
Paper Ageing: The Effect of Paper Chemical Composition on Hydrolysis and Oxidation

 ,
,
Abstract
1. Introduction
- the extent to which increased humidity could affect the changes in the cellulose structure;
- the Kappa numbers for which the instrumental methods are applicable.
2. Materials and Methods
2.1. Cellulosic Pulps
2.2. Nomenclature of the Paper Samples and Sheets
2.3. Ageing Tests
2.4. Characterisation of Cellulose in the Paper Sheets
2.4.1. Determination of the Molecular Weight Distributions by SEC
2.4.2. Diffuse Reflectance Infrared Fourier-Transform Spectroscopy (DRIFTS)
2.4.3. UV–Vis Spectroscopy
2.4.4. X-ray Diffraction (XRD)
3. Results and Discussion
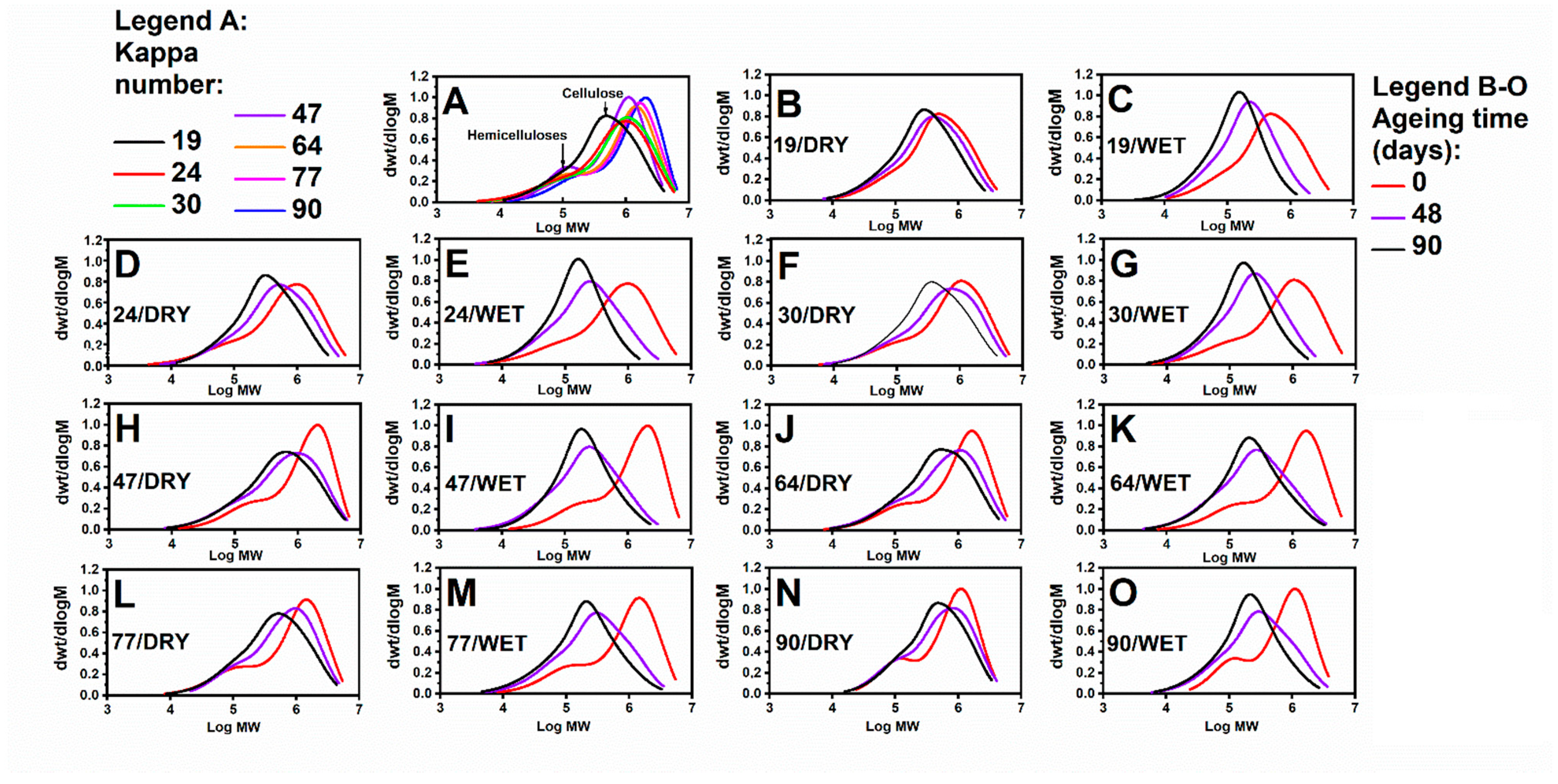
3.1. Impact of Accelerated Ageing on the Cellulose Depolymerisation Process: SEC Analysis
