
Sustainable Esterification of a Soda Lignin with Phloretic Acid


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
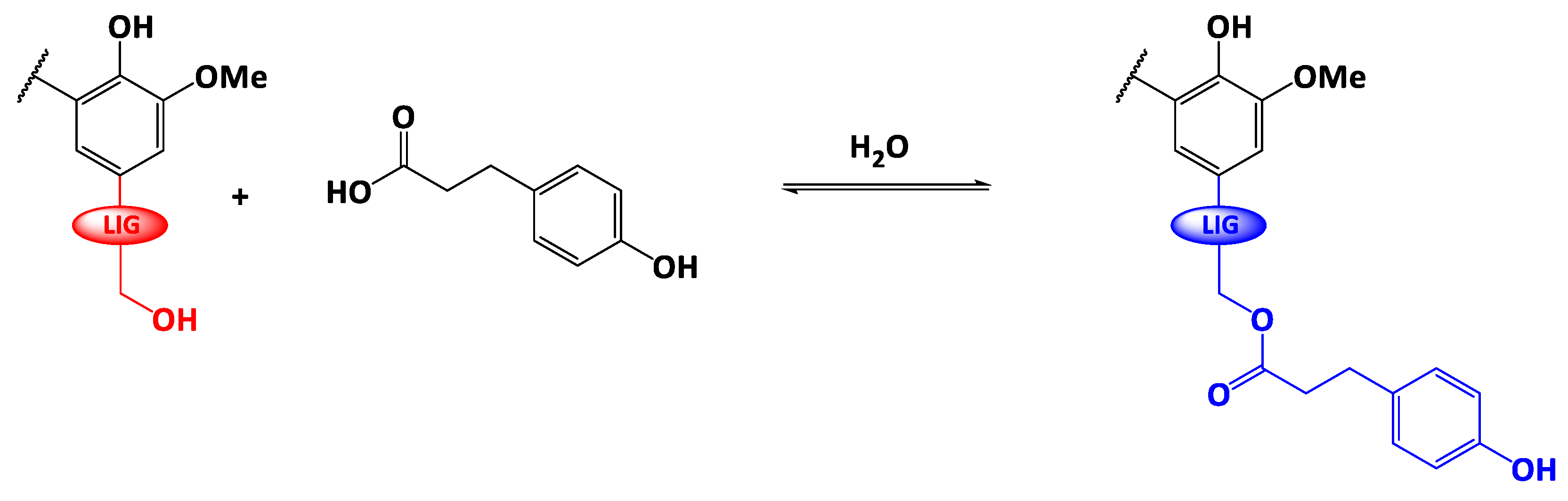
2.2. Esterification of Lignin
- the reaction time (t)
- the molar ratio between the carboxylic acid units from PA (nCOOH_PA) and the lignin aliphatic ‒OH groups determined by 31P NMR (nOHali_P2400) (n)
- the catalyst loading expressed as weight percentage (wt.%), relative to the initial amount of lignin (c)
