
Effect of the Molecular Structure of TPU on the Cellular Structure of Nanocellular Polymers Based on PMMA/TPU Blends

 ,
, 
 ,
, 

Abstract
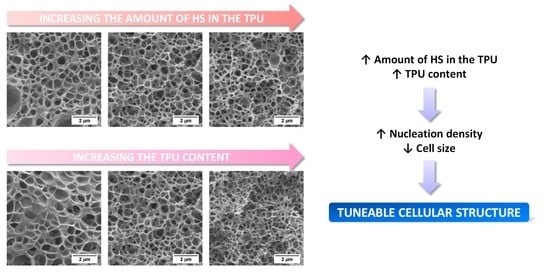
:
1. Introduction
2. Materials and Methods
2.1. Materials
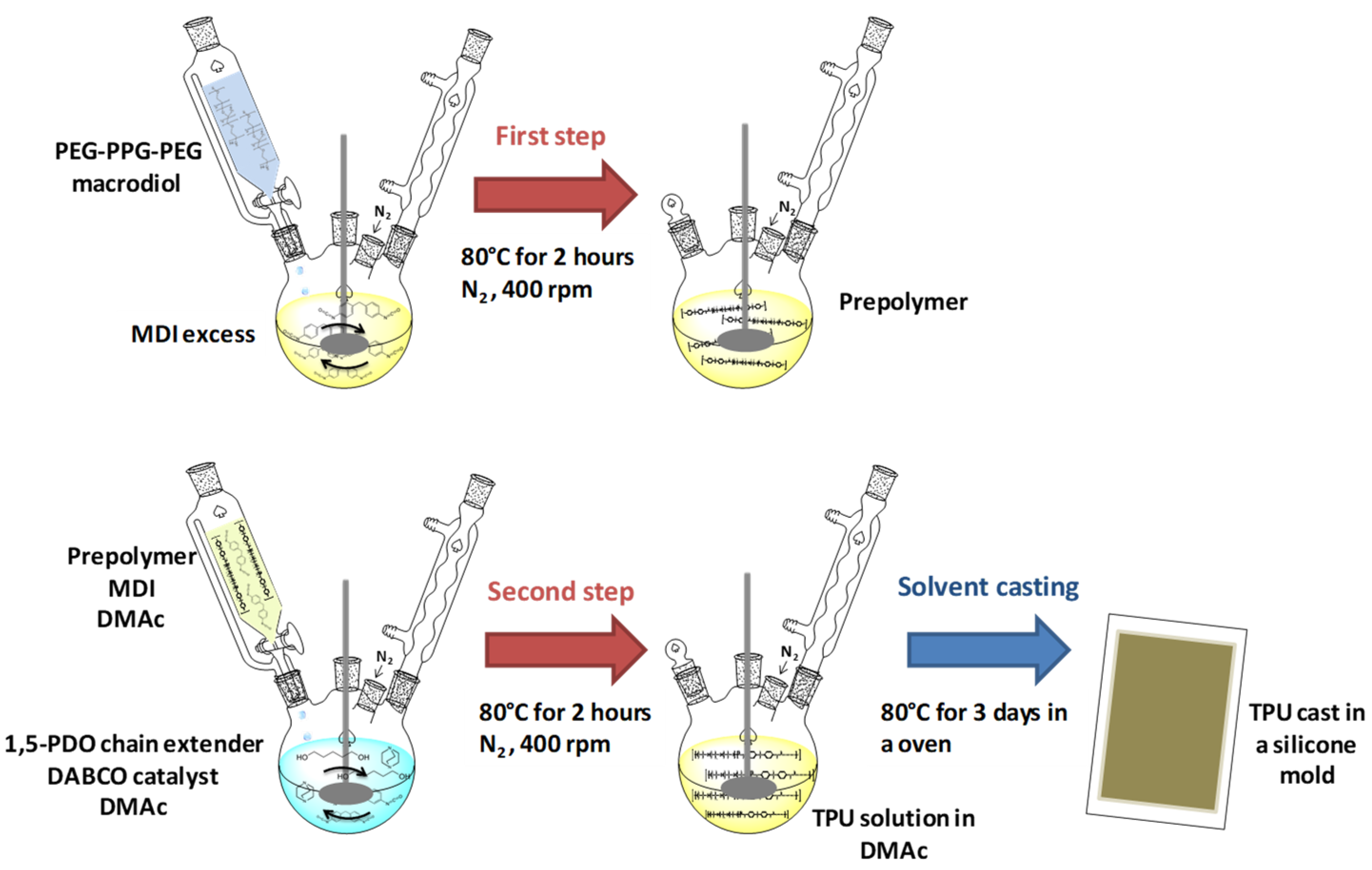
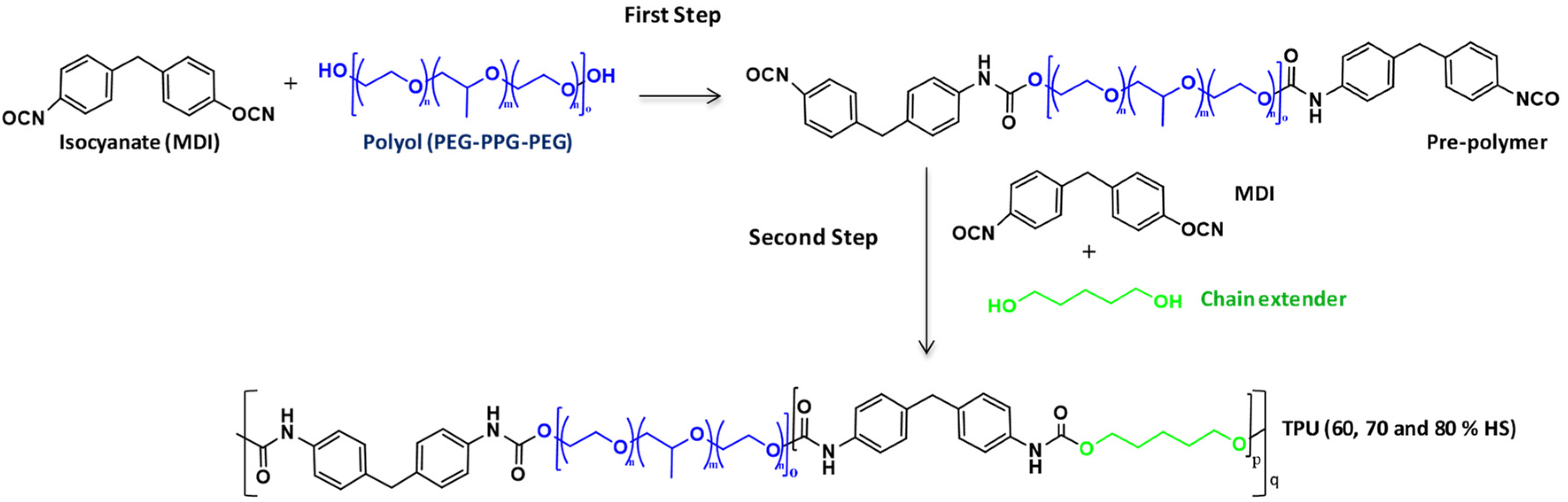
2.2. Synthesis of TPU Grades
2.3. Solid Blends Production
2.4. Gas Dissolution Foaming Experiments
2.5. Characterization of TPU Grades
2.5.1. Density
2.5.2. Gel Permeation Chromatography
2.5.3. Differential Scanning Calorimetry
2.5.4. Shore Hardness
2.6. Characterization of PMMA/TPU Blends and Cellular Materials
2.6.1. PMMA/TPU Blends Nanostructuration
2.6.2. Density
2.6.3. Cellular Structure
3. Results and Discussion
3.1. Characterization of the TPU Grades
3.2. Nanostructuration of the PMMA/TPU Blends
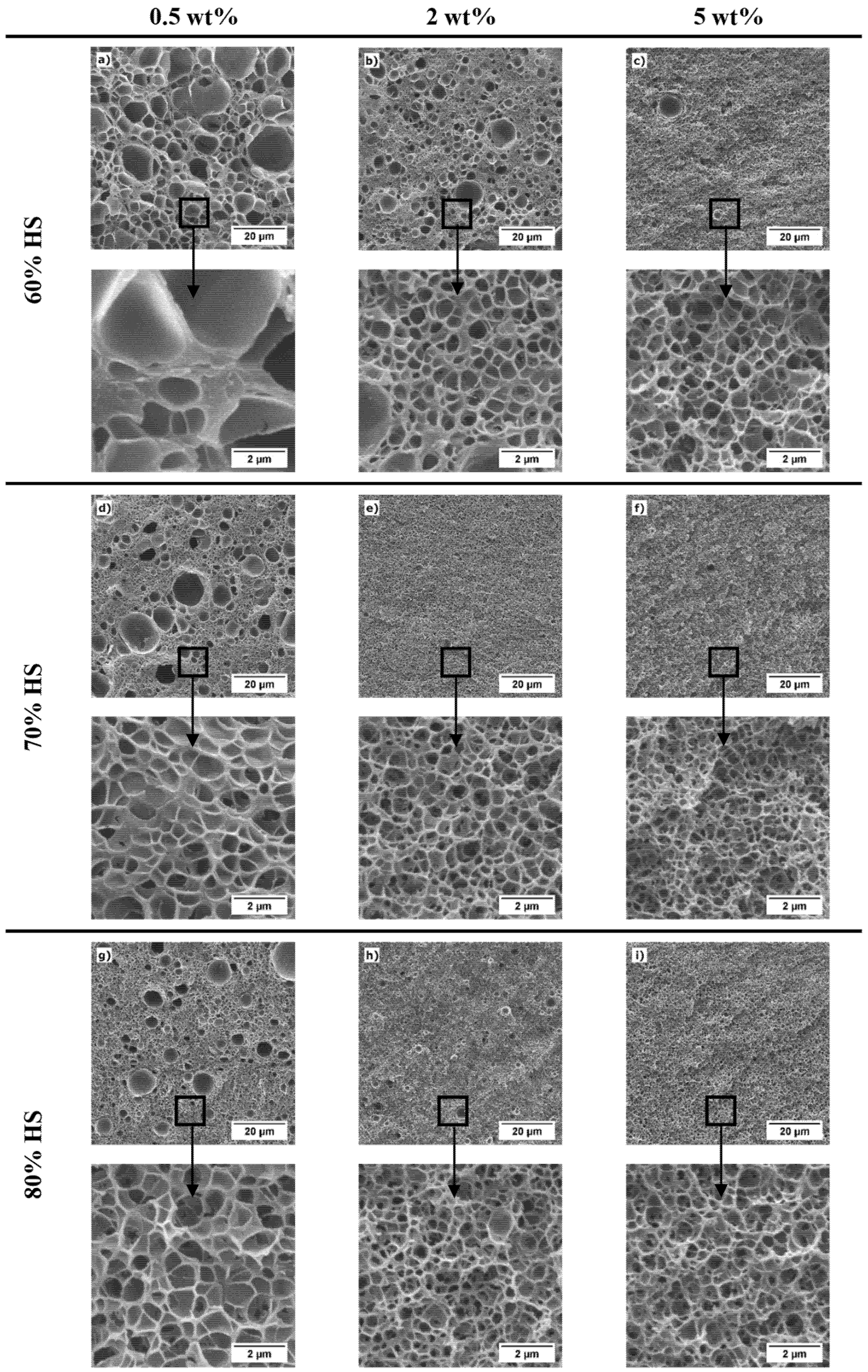
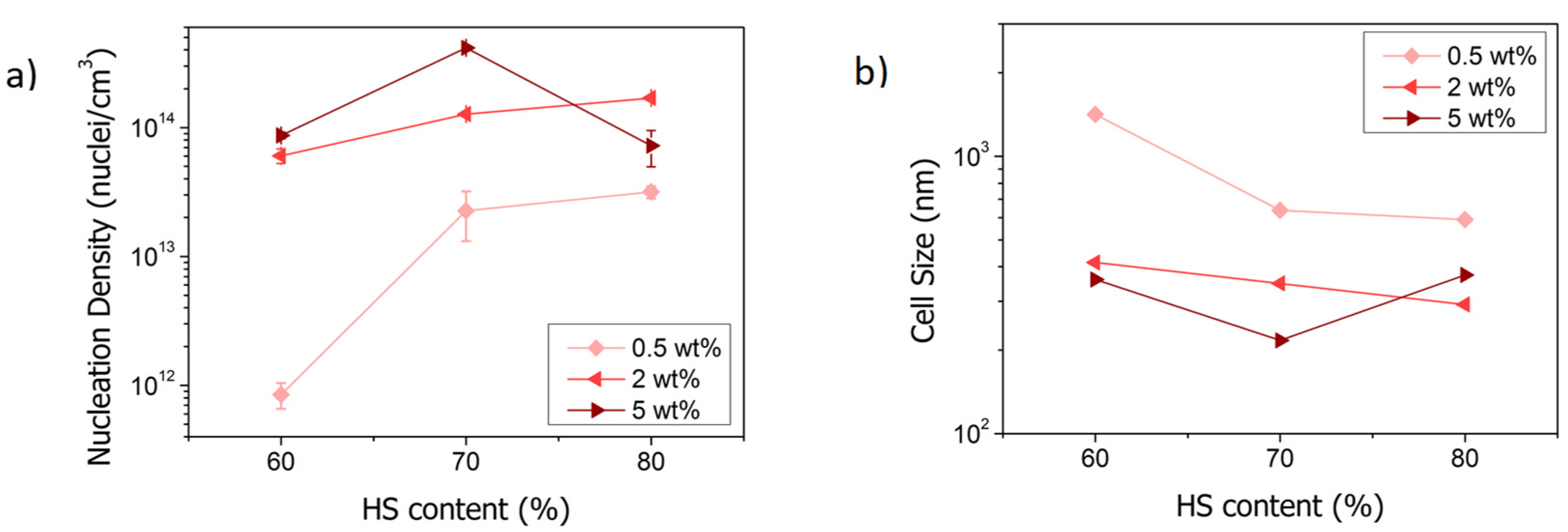
3.3. Effect of the Concentration of TPU and of the Fraction of HS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martín-de León, J.; Bernardo, V.; Rodríguez-Pérez, M. Nanocellular Polymers: The Challenge of Creating Cells in the Nanoscale. Materials 2019, 12, 797. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Zhu, C.-Y.; Key, X.-P.Z. A theoretical and numerical study on the gas-contributed thermal conductivity in aerogel. Int. J. Heat Mass Transf. 2017, 108, 1982–1990. [Google Scholar] [CrossRef]
- Notario, B.; Pinto, J.; Rodriguez-Perez, M.A. Nanoporous polymeric materials: A new class of materials with enhanced properties. Prog. Mater. Sci. 2016, 78–79, 93–139. [Google Scholar] [CrossRef]
- Notario, B.; Pinto, J.; Solorzano, E.; de Saja, J.A.; Dumon, M.; Rodríguez-Pérez, M.A. Experimental validation of the Knudsen effect in nanocellular polymeric foams. Polymer 2015, 56, 57–67. [Google Scholar] [CrossRef]