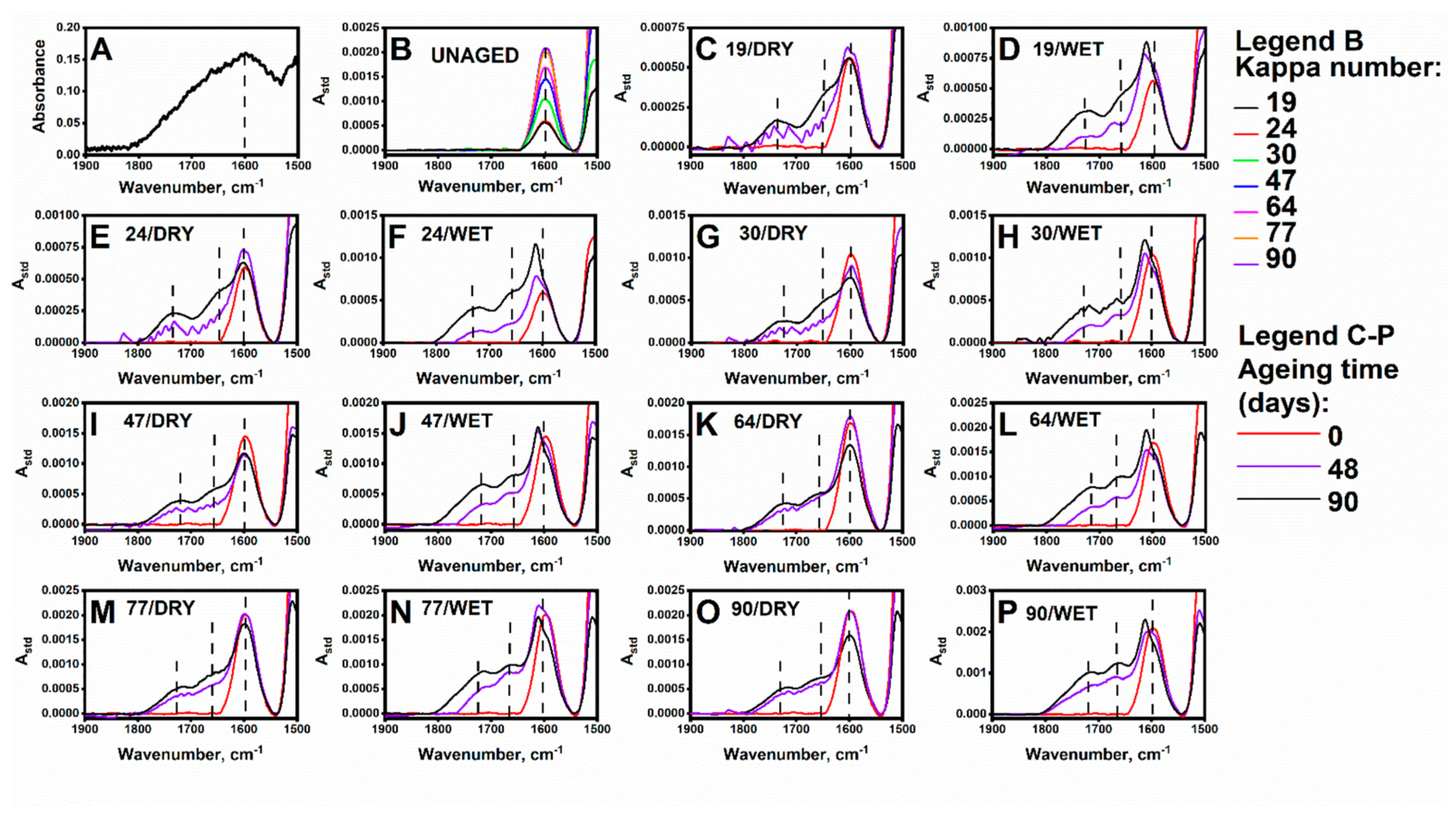
3.2. Impact of Accelerated Ageing on Cellulose and the Lignin Oxidation Process: DRIFT
3.3. Impact of Accelerated Ageing on the Cellulose Oxidation Process: UV–Vis Analysis
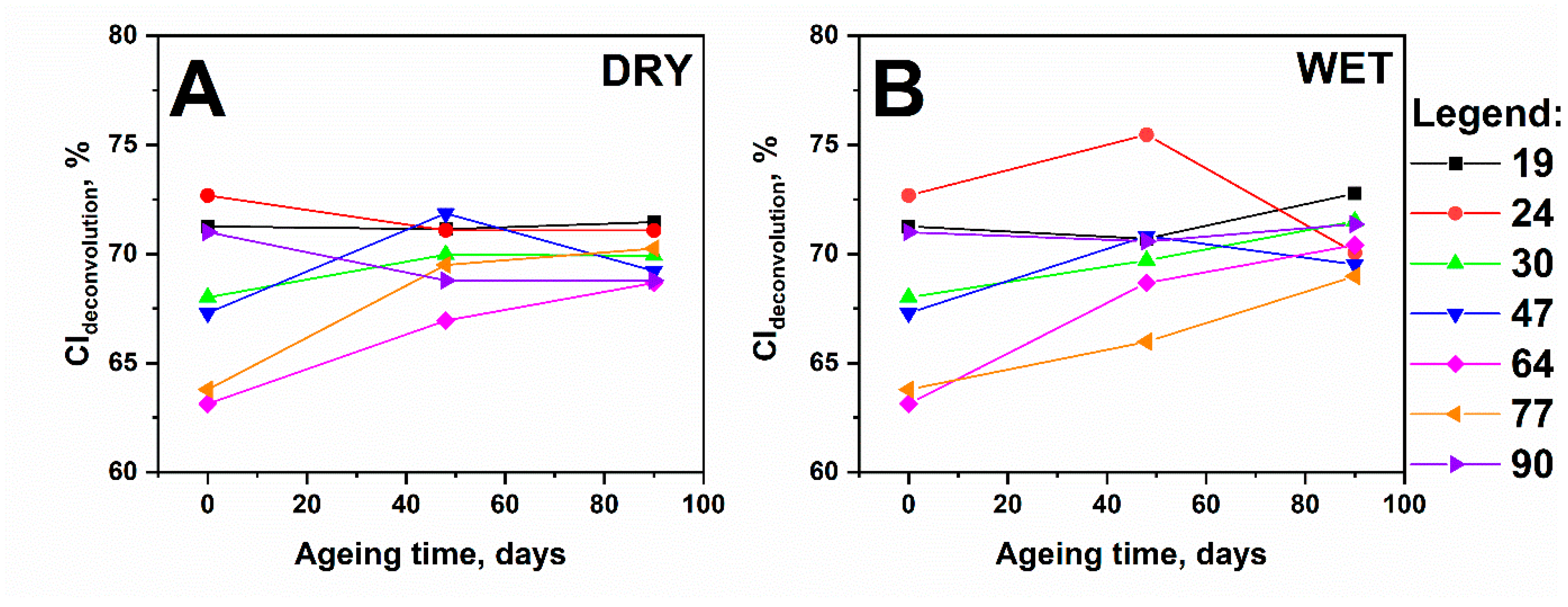
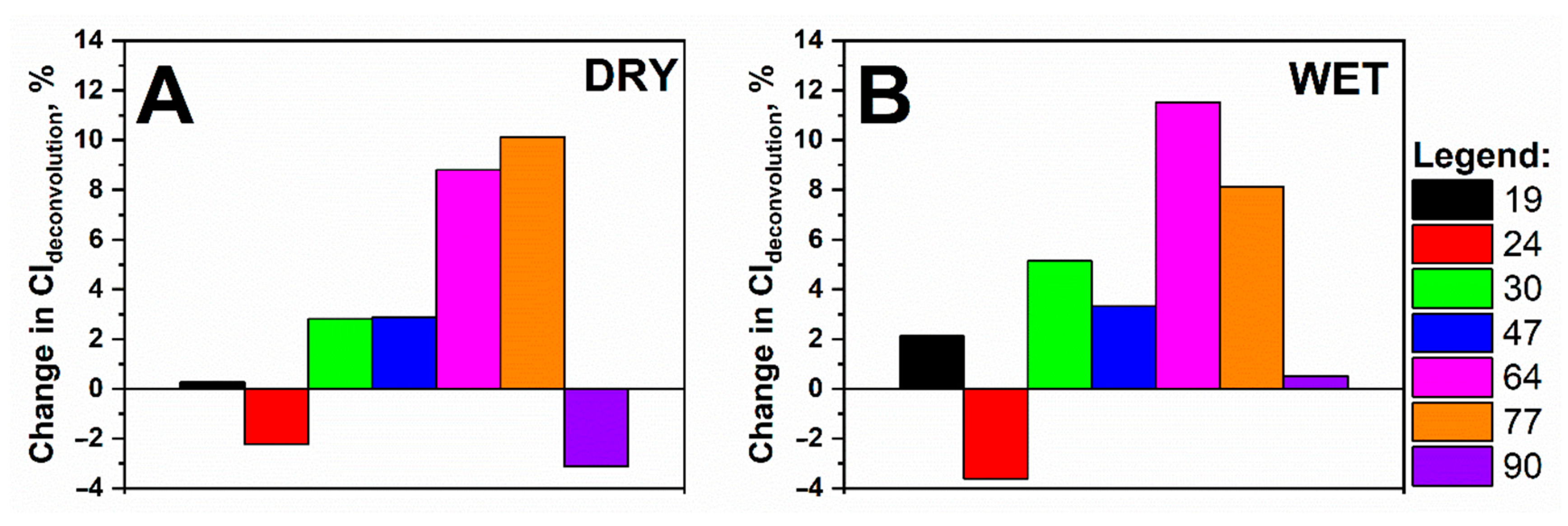
3.4. Impact of Accelerated Ageing on the Crystallinity of Cellulose
- CIheight, calculated as the ratio of the peak height from the reflex (002) to the minimum height between the reflections (002) and (10͞1), is proportional to the amount of the amorphous phase, obtained by the formula: CIheight = (I002 − Iamorphous)/I002 × 100%;
- CIdeconvolution, calculated as the percentage of the peak areas were derived from the crystalline phase, (101), (10͞1), (002), (040), on the total surface of all the fitted Gaussian curves.
- This factor did not accurately reflect the amount of the amorphous phase that was responsible for the formation of a wide peak with a maximum of about 21.9° while the intensity of the minimum, at about 18.5°, was employed to calculate CIheight. An underestimation of the amount of amorphous phase is associated with the overlap of the reflex, derived from the amorphous phase with the most intense peak, derived from the crystalline phase (002) at about 22.6°. As a result, the sensitivity of the intensity of the measured Iamorphous to changes in the amount of the amorphous phase was low and further weakened by the contribution of the peak (10͞1) to the value of Iamorphous. Accordingly, the CIheight index values, compared to other crystallinity indexes, were not very accurate.
- The number of crystalline phases was calculated based on the intensity of the peak (002): one of the four, observed on the diffractograms. In the case of the relative differences in the intensities of the reflections from the walls, (101), (10͞1), (002), (040), this could be a source of error.
- The reflections on the diffractograms of the lignocellulose samples were characterised by a large width, which changed with the progression of degradation. It also depended on the size of the crystallites and may vary under different measuring conditions. Therefore, the estimation of the amount of crystalline and amorphous phases in cellulose should depend on the calculation of the peak areas derived from the individual phase reflections and not on the comparison of the relative peak heights (expressed as intensities).