2.3. Equipments and Characterization
3. Results
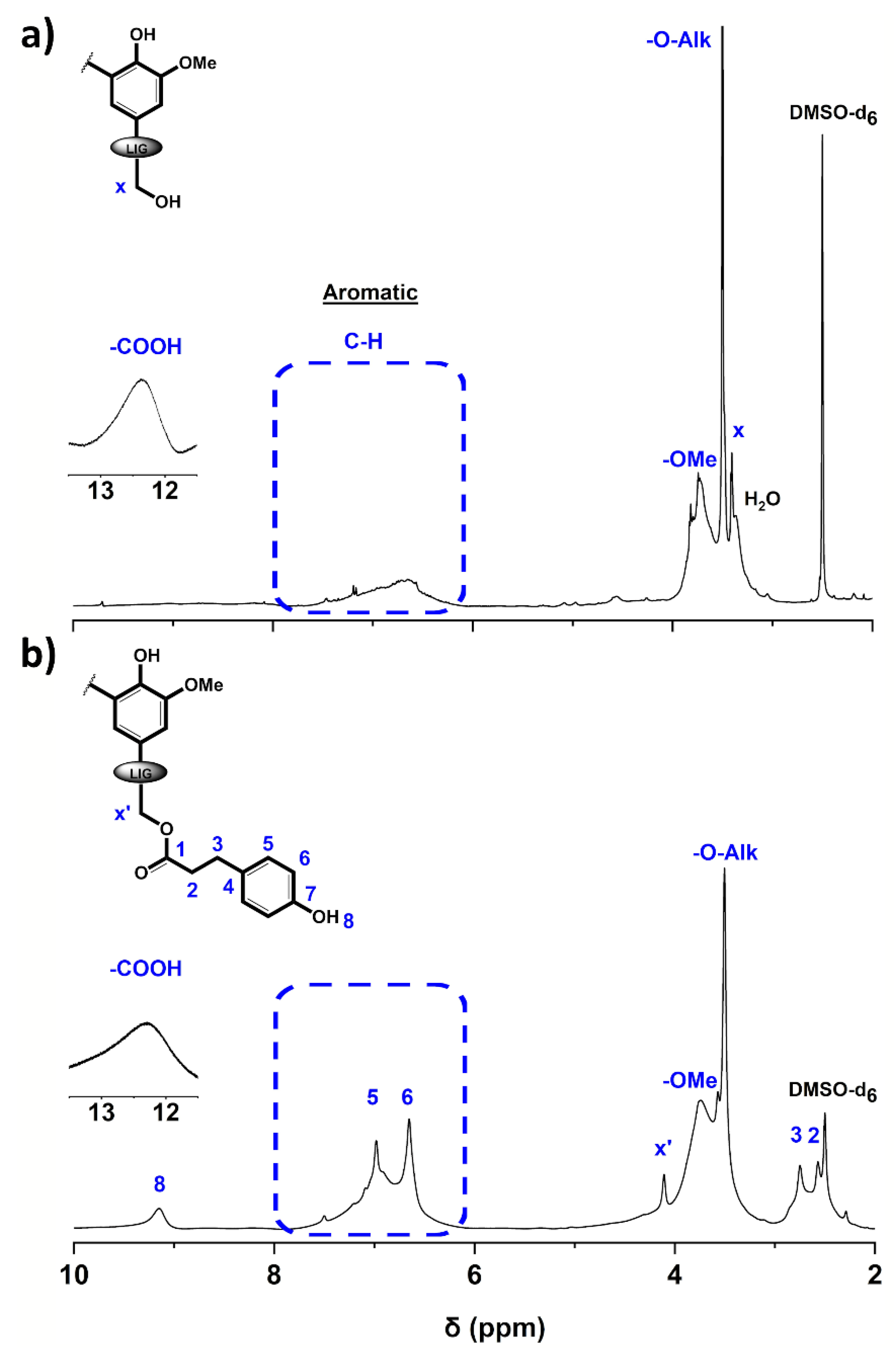
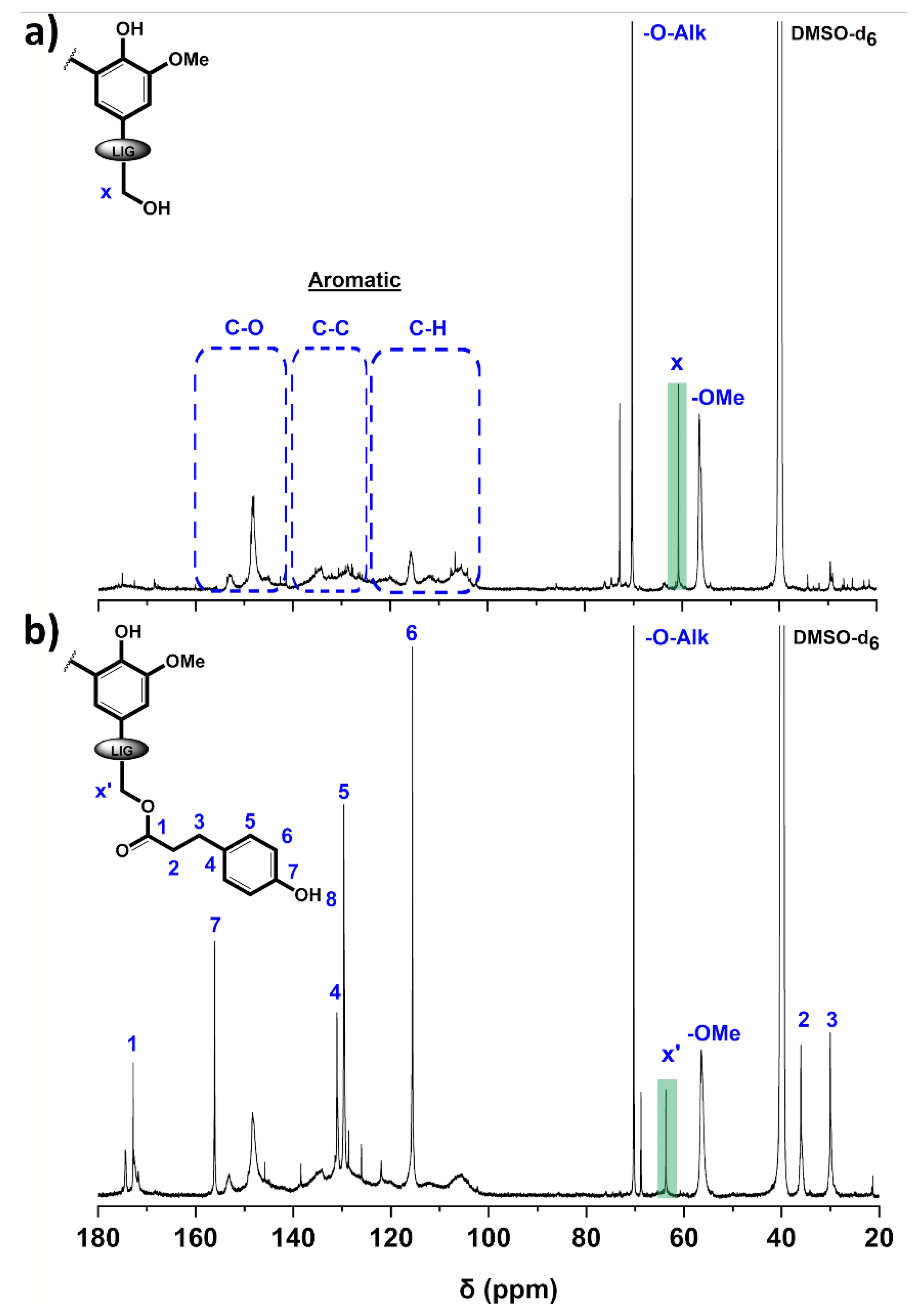
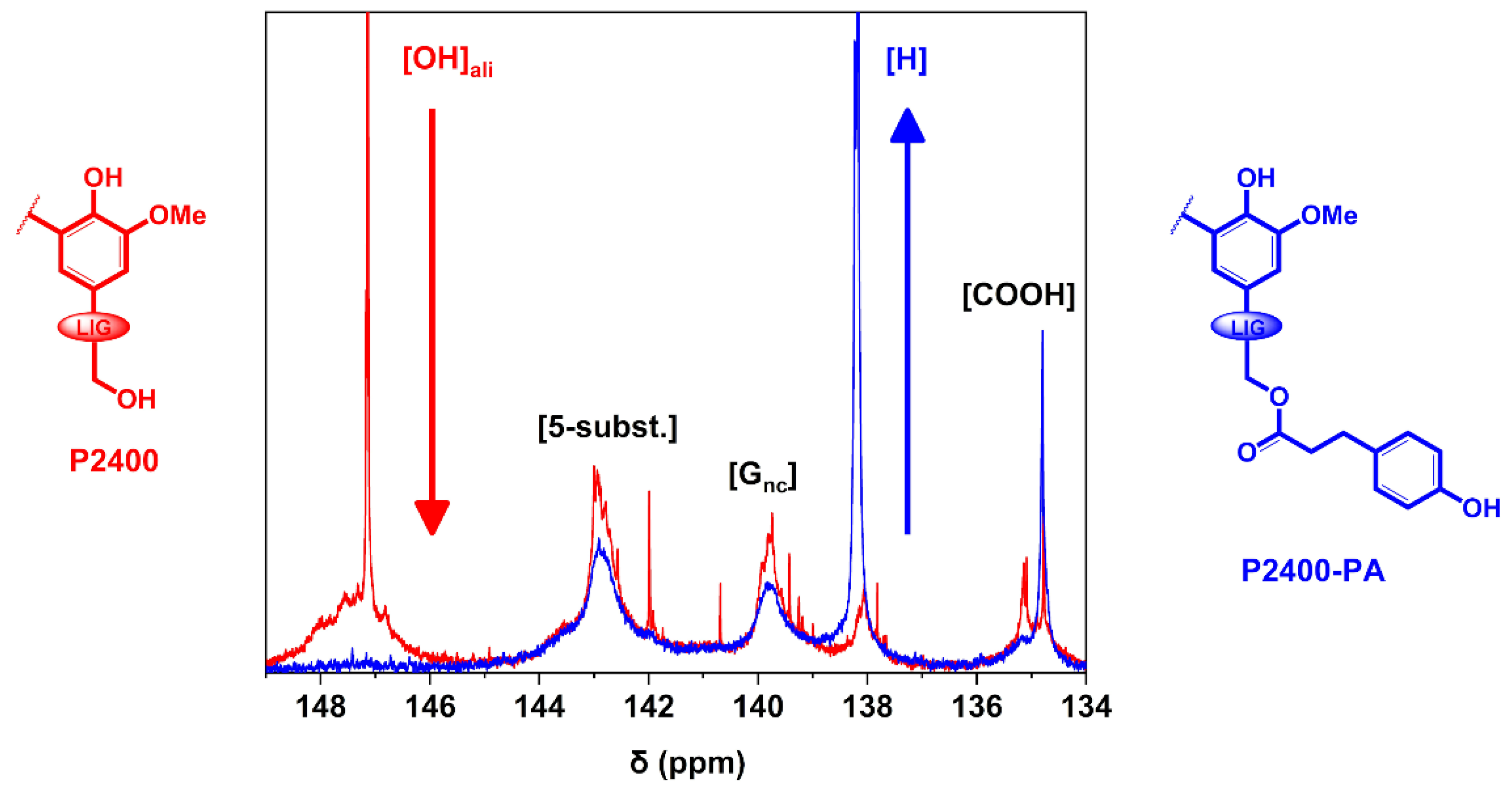
3.1. Structural Characterization of the Esterified Lignin
3.2. Optimization of the Fischer Esterification of Protobind® Lignin
- when a stoichiometric amount of reactants is used (n = 1; Table 4, rows 1,2, 5 and 6), the increase in the amount of [H] units does not exceed 28%,
- when an excess of PA is used (n = 5; Table 3, rows 3, 4, 7, 8), higher conversion yields of aliphatic ‒OH groups (~90%) and amount of [H] units are reached (~50% and 65 % for t = 12 and 48 h, respectively),
- for n = 1, the amount of [H] units is equivalent to ~ 0.9 and ~1.1 mmol g−1 (c = 0.5 and 2.5 wt.%, respectively) independent of the reaction time,
- for an excess of PA (n = 5), this amount is equivalent to ~1.6 and 1.8 mmol g−1 (t = 12 and 48, respectively) regardless of the catalyst loading.
