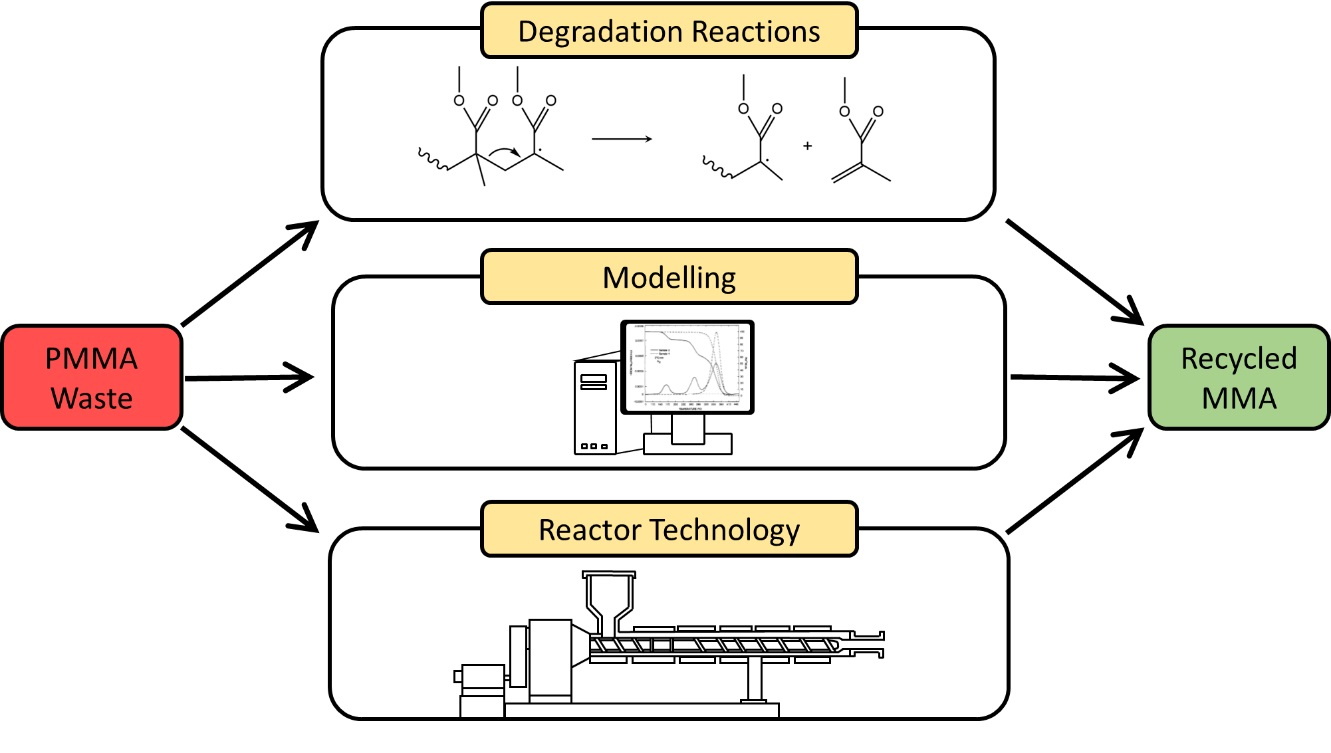
Progress in Reaction Mechanisms and Reactor Technologies for Thermochemical Recycling of Poly(methyl methacrylate)

 , , , ,
, , , , 
 and
and 
Abstract

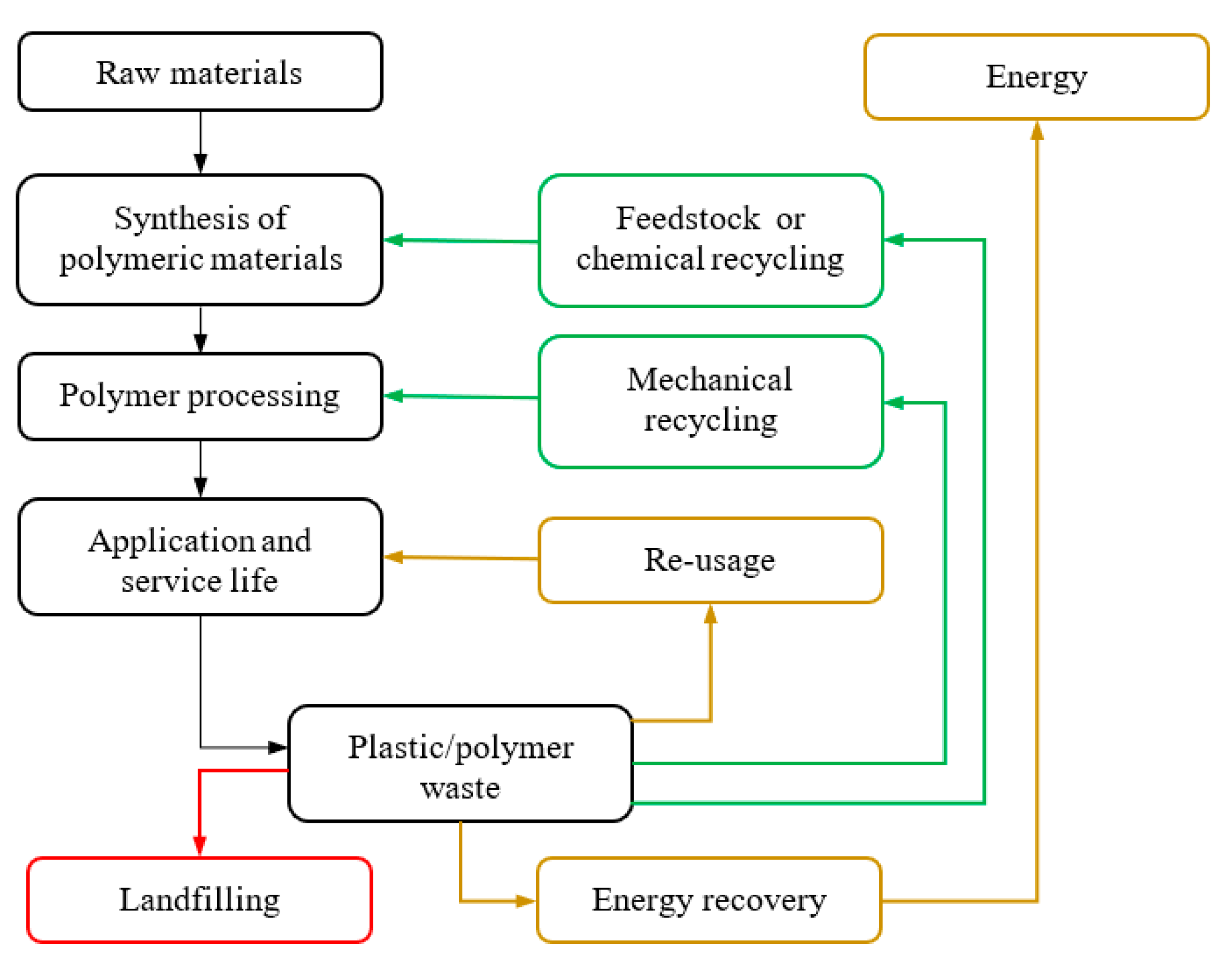
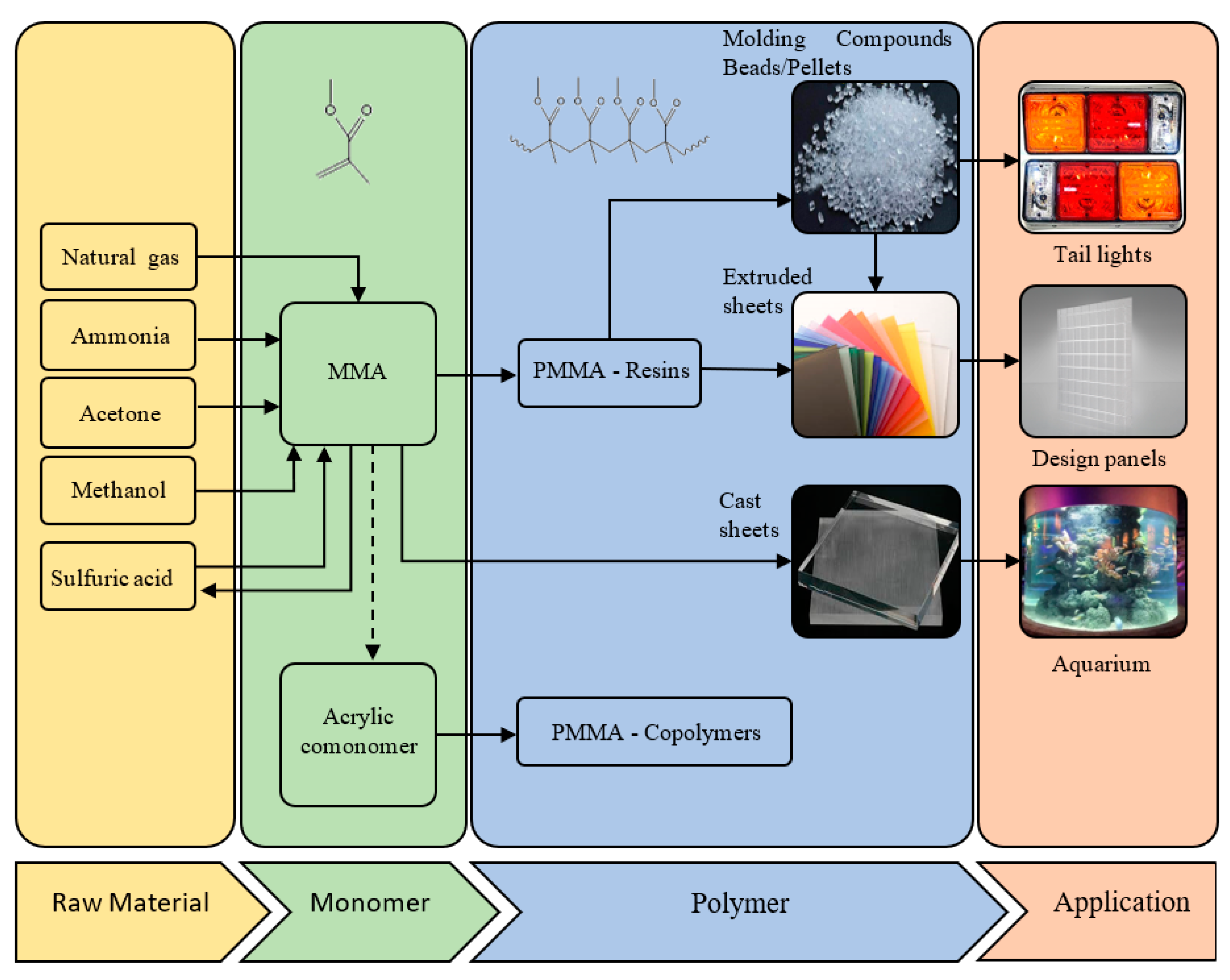
1. Introduction
2. Radical and Anionic Polymerization Reaction Mechanisms for PMMA Synthesis
2.1. Radical Polymerization Mechanism
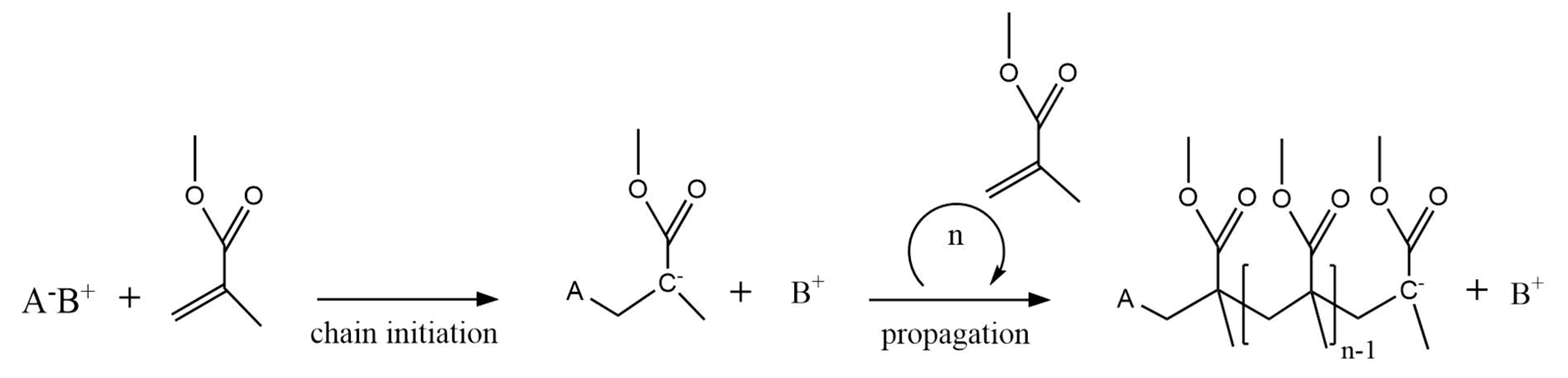
2.2. Anionic Polymerization Mechanism
3. Radical Reaction Mechanisms for Thermal PMMA Chemical Recycling
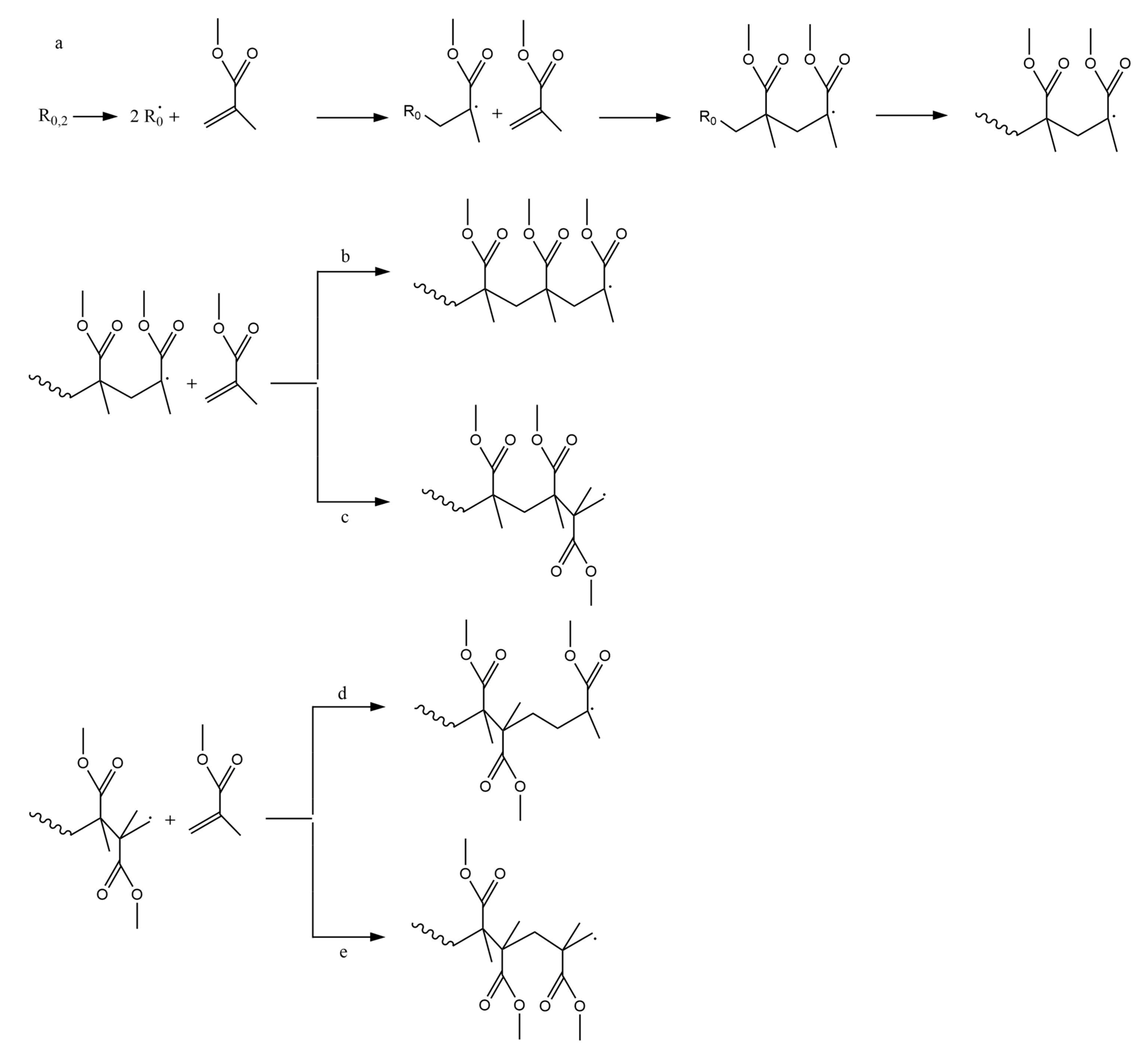
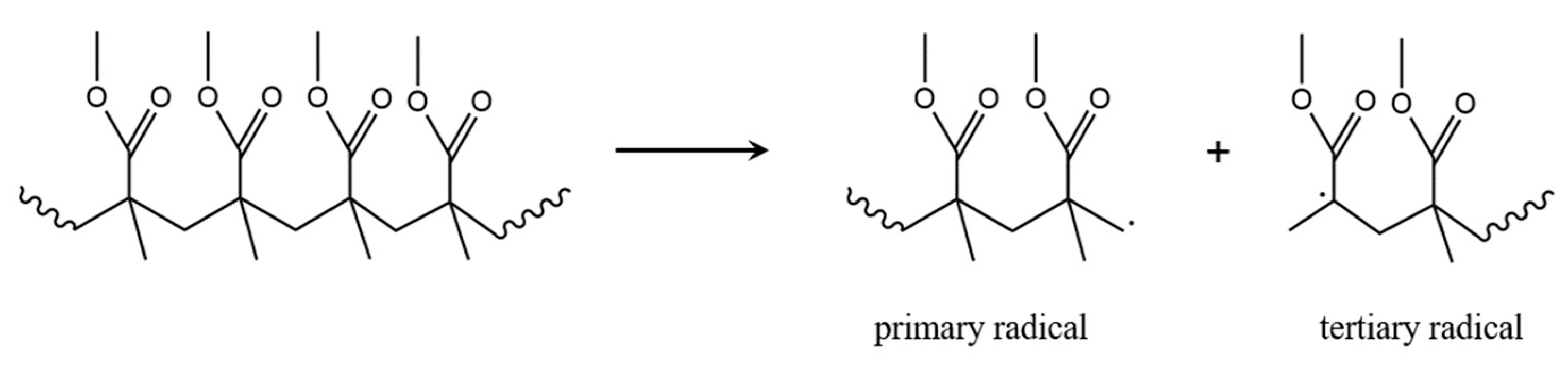
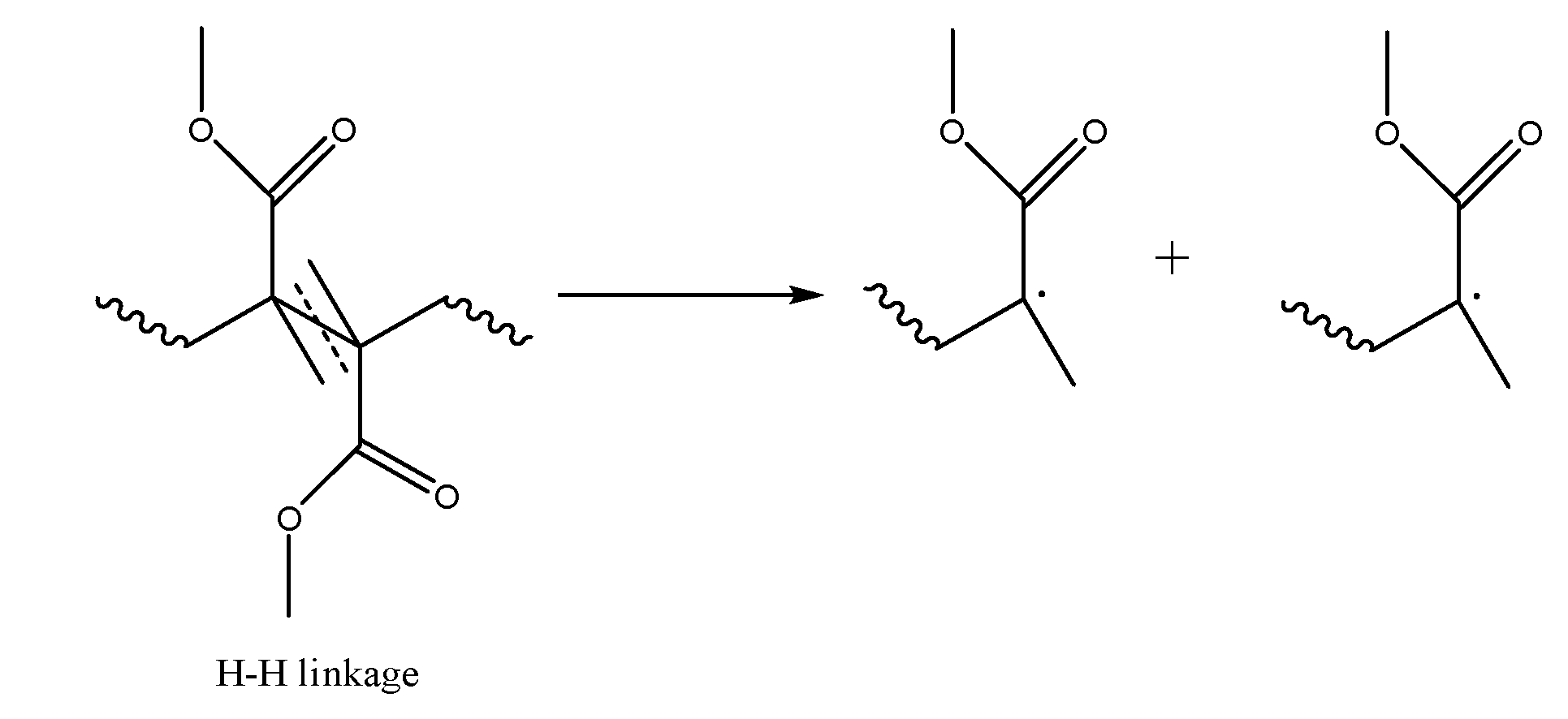
3.1. Initiation by Head-Tail Fission
3.2. Initiation by Head-Head Fission
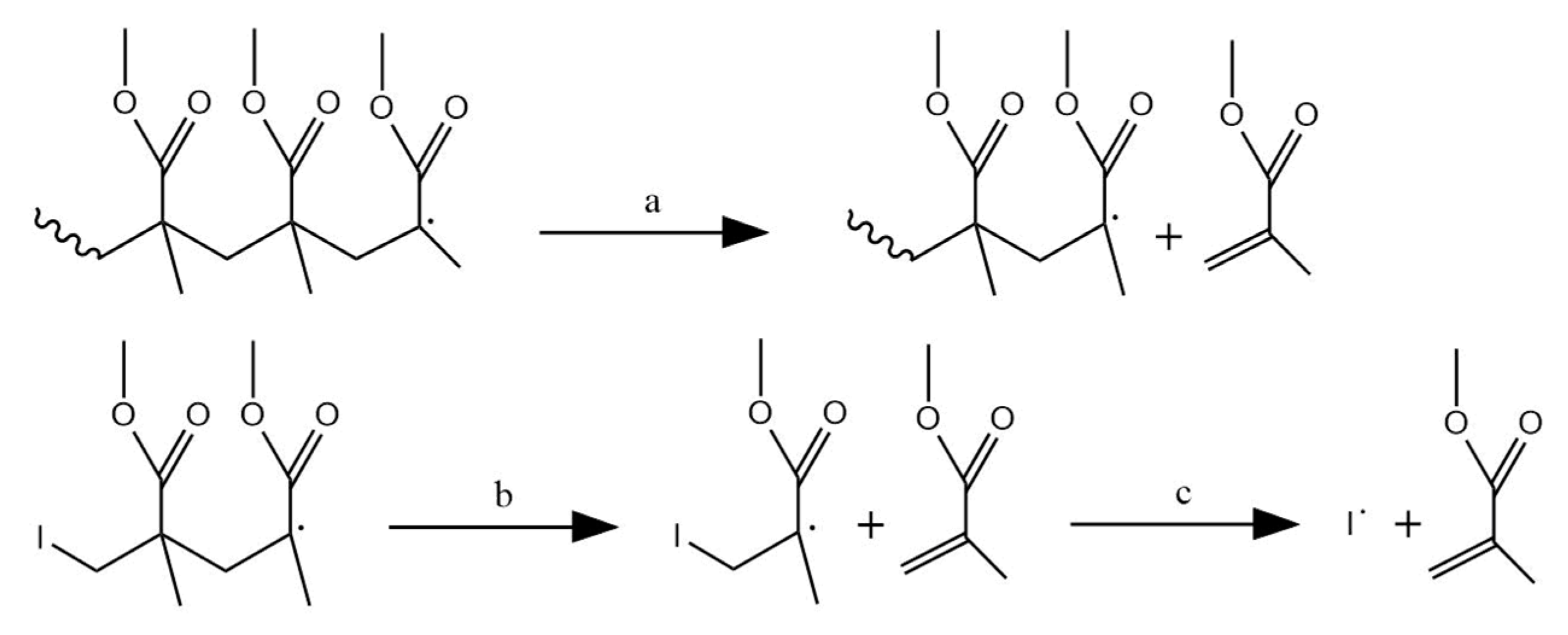
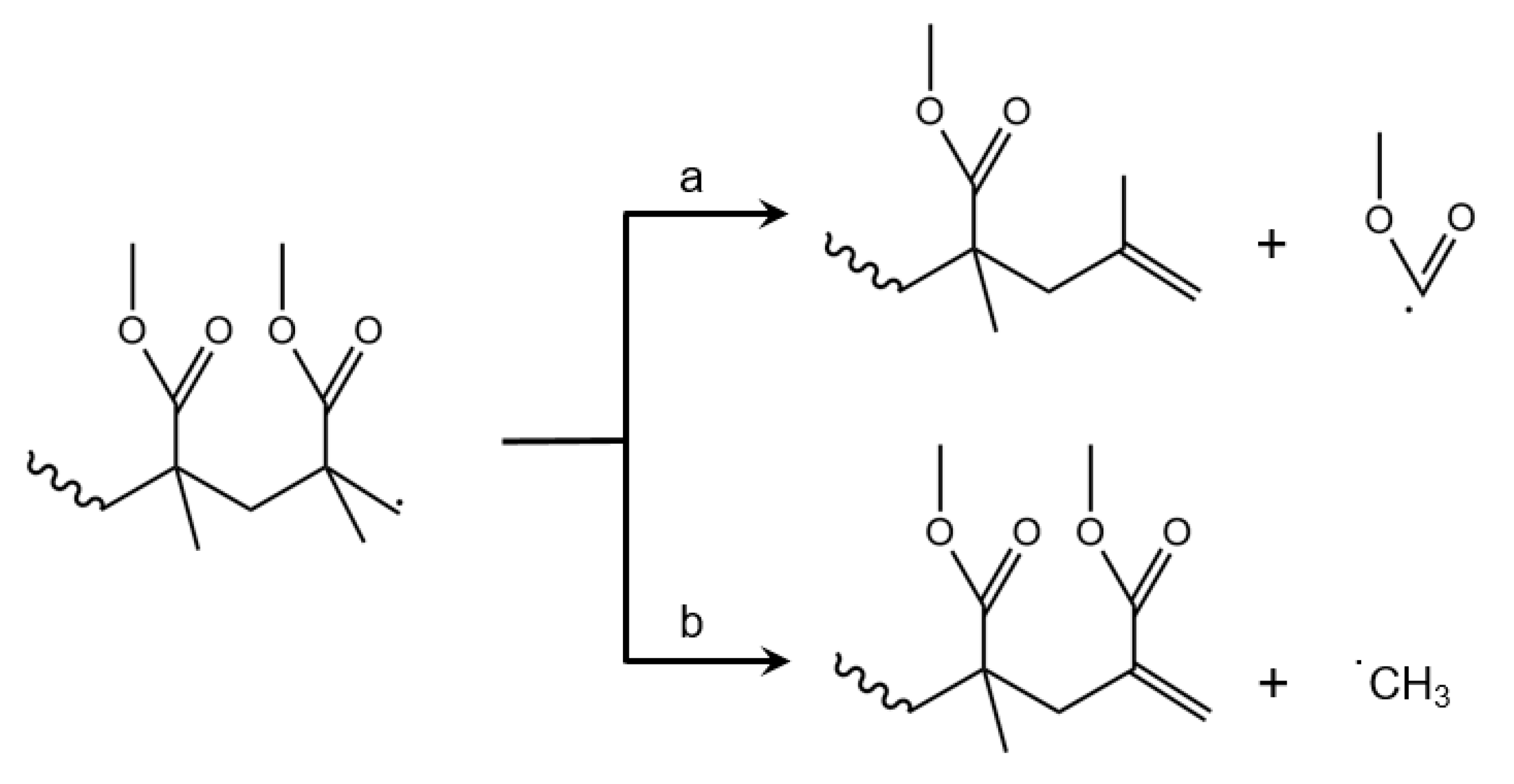
3.3. Initiation by Side-Group Fission
3.4. Initialization Involving Transfer Reactions or Interactions
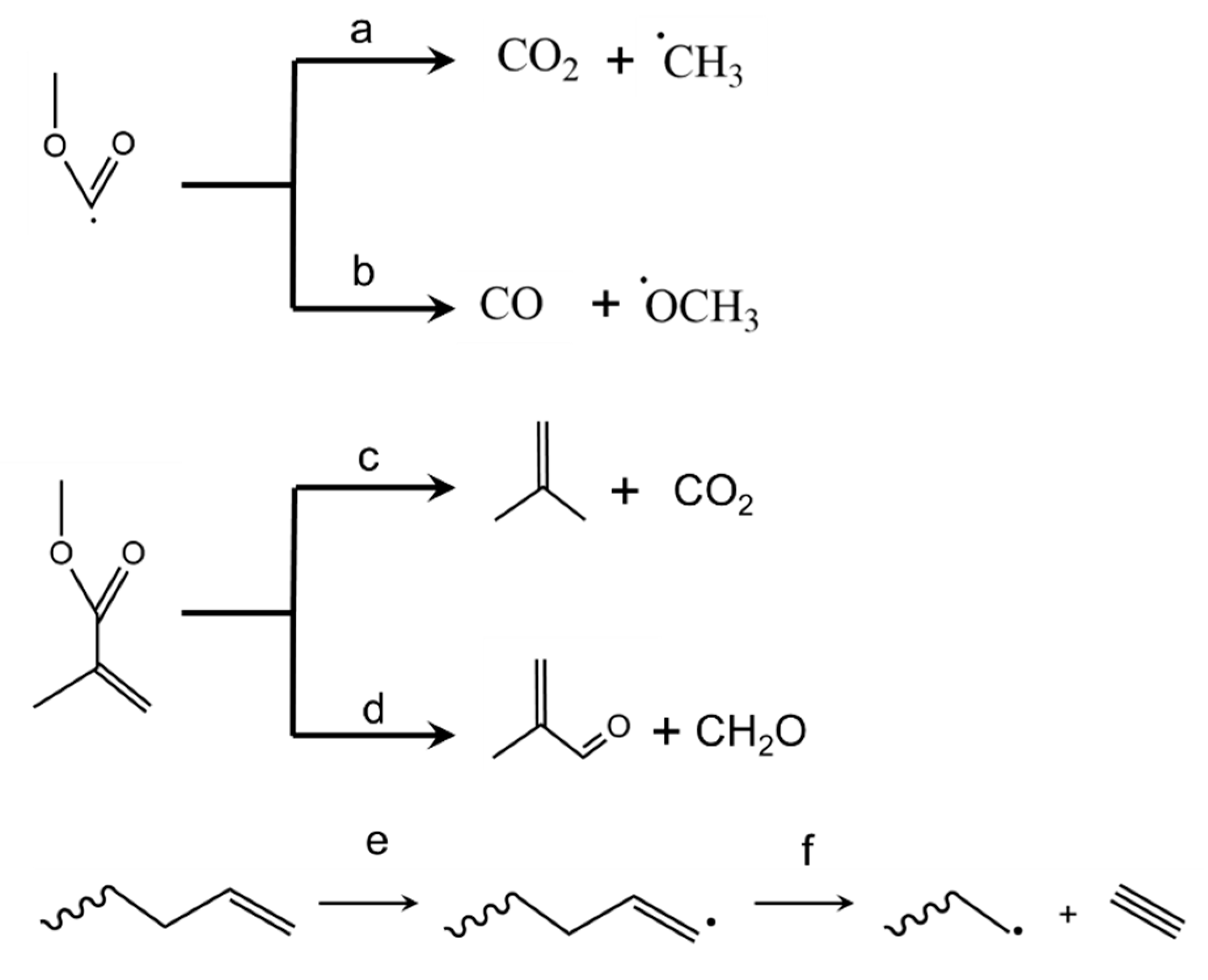
3.5. Depropagation through End-Chain β-Scission
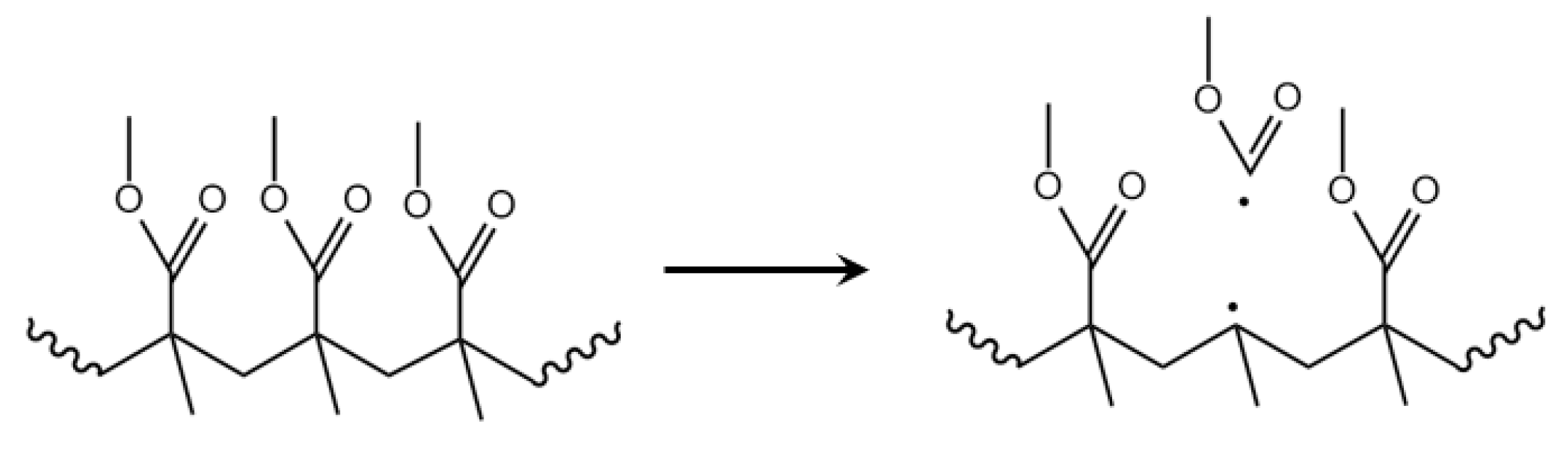
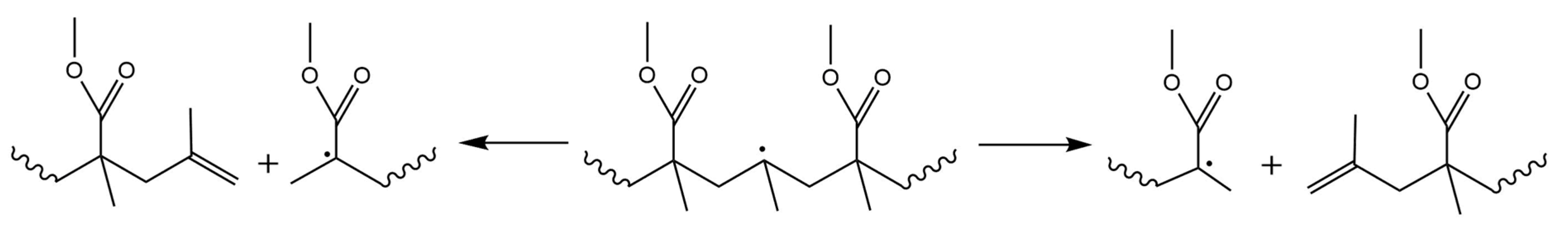
3.6. Depropagation through Mid-Chain β-Scission
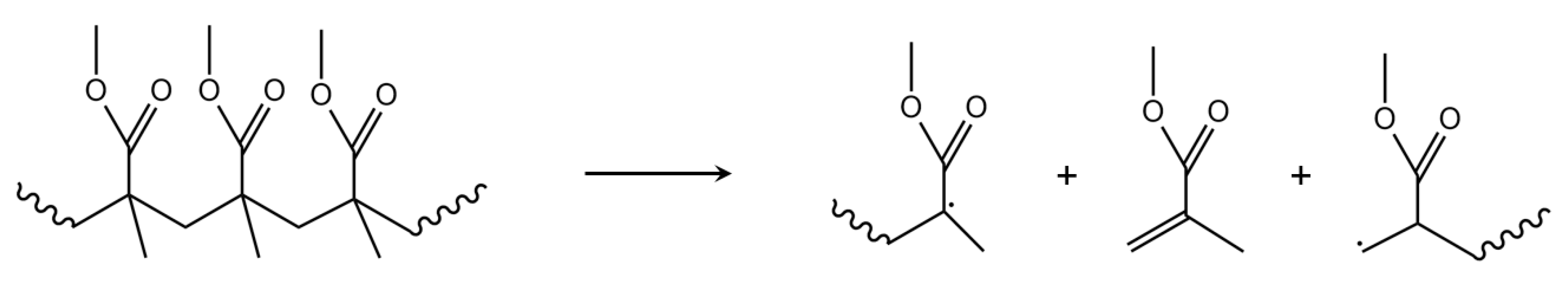
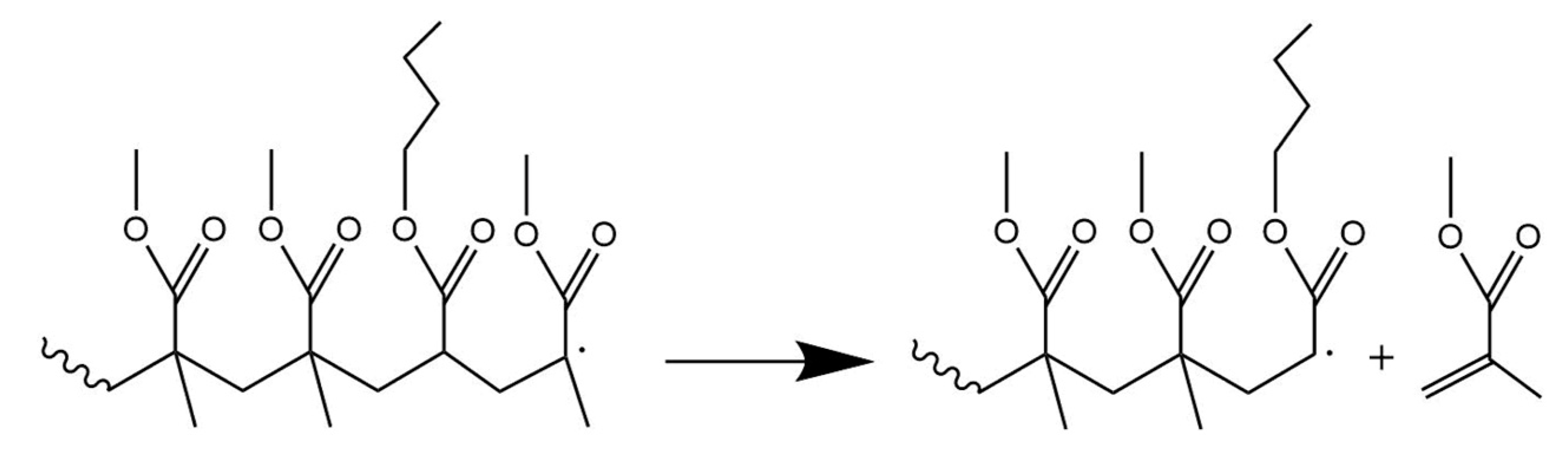
3.7. Depropagation through Side Group β-Scission
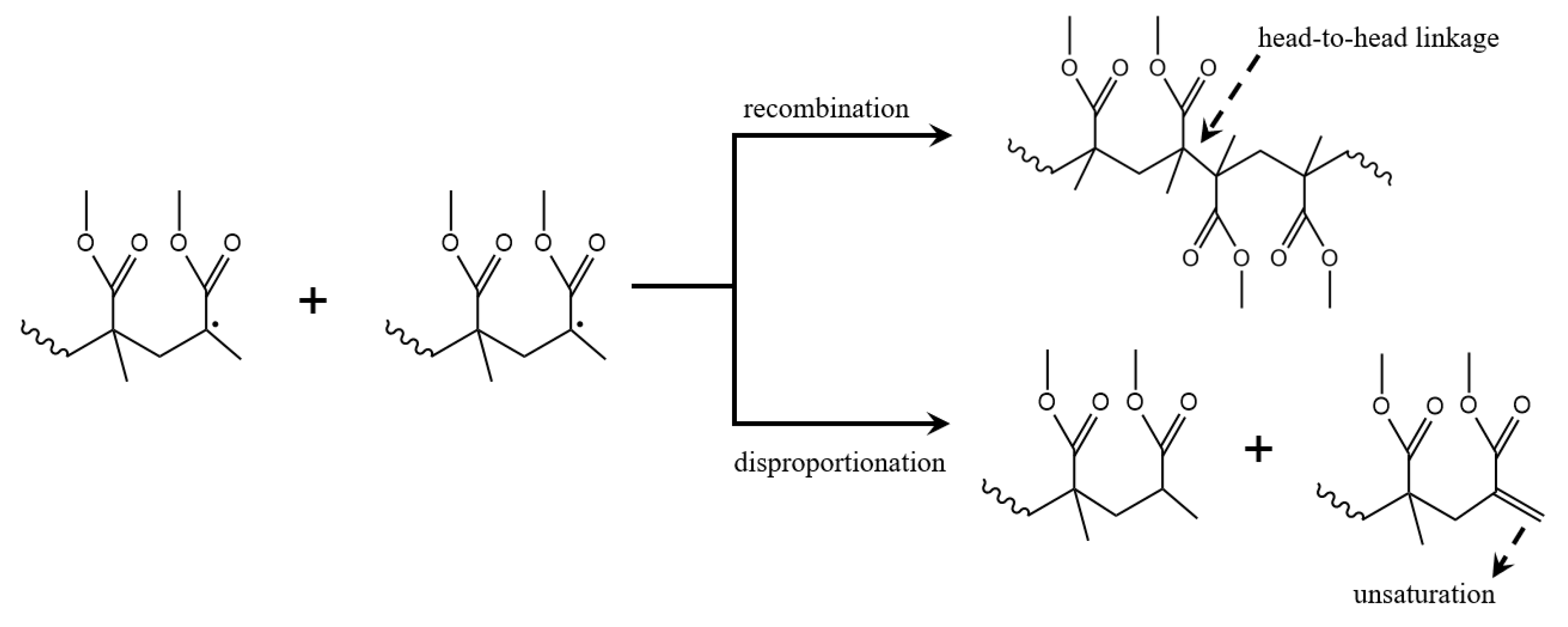
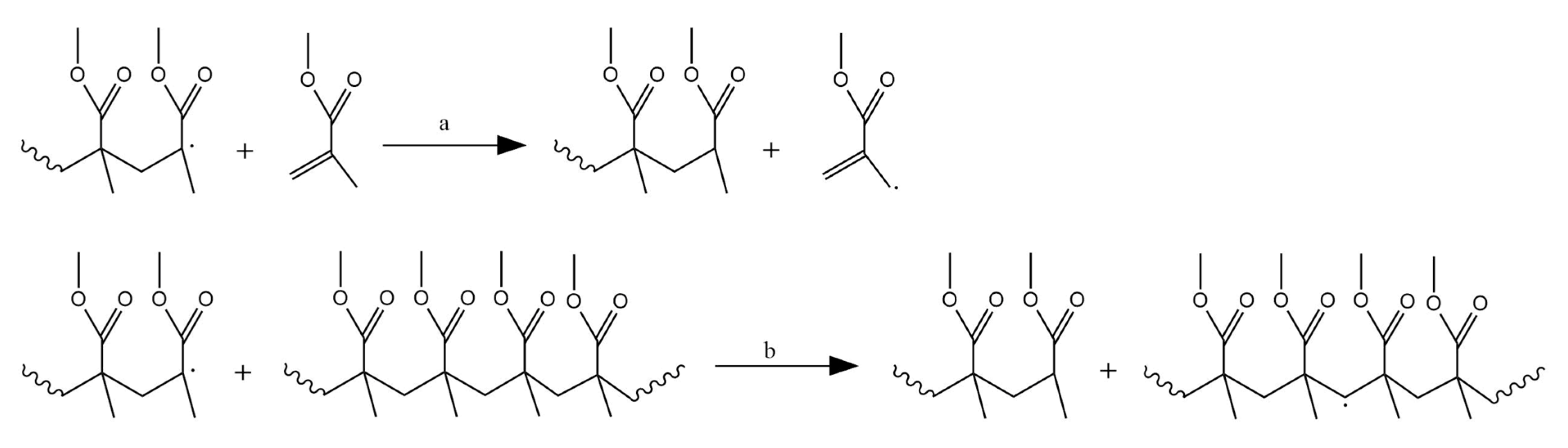
3.8. Termination Reactions
3.9. Side Reactions Blocking MMA Formation
4. From Degradation Reaction Mechanisms for PMMA to MMA-Rich Copolymers with Additives
5. Lab Scale Kinetic Modeling Descriptions for Chemical Recycling of PMMA and Related Copolymers
5.1. Global Conversion Based Models
5.1.1. Differential and Integrated Overall Rate Laws
