
Durable Modification of Wood by Benzoylation—Proof of Covalent Bonding by Solution State NMR and DOSY NMR Quick-Test †



Abstract
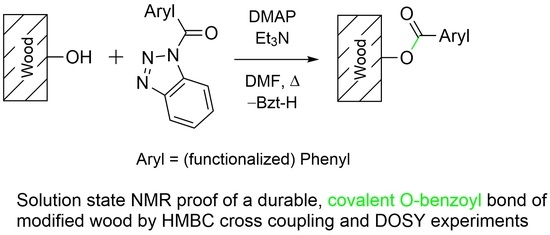
:
1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Chemicals, Solvents, and Wood Materials
2.3. Synthesis of 1H-benzotriazol-1-yl(phenyl)methanone-α-13C

2.4. Synthesis of 1H-benzotriazol-1-yl[3,5-bis(trifluoromethyl)phenyl]methanone
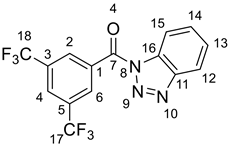
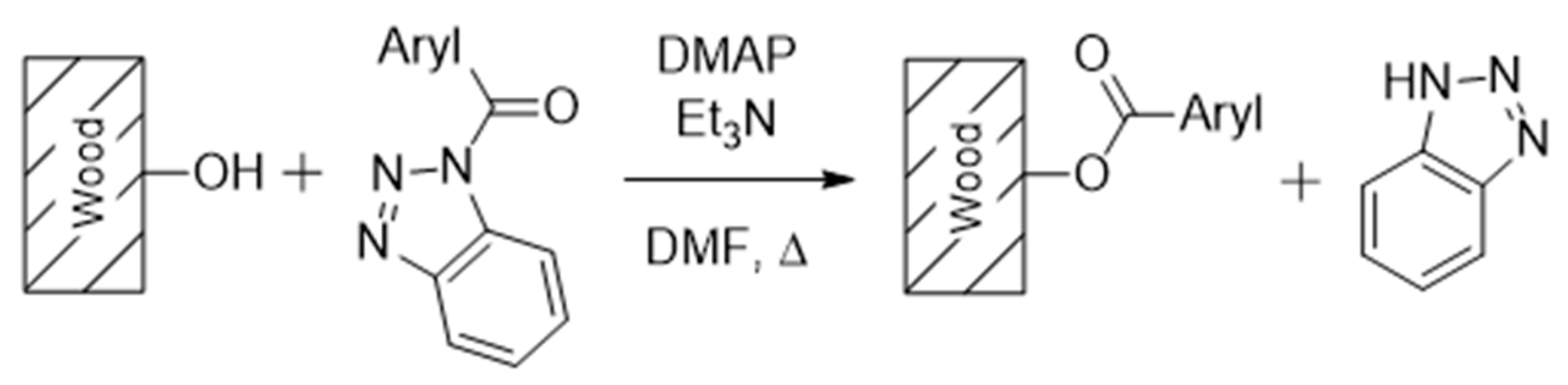
2.5. Chemical Wood Modification Procedure
2.6. Wood Sample Preparation for Solution State NMR
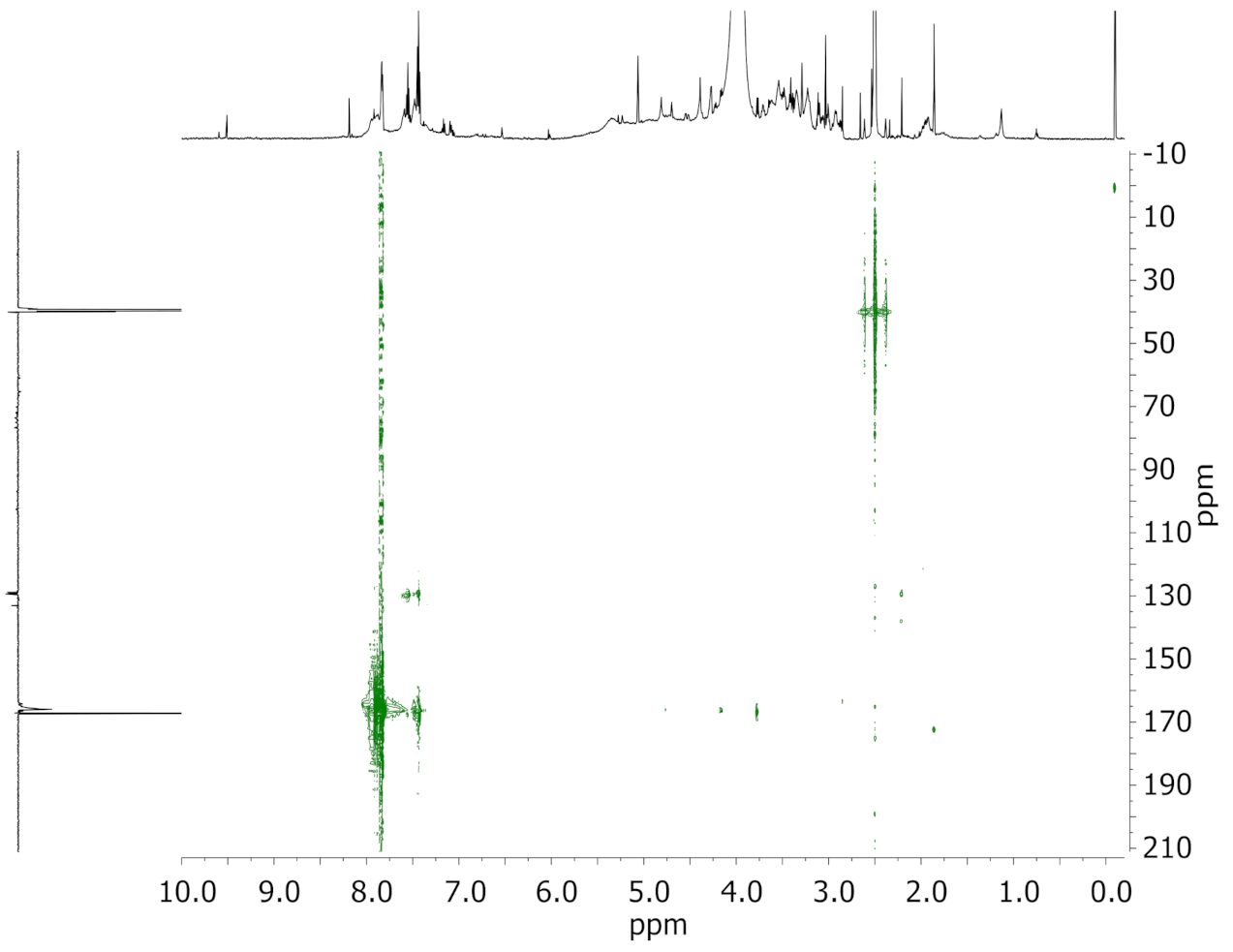
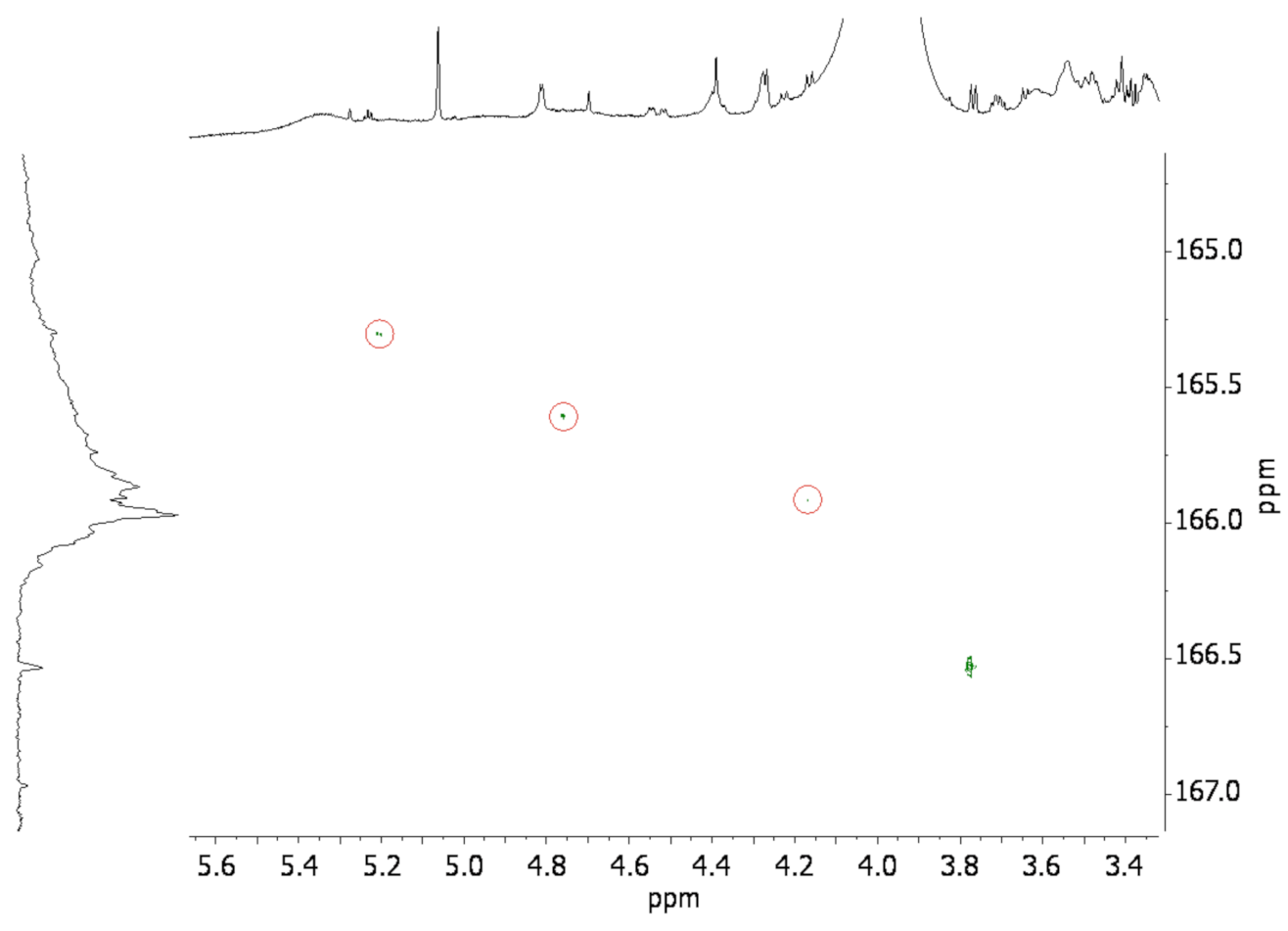
2.7. Solution State 1D and 2D Correlation NMR Spectroscopy
2.8. DOSY Spectroscopy
3. Results and Discussion
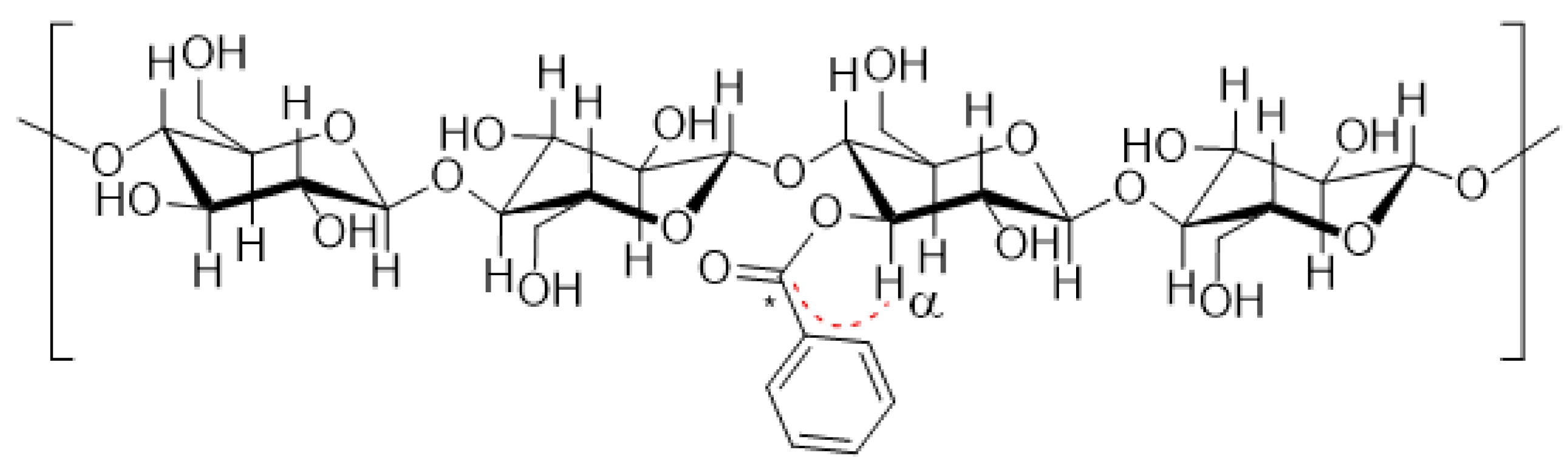
3.1. Wood Modification: Chemical Background and Precursor Synthesis
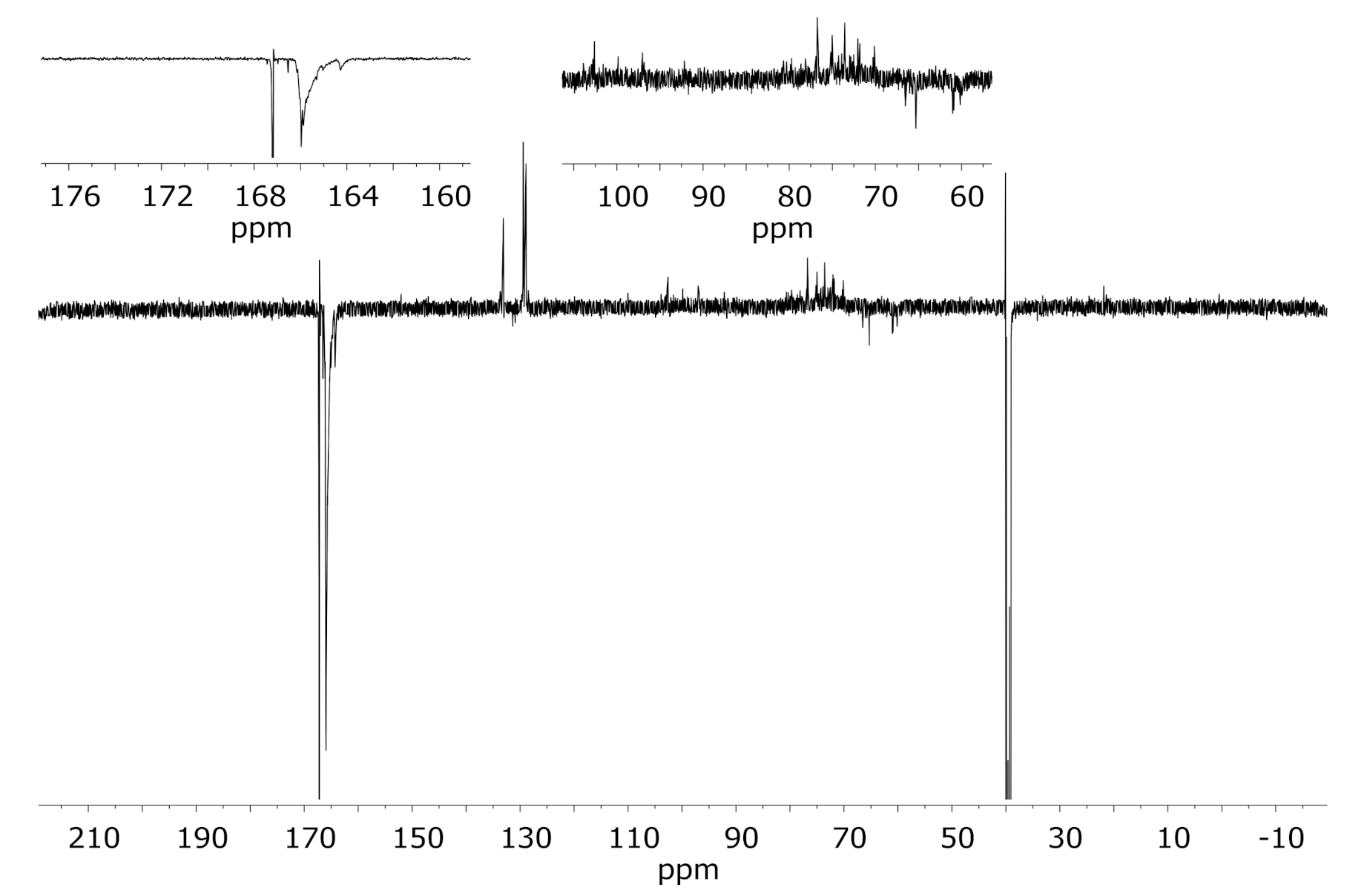
3.2. Solution State NMR of Modified Wood Samples
3.3. Comparison to Corresponding Spectra of Cellulose Triacetate as a Similarly Esterified Model Compound
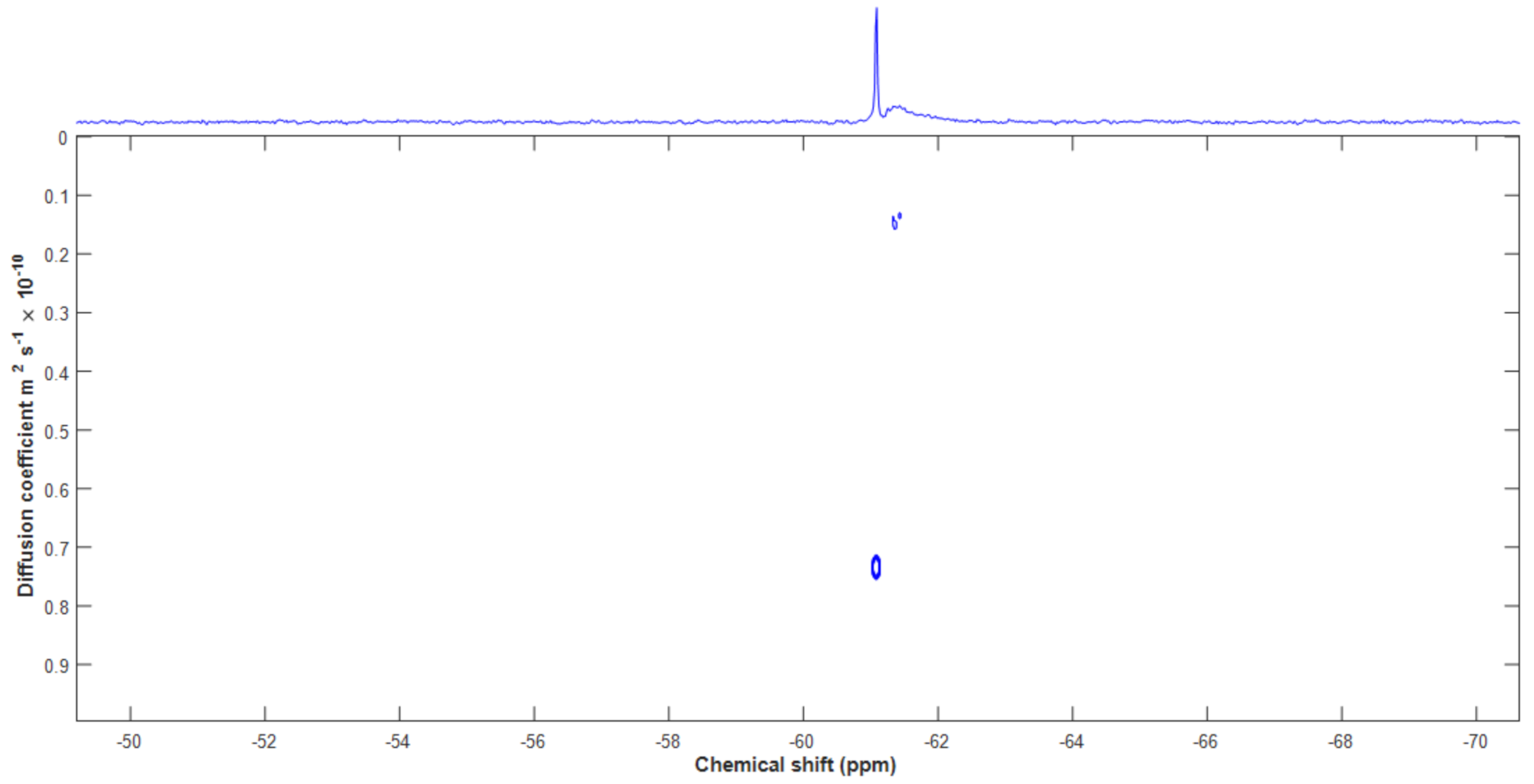
3.4. Diffusion Ordered NMR Spectroscopy (DOSY)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, C.A.S. Wood Modification. Chemical, Thermal and Other Processes; John Wiley & Sons: Chichester, UK; Hoboken, NJ, USA, 2006. [Google Scholar]
- Rowell, R.M. Handbook of Wood Chemistry and Wood Composites; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Namyslo, J.C.; Kaufmann, D.E. Chemical improvement of surfaces. Part 1: Novel functional modification of wood with covalently bound organoboron compounds. Holzforschung 2009, 63, 627–632. [Google Scholar] [CrossRef]
- Drafz, M.H.H.; Dahle, S.; Maus-Friedrichs, W.; Namyslo, J.C.; Kaufmann, D.E. Chemical improvement of surfaces. Part 2: Permanent hydrophobization of wood by covalently bonded fluoroorganyl substituents. Holzforschung 2012, 66, 727–733. [Google Scholar] [CrossRef]
- Namyslo, J.C.; Kaufmann, D.E.; Mai, C.; Militz, H. Chemical Improvement of surfaces. Part 3: Covalent modification of Scots pine sapwood with substituted benzoates providing resistance to Aureobasidium pullulans staining fungi. Holzforschung 2015, 69, 595–601. [Google Scholar] [CrossRef]
- Kaldun, C.; Dahle, S.; Maus-Friedrichs, W.; Namyslo, J.C.; Kaufmann, D.E. Chemical improvement of surfaces. Part 4: Significantly enhanced hydrophobicity of wood by covalent modification with p-silyl-functionalized benzoates. Holzforschung 2016, 70, 4114–4119. [Google Scholar] [CrossRef]
- Kaldun, C.; Söftje, M.; Namyslo, J.C.; Kaufmann, D.E. Chemical improvement of surfaces. Part 5: Surfactants as structural lead for wood hydrophobization—Covalent modification with p-alkylated benzoates. Holzforschung 2020, 74, 703–713. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Tapken, M.; Namyslo, J.C.; Kaufmann, D.E. Chemical improvement of surfaces. Part 6: Enhanced flame retardancy of Scots pine sapwood by covalent modification with phosphorus and boron functionalized benzoates. Holzforschung 2021, 75, 83–90. [Google Scholar] [CrossRef]