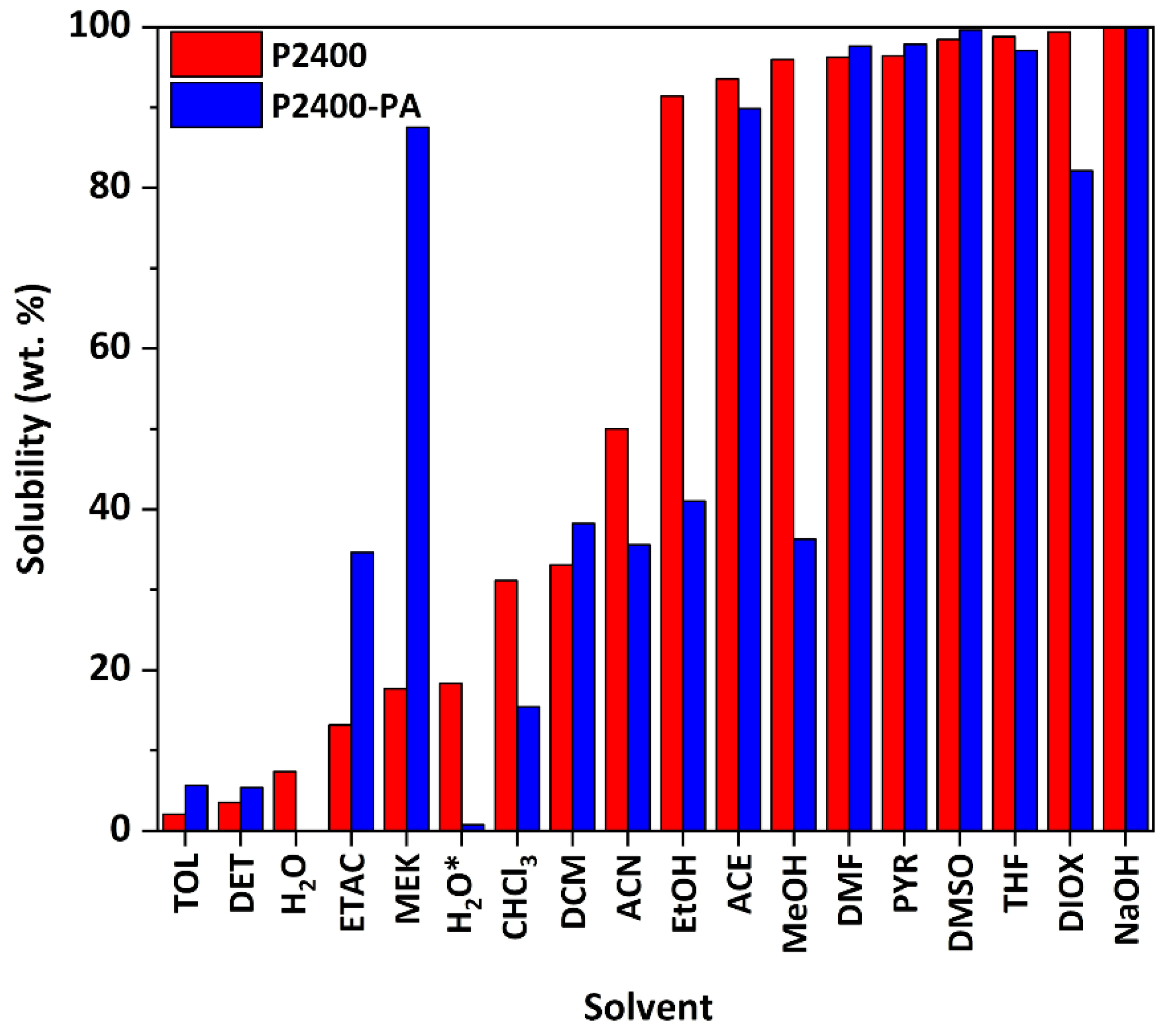
3.3. Physicochemical Properties of Esterified Lignin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Spekreijse, J.; Lammens, T.; Parisi, C.; Ronzon, T.; Vis, M. Insights into the European Market for Bio-Based Chemicals; Publications office of the EU: Luxembourg, 2019. [Google Scholar]
- Calvo-Flores, F.G.; Dobado, J.; Isac-García, J.; Martin-Martinez, F. Lignin and Lignans as Renewable Raw Materials: Chemistry, Technology and Applications; Wiley: Chichester, UK, 2015. [Google Scholar]
- Strassberger, Z.; Tanase, S.; Rothenberg, G. The pros and cons of lignin valorisation in an integrated biorefinery. RSC Adv. 2014, 4, 25310–25318. [Google Scholar] [CrossRef]
- Saratale, R.G.; Saratale, G.D.; Ghodake, G.; Cho, S.-K.; Kadam, A.; Kumar, G.; Jeon, B.-H.; Pant, D.; Bhatnagar, A.; Shin, H.S. Wheat straw extracted lignin in silver nanoparticles synthesis: Expanding its prophecy towards antineoplastic potency and hydrogen peroxide sensing ability. Int. J. Biol. Macromol. 2019, 128, 391–400. [Google Scholar] [CrossRef]
- Ganesh Saratale, R.; Cho, S.-K.; Dattatraya Saratale, G.; Kadam, A.A.; Ghodake, G.S.; Kumar, M.; Naresh Bharagava, R.; Kumar, G.; Su Kim, D.; Mulla, S.I.; et al. A comprehensive overview and recent advances on polyhydroxyalkanoates (PHA) production using various organic waste streams. Bioresour. Technol. 2021, 325, 124685. [Google Scholar] [CrossRef]
- Upton, B.M.; Kasko, A.M. Strategies for the Conversion of Lignin to High-Value Polymeric Materials: Review and Perspective. Chem. Rev. 2016, 116, 2275–2306. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Pu, Y.; Ragauskas, A.; Yang, B. From Lignin to Valuable Products–Strategies, Challenges, and Prospects. Bioresour. Technol. 2018, 271, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Laurichesse, S.; Avérous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Kai, D.; Tan, M.J.; Chee, P.L.; Chua, Y.K.; Yap, Y.L.; Loh, X.J. Towards lignin-based functional materials in a sustainable world. Green Chem. 2016, 18, 1175–1200. [Google Scholar] [CrossRef]
- Alonso, M.V.; Oliet, M.; Rodríguez, F.; García, J.; Gilarranz, M.A.; Rodríguez, J.J. Modification of ammonium lignosulfonate by phenolation for use in phenolic resins. Bioresour. Technol. 2005, 96, 1013–1018. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, J.; Du, X.; Hu, Z.; Chang, H.-m.; Jameel, H. Phenolation to Improve Lignin Reactivity toward Thermosets Application. ACS Sustain. Chem. Eng. 2018, 6, 5504–5512. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, X.; Lin, J.; Zhao, G.; Chang, H.-m.; Jameel, H. Reactivity improvement by phenolation of wheat straw lignin isolated from a biorefinery process. New J. Chem. 2019, 43, 2238–2246. [Google Scholar] [CrossRef]
- Yang, S.; Wen, J.-L.; Yuan, T.-Q.; Sun, R.-C. Characterization and phenolation of biorefinery technical lignins for lignin–phenol–formaldehyde resin adhesive synthesis. RSC Adv. 2014, 4, 57996–58004. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Yuan, T.-Q.; Sun, R.-C. Lignin–phenol–formaldehyde resin adhesives prepared with biorefinery technical lignins. J. Appl. Polym. Sci. 2015, 132, 42493. [Google Scholar] [CrossRef]