5.1.2. Example of Thermal Degradation Process with Superposition of Overall Degradation Steps
5.1.3. Single Versus Multiphase Kinetics
5.2. Global Chain Length Based Models
5.3. Elementary Reaction Step Based Models
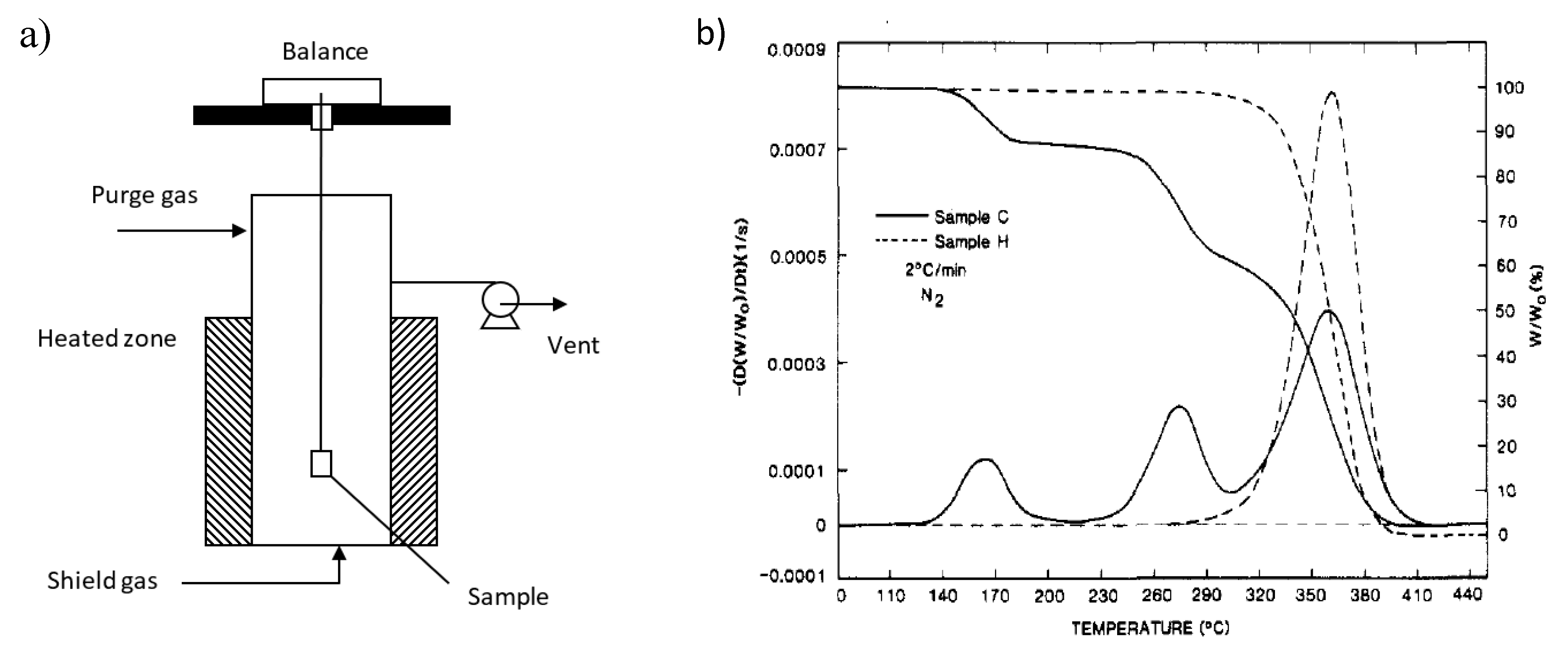
6. Reactor Technologies
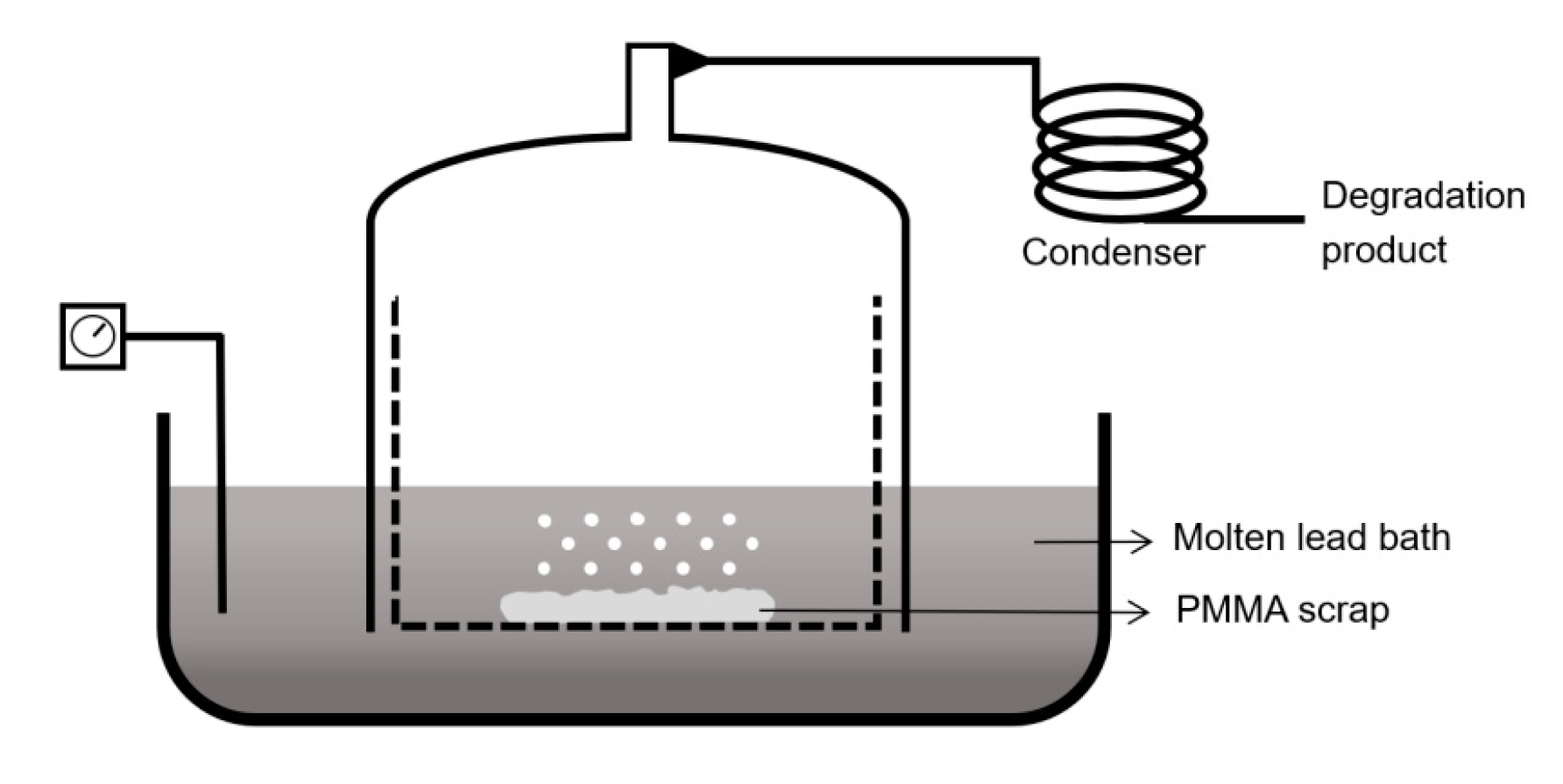
6.1. Molten Metal-Bath Reactor
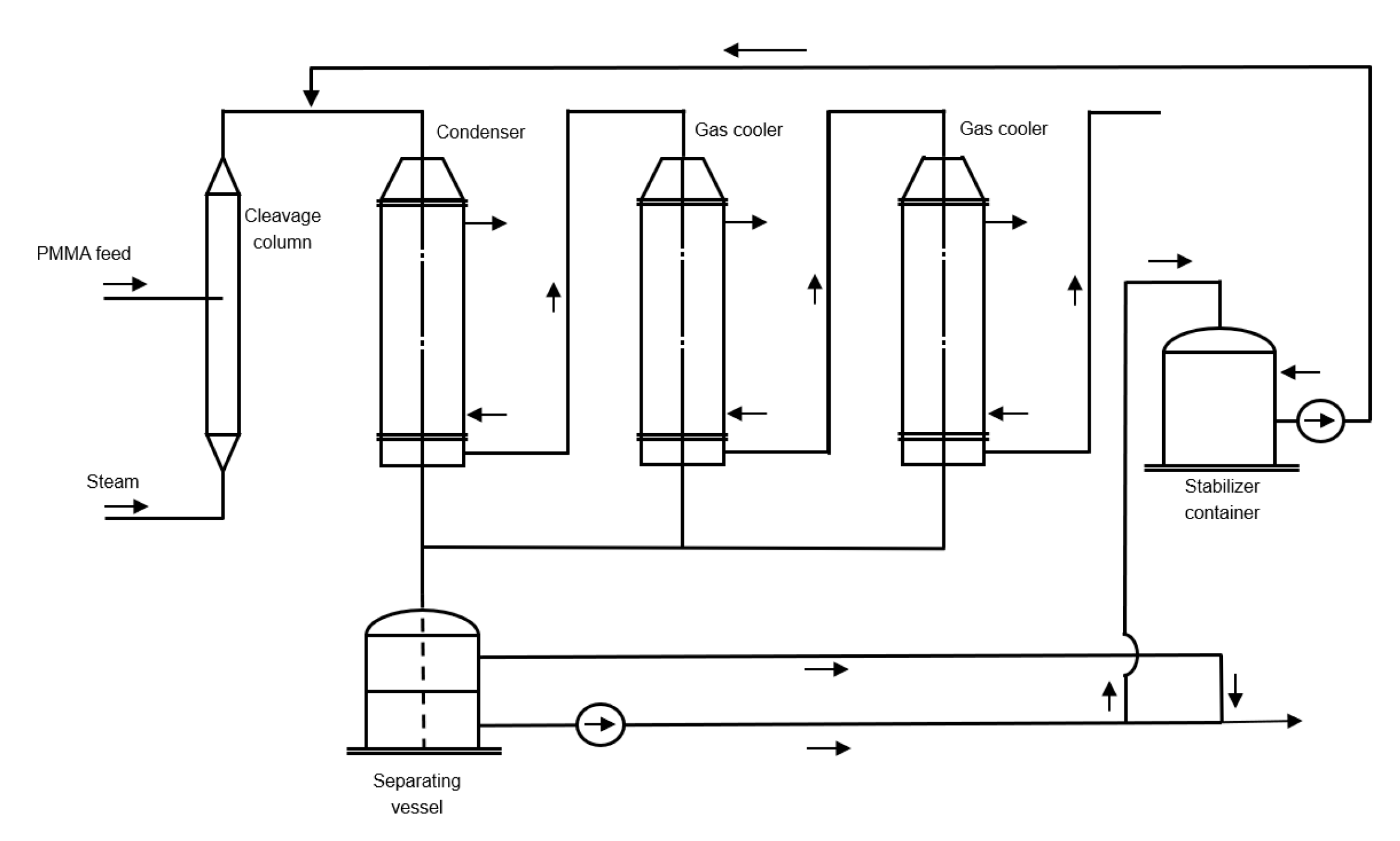
6.2. Counter Current Reactor
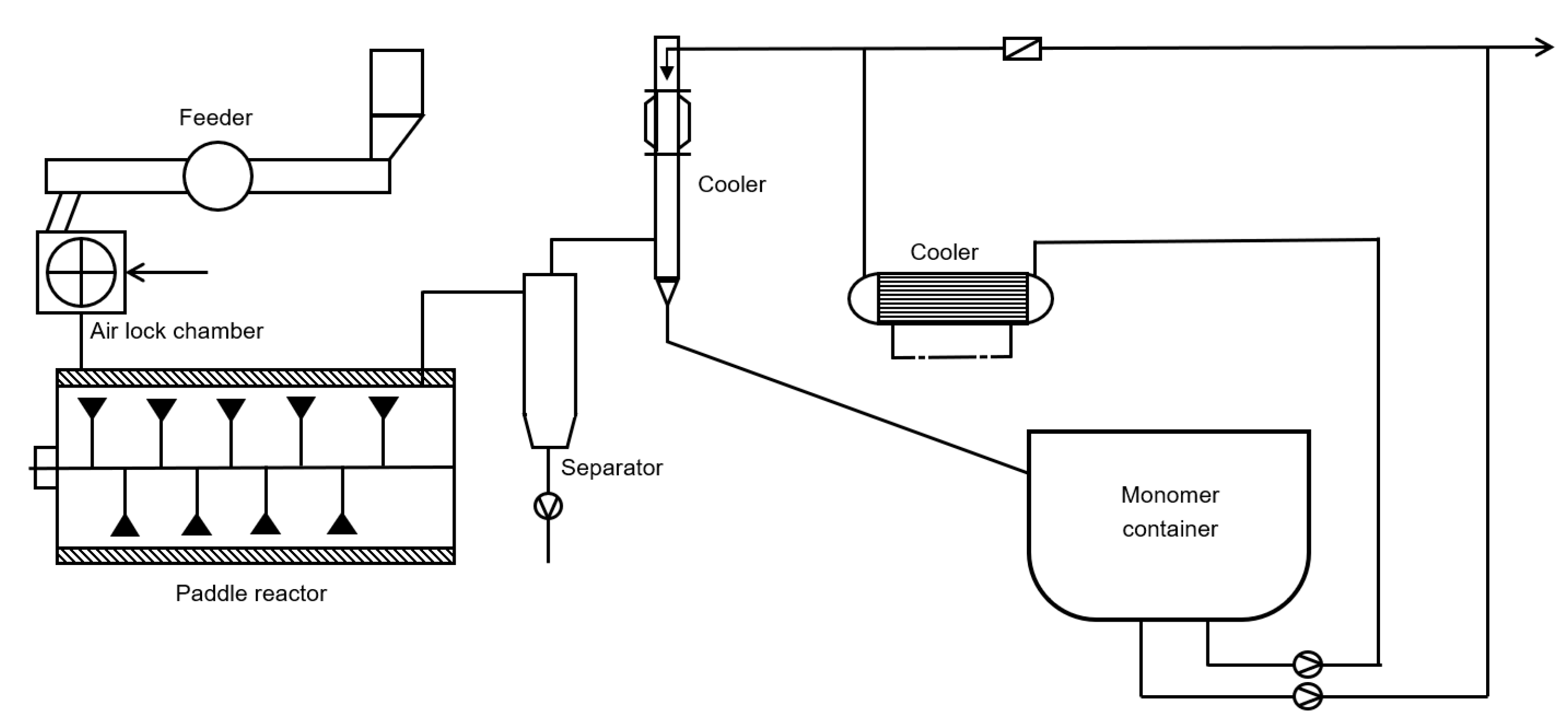
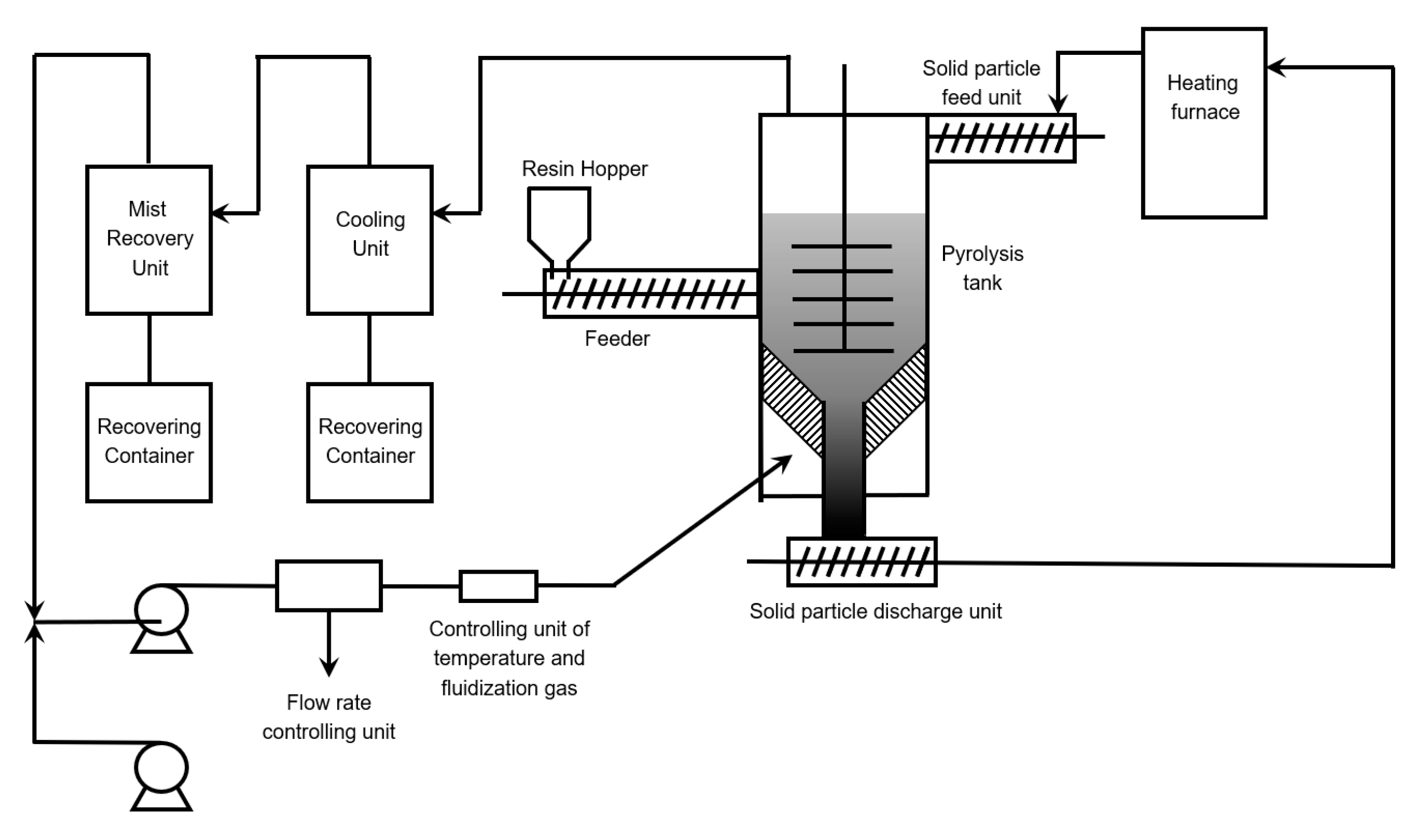
6.3. Paddle Reactor
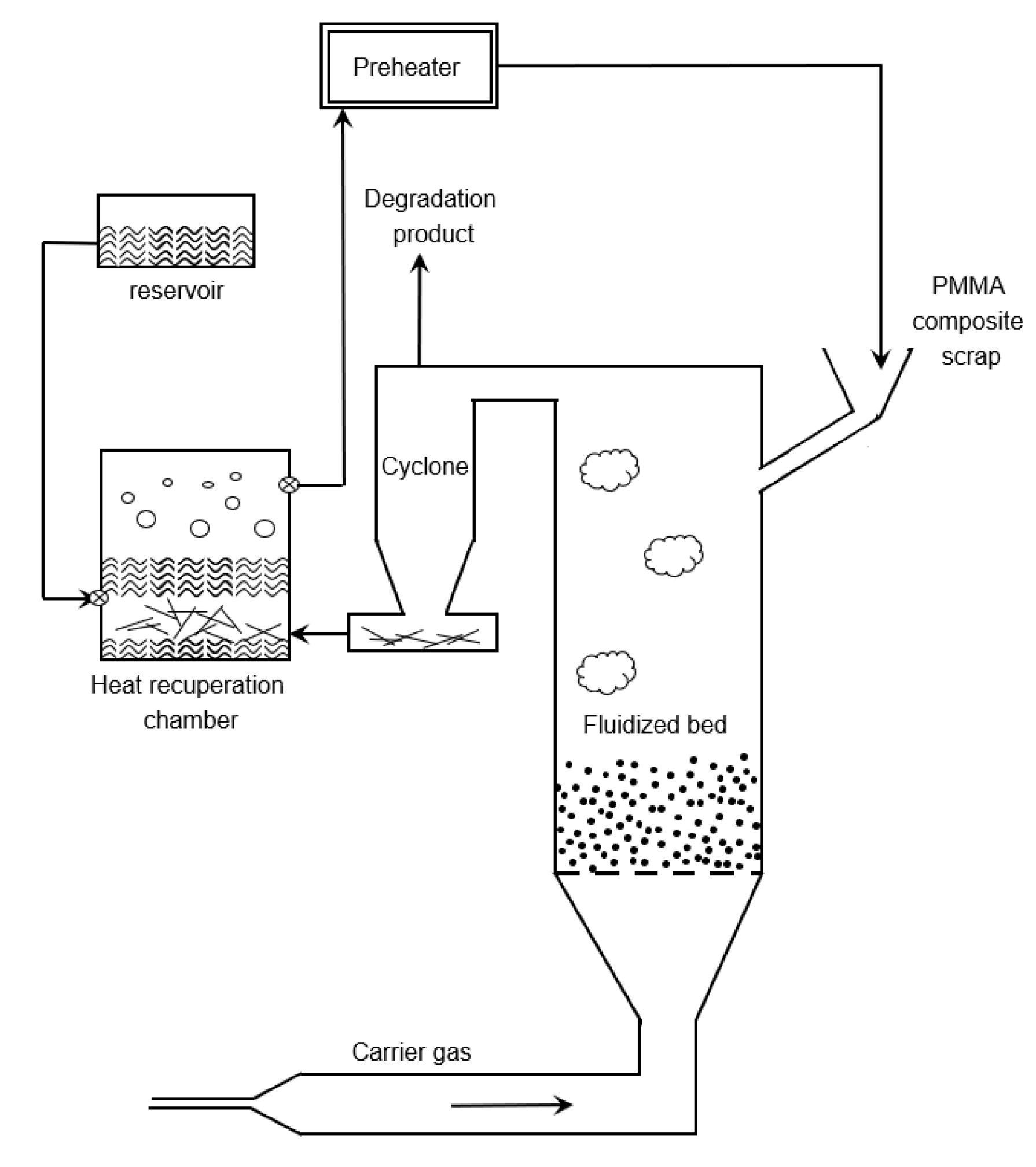
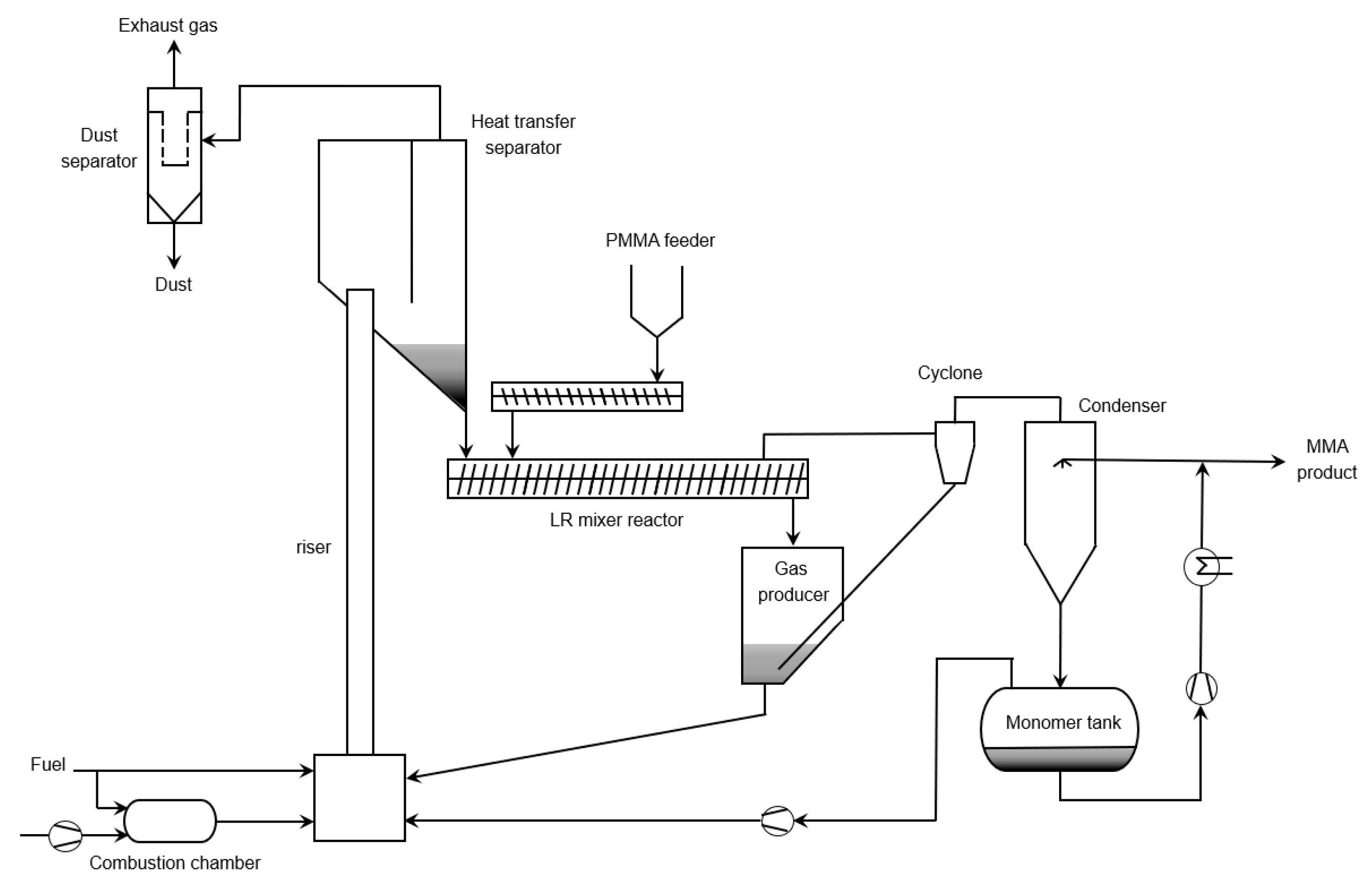
6.4. Fluidized Bed Reactor
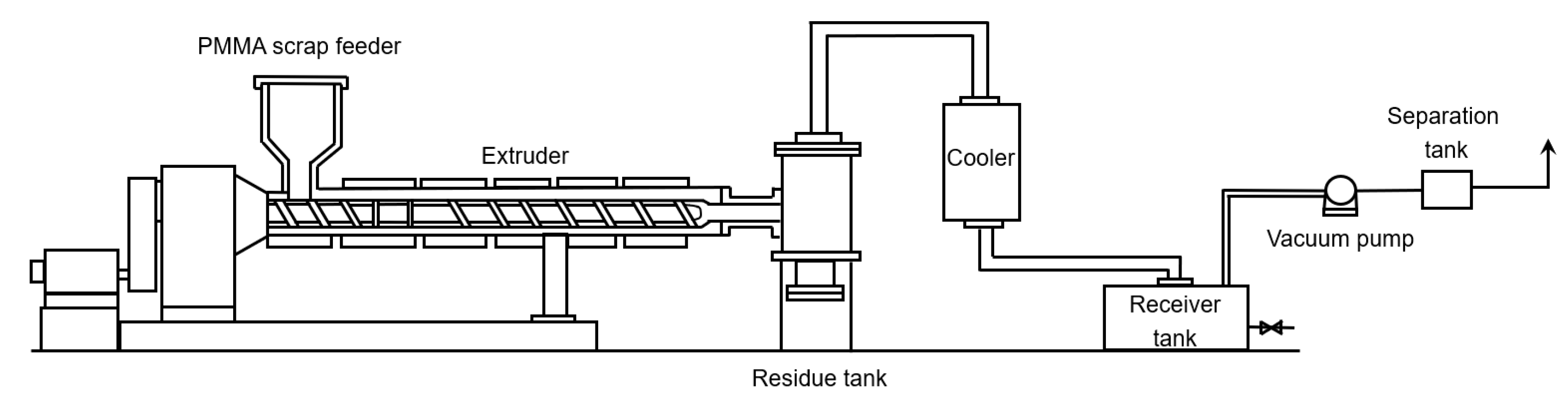
6.5. Extrusion Based Reactors
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- European Commission. A European Strategy for Plastics in a Circular Economy; European Commission, Ed.; European Commission: New York, NY, USA, 2015; p. 24. [Google Scholar]
- PlasticsEurope. PlasticsEurope Annual Review 2017–2018; PlasticsEurope AISBL, Ed.; PlasticsEurope: Brussels, Belgium, 2018. [Google Scholar]
- Vilaplana, F.; Karlsson, S. Quality concepts for the improved use of recycled polymeric materials: A review. Macromol. Mater. Eng. 2008, 293, 274–297. [Google Scholar] [CrossRef]
- Al-Salem, S.; Lettieri, P.; Baeyens, J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manag. 2009, 29, 2625–2643. [Google Scholar] [CrossRef] [PubMed]
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manag. 2017, 69, 24–58. [Google Scholar] [CrossRef] [PubMed]
- Broadbelt, L.J.; Stark, S.M.; Klein, M.T. Computer generated pyrolysis modeling: On-the-fly generation of species, reactions, and rates. Ind. Eng. Chem. Res. 1994, 33, 790–799. [Google Scholar] [CrossRef]