- Söftje, M.; Acker, S.; Plarre, R.; Namyslo, J.C.; Kaufmann, D.E. Chemical improvement of surfaces. Part 7: Novel nicotinoid structures for covalent modification of wood: An environmentally friendly way for its protection against insects. RSC Adv. 2020, 10, 15726–15733. [Google Scholar] [CrossRef]
- Hill, C.A.S.; Jones, D. Dimensional Changes in corsican pine sapwood due to chemical modification with linear chain anhydrides. Holzforschung 1999, 53, 267–271. [Google Scholar] [CrossRef]
- Wienhaus, O.; Niemz, P.; Fabian, J. Untersuchungen zur Holzartendifferenzierung mit Hilfe der Infrarotspektroskopie. Teil 1. Holzforsch. Holzverwert. 1988, 40, 1201–1225. [Google Scholar]
- Niemz, P.; Wienhaus, O.; Schaarschmidt, K.; Ramin, R. Untersuchungen zur Holzartendifferenzierung mit Hilfe der Infrarotspektroskopie, Teil 2. Holzforsch. Holzverwert. 1989, 41, 22–26. [Google Scholar]
- Zollfrank, C.; Wegener, G. FTIR microscopy and ultrastructural investigation of silylated solid wood. Holzforschung 2002, 56, 39–42. [Google Scholar] [CrossRef]
- Tjeerdsma, B.F.; Militz, H. Chemical changes in hydrothermal treated wood: FTIR analysis of combined hydrothermal and dry heat-treated wood. Holz Roh Werkst. 2005, 63, 102–111. [Google Scholar] [CrossRef]
- Stefke, B.; Windeisen, E.; Schwanninger, M.; Hinterstoisser, B. Determination of the weight percentage gain and of the acetyl group content of acetylated wood by means of different infrared spectroscopic methods. Anal. Chem. 2008, 80, 1272–1279. [Google Scholar] [CrossRef]
- Hyberts, S.G.; Milbradt, A.G.; Wagner, A.B.; Arthanari, H.; Wagner, G. Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 2012, 52, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Hyberts, S.G.; Arthanari, H.; Robson, S.A.; Wagner, G. Perspectives in magnetic resonance: NMR in the Post-FFT Era. J. Magn. Reson. 2014, 241, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, C.P.; Prestel, A.; Teilum, K. Software for reconstruction of nonuniformly sampled NMR data. Magn. Reson. Chem. 2021, 59, 315–323. [Google Scholar] [CrossRef]
- Stilbs, P. Molecular Self-diffusion coefficients in fourier transform nuclear magnetic resonance spectrometric analysis of complex mixtures. Anal. Chem. 1981, 53, 2135–2137. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S., Jr. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part 1. Basic theory. Concepts Magn. Reson. 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Pelta, M.D.; Barjat, H.; Morris, G.A.; Davis, A.L.; Hammond, S.J. Pulse sequences for high-resolution diffusion-ordered spectroscopy (HR-DOSY). Magn. Reson. Chem. 1998, 36, 706–714. [Google Scholar] [CrossRef]
- Johnson, C.S., Jr. Diffusion ordered nuclear magnetic resonance spectroscopy: Principles and applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Morris, G.A. Encyclopedia of NMR; Grant, D.M., Harris, R.K., Eds.; Wiley: New York, NY, USA, 2002; Volume 9, pp. 35–44. [Google Scholar]
- Pelta, M.D.; Morris, G.A.; Stchedroff, M.J.; Hammond, S.J. A one-shot sequence for high-resolution diffusion-ordered spectroscopy. Magn. Reson. Chem. 2002, 40, S147–S152. [Google Scholar] [CrossRef]