- Podschun, J.; Saake, B.; Lehnen, R. Reactivity enhancement of organosolv lignin by phenolation for improved bio-based thermosets. Eur. Polym. J. 2015, 67, 1–11. [Google Scholar] [CrossRef]
- Podschun, J.; Stücker, A.; Saake, B.; Lehnen, R. Structure–Function Relationships in the Phenolation of Lignins from Different Sources. ACS Sustain. Chem. Eng. 2015, 3, 2526–2532. [Google Scholar] [CrossRef]
- Çetin, N.S.; Özmen, N. Use of organosolv lignin in phenol–formaldehyde resins for particleboard production: I. Organosolv lignin modified resins. Int. J. Adhes. Adhes. 2002, 22, 477–480. [Google Scholar] [CrossRef]
- Hu, L.; Zhou, Y.; Zhang, M.; Liu, R. Characterization and properties of a lignosulfonate-based phenolic foam. Bioresources 2011, 7, 554–564. [Google Scholar]
- Abarro, G.J.; Podschun, J.; Diaz, L.J.; Ohashi, S.; Saake, B.; Lehnen, R.; Ishida, H. Benzoxazines with enhanced thermal stability from phenolated organosolv lignin. RSC Adv. 2016, 6, 107689–107698. [Google Scholar] [CrossRef]
- Tan, T.T.M. Cardanol–lignin-based polyurethanes. Polym. Int. 1996, 41, 13–16. [Google Scholar] [CrossRef]
- Zhao, B.Y.; Hu, K.A.; Wu, R.J. Primary study on the phenolic modification of sodium lignosulfonate. Polym. Eng. Sci. 2000, 16, 158–161. [Google Scholar]
- Hoffmann, A.; Nong, J.P.; Porzel, A.; Bremer, M.; Fischer, S. Modification of Lignoboost Kraft Lignin from softwoods with dihydroxybenzenes. React. Funct. Polym. 2019, 142, 112–118. [Google Scholar] [CrossRef]
- Lewis, H.F.; Brauns, F.E.; Buchanan, M.A.; Brookbank, E.B. Lignin Esters of Mono- and Dibasic Aliphatic Acids. Ind. Eng. Chem. Res. 1943, 35, 1113–1117. [Google Scholar] [CrossRef]
- Xiao, B.; Sun, X.F.; Sun, R. The chemical modification of lignins with succinic anhydride in aqueous systems. Polym. Degrad. Stab. 2001, 71, 223–231. [Google Scholar] [CrossRef]
- Fox, S.; McDonald, A. Chemical and Thermal Characterization of Three Industrial Lignins and Their Corresponding Lignin Esters. BioResources 2010, 5, 990–1009. [Google Scholar]
- Sailaja, R.R.N.; Deepthi, M.V. Mechanical and thermal properties of compatibilized composites of polyethylene and esterified lignin. Mater. Des. 2010, 31, 4369–4379. [Google Scholar] [CrossRef]
- Cachet, N.; Camy, S.; Benjelloun-Mlayah, B.; Condoret, J.-S.; Delmas, M. Esterification of organosolv lignin under supercritical conditions. Indus. Crops Prod. 2014, 58, 287–297. [Google Scholar] [CrossRef]
- Thiebaud, S.; Borredon, M.E. Solvent-free wood esterification with fatty acid chlorides. Bioresour. Technol. 1995, 52, 169–173. [Google Scholar] [CrossRef]
- Thiebaud, S.; Borredon, M.E.; Baziard, G.; Senocq, F. Properties of wood esterified by fatty-acid chlorides. Bioresour. Technol. 1997, 59, 103–107. [Google Scholar] [CrossRef]
- Bonini, C.; D’Auria, M.; Emanuele, L.; Ferri, R.; Pucciariello, R.; Sabia, A. Polyurethanes and polyesters from lignin. J. Appl. Polym. Sci. 2005, 98, 1451–1456. [Google Scholar] [CrossRef]
- Laurichesse, S.; Huillet, C.; Avérous, L. Original polyols based on organosolv lignin and fatty acids: New bio-based building blocks for segmented polyurethane synthesis. Green Chem. 2014, 16, 3958–3970. [Google Scholar] [CrossRef]