- Lopez, G.; Artetxe, M.; Amutio, M.; Elordi, G.; Aguado, R.; Olazar, M.; Bilbao, J. Recycling poly-(methyl methacrylate) by pyrolysis in a conical spouted bed reactor. Chem. Eng. Process. Process Intensif. 2010, 49, 1089–1094. [Google Scholar] [CrossRef]
- López, G.; Olazar, M.; Aguado, R.; Bilbao, J. Continuous pyrolysis of waste tyres in a conical spouted bed reactor. Fuel 2010, 89, 1946–1952. [Google Scholar] [CrossRef]
- Hernández-Ortiz, J.C.; Van Steenberge, P.H.; Duchateau, J.N.; Toloza, C.; Schreurs, F.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. A two-phase stochastic model to describe mass transport and kinetics during reactive processing of polyolefins. Chem. Eng. J. 2019, 377, 119980. [Google Scholar] [CrossRef]
- Hernández-Ortiz, J.C.; Van Steenberge, P.H.; Reyniers, M.F.; Marin, G.B.; D’hooge, D.R.; Duchateau, J.N.; Remerie, K.; Toloza, C.; Vaz, A.L.; Schreurs, F. Modeling the reaction event history and microstructure of individual macrospecies in postpolymerization modification. AIChE J. 2017, 63, 4944–4961. [Google Scholar] [CrossRef]
- Yoshioka, T.; Grause, G.; Eger, C.; Kaminsky, W.; Okuwaki, A. Pyrolysis of poly (ethylene terephthalate) in a fluidised bed plant. Polym. Degrad. Stab. 2004, 86, 499–504. [Google Scholar] [CrossRef]
- Al-Salem, S.; Lettieri, P.; Baeyens, J. The valorization of plastic solid waste (PSW) by primary to quaternary routes: From re-use to energy and chemicals. Prog. Energy Combust. Sci. 2010, 36, 103–129. [Google Scholar] [CrossRef]
- Gigmes, D.; Van Steenberge, P.H.; Siri, D.; D’hooge, D.R.; Guillaneuf, Y.; Lefay, C. Simulation of the degradation of cyclic ketene acetal and vinyl-based copolymers synthesized via a radical process: Influence of the reactivity ratios on the degradability properties. Macromol. Rapid Commun. 2018, 39, 1800193. [Google Scholar] [CrossRef]
