On Complex Coacervate Core Micelles: Structure-Function Perspectives




Abstract
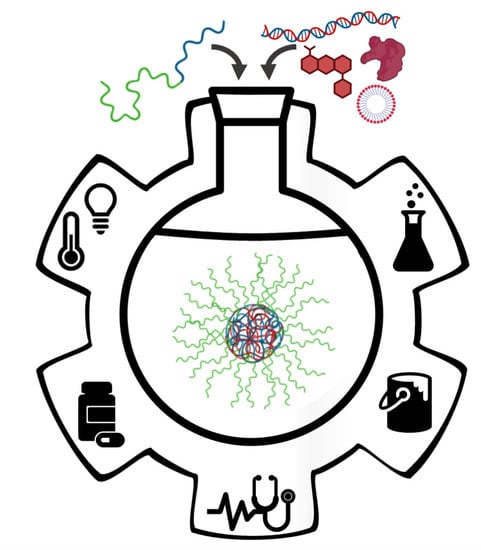
1. Introduction
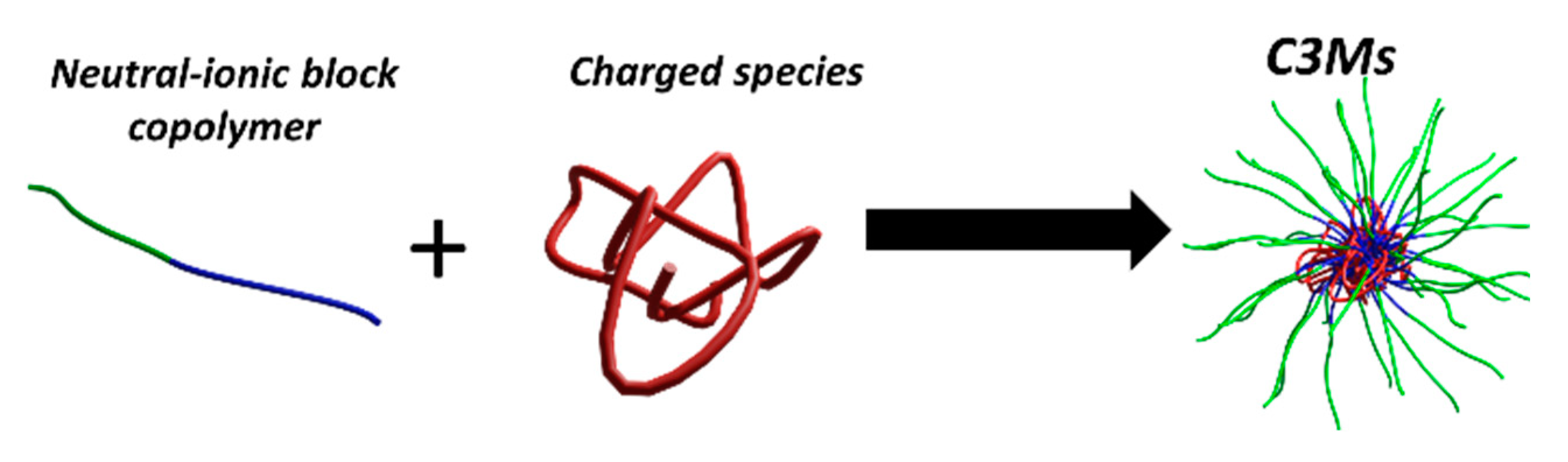
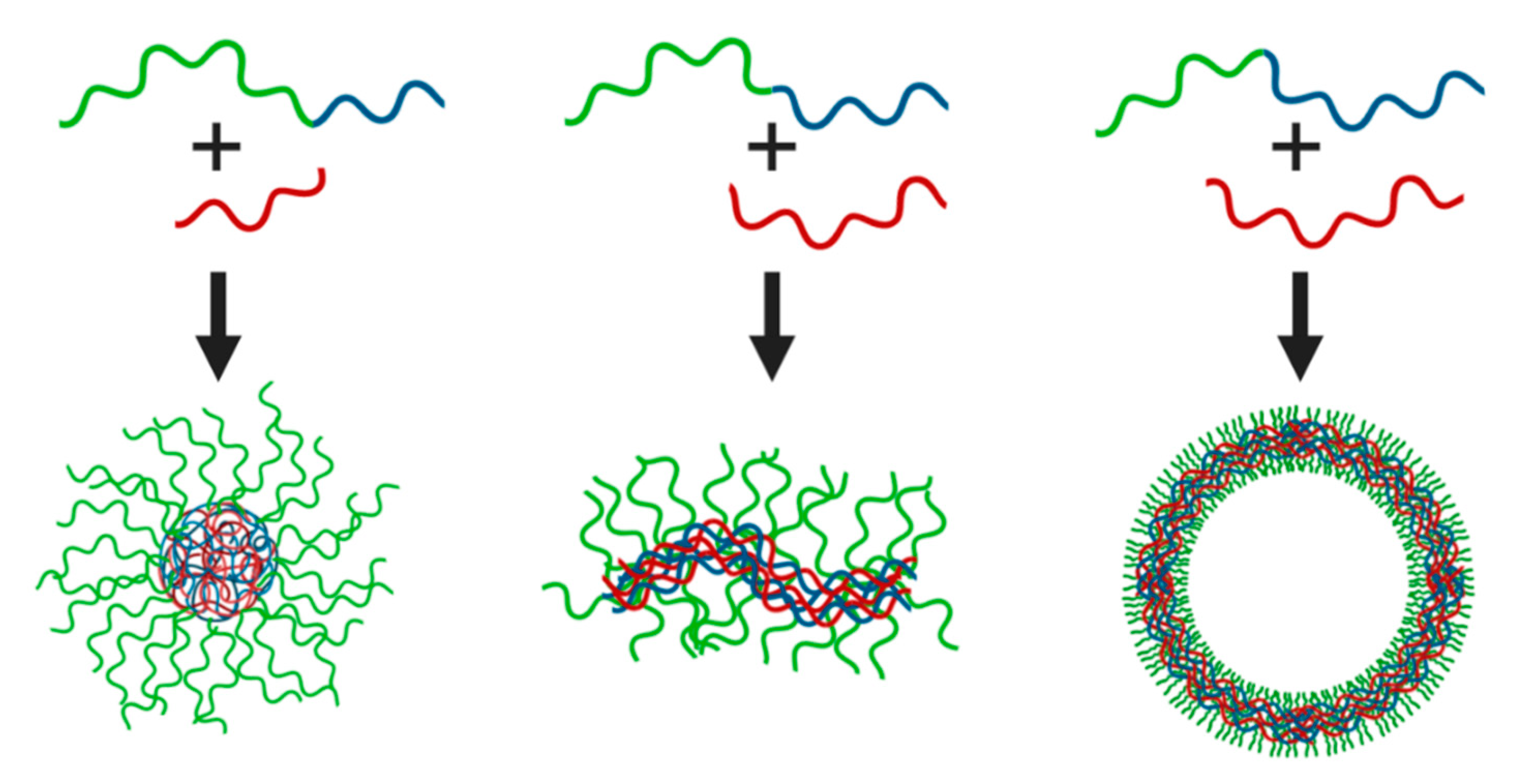
2. Fundamentals
3. Biotechnological Applications of C3Ms
3.1. Polynucleotide-Based C3Ms
3.2. Encapsulation of Proteins in C3Ms
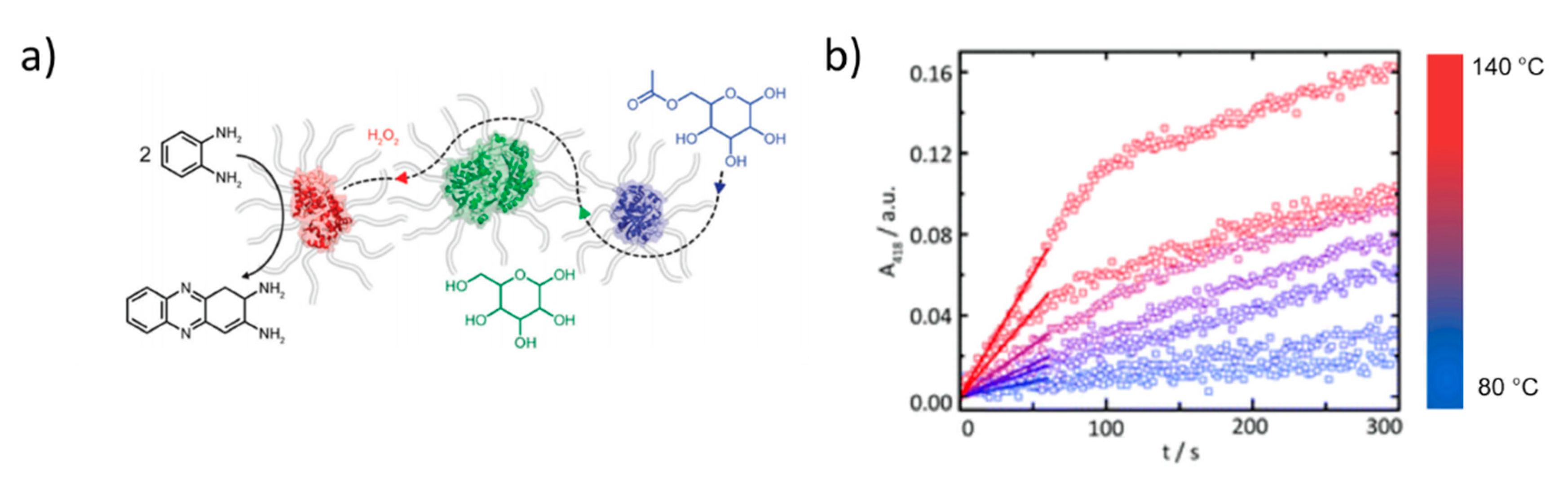
Strategies for Enhanced Protein Encapsulation and Stabilization
3.3. C3Ms as Nano-Compartments for Small Ionic Therapeutics and Theragnostic
4. Other Technological Applications
4.1. C3Ms as Nanoreactors and Templates
4.2. C3Ms Based on Bio-Inspired Polymer Design
5. Conclusions and Outlook
Funding
Conflicts of Interest
Abbreviations
| Acronym | Full Name | Structure | Properties |
| Chitosan | poly(d-glucosamine) |  | Biobased polysaccharide |
| Chol | Cholesterol |  | Most abundant sterol in animals |
| cPF | poly[9,9′-bis(3′-sodium propanoate)fluoren-2,7-yl] |  | Conjugated, solvency-dependent fluorescence emission |
| DTS | 2,2′-dithiodisuccinic acid |  | Oligoanionic species; disulfide bond cleaved under reductive conditions |
| 2IT | 2-iminothiolane |  | Thiolating agent, reactive towards primary amines, such as in lysine |
| PAA | poly(acrylic acid) |  | Weak polyanion |
| PAAm | poly(acrylamide) |  | Water-soluble, providing steric stabilization |
| PAAOA | poly(acryloylaminooctanoic acid) |  | Charged at basic pH; amphiphilic when protonated at acidic pH |
| PAAVA | poly(acryloylaminovaleric acid) |  | Charged at basic pH; amphiphilic when protonated at acidic pH |
| PAMPS | poly(2-acrylamido-2-methylpropane sulfonic acid) |  | Strong polyanion, can form gels |
| PAPA | poly(aminopalmitic acid) |  | Lipid-like, hydrophobic side chain |
| PAPTAC | poly(3-acrylamidopropyl trimethylammonium chloride) |  | Strong polycation |
| PAsp | poly(l-aspartic acid) |  | Weak anionic polypeptide |
| PAsp(DET) | poly[N-{N-(2-aminoethyl)-2-aminoethyl} aspartamide] |  | Cationized polypeptide, destabilization of the endosomal membrane |
| PBP | poly(benzophenone methacrylate) |  | Photo-cross-linkable sidechain |
| PCEA | poly(carboxyethyl acrylate) |  | Weak polyelectrolyte |
| PCL | poly(caprolactone) |  | Biocompatible and relatively slowly degraded in the body |
| PDMAEMA | poly(dimethylaminoethyl methacrylate) |  | Strong polycation |
| PEG or PEO | poly(ethylene glycol) or poly(ethylene oxide) |  | Biocompatible, providing steric stabilization |
| PEI | poly(ethylene imine) |  | Polycation, can also be branched; high transfection efficiency but also high toxicity |
| P[F]BA | 4-carboxy[-3-fluoro]phenylboronic acid |  | Reversibly cross-linkable functional group |
| PGA | poly(glycolic acid) |  | Biodegradable polymer used for example for sutures |
| PGAA | poly(glycoamidoamine) |  | Reduced immune response and cytotoxicity and enhanced transfection efficiency; sugars and length of oligoamine can be varied (example shown) |
| PGlu | poly(glutamic acid) |  | Anionic polypeptide |
| PGluPBA | poly(glutamicamidophenylboronic acid) |  | Forms reversible, covalent complexes; Lewis acid |
| PHis | poly(histidine) |  | Provides efficient endosomal escape |
| PLL or PLys | poly(l-lysine) |  | Cationic polypeptide |
| PLLCA | poly(ε-3,4-dihydroxyphenylcarboxyl-l-lysine) |  | Catechol group forms boronate ester with boronic acid under mild conditions |
| PMA | poly(methacrylic acid) |  | Weak polyanion |
| PM2VP | poly(N-methyl-2-vinylpyridinium iodide) |  | Strong polycation, degree of quaternization can yield varying charge densities |
| PM4VP | poly(N-methyl-4-vinylpyridinium iodide) |  | Strong polycation, degree of quaternization can yield varying charge densities |
| PmDOPA | poly(N-methacryloyl-3,4-dihydroxy-l-phenylalanine) |  | Catechol group provides adhesion to inorganic substrates (glass, metals, metal oxides) |
| PNIPAM | poly(N-isopropyl acrylamide) |  | Thermoresponsive; LCST polymer forms a hydrophobic barrier at body temperature |
| PnPrOx | poly(2-n-propyl-2-oxazoline) |  | Thermoresponsive; LCST polymer forms a hydrophobic barrier at body temperature |
| POEGMA | poly(oligo(ethylene glycol) methyl ether methacrylate) |  | Biocompatible; provides steric stabilization and anti-fouling |
| PSS | poly(styrene sulfonate) sodium salt |  | Strong polyanion |
| PTreA | poly(trehalose propylacrylamide) |  | Sugar moieties recognized by receptors |
| PVA | poly(vinyl alcohol) |  | Ice-binding; inhibits ice recrystallization |
| (I)PEC | (inter)polyelectrolyte complex | ||
| ATP | adenosine triphosphate | ||
| BIC | block ionomer complex | ||
| C3M | complex coacervate core micelle | ||
| dsDNA | double-stranded DNA | ||
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide | ||
| EPR | enhanced permeability and retention | ||
| GFP | green fluorescent protein | ||
| GOx | glucose oxidase | ||
| HRP | horseradish peroxidase | ||
| mRNA | messenger RNA | ||
| NP | nanoparticle | ||
| NPC | nuclear pore complex | ||
| OPH | organophosphate hydrolase | ||
| pDNA | plasmid DNA | ||
| PDT | photodynamic therapy | ||
| PIC | polyion complex | ||
| PIESA | polymerization-induced electrostatic self-assembly | ||
| PTT | photothermal therapy | ||
| RAFT | reversible addition-fragmentation chain-transfer | ||
| siRNA | small interfering RNA | ||
| ssDNA | single-stranded DNA |
References
- Zhao, L.; Skwarczynski, M.; Toth, I. Polyelectrolyte-Based Platforms for the Delivery of Peptides and Proteins. ACS Biomater. Sci. Eng. 2019, 5, 4937–4950. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Polyion Complex Micelle Formation From Double-hydrophilic Block Copolymers Composed of Charged and Non-Charged Segments in Aqueous Media. Polym. J. 2018, 50, 95–100. [Google Scholar] [CrossRef]
- Blocher McTigue, W.C.; Perry, S.L. Protein Encapsulation Using Complex Coacervates: What Nature Has to Teach Us. Small 2020, 16, 1907671. [Google Scholar] [CrossRef]
- Gao, S.; Holkar, A.; Srivastava, S. Protein–Polyelectrolyte Complexes and Micellar Assemblies. Polymers 2019, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Stenzel, M.H. Polyion Complex Micelles for Protein Delivery. Aust. J. Chem. 2018, 71, 768. [Google Scholar] [CrossRef]
- Horn, J.M.; Kapelner, R.A.; Obermeyer, A.C. Macro- and Microphase Separated Protein-Polyelectrolyte Complexes: Design Parameters and Current Progress. Polymers 2019, 11, 578. [Google Scholar] [CrossRef]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef]
- Tang, Y.; Duan, J.; Wu, J. A Laser Light Scattering Study of Complex Formation Between Soybean Peroxidase and poly(N-Isopropylacrylamide-co-Sodium Styrene Sulfonate). Colloids Surfaces A Physicochem. Eng. Asp. 2012, 395, 82–87. [Google Scholar] [CrossRef]
- Dähling, C.; Lotze, G.; Drechsler, M.; Mori, H.; Pergushov, D.V.; Plamper, F.A. Temperature-Induced Structure Switch in Thermo-Responsive Micellar Interpolyelectrolyte Complexes: Toward Core-Shell-Corona and Worm-Like Morphologies. Soft Matter 2016, 12, 5127–5137. [Google Scholar] [CrossRef]
- Plamper, F.A.; Gelissen, A.P.; Timper, J.; Wolf, A.; Zezin, A.B.; Richtering, W.; Tenhu, H.; Simon, U.; Mayer, J.; Borisov, O.V.; et al. Spontaneous Assembly of Miktoarm Stars into Vesicular Interpolyelectrolyte Complexes. Macromol. Rapid Commun. 2013, 34, 855–860. [Google Scholar] [CrossRef]
- Serefoglou, E.; Oberdisse, J.; Staikos, G. Characterization of the Soluble Nanoparticles Formed Through Coulombic Interaction of Bovine Serum Albumin with Anionic Graft Copolymers at Low pH. Biomacromolecules 2007, 8, 1195–1199. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Voets, I.K.; de Keizer, A.; Cohen Stuart, M.A. Complex Coacervate Core Micelles. Adv. Colloid Interface Sci. 2009, 147–148, 300–318. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Miyata, K.; Osada, K.; Christie, R.J.; Sanjoh, M.; Li, W.; Fukushima, S.; Ishii, T.; Kano, M.R.; Nishiyama, N.; et al. Polyplex Micelles Prepared from ω-Cholesteryl PEG-Polycation Block Copolymers for Systemic Gene Delivery. Biomaterials 2011, 32, 652–663. [Google Scholar] [CrossRef]