- Gordobil, O.; Robles, E.; Egüés, I.; Labidi, J. Lignin-ester derivatives as novel thermoplastic materials. RSC Adv. 2016, 6, 86909–86917. [Google Scholar] [CrossRef]
- Gordobil, O.; Herrera, R.; Llano-Ponte, R.; Labidi, J. Esterified organosolv lignin as hydrophobic agent for use on wood products. Prog. Org. Coat. 2017, 103, 143–151. [Google Scholar] [CrossRef]
- Fang, R.; Cheng, X.S.; Lin, W.S. Preparation and application of dimer acid/lignin graft copolymer. BioResources 2011, 6, 2874–2884. [Google Scholar]
- Sivasankarapillai, G.; McDonald, A.G. Synthesis and properties of lignin-highly branched poly (ester-amine) polymeric systems. Biomass Bioenergy 2011, 35, 919–931. [Google Scholar] [CrossRef]
- Sivasankarapillai, G.; McDonald, A.; Li, H. Lignin valorization by forming toughened lignin-co-polymers: Development of hyperbranched prepolymers for cross-linking. Biomass Bioenergy 2012, 47, 99–108. [Google Scholar] [CrossRef]
- Liu, L.-Y.; Hua, Q.; Renneckar, S. A simple route to synthesize esterified lignin derivatives. Green Chem. 2019, 21, 3682–3692. [Google Scholar] [CrossRef]
- Liu, L.-Y.; Cho, M.; Sathitsuksanoh, N.; Chowdhury, S.; Renneckar, S. Uniform Chemical Functionality of Technical Lignin Using Ethylene Carbonate for Hydroxyethylation and Subsequent Greener Esterification. ACS Sustain. Chem. Eng. 2018, 6, 12251–12260. [Google Scholar] [CrossRef]
- Picinelli, A.; Dapena, E.; Mangas, J.J. Polyphenolic Pattern in Apple Tree Leaves in Relation to Scab Resistance. A Preliminary Study. J. Agric. Food. Chem. 1995, 43, 2273–2278. [Google Scholar] [CrossRef]
- Trejo-Machin, A.; Verge, P.; Puchot, L.; Quintana, R. Phloretic acid as an alternative to the phenolation of aliphatic hydroxyls for the elaboration of polybenzoxazine. Green Chem. 2017, 19, 5065–5073. [Google Scholar] [CrossRef]
- Lora, J.H. Characteristics, Industrial Sources, and Utilization of Lignins from Non-Wood Plants. In Chemical Modification, Properties, and Usage of Lignin; Hu, T.Q., Ed.; Springer: Boston, MA, USA, 2002. [Google Scholar]
- Lora, J.H.; Glasser, W.G. Recent Industrial Applications of Lignin: A Sustainable Alternative to Nonrenewable Materials. J. Polym. Environ. 2002, 10, 39–48. [Google Scholar] [CrossRef]
- Granata, A.; Argyropoulos, D.S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a Reagent for the Accurate Determination of the Uncondensed and Condensed Phenolic Moieties in Lignins. J. Agric. Food. Chem. 1995, 43, 1538–1544. [Google Scholar] [CrossRef]
- Zawadzki, M.; Ragauskas, A. N-Hydroxy Compounds as New Internal Standards for the 31P-NMR Determination of Lignin Hydroxy Functional Groups. Holzforschung 2001, 55, 283–285. [Google Scholar] [CrossRef]
- Balakshin, M.; Capanema, E. On the Quantification of Lignin Hydroxyl Groups with 31P and 13C NMR Spectroscopy. J. Wood Chem. Technol. 2015, 35, 220–237. [Google Scholar] [CrossRef]
- Capanema, E.; Balakshin, M.; Kadla, J. A Comprehensive Approach for Quantitative Lignin Characterization by NMR Spectroscopy. J. Agric. Food. Chem. 2004, 52, 1850–1860. [Google Scholar] [CrossRef]
- Tran, F.; Lancefield, C.S.; Kamer, P.C.J.; Lebl, T.; Westwood, N.J. Selective modification of the β–β linkage in DDQ-treated Kraft lignin analysed by 2D NMR spectroscopy. Green Chem. 2015, 17, 244–249. [Google Scholar] [CrossRef]