- Pinto, J.; Notario, B.; Verdejo, R.; Dumon, M.; Costeux, S.; Rodriguez-Perez, M.A. Molecular confinement of solid and gaseous phases of self-standing bulk nanoporous polymers inducing enhanced and unexpected physical properties. Polymer 2017, 113, 27–33. [Google Scholar] [CrossRef]
- Notario, B.; Pinto, J.; Rodríguez-Pérez, M.A. Towards a new generation of polymeric foams: PMMA nanocellular foams with enhanced physical properties. Polymer 2015, 63, 116–126. [Google Scholar] [CrossRef]
- Xiang, B.; Jia, Y.; Lei, Y.; Zhang, F.; He, J.; Liu, T.; Luo, S. Mechanical properties of microcellular and nanocellular silicone rubber foams obtained by supercritical carbon dioxide. Polym. J. 2019, 51, 559–568. [Google Scholar] [CrossRef]
- Martín-de León, J.; Bernardo, V.; Rodríguez-Pérez, M.Á. Key Production Parameters to Obtain Transparent Nanocellular PMMA. Macromol. Mater. Eng. 2017, 302, 1700343. [Google Scholar] [CrossRef]
- Martín-de León, J.; Pura, J.L.; Bernardo, V.; Rodríguez-Pérez, M.Á. Transparent nanocellular PMMA: Characterization and modeling of the optical properties. Polymer 2019, 170, 16–23. [Google Scholar] [CrossRef]
- Pinto, J.; Dumon, M.; Rodriguez-Perez, M.A.; Garcia, R.; Dietz, C. Block Copolymers Self-Assembly Allows Obtaining Tunable Micro or Nanoporous Membranes or Depth Filters Based on PMMA; Fabrication Method and Nanostructures. J. Phys. Chem. C 2014, 118, 4656–4663. [Google Scholar] [CrossRef]
- Lu, G.Q.; Zhao, X.S. Nanoporous Materials—An Overview. In Nanoporous Materials: Science and Engineering; Imperial College Press: London, UK, 2004; pp. 1–13. [Google Scholar]
- Wang, G.; Zhao, G.; Zhang, L.; Mu, Y.; Park, C.B. Lightweight and tough nanocellular PP/PTFE nanocomposite foams with defect-free surfaces obtained using in situ nanofibrillation and nanocellular injection molding. Chem. Eng. J. 2018, 350, 1–11. [Google Scholar] [CrossRef]