- Oba, M.; Vachutinsky, Y.; Miyata, K.; Kano, M.R.; Ikeda, S.; Nishiyama, N.; Itaka, K.; Miyazono, K.; Koyama, H.; Kataoka, K. Antiangiogenic Gene Therapy of Solid Tumor by Systemic Injection of Polyplex Micelles Loading Plasmid DNA Encoding Soluble flt-1. Mol. Pharm. 2010, 7, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Pippa, N.; Kalinova, R.; Dimitrov, I.; Pispas, S.; Demetzos, C. Insulin/Poly(Ethylene Glycol)-block-Poly(l-Lysine) Complexes: Physicochemical Properties and Protein Encapsulation. J. Phys. Chem. B 2015, 119, 6813–6819. [Google Scholar] [CrossRef] [PubMed]
- Gradzielski, M.; Hoffmann, I. Polyelectrolyte-Surfactant Complexes (PESCs) Composed of Oppositely Charged Components. Curr. Opin. Colloid Interface Sci. 2018, 35, 124–141. [Google Scholar] [CrossRef]
- Facciotti, C.; Saggiomo, V.; van Hurne, S.; Bunschoten, A.; Kaup, R.; Velders, A.H. Oxidant-Responsive Ferrocene-Based Cyclodextrin Complex Coacervate Core Micelles. Supramol. Chem. 2020, 32, 30–38. [Google Scholar] [CrossRef]
- Wang, J.; Guan, W.; Tan, T.; Saggiomo, V.; Cohen Stuart, M.A.; Velders, A.H. Response of Metal-Coordination-Based Polyelectrolyte Complex Micelles to Added Ligands and Metals. Soft Matter 2020, 16, 2953–2960. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Q.; Chang, H.; Cheng, Y. Surface-Engineered Dendrimers in Gene Delivery. Chem. Rev. 2015, 115, 5274–5300. [Google Scholar] [CrossRef]
- Pergushov, D.V.; Müller, A.H.E.; Schacher, F.H. Micellar interpolyelectrolyte complexes. Chem. Soc. Rev. 2012, 41, 6888–6901. [Google Scholar] [CrossRef]
- Voets, I.K.; Fokkink, R.; Hellweg, T.; King, S.M.; De Waard, P.; De Keizer, A.; Cohen Stuart, M.A. Spontaneous Symmetry Breaking: Formation of Janus Micelles. Soft Matter 2009, 5, 999–1005. [Google Scholar] [CrossRef]
- Betthausen, E.; Drechsler, M.; Förtsch, M.; Pergushov, D.V.; Schacher, F.H.; Müller, A.H.E. Stimuli-Responsive Micellar Interpolyelectrolyte Complexes-Control of Micelle Dynamics via Core Crosslinking. Soft Matter 2012, 8, 10167–10177. [Google Scholar] [CrossRef]
- Synatschke, C.V.; Schacher, F.H.; Förtsch, M.; Drechsler, M.; Müller, A.H.E. Double-Layered Micellar Interpolyelectrolyte Complexes—How Many Shells to a Core? Soft Matter 2011, 7, 1714–1725. [Google Scholar] [CrossRef]
- Voets, I.K.; Moll, P.M.; Aqil, A.; Jérôme, C.; Detrembleur, C.; De Waard, P.; De Keizer, A.; Cohen Stuart, M.A. Temperature responsive complex coacervate core micelles with a PEO and PNIPAAm corona. J. Phys. Chem. B 2008, 112, 10833–10840. [Google Scholar] [CrossRef] [PubMed]
- Van der Gucht, J.; Spruijt, E.; Lemmers, M.; Cohen Stuart, M.A. Polyelectrolyte complexes: Bulk phases and colloidal systems. J. Colloid Interface Sci. 2011, 361, 407–422. [Google Scholar] [CrossRef]
- Sadman, K.; Wang, Q.; Chen, Y.; Keshavarz, B.; Jiang, Z.; Shull, K.R. Influence of Hydrophobicity on Polyelectrolyte Complexation. Macromolecules 2017, 50, 9417–9426. [Google Scholar] [CrossRef]
- Van Hees, I.A.; Swinkels, P.J.M.; Fokkink, R.G.; Velders, A.H.; Voets, I.K.; van der Gucht, J.; Kamperman, M. Self-Assembly of Oppositely Charged Polyelectrolyte Block Copolymers Containing Short Thermoresponsive Blocks. Polym. Chem. 2019, 10, 3127–3134. [Google Scholar] [CrossRef]
- Fehér, B.; Zhu, K.; Nyström, B.; Varga, I.; Pedersen, J.S. Effect of Temperature and Ionic Strength on Micellar Aggregates of Oppositely Charged Thermoresponsive Block Copolymer Polyelectrolytes. Langmuir 2019, 35, 13614–13623. [Google Scholar] [CrossRef]
- Van Der Kooij, H.M.; Spruijt, E.; Voets, I.K.; Fokkink, R.; Cohen Stuart, M.A.; Van Der Gucht, J. On the Stability and Morphology of Complex Coacervate Core Micelles: From Spherical to Wormlike Micelles. Langmuir 2012, 28, 14180–14191. [Google Scholar] [CrossRef]
- Yoon, H.; Dell, E.J.; Freyer, J.L.; Campos, L.M.; Jang, W.D. Polymeric Supramolecular Assemblies Based on Multivalent Ionic Interactions for Biomedical Applications. Polymer 2014, 55, 453–464. [Google Scholar] [CrossRef]
- Anraku, Y.; Kishimura, A.; Kamiya, M.; Tanaka, S.; Nomoto, T.; Toh, K.; Matsumoto, Y.; Fukushima, S.; Sueyoshi, D.; Kano, M.R.; et al. Systemically Injectable Enzyme-Loaded Polyion Complex Vesicles as in Vivo Nanoreactors Functioning in Tumors. Angew. Chem. Int. Ed. 2016, 55. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Dahlman, J.E.; Langer, R. Emerging Frontiers in Drug Delivery. J. Am. Chem. Soc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gary, E.N.; Weiner, D.B. DNA Vaccines: Prime Time is Now. Curr. Opin. Immunol. 2020, 65, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Wahlich, J.; Desai, A.; Greco, F.; Hill, K.; Jones, A.T.; Mrsny, R.J.; Pasut, G.; Perrie, Y.; Seib, F.P.; Seymour, L.W.; et al. Nanomedicines for the Delivery of Biologics. Pharmaceutics 2019, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Pisal, D.S.; Kosloski, M.P.; Balu-Iyer, S.V. Delivery of Therapeutic Proteins. J. Pharm. Sci. 2010, 99, 2557–2575. [Google Scholar] [CrossRef]
- Aied, A.; Greiser, U.; Pandit, A.; Wang, W. Polymer Gene Delivery: Overcoming the Obstacles. Drug Discov. Today 2013, 18, 1090–1098. [Google Scholar] [CrossRef]
- Christie, R.J.; Matsumoto, Y.; Miyata, K.; Nomoto, T.; Fukushima, S.; Osada, K.; Halnaut, J.; Pittella, F.; Kim, H.J.; Nishiyama, N.; et al. Targeted Polymeric Micelles for siRNA Treatment of Experimental Cancer by Intravenous Injection. ACS Nano 2012, 6, 5174–5189. [Google Scholar] [CrossRef]
- Oishi, M.; Nagasaki, Y.; Itaka, K.; Nishiyama, N.; Kataoka, K. Lactosylated Poly(Ethylene Glycol)-siRNA Conjugate Through Acid-Labile β-Thiopropionate Linkage to Construct pH-Sensitive Polyion Complex Micelles Achieving Enhanced Gene Silencing in Hepatoma Cells. J. Am. Chem. Soc. 2005, 127, 1624–1625. [Google Scholar] [CrossRef]
- Qian, Y.; Zha, Y.; Feng, B.; Pang, Z.; Zhang, B.; Sun, X.; Ren, J.; Zhang, C.; Shao, X.; Zhang, Q.; et al. PEGylated Poly(2-(Dimethylamino) Ethyl Methacrylate)/DNA Polyplex Micelles Decorated with Phage-Displayed TGN Peptide for Brain-Targeted Gene Delivery. Biomaterials 2013, 34, 2117–2129. [Google Scholar] [CrossRef]
- Burke, R.S.; Pun, S.H. Extracellular Barriers to in Vivo PEI and PEGylated PEI Polyplex-Mediated Gene Delivery to the Liver. Bioconjug. Chem. 2008, 19, 693–704. [Google Scholar] [CrossRef]
- Wen, H.; Yu, Q.; Yin, Y.; Pan, W.; Yang, S.; Liang, D. Shear Effects on Stability of DNA Complexes in the Presence of Serum. Biomacromolecules 2017, 18, 3252–3259. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Ishii, T.; Matsumoto, A.; Miyata, K.; Miyahara, Y.; Kataoka, K. A Phenylboronate-Functionalized Polyion Complex Micelle for ATP-Triggered Release of siRNA. Angew. Chem. Int. Ed. 2012, 51, 10751–10755. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Christie, R.J.; Nishiyama, N.; Miyata, K.; Ishii, A.; Oba, M.; Koyama, H.; Yamasaki, Y.; Kataoka, K. Environment-Responsive Block Copolymer Micelles with a Disulfide Cross-Linked Core for Enhanced siRNA Delivery. Biomacromolecules 2009, 10, 119–127. [Google Scholar] [CrossRef]
- Heller, P.; Hobernik, D.; Lächelt, U.; Schinnerer, M.; Weber, B.; Schmidt, M.; Wagner, E.; Bros, M.; Barz, M. Combining Reactive Triblock Copolymers with Functional Cross-Linkers: A Versatile Pathway to Disulfide Stabilized-Polyplex Libraries and their Application as pDNA Vaccines. J. Control Release 2017, 258, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ishii, T.; Zheng, M.; Watanabe, S.; Toh, K.; Matsumoto, Y.; Nishiyama, N.; Miyata, K.; Kataoka, K.; Kim, H.J.; et al. Multifunctional Polyion Complex Micelle Featuring Enhanced Stability, Targetability, and Endosome Escapability for Systemic siRNA Delivery to Subcutaneous Model of Lung Cancer. Drug Deliv. Transl. Res 2014, 4, 50–60. [Google Scholar] [CrossRef]
- Kim, H.J.; Ishii, A.; Miyata, K.; Lee, Y.; Wu, S.; Oba, M.; Nishiyama, N.; Kataoka, K. Introduction of Stearoyl Moieties into a Biocompatible Cationic Polyaspartamide Derivative, PAsp(DET), with Endosomal Escaping Function for Enhanced siRNA-Mediated Gene Knockdown. J. Control Release 2010, 145, 141–148. [Google Scholar] [CrossRef]