- McClelland, D.J.; Motagamwala, A.H.; Li, Y.; Rover, M.R.; Wittrig, A.M.; Wu, C.; Buchanan, J.S.; Brown, R.C.; Ralph, J.; Dumesic, J.A.; et al. Functionality and molecular weight distribution of red oak lignin before and after pyrolysis and hydrogenation. Green Chem. 2017, 19, 1378–1389. [Google Scholar] [CrossRef]
- Sameni, J.; Krigstin, S.; Sain, M. Solubility of Lignin and Acetylated Lignin in Organic Solvents. Bioresources 2017, 12, 1548–1565. [Google Scholar] [CrossRef]
- Hansen, C. Hansen Solubility Parameters: A User’s Handbook; Taylor & Francis: New York, NY, USA, 2012. [Google Scholar]
- Balakshin, M.; Capanema, E.; Santos, R.; Chang, H.-M.; Jameel, H. Structural analysis of hardwood native lignins by quantitative 13C NMR spectroscopy. Holzforschung 2015, 70, 95–108. [Google Scholar] [CrossRef]
- Ahvazi, B.; Wojciechowicz, O.; Xu, P.; Ngo, T.; Hawari, J. Formation of Ligno-Polyols: Fact or Fiction. Bioresources 2017, 12, 6629–6655. [Google Scholar] [CrossRef][Green Version]
- Holtman, K.; Chang, H.-M.; Jameel, H.; Kadla, J. Quantitative 13 C NMR Characterization of Milled Wood Lignins Isolated by Different Milling Techniques. J. Wood Chem. Technol. 2006, 26, 21–34. [Google Scholar] [CrossRef]
- Del Río, J.C.; Rencoret, J.; Prinsen, P.; Martínez, Á.T.; Ralph, J.; Gutiérrez, A. Structural Characterization of Wheat Straw Lignin as Revealed by Analytical Pyrolysis, 2D-NMR, and Reductive Cleavage Methods. J. Agric. Food Chem. 2012, 60, 5922–5935. [Google Scholar] [CrossRef]
- Wen, J.-L.; Sun, S.-L.; Xue, B.-L.; Sun, R.-C. Recent Advances in Characterization of Lignin Polymer by Solution-State Nuclear Magnetic Resonance (NMR) Methodology. Materials 2013, 6, 359–391. [Google Scholar] [CrossRef]
- Strassberger, Z.; Prinsen, P.; Klis, F.v.d.; van Es, D.S.; Tanase, S.; Rothenberg, G. Lignin solubilisation and gentle fractionation in liquid ammonia. Green Chem. 2015, 17, 325–334. [Google Scholar] [CrossRef]
- Constant, S.; Wienk, H.L.J.; Frissen, A.E.; Peinder, P.d.; Boelens, R.; van Es, D.S.; Grisel, R.J.H.; Weckhuysen, B.M.; Huijgen, W.J.J.; Gosselink, R.J.A.; et al. New insights into the structure and composition of technical lignins: A comparative characterisation study. Green Chem. 2016, 18, 2651–2665. [Google Scholar] [CrossRef]
- Funaoka, M.; Kako, T.; Abe, I. Condensation of lignin during heating of wood. Wood Sci. Technol. 1990, 24, 277–288. [Google Scholar] [CrossRef]
- Demirbaş, A. Mechanisms of liquefaction and pyrolysis reactions of biomass. Energy Convers. Manage. 2000, 41, 633–646. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Hwang, H.; Oh, S.; Kim, Y.-S.; Kim, U.-J.; Choi, J.W. Investigation of structural modification and thermal characteristics of lignin after heat treatment. Int. J. Biol. Macromol. 2014, 66, 57–65. [Google Scholar] [CrossRef]
- Ibrahim, M.; Iqbal, A.; Shen, C.; Bhawani, S.; Adam, F. Synthesis of lignin based composites of TiO2 for potential application as radical scavengers in sunscreen formulation. BMC Chem. 2019, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Faix, O. Fourier Transform Infrared Spectroscopy. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 233–241. [Google Scholar]