- Brand, T.; Cabrita, E.J.; Berger, S. Intermolecular interaction as investigated by NOE and diffusion studies. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 46, 159–196. [Google Scholar] [CrossRef]
- Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Diffusion Ordered NMR Spectroscopy (DOSY). Supramolecular Chemistry: From Molecules to Nanomaterials; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Power, J.E.; Foroozandeh, M.; Moutzouri, P.; Adams, R.W.; Nilsson, M.; Coombes, S.R.; Phillips, A.R.; Morris, G.A. Very broadband diffusion-ordered NMR spectroscopy: 19F DOSY. Chem. Commun. 2016, 52, 6892–6894. [Google Scholar] [CrossRef]
- Sulaeva, I.; Sumerskii, I.; Bacher, M.; Zinovyev, G.; Henniges, U.; Rosenau, T.; Potthast, A. Comparing different approaches to measure molar mass of lignin: SEC, DOSY and AsFIFFF. In Proceedings of the 249th ACS National Meeting & Exposition, Denver, CO, USA, 22–26 March 2015. CELL-72. [Google Scholar]
- Montgomery, J.R.D.; Lancefield, C.S.; Miles-Barrett, D.M.; Ackermann, K.; Bode, B.E.; Westwood, N.J.; Lebl, T. Fractionation and DOSY NMR as analytical tools: From model polymers to a technical lignin. ACS Omega 2017, 2, 8466–8474. [Google Scholar] [CrossRef] [Green Version]
- Cornejo, A.; García-Yoldi, I.; Alegria-Dallo, I.; Galilea-Gonzalo, R.; Hablich, K.; Sánchez, D.; Otazu, E.; Funcia, I.; Gil, M.J.; Martínez-Merino, V. Systematic diffusion-ordered spectroscopy for the selective determination of molecular weight in real lignins and fractions arising from base-catalyzed depolymerization reaction mixtures. ACS Sustain. Chem. Eng. 2020, 8, 8638–8647. [Google Scholar] [CrossRef]
- King, A.W.T.; Jalomäki, J.; Granström, M.; Argyropoulos, D.S.; Heikkinen, S.; Kilpeläinen, I. A new method for rapid degree of substitution and purity determination of chloroform-soluble cellulose esters, using 31P NMR. Anal. Methods 2010, 2, 1499–1505. [Google Scholar] [CrossRef]
- Ohkoshi, M.; Kato, A. Determination of substituent distribution of DMSO-soluble portion of acetylated wood meal by 13C-NMR spectroscopy. Mokuzai Gakkaishi 1993, 39, 849–854. [Google Scholar]
- Ohkoshi, M.; Kato, A.; Hayashi, N. 13C-NMR analysis of acetyl groups in acetylated wood I. Acetyl groups in cellulose and hemicellulose. Mokuzai Gakkaishi 1997, 43, 327–336. [Google Scholar]
- Ohkoshi, M.; Kato, A. 13C-NMR analysis of acetyl groups in acetylated wood II. Acetyl groups in lignin. Mokuzai Gakkaishi 1997, 43, 364–369. [Google Scholar]
- Ohkoshi, M.; Kato, A.; Suzuki, K.; Hayashi, N.; Ishihara, M. Characterization of acetylated wood decayed by brown-rot and white-rot fungi. J. Wood Sci. 1999, 45, 69–75. [Google Scholar] [CrossRef]
- Moulthrop, J.S.; Swatloski, R.P.; Moyna, G.; Rogers, R.D. High-resolution 13C NMR studies of cellulose and cellulose oligomers in ionic liquid solutions. Chem. Commun. 2005, 1557–1559. [Google Scholar] [CrossRef]
- Lu, F.; Ralph, J. Non-degradative dissolution and acetylation of ball-milled plant cell walls: High-resolution solution-state NMR. Plant J. 2003, 35, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Ralph, J.; Lu, F. Cryoprobe 3D NMR of acetylated ball-milled pine cell walls. Org. Biomol. Chem. 2004, 2, 2714–2715. [Google Scholar] [CrossRef]