- Agarwal, S.; Kumar, R.; Kissel, T.; Reul, R. Synthesis of degradable materials based on caprolactone and vinyl acetate units using radical chemistry. Polym. J. 2009, 41, 650. [Google Scholar] [CrossRef]
- Undin, J.; Finne-Wistrand, A.; Albertsson, A.-C. Adjustable degradation properties and biocompatibility of amorphous and functional poly (ester-acrylate)-based materials. Biomacromolecules 2014, 15, 2800–2807. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Lenz, R.W. Synthesis, characterization, and hydrolytic degradation of copolymers of 2-methylene-1, 3-dioxepane with ethylene and with styrene. J. Environ. Polym. Degrad. 1998, 6, 23–29. [Google Scholar] [CrossRef]
- Guimard, N.K.; Oehlenschlaeger, K.K.; Zhou, J.; Hilf, S.; Schmidt, F.G.; Barner-Kowollik, C. Current trends in the field of self-healing materials. Macromol. Chem. Phys. 2012, 213, 131–143. [Google Scholar] [CrossRef]
- Froimowicz, P.; Frey, H.; Landfester, K. Towards the generation of self-healing materials by means of a reversible photo-induced approach. Macromol. Rapid Commun. 2011, 32, 468–473. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Hirao, M.; Ookubo, T.; Sasaki, A. Design of recycling system for poly(methyl methacrylate) (PMMA). Part 1: Recycling scenario analysis. Int. J. Life Cycle Assess. 2014, 19, 120–129. [Google Scholar] [CrossRef]
- Albrecht, K.; Stickler, M.; Rhein, T. Polymethacrylates. Ullmann’s Encycl. Ind. Chem. 2013. [Google Scholar] [CrossRef]
- Hansjörg Sinn, H.; Kaminsky, W.; Janning, J. Verarbeitung von kunststoffmüll und altreifen zu chemie-rohstoffen, besonders durch pyrolyse. Angew. Chem. 1976, 88, 737–750. [Google Scholar] [CrossRef]