- Schorr, D.; Diouf, P.N.; Stevanovic, T. Evaluation of industrial lignins for biocomposites production. Indus. Crops Prod. 2014, 52, 65–73. [Google Scholar] [CrossRef]
- Sahoo, S.; Seydibeyoğlu, M.Ö.; Mohanty, A.K.; Misra, M. Characterization of industrial lignins for their utilization in future value added applications. Biomass Bioenergy 2011, 35, 4230–4237. [Google Scholar] [CrossRef]
- Van Krevelen, D.W. Some basic aspects of flame resistance of polymeric materials. Polymer 1975, 16, 615–620. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Parameters | Symbol | Level | |
|---|---|---|---|
| −1 | +1 | ||
| Time (h) | t | 12 | 48 |
| Molar ratio (nCOOH_PA/nOHali_P2400 a) | n | 1 | 5 |
| Catalyst loading (wt.%) | c | 0.5 | 2.5 |
| Sample | [COOH] | [5-subst.] | [Gnc] | [H] | [OH]aro | [OH]ali |
|---|---|---|---|---|---|---|
| P2400 | 0.78 ± 0.04 | 2.12 ± 0.23 | 1.29 ± 0.22 | 0.53 ± 0.07 | 3.94 ± 0.51 | 2.00 ± 0.23 |
| P2400-PA | 0.81 ± 0.03 | 1.79 ± 0.09 | 1.03 ± 0.07 | 1.81 ± 0.07 | 4.63 ± 0.22 | 0.14 ± 0.02 |
| Sample | C (%) | H (%) | N (%) | S (%) | O (%) |
|---|---|---|---|---|---|
| P2400 | 64.44 ± 0.01 | 6.06 ± 0.14 | 0.56 ± 0.05 | 0.63 ± 0.03 | 28.31 |
| P2400-PA | 67.20 ± 0.04 | 5.68 ± 0.16 | 0.65 ± 0.03 | 0.58 ± 0.01 | 25.89 |
| Run | t (h) | n | c (wt.%) | a [OH]ali (mmol g−1) | b Conversion (%) | a [H] (mmol g−1) | c Response (%) |
|---|---|---|---|---|---|---|---|
| EL-1 | 12 | 1 | 0.5 | 0.61 ± 0.03 | 69.5 | 0.87 ± 0.02 | 17.0 |
| EL-2 | 48 | 1 | 0.5 | 0.40 ± 0.06 | 80.0 | 0.96 ± 0.05 | 21.5 |
| EL-3 | 12 | 5 | 0.5 | 0.33 ± 0.01 | 83.5 | 1.58 ± 0.08 | 52.5 |
| EL-4 | 48 | 5 | 0.5 | 0.14 ± 0.02 | 93.0 | 1.78 ± 0.10 | 62.5 |
| EL-5 | 12 | 1 | 2.5 | 0.51 ± 0.01 | 74.5 | 1.09 ± 0.17 | 28.0 |
| EL-6 | 48 | 1 | 2.5 | 0.26 ± 0.08 | 87.0 | 1.04 ± 0.08 | 25.5 |
| EL-7 | 12 | 5 | 2.5 | 0.19 ± 0.12 | 90.5 | 1.56 ± 0.12 | 51.5 |
| EL-8 | 48 | 5 | 2.5 | 0.14 ± 0.02 | 93.0 | 1.81 ± 0.07 | 64.0 |
| Sample | a δD (MPa1/2) | a δP (MPa1/2) | a δH (MPa1/2) | bMn (g mol−1) | cMw (g mol−1) | dĐ |
|---|---|---|---|---|---|---|
| P2400 | 18.0 | 9.4 | 16.6 | 575 | 4323 | 7.5 |
| P2400-PA | 17.1 | 13.1 | 12.5 | 1570 | 8546 | 5.4 |
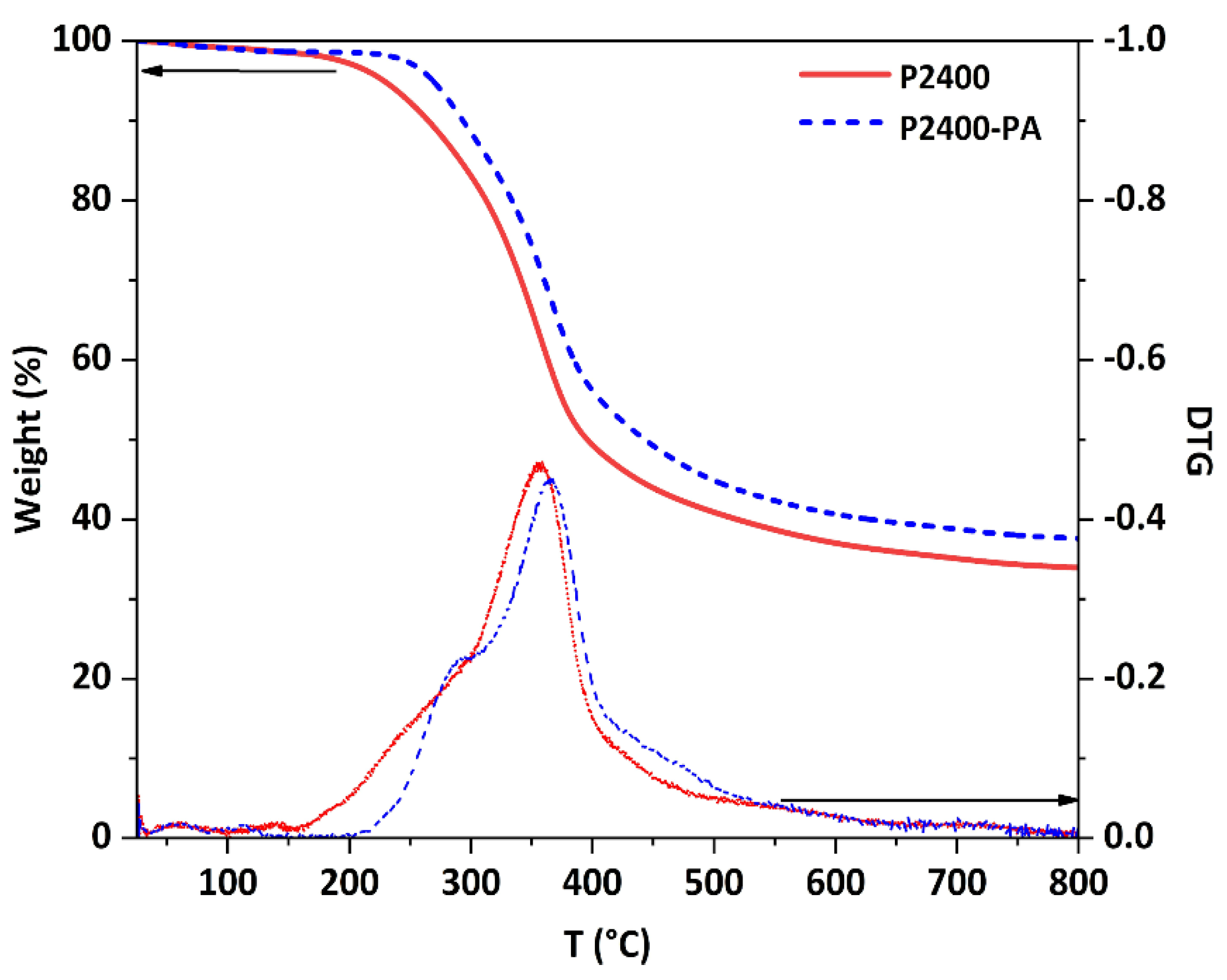
| Sample | aTonset (°C) | a,bTmax (°C) | c Char (%) | d LOI (%) | eTg (°C) |
|---|---|---|---|---|---|
| P2400 | 157 | 359 | 33.9 | 31.1 | 92 |
| P2400-PA | 220 | 367 | 37.6 | 32.5 | 112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adjaoud, A.; Dieden, R.; Verge, P. Sustainable Esterification of a Soda Lignin with Phloretic Acid. Polymers 2021, 13, 637. https://doi.org/10.3390/polym13040637
Adjaoud A, Dieden R, Verge P. Sustainable Esterification of a Soda Lignin with Phloretic Acid. Polymers. 2021; 13(4):637. https://doi.org/10.3390/polym13040637
Chicago/Turabian StyleAdjaoud, Antoine, Reiner Dieden, and Pierre Verge. 2021. "Sustainable Esterification of a Soda Lignin with Phloretic Acid" Polymers 13, no. 4: 637. https://doi.org/10.3390/polym13040637
APA StyleAdjaoud, A., Dieden, R., & Verge, P. (2021). Sustainable Esterification of a Soda Lignin with Phloretic Acid. Polymers, 13(4), 637. https://doi.org/10.3390/polym13040637