- Costeux, S. CO2-blown nanocellular foams. J. Appl. Polym. Sci. 2014, 131, 41293. [Google Scholar] [CrossRef]
- Martín-de León, J.; Bernardo, V.; Rodríguez-Pérez, M. Low Density Nanocellular Polymers Based on PMMA Produced by Gas Dissolution Foaming: Fabrication and Cellular Structure Characterization. Polymers 2016, 8, 265. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Nicolae, A.; Kumar, V. Solid-state poly(methyl methacrylate) (PMMA) nanofoams. Part II: Low-temperature solid-state process space using CO2 and the resulting morphologies. Polymer 2015, 70, 231–241. [Google Scholar] [CrossRef]
- Yoon, T.J.; Kong, W.; Kwon, D.E.; Park, B.K.; Lee, W.I.; Lee, Y.W. Preparation of solid-state micro- and nanocellular acrylonitrile-butadiene-styrene (ABS) foams using sub- and supercritical CO2 as blowing agents. J. Supercrit. Fluids 2017, 124, 30–37. [Google Scholar] [CrossRef]
- Costeux, S.; Zhu, L. Low density thermoplastic nanofoams nucleated by nanoparticles. Polymer 2013, 54, 2785–2795. [Google Scholar] [CrossRef]
- Bernardo, V.; Martín-de León, J.; Laguna-Gutiérrez, E.; Rodríguez-Pérez, M.Á. PMMA-sepiolite nanocomposites as new promising materials for the production of nanocellular polymers. Eur. Polym. J. 2017, 96, 10–26. [Google Scholar] [CrossRef]
- Xiao, S.P.; Huang, H.X. Generation of nanocellular TPU/reduced graphene oxide nanocomposite foams with high cell density by manipulating viscoelasticity. Polymer 2019, 183, 121879. [Google Scholar] [CrossRef]
- Jahani, D.; Azimi, H.; Nazari, A. An experimental study on the micro- And nanocellular foaming of polystyrene/poly(methyl methacrylate) blend composites. J. Polym. Eng. 2019, 39, 926–933. [Google Scholar] [CrossRef]