- Osawa, S.; Osada, K.; Hiki, S.; Dirisala, A.; Ishii, T.; Kataoka, K. Polyplex Micelles with Double-Protective Compartments of Hydrophilic Shell and Thermoswitchable Palisade of Poly(oxazoline)-Based Block Copolymers for Promoted Gene Transfection. Biomacromolecules 2016, 17, 354–361. [Google Scholar] [CrossRef]
- Nelson, C.E.; Kintzing, J.R.; Hanna, A.; Shannon, J.M.; Gupta, M.K.; Duvall, C.L. Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity in Vivo. ACS Nano 2013, 7, 8870–8880. [Google Scholar] [CrossRef]
- Takemoto, H.; Miyata, K.; Hattori, S.; Ishii, T.; Suma, T.; Uchida, S.; Nishiyama, N.; Kataoka, K. Acidic pH-Responsive siRNA Conjugate for Reversible Carrier Stability and Accelerated Endosomal Escape with Reduced IFNα-Associated Immune Response. Angew. Chem. Int. Ed. 2013, 52, 6218–6221. [Google Scholar] [CrossRef]
- Mok, H.; Lee, S.H.; Park, J.W.; Park, T.G. Multimeric Small Interfering Ribonucleic Acid for Highly Efficient Sequence-Specific Gene Silencing. Nat. Mater. 2010, 9, 272–278. [Google Scholar] [CrossRef]
- Takemoto, H.; Ishii, A.; Miyata, K.; Nakanishi, M.; Oba, M.; Ishii, T.; Yamasaki, Y.; Nishiyama, N.; Kataoka, K. Polyion Complex Stability and Gene Silencing Efficiency with a siRNA-Grafted Polymer Delivery System. Biomaterials 2010, 31, 8097–8105. [Google Scholar] [CrossRef] [PubMed]
- Osada, K.; Shiotani, T.; Tockary, T.A.; Kobayashi, D.; Oshima, H.; Ikeda, S.; Christie, R.J.; Itaka, K.; Kataoka, K. Enhanced Gene Expression Promoted by the Quantized Folding of pDNA Within Polyplex Micelles. Biomaterials 2012, 33, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Q.; Zha, Z.; Li, H.; Toh, K.; Dirisala, A.; Matsumoto, Y.; Osada, K.; Kataoka, K.; Ge, Z. Ternary Polyplex Micelles with PEG Shells and Intermediate Barrier to Complexed DNA Cores For Efficient Systemic Gene Delivery. J. Control Release 2015, 209, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In Vitro Cytotoxicity Testing of Polycations: Influence of Polymer Structure on Cell Viability and Hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Wagner, M.; Rinkenauer, A.C.; Schallon, A.; Schubert, U.S. Opposites Attract: Influence of the Molar Mass of Branched Poly(Ethylene Imine) on Biophysical Characteristics of siRNA-Based Polyplexese. RSC Adv. 2013, 3, 12774. [Google Scholar] [CrossRef]
- Cai, J.; Yue, Y.; Rui, D.; Zhang, Y.; Liu, S.; Wu, C. Effect of Chain Length on Cytotoxicity and Endocytosis of Cationic Polymers. Macromolecules 2011, 44, 2050–2057. [Google Scholar] [CrossRef]
- Fichter, K.M.; Ingle, N.P.; McLendon, P.M.; Reineke, T.M. Polymeric Nucleic Acid Vehicles Exploit Active Interorganelle Trafficking Mechanisms. ACS Nano 2013, 7, 347–364. [Google Scholar] [CrossRef]
- Lee, C.-C.; Liu, Y.; Reineke, T.M. Glucose-Based Poly(ester amines): Synthesis, Degradation, and Biological Delivery. ACS Macro Lett. 2012, 1, 1388–1392. [Google Scholar] [CrossRef]
- Taori, V.P.; Lu, H.; Reineke, T.M. Structure–Activity Examination of Poly(glycoamidoguanidine)s: Glycopolycations Containing Guanidine Units for Nucleic Acid Delivery. Biomacromolecules 2011, 12, 2055–2063. [Google Scholar] [CrossRef]
- Van Bruggen, C.; Hexum, J.K.; Tan, Z.; Dalal, R.J.; Reineke, T.M. Nonviral Gene Delivery with Cationic Glycopolymers. Acc. Chem. Res. 2019, 1347–1358. [Google Scholar] [CrossRef]
- Liu, Y.; Reineke, T.M. Degradation of Poly(glycoamidoamine) DNA Delivery Vehicles: Polyamide Hydrolysis at Physiological Conditions Promotes DNA Release. Biomacromolecules 2010, 11, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; May, S. Release of Cationic Polymer-DNA Complexes from the Endosome: A Theoretical Investigation of the Proton Sponge Hypothesis. J. Chem. Phys. 2008, 129, 185105. [Google Scholar] [CrossRef] [PubMed]
- Wojnilowicz, M.; Glab, A.; Bertucci, A.; Caruso, F.; Cavalieri, F. Super-resolution Imaging of Proton Sponge-Triggered Rupture of Endosomes and Cytosolic Release of Small Interfering RNA. ACS Nano 2019, 13, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Lungwitz, U.; Breunig, M.; Blunk, T.; Göpferich, A. Polyethylenimine-Based Non-Viral Gene Delivery Systems. Eur. J. Pharm. Biopharm. 2005, 60, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, A.; Kouhbanani, M.A.J.; Beheshtkhoo, N.; Beigi, V.; Mousavi, S.M.; Hashemi, S.A.R.; Karimi Zade, A.; Amani, A.M.; Savardashtaki, A.; Mirzaei, E.; et al. Polyethylenimine-Based Nanocarriers in Co-Delivery of Drug and Gene: A Developing Horizon. Nano Rev. Exp. 2018, 9, 1488497. [Google Scholar] [CrossRef]
- Najafi, H.; Abolmaali, S.S.; Owrangi, B.; Ghasemi, Y.; Tamaddon, A.M. Serum Resistant and Enhanced Transfection of Plasmid DNA by PEG-Stabilized Polyplex Nanoparticles of L-Histidine Substituted Polyethyleneimine. Macromol. Res. 2015, 23, 618–627. [Google Scholar] [CrossRef]
- Uchida, H.; Miyata, K.; Oba, M.; Ishii, T.; Suma, T.; Itaka, K.; Nishiyama, N.; Kataoka, K. Odd-Even Effect of Repeating Aminoethylene Units in the Side Chain of N-Substituted Polyaspartamides on Gene Transfection Profiles. J. Am. Chem. Soc. 2011, 133, 15524–15532. [Google Scholar] [CrossRef]
- Itaka, K.; Ishii, T.; Hasegawa, Y.; Kataoka, K. Biodegradable Polyamino Acid-Based Polycations as Safe and Effective Gene Carrier Minimizing Cumulative Toxicity. Biomaterials 2010, 31, 3707–3714. [Google Scholar] [CrossRef]
- Suma, T.; Miyata, K.; Anraku, Y.; Watanabe, S.; Christie, R.J.; Takemoto, H.; Shioyama, M.; Gouda, N.; Ishii, T.; Nishiyama, N.; et al. Smart Multilayered Assembly for Biocompatible siRNA Delivery Featuring Dissolvable Silica, Endosome-Disrupting Polycation, and Detachable PEG. ACS Nano 2012, 6, 6693–6705. [Google Scholar] [CrossRef]
- Sun, C.Y.; Shen, S.; Xu, C.F.; Li, H.J.; Liu, Y.; Cao, Z.T.; Yang, X.Z.; Xia, J.X.; Wang, J. Tumor Acidity-Sensitive Polymeric Vector for Active Targeted siRNA Delivery. J. Am. Chem. Soc. 2015, 137, 15217–15224. [Google Scholar] [CrossRef]
- Champion, J.A.; Katare, Y.K.; Mitragotri, S. Particle shape: A New Design Parameter for Micro- and Nanoscale Drug Delivery Carriers. J. Control Release 2007, 121, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Dalhaimer, P.; Cai, S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D.E. Shape Effects of Filaments Versus Spherical Particles in Flow and Drug Delivery. Nat. Nanotechnol. 2007, 2, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Dirisala, A.; Osada, K.; Chen, Q.; Tockary, T.A.; Machitani, K.; Osawa, S.; Liu, X.; Ishii, T.; Miyata, K.; Oba, M.; et al. Optimized Rod Length of Polyplex Micelles for Maximizing Transfection Efficiency and their Performance in Systemic Gene Therapy Against Stroma-Rich Pancreatic Tumors. Biomaterials 2014, 35, 5359–5368. [Google Scholar] [CrossRef]
- Marras, A.E.; Vieregg, J.R.; Ting, J.M.; Rubien, J.D.; Tirrell, M.V. Polyelectrolyte Complexation of Oligonucleotides by Charged Hydrophobic—Neutral Hydrophilic Block Copolymers. Polymers 2019, 11, 83. [Google Scholar] [CrossRef] [PubMed]
- Lueckheide, M.; Vieregg, J.R.; Bologna, A.J.; Leon, L.; Tirrell, M.V. Structure–Property Relationships of Oligonucleotide Polyelectrolyte Complex Micelles. Nano Lett. 2018, 18, 7111–7117. [Google Scholar] [CrossRef]
- Hud, N.V.; Downing, K.H. Cryoelectron Microscopy of λ Phage DNA Condensates in Vitreous Ice: The Fine Structure of DNA Toroids. Proc. Natl. Acad. Sci. USA 2001, 98, 14925–14930. [Google Scholar] [CrossRef]
- Shen, L.; Li, Y.; Lu, Q.; Qi, X.; Wu, X.; Zhou, Z.; Shen, J. Directed Arrangement of siRNA via Polymerization-Induced Electrostatic Self-Assembly. Chem. Commun. 2020, 56, 2411–2414. [Google Scholar] [CrossRef]
- Boylan, N.J.; Suk, J.S.; Lai, S.K.; Jelinek, R.; Boyle, M.P.; Cooper, M.J.; Hanes, J. Highly Compacted DNA Nanoparticles With low MW PEG Coatings: In Vitro, ex Vivo and in Vivo Evaluation. J. Control Release 2012, 157, 72–79. [Google Scholar] [CrossRef]
- Jiang, X.; Qu, W.; Pan, D.; Ren, Y.; Williford, J.M.; Cui, H.; Luijten, E.; Mao, H.Q. Plasmid-Templated Shape Control of Condensed DNA-Block Copolymer Nanoparticles. Adv. Mater. 2013, 25, 227–232. [Google Scholar] [CrossRef]
- Li, Y.; Osada, K.; Chen, Q.; Tockary, T.A.; Dirisala, A.; Takeda, K.M.; Uchida, S.; Nagata, K.; Itaka, K.; Kataoka, K. Toroidal Packaging of pDNA into Block Ionomer Micelles Exerting Promoted in Vivo Gene Expression. Biomacromolecules 2015, 16, 2664–2671. [Google Scholar] [CrossRef]
