Polymers through Coarse-Grained Computational Studies. Phys. Rev. Lett. 2016, 116, 058302. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Bedrov, D.; Li, L.; Byutner, O. A molecular dynamics simulation study of the viscoelastic properties of polymer nanocomposites. J. Chem. Phys. 2002, 117, 9478–9489. [Google Scholar] [CrossRef]
- Wang, M.; Galpaya, D.; Lai, Z.B.; Xu, Y.; Yan, C. Surface functionalization on the thermal conductivity of graphene–polymer nanocomposites. Int. J. Smart. Nano Mater. 2014, 5, 123–132. [Google Scholar] [CrossRef]
- Liu, J.; Cao, D.; Zhang, L. Molecular Dynamics Study on Nanoparticle Diffusion in Polymer Melts: A Test of the Stokes-Einstein Law. J. Phys. Chem. C 2008, 112, 6653–6661. [Google Scholar] [CrossRef]
- Hagita, K.; Morita, H.; Doi, M.; Takano, H. Coarse-Grained Molecular Dynamics Simulation of Filled Polymer Nanocomposites under Uniaxial Elongation. Macromolecules 2016, 49, 1972–1983. [Google Scholar] [CrossRef]
- Song, J.; Hsu, D.D.; Shull, K.R.; Phelan, F.R.; Douglas, J.F.; Xia, W.; Keten, S. Energy Renormalization Method for the Coarse-Graining of Polymer Viscoelasticity. Macromolecules 2018, 51, 3818–3827. [Google Scholar] [CrossRef] [PubMed]
- Henry, M.M.; Thomas, S.; Alberts, M.; Estridge, C.E.; Farmer, B.; McNair, O.; Jankowski, E. General-Purpose Coarse-Grained Toughened Thermoset Model for 44DDS/DGEBA/PES. Polymers 2020, 12, 2547. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Smith, J.S.; Bedrov, D.; Smith, G.D. A molecular dynamics simulation study of nanoparticle interactions in a model polymer-nanoparticle composite. Compos. Sci. Technol. 2003, 63, 1599–1605. [Google Scholar] [CrossRef]
- Molinari, N.; Sutton, A.P.; Mostofi, A.A. Mechanisms of reinforcement in polymer nanocomposites. Phys. Chem. Chem. Phys. 2018, 20, 23085–23094. [Google Scholar] [CrossRef]
- Trazkovich, A.J.; Wendt, M.F.; Hall, L.M. Effect of Copolymer Sequence on Local Viscoelastic Properties near a Nanoparticle. Macromolecules 2019, 52, 513–527. [Google Scholar] [CrossRef]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of Repulsive Forces in Determining the Equilibrium Structure of Simple Liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Schultz, A.J.; Hall, C.K.; Genzer, J. Computer simulation of copolymer phase behavior. J. Chem. Phys. 2002, 117, 10329–10338. [Google Scholar] [CrossRef]
- Schultz, A.J.; Hall, C.K.; Genzer, J. Computer Simulation of Block Copolymer/Nanoparticle Composites. Macromolecules 2005, 38, 3007–3016. [Google Scholar] [CrossRef]
- He, L.; Zhang, L.; Liang, H. The Effects of Nanoparticles on the Lamellar Phase Separation of Diblock Copolymers. J. Phys. Chem. B 2008, 112, 4194–4203. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Large-Scale Atomic/Molecular Massively Parallel Simulator (LAMMPS), Stable Release 11 August 2017. Available online: lammps.sandia.gov (accessed on 24 March 2021).
- Barkla High Performance Computing Facility; Computing Services Department, University of Liverpool: Liverpool, UK, 2020.