- Kaminsky, W.; Sinn, H.; Janning, J. Technische prototypen für die altreifen-und kunststoff-pyrolyse. Chem. Ing. Tech. 1979, 51, 419–429. [Google Scholar] [CrossRef]
- Kaminsky, W. Pyrolysis of plastic waste and scrap tyres in a fluid bed reactor. Resour. Recovery Conserv. 1980, 5, 205–216. [Google Scholar] [CrossRef]
- CEFIC. Methyl Methacrylate (MMA): Eco-Profiles and Environmental Product Declarations of the European Plastics Manufacturers; CEFIC: Brussels, Belgium, 2013. [Google Scholar]
- Heijden, S.V.D.; Dubois, J.-L.; Bossard, M. New Innovative Process by MMAtwo for Recycling End-of-Life PMMA Waste. 2018. Available online: https://www.mmatwo.eu/wp-content/uploads/2019/01/Press-release-MMAtwo_ENG_VF.pdf (accessed on 26 July 2020).
- Dubois, J.-L.; New Innovative Process for Recycling End-of-Life PMMA Waste. MMAtwo Second Generation Methyl Methacrylate. 2019. Available online: https://www.researchgate.net/publication/341655536_New_innovative_process_for_recycling_end-of-life_PMMA_waste_MMAtwo_second_generation_Methyl_Methacrylate (accessed on 26 July 2020).
- Kashiwagi, T.; Inabi, A.; Hamins, A. Behavior of primary radicals during thermal degradation of poly (methyl methacrylate). Polym. Degrad. Stab. 1989, 26, 161–184. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Inaba, A.; Brown, J.E.; Hatada, K.; Kitayama, T.; Masuda, E. Effects of weak linkages on the thermal and oxidative degradation of poly (methyl methacrylates). Macromolecules 1986, 19, 2160–2168. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Hirata, T.; Brown, J.E. Thermal and oxidative degradation of poly (methyl methacrylate) molecular weight. Macromolecules 1985, 18, 131–138. [Google Scholar] [CrossRef]
- Manring, L.E. Thermal degradation of poly (methyl methacrylate). 2. Vinyl-terminated polymer. Macromolecules 1989, 22, 2673–2677. [Google Scholar] [CrossRef]