- Rinkenauer, A.C.; Schubert, S.; Traeger, A.; Schubert, U.S. The Influence of Polymer Architecture on in Vitro pDNA Transfection. J. Mater. Chem. B 2015, 3, 7477–7493. [Google Scholar] [CrossRef] [PubMed]
- Aono, R.; Yuba, E.; Harada, A.; Kono, K. Nanofiber Polyplex Formation Based on the Morphology Elongation by the Intrapolyplex PEG Crowding Effect. ACS Macro Lett. 2014, 3, 333–336. [Google Scholar] [CrossRef]
- Feiner-Gracia, N.; Olea, R.A.; Fitzner, R.; El Boujnouni, N.; Van Asbeck, A.H.; Brock, R.; Albertazzi, L. Super-resolution Imaging of Structure, Molecular Composition, and Stability of Single Oligonucleotide Polyplexes. Nano Lett. 2019, 19, 2784–2792. [Google Scholar] [CrossRef]
- Riera, R.; Feiner-Gracia, N.; Fornaguera, C.; Cascante, A.; Borrós, S.; Albertazzi, L. Tracking the DNA Complexation State of pBAE Polyplexes in Cells with Super Resolution Microscopy. Nanoscale 2019, 11, 17869–17877. [Google Scholar] [CrossRef] [PubMed]
- Grandinetti, G.; Reineke, T.M. Exploring the Mechanism of Plasmid DNA Nuclear Internalization with Polymer-Based Vehicles. Mol. Pharm. 2012. [Google Scholar] [CrossRef]
- Grosse, S.; Thévenot, G.; Monsigny, M.; Fajac, I. Which Mechanism for Nuclear Import of Plasmid DNA Complexed with Polyethylenimine Derivatives? J. Gene Med. 2006, 8, 845–851. [Google Scholar] [CrossRef]
- Tan, Z.; Jiang, Y.; Zhang, W.; Karls, L.; Lodge, T.P.; Reineke, T.M. Polycation Architecture and Assembly Direct Successful Gene Delivery: Micelleplexes Outperform Polyplexes via Optimal DNA Packaging. J. Am. Chem. Soc. 2019, 141, 15804–15817. [Google Scholar] [CrossRef]
- Kim, B.; Lam, C.N.; Olsen, B.D. Nanopatterned Protein Films Directed by Ionic Complexation with Water-Soluble Diblock Copolymers. Macromolecules 2012, 45, 4572–4580. [Google Scholar] [CrossRef]
- Cummings, C.S.; Obermeyer, A.C. Phase Separation Behavior of Supercharged Proteins and Polyelectrolytes. Biochemistry 2018, 57, 314–323. [Google Scholar] [CrossRef]
- Obermeyer, A.C.; Mills, C.E.; Dong, X.-H.; Flores, R.J.; Olsen, B.D. Complex Coacervation of Supercharged Proteins with Polyelectrolytes. Soft Matter 2016, 12, 3570–3581. [Google Scholar] [CrossRef]
- Nolles, A.; Westphal, A.H.; Kleijn, J.M.; van Berkel, W.J.H.; Borst, J.W. Colorful Packages: Encapsulation of Fluorescent Proteins in Complex Coacervate Core Micelles. Int. J. Mol. Sci. 2017, 18, 1557. [Google Scholar] [CrossRef] [PubMed]
- Karayianni, M.; Pispas, S. Complexation of Stimuli-Responsive Star-Like Amphiphilic Block Polyelectrolyte Micelles with Lysozyme. Soft Matter 2012, 8, 8758–8769. [Google Scholar] [CrossRef]
- Xu, Y.; Mazzawi, M.; Chen, K.; Sun, L.; Dubin, P.L. Protein Purification by Polyelectrolyte Coacervation: Influence of Protein Charge Anisotropy on Selectivity. Biomacromolecules 2011, 12, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Pathak, J.; Rawat, K.; Bohidar, H.B. Is Surface Patch Binding Between Proteins Symmetric About Isoelectric pH? RSC Adv. 2014. [Google Scholar] [CrossRef]
- Lindhoud, S.; de Vries, R.; Willem Norde, A.; Cohen Stuart, M.A. Structure and Stability of Complex Coacervate Core Micelles with Lysozyme. Biomacromolecules 2007. [Google Scholar] [CrossRef]
- Lindhoud, S.; Norde, W.; Cohen Stuart, M.A. Effects of Polyelectrolyte Complex Micelles and Their Components on the Enzymatic Activity of Lipase. Langmuir 2010, 26, 9802–9808. [Google Scholar] [CrossRef]
- Lindhoud, S.; De Vries, R.; Schweins, R.; Cohen Stuart, M.A.; Norde, W. Salt-induced release of lipase from polyelectrolyte complex micelles. Soft Matter 2009, 5, 242–250. [Google Scholar] [CrossRef]
- Black, K.A.; Priftis, D.; Perry, S.L.; Yip, J.; Byun, W.Y.; Tirrell, M. Protein Encapsulation via Polypeptide Complex Coacervation. ACS Macro Lett. 2014, 3, 1088–1091. [Google Scholar] [CrossRef]
- Mills, C.E.; Obermeyer, A.; Dong, X.; Walker, J.; Olsen, B.D. Complex Coacervate Core Micelles for the Dispersion and Stabilization of Organophosphate Hydrolase in Organic Solvents. Langmuir 2016, 32, 13367–13376. [Google Scholar] [CrossRef]
- Krishnan, Y.; Rees, H.A.; Rossitto, C.P.; Kim, S.-E.; Hung, H.-H.K.; Frank, E.H.; Olsen, B.D.; Liu, D.R.; Hammond, P.T.; Grodzinsky, A.J. Green Fluorescent Proteins Engineered for Cartilage-Targeted Drug Delivery: Insights for Transport into Highly Charged Avascular Tissues. Biomaterials 2018, 183, 218–233. [Google Scholar] [CrossRef]
- Lee, Y.; Ishii, T.; Kim, H.J.; Nishiyama, N.; Hayakawa, Y.; Itaka, K.; Kataoka, K. Efficient Delivery of Bioactive Antibodies into the Cytoplasm of Living Cells by Charge-Conversional Polyion Complex Micelles. Angew. Chem. Int. Ed. 2010, 49, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Miura, Y.; Ishii, T.; Mutaf, O.F.; Nishiyama, N.; Cabral, H.; Kataoka, K. Intracellular Delivery of Charge-Converted Monoclonal Antibodies by Combinatorial Design of Block/Homo Polyion Complex Micelles. Biomacromolecules 2016, 17, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Fan, Q.; Wang, H.; Cheng, Y. Polymers for Cytosolic Protein Delivery. Biomaterials 2019, 218, 119358. [Google Scholar] [CrossRef] [PubMed]
- Mout, R.; Ray, M.; Tay, T.; Sasaki, K.; Yesilbag Tonga, G.; Rotello, V.M. General Strategy for Direct Cytosolic Protein Delivery via Protein–Nanoparticle Co-engineering. ACS Nano 2017, 11, 6416–6421. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic Lipid-Mediated Delivery of Proteins Enables Efficient Protein-Based Genome Editing in Vitro and in Vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef]
- Simon, J.R.; Carroll, N.J.; Rubinstein, M.; Chilkoti, A.; López, G.P. Programming Molecular Self-Assembly of Intrinsically Disordered Proteins Containing Sequences of Low Complexity. Nat. Chem. 2017, 9, 509–515. [Google Scholar] [CrossRef]
- Kapelner, R.A.; Obermeyer, A.C. Ionic Polypeptide Tags for Protein Phase Separation. Chem. Sci. 2019, 10, 2700–2707. [Google Scholar] [CrossRef]
- Postupalenko, V.; Desplancq, D.; Orlov, I.; Arntz, Y.; Spehner, D.; Mely, Y.; Klaholz, B.P.; Schultz, P.; Weiss, E.; Zuber, G. Protein Delivery System Containing a Nickel-Immobilized Polymer for Multimerization of Affinity-Purified His-Tagged Proteins Enhances Cytosolic Transfer. Angew. Chem. Int. Ed. 2015, 54, 10583–10586. [Google Scholar] [CrossRef]
- Li, K.; Chen, F.; Wang, Y.; Stenzel, M.H.; Chapman, R. Polyion Complex Micelles for Protein Delivery Benefit from Flexible Hydrophobic Spacers in the Binding Group. Macromol. Rapid Commun. 2020, 2000208. [Google Scholar] [CrossRef]
- Qiu, M.; Zhang, Z.; Wei, Y.; Sun, H.; Meng, F.; Deng, C.; Zhong, Z. Small-Sized and Robust Chimaeric Lipopepsomes: A Simple and Functional Platform with High Protein Loading for Targeted Intracellular Delivery of Protein Toxin in Vivo. Chem. Mater. 2018, 30, 6831–6838. [Google Scholar] [CrossRef]
- Tan, Z.; Jiang, Y.; Ganewatta, M.S.; Kumar, R.; Keith, A.; Twaroski, K.; Pengo, T.; Tolar, J.; Lodge, T.P.; Reineke, T.M. Block Polymer Micelles Enable CRISPR/Cas9 Ribonucleoprotein Delivery: Physicochemical Properties Affect Packaging Mechanisms and Gene Editing Efficiency. Macromolecules 2019, 52, 8197–8206. [Google Scholar] [CrossRef]
- Klyachko, N.L.; Manickam, D.S.; Brynskikh, A.M.; Uglanova, S.V.; Li, S.; Higginbotham, S.M.; Bronich, T.K.; Batrakova, E.V.; Kabanov, A.V. Cross-Linked Antioxidant Nanozymes for Improved Delivery to CNS. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Klyachko, N.L.; Haney, M.J.; Zhao, Y.; Manickam, D.S.; Mahajan, V.; Suresh, P.; Hingtgen, S.D.; Mosley, R.L.; Gendelman, H.E.; Kabanov, A.V.; et al. Macrophages Offer a Paradigm Switch for CNS Delivery of Therapeutic Proteins. Nanomedicine 2014, 9, 1403–1422. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, M.J.; Murthy, N. Disulfide-Crosslinked Polyion Micelles for Delivery of Protein Therapeutics. Ann. Biomed. Eng. 2009, 37, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, Y.; Zhang, J.; Gao, H.; Liu, G.; Ma, R.; An, Y.; Kong, D.; Shi, L. pH/Sugar Dual Responsive Core-Cross-Linked PIC Micelles for Enhanced Intracellular Protein Delivery. Biomacromolecules 2013, 14, 3434–3443. [Google Scholar] [CrossRef] [PubMed]