- Shinoda, W.; Shiga, M.; Mikami, M. Rapid estimation of elastic constants by molecular dynamics simulation under constant stress. Phys. Rev. B 2004, 69, 134103. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Alejandre, J.; López-Rendón, R.; Jochim, A.L.; Martyna, G.J. A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble. J. Phys. Math. Gen. 2006, 39, 5629–5651. [Google Scholar] [CrossRef]
- Gam, S.; Meth, J.S.; Zane, S.G.; Chi, C.; Wood, B.A.; Winey, K.I.; Clarke, N.; Composto, R.J. Polymer diffusion in a polymer nanocomposite: Effect of nanoparticle size and polydispersity. Soft Matter 2012, 8, 6512–6520. [Google Scholar] [CrossRef]
- Hales, T.; Adams, M.; Bauer, G.; Dang, T.D.; Harrison, J.; Hoang, L.T.; Kaliszyk, C.; Magron, V.; McLaughlin, S.; Nguyen, T.; et al. A formal proof of the Kepler Conjecture. Forum Math. Pi 2017, 5, e2. [Google Scholar] [CrossRef]
- Burgos-Mármol, J.J. Molecular Simulation of Polymer Nanocomposites. Ph.D. Thesis, The University of Manchester, Manchester, UK, 2017. [Google Scholar]
- Weisstein, E.W. Sphere-Sphere Intersection. From Mathworld—A Wolfram Web Resource. Available online: https://mathworld.wolfram.com/Sphere-SphereIntersection.html (accessed on 24 March 2021).
- Burgos-Mármol, J.J. LamAnalysis: Software toolkit for Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer. Zenodo 2021. [Google Scholar] [CrossRef]
- Morillo, N.; Patti, A.; Cuetos, A. Brownian dynamics simulations of oblate and prolate colloidal particles in nematic liquid crystals. J. Chem. Phys. 2019, 150, 204905. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.P.; McDonald, I.R. Chapter 11—Molecular Liquids, 4th ed.; Academic Press: Oxford, UK, 2013; pp. 455–510. [Google Scholar] [CrossRef]
- Chiu, J.J.; Kim, B.J.; Yi, G.R.; Bang, J.; Kramer, E.J.; Pine, D.J. Distribution of Nanoparticles in Lamellar Domains of Block Copolymers. Macromolecules 2007, 40, 3361–3365. [Google Scholar] [CrossRef]
- Burgos-Mármol, J.J.; Patti, A. Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer (Dataset 1 of 2). Zenodo 2021. [Google Scholar] [CrossRef]
- Burgos-Mármol, J.J.; Patti, A. Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer (Dataset 2 of 2). Zenodo 2021. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bead | Non-Bonded | Bonded | ||||
|---|---|---|---|---|---|---|
| h | h | 1 | 0 | 30 | 1 | |
| h | t | 52 | 0 | 30 | 1 | |
| t | t | 1 | 0 | 30 | 1 | |
| H | H | 52 | 2 | 15 | 3 | |
| H | T | 52 | 2 | 15 | 3 | |
| H | 0 | 52 | 2 | 15 | 3 | |
| T | T | 52 | 2 | 15 | 3 | |
| T | 0 | 52 | 2 | 15 | 3 | |
| 0 | 0 | 52 | 2 | 15 | 3 | |
| H | h | 1 | 1 | |||
| H | t | 52 | 1 | |||
| T | h | 52 | 1 | |||
| T | t | 1 | 1 | |||
| 0 | h | 1 | 1 | |||
| 0 | t | 1 | 1 | |||
| System | n | ND Type | ||
|---|---|---|---|---|
| PNC | 12,696 | 0 | 0 | |
| PNC | 12,696 | 00 | 1 | 96 |
| PNC | 12,696 | HH | 1 | 96 |
| PNC | 12,696 | H0 | 1 | 96 |
| PNC | 12,696 | HT | 1 | 96 |
| PNC | 12,696 | 00 | 5 | 510 |
| PNC | 12,696 | HH | 5 | 510 |
| PNC | 12,696 | H0 | 5 | 510 |
| PNC | 12,696 | HT | 5 | 510 |
| PNC | 12,696 | 00 | 10 | 1080 |
| PNC | 12,696 | HH | 10 | 1080 |
| PNC | 12,696 | H0 | 10 | 1080 |
| PNC | 12,696 | HT | 10 | 1080 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgos-Mármol, J.J.; Patti, A. Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer. Polymers 2021, 13, 1524. https://doi.org/10.3390/polym13091524
Burgos-Mármol JJ, Patti A. Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer. Polymers. 2021; 13(9):1524. https://doi.org/10.3390/polym13091524
Chicago/Turabian StyleBurgos-Mármol, J. Javier, and Alessandro Patti. 2021. "Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer" Polymers 13, no. 9: 1524. https://doi.org/10.3390/polym13091524
APA StyleBurgos-Mármol, J. J., & Patti, A. (2021). Molecular Dynamics of Janus Nanodimers Dispersed in Lamellar Phases of a Block Copolymer. Polymers, 13(9), 1524. https://doi.org/10.3390/polym13091524