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strlič, M.; Kolar, J.; Scholten, S. Paper and durability. In Ageing and Stabilisation of Paper; Strlič, M., Kolar, J., Eds.; Narodna in univerzitetna knjižnica: Ljubljana, Slovenia, 2005. [Google Scholar]
- Vizárová, K.; Kirschnerová, S.; Kačík, F.; Briškárová, A.; Šutý, Š.; Katuščák, S. Relationship between the decrease of degree of polymerisation of cellulose and the loss of groundwood pulp paper mechanical properties during accelerated ageing. Chem. Pap. 2012, 66, 1124–1129. [Google Scholar] [CrossRef]
- Banik, G.; Brückle, G. Paper and Water: A Guide for Conservators; Butterworth-Heinemann: Oxford, UK, 2011. [Google Scholar]
- Mleziva, M.M.; Wang, J.H. Polymers for a sustainable environment and green energy. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Strlič, M.; Kolar, J.; Kočar, D.; Rychlý, J. Theromo-oxidative degradation. In Aging and Stabilization of Paper; Strlič, M., Kolar, J., Eds.; National and University Library: Ljubljana, Slovenia, 2004; pp. 101–120. [Google Scholar]
- Knott, B.C.; Momeni, M.H.; Crowley, M.F.; Mackenzie, L.F.; Götz, A.W.; Sandgren, M.; Withers, S.G.; Ståhlberg, J.; Beckham, G.T. The mechanism of cellulose hydrolysis by a two-step, retaining cellobiohydrolase elucidated by structural and transition path sampling studies. J. Am. Chem. Soc. 2014, 136, 321–329. [Google Scholar] [CrossRef]
- Ofem, M.I.; Ene, E.B.; Ubi, P.A.; Odey, S.O.; Fakorede, D.O. Properties of cellulose reinforced composites: A review. Niger. J. Technol. 2020, 39, 386–402. [Google Scholar] [CrossRef]
- Calvini, P.; Gorassini, A.; Merlani, A.L. Autocatalytic degradation of cellulose paper in sealed vessels. Restaurator 2007, 28, 47–54. [Google Scholar] [CrossRef]
- Calvini, P.; Gorassini, A. On the rate of paper degradation: Lessons from the past. Restaurator 2006, 27, 275–290. [Google Scholar] [CrossRef]
- Jönsson, L.J.; Martín, C. Pretreatment of lignocellulose: Formation of inhibitory by-products and strategies for minimizing their effects. Bioresour. Technol. 2016, 199, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Uesaka, T.; Gurnagul, N. Prediction of paper permanence by accelerated aging I. Kinetic analysis of the aging process. Cellulose 1996, 3, 243–267. [Google Scholar] [CrossRef]
- Tumkur, P.P.; Bayon, N.N.; Gunasekaran, N.K.; Lamani, B.R.; Prabhakaran, K.; Hall, J.C.; Ramesh, G.T. Evaluation of cellulose nanoparticles synthesized by acid hydrolysis for safe applications. J. Nano. Sci. Res. Rep. 2020, 2, 1–5. [Google Scholar] [CrossRef]
- Hon, D.; Shiraishi, N. Wood and Cellulose Chemistry, 2nd ed.; Marcel Dekker: New York, NY, USA, 2001. [Google Scholar]
- Barański, A. Ageing kinetics of cellulose and paper. Restaurator 2002, 23, 77–88. [Google Scholar] [CrossRef]
- Łojewska, J.; Miśkowiec, P.; Proniewicz, L.M. Oxidative and hydrolytic path of paper degradation studied by in-situ FTIR transmission spectroscopy. In Proceedings of the International Conference “Durability of Paper and Writing”, Ljubljana, Slovenia, 24–25 November 2004. [Google Scholar]
- Barański, A.; Łagan, J.M.; Łojewski, T. Acid-catalysed degradation. In Ageing and Stabilisation of Paper; Strlič, M., Kolar, J., Eds.; Narodna in Univerzitetna Knjižnica: Ljubljana, Slovenia, 2005. [Google Scholar]
- Łojewska, J.; Missori, M.; Lubańska, A.; Grimaldi, P.; Ziȩba, K.; Proniewicz, L.M.; Castellano, A.C. Carbonyl groups development on degraded cellulose. Correlation between spectroscopic and chemical results. Appl. Phys. A 2007, 89, 883–887. [Google Scholar] [CrossRef]