- Tao, A.; Huang, G.L.; Igarashi, K.; Hong, T.; Liao, S.; Stellacci, F.; Matsumoto, Y.; Yamasoba, T.; Kataoka, K.; Cabral, H. Polymeric Micelles Loading Proteins through Concurrent Ion Complexation and pH-Cleavable Covalent Bonding for In Vivo Delivery. Macromol. Biosci. 2020, 20, 1900161. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, Y.T.; Cao, C.; Oliver, J.D.; Stenzel, M.H.; Chapman, R. Polyion Complex-Templated Synthesis of Cross-Linked Single-Enzyme Nanoparticles. Macromolecules 2020, 53, 5487–5496. [Google Scholar] [CrossRef]
- Farrugia, T.; Perriman, A.W.; Sharma, K.P.; Mann, S. Multi-Enzyme Cascade Reactions Using Protein-Polymer Surfactant Self-Standing Films. Chem. Commun. 2017, 53, 2094–2097. [Google Scholar] [CrossRef]
- Sureka, H.V.; Obermeyer, A.C.; Flores, R.J.; Olsen, B.D. Catalytic Biosensors from Complex Coacervate Core Micelle (C3M) Thin Films. ACS Appl. Mater. Interfaces 2019, 11, 32354–32365. [Google Scholar] [CrossRef]
- Nolles, A.; Van Dongen, N.J.E.; Westphal, A.H.; Visser, A.J.W.G.; Kleijn, J.M.; Van Berkel, W.J.H.; Borst, J.W. Encapsulation into Complex Coacervate Core Micelles Promotes EGFP Dimerization. Phys. Chem. Chem. Phys. 2017, 19, 11380–11389. [Google Scholar] [CrossRef]
- Kawamura, A.; Yoshioka, Y.; Harada, A.; Kono, K. Acceleration of Enzymatic Reaction of Trypsin Through the Formation of Water-Soluble Complexes with Poly(Ethylene Glycol)-Block-Poly(α,β-Aspartic Acid). Biomacromolecules 2005, 6, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Yoshioka, Y.; Kawamura, A.; Kojima, C.; Kono, K. Effect of Polycarboxylate Blocks on the Amidase Activity of Trypsin through Complexation with PEG/Polycarboxylate Block Ionomers. Macromol. Biosci. 2007, 7, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Kataoka, K. On-off Control of Enzymatic Activity Synchronizing With Reversible Formation of Supramolecular Assembly from Enzyme and Charged Block Copolymers. J. Am. Chem. Soc. 1999, 121, 9241–9242. [Google Scholar] [CrossRef]
- Perriman, A.W.; Brogan, A.P.S.; Cölfen, H.; Tsoureas, N.; Owen, G.R.; Mann, S. Reversible Dioxygen Binding in Solvent-Free Liquid Myoglobin. Nat. Chem. 2010, 2, 622–626. [Google Scholar] [CrossRef]
- Sharma, K.P.; Zhang, Y.; Thomas, M.R.; Brogan, A.P.S.; Perriman, A.W.; Mann, S. Self-Organization of Glucose Oxidase-Polymer Surfactant Nanoconstructs in Solvent-Free Soft Solids and Liquids. J. Phys. Chem. B 2014, 118, 11573–11580. [Google Scholar] [CrossRef]
- Perriman, A.W.; Cölfen, H.; Hughes, R.W.; Barrie, C.L.; Mann, S. Solvent-Free Protein Liquids and Liquid Crystals. Angew. Chem. Int. Ed. 2009, 48, 6242–6246. [Google Scholar] [CrossRef]
- Perriman, A.W.; Mann, S. Liquid Proteins-A new Frontier for Biomolecule-Based Nanoscience. ACS Nano 2011, 5, 6085–6091. [Google Scholar] [CrossRef]
- Atkins, D.L.; Berrocal, J.A.; Mason, A.F.; Voets, I.K. Tandem Catalysis in Multicomponent Solvent-Free Biofluids. Nanoscale 2019, 11. [Google Scholar] [CrossRef]
- Lopez-Blanco, R.; Fernandez-Villamarin, M.; Jatunov, S.; Novoa-Carballal, R.; Fernandez-Megia, E. Polysaccharides Meet Dendrimers to Fine-Tune the Stability and Release Properties of Polyion Complex Micelles. Polym. Chem. 2019, 10, 4709–4717. [Google Scholar] [CrossRef]
- Du, Y.; Yan, W.; Lian, H.; Xiang, C.; Duan, L.; Xiao, C. 2,2′-Dithiodisuccinic Acid-Stabilized Polyion Complex Micelles for pH and Reduction Dual-Responsive Drug Delivery. J. Colloid Interface Sci. 2018, 522, 74–81. [Google Scholar] [CrossRef]
- Fernandez-Villamarin, M.; Sousa-Herves, A.; Porto, S.; Guldris, N.; Martínez-Costas, J.; Riguera, R.; Fernandez-Megia, E. A dendrimer-Hydrophobic Interaction Synergy Improves the Stability of Polyion Complex Micelles. Polym. Chem. 2017, 8, 2528–2537. [Google Scholar] [CrossRef]
- Ramasamy, T.; Poudel, B.K.; Ruttala, H.; Choi, J.Y.; Hieu, T.D.; Umadevi, K.; Youn, Y.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Cationic Drug-Based Self-Assembled Polyelectrolyte Complex Micelles: Physicochemical, Pharmacokinetic, and Anticancer Activity Analysis. Colloids Surfaces B Biointerfaces 2016, 146, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ye, L.; Zhang, A.; Feng, Z. The Preparation and Morphology Control of Heparin-Based pH Sensitive Polyion Complexes and their Application as Drug Carriers. Carbohydr. Polym. 2019, 211, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lord, M.S.; Stenzel, M.H. A Polyion Complex Micelle with Heparin for Growth Factor Delivery and Uptake into Cells. J. Mater. Chem. B 2013, 1, 1635–1643. [Google Scholar] [CrossRef]
- Karanikolopoulos, N.; Pitsikalis, M.; Hadjichristidis, N.; Georgikopoulou, K.; Calogeropoulou, T.; Dunlap, J.R. PH-Responsive Aggregates From Double Hydrophilic Block Copolymers Carrying Zwitterionic Groups. Encapsulation of Antiparasitic Compounds for the Treatment of Leishmaniasis. Langmuir 2007, 23, 4214–4224. [Google Scholar] [CrossRef]
- Li, Y.; Ikeda, S.; Nakashima, K.; Nakamura, H. Nanoaggregate Formation of Poly(Ethylene Oxide)-b-Polymethacrylate Copolymer Induced by Cationic Anesthetics Binding. Colloid Polym. Sci. 2003, 281, 562–568. [Google Scholar] [CrossRef]
- Ramasamy, T.; Kim, J.H.; Choi, J.Y.; Tran, T.H.; Choi, H.G.; Yong, C.S.; Kim, J.O. pH Sensitive Polyelectrolyte Complex Micelles for Highly Effective Combination Chemotherapy. J. Mater. Chem. B 2014, 2, 6324–6333. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, J.; Ding, P.; Guo, X.; Cohen Stuart, M.A.; Wang, J. Functional Polyion Complex Vesicles Enabled by Supramolecular Reversible Coordination Polyelectrolytes. Angew. Chem. Int. Ed. 2019, 58, 8494–8498. [Google Scholar] [CrossRef]
- Pothayee, N.; Pothayee, N.; Jain, N.; Hu, N.; Balasubramaniam, S.; Johnson, L.M.; Davis, R.M.; Sriranganathan, N.; Riffle, J.S. Magnetic Block Ionomer Complexes for Potential Dual Imaging and Therapeutic Agents. Chem. Mater. 2012, 24, 2056–2063. [Google Scholar] [CrossRef]
- Pothayee, N.; Pothayee, N.; Hu, N.; Zhang, R.; Kelly, D.F.; Koretsky, A.P.; Riffle, J.S. Manganese Graft Ionomer Complexes (MaGICs) for Dual Imaging and Chemotherapy. J. Mater. Chem. B 2014, 2, 1087–1099. [Google Scholar] [CrossRef]
- Jian, W.H.; Yu, T.W.; Chen, C.J.; Huang, W.C.; Chiu, H.C.; Chiang, W.H. Indocyanine Green-Encapsulated Hybrid Polymeric Nanomicelles for Photothermal Cancer Therapy. Langmuir 2015, 31, 6202–6210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jang, W.D.; Nishiyama, N.; Kishimura, A.; Kawauchi, S.; Morimoto, Y.; Miake, S.; Yamashita, T.; Kikuchi, M.; Aida, T.; et al. Dendrimer Generation Effects on Photodynamic Efficacy of Dendrimer Porphyrins and Dendrimer-Loaded Supramolecular Nanocarriers. Chem. Mater. 2007, 19, 5557–5562. [Google Scholar] [CrossRef]
- Park, W.; Park, S.J.; Na, K. The Controlled Photoactivity of Nanoparticles Derived from Ionic Interactions Between a Water Soluble Polymeric Photosensitizer and Polysaccharide Quencher. Biomaterials 2011, 32, 8261–8270. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, N.; Nakagishi, Y.; Morimoto, Y.; Lai, P.S.; Miyazaki, K.; Urano, K.; Horie, S.; Kumagai, M.; Fukushima, S.; Cheng, Y.; et al. Enhanced Photodynamic Cancer Treatment by Supramolecular Nanocarriers Charged with Dendrimer Phthalocyanine. J. Control Release 2009, 133, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Nukolova, N.V.; Yang, Z.; Kim, J.O.; Kabanov, A.V.; Bronich, T.K. Polyelectrolyte Nanogels Decorated with Monoclonal Antibody for Targeted Drug Delivery. React. Funct. Polym. 2011, 71, 315–323. [Google Scholar] [CrossRef]
- Danhier, F. To Exploit the Tumor Microenvironment: Since the EPR Effect Fails in the Clinic, What is the Future of Nanomedicine? J. Control Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer Nanotechnology: The impact of Passive and Active Targeting in the Era of Modern Cancer Biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and Fallacy. J. Control Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Ahn, J.; Miura, Y.; Yamada, N.; Chida, T.; Liu, X.; Kim, A.; Sato, R.; Tsumura, R.; Koga, Y.; Yasunaga, M.; et al. Antibody Fragment-Conjugated Polymeric Micelles Incorporating Platinum Drugs for Targeted Therapy of Pancreatic Cancer. Biomaterials 2015, 39, 23–30. [Google Scholar] [CrossRef]