- Jiang, N.; Pu, Y.; Ragauskas, A.J. Rapid determination of lignin content via direct dissolution and 1H NMR analysis of plant cell walls. ChemSusChem 2010, 3, 1285–1289. [Google Scholar] [CrossRef]
- Kim, H.; Ralph, J. Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d6/pyridine-d5. Org. Biomol. Chem. 2010, 8, 576–591. [Google Scholar] [CrossRef]
- Mansfield, S.D.; Kim, H.; Lu, F.; Ralph, J. Whole plant cell wall characterizationS using solution-state 2D NMR. Nat. Protoc. 2012, 7, 1579–1589. [Google Scholar] [CrossRef]
- Cheng, K.; Sorek, H.; Zimmermann, H.; Wemmer, D.E.; Pauly, M. Solution-state 2D NMR spectroscopy of plant cell walls enabled by a dimethylsulfoxide-d6/1-ethyl-3-methylimidazolium acetate solvent. Anal. Chem. 2013, 85, 3213–3221. [Google Scholar] [CrossRef]
- Fukagawa, N.; Meshitsuka, G.; Ishizu, A. A two-dimensional NMR study of birch milled wood lignin. J. Wood Chem. Technol. 1991, 11, 373–396. [Google Scholar] [CrossRef]
- Landucci, L.L.; Deka, G.C.; Roy, D.N. A 13 C NMR study of milled wood lignins from hybrid salix clones. Holzforschung 1992, 46, 505–511. [Google Scholar] [CrossRef]
- Li, S.; Lundquist, K. A new method for the analysis of phenolic groups in lignins by 1H NMR spectroscopy. Nord. Pulp Pap. Res. J. 1994, 9, 191–195. [Google Scholar] [CrossRef]
- Galkin, S.; Ämmälahti, E.; Kilpeläinen, I.; Brunow, G.; Hatakka, A. Characterisation of Milled Wood Lignin from Reed Canary Grass (Phalaris arundinacea). Holzforschung 1997, 51, 130–134. [Google Scholar] [CrossRef]
- Li, S.; Lundquist, K. Analysis of hydroxyl groups in lignins by 1H NMR spectrometry. Nord. Pulp Pap. Res. J. 2001, 16, 63–67. [Google Scholar] [CrossRef]
- Heikkinen, S.; Toikka, M.M.; Karhunen, P.T.; Kilpeläinen, I.A. Quantitative 2D HSQC (Q-HSQC) via suppression of j-dependence of polarization transfer in NMR spectroscopy: Application to wood lignin. J. Am. Chem. Soc. 2003, 125, 4362–4367. [Google Scholar] [CrossRef] [PubMed]
- Holtman, K.M.; Chang, H.-M.; Kadla, J.F. An NMR comparison of the whole lignin from milled wood, MWL, and REL dissolved by the DMSO/NMI procedure. J. Wood Chem. Technol. 2007, 27, 179–200. [Google Scholar] [CrossRef]
- Crestini, C.; Melone, F.; Sette, M.; Saladino, R. Milled wood lignin: A linear oligomer. Biomacromolecules 2011, 12, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Rencoret, J.; del Río, J.C.; Gutiérrez, A.; Martínez, Á.T.; Li, S.; Parkas, J.; Lundquist, K. Origin of the acetylated structures present in white birch (Betula pendula Roth) milled wood lignin. Wood Sci. Technol. 2012, 46, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Capanema, E.A.; Balakshin, M.Y.; Chen, C.-L.; Gratzl, J.S.; Gracz, H. Structural analysis of residual and technical lignins by 1H-13C correlation 2D NMR-spectroscopy. Holzforschung 2001, 55, 302–308. [Google Scholar] [CrossRef]
- Pinto, P.C.; Evtuguin, D.V.; Pascoal Neto, C.; Silvestre, A.J.D.; Amado, F.M.L. Behavior of eucalyptus globulus lignin during kraft pulping. II. Analysis by NMR, ESI/MS, AND GPC. J. Wood Chem. Technol. 2002, 22, 109–125. [Google Scholar] [CrossRef]