- Łojewska, J.; Lubańska, A.; Łojewski, T.; Miśkowiec, P.; Proniewicz, L.M. Kinetic approach to degradation of paper. In situ FTIR transmission studies on hydrolysis and oxidation. e-Preserv. Sci. 2005, 2, 1–12. [Google Scholar]
- Łojewska, J.; Lubańska, A.; Miśkowiec, P.; Łojewski, T.; Proniewicz, L.M. FTIR in situ transmission studies on the kinetics of paper degradation via hydrolytic and oxidative reaction paths. Appl. Phys. A 2006, 83, 597–603. [Google Scholar] [CrossRef]
- Kato, K.L.; Cameron, R.E. A review of the relationship between thermally-accelerated ageing of paper and hornification. Cellulose 1999, 6, 3–40. [Google Scholar] [CrossRef]
- Łojewski, T.; Zięba, K.; Knapik, A.; Bagniuk, J.; Lubańska, A.; Łojewska, J. Evaluating paper degradation progress. Cross-linking between chromatographic, spectroscopic and chemical results. Appl. Phys. A 2010, 100, 809–821. [Google Scholar] [CrossRef]
- Zervos, S. Natural and accelerated aging of cellulose and paper: A literature review. In Cellulose: Structure and Properties Derivatives and Industrial Uses; Lejeune, A., Deprez, T., Eds.; Nova Publishing: New York, NY, USA, 2010; pp. 155–203. [Google Scholar]
- Łojewska, J.; Miśkowiec, P.; Łojewski, T.; Proniewicz, L.M. Cellulose oxidative and hydrolytic degradation: In situ FTIR approach. Polym. Degrad. Stabil. 2005, 88, 512–520. [Google Scholar] [CrossRef]
- Calvini, P. The role of the ekenstam equation on the kinetics of cellulose hydrolytic degradation. Cellulose 2012, 19, 313–318. [Google Scholar] [CrossRef]
- Łojewski, T.; Miśkowiec, P.; Missori, M.; Lubańska, A.; Łojewska, J. FTIR and UV/vis as methods for evaluation of oxidative degradation of model paper: DFT approach for carbonyl vibrations. Carbohydr. Polym. 2010, 82, 370–375. [Google Scholar] [CrossRef]
- Baty, J.W.; Maitland, C.L.; Minter, W.; Hubbe, M.A.; Jordan-Mowery, S.K. Deacidification for the coservation and preservation of paper-based works: A review. BioResources 2010, 5, 1955–2023. [Google Scholar] [CrossRef]
- Ciglanská, M.; Jančovičová, V.; Havlínová, B.; Machatová, Z.; Brezová, V. The influence of pollutants on accelerated ageing of parchment with iron gall inks. J. Cult. Herit. 2014, 15, 373–381. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, Y.; Tan, W.; Liu, C. Preservation of aged paper using borax in alcohols and the supercritical carbon dioxide system. J. Cult. Herit. 2013, 14, 16–22. [Google Scholar] [CrossRef]
- Gregorova, A.; Lahti, J.; Schennach, R.; Stelzer, F. Humidity response of Kraft papers determined by dynamic mechanical analysis. Thermochim. Acta 2013, 570, 33–40. [Google Scholar] [CrossRef]
- Sandy, M.; Manning, A.; Bollet, F. Changes in the tensile properties of paper in response to fluctuating relative humidity—Relevance to paper conservation. Int. Circ. Graphic Educ. Res. 2009, 2, 6–14. [Google Scholar]
- Barclay, L.R.C.; Xi, F.; Norris, J.Q. Antioxidant properties of phenolic lignin model compounds. J. Wood Chem. Technol. 1997, 17, 73–90. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Rye, C.S.; Gurnagul, N. Lignin inhibits autoxidative degradation of cellulose. Polym. Degrad. Stab. 1995, 49, 291–297. [Google Scholar] [CrossRef]
- Małachowska, E.; Dubowik, M.; Boruszewski, P.; Łojewska, Ł.; Przybysz, P. Influence of lignin content in cellulose pulp on paper durability. Sci. Rep. 2020, 10, 19998. [Google Scholar] [CrossRef] [PubMed]
- Modrzejewski, K.; Olszewski, J.; Rutkowski, J. Analysis in Papermaking Industry; Editorial Office of the Lodz University of Technology: Lodz, Poland, 1969; pp. 56–70, 158. [Google Scholar]