- Manring, L.E.; Sogah, D.Y.; Cohen, G.M. Thermal degradation of poly (methyl methacrylate). 3. Polymer with head-to-head linkages. Macromolecules 1989, 22, 4652–4654. [Google Scholar] [CrossRef]
- Manring, L.E. Thermal degradation of poly (methyl methacrylate). 4. Random side-group scission. Macromolecules 1991, 24, 3304–3309. [Google Scholar] [CrossRef]
- Manring, L.E. Thermal degradation of saturated poly (methyl methacrylate). Macromolecules 1988, 21, 528–530. [Google Scholar] [CrossRef]
- Ali, U.; Karim, K.J.B.A.; Buang, N.A. A review of the properties and applications of poly (methyl methacrylate)(PMMA). Polym. Rev. 2015, 55, 678–705. [Google Scholar] [CrossRef]
- Cefic, T.M.S.G.M.O. Frequent Asked Questions. Available online: https://www.pmma-online.eu/pmma-science/faq/ (accessed on 2 June 2020).
- Mahboub, M.J.D.; Dubois, J.-L.; Cavani, F.; Rostamizadeh, M.; Patience, G.S. Catalysis for the synthesis of methacrylic acid and methyl methacrylate. Chem. Soc. Rev. 2018, 47, 7703–7738. [Google Scholar] [CrossRef]
- Gail, E.; Gos, S.; Kulzer, R.; Lorösch, J.; Rubo, A.; Sauer, M.; Kellens, R.; Reddy, J.; Steier, N.; Hasenpusch, W. Cyano compounds, inorganic. Ullmann’s Encycl. Ind. Chem. 2000, 10, 38. [Google Scholar]
- Wang, Z. Andrussow process. Compr. Org. Name React. Reag. 2010, 1, 80–82. [Google Scholar] [CrossRef]
- Ahn, S.; White, J. Influence of low molecular weight additives on die extrusion rheometer flow of thermoplastic melts. Int. Polym. Process. 2003, 18, 243–251. [Google Scholar] [CrossRef]
- PMMA/Acrylic Sustainable Solutions. Available online: https://www.pmma-online.eu/pmma-science/types-and-production/ (accessed on 11 February 2020).