- Ekholm, F.S.; Ardá, A.; Eklund, P.; André, S.; Gabius, H.-J.; Jiménez-Barbero, J.; Leino, R. Studies related to Norway spruce galactoglucomannanes: Chemical synthesis, conformation analysis, NMR spectroscopic characterization, and Molecular recognition of model compounds. Chem. Eur. J. 2012, 18, 14392–14405. [Google Scholar] [CrossRef]
- Pu, Y.; Jiang, N.; Ragauskas, A.J. Ionic liquid as a green solvent for lignin. J. Wood Chem. Technol. 2007, 27, 23–33. [Google Scholar] [CrossRef]
- Kilpeläinen, I.; Xie, H.; King, A.; Granstrom, M.; Heikkinen, S.; Argyropoulos, D.S. Dissolution of wood in ionic liquids. J. Agric. Food Chem. 2007, 55, 9142–9148. [Google Scholar] [CrossRef]
- Xie, H.; King, A.; Kilpeläinen, I.; Granström, M.; Argyropoulos, D.S. Thorough chemical modification of wood-based lignocellulosic materials in ionic liquids. Biomacromolecules 2007, 8, 3740–3748. [Google Scholar] [CrossRef]
- Qu, C.; Kishimoto, T.; Kishino, M.; Hamada, M.; Nakajima, N. Heteronuclear single-quantum coherence nuclear magnetic resonance (HSQC NMR) characterization of acetylated fir (Abies sachallnensis MAST). Wood regenerated from ionic liquid. J. Agric. Food Chem. 2011, 59, 5382–5389. [Google Scholar] [CrossRef]
- Giernoth, R.; Bankmann, D.; Schlörer, N. High performance NMR in ionic liquids. Green Chem. 2005, 7, 279–282. [Google Scholar] [CrossRef]
- Giernoth, R. NMR spectroscopy in ionic liquids. Top. Curr. Chem. 2009, 290, 263–283. [Google Scholar]
- Yelle, D.J.; Ralph, J.; Frihart, C.R. Characterization of nonderivatized plant cell walls using high-resolution solution-state NMR spectroscopy. Magn. Res. Chem. 2008, 46, 508–517. [Google Scholar] [CrossRef]
- Yoneda, Y.; Ebner, G.; Takano, T.; Nakatsubo, F.; Potthast, A.; Rosenau, T. Synthesis of the perdeuterated cellulose solvents 1-ethyl-3-methyl imidazolium acetate (EMIM-OAc-d14) and 1-butyl-3-methylimidazolium acetate (BMIM-OAc-d18) and of 2-13C-butyl-3-methylimidazolium acetate. J. Label. Compd. Radiopharm. 2009, 52, 223–226. [Google Scholar] [CrossRef]
- Akim, L.G.; Argyropoulos, D.S.; Jouanin, L.; Leplé, J.C.; Pilate, G.; Pollet, B.; Lapierre, C. Quantitative 31P NMR spectroscopy of lignins from transgenic poplars. Holzforschung 2001, 55, 386–390. [Google Scholar] [CrossRef]
- Nimz, H.H.; Robert, D.; Faix, O.; Nemr, M. Carbon-13 NMR spectra of lignins, 8: Structural differences between lignins of hardwoods, softwoods, grasses and compression wood. Holzforschung 1981, 35, 16–26. [Google Scholar] [CrossRef]
- Gagnaire, D. Recent developments in wood chemistry. Actual. Chim. 1984, 10, 30–34. [Google Scholar]
- Gil, A.M.; Pascoal Neto, C. Solid-state NMR studies of wood and other lignocellulosic materials. Annu. Rep. NMR Spectrosc. 1999, 37, 75–117. [Google Scholar]
- Frazier, C.E.; Ni, J.; Schmidt, R.G. Applications of NMR spectroscopy to the analysis of wood/adhesive bondlines. Adv. Lignocellul. Charact. 1999, 145–156. [Google Scholar]
- Atalla, R.H.; Vander Hart, D.L. The role of solid state 13C NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl. Magn. Reson. 1999, 15, 1–9. [Google Scholar] [CrossRef]