- Bae, Y.; Jang, W.D.; Nishiyama, N.; Fukushima, S.; Kataoka, K. Multifunctional Polymeric Micelles with Folate-Mediated Cancer Cell Targeting and pH-Triggered Drug Releasing Properties for Active Intracellular Drug Delivery. Mol. Biosyst. 2005, 1, 242–250. [Google Scholar] [CrossRef]
- Zhang, X.X.; Eden, H.S.; Chen, X. Peptides in Cancer Nanomedicine: Drug Carriers, Targeting Ligands and Protease Substrates. J. Control Release 2012, 159, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Voeikov, R.; Abakumova, T.; Grinenko, N.; Melnikov, P.; Bespalov, V.; Stukov, A.; Chekhonin, V.; Klyachko, N.; Nukolova, N. Dioxadet-loaded Nanogels as a Potential Formulation for Glioblastoma Treatment. J. Pharm. Investig. 2017, 47, 75–83. [Google Scholar] [CrossRef]
- Cingil, H.E.; Meertens, N.C.H.; Voets, I.K. Temporally Programmed Disassembly and Reassembly of C3Ms. Small 2018, 14, 1802089. [Google Scholar] [CrossRef] [PubMed]
- Sproncken, C.C.M.; Gumí-Audenis, B.; Panzarasa, G.; Voets, I.K. Two-Stage Polyelectrolyte Assembly Orchestrated by a Clock Reaction. ChemSystemsChem 2020, syst.202000005. [Google Scholar] [CrossRef]
- Nishiyama, N.; Morimoto, Y.; Jang, W.D.; Kataoka, K. Design and Development of Dendrimer Photosensitizer-Incorporated Polymeric Micelles for Enhanced Photodynamic Therapy. Adv. Drug Deliv. Rev. 2009, 61, 327–338. [Google Scholar] [CrossRef]
- Stapert, H.R.; Nishiyama, N.; Jiang, D.L.; Aida, T.; Kataoka, K. Polyion Complex Micelles Encapsulating Light-Harvesting Ionic Dendrimer Zinc Porphyrins. Langmuir 2000, 16, 8182–8188. [Google Scholar] [CrossRef]
- Tangso, K.J.; Lindberg, S.; Hartley, P.G.; Knott, R.; Spicer, P.; Boyd, B.J. Formation of Liquid-Crystalline Structures in the Bile Salt-Chitosan System and Triggered Release from Lamellar Phase Bile Salt-Chitosan Capsules. ACS Appl. Mater. Interfaces 2014, 6, 12363–12371. [Google Scholar] [CrossRef]
- Seyrig, C.; Le Griel, P.; Cowieson, N.; Perez, J.; Baccile, N. Synthesis of Multilamellar Walls Vesicles Polyelectrolyte Surfactant Complexes From pH-Stimulated Phase Transition Using Microbial Biosurfactants. J. Colloid Interface Sci. 2020. [Google Scholar] [CrossRef]
- Bronich, T.K.; Nehls, A.; Eisenberg, A.; Kabanov, V.A.; Kabanov, A.V. Novel Drug Delivery Systems Based on The Complexes of Block Ionomers and Surfactants of Opposite Charge. Colloids Surfaces B Biointerfaces 1999, 16, 243–251. [Google Scholar] [CrossRef]
- Oh, K.T.; Bronich, T.K.; Bromberg, L.; Hatton, T.A.; Kabanov, A.V. Block ionomer Complexes as Prospective Nanocontainers for Drug Delivery. J. Control Release 2006, 115, 9–17. [Google Scholar] [CrossRef]
- Tam, K.C.; Wyn-Jones, E. Insights on Polymer Surfactant Complex Structures During the Binding of Surfactants to Polymers as Measured by Equilibrium and Structural Techniques. Chem. Soc. Rev. 2006, 35, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, E.; Llamas, S.; Maestro, A.; Fernández-Peña, L.; Akanno, A.; Miller, R.; Ortega, F.; Rubio, R.G. Polymer-Surfactant Systems in Bulk and at Fluid Interfaces. Adv. Colloid Interface Sci. 2016, 233, 38–64. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, C.; Obiols-Rabasa, M.; Piculell, L. Water-Insoluble Surface Coatings of Polyion-Surfactant Ion Complex Salts Respond to Additives in a Surrounding Aqueous Solution. Langmuir 2015, 31, 6487–6496. [Google Scholar] [CrossRef] [PubMed]
- Vitorazi, L.; Berret, J.F.; Loh, W. Self-Assembly of Complex Salts of Cationic Surfactants and Anionic-Neutral Block Copolymers. Dispersions with Liquid-Crystalline Internal Structure. Langmuir 2013, 29, 14024–14033. [Google Scholar] [CrossRef]
- Guzmán, E.; Fernández-Peña, L.; Ortega, F.; Rubio, R.G. Equilibrium and Kinetically Trapped Aggregates in Polyelectrolyte–Oppositely Charged Surfactant Mixtures. Curr. Opin. Colloid Interface Sci. 2020, 48, 91–108. [Google Scholar] [CrossRef]
- Simon, M.; Schneck, E.; Noirez, L.; Rahn, S.; Davidovich, I.; Talmon, Y.; Gradzielski, M. Effect of Polymer Architecture on the Phase Behavior and Structure of Polyelectrolyte/Microemulsion Complexes (PEMECs). Macromolecules 2020, 53, 4055–4067. [Google Scholar] [CrossRef]
- Simon, M.; Krause, P.; Chiappisi, L.; Noirez, L.; Gradzielski, M. Structural Control of Polyelectrolyte/Microemulsion Droplet Complexes (PEMECs) with Different Polyacrylates. Chem. Sci. 2019, 10, 385–397. [Google Scholar] [CrossRef]
- Bourouina, N.; Cohen Stuart, M.A.; Mieke Kleijn, J. Complex Coacervate Core Micelles as Diffusional Nanoprobes. Soft Matter 2014, 10, 320–331. [Google Scholar] [CrossRef]
- Bourouina, N.; De Kort, D.W.; Hoeben, F.J.M.; Janssen, H.M.; Van As, H.; Hohlbein, J.; Van Duynhoven, J.P.M.; Kleijn, J.M. Complex Coacervate Core Micelles with Spectroscopic Labels for Diffusometric Probing of Biopolymer Networks. Langmuir 2015, 31, 12635–12643. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Ding, P.; Zhou, W.; Li, Y.; Drechsler, M.; Guo, X.; Cohen Stuart, M.A. A Supramolecular Crosslinker To Give Salt-Resistant Polyion Complex Micelles and Improved MRI Contrast Agents. Angew. Chem. Int. Ed. 2018, 57, 12680–12684. [Google Scholar] [CrossRef]
- Wang, J.; Velders, A.H.; Gianolio, E.; Aime, S.; Vergeldt, F.J.; Van As, H.; Yan, Y.; Drechsler, M.; De Keizer, A.; Cohen Stuart, M.A.; et al. Controlled Mixing of Lanthanide(iii) Ions in Coacervate Core Micelles. Chem. Commun. 2013, 49, 3736–3738. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Ding, P.; Nie, X.; Cohen Stuart, M.A.; Wang, J. Europium Based Coordination Polyelectrolytes Enable Core–Shell–Corona Micelles as Luminescent Probes. Soft Matter 2020, 16, 5727–5733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Li, Y.; Zhao, H.; Wan, X. Hybrid Assemblies of Eu-Containing Polyoxometalates and Hydrophilic Block Copolymers with Enhanced Emission in Aqueous Solution. Angew. Chem. Int. Ed. 2012, 51, 4598–4602. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Lee, S.H.; Lee, S.; Choi, S.H.; Hawker, C.J.; Kim, B.S. Highly Stable Au Nanoparticles with Double Hydrophilic Block Copolymer Templates: Correlation Between Structure and Stability. Polym. Chem. 2017, 8, 4528–4537. [Google Scholar] [CrossRef]
- Lemmers, M.; Sprakel, J.; Voets, I.K.; Van Der Gucht, J.; Cohen Stuart, M.A. Multiresponsive Reversible Gels Based on Charge Driven Assembly. Angew. Chem. Int. Ed. 2010, 49, 708–711. [Google Scholar] [CrossRef]
- Sproncken, C.C.M.; Surís-Valls, R.; Cingil, H.E.; Detrembleur, C.; Voets, I.K. Complex Coacervate Core Micelles Containing Poly(vinyl alcohol) Inhibit Ice Recrystallization. Macromol. Rapid Commun. 2018, 39, 1700814. [Google Scholar] [CrossRef]
- Ten Hove, J.B.; Wang, J.; Van Oosterom, M.N.; Van Leeuwen, F.W.B.; Velders, A.H. Size-Sorting and Pattern Formation of Nanoparticle-Loaded Micellar Superstructures in Biconcave Thin Films. ACS Nano 2017, 11, 11225–11231. [Google Scholar] [CrossRef]
- Aloi, A.; Guibert, C.; Olijve, L.L.C.; Voets, I.K. Morphological Evolution of Complex Coacervate Core Micelles Revealed by iPAINT Microscopy. Polymer 2016, 107, 450–455. [Google Scholar] [CrossRef]
- Cingil, H.E.; Boz, E.B.; Wang, J.; Cohen Stuart, M.A.; Sprakel, J. Probing Nanoscale Coassembly with Dual Mechanochromic Sensors. Adv. Funct. Mater. 2016, 26, 1420–1427. [Google Scholar] [CrossRef]
- Bastakoti, B.P.; Guragain, S.; Yusa, S.I.; Nakashima, K. Novel Synthesis Route for Ag@SiO2 Core-Shell Nanoparticles via Micelle Template of Double Hydrophilic Block Copolymer. RSC Adv. 2012, 2, 5938–5940. [Google Scholar] [CrossRef]
- Seo, E.; Ko, S.J.; Min, S.H.; Kim, J.Y.; Kim, B.S. Plasmonic Transition via Interparticle Coupling of Au@Ag Core-Shell Nanostructures Sheathed in Double Hydrophilic Block Copolymer for High-Performance Polymer Solar Cell. Chem. Mater. 2015, 27, 4789–4798. [Google Scholar] [CrossRef]
- Bastakoti, B.P.; Li, Y.; Guragain, S.; Alshehri, S.M.; Shiddiky, M.J.A.; Liu, Z.; Shim, K.; Kim, J.H.; Hossain, M.S.A.; Malgras, V.; et al. Formation of Mesopores Inside Platinum Nanospheres by Using Double Hydrophilic Block Copolymers. Mater. Lett. 2016, 182, 190–193. [Google Scholar] [CrossRef]