- Fierens, S.K.; Van Steenberge, P.H.; Vermeire, F.; Reyniers, M.F.; Marin, G.B.; D’hooge, D.R. An evaluation of the impact of SG1 disproportionation and the addition of styrene in NMP of methyl methacrylate. AIChE J. 2018, 64, 2545–2559. [Google Scholar] [CrossRef]
- Hutchinson, R.A.; Penlidis, A. Free-radical polymerization: Homogeneous systems. Polym. React. Eng. 2007, 118–178. [Google Scholar] [CrossRef]
- Moad, G.; Solomon, D.H. The Chemistry of Radical Polymerization, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 233–278. [Google Scholar]
- Nuyken, O. Polymers of acrylic acid, methacrylic acid, maleic acid and their derivatives. In Handbook of Polymer Synthesis; CRC Press: Boca Raton, FL, USA, 2004; pp. 253–344. [Google Scholar]
- Thieo Hogen-Esch, A.Z. Narrow Molecular Weight Distribution Block Copolymers by Metal-Free Anionic Polymerization in the Presence of a Phosphonium Cation. WIPO Patent WO1996028487A2, 19 September 1996. [Google Scholar]
- Marien, Y.W.; Van Steenberge, P.H.; Kockler, K.B.; Barner-Kowollik, C.; Reyniers, M.-F.; Marin, G.B.; D’hooge, D.R. Estimating the photodissociation quantum yield from PLP-SEC peak heights. Polym. Chem. 2017, 8, 3124–3128. [Google Scholar] [CrossRef]
- Russell, G.T.; Napper, D.H.; Gilbert, R.G. Initiator efficiencies in high-conversion bulk polymerizations. Macromolecules 1988, 21, 2141–2148. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Davis, T.P. Handbook of Radical Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Odian, G. Principles of Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 209–235. [Google Scholar]
- Elvers, B.; Ullmann, F. Ullmann’s Polymers and Plastics: Products and Processes; Wiley-VCH Verlag GmbH & Company KGaA: Weinheim, Germany, 2016; pp. 265–315. [Google Scholar]
- Achilias, D.S. Chemical recycling of poly (methyl methacrylate) by pyrolysis. Potential use of the liquid fraction as a raw material for the reproduction of the polymer. Eur. Polym. J. 2007, 43, 2564–2575. [Google Scholar] [CrossRef]
- Fierens, S.K.; D’hooge, D.R.; Van Steenberge, P.H.; Reyniers, M.-F.; Marin, G.B. MAMA-SG1 initiated nitroxide mediated polymerization of styrene: From Arrhenius parameters to model-based design. Chem. Eng. J. 2015, 278, 407–420. [Google Scholar] [CrossRef]
- Desmet, G.B.; Marien, Y.W.; Van Steenberge, P.H.; D’hooge, D.R.; Reyniers, M.-F.; Marin, G.B. Ab initio based kinetic Monte Carlo analysis to unravel the propagation kinetics in vinyl acetate pulsed laser polymerization. Polym. Chem. 2017, 8, 7143–7150. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yamago, S. Termination mechanism in the radical polymerization of methyl methacrylate and styrene determined by the reaction of structurally well-defined polymer end radicals. Macromolecules 2015, 48, 6450–6456. [Google Scholar] [CrossRef]
- Achilias, D.S. A review of modeling of diffusion controlled polymerization reactions. Macromol. Theory Simul. 2007, 16, 319–347. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Reyniers, M.F.; Marin, G.B. The crucial role of diffusional limitations in controlled radical polymerization. Macromol. React. Eng. 2013, 7, 362–379. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Van Steenberge, P.H.; Reyniers, M.-F.; Marin, G.B. The strength of multi-scale modeling to unveil the complexity of radical polymerization. Prog. Polym. Sci. 2016, 58, 59–89. [Google Scholar] [CrossRef]
- Johnston-Hall, G.; Stenzel, M.H.; Davis, T.P.; Barner-Kowollik, C.; Monteiro, M.J. Chain length dependent termination rate coefficients of methyl methacrylate (MMA) in the gel regime: Accessing kti, i using reversible addition-fragmentation chain transfer (RAFT) polymerization. Macromolecules 2007, 40, 2730–2736. [Google Scholar] [CrossRef]
- Derboven, P.; D’hooge, D.R.; Stamenovic, M.M.; Espeel, P.; Marin, G.B.; Du Prez, F.E.; Reyniers, M.-F. Kinetic modeling of radical thiol–ene chemistry for macromolecular design: Importance of side reactions and diffusional limitations. Macromolecules 2013, 46, 1732–1742. [Google Scholar] [CrossRef]
- Barner-Kowollik, C.; Russell, G.T. Chain-length-dependent termination in radical polymerization: Subtle revolution in tackling a long-standing challenge. Prog. Polym. Sci. 2009, 34, 1211–1259. [Google Scholar] [CrossRef]
- Zhou, Y.N.; Luo, Z.H. State-of-the-art and progress in method of moments for the model-based reversible-deactivation radical polymerization. Macromol. React. Eng. 2016, 10, 516–534. [Google Scholar] [CrossRef]
- De Keer, L.; Van Steenberge, P.H.; Reyniers, M.F.; Marin, G.B.; Hungenberg, K.D.; Seda, L.; D’hooge, D.R. A complete understanding of the reaction kinetics for the industrial production process of expandable polystyrene. AIChE J. 2017, 63, 2043–2059. [Google Scholar] [CrossRef]
- Van Steenberge, P.H.; Hutchinson, R.A. Design of 2-hydroxyethyl methacrylate-functional macromonomer dispersants by semi-batch cobalt chain transfer polymerization. AIChE J. 2019, 65, e16723. [Google Scholar] [CrossRef]
- Heuts, J.P.; Russell, G.T. The nature of the chain-length dependence of the propagation rate coefficient and its effect on the kinetics of free-radical polymerization. 1. Small-molecule studies. Eur. Polym. J. 2006, 42, 3–20. [Google Scholar] [CrossRef]
- Achilias, D.; Kiparissides, C. Development of a general mathematical framework for modeling diffusion-controlled free-radical polymerization reactions. Macromolecules 1992, 25, 3739–3750. [Google Scholar] [CrossRef]
- Fierens, S.K.; Van Steenberge, P.H.; Reyniers, M.F.; Marin, G.B.; D’hooge, D.R. How penultimate monomer unit effects and initiator influence ICAR ATRP of n-butyl acrylate and methyl methacrylate. AIChE J. 2017, 63, 4971–4986. [Google Scholar] [CrossRef]
- D’hooge, D.R.; Van Steenberge, P.H.; Derboven, P.; Reyniers, M.-F.; Marin, G.B. Model-based design of the polymer microstructure: Bridging the gap between polymer chemistry and engineering. Polym. Chem. 2015, 6, 7081–7096. [Google Scholar] [CrossRef]
- Coote, M.L.; Davis, T.P.; Radom, L. Conformational dependence of the penultimate unit effect in free-radical copolymerization. Macromolecules 1999, 32, 5270–5276. [Google Scholar] [CrossRef]
- Fierens, S.; Van Steenberge, P.; Reyniers, M.-F.; D’hooge, D.; Marin, G. Analytical and advanced kinetic models for characterization of chain-growth copolymerization: The state-of-the-art. React. Chem. Eng. 2018, 3, 128–145. [Google Scholar] [CrossRef]
- Rimez, B.; Rahier, H.; Van Assche, G.; Artoos, T.; Biesemans, M.; Van Mele, B. The thermal degradation of poly (vinyl acetate) and poly (ethylene-co-vinyl acetate), Part I: Experimental study of the degradation mechanism. Polym. Degrad. Stab. 2008, 93, 800–810. [Google Scholar] [CrossRef]
- Hirata, T.; Kashiwagi, T.; Brown, J.E. Thermal and oxidative degradation of poly (methyl methacrylate): Weight loss. Macromolecules 1985, 18, 1410–1418. [Google Scholar] [CrossRef]
- MacCallum, J. The thermal degradation of poly (methyl methacrylate). Die Makromol. Chem. Macromol. Chem. Phys. 1965, 83, 137–147. [Google Scholar] [CrossRef]
- Zeng, W.; Li, S.; Chow, W. Review on chemical reactions of burning poly (methyl methacrylate) PMMA. J. Fire Sci. 2002, 20, 401–433. [Google Scholar] [CrossRef]
- Stoliarov, S.I.; Westmoreland, P.R.; Nyden, M.R.; Forney, G.P. A reactive molecular dynamics model of thermal decomposition in polymers: I. Poly (methyl methacrylate). Polymer 2003, 44, 883–894. [Google Scholar] [CrossRef]
- Madorsky, S. Rates and activation energies of thermal degradation of styrene and acrylate polymers in a vacuum. J. Polym. Sci. 1953, 11, 491–506. [Google Scholar] [CrossRef]
- Jellinek, H.; Clark, J. A New Technique for the Study of High-polymer Degradation Reactions. Can. J. Chem. 1963, 41, 355–362. [Google Scholar] [CrossRef]
- Lomakin, S.; Brown, J.E.; Breese, R.S.; Nyden, M.R. An investigation of the thermal stability and char-forming tendency of cross-linked poly (methyl methacrylate). Polym. Degrad. Stab. 1993, 41, 229–243. [Google Scholar] [CrossRef]
- Burge, S.; Tipper, C. The burning of polymers. Combust. Flame 1969, 13, 495–505. [Google Scholar] [CrossRef]
- Forman, R.; Mackinnon, H.; Ritchie, P. Studies in pyrolysis. Part XXV. Acrylic, methacrylic, and crotonic acid, and some derivatives: Novel decarbonylation of αβ-unsaturated carboxylic acids. J. Chem. Soc. C Org. 1968, 2013–2016. [Google Scholar] [CrossRef]
- McNeill, I.C. Products of thermal degradation in polymer systems based on Methyl Methacrylate. Int. J. Polym. Mater. 1994, 24, 31–45. [Google Scholar] [CrossRef]
- Özlem-Gundogdu, S.; Gurel, E.A.; Hacaloglu, J. Pyrolysis of of poly (methy methacrylate) copolymers. J. Anal. Appl. Pyrolysis 2015, 113, 529–538. [Google Scholar] [CrossRef]
- Szabo, E.; Olah, M.; Ronkay, F.; Miskolczi, N.; Blazso, M. Characterization of the liquid product recovered through pyrolysis of PMMA–ABS waste. J. Anal. Appl. Pyrolysis 2011, 92, 19–24. [Google Scholar] [CrossRef]
- McNeill, I. A study of the thermal degradation of methyl methacrylate polymers and copolymers by thermal volatilization analysis. Eur. Polym. J. 1968, 4, 21–30. [Google Scholar] [CrossRef]
- McNeill, I.; Straiton, T. Thermal degradation of copolymers of 36Cl-vinyl chloride and methyl methacrylate. Eur. Polym. J. 1979, 15, 1043–1049. [Google Scholar] [CrossRef]
- McNeill, I.; Jamieson, A.; Tosh, D.; McClune, J. The thermal degradation of copolymers of vinyl acetate with methyl methacrylate and other monomers. Eur. Polym. J. 1976, 12, 305–312. [Google Scholar] [CrossRef]
- Jamieson, A.; McNeill, I. The thermal degradation of copolymers of methyl methacrylate with methacrylic acid. Eur. Polym. J. 1974, 10, 217–225. [Google Scholar] [CrossRef]
- Kryven, I.; Iedema, P.D. Deterministic modeling of copolymer microstructure: Composition drift and sequence patterns. Macromol. React. Eng. 2015, 9, 285–306. [Google Scholar] [CrossRef]
- Wang, L.; Broadbelt, L.J. Explicit sequence of styrene/methyl methacrylate gradient copolymers synthesized by forced gradient copolymerization with nitroxide-mediated controlled radical polymerization. Macromolecules 2009, 42, 7961–7968. [Google Scholar] [CrossRef]
- Pladis, P.; Kiparissides, C. A comprehensive model for the calculation of molecular weight long-chain branching distribution in free-radical polymerizations. Chem. Eng. Sci. 1998, 53, 3315–3333. [Google Scholar] [CrossRef]
- Delgadillo-Velazquez, O.; Vivaldo-Lima, E.; Quintero-Ortega, I.A.; Zhu, S.P. Effects of diffusion-controlled reactions on atom-transfer radical polymerization. AIChE J. 2002, 48, 2597–2608. [Google Scholar] [CrossRef]
- Butte, A.; Storti, G.; Morbidelli, M. Miniemulsion living free radical polymerization of styrene. Macromolecules 2000, 33, 3485–3487. [Google Scholar] [CrossRef]
- Plessis, C.; Arzamendi, G.; Leiza, J.R.; Schoonbrood, H.A.S.; Charmot, D.; Asua, J.M. Modeling of seeded semibatch emulsion polymerization of n-BA. Ind. Eng. Chem. Res. 2001, 40, 3883–3894. [Google Scholar] [CrossRef]
- Holland, B.; Hay, J. The effect of polymerisation conditions on the kinetics and mechanisms of thermal degradation of PMMA. Polym. Degrad. Stab. 2002, 77, 435–439. [Google Scholar] [CrossRef]
- Kiparissides, C. Polymerization reactor modeling: A review of recent developments and future directions. Chem. Eng. Sci. 1996, 51, 1637–1659. [Google Scholar] [CrossRef]
- Kruse, T.M.; Wong, H.-W.; Broadbelt, L.J. Mechanistic modeling of polymer pyrolysis: Polypropylene. Macromolecules 2003, 36, 9594–9607. [Google Scholar] [CrossRef]
- De Rybel, N.; Van Steenberge, P.H.; Reyniers, M.-F.; D’hooge, D.R.; Marin, G.B. Interplay of head, tail, and mid-chain radicals in bulk free-radical and reversible degenerative addition fragmentation chain-transfer polymerizations of vinyl acetate. Macromolecules 2019, 52, 4555–4569. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.; Pich, A.; D’hooge, D.R. Coupled stochastic simulation of the chain length and particle size distribution in miniemulsion radical copolymerization of styrene and N-vinylcaprolactam. React. Chem. Eng. 2019, 4, 1935–1947. [Google Scholar] [CrossRef]
- Ali Parsa, M.; Kozhan, I.; Wulkow, M.; Hutchinson, R.A. Modeling of functional group distribution in copolymerization: A comparison of deterministic and stochastic approaches. Macromol. Theory Simul. 2014, 23, 207–217. [Google Scholar] [CrossRef]