- Forest, C.; Chaumont, P.; Cassagnau, P.; Swoboda, B.; Sonntag, P. CO2 Nano-foaming of nanostructured PMMA. Polymer 2015, 58, 76–87. [Google Scholar] [CrossRef]
- Nofar, M.; Büşra Küçük, E.; Batı, B. Effect of hard segment content on the microcellular foaming behavior of TPU using supercritical CO2. J. Supercrit. Fluids 2019, 153, 104590. [Google Scholar] [CrossRef]
- Liu, S.; Yin, S.; Duvigneau, J.; Vancso, G.J. Bubble Seeding Nanocavities: Multiple Polymer Foam Cell Nucleation by Polydimethylsiloxane-Grafted Designer Silica Nanoparticles. ACS Nano 2020, 14, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, Y.; Yang, G.; Yang, Q. Nucleation thermodynamics inside micro/nanocavity. J. Mater. Sci. Technol. 2008, 24, 183–186. [Google Scholar]
- Yeh, S.-K.; Chen, Y.-R.; Kang, T.-W.; Tseng, T.-J.; Peng, S.-P.; Chu, C.-C.; Rwei, S.-P.; Guo, W.-J. Different approaches for creating nanocellular TPU foams by supercritical CO2 foaming. J. Polym. Res. 2018, 25, 30. [Google Scholar] [CrossRef]
- Ni, J.; Yu, K.; Zhou, H.; Mi, J.; Chen, S.; Wang, X. Morphological evolution of PLA foam from microcellular to nanocellular induced by cold crystallization assisted by supercritical CO2. J. Supercrit. Fluids 2020, 158, 104719. [Google Scholar] [CrossRef]
- Bernardo, V.; Martin-de Leon, J.; Laguna-Gutierrez, E.; Catelani, T.; Pinto, J.; Athanassiou, A.; Rodriguez-Perez, M.A. Understanding the role of MAM molecular weight in the production of PMMA/MAM nanocellular polymers. Polymer 2018, 153, 262–270. [Google Scholar] [CrossRef]
- Bernardo, V.; Martin-de Leon, J.; Pinto, J.; Catelani, T.; Athanassiou, A.; Rodriguez-Perez, M.A. Low-density PMMA/MAM nanocellular polymers using low MAM contents: Production and characterization. Polymer 2019, 163, 115–124. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, J.; Mark, L.H.; Wang, G.; Yu, K.; Wang, C.; Park, C.B.; Zhao, G. Ultra-tough and super thermal-insulation nanocellular PMMA/TPU. Chem. Eng. J. 2017, 325, 632–646. [Google Scholar] [CrossRef]