- Łojewski, T.; Zięba, K.; Kołodziej, A.; Łojewska, J. Following cellulose depolymerization in paper: Comparison of size exclusion chromatography techniques. Cellulose 2011, 18, 1349–1363. [Google Scholar] [CrossRef]
- Łojewski, T.; Zięba, K.; Łojewska, J. Size exclusion chromatography and viscometry in paper degradation studies. New Mark-Houwink coefficients for cellulose in cupriethylenediamine. J. Chromatogr. A 2010, 1217, 6462–6468. [Google Scholar] [CrossRef]
- Pawcenis, D.; Thomas, J.L.; Łojewski, T.; Milczarek, J.M.; Łojewska, J. Towards determination of absolute molar mass of cellulose polymer by size exclusion chromatography with mulitple angle laser light scattering detection. J. Chromatogr. A 2015, 1409, 53–59. [Google Scholar] [CrossRef]
- Xu, F.; Yu, J.; Tesso, T.; Dowell, F.; Wang, D. Qualitative and quantitative analysis of lignocellulosic biomass using infrared techniques: A mini review. Appl. Energy 2013, 104, 801–809. [Google Scholar] [CrossRef]
- Lähdetie, A.; Liitiä, T.; Tamminen, T.; Jääskeläinen, A.S. Reflectance UV-Vis and UV resonance Raman spectroscopy in characterization of kraft pulps. BioRes. 2009, 4, 1600–1619. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Heitner, C. Use of diffuse reflectance UV-Visible spectroscopy to characterize chromophores in wood fibers. In Advances in Lignocellulosics Characterization; Argyropoulos, D., Ed.; TAPPI Press: Atlanta, GA, USA, 1999; pp. 179–199. [Google Scholar]
- Tribulová, T.; Kacík, F.; Evtuguin, D.; Cabalová, I. Assessment of chromophores in chemically treated and aged wood by Uv-vis diffuse reflectance spectroscopy. Cell. Chem. Technol. 2016, 50, 659–667. [Google Scholar]
- Park, S.; Baker, J.O.; Himmel, M.E.; Parilla, P.A.; Johnson, D.K. Cellulose crystallinity index: Measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 2010, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Segal, L.; Creely, J.J.; Martin, A.E.; Conrad, C.M. An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Tex. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Thygesen, A.; Oddershede, J.; Lilholt, H.; Thomsen, A.B.; Ståhl, K. On the determination of crystallinity and cellulose content in plant fibres. Cellulose 2005, 12, 563–576. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Percentage Fall of DP upon Ageing | ||||
|---|---|---|---|---|
| Kappa Number [–] | After 48 Days | After 90 Days | ||
| DRY | WET | DRY | WET | |
| 19 | 21.35 | 58.30 | 36.28 | 73.09 |
| 24 | 30.49 | 71.60 | 46.66 | 77.74 |
| 30 | 17.70 | 68.45 | 44.35 | 77.91 |
| 47 | 36.36 | 75.68 | 42.29 | 81.48 |
| 64 | 28.80 | 67.58 | 40.24 | 72.96 |
| 77 | 23.40 | 58.30 | 36.23 | 70.39 |
| 90 | 12.76 | 44.11 | 24.22 | 62.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Małachowska, E.; Pawcenis, D.; Dańczak, J.; Paczkowska, J.; Przybysz, K. Paper Ageing: The Effect of Paper Chemical Composition on Hydrolysis and Oxidation. Polymers 2021, 13, 1029. https://doi.org/10.3390/polym13071029
Małachowska E, Pawcenis D, Dańczak J, Paczkowska J, Przybysz K. Paper Ageing: The Effect of Paper Chemical Composition on Hydrolysis and Oxidation. Polymers. 2021; 13(7):1029. https://doi.org/10.3390/polym13071029
Chicago/Turabian StyleMałachowska, Edyta, Dominika Pawcenis, Jacek Dańczak, Joanna Paczkowska, and Kamila Przybysz. 2021. "Paper Ageing: The Effect of Paper Chemical Composition on Hydrolysis and Oxidation" Polymers 13, no. 7: 1029. https://doi.org/10.3390/polym13071029
APA StyleMałachowska, E., Pawcenis, D., Dańczak, J., Paczkowska, J., & Przybysz, K. (2021). Paper Ageing: The Effect of Paper Chemical Composition on Hydrolysis and Oxidation. Polymers, 13(7), 1029. https://doi.org/10.3390/polym13071029