- Busch, M. Modeling kinetics and structural properties in high-pressure fluid-phase polymerization. Macromol. Theory Simul. 2001, 10, 408–429. [Google Scholar] [CrossRef]
- Kryven, I.; Zhao, Y.R.; McAuley, K.B.; Iedema, P. A deterministic model for positional gradients in copolymers. Chem. Eng. Sci. 2018, 177, 491–500. [Google Scholar] [CrossRef]
- Van Steenberge, P.; D’hooge, D.; Reyniers, M.-F.; Marin, G. Improved kinetic Monte Carlo simulation of chemical composition-chain length distributions in polymerization processes. Chem. Eng. Sci. 2014, 110, 185–199. [Google Scholar] [CrossRef]
- Das, P.; Tiwari, P. Thermal degradation kinetics of plastics and model selection. Thermochim. Acta 2017, 654, 191–202. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Popescu, C.; Sbirrazzuoli, N. ICTAC Kinetics Committee recommendations for performing kinetic computations on thermal analysis data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Pérez-Maqueda, L.A.; Sánchez-Jiménez, P.E.; Perejón, A.; García-Garrido, C.; Criado, J.M.; Benítez-Guerrero, M. Scission kinetic model for the prediction of polymer pyrolysis curves from chain structure. Polym. Test. 2014, 37, 1–5. [Google Scholar] [CrossRef]
- Aboulkas, A.; El Bouadili, A. Thermal degradation behaviors of polyethylene and polypropylene. Part I: Pyrolysis kinetics and mechanisms. Energy Convers. Manag. 2010, 51, 1363–1369. [Google Scholar] [CrossRef]
- Friedman, H.L. Kinetics of thermal degradation of char-forming plastics from thermogravimetry. Application to a phenolic plastic. J. Polym. Sci. Part C Polym. Symp. 1964, 6, 183–195. [Google Scholar] [CrossRef]
- Tiwari, P.; Deo, M. Detailed kinetic analysis of oil shale pyrolysis TGA data. AIChE J. 2012, 58, 505–515. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. General treatment of the thermogravimetry of polymers. J. Res. Nat. Bur. Stand. 1966, 70, 487–523. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J. The isoconversional method for determination of energy of activation at constant heating rates: Corrections for the Doyle approximation. J. Therm. Anal. Calorim. 1983, 27, 95–102. [Google Scholar] [CrossRef]
- Akahira, T.; Sunose, T. Method of determining activation deterioration constant of electrical insulating materials. Res. Rep. Chiba Inst. Technol. 1971, 16, 22–31. [Google Scholar]
- Starink, M. The determination of activation energy from linear heating rate experiments: A comparison of the accuracy of isoconversion methods. Thermochim. Acta 2003, 404, 163–176. [Google Scholar] [CrossRef]
- Ferriol, M.; Gentilhomme, A.; Cochez, M.; Oget, N.; Mieloszynski, J. Thermal degradation of poly (methyl methacrylate)(PMMA): Modelling of DTG and TG curves. Polym. Degrad. Stab. 2003, 79, 271–281. [Google Scholar] [CrossRef]
- Lyon, R.E. An integral method of nonisothermal kinetic analysis. Thermochim. Acta 1997, 297, 117–124. [Google Scholar] [CrossRef]
- Korobeinichev, O.; Paletsky, А.; Gonchikzhapov, M.; Glaznev, R.; Gerasimov, I.; Naganovsky, Y.; Shundrina, I.; Snegirev, A.Y.; Vinu, R. Kinetics of thermal decomposition of PMMA at different heating rates and in a wide temperature range. Thermochim. Acta 2019, 671, 17–25. [Google Scholar] [CrossRef]
- Fateh, T.; Richard, F.; Rogaume, T.; Joseph, P. Experimental and modelling studies on the kinetics and mechanisms of thermal degradation of polymethyl methacrylate in nitrogen and air. J. Anal. Appl. Pyrolysis 2016, 120, 423–433. [Google Scholar] [CrossRef]
- Jellinek, H.; Luh, M.D. Thermal degradation of isotactic and syndiotactic poly (methyl methacrylate). J. Phys. Chem. 1966, 70, 3672–3680. [Google Scholar] [CrossRef]
- Kruse, T.M.; Woo, O.S.; Broadbelt, L.J. Detailed mechanistic modeling of polymer degradation: Application to polystyrene. Chem. Eng. Sci. 2001, 56, 971–979. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Holland, B.; Hay, J.N. The kinetics and mechanisms of the thermal degradation of poly (methyl methacrylate) studied by thermal analysis-Fourier transform infrared spectroscopy. Polymer 2001, 42, 4825–4835. [Google Scholar] [CrossRef]
- Lehrle, R.S.; Peakman, R.E.; Robb, J.C. Pyrolysis-gas-liquid-chromatography utilised for a kinetic study of the mechanisms of initiation and termination in the thermal degradation of polystyrene. Eur. Polym. J. 1982, 18, 517–529. [Google Scholar] [CrossRef]
- Da Ros, S.; Braido, R.S.; e Castro, N.L.D.S.; Brandão, A.L.; Schwaab, M.; Pinto, J.C. Modelling the chemical recycling of crosslinked poly (methyl methacrylate): Kinetics of depolymerisation. J. Anal. Appl. Pyrolysis 2019, 144, 104706. [Google Scholar] [CrossRef]
- Smolders, K.; Baeyens, J. Thermal degradation of PMMA in fluidised beds. Waste Manag. 2004, 24, 849–857. [Google Scholar] [CrossRef]
- Barlow, A.; Lehrle, R.; Robb, J.; Sunderland, D. Polymethylmethacrylate degradation—Kinetics and mechanisms in the temperature range 340 to 460 C. Polymer 1967, 8, 537–545. [Google Scholar] [CrossRef]
- Staggs, J. Modelling end-chain scission and recombination of linear polymers. Polym. Degrad. Stab. 2004, 85, 759–767. [Google Scholar] [CrossRef]
- Staggs, J. Modelling random scission of linear polymers. Polym. Degrad. Stab. 2002, 76, 37–44. [Google Scholar] [CrossRef]
- Segui, E.D.; Alarcon, B.C. Process and Device for the Regeneration of Monomers Starting from Polymethacrylate and, More Especially, Methyl Polymethacrylate. U.S. Patent US2858255A, 28 October 1958. [Google Scholar]
- Vaughan, P.W.; Highgate, D.J. Depolymerisation. U.S. Patent US5663420A, 2 September 1997. [Google Scholar]
- Miller, I.; Arthur, L.B.; Jackson, H. Art of Reclaiming Plastic Scrap. U.S. Patent US2470361A, 17 May 1945. [Google Scholar]
- Takeichi Tatsumi; Hiroshi Yoshihara; Uesaka, G. Method for Depolymerizing Thermoplastic Resins by Liquid Heat Transfer Media. U.S. Patent US3886202A, 27 May 1974.
- Mannsfeld, S.-P.; Paulsen, K.-J.; Buchholz, E. Production of Monomeric Cleavage Products by Thermal Decomposition of Polymers. U.S. Patent US3494958A, 10 February 1970. [Google Scholar]
- Egbert, S.; Mojmir, R. Depolymerization Method and Device. U.S. Patent US20060205845A1, 14 September 2006. [Google Scholar]
- Sasaki, A.; Kikuya, N.; Ookubo, T.; Hayashida, M. Recovery Method of Pyrolysis Product of Resin. U.S. Patent US8304573B2, 6 November 2012. [Google Scholar]
- Kaminsky, W.; Franck, J. Monomer recovery by pyrolysis of poly (methyl methacrylate)(PMMA). J. Anal. Appl. Pyrolysis 1991, 19, 311–318. [Google Scholar] [CrossRef]
- Kaminsky, W.; Eger, C. Pyrolysis of filled PMMA for monomer recovery. J. Anal. Appl. Pyrolysis 2001, 58, 781–787. [Google Scholar] [CrossRef]
- Kaminsky, W.; Predel, M.; Sadiki, A. Feedstock recycling of polymers by pyrolysis in a fluidised bed. Polym. Degrad. Stab. 2004, 85, 1045–1050. [Google Scholar] [CrossRef]
- Kang, B.-S.; Kim, S.G.; Kim, J.-S. Thermal degradation of poly (methyl methacrylate) polymers: Kinetics and recovery of monomers using a fluidized bed reactor. J. Anal. Appl. Pyrolysis 2008, 81, 7–13. [Google Scholar] [CrossRef]
- Dubois, J.-L. Method for recycling composite materials with an improved energy balance. WIOP Patent WO2019207259A1, 31 October 2019. [Google Scholar]
- Weiss, H.-J.; Schmalfeld, J.; Zentner, U.; Groschang, T.; Gropp, U.; Fuss, W.; Goedecke, R.; Schöla, E. Method for Depolymerizing Polymethylmethacrylate. U.S. Patent US6469203B1, 31 October 2002. [Google Scholar]
- Olazar, M.; San José, M.; Aguayo, A.; Arandes, J.; Bilbao, J. Pressure drop in conical spouted beds. Chem. Eng. J. 1993, 51, 53–60. [Google Scholar] [CrossRef]
- Koyanagi, K.; Masaki, T.; Morooka, S.; Saito, H. Continuous Regeneration Method and Apparatus the Acrylic Resin. Japan Patent JP3410343B2, 26 May 1997. [Google Scholar]
- Mueller, M.; Dr Wenzel, F. Verfahren und Vorrichtung zur Thermischen Depolymerisation von Polymeren. Germany Patent DE3146194A1, 26 May 1983. [Google Scholar]
- Breyer, K.; Enders, M.; Weyers, J. Verfahren zur Depolymerisation von Polymethylmethacrylat (PMMA) unter Einwirkung von Wärme und unter Verwendung von Zusatzstoffen. Germany Patent DE19729065A1, 14 January 1999. [Google Scholar]
- Wieme, T.; Tang, D.; Delva, L.; D’hooge, D.R.; Cardon, L. The relevance of material and processing parameters on the thermal conductivity of thermoplastic composites. Polym. Eng. Sci. 2018, 58, 466–474. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | f(α) | g(α) |
|---|---|---|
| Power law (P2) | 2 α1/2 | α ½ |
| Power law (P3) | 3 α2/3 | α 1/3 |
| Power law (P4) | 4 α 3/4 | α ¼ |
| Avarami-Erofe’ve (A2) | 2(1 − α)[−ln(1 − α)] 1/2 | [−ln(1 − α)]1/2 |
| Avarami-Erofe’ve (A3) | 3(1 − α)[−ln(1 − α)] 2/3 | [−ln(1 − α)]1/3 |
| Avarami-Erofe’ve (A4) | 4(1 − α)[−ln(1 − α)] 3/4 | [−ln(1 − α)]1/4 |
| Contracting sphere (R2) | 2(1 − α) 1/2 | [1 − (1 − α)1/2] |
| Contracting sphere (R3) | 3(1 − α) 2/3 | [1 − (1 − α)1/3] |
| One-dimensional diffusion | 1/2 α | α2 |
| two-dimensional diffusion | [−ln(1 − α)] −1 | [(1 − α)ln(1 − α)] + α |
| three-dimensional diffusion | 3(1 − α)2/3/[2(1 − (1 − α)1/3)] | [1 − (1 − α)2/3]2 |
| Ginstling-Brounshtein | 3/2((1 − α)−1/3 − 1) | 1 − (2α/3) − (1 − α)2/3 |
| First-order | 1 − α | −ln(1 − α) |
| Second-order | (1 − α)2 | (1 − α)−1 − 1 |
| Third-order | (1 − α)3 | [(1 − α)−2 − 1]/2 |
| Method | B | C | Expression | Reference |
|---|---|---|---|---|
| Ozawa Flynn Wall | 0 | 1.052 | [108,109] | |
| Kissinger Akahira Sunose | 2 | 1 | [110] | |
| Starink | 1.92 | 1.0008 | [111] |
| Step | Mm | Ei (kJ mol−1) | Log(Ai) | ni | ri |
|---|---|---|---|---|---|
| 1 | Low | 182.5 ± 3.4 | 20.817 ± 0.376 | 2.06 ± 0.46 | 0.041 ± 0.012 |
| High | 190.0 ± 1.2 | 21.887 ± 0.211 | 1.90 ± 0.31 | 0.030 ± 0.011 | |
| 2 | Low | 265.7 ± 10.8 | 27.828 ± 0.519 | 7.59 ± 1.82 | 0.039 ± 0.027 |
| High | 263.7 ± 5.2 | 27.001 ± 0.720 | 2.19 ± 0.31 | 0.019 ± 0.015 | |
| 3 | Low | 124.8 ± 3.3 | 10.560 ± 0.102 | 1.53 ± 0.21 | 0.143 ± 0.032 |
| High | 118.9 ± 1.6 | 10.657 ± 0.085 | 1.30 ± 0.19 | 0.337 ± 0.033 | |
| 4 | Low | 200.4 ± 0.3 | 16.059 ± 0.023 | 1.21 ± 0.04 | 0.777 ± 0.041 |
| High | 199.2 ± 04 | 15.976 ± 0.023 | 1.21 ± 0.06 | 0.614 ± 0.017 |
| Step | Log(Ai) | Ei | ni | ||
|---|---|---|---|---|---|
| 1 | 16.5 | 158 | 3.9 | 0.98 | |
| 2 | 10.8 | 154 | 0.85 | 0.60 | |
| 3 | 17.0 | 161 | 1 | 0.17 | |
| 4 | 14.3 | 215 | 0.83 | 0.02 |
| Model with Independent Reactions | Model with Consecutive Reactions |
|---|---|
| Mechanism | Reaction Steps | Reaction Rates | Initial Conditions |
|---|---|---|---|
| 1 | Random fission followed by complete depropagation | T = 700–730 K DP = 105–570 | |
| 2 | Random fission, depropagation and termination by disproportionation | T = 700–730 K DP = 570–2970 | |
| 3 | End-chain fission, depropagation and termination by disproportionation | T = 600–670 K |
| Mechanism | Overall Rate Coefficient | Activation Energy a |
|---|---|---|
| 1 | ||
| 2 | ||
| 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moens, E.K.C.; De Smit, K.; Marien, Y.W.; Trigilio, A.D.; Van Steenberge, P.H.M.; Van Geem, K.M.; Dubois, J.-L.; D’hooge, D.R. Progress in Reaction Mechanisms and Reactor Technologies for Thermochemical Recycling of Poly(methyl methacrylate). Polymers 2020, 12, 1667. https://doi.org/10.3390/polym12081667
Moens EKC, De Smit K, Marien YW, Trigilio AD, Van Steenberge PHM, Van Geem KM, Dubois J-L, D’hooge DR. Progress in Reaction Mechanisms and Reactor Technologies for Thermochemical Recycling of Poly(methyl methacrylate). Polymers. 2020; 12(8):1667. https://doi.org/10.3390/polym12081667
Chicago/Turabian StyleMoens, Eli K.C., Kyann De Smit, Yoshi W. Marien, Alessandro D. Trigilio, Paul H.M. Van Steenberge, Kevin M. Van Geem, Jean-Luc Dubois, and Dagmar R. D’hooge. 2020. "Progress in Reaction Mechanisms and Reactor Technologies for Thermochemical Recycling of Poly(methyl methacrylate)" Polymers 12, no. 8: 1667. https://doi.org/10.3390/polym12081667
APA StyleMoens, E. K. C., De Smit, K., Marien, Y. W., Trigilio, A. D., Van Steenberge, P. H. M., Van Geem, K. M., Dubois, J.-L., & D’hooge, D. R. (2020). Progress in Reaction Mechanisms and Reactor Technologies for Thermochemical Recycling of Poly(methyl methacrylate). Polymers, 12(8), 1667. https://doi.org/10.3390/polym12081667