- Seo, E.; Kim, J.; Hong, Y.; Kim, Y.S.; Lee, D.; Kim, B.S. Double hydrophilic Block Copolymer Templated Au Nanoparticles with Enhanced Catalytic Activity Toward Nitroarene Reduction. J. Phys. Chem. C 2013, 117, 11686–11693. [Google Scholar] [CrossRef]
- Kim, A.; Sharma, B.; Kim, B.S.; Park, K.H. Double-Hydrophilic Block Copolymer Nanoreactor for the Synthesis of Copper Nanoparticles and for Application in Click Chemistry. J. Nanosci. Nanotechnol. 2011, 11, 6162–6166. [Google Scholar] [CrossRef] [PubMed]
- Layrac, G.; Destarac, M.; Gérardin, C.; Tichit, D. Highly Stable Layered Double Hydroxide Colloids: A Direct Aqueous Synthesis Route From hHbrid Polyion Complex Micelles. Langmuir 2014, 30, 9663–9671. [Google Scholar] [CrossRef]
- Layrac, G.; Harrisson, S.; Destarac, M.; Gérardin, C.; Tichit, D. Comprehensive Study of the Formation of Stable Colloids of Cu/Al Layered Double Hydroxide Assisted by Double Hydrophilic Block Copolymers. Appl. Clay Sci. 2020, 193, 105673. [Google Scholar] [CrossRef]
- Bastakoti, B.P.; Sukegawa, H.; Wu, K.C.-W.; Yamauchi, Y. Synthesis of Porous Iron Oxide Microspheres by a Double Hydrophilic Block Copolymer. RSC Adv. 2014, 4, 9986. [Google Scholar] [CrossRef]
- Yu, S.H.; Cölfen, H.; Fischer, A. High Quality CeO2 Nanocrystals Stabilized by a Double Hydrophilic Block Copolymer. Colloids Surfaces A Physicochem. Eng. Asp. 2004, 243, 49–52. [Google Scholar] [CrossRef]
- Seo, E.; Lee, T.; Lee, K.T.; Song, H.K.; Kim, B.S. Versatile Double Hydrophilic Block Copolymer: Dual Role as Synthetic Nanoreactor and Ionic and Electronic Conduction Layer for Ruthenium Oxide Nanoparticle Supercapacitors. J. Mater. Chem. 2012, 22, 11598–11604. [Google Scholar] [CrossRef][Green Version]
- Tarasov, K.; Houssein, D.; Destarac, M.; Marcotte, N.; Gérardin, C.; Tichit, D. Stable Aqueous Colloids of ZnS Quantum Dots Prepared Using Double Hydrophilic Block Copolymers. New J. Chem. 2013, 37, 508–514. [Google Scholar] [CrossRef]
- Qi, L.; Cölfen, H.; Antonietti, M. Synthesis and Characterization of CdS Nanoparticles Stabilized by Double-Hydrophilic Block Copolymers. Nano Lett. 2001, 1, 61–65. [Google Scholar] [CrossRef]
- Uchman, M.; Procházka, K.; Gatsouli, K.; Pispas, S.; Špírková, M. CdS-Containing Nano-Assemblies of Double Hydrophilic Block Copolymers in Water. Colloid Polym. Sci. 2011, 289, 1045–1053. [Google Scholar] [CrossRef]
- Hu, J.J.; Hsieh, Y.H.; Jan, J.S. Polyelectrolyte Complex-Silica Hybrid Colloidal Particles Decorated with Different Polyelectrolytes. J. Colloid Interface Sci. 2015, 438, 94–101. [Google Scholar] [CrossRef]
- Guo, X.H.; Yu, S.H. Controlled Mineralization of Barium Carbonate Mesocrystals in a Mixed Solvent and at the Air/Solution Interface Using a Double Hydrophilic Block Copolymer as a Crystal Modifier. Cryst. Growth Des. 2007, 7, 354–359. [Google Scholar] [CrossRef]
- Cölfen, H.; Qi, L. A Systematic Examination of the Morphogenesis of Calcium Carbonate in the Presence of a Double-Hydrophilic Block Copolymer. Chem. Eur. J. 2001, 7, 106–116. [Google Scholar] [CrossRef]
- An, Z.; Shi, Q.; Tang, W.; Tsung, C.K.; Hawker, C.J.; Stucky, G.D. Facile RAFT Precipitation Polymerization for the Microwave-Assisted Synthesis of Well-Defined, Double Hydrophilic Block Copolymers and Nanostructured Hydrogels. J. Am. Chem. Soc. 2007, 129, 14493–14499. [Google Scholar] [CrossRef] [PubMed]
- Maggi, F.; Ciccarelli, S.; Diociaiuti, M.; Casciardi, S.; Masci, G. Chitosan Nanogels by Template Chemical Cross-Linking in Polyion Complex Micelle Nanoreactors. Biomacromolecules 2011, 12, 3499–3507. [Google Scholar] [CrossRef] [PubMed]
- De Santis, S.; Diociaiuti, M.; Cametti, C.; Masci, G. Hyaluronic Acid and Alginate Covalent Nanogels by Template Cross-Linking in Polyion Complex Micelle Nanoreactors. Carbohydr. Polym. 2014, 101, 96–103. [Google Scholar] [CrossRef]
- Zhang, D.; Qi, L.; Ma, J.; Cheng, H. Formation of Silver Nanowires in Aqueous Solutions of a Double-Hydrophilic Block Copolymer. Chem. Mater. 2001, 13, 2753–2755. [Google Scholar] [CrossRef]
- Voets, I.K.; de Keizer, A.; Frederik, P.M.; Jellema, R.; Cohen Stuart, M.A. Environment-Sensitive Stabilisation of Silver Nanoparticles in Aqueous Solutions. J. Colloid Interface Sci. 2009, 339, 317–324. [Google Scholar] [CrossRef]
- Voets, I.K.; de Keizer, A.; Leermakers, F.A.M.; Debuigne, A.; Jérôme, R.; Detrembleur, C.; Cohen Stuart, M.A. Electrostatic Hierarchical Co-Assembly in Aqueous Solutions of Two Oppositely Charged Double Hydrophilic Diblock Copolymers. Eur. Polym. J. 2009, 45, 2913–2925. [Google Scholar] [CrossRef]
- Voets, I.K.; De Keizer, A.; De Waard, P.; Frederik, P.M.; Bomans, P.H.H.; Schmalz, H.; Walther, A.; King, S.M.; Leermakers, F.A.M.; Cohen Stuart, M.A. Double-Faced Micelles From Water-Soluble Polymers. Angew. Chem. Int. Ed. 2006, 45, 6673–6676. [Google Scholar] [CrossRef] [PubMed]
- Bayati, S.; Bergquist, K.-E.; Zhu, K.; Nyström, B.; Skov Pedersen, J.; Galantini, L.; Schillén, K. Mixed Micelles of Oppositely Charged Poly( N-Isopropylacrylamide) Diblock Copolymers. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 1457–1470. [Google Scholar] [CrossRef]
- Biggs, C.I.; Bailey, T.L.; Graham, B.; Stubbs, C.; Fayter, A.; Gibson, M.I. Polymer Mimics of Biomacromolecular Antifreezes. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Hofs, B.; Brzozowska, A.; de Keizer, A.; Norde, W.; Cohen Stuart, M.A. Reduction of Protein Adsorption to a Solid Surface by a Coating Composed of Polymeric Micelles With a Glass-Like Core. J. Colloid Interface Sci. 2008, 325, 309–315. [Google Scholar] [CrossRef]
- Voets, I.K.; De Vos, W.M.; Hofs, B.; De Keizer, A.; Cohen Stuart, M.A.; Steitz, R.; Lott, D. Internal Structure of a Thin Film of Mixed Polymeric Micelles on a Solid/Liquid Interface. J. Phys. Chem. B 2008, 112, 6937–6945. [Google Scholar] [CrossRef]
- Brzozowska, A.M.; De Keizer, A.; Norde, W.; Detrembleur, C.; Cohen Stuart, M.A. Grafted Block Complex Coacervate Core Micelles and Their Effect on Protein Adsorption on Silica and Polystyrene. Colloid Polym. Sci. 2010, 288, 1081–1095. [Google Scholar] [CrossRef]
- Brzozowska, A.M.; Spruijt, E.; de Keizer, A.; Cohen Stuart, M.A.; Norde, W. On the Stability of the Polymer Brushes Formed by Adsorption of Ionomer Complexes on Hydrophilic and Hydrophobic Surfaces. J. Colloid Interface Sci. 2011, 353, 380–391. [Google Scholar] [CrossRef]
- Falentin-Daudré, C.; Faure, E.; Svaldo-Lanero, T.; Farina, F.; Jérôme, C.; Van De Weerdt, C.; Martial, J.; Duwez, A.S.; Detrembleur, C. Antibacterial Polyelectrolyte Micelles for Coating Stainless Steel. Langmuir 2012, 28, 7233–7241. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | ||||
| Block Copolymer | Oppositely Charged Species | Application | Size (Morphology) | [REF] |
| PEG-b-pAsp(DET)-Chol | pDNA | Gene delivery | 120 nm (ND) | [13] |
| PEG-b-P(Lys-co-Lys(2IT)) | pDNA | Gene delivery | 100–150 nm (ND) | [14] |
| cRGD-PEG-b-p(Lys-co-Lys(2IT)) | siRNA | Gene silencing | 20 nm (S) | [37] |
| Lac-PEG-b-siRNA | PLL | Gene silencing | 120 nm (S) | [38] |
| TGN-PEG-b-PDMAEMA | pDNA | Gene delivery | 80 nm (S) | [39] |
| PEG-b-PEI | pDNA | Gene delivery | 100–150 nm (ND) | [40] |
| PEG-b-p(Lys-co-Lys(FPBA)) | siRNA | Gene silencing | 60–80 nm (ND) | [42] |
| PEG-PLL | siRNA | Gene silencing | 60 nm (S) | [43] |
| cRGD-PEG-b-PAsp(TEP) tetraethylenepentamine | siRNA | Gene silencing | 50 nm (ND) | [45] |
| PEtOx-b-PnPrOx-b-PLL | pDNA | Gene delivery | 100 nm (C) | [47] |
| PEG-b-(DMAEMA-co-BMA) | siRNA | Gene silencing | 30 nm (S) | [48] |
| PEG-b-PLL | pDNA | Gene delivery | 70–300 nm (C & S) | [52] |
| PEG-b-PAsp(DET) + PNIPAM-b-PAsp(DET) | pDNA | Gene delivery | 70–90 nm (C) | [53] |
| Poly(galactaramidopentaethylenetetramine) | pDNA | Gene delivery | ND | [57] |
| Polyester-based glycopolycation | pDNA | Gene delivery | 50–70 nm (ND) | [58] |
| poly(glycoamidoguanidine)s | pDNA | Gene delivery | 60–200 nn (ND) | [59] |
| Poly(glycoamidoamine)s | pDNA | Gene delivery | ND | [61,85] |