- Bernardo, V.; Martin-de Leon, J.; Sanchez-Calderon, I.; Laguna-Gutierrez, E.; Rodriguez-Perez, M.A. Nanocellular Polymers with a Gradient Cellular Structure Based on Poly(methyl methacrylate)/Thermoplastic Polyurethane Blends Produced by Gas Dissolution Foaming. Macromol. Mater. Eng. 2019, 305, 1900428. [Google Scholar] [CrossRef]
- Ghaffari Mosanenzadeh, S.; Naguib, H.E.; Park, C.B.; Atalla, N. Design and development of novel bio-based functionally graded foams for enhanced acoustic capabilities. J. Mater. Sci. 2015, 50, 1248–1256. [Google Scholar] [CrossRef]
- Monnereau, L.; Urbanczyk, L.; Thomassin, J.-M.; Pardoen, T.; Bailly, C.; Huynen, I.; Jérôme, C.; Detrembleur, C. Gradient foaming of polycarbonate/carbon nanotube based nanocomposites with supercritical carbon dioxide and their EMI shielding performances. Polymer 2015, 59, 117–123. [Google Scholar] [CrossRef]
- Pinto, J.; Morselli, D.; Bernardo, V.; Notario, B.; Fragouli, D.; Rodriguez-Perez, M.A.; Athanassiou, A. Nanoporous PMMA foams with templated pore size obtained by localized in situ synthesis of nanoparticles and CO 2 foaming. Polymer 2017, 124, 176–185. [Google Scholar] [CrossRef]
- Trofa, M.; Di Maio, E.; Maffettone, P.L. Multi-graded foams upon time-dependent exposition to blowing agent. Chem. Eng. J. 2019, 362, 812–817. [Google Scholar] [CrossRef]
- Kumar, V.; Suh, N.P. A process for making microcellular thermoplastic parts. Polym. Eng. Sci. 1990, 30, 1323–1329. [Google Scholar] [CrossRef]
- Pinto, J.; Dumon, M.; Pedros, M.; Reglero, J.; Rodriguez-Perez, M.A. Nanocellular CO2 foaming of PMMA assisted by block copolymer nanostructuration. Chem. Eng. J. 2014, 243, 428–435. [Google Scholar] [CrossRef]