- Maunu, S.L. NMR studies of wood and wood products. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 40, 151–174. [Google Scholar] [CrossRef]
- Wikberg, H.; Maunu, S.L. Characterisation of thermally modified hard- and softwoods by 13C CPMAS NMR. Carbohydr. Polym. 2004, 58, 461–466. [Google Scholar] [CrossRef]
- Bardet, M.; Gerbaud, G.; Giffard, M.; Doan, C.; Hediger, S.; Pape, L.L. 13C high-resolution solid-state NMR for structural elucidation of archaeological woods. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 55, 199–214. [Google Scholar] [CrossRef]
- Ralph, J.; Landucci, L.L. NMR of lignins. In Lignin and Lignans; Advances in Chemistry; Heitner, C., Dimmel, D.R., Schmidt, J.A., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 137–234. [Google Scholar]
- Terrett, O.M.; Lyczakowski, J.J.; Dinu Iuga, L.Y.; Franks, W.T.; Brown, S.P.; Dupree, R.; Dupree, P. Molecular architecture of softwood revealed by solid-state NMR. Nat. Commun. 2019, 10, 4978. [Google Scholar] [CrossRef] [Green Version]
- Bardet, M.; Gagnaire, D.; Nardin, R.; Robert, D.; Vincendon, M. Use of carbon-13 enriched wood for structural NMR investigation of wood and wood components, cellulose and lignin, in solid and in solution. Holzforschung 1986, 40, 17–24. [Google Scholar]
- Hawkes, G.E.; Smith, C.Z.; Utley, J.H.P.; Vargas, R.R.; Viertler, H. A comparison of solution and solid state 13 C NMR spectra of lignins and lignin model compounds. Holzforschung 1993, 47, 302–312. [Google Scholar] [CrossRef]
- Pu, Y.; Hallac, B.; Ragauskas, A.J. Plant biomass characterization: Application of solution and solid-state NMR spectroscopy. In Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals; Wyman, C.E., Ed.; John Wiley & Sons: Chichester, UK, 2013; pp. 369–390. [Google Scholar]
- Foston, M.; Samuel, R.; He, J.; Ragauskas, A.J. A review of whole cell wall NMR by the direct-dissolution of biomass. Green Chem. 2016, 18, 608–621. [Google Scholar] [CrossRef]
- Aoki, D.; Nomura, K.; Hashiura, M.; Imamura, Y.; Miyata, S.; Terashima, N.; Matsushita, Y.; Nishimura, H.; Watanabe, T.; Katahira, M.; et al. Evaluation of ring-5 structures of guaiacyl lignin in Ginkgo biloba L. using solid- and liquid-state 13C NMR difference spectroscopy. Holzforschung 2019, 73, 1083–1092. [Google Scholar] [CrossRef]
- Happs, R.M.; Addison, B.; Doeppke, C.; Donohoe, B.S.; Davis, M.F.; Harman-Ware, A.E. Comparison of methodologies used to determine aromatic lignin unit ratios in lignocellulosic biomass. Biotechnol. Biofuels 2021, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Schledorn, M.; Malär, A.A.; Torosyan, A.; Penzel, S.; Klose, D.; Oss, A.; Org, M.-L.; Wang, S.; Lecoq, L.; Cadalbert, R.; et al. Protein NMR Spectroscopy at 150 kHz Magic—Angle Spinning Continues To Improve Resolution and Mass Sensitivity. ChemBioChem 2020, 21, 2540–2548. [Google Scholar] [CrossRef]
- Successful Installation of World’s First 1.2 GHz NMR System Enables Novel Functional Structural Biology Research. Available online: https://www.bruker.com/en/news-and-events/news/2020/successful-installation-of-worlds-first-12-ghz-NMR-system-enables-novel-functional-structural-biology-research.html (accessed on 5 May 2021).