| PEG-b-PEI | pDNA | Gene delivery | 150 nm (ND) | [66] |
| PEG-b-Arg-b-PCL (polycaprolactone) | siRNA | Gene silencing | 100 nm (V) | [70] |
| PEG-b-PLL | pDNA | Gene delivery | 100–600 nm (C) | [73] |
| PEG-PLL | ssDNA & dsDNA | Fundamental | 10–20 nm (S & C) | [75] |
| PEG-b-PAPTAC ((3-Acrylamidopropyl)trimethylammonium) | siRNA | Fundamental | (S, C & L) | [77] |
| PEG-b-PLL | pDNA | Gene delivery | 200–350 nm | [78] |
| PEG-b-PAsp(DET) | pDNA | Gene delivery | 80–600 nm (T, C) | [80] |
| maPEG-b-PLL (ma is multiarm) | pDNA | Gene delivery | 200–900 nm (C) | [82] |
| PEG-b-PDMAEMA, PEG-b-PDMAEMA-b-PnBMA & PDMAEMA-b-PnBMA | pDNA | Gene delivery | 150–100 nm (UD) | [87] |
| (B) | ||||
| Block Copolymer | Oppositely Charged Species | Application | Size (Morphology) | [REF] |
| PEG-b-PLL | Insulin | Protein delivery | 60–200 nm (ND) | [15] |
| PEG-b-PMVP | Cyclodextrin-ferrocene host-guest | Fundamental | 60 nm (S) | [17] |
| PEG-b-pAsp | PAsp(DET) + β-gal | Protein delivery | 100 nm (V) | [31] |
| PNIPAM-b-PDMAEA | mCherry | Nanostructured film | 20–50 nm (ND) | [88] |
| PEG-b-P2MVP | Fluorescent proteins (SBFP2, mTurquoise2, mEGFP, SYFP2, mKO2, TagRFP, mCherry) | Fluorescent probes | ca. 60 nm (ND) | [91] |
| PAAm-b-PAA | PDMAEMA + Lysozyme | Fundamental | 60–80 nm (E & S) | [95] |
| PEG-b-P2MVP | PAA + Lipase | Fundamental | ca. 50 nm (S) | [96] |
| PEG-b-P2MVP | PAA + Lipase | Fundamental | 40 nm (S) | [97] |
| POEGMA-b-qP4VP | PAA + Organophosphate Hydrolase | Enzymatic reactions in organic solvents | 50–90 nm (S) | [99] |
| PEG-b-pAsp(DET) | Supercharged IgG | Monoclonal antibody delivery | ca. 100 nm (ND) | [101] |
| PEG-b-pAsp(DET) | Supercharged IgG + PAsp (DET) | Monoclonal antibody delivery | 100–200 nm (ND) | [102] |
| POEGMA-b-PAA, POEGMA-b-PCEA, POEGMA-b-PAAVA, POEGMA-b-PAAOA | Lysozyme | Protein delivery | 30–100 nm (ND) | [109] |
| PEG-b-PAPA-b-PAsp | Cytocrome C | Protein delivery | 90 nm (V) | [110] |
| PEG-b-PDMAEMA-b-PnBMA | Cas9 protein | Gene editing | 60–80 nm (ND) | [111] |
| PEG-b-PEI and PEG-b-PLL | Superoxide Dismutase and Catalase | Protein delivery in the central nervous system | 70–170 nm (S) | [112] |
| PEG-b-PEI | Catalase | Protein delivery in the central nervous system | 200 nm (ND) | [113] |
| PEG-b-PLL | Ovalbumin and Catalase + DNA | Vaccine delivery | 130 nm (S) | [114] |
| PEG-b-P(Glu-co-GluPBA) + PEG-b-P(Lys-co-LysCA) | Insulin and Cytochrome C | Protein delivery | 80–120 nm (S) | [115] |
| PEG-PLL and | Myoglobin and supercharged Myoglobin | Protein delivery | 40 nm (ND) | [116] |
| PEG-b-(DMAPA-co-TreA) | Glucose Oxidase and Horseradish Peroxidase | Fundamental | ca. 10 nm (S) | [117] |
| Oxidized Brij | Supercharged Β-glucosidase, Supercharged Glucose Oxidase, Supercharged Horseradish Peroxidase | Enzymatic self-standing films | ND | [118] |
| (POEGMA-r-BP)-b-qP4VP | Alkaline Phosphatase | Enzymatic film | ca. 100 nm (ND) | [119] |
| PEG-b-P2VP | EGFP | Fundamental | 60 nm | [120] |
| PEG-b-pAsp | Trypsin | Fundamental | 70–100 nm (ND) | [121] |
| PEG-b-PAA, PEG-b-PGA, PEG-b-PMA | Trypsin | Fundamental | ND | [122] |
| PEG-b-pAsp | Lysozyme | Fundamental | 55 nm (ND) | [123] |
| Oxidized Brij | Supercharged Myoglobin | Fundamental | ND | [124] |
| Oxidized Brij | Supercharged Glucose Oxidase | Fundamental | ND | [125] |
| 4-nonylphenyl-3-sulfopropyl ether | Supercharged Ferritin | Fundamental | ND | [126] |
| Oxidized Brij | Supercharged Horseradish Peroxidase, Supercharged Glucose oxidase, Supercharged Lipase | Fundamental | ca. 4 nm | [128] |
| (C) | ||||
| Block Copolymer | Oppositely Charged Species | Application | Size (Morphology) | [REF] |
| PEG-b-P2MVP | Metallic complexes | Fundamental | 60 nm (ND) | [18] |
| PEG-b-PLL | DTS + Doxorubicin | Drug delivery | 150 nm (S) | [130] |
| PEG-b-PAA | Doxorubicin; mitoxantrone | Drug delivery | 200 nm (S) | [132] |
| POEGMA-b-PqDMAEMA | Heparin | Drug delivery | <50 nm (S) | [134] |
| PEO-b-PDMAEMA | alkyl phosphobetaines | Drug delivery | ND | [135] |
| PEO-b-PMA | dibucaine, tetracaine, and procaine | Anaesthetic delivery | 100 nm (S) | [136] |
| PEO-b-PAA | Doxorubicin; mitoxantrone | Drug delivery | 200 nm (S) | [137] |
| PEO-b-PAA | Gentamicin and magnetite | Drug loading and contrast agent | 80 nm (S) | [139] |
| POEGMA-co-PBAPMA | Manganese complex + Doxorubicin | Contrast agent or drug delivery | 50–100 nm (S) | [140] |
| PEG-b-PLGA | ICG-PEI complex | Photothermal therapy | 150 nm (S) | [141] |
| PEG-b-PLL | porphyrin dendrimer | Photodynamic therapy | 40–120 nm (S) | [142] |
| PEG-b-bPEI | Chondroitin sulfate | Photodynamic therapy | 150 nm (S) | [143] |
| PEG-b-PLL | Phthalocyanine dendrimer | Photodynamic therapy | 50 nm (S) | [144] |
| PEG-b-PMA | Ca2+, crosslink, remove calcium | Monoclonal antibody carrier | 150 nm (S) | [145] |
| PEG-b-PGlu | oxaliplatin (platinum drugs) | Drug delivery | 30 nm (S) | [149] |
| PEG-b-PMA | Dioxadet | Drug delivery | 120 nm (S) | [152] |
| PEG-b-PM2VP | Carboxylated polyfluorene-Doxorubicin complex | Drug delivery | 150 nm (E) | [153] |
| PEG-b-PLys | Zn-porphyrin dendrimer | Photodynamic therapy | 50 nm (S) | [156] |
| PEG-b-PMA | Doxorubicin | Drug delivery | ND | [159] |
| PEG-b-PMA | Dye or isotope labelled PAH | Diffusional nanoprobe | 10–20 nm (S) | [168,169] |
| PEG-b-PM2VP | Manganese dipicolinic acid complexes | Contrast agent | 25 nm (S) | [170] |
| (D) | ||||
| Block Copolymer | Oppositely Charged Species | Application | Size (Morphology) | [REF] |
| PEG-b-PM2VP | Lanthanide complexes | Contrast agent | 20 nm (S) | [171] |
| PEG-b-PM2VP-b-PS | Europium complexes | Ion-specific sensor | 90 nm (S) | [172] |
| PEG-b-PDMAEMA | Eu polyoxometalates | Labelling and imaging | 80 nm (S) | [173] |
| PEG-b-PAA | Au precursor | Templated nanoparticle formation | 100 nm (S) | [174] |
| PVA-b-PAA | PM4VP | Ice growth inhibitor | 100 nm (S) | [176] |
| PEG-b-PM2VP | cPF-alt-PBT | Mechanochromic sensor | 60 nm (S) | [179] |
| PEG-b-PAA | Au precursor | Gold-silver core-shell nanoparticle formation | 50–200 nm (S) | [181] |
| PEG-b-PVP | PtCl42- | Mesoporous Pt particle formation | 25 nm (S) | [182] |
| PEG-b-PAA | Au precursor | Catalysis | 10 nm (S) | [183] |
| PAAm-b-PAA | Mg2+ and Al3+ | Formation of stable layered metal oxides | 20 nm (S) | [185] |
| PAAm-b-PAA | Cu2+ and Al3+ | Formation of stable layered metal oxides | 50 nm (S) | [186] |
| PEG-b-PAA | Fe2O3 | Porous nanocarrier, e.g., for drug molecules | 200 nm (S) | [187] |
| PEG-b-PMA | (NH4)2Ce(NO3)6 | CeO2 production for catalysis | ND | [188] |
| PEG-b-PAA | Ru3+ | Formation of RuO2 as supercapacitor | ND | [189] |
| PAAm-b-PAA | Zn2+ | Production of ZnS for optoelectronics | ND | [190] |
| PEG-PSCI | Cd2+ | Production of stable CdS quantum dots | ND | [192] |
| PEG-b-PAMPS | Chitosan | Nanogel templating and formation | 50 nm (S) | [197] |
| PEG-b-PAPTAC or PNIPAM-b-PAPTAC | Hyaluronic acid or alginate | Nanogel templating and formation | 50–300 nm (S) | [198] |
| PEG-b-PAETB | PEG-b-PCETB | Reduction of protein adsorption on C3M-coated substrate | 30 nm (S) | [205] |
| PPEGMA-b-PAA | PM2VP | Reduction of protein adsorption on silica and PS | 200 nm (multi-micelle aggregates) | [207] |
| PmDOPA-PDMAEMA | PSS | Antimicrobial film formation by reduction of silver ions in the complexes | 90 nm (S) | [209] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magana, J.R.; Sproncken, C.C.M.; Voets, I.K. On Complex Coacervate Core Micelles: Structure-Function Perspectives. Polymers 2020, 12, 1953. https://doi.org/10.3390/polym12091953
Magana JR, Sproncken CCM, Voets IK. On Complex Coacervate Core Micelles: Structure-Function Perspectives. Polymers. 2020; 12(9):1953. https://doi.org/10.3390/polym12091953
Chicago/Turabian StyleMagana, Jose Rodrigo, Christian C. M. Sproncken, and Ilja K. Voets. 2020. "On Complex Coacervate Core Micelles: Structure-Function Perspectives" Polymers 12, no. 9: 1953. https://doi.org/10.3390/polym12091953
APA StyleMagana, J. R., Sproncken, C. C. M., & Voets, I. K. (2020). On Complex Coacervate Core Micelles: Structure-Function Perspectives. Polymers, 12(9), 1953. https://doi.org/10.3390/polym12091953