- Tammaro, D.; Iannace, S.; Di Maio, E. Insight into bubble nucleation at high-pressure drop rate. J. Cell. Plast. 2017, 53, 551–560. [Google Scholar] [CrossRef]
- UNE-EN ISO 868:2003. Determinación de la Dureza de Indentación por Medio de un Durómetro (Dureza Shore); Plásticos y ebonita; AENOR: Madrid, Spain, 2003; p. 11. [Google Scholar]
- Pinto, J.; Solórzano, E.; Rodriguez-Perez, M.A.; de Saja, J.A. Characterization of the cellular structure based on user-interactive image analysis procedures. J. Cell. Plast. 2013, 49, 555–575. [Google Scholar] [CrossRef]
- Kumar, V. Process synthesis for manufacturing microcellular thermoplastic parts. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 1988. [Google Scholar]
- Prisacariu, C. Polyurethane Elastomers; Springer: Wien, Austria; New York, NY, USA, 2011. [Google Scholar]
- Wu, S. Formation of dispersed phase in incompatible polymer blends: Interfacial and rheological effects. Polym. Eng. Sci. 1987, 27, 335–343. [Google Scholar] [CrossRef]
- Wong, A.; Guo, Y.; Parka, C.B. Fundamental mechanisms of cell nucleation in polypropylene foaming with supercritical carbon dioxide—Effects of extensional stresses and crystals. J. Supercrit. Fluids 2013, 79, 142–151. [Google Scholar] [CrossRef]
- Lips, P.A.M.; Velthoen, I.W.; Dijkstra, P.J.; Wessling, M.; Feijen, J. Gas foaming of segmented poly(ester amide) films. Polymer 2005, 46, 9396–9403. [Google Scholar] [CrossRef]
- Mihai, M.; Huneault, M.A.; Favis, B.D. Rheology and extrusion foaming of chain-branched poly(lactic acid). Polym. Eng. Sci. 2010, 50, 629–642. [Google Scholar] [CrossRef]
- Taki, K.; Kitano, D.; Ohshima, M. Effect of growing crystalline phase on bubble nucleation in poly(L -lactide)/CO2 batch foaming. Ind. Eng. Chem. Res. 2011, 50, 3247–3252. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPU GRADES | FIRST STEP | SECOND STEP | |||