- Wang, Z.; Yokoyama, T.; Chang, H.-M.; Matsumoto, Y. Dissolution of beech and spruce milled woods in LiCl/DMSO. J. Agric. Food Chem. 2009, 57, 6167–6170. [Google Scholar] [CrossRef]
- Drafz, M.H.H. Synthese und Analytik Neuartig Modifizierter Holzoberflächen; Clausthal University of Technology: Clausthal-Zellerfeld, Germany, 2014. [Google Scholar]
- Holz, M.; Weingärtner, H. Calibration in accurate spin-echo self-diffusion measurements using 1H and less-common nuclei. J. Magn. Reson. 1991, 92, 115–125. [Google Scholar] [CrossRef]
- Lounila, J.; Oikarinen, K.; Ingman, P.; Jokisaari, J. Effects of thermal convection on NMR and their elimination by sample rotation. J. Magn. Reson. A 1996, 118, 50–54. [Google Scholar] [CrossRef]
- Nakamura, I.; Nemoto, T.; Shiraiwa, N.; Terada, M. Palladium-catalyzed indolization of N-aroylbenzotriazoles with disubstituted alkynes. Org. Lett. 2009, 11, 1055–1058. [Google Scholar] [CrossRef]
- Hill, C.A.S.; Jones, D. The Dimensional stabilisation of Corsican pine sapwood by reaction with carboxylic acid anhydrides. The effect of chain length. Holzforschung 1996, 50, 457–462. [Google Scholar] [CrossRef]
- Kerssebaum, R. DOSY and Diffusion by NMR. A Tutorial for TopSpin 2.0; Bruker BioSpin GmbH: Rheinstetten, Germany, 2002. [Google Scholar]
- Nilsson, M. The DOSY Toolbox: A new tool for processing PFG NMR diffusion data. J. Magn. Reson. 2009, 200, 296–302. [Google Scholar] [CrossRef]
- Cabrita, E.J.; Berger, S. DOSY studies of hydrogen bond association: Tetramethylsilane as a reference compound for diffusion studies. Magn. Reson. Chem. 2001, 39, S142–S148. [Google Scholar] [CrossRef]
- Phuong, L.X.; Takayama, M.; Shida, S.; Matsumoto, Y.; Aoyagi, T. Determination of the accessible hydroxyl groups in heat-treated Styrax tonkinensis (Pierre) Craib ex Hartwich wood by hydrogen-deuterium exchange and 2H NMR spectroscopy. Holzforschung 2007, 61, 488–491. [Google Scholar] [CrossRef]
- Burger, R.; Bigler, P. DEPTQ: Distorsionless enhancement by polarization transfer including the detection of quaternary nuclei. J. Magn. Res. 1998, 135, 529–534. [Google Scholar] [CrossRef]
- Bigler, P.; Kümmerle, R.; Bermel, W. Multiplicity editing including quaternary carbons: Improved performance for the 13C-DEPTQ pulse sequence. Magn. Reson. Chem. 2007, 45, 469–472. [Google Scholar] [CrossRef]
- Cicero, D.O.; Barbato, G.; Bazzo, R. Sensitivity enhancement of a Two-dimensional experiment for the measurement of heteronuclear long-range coupling constants, by a new scheme of coherence selection by gradients. J. Magn. Reson. 2001, 148, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Claridge, T.D.W.; Perez-Victoria, I. Enhanced 13C resolution in semi-selective HMBC: A band-selective, constant-time HMBC for complex organic structure elucidation by NMR. Org. Biomol. Chem. 2003, 1, 3632–3634. [Google Scholar] [CrossRef]
- ACD/C+H NMR Predictors and DB 2020; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2020; Available online: http://www.acdlabs.com.