|---|---|---|---|---|---|
| MDI Isocyanate | PEG-PPG-PEG Macrodiol | PREPOLYMER | MDI Isocyanate | 1,5-PDO Chain Extender | |
| 60% HS | 64.2 g, 0.257 mol | 85.8 g, 0.043 mol | 69.9 g | 13.4 g, 0.054 mol | 16.7 g, 0.160 mol |
| 70% HS | 64.2 g, 0.257 mol | 85.8 g, 0.043 mol | 52.5 g | 28.3 g, 0.113 mol | 19.2 g, 0.184 mol |
| 80% HS | 64.2 g, 0.257 mol | 85.8 g, 0.043 mol | 35.0 g | 42.6 g, 0.170 mol | 22.4 g, 0.215 mol |
| Description | ρ (g/cm3) | Tg,SS (°C) | Tg,HS (°C) | Tm (°C) | Xc (%) | Hardness (Shore D) | Mn (g/mol) | Mw (g/mol) | Polydispersity Index |
|---|---|---|---|---|---|---|---|---|---|
| 60% HS | 1.192 | −43.3 | 55.5 | 177.5 | 10.6 | 45.6 ± 0.5 | 8552 ± 1602 | 18,325 ± 535 | 2.18 ± 0.32 |
| 70% HS | 1.204 | - | 55.5 | 183.5 | 12.8 | 63.2 ± 0.4 | 14,043 ± 1162 | 31,449 ± 128 | 2.25 ± 0.20 |
| 80% HS | 1.217 | - | 51.3 | 183.4 | 13.4 | 72.4 ± 0.5 | 15,086 ± 2126 | 32,603 ± 4825 | 2.16 ± 0.02 |
| Material | ϕd (nm) | SD/ϕd | Nd (Domains/cm3) |
|---|---|---|---|
| 5_60HS | 160 | 0.33 | 2.3 · 1013 |
| 5_70HS | 104 | 0.19 | 4.0 · 1013 |
| 5_80HS | 95 | 0.15 | 1.2 · 1014 |
| Sample ID | ρr,g | ρr,c |
|---|---|---|
| PMMA | 0.147 ± 0.001 | 0.175 ± 0.002 |
| 0.5_60HS | 0.143 ± 0.001 | 0.206 ± 0.002 |
| 2_60HS | 0.138 ± 0.001 | 0.200 ± 0.001 |
| 5_60HS | 0.148 ± 0.001 | 0.174 ± 0.002 |
| 0.5_70HS | 0.123 ± 0.001 | 0.177 ± 0.001 |
| 2_70HS | 0.107 ± 0.001 | 0.156 ± 0.005 |
| 5_70HS | 0.139 ± 0.001 | 0.171 ± 0.002 |
| 0.5_80HS | 0.110 ± 0.001 | 0.153 ± 0.001 |
| 2_80HS | 0.134 ± 0.001 | 0.191 ± 0.001 |
| 5_80HS | 0.152 ± 0.001 | 0.202 ± 0.001 |
| Sample ID | N0 (Nulcei/cm3) | ϕ3D (nm) | SD/ϕ3D | Bimodality | ||
|---|---|---|---|---|---|---|