- Kalinowski, H.-O.; Berger, S.; Braun, S. 13C-NMR-Spektroskopie. 200 Tabellen; Thieme: Stuttgart, Germany, 1984. [Google Scholar]
- Barrelle, M. A new method for the quantitative 19F NMR spectroscopic analysis of hydroxyl groups in lignins. Holzforschung 1993, 47, 261–267. [Google Scholar] [CrossRef]
- Barrelle, M. Improvements in the structural investigation of lignins by 19F NMR spectroscopy. J. Wood Chem. Technol. 1995, 15, 179–188. [Google Scholar] [CrossRef]
- Ahvazi, B.C.; Argyropoulos, D.S. 19F nuclear magnetic resonance spectroscopy for the elucidation of carbonyl groups in lignins.1. Model compounds. J. Agric. Food Chem. 1996, 44, 2167–2175. [Google Scholar] [CrossRef]
- Ahvazi, B.C.; Crestini, C.; Argyropoulos, D.S. 19F nuclear magnetic resonance spectroscopy for the quantitative detection and classification of carbonyl groups in lignins. J. Agric. Food Chem. 1999, 47, 190–201. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef] [Green Version]
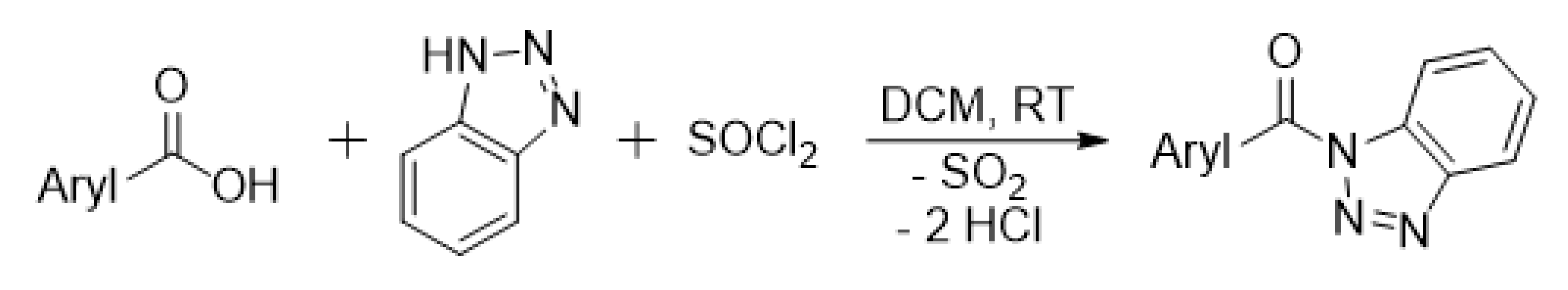




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
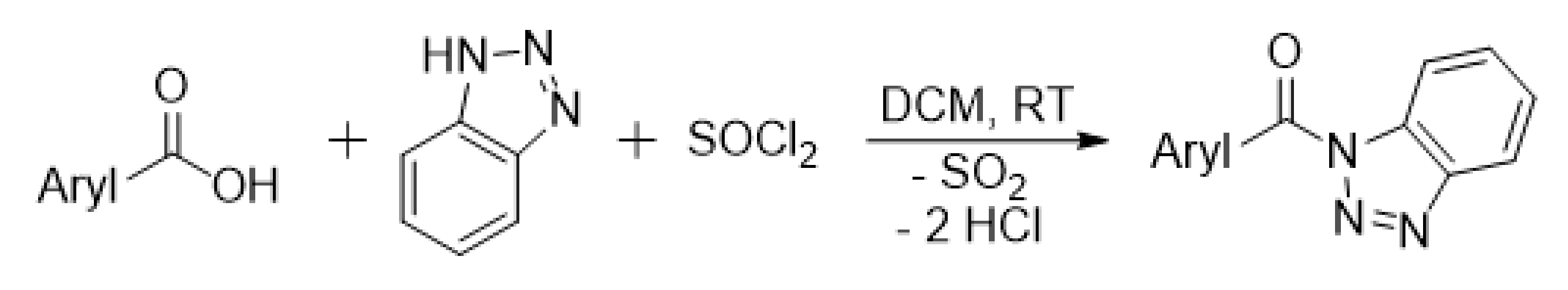{kind=link}
| Entry | Compound | Solvent | Diffusion Coefficient D [10−10 m2∙s−1] |
|---|---|---|---|
| 1 | 3,5-(CF3)2C6H3-COO-wood a | LiCl/DMSO-d6 | 0.120 ± 0.012 b,d |
| 2 | 0.121 ± 0.005 c | ||
| 3 | cellulose triacetate | LiCl/DMSO-d6 | 0.028 ± 0.002 c |
| 4 | DMSO-d6 | 0.081 ± 0.002 c | |
| 5 | tetramethylsilane (TMS) | LiCl/DMSO-d6 | 2.33 ± 0.04 c |
| 6 | DMSO-d6 | 5.85 ± 0.02 c | |
| 7 | 3,5-(CF3)2C6H3-COOH | LiCl/DMSO-d6 | 0.71 ± 0.00(6) b |
| 8 | DMSO-d6 | 3.59 ± 0.04 b | |
| 9 | 3,5-(CF3)2C6H3-COOMe | LiCl/DMSO-d6 | 1.22 ± 0.06 b |
| 10 | DMSO-d6 | 4.45 ± 0.10 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Namyslo, J.C.; Drafz, M.H.H.; Kaufmann, D.E. Durable Modification of Wood by Benzoylation—Proof of Covalent Bonding by Solution State NMR and DOSY NMR Quick-Test. Polymers 2021, 13, 2164. https://doi.org/10.3390/polym13132164
Namyslo JC, Drafz MHH, Kaufmann DE. Durable Modification of Wood by Benzoylation—Proof of Covalent Bonding by Solution State NMR and DOSY NMR Quick-Test. Polymers. 2021; 13(13):2164. https://doi.org/10.3390/polym13132164
Chicago/Turabian StyleNamyslo, Jan C., Martin H. H. Drafz, and Dieter E. Kaufmann. 2021. "Durable Modification of Wood by Benzoylation—Proof of Covalent Bonding by Solution State NMR and DOSY NMR Quick-Test" Polymers 13, no. 13: 2164. https://doi.org/10.3390/polym13132164
APA StyleNamyslo, J. C., Drafz, M. H. H., & Kaufmann, D. E. (2021). Durable Modification of Wood by Benzoylation—Proof of Covalent Bonding by Solution State NMR and DOSY NMR Quick-Test. Polymers, 13(13), 2164. https://doi.org/10.3390/polym13132164