| Amicrocells (%) | ϕmicrocells (µm) | SD/ϕmicrocells | ||||
| 0.5_60HS | (8.5 ± 1.9) · 1011 | 1417 | 1.08 | - | - | - |
| 2_60HS | (6.0 ± 0.8) · 1013 | 414 | 0.47 | 33 | 4.4 | 0.75 |
| 5_60HS | (8.7 ± 0.5) · 1013 | 359 | 0.64 | - | - | - |
| 0.5_70HS | (2.3 ± 0.9) · 1013 | 638 | 0.52 | 28 | 5.8 | 0.63 |
| 2_70HS | (1.3 ± 0.0) · 1014 | 348 | 0.52 | - | - | - |
| 5_70HS | (4.1 ± 0.1) · 1014 | 217 | 0.57 | - | - | - |
| 0.5_80HS | (3.2 ± 0.3) · 1013 | 591 | 0.48 | 13 | 5.8 | 0.50 |
| 2_80HS | (1.7 ± 0.0) · 1014 | 292 | 0.49 | 3 | 3.5 | 0.37 |
| 5_80HS | (7.2 ± 2.2) · 1013 | 373 | 0.62 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Calderón, I.; Bernardo, V.; Santiago-Calvo, M.; Naji, H.; Saiani, A.; Rodríguez-Pérez, M.Á. Effect of the Molecular Structure of TPU on the Cellular Structure of Nanocellular Polymers Based on PMMA/TPU Blends. Polymers 2021, 13, 3055. https://doi.org/10.3390/polym13183055
Sánchez-Calderón I, Bernardo V, Santiago-Calvo M, Naji H, Saiani A, Rodríguez-Pérez MÁ. Effect of the Molecular Structure of TPU on the Cellular Structure of Nanocellular Polymers Based on PMMA/TPU Blends. Polymers. 2021; 13(18):3055. https://doi.org/10.3390/polym13183055
Chicago/Turabian StyleSánchez-Calderón, Ismael, Victoria Bernardo, Mercedes Santiago-Calvo, Haneen Naji, Alberto Saiani, and Miguel Ángel Rodríguez-Pérez. 2021. "Effect of the Molecular Structure of TPU on the Cellular Structure of Nanocellular Polymers Based on PMMA/TPU Blends" Polymers 13, no. 18: 3055. https://doi.org/10.3390/polym13183055
APA StyleSánchez-Calderón, I., Bernardo, V., Santiago-Calvo, M., Naji, H., Saiani, A., & Rodríguez-Pérez, M. Á. (2021). Effect of the Molecular Structure of TPU on the Cellular Structure of Nanocellular Polymers Based on PMMA/TPU Blends. Polymers, 13(18), 3055. https://doi.org/10.3390